

Short abstract
MyD88 signaling in monocytes/macrophages sustains disease progression in the IMQ‐induced mouse model of psoriasis.
Keywords: neutrophils, monocytes/macrophages, skin inflammation
Abstract
Psoriasis is a chronic skin disease associated with deregulated activation of immune cells and keratinocytes. In this study, we used the imiquimod (IMQ)‐induced mouse model of psoriasis to dissect better the contribution of hematopoietic and skin‐resident stromal cells to psoriasis development. The comparison of disease development in mice carrying the hematopoietic cell‐specific deletion of MyD88 (Myd88fl/flVav‐cre+ mice) with mice carrying the total MyD88 deficiency (Myd88 −/− mice), we show that the progression of skin and systemic inflammation, as well as of epidermal thickening, was completely dependent on MyD88 expression in hematopoietic cells. However, both Myd88−/− mouse strains developed some degree of epidermal thickening during the initial stages of IMQ‐induced psoriasis, even in the absence of hematopoietic cell activation and infiltration into the skin, suggesting a contribution of MyD88‐independent mechanisms in skin‐resident stromal cells. With the use of conditional knockout mouse strains lacking MyD88 in distinct lineages of myeloid cells (Myd88fl/flLysM‐cre+ and Myd88fl/flMRP8‐cre+ mice), we report that MyD88 signaling in monocytes and Mϕ, but not in neutrophils, plays an important role in disease propagation and exacerbation by modulating their ability to sustain γδ T cell effector functions via IL‐1β and IL‐23 production. Overall, these findings add new insights into the specific contribution of skin‐resident stromal vs. hematopoietic cells to disease initiation and progression in the IMQ‐induced mouse model of psoriasis and uncover a potential novel pathogenic role for monocytes/Mϕ to psoriasis development.
Abbreviations
- AMP
antimicrobial protein
- CD62L
cluster of differentiation 62 ligand
- CRAMP
cathelicidin antimicrobial peptide
- DC
dendritic cell
- IHC
immunohistochemistry
- IMQ
imiquimod
- K16
keratin‐16
- LCN2
lipocalin‐2
- LysM
lysozyme M
- MHCII
MHC class II
- MNE
mean normalized expression
- MRP8
myeloid‐related protein 8
- Myd88−/−
total knockout (deficiency) for MyD88
- Myd88fl/flVav‐cre+
hematopoietic cell‐specific deletion of MyD88
- Pam3CSK4
palmitoyl‐3‐cysteine‐serine‐lysine‐4
- qRT‐PCR
quantitative RT‐PCR
- RPL32
ribosomal protein L32
- S100A7
S100 calcium‐binding protein A7
Introduction
Psoriasis is a common, chronic autoimmune skin disease, whose multifactorial pathogenesis is thought to result from a combination of genetic, environmental, and immunologic factors [1, 2–3]. Recent progress in the field has revealed that dysregulated interaction between immune cells (particularly T cells and DCs [1, 4, 5]) and keratinocytes is crucial in driving inflammatory processes involved in the development and maintenance of the disease [1, 6, 7–8]. By contrast, although innate immune cells, including neutrophils, monocytes, and Mϕ, have also been shown to infiltrate the psoriatic plaques and to display abnormal functions in psoriatic patients, their role in disease pathogenesis has been poorly investigated [1, 7, 9].
The IMQ‐induced mouse model of psoriasis, which consists of the topical administration of Aldara cream [containing the TLR7/8 ligand IMQ (5%); Meda AB, Solna, Sweden] to the ear or shaved back of mice, is currently used broadly to elucidate pathogenic mechanisms involved in psoriasis development, as well as to evaluate possible new therapies for this disease [10, 11–12]. Repeated applications of IMQ‐containing cream rapidly induce skin inflammation in mice with remarkable pathologic and histologic resemblance to human psoriasis, including the development of skin erythema and scaling, epidermal thickening (acanthosis), altered keratinocyte differentiation, neoangiogenesis, and skin infiltration of immune cells [10]. Interestingly, the involvement of a deregulated IL‐23/IL‐17 axis and the overproduction of other inflammatory cytokines, such as IL‐1, IL‐36, and IL‐22, which are known to trigger pivotal pathogenic pathways involved in human psoriasis, appear to be mirrored in the IMQ‐induced psoriasis [10, 11, 13, 14, 15, 16, 17–18]. Among the cellular mediators, the crucial role of DCs and T cells (mostly γδ T cells) in the development of IMQ‐induced psoriasis has been elegantly demonstrated in several recent papers [13, 16, 19, 20, 21–22]. By contrast, B cell‐derived IL‐10 has been suggested to play a protective role in the same model [23, 24]. In terms of mechanisms of action, IMQ‐induced psoriasiform skin inflammation primarily involves TLR7‐ and MyD88‐dependent signaling [10], even though a contribution of MyD88‐independent mechanisms (e.g., inflammasome activation by the vehicle) to this disease model has also been reported [25]. However, the specific contribution of MyD88 signaling in skin‐resident stromal cells vs. hematopoietic cells to the development of IMQ‐induced psoriasiform skin inflammation has been poorly investigated. Among hematopoietic cells, 2 studies have investigated the role of MyD88 signaling in DCs in this model [20, 26] but not in other myeloid cell types, such as neutrophils and monocytes/Mϕ.
Herein, to investigate better the role of MyD88 signaling in skin‐resident stromal cells vs. hematopoietic cells and, in particular, myeloid cells, we took advantage of a recent mouse model in which specific ablation of the mouse Myd88 gene in certain cell types can be engineered using Cre‐lox technology [27]. Specifically, we compared the development of IMQ‐induced psoriasiform skin inflammation in Myd88 −/− mice with mice carrying the conditional (floxed) Myd88 allele, crossed with mice that express Cre in all hematopoietic cells (Vav‐cre mice) or in distinct lineages of myeloid cells (MRP8‐cre mice or LysM‐cre mice) [28]. Our results provide novel insights into the role of MyD88 signaling in hematopoietic cells, with a particular focus on innate myeloid cells, to psoriasis development.
MATERIALS AND METHODS
Mice
Myd88fl/fl (Myd88fl/fl‐cre−), Myd88+/+Vav‐cre+, Myd88fl/flVav‐cre+, Myd88+/+LysM‐cre+ Myd88fl/flLysM‐cre+, Myd88+/+MRP8‐cre+, and Myd88fl/flMRP8‐cre+ mice were described previously [27, 28, 29, 30–31]. Myd88−/− mice were a gift from Francesca Granucci (Universita Di Milano‐Bicocca, Milan, Italy). C57BL/6 mice and Ccr2 −/− were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Most of the experiments (see Results section for exceptions) were done by comparing the following: Myd88fl/fl (Myd88fl/fl‐cre−) and Myd88+/+‐cre+ with Myd88fl/flVav‐cre+, Myd88fl/flLysM‐cre+, or Myd88fl/flMRP8‐cre+ mice and C57BL/6 with Myd88 −/− or Ccr2 −/− mice. As all control mice used (e.g., Myd88fl/fl, Myd88+/+‐cre+, and C57BL/6) showed similar results, data from these groups were pooled and defined as the “control group.” All mice used in this study were back crossed at least 9 generations onto the C57BL/6 background and cohoused in a specific pathogen‐free facility. All mouse experiments were carried out in accordance with guidelines prescribed by the Ethics Committee for the use of laboratory animals for research purposes at the University of Verona and by the Italian Ministry of Health.
IMQ‐induced psoriasis model
For induction of psoriasis‐like skin inflammation, mice at 8–12 wk of age received a daily topical dose of 62.5 mg commercially available IMQ cream (5%; Aldara cream, Meda AB) or control cream (Vaseline) on their shaved backs for 6 consecutive d, as described previously [10]. On the seventh day, back skin was isolated, and ½ was fixed in 10% formaldehyde for histopathology analysis, whereas the other ½ was finely chopped and stored in RNAlater (Thermo Fisher Scientific, Waltham, MA, USA) for qRT‐PCR or digested to achieve single‐cell suspensions for flow cytometry analysis (see Supplemental information, Extended Methods).
Skin histology and IHC
Dorsal skin samples (3 mm) were obtained by a transversal cut of the central skin area, fixed in 10% neutral‐buffered formalin, embedded in paraffin blocks, cut into a 4 µm‐thick cross‐section, and stained with H&E by using a tissue stainer TST 44C (Medite, Burgdorf, Germany). Epidermal thickness for each mouse was determined by measuring the interfollicular distance in 6 random fields per 1 skin section per mouse in a blinded manner. The mean thickness was then calculated. For CD45 and K16 immunohistochemical staining, 4 µm formalin‐fixed, paraffin‐embedded tissue sections were stained after appropriate antigen retrieval with rat anti‐mouse CD45 (30‐F11; BD Biosciences, San Jose, CA, USA) or KRT16 (R20‐S; Abnova, Taipei City, Taiwan), followed by rat‐on‐mouse polymer HRP‐linked (Biocare Medical, Pacheco, CA, USA) or Dako EnVision rabbit HRP‐linked (Agilent Technologies, Santa Clara, CA, USA). Slides were developed by diaminobenzidine and then counterstained with hematoxylin. Slides were photographed using the DP73 Olympus digital camera mounted on an Olympus BX60 microscope and resized using Adobe Photoshop.
Isolation of peritoneal monocytes/Mϕ, neutrophils, and splenic DCs
Peritoneal exudates were recovered 16 h after i.p. injection of Bio‐Gel P Polyacrylamide Beads (Bio‐Rad Laboratories, Hercules, CA, USA). Single‐cell suspensions of peritoneal exudates or spleen were incubated with anti‐CD45, anti‐CD11b, anti‐Ly6G, anti‐MHCII, and anti‐CD11c mAb, as described in the flow cytometry section (see Supplemental information, Extended Methods). CD11b+Ly6G−CD11clow/− monocytes/Mϕ, CD11bhighLy6Ghigh neutrophils, and CD11c+/highMHCIIhigh DCs were sorted using a FACSAria II flow cytometer (>99.0% purity; BD Biosciences; Supplemental Fig. 1A) [unpublished results]. Purified peritoneal monocytes/Mϕ (2 × 106/ml) and neutrophils were suspended at 2 × 106/ml or 5 × 106/ml, respectively, in RPMI‐1640 medium, supplemented with 10% FBS, 1% ultraglutamine, and 1% penicillin/streptomycin (BioWhittaker, Lonza, Walkersville, MD, USA) and cultured (at 37°C, 5% CO2), with or without 25 μM IMQ (InvivoGen, San Diego, CA, USA) or 10 μg/ml Pam3CSK4 (InvivoGen), for 16 h. Supernatants were then harvested for measurement of IL‐23, IL‐1β, ΤΝF‐α, and CXCL2 by using specific ELISA kits (R&D Systems, Bio‐Techne, Minneapolis, MN, USA, or eBioscience, San Diego, CA, USA).
Proliferation and IL‐17A production by γδ T cells
γδ T Cells were isolated from single‐cell suspensions of mouse spleen and lymph nodes using the TCRγ/δ+ T Cell Isolation Kit (>85.5% purity; Miltenyi Biotec, San Diego, CA, USA; Supplemental Fig. 1B). In selected experiments, γδ and αβ T cells were sorted (>99.0% purity) using a FACSAria II flow cytometer (BD Biosciences). Proliferation assays were performed in 96‐well plates, precoated (for 1 h at 37°C) with 1 µg/ml anti‐CD3 mAb (G23‐8; eBioscience). γδ T cells/well (1 × 105) or αβ T cells/well and 2 µg/ml anti‐CD28 mAb (B122; eBioscience) were then added to the plates (at 37°C, 5% CO2) in the presence or absence of 10 ng/ml IL‐1β (eBioscience) plus 100 ng/ml IL‐23 (eBioscience) or supernatants from IMQ‐stimulated monocytes/Mϕ (added as 1–4 dilutions) in the presence or absence of 0.5 μg/ml neutralizing anti‐IL‐1β mAb (eBioscience) and/or 1 μg/ml neutralizing anti‐IL‐23 p19 mAb (eBioscience; or their relevant isotype control mAb). Following a 92 h incubation, BrdU was added to the cocultures for an additional 4 h; supernatants were then harvested for measurement of IL‐17A by using a specific ELISA kit (R&D Systems, Bio‐Techne), and proliferation was determined by BrdU incorporation by ELISA (Cell Proliferation ELISA, BrdU; Roche), following the manufacturer's protocol. T Cell proliferation is expressed as fold increase of the absorbance value (at 450 nm; revealing the amount of BrdU incorporated in proliferating T cells) of each experimental condition over the absorbance value of anti‐CD3/CD28‐stimulated T cells.
Statistical analysis
Data were expressed as the means ± sd and analyzed using GraphPad Prism Version 5 software (GraphPad Software, La Jolla, CA, USA). The comparison of variables was performed using 2‐tailed Student t‐ test (for comparison between 2 groups) or a 1‐way ANOVA with Bonferroni's post‐test (used for multiple comparisons) or Dunnett's post‐test (when multiple comparisons with the control group were made). P < 0.05 was considered significant, and symbols indicate significant increases (see figure legends).
RESULTS
MyD88 expression in hematopoietic cells is crucial for the progression but not the initiation of skin inflammation and epidermal thickening in IMQ‐induced psoriasis
To investigate the specific contribution of MyD88‐dependent signaling pathways occurring in hematopoietic cells to the development of psoriasis‐like skin lesions in response to topical IMQ treatment (Aldara), we compared disease development in Myd88fl/flVav‐cre+ mice vs. Myd88 −/− mice and control mice. As control for Aldara, we used Vaseline treatment [10]. As there was no difference between Vaseline‐treated control and Myd88‐deficient mouse strains, we pooled together these 2 groups of data in a single reference group throughout the manuscript. By contrast, we found that all mouse strains developed an increase of epidermal thickening compared with Vaseline‐treated mice, within 3 d of IMQ treatment ( Fig. 1A ). Interestingly, we observed only a mild reduction of epidermal thickening in both Myd88fl/flVav‐cre+ and Myd88 −/− mice compared with IMQ‐treated control strains at these initial stages of disease development (Fig. 1A). However, both Myd88fl/flVav‐cre+ and Myd88 −/− mice similarly failed to develop the progressive increase in epidermal thickening observed in control mice upon consecutive IMQ treatments (up to 6 d; Fig. 1A and B). Accordingly, the expression of K16 (a marker often used, proving a keratinocyte hyperproliferation in psoriasis) was reduced in both Myd88fl/flVav‐cre+ and Myd88 −/− mice after 6 d of IMQ treatment, as revealed by IHC staining (Fig. 1C). Furthermore, in the skin of IMQ‐treated, but not Vaseline‐treated, control mice, there was a strong infiltration of CD45+ cells, as revealed by both flow cytometry (Fig. 1D and Supplemental Fig. 2A) and IHC (Fig. 1E). A more detailed characterization of the CD45+ infiltrate revealed that compared with Vaseline‐treated mice, in the skin of control mice, there were increased CD11b+ cells, which included neutrophils (CD11bhighLy6Ghigh cells), monocytes (CD11bhighLy6G−CD11clow/−MHCIIlow/− cells, among which ∼45% were Ly6Chigh inflammatory monocytes), and Mϕ (CD11bhighLy6G−CD11clow/−MHCIIhigh cells; Fig. 2 and Supplemental Fig. 2B and C). Such an infiltrate was evident as early as after 3 d and progressively increased during the 6 d of IMQ treatment (Fig. 2). We also observed a strong increase of dermal γδ TCRlow T cells, which are the main pathologic γδ T cell subset implicated in this model [19, 32] but only after 6 d of IMQ treatment (Fig. 2 and Supplemental Fig. 2D). αβ TCR+ T Cells and DCs (CD11c+/highMHCIIhigh, which included all of the main pathologic DC subsets described in this model, such as Langerin+, conventional, and infiltrating monocyte‐derived DCs [20, 21–22, 32]) also infiltrated the skin of IMQ‐treated control mice, although to a lower extent (Fig. 2 and Supplemental Fig. 2E and F). Importantly, none of these CD45+ inflammatory cell populations significantly infiltrated the skin of IMQ‐treated Myd88fl/flVav‐cre+ or Myd88 −/− mice, either at 3 or 6 d (Figs. 1D and E and 2).
Figure 1.

Myd88fl/flVav‐cre+ and Myd88 −/− mice manifest a partial protection from the development of epidermal thickening in response to IMQ treatment, despite the strong reduction of skin infiltration by CD45+ cells.
Dorsal skin of control (which included regular B6, Myd88fl/fl, and Myd88+/+Vav‐cre+ mouse strains), Myd88fl/flVav‐cre+, and Myd88 −/− mice were topically treated with Vaseline or IMQ‐containing cream (Aldara) for 3 or 6 consecutive d. (A) The height of epidermal hyperplasia (epidermal thickening, indicated by an arrow in (B)) was measured in interfollicular epidermis on H&E‐stained slides by light microscopic evaluation. Data are pooled from 3 separate time course experiments and are expressed as means ± sd (n = 6–14 mice per time point). (B) Representative H&E staining of dorsal skin of control, Myd88fl/flVav‐cre+, or Myd88 −/− mice treated with Vaseline or IMQ for 6 d. Original magnification, ×100; original scale bar, 200 μm. (C and E) Immunohistochemical detection of K16 (C) and CD45 (E) expression in dorsal skin from control, Myd88fl/flVav‐cre+ or Myd88 −/− mice treated with Vaseline or IMQ for 6 d. Original magnification, ×100; original scale bar, 200 μm. (D) Total skin was digested and analyzed by flow cytometry. The total number of CD45+ cells in 2 × 2 cm of dorsal skin is reported. Data are pooled from 3 separate time course experiments and are expressed as means ± sd (n = 6–14 mice per time point). Statistical differences of IMQ‐treated control or Myd88−/− mouse strains vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flVav‐cre+ or Myd88 −/− mice (*) are reported. #/*P ≤ 0.05; ##/**P ≤ 0.01; ###/***P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
Figure 2.

Skin infiltration of inflammatory cells in IMQ‐treated mice depends on MyD88 expression in hematopoietic cells.
The dorsal skin of control, Myd88fl/flVav‐cre+, and Myd88 −/− mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 3 or 6 consecutive d. Total skin (2 × 2 cm) was digested and analyzed by flow cytometry. The total number of CD11b+ cells, neutrophils (CD11bhighLy6Ghigh cells), monocytes/Mϕ (CD11bhighLy6G−CD11clow/−MHCIIlow/− cells plus CD11bhighLy6G−CD11clow/−MHCIIhigh cells), dermal γδ TCRlow T cells, αβ TCR+ T cells, and DCs (CD11c+/highMHCIIhigh) is reported. Data are pooled from 3 separate time course experiments and are expressed as means ± sd (n = 6–14 mice per time‐point). Statistical differences of IMQ‐treated control or MyD88‐deficient mouse strains vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flVav‐cre+ or Myd88 −/− mice (*) are reported. *P ≤ 0.05; ##/**P ≤ 0.01; ###/***P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
We next performed a gene‐expression analysis, using qRT‐PCR on inflammatory skin samples, for a number of inflammatory cytokines, chemokines, AMPs (or alarmins), and other inflammatory molecules whose expression has been described to be up‐regulated in the skin of IMQ‐treated mice. Consistent with the initial increase of epidermal thickening, the expression of K16 and IL‐1α and IL‐36α and cathelicidin (CRAMP) was not significantly reduced in Myd88fl/flVav‐cre+ or Myd88 −/− mice compared with IMQ‐treated control mice after 3 d of IMQ treatment ( Fig. 3 ). However, the expression of these molecules was reduced significantly in Myd88fl/flVav‐cre+ and Myd88 −/− mice after 6 d of IMQ treatment (Fig. 3). Furthermore, the expression of several other skin‐associated psoriatic genes, such as S100A7/psoriasin and Lcn2 [33], as well as of IL‐17A, IL‐22, IL‐6, IL‐23, IL‐1β, and CXCL1, was strongly reduced in IMQ‐treated Myd88fl/flVav‐cre+ and Myd88 −/− mice compared with IMQ‐treated control mice after both 3 and 6 d of IMQ treatment (Fig. 3). Overall, these data suggest that MyD88‐dependent signaling in hematopoietic cells, but not in stromal cells, is crucial for sustaining the progressive skin inflammation typically observed in IMQ‐induced psoriasis, including the recruitment of inflammatory immune cells, the expression of inflammatory molecules, and the full development of epidermal thickening. Data also suggest that the activation of MyD88‐independent pathways, likely in skin‐resident stromal cells, plays instead a major role in inducing the initial development of epidermal thickening observed up to 3 d of IMQ treatment.
Figure 3.

Expression of inflammatory molecule mRNAs in the skin of control, Myd88fl/flVav‐cre+, or Myd88 −/− mice in response to IMQ treatment.
The dorsal skin of control, Myd88fl/flVav‐cre+, and Myd88 −/− mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 3 or 6 consecutive d. Total skin RNA was extracted and reverse transcribed. mRNA expression of the indicated genes for IMQ‐treated control, Myd88fl/flVav‐cre+, or Myd88 −/− mice is displayed as fold changes of MNE units (after RPL32 normalization) over Vaseline‐treated control. Data are pooled from 2 separate time course experiments and are expressed as means ± sd (n = 6–10 mice per time point). Statistical differences of IMQ‐treated control vs. IMQ‐treated Myd88fl/flVav‐cre+ and Myd88 −/− mice are reported. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
MyD88 expression in hematopoietic cells is essential for immune cell expansion and activation in the draining lymph nodes of IMQ‐treated mice
We next characterized the specific contribution of MyD88‐dependent signaling pathways in hematopoietic cells to the development of systemic immune cell activation in IMQ‐induced psoriasis. Flow cytometry analysis of the draining lymph nodes, performed in control mice after 6 d of IMQ treatment, revealed an expansion and activation of αβ and γδ TCR+ T cells (activation being revealed as differentiation into CD44highCD62Llow effector cells; Supplemental Fig. 3A and B), as well as of DCs (whose activation status was assessed by measuring the up‐regulation of MHCII expression; Supplemental Fig. 3C), which were, however, completely absent in both Myd88fl/flVav‐cre+ and Myd88 −/− mice ( Fig. 4A–C ). Interestingly, there was also a strong expansion and activation of CD11b+ myeloid cell populations in the draining lymph nodes of control mice, including neutrophils and monocytes (whose activation status was assessed by verifying the number of CD62Llow/− cells; Supplemental Fig. 3D and E), as well as Mϕ (whose activation status was assessed by measuring their up‐regulation of CD86 expression; Supplemental Fig. 3F and Fig. 4D–F). Similarly to what was observed in the skin, ∼35% of the expanded monocytes were Ly6Chigh inflammatory monocytes (Supplemental Fig. 3E). Importantly, the expansion and activation of these CD11b+ myeloid cell populations were absent in both Myd88fl/flVav‐cre+ and Myd88 −/− mice after both 3 and 6 d after IMQ treatment (Figure 4D–F).
Figure 4.

Analysis of inflammatory cell infiltration in the draining lymph nodes of IMQ‐treated control, Myd88fl/flVav‐cre+, or Myd88 −/− mice.
The dorsal skin of control, Myd88fl/flVav‐cre+, and Myd88 −/− mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 6 consecutive d. Draining lymph nodes were collected and analyzed by flow cytometry. Panels report the number of total and effector (CD44highCD62Llow) αβ TCR+ T cells (A), the number of total and effector (CD44highCD62Llow) γδ TCR+ T cells (B), the total number and activation [revealed as increased in MHCII mean fluorescence intensity (MFI) values] of CD11c+/highMHCIIhigh DCs (C), the number of total and activated (CD62Llow) CD11bhighLy6Ghigh neutrophils (D), the number of total and activated (CD62Llow) CD11bhighLy6G−CD11clow/−MHCIIlow/− monocytes (E), and the total number and activation (revealed as increased in CD86 MFI values) of CD11bhighLy6G−CD11clow/−MHCIIhigh Mϕ (F). Data are pooled from 3 separate experiments and are expressed as means ± sd (n = 8–14 mice). Statistical differences of IMQ‐treated control or Myd88−/− mouse strains vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flVav‐cre+ or Myd88 −/− mice (*) are reported. #/*P ≤ 0.05; ##/**P ≤ 0.01; ###/***P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
Overall, data demonstrate that the development of systemic immune cell expansion and activation in the IMQ‐induced mouse model of psoriasis are completely dependent on MyD88 signaling in hematopoietic cells.
MyD88 signaling in monocytes/Mϕ contributes to the progression of skin and systemic inflammation in IMQ‐induced psoriasis
To determine the role of MyD88‐dependent signaling in individual myeloid cell lineages, we investigated IMQ‐induced psoriasis in both Myd88fl/flMRP8‐cre+ mice (carrying the specific deletion of MyD88 in neutrophils [28, 30, 31]) and Myd88fl/flLysM‐cre+ mice (carrying the specific deletion of MyD88 in neutrophils, monocytes, and Mϕ [28, 29, 31, 34]), as these are the cell types that massively expand and infiltrate the inflamed skin in this model (Figs. 2 and 4D–F). The specificity and efficiency of MRP8‐cre‐mediated deletion of the Myd88fl/fl allele in neutrophils compared with other myeloid cell populations were proved by various experiments. We [30, 31] analyzed the expression of the internal ribosome entry site‐GFP constructs contained within the MRP8‐cre by flow cytometry and ascertained that whereas 90% of CD11bhighLy6Ghigh neutrophils expressed GFP, <20% of monocytes/Mϕ or DCs were GFP+ (Supplemental Fig. 4A). Likewise, intracellular detection of MyD88 by flow cytometry proved the specificity of the deletion of the MyD88fl/fl allele in neutrophils by Cre recombination under the control of the MRP8 promoter (Supplemental Fig. 4B). In addition, measurement of MyD88 mRNA levels in purified cells confirmed that ∼80% of the Myd88fl/fl allele was deleted in neutrophils, whereas no significant deletion was evident in monocytes/Mϕ or DCs (Supplemental Fig. 4C). Finally, we found that CXCL2 and/or TNF‐α production by Pam3CSK4‐stimulated neutrophils (Supplemental Fig. 4D) but not monocytes/Mϕ (Supplemental Fig. 4D and E) or DCs (Supplemental Fig. 4E) was impaired. In these experiments, Pam3CSK4 was chosen as a common agonist for all myeloid cell types, as in line with what was reported previously [35, 36–37], we did not observe any production of TNF‐α and/or CXCL2 by IMQ‐stimulated neutrophils [unpublished results]. Consistent with the latter observations, initial experiments revealed that no significant differences were present in Myd88fl/flMRP8‐cre+ mice compared with control mice at the skin or systemic level after neither 3 nor 6 d of IMQ treatment ( Fig. 5A and B ) [unpublished results]. Therefore, we next investigated disease development in IMQ‐treated Myd88fl/flLysM‐cre+ mice, reasoning that as Myd88fl/flMRP8‐cre+ mice did not show any phenotype, eventual differences observed between Myd88fl/flLysM‐cre+ and control mice in response to IMQ treatment could be attributed to the specific contribution of MyD88‐dependent signaling in monocyte/Mϕ. Intracellular detection of MyD88 by flow cytometry proved the specificity of the deletion of the MyD88fl/fl allele in neutrophils and monocytes/Mϕ by Cre recombination under the control of the LysM promoter (Supplemental Fig. 5A). The efficiency of LysM‐cre‐mediated deletion of the Myd88fl/fl allele in neutrophils and monocytes/Mϕ compared with DCs was calculated by measuring MyD88 mRNA levels in purified cells (Supplemental Fig. 5B). We found that whereas >65% of the Myd88fl/fl allele was deleted in both neutrophils and monocytes/Mϕ, no significant deletion was observed in DCs (Supplemental Fig. 5B). We also ascertained that CXCL2 and/or TNF‐α production was impaired in Pam3CSK4‐stimulated neutrophils and monocytes/Mϕ (Supplemental Fig. 5C and D) but normal in Pam3CSK4‐stimulated DCs (Supplemental Fig. 5D). Consistent with the data obtained with Myd88fl/flVav‐cre+ and Myd88 −/− mice (Fig. 1A), a mild but still significant reduction in epidermal thickening (Fig. 5C and D) and K16 expression (Fig. 5E) was observed only after 6 d of IMQ treatment in Myd88fl/flLysM‐cre+ mice compared with control mice, accompanied, however, by a strong reduction of CD45+ cell infiltration in the skin that was more evident after 6 rather than 3 d of IMQ treatment (Fig. 5F and G). A detailed flow cytometry analysis of total skin cells revealed that among the CD45+ cell types infiltrating the skin of Myd88fl/flLysM‐cre+ mice, there was a selective reduction of only monocytes/Mϕ (already evident after 3 d of IMQ treatment) and dermal γδ TCRlow T cells ( Fig. 6 ) but not of neutrophils, αβ T cells, and DCs at any of the time point tested (Fig. 6). qRT‐PCR analysis of the skin revealed that in line with the data on the epidermal thickening (Fig. 5C), the mRNA expression of several AMPs, psoriatic genes, and cytokines was reduced significantly only after 6 d of IMQ treatment ( Fig. 7 ). However, the expression of IL‐1α and IL‐36α, whose production is mostly sustained by skin‐resident stromal cells and DCs [16, 25], was not reduced significantly in Myd88fl/flLysM‐cre+ mice after 3 or 6 d of IMQ treatment (Fig. 7). Notably, a similar specific reduction in the expansion and activation of γδ T cells and monocytes/Mϕ was also observed in the draining lymph nodes of Myd88fl/flLysM‐cre+ mice after 6 d of IMQ treatment ( Fig. 8 ). In the latter tissues of Myd88fl/flLysM‐cre+ mice, we also observed a significantly reduced expression ( Fig. 9A ) and/or production (Fig. 9B and C) of IL‐17A and IL‐22 by γδ T cells compared with that occurring in control mice after 6 d of IMQ treatment.
Figure 5.

Myd88fl/flLysM‐cre+ but not Myd88fl/flMRP8‐cre+ mice manifest a reduced development of epidermal thickening and CD45+ cell infiltration into the skin after 6 d of IMQ treatment.
The dorsal skin of control (which included Myd88fl/fl, Myd88 +/+ MRP8‐cre+, and Myd88 +/+ LysM‐cre+ mouse strains), Myd88fl/flMRP8‐cre+ mice, and Myd88fl/flLysM‐cre+ mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 3 or 6 consecutive d. (A and C) The height of epidermal hyperplasia (epidermal thickening, indicated by an arrow, in D) was measured in interfollicular epidermis on H&E‐stained slides by light microscopic evaluation. (B and F) Total skin was digested and analyzed by flow cytometry. The total number of CD45+ cells in 2 × 2 cm of dorsal skin is reported. Data are pooled from 2 separate experiments performed after 6 d of IMQ treatment and expressed as means ± sd (n = 6–10 mice) for Myd88fl/flMRP8‐cre+ mice (A and B) and 3 separate time course experiments and expressed as means ± sd (n = 9–17 mice per time point) for Myd88fl/flLysM‐cre+ mice (C and F). (D) Representative H&E staining of dorsal skin of control or Myd88fl/flLysM‐cre+ mice treated with Vaseline or IMQ for 6 d. Original magnification, ×100; original scale bar, 200 μm. (E and G) Immunohistochemical detection of K16 (E) and CD45 (G) expression in dorsal skin from control or Myd88fl/flLysM‐cre+ mice treated with Vaseline or IMQ for 6 d. Original magnification, ×100; original scale bars, 200 μm. Statistical differences of IMQ‐treated control or Myd88fl/flLysM‐cre + mice vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flLysM‐cre+ mice (*) are reported. #/*P ≤ 0.05; ##/**P ≤ 0.01; ###P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
Figure 6.

The infiltration of monocytes/Mϕ and γδ TCRlow dermal T cells is reduced in the skin of IMQ‐treated mice Myd88fl/flLysM‐cre+ mice.
The dorsal skin of control and Myd88fl/flLysM‐cre+ mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 3 or 6 consecutive d. Total skin (2 × 2 cm) was digested and analyzed by flow cytometry. The total number of CD11b+ cells, neutrophils (CD11bhighLy6Ghigh cells), monocytes/Mϕ (CD11bhighLy6G−CD11clow/−MHCIIlow/− cells plus CD11bhighLy6G−CD11clow/−MHCIIhigh cells), dermal γδ TCRlow T cells, αβ TCR+ T cells, and DCs (CD11c+/highMHCIIhigh) is reported. Data are pooled from 3 separate time course experiments and are expressed as means ± sd (n = 6–12 mice per time point). Statistical differences of IMQ‐treated control or Myd88fl/flLysM‐cre+ mice vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flLysM‐cre+ mice (*) are reported. #/*P ≤ 0.05; ##/**P ≤ 0.01; ###P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
Figure 7.
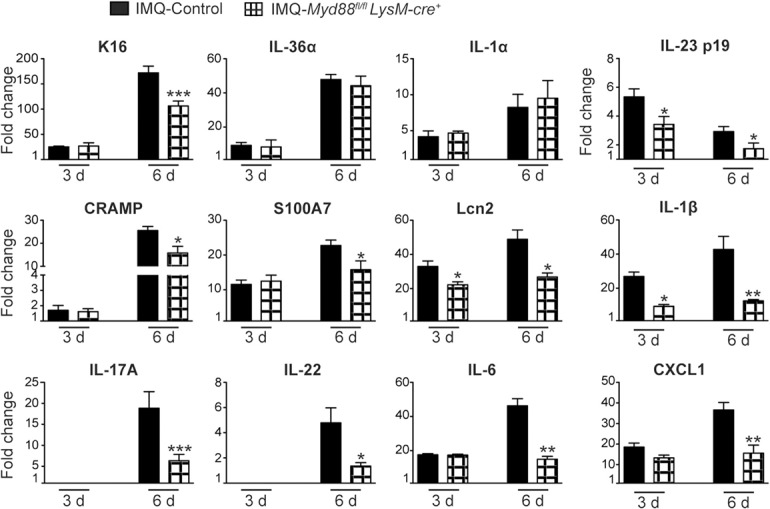
Gene‐expression analysis of inflammatory molecules in the skin of control or Myd88fl/flLysM‐cre+ mice in response to IMQ treatment.
The dorsal skin of control and Myd88fl/flLysM‐cre+ mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 3 or 6 consecutive d. Total skin RNA was extracted and reverse transcribed. mRNA expression of the indicated genes for IMQ‐treated control or Myd88fl/flLysM‐cre+ mice is displayed as fold changes of MNE units (after RPL32 normalization) over Vaseline‐treated control. Data are pooled from 2 separate time course experiments and are expressed as means ± sd (n = 6–10 mice per time point). Statistical differences of IMQ‐treated control vs. Myd88fl/flLysM‐cre+ mice are reported. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 by t test.
Figure 8.

Infiltration of inflammatory cells in the draining lymph nodes of IMQ‐treated control or Myd88fl/flLysM‐cre+ mice.
The dorsal skin of control and Myd88fl/flLysM‐cre+ mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 6 consecutive d. Draining lymph nodes were collected and analyzed by flow cytometry. Panels report the number of total and effector (CD44highCD62Llow) αβ TCR+ T cells (A), the number of total and effector (CD44highCD62Llow) γδ TCR+ T cells (B), the total number and activation (revealed as increased in MHCII MFI values) of CD11c+/highMHCIIhigh DCs (C), the number of total and activated (CD62Llow) CD11bhighLy6Ghigh neutrophils (D), the number of total and activated (CD62Llow) CD11bhighLy6G−CD11clow/−MHCIIlow/− monocytes (E), and the total number and activation (revealed as increased in CD86 MFI values) of CD11bhighLy6G−CD11clow/−MHCIIhigh Mϕ (F). Data are pooled from 3 separate experiments and are expressed as means ± sd (n = 6–12 mice). Statistical differences of IMQ‐treated control or Myd88fl/flLysM‐cre+ mice vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flLysM‐cre+ mice (*) are reported. #/*P ≤ 0.05; ##/**P ≤ 0.01; ###/***P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test.
Figure 9.

IL‐17A and IL‐22 mRNA expression and production are reduced in the draining lymph nodes of IMQ‐treated control or Myd88fl/flLysM‐cre+ mice.
The dorsal skin of control and Myd88fl/flLysM‐cre+ mice was topically treated with Vaseline or IMQ‐containing cream (Aldara) for 6 consecutive d. (A) Total RNA was extracted from draining lymph nodes and reverse transcribed. mRNA expression of IL‐17A and IL‐22 for IMQ‐treated control or Myd88fl/flLysM‐cre+ mice is displayed as fold changes of MNE units (after RPL32 normalization) over Vaseline‐treated control. Data are pooled from 2 separate experiments and are expressed as means ± sd (n = 6–10 mice). Statistical differences of IMQ‐treated control vs. IMQ‐treated Myd88fl/flLysM‐cre+ mice (*) are reported. (B and C) Draining lymph nodes were collected and analyzed by flow cytometry. (B) Representative FACS plots showing the frequencies of IL‐17A‐producing γδ TCR+ T cells in Vaseline or IMQ‐treated control or Myd88fl/flLysM‐cre+ mice. (C) Frequencies of IL‐17A‐producing γδ TCR+ T cells in Vaseline or IMQ‐treated control or Myd88fl/flLysM‐cre+ mice. Data are pooled from 2 separate experiments and are expressed as means ± sd (n = 4–6 mice). Statistical differences of IMQ‐treated control or Myd88fl/flLysM‐cre+ mice vs. Vaseline‐treated mice (#) and IMQ‐treated control vs. IMQ‐treated Myd88fl/flLysM‐cre+ mice (*) are reported. *P ≤ 0.05; **P ≤ 0.01 by t test (A) or *P ≤ 0.05; ###P ≤ 0.001 by 1‐way ANOVA with Bonferroni's post‐test (C).
Finally, as controversial data exist on the role of Ly6Chigh inflammatory monocytes in the IMQ‐induced model of psoriasis [22, 38], we decided to test disease development in Ccr2 −/− mice under our experimental conditions. As shown in Supplemental Fig. 5E and F, we found no differences between IMQ‐treated Ccr2 −/− mice and control mice, therefore, excluding a specific role of Ly6Chigh inflammatory monocytes in this psoriasis model.
Altogether, data suggest that MyD88 signaling pathways, occurring in monocytes/Mϕ but not neutrophils, contribute to the sustainment and exacerbation of skin and systemic inflammation occurring in the IMQ‐induced mouse model of psoriasis.
IMQ‐activated monocytes/Mϕ sustain the proliferation of and IL‐17A production by γδ T cells via MyD88‐dependent production of IL‐1β and IL‐23
Based on previous findings [13], highlighting their crucial role of IL‐23 and IL‐1β in sustaining the proliferation and IL‐17A production by γδ T cells under inflammatory conditions, we tested whether an altered production of these cytokines by Myd88−/− monocytes/Mϕ might be responsible for the impaired responses by γδ T cells observed in Myd88fl/flLysM‐cre+ mice. For such a purpose, we isolated peritoneal monocytes/Mϕ and neutrophils from control and Myd88fl/flVav‐cre+ or Myd88 −/− mice (to use cells carrying a 100% deletion of MyD88 [27]; Supplemental Fig. 1A) and cultured these cells for 16 h in the absence or presence of 25 μM IMQ. As expected, we found that whereas very low amounts of IL‐1β and IL‐23 were present in the culture supernatants of resting cells, they increased significantly upon monocytes/Mϕ stimulation with IMQ in a MyD88‐dependent manner ( Fig. 10A ). No IL‐1β and IL‐23 production was instead observed in IMQ‐stimulated peritoneal neutrophils (Fig. 10A). γδ T Cells isolated from the spleen and lymph nodes of control mice (Supplemental Fig. 1B) confirmed, as demonstrated previously, their ability to proliferate and produce IL‐17A in response to anti‐CD3/CD28 antibodies in a manner strongly enhanced by the presence of 100 ng/ml IL‐23 and 10 ng/ml IL‐1β (Fig. 10B and C, left). This effect was abrogated completely in the presence of neutralizing, but not isotype control, mAb toward these cytokines (Fig. 10B and C, left) [unpublished results]. Neither increased proliferation nor IL‐17 production was observed in CD3/CD28‐stimulated αβ T cells treated with IL‐23 and IL‐1β (Fig. 10B and C, left). Thus, we cultured CD3/CD28‐incubated γδ T cells with supernatants from IMQ‐treated control or Myd88−/− monocytes/Mϕ in the presence or absence of neutralizing antibodies toward IL‐23 and/or IL‐1β (or relative isotype control antibodies; Fig. 10B and C, right) [unpublished results]. Notably, we found that supernatants from IMQ‐treated control, but not Myd88−/− monocytes/Mϕ, enhanced the proliferation and IL‐17A production by CD3/CD28‐stimulated γδ T cells (Fig. 10B and C, right). The latter function was partially reversed by the addition of anti‐IL‐23 or anti‐IL‐1β antibodies added alone (with anti‐IL‐1β antibodies more effective) but was completely reverted only when the 2 neutralizing antibodies were added together (Fig. 10B and C, right). No effects of isotype control antibodies were observed [unpublished observations]. Overall, these findings reveal a possible important role of monocytes/Mϕ‐derived IL‐β and IL‐23 in sustaining the proliferation and IL‐17A production by γδ T cells.
Figure 10.

IL‐1β and IL‐23 are produced in a MyD88‐dependent manner by IMQ‐activated monocytes/Mϕ and induce proliferation of and IL‐17A production by γδ T cells.
(A) Peritoneal monocytes/Mϕ or neutrophils from control or Myd88fl/flVav‐cre+ or Myd88 −/− mice were isolated and cultured for 16 h in the absence or presence of 25 μM IMQ. The amount of IL‐23 and IL‐1β in the culture medium was measured by specific ELISA. Means ± sd of data from 3 independent experiments are reported. Statistical differences of untreated control vs. IMQ‐treated control monocytes/Mϕ are reported. (B and C) CD3/CD28‐stimulated αβ or γδ T cells (left) and CD3/CD28‐stimulated γδ T cells (right) were cultured for 96 h in the absence or presence of 100 ng/ml IL‐23 plus 10 ng/ml IL‐1β (left) or of supernatant (Supnt) from IMQ‐stimulated control or Myd88−/− monocytes/Mϕ, with or without anti (α)‐IL‐23 (1 μg/ml) and anti‐IL‐1β (0.5 μg/ml) mAb used in single or in combination (right). T Cell proliferation was measured by BrdU incorporation (and expressed as fold increase over CD3/CD28‐stimulated T cells; see Supplemental Methods), whereas T cell‐derived IL‐17A was measured in culture supernatants by ELISA. rIL, Recombinant IL. Means ± sd of data from 3 independent experiments are reported. Statistical differences of the effects of stimulation of γδ T cells by IL‐23 plus IL‐1β or supernatants from IMQ‐treated control monocytes/Mϕ in the presence or absence of anti‐IL‐1β and/or anti IL‐23 mAb are reported. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 by 1‐way ANOVA with Dunnett's post‐test.
DISCUSSION
It is widely recognized that complex interactions between keratinocytes and immune cells contribute to psoriasis pathogenesis [1, 2–3, 39], as also emerging from the IMQ‐induced mouse model of acute skin inflammation [11, 12]. Despite the number of publications that have focused on the pathologic mechanisms at the basis of the IMQ‐induced skin inflammation, confused data still exist on the specific contribution of MyD88‐dependent vs. MyD88‐independent pathways activated during the disease. In this study, we found that mice carrying either Myd88 −/− or Myd88fl/flVav‐cre+ both develop some degree of epidermal thickening during the initial stages of IMQ‐induced psoriasis, even in the complete absence of immune cell activation and skin infiltration. Therefore, these observations would suggest a contribution of MyD88‐independent mechanisms to the initial keratinocyte hyperproliferation, likely occurring in skin‐resident stromal cells. However, MyD88 signaling in hematopoietic is required to sustain immune cell activation and the full development of skin and systemic inflammation responsible for the progressive epidermal thickening observed in response to consecutive topical IMQ application. In particular, we found that MyD88 signaling occurring in monocytes/Mϕ but not in neutrophils may contribute to the progressive development of skin inflammation, which involves inflammatory cell infiltration and cytokine/psoriatic gene overexpression more than epidermal thickening. Such an effect seems to be mediated by the ability of IMQ‐stimulated monocytes/Mϕ to sustain the functions of γδ T cells (key effector components of psoriatic inflammation) via MyD88‐dependent IL‐23 and IL‐1β production. Few studies have investigated the role of TRL7‐ and MyD88‐dependent signaling in IMQ‐induced psoriasis [15, 25, 40]. Surprisingly, whereas skin and systemic inflammation were reported to be abolished completely in TLR7 −/− and Myd88 −/− mice, development of epidermal thickening was shown to be absent completely only in Myd88−/− mice [15, 25, 40]. Our results differ from those by Walter et al. [25] and Rabeony et al. [15], as we found that mice carrying either Myd88 −/− or Myd88fl/flVav‐cre+ both develop some degree of epidermal thickening. We also found that differently from other inflammatory mediators and cytokines, IL‐1α and IL‐36α—cytokines that have been shown to be crucial in driving epidermal thickening in this model [17]—could still be normally produced in both Myd88−/− mice within the first 3 d of IMQ treatment. Accordingly, some production of IL‐36α and other inflammatory cytokines in response to IMQ treatment in Myd88 −/− mice was reported also in the studies by Rabeony et al. [15] and Walter et al. [25]. In our hands, the expression of these cytokines perfectly correlated with the enhanced expression of K16—a marker of keratinocyte proliferation—observed in both Myd88−/− strains up to the first 3 d of IMQ treatment. Therefore, we would envision that the initial epidermal thickening, observed in response to IMQ treatment in both Myd88fl/flVav‐cre+ and Myd88 −/− mice, is likely caused by the MyD88‐independent IL‐1α and IL‐36α production by skin‐resident stromal cells, occurring even in the absence of immune cell activation. It remains to be clarified, however, which are the mechanisms responsible for this rapid MyD88‐independent IL‐1α and IL‐36α production by skin‐resident stromal cells. A role for TRL7‐independent IMQ induction of both keratinocyte activation and cytokine production [41, 42], including that of TNF‐α, which triggers IL‐36α production by keratinocytes [43], might not be excluded. Overall, these observations might also explain why, for example, mice deficient for cytokines that are crucial for disease progression and are directly produced in a MyD88‐dependent manner by hematopoietic cells in response to IMQ treatment, such as, for example, IL‐23, are only partially protected from epidermal thickening, despite the strong impairment observed in hematopoietic cell activation and skin infiltration [10, 16, 19, 21].
Differently from what observed at the early stages, we found that the progressive devolvement of psoriasis‐like lesions and epidermal thickening, occurring after a number of days following consecutive IMQ treatments, is instead completely dependent on hematopoietic cell activation and cytokine production in a MyD88‐dependent fashion. In line with these observations, the crucial role of MyD88 signaling in IMQ‐induced production of IL‐23 by DCs [20, 21], which in turn, induces IL‐17A and IL‐22 production by γδT cells, has been described previously in this model [13, 19]. IL‐17A and IL‐22 are then involved in sustaining IL‐36α production by keratinocytes [43]. Furthermore, IMQ can also directly induce IL‐1α/β and IL‐36α production by DCs in a MyD88‐dependent manner [16]. Therefore, it is not surprising that at late stages of disease development, when activated immune cells infiltrate the inflamed skin, the contribution of cytokine production by DCs and γδT cells becomes more relevant to sustain disease progression. In this context, our data reveal, for the first time, a possible additional, important role in IMQ‐induced psoriasis by MyD88 signaling occurring in monocytes and Mϕ, as required to sustain their production of IL‐23 and IL‐1β and the consequent proliferation and IL‐17A production by γδT cells observed in vitro and in vivo. However, this impairment of MyD88 signaling in monocytes and Mϕ affected more significantly systemic and skin inflammation than epidermal thickening, which as highlighted before, is probably sustained by the normal production of IL‐1α and IL‐36α, observed even at later time points in IMQ‐treated Myd88fl/flLysM‐cre+ mice. Our findings are supported by other studies in which a role of Mϕ in the IMQ‐induced or in other mouse models of psoriasiform skin inflammation has been reported [22, 44, 45–46]. In particular, the study by Morimura et al. [46] has shown that CX3CR1‐deficient mice display reduced disease development in the IMQ model of psoriasis as a result of a reduced expansion and cytokine production of inflammatory cytokines by inflammatory Mϕ (classically activated). Likewise, the depletion of inflammatory monocytes by the use of anti‐Gr1 (RB6‐8C5) antibodies or the use of Ccr2 −/− mice very recently has been shown to protect from IMQ‐induced psoriasis development by reducing the number of monocyte‐derived inflammatory DCs infiltrating in the skin and the production of inflammatory cytokines [22]. However, a similar protection from IMQ‐induced psoriasis in Ccr2 −/− mice was observed neither by us nor in the study by Sumida et al. [38]. These apparent discrepancies might be, at least in part, explained by the fact that the different models of monocytes/macrophage depletion used (e.g., use of Cxcr3− / − or Ccr2 −/− mice or depletion by anti‐Gr1 injections) might have targeted, more or less effectively, different populations of monocytes/Mϕ [22, 38, 46]. Our experimental choice to use Myd88fl/flLysM‐cre+ mice was dictated by the need to obtain a conditional MyD88 targeting in all of the Ly6Clow and Ly6Chigh monocyte and Mϕ populations that were found to be expanded and activated in this model, without preferentially targeting only the inflammatory Ly6Chigh monocyte/macrophage populations, as it happens, for example, in Ccr2 −/− mice, or by performing anti‐Gr1 (RB6‐8C5) depletion. Finally, our data exclude that in this IMQ‐induced model of psoriasis, MyD88 signaling in neutrophils plays a role. This is in line with the poor responsiveness of neutrophils to IMQ [35, 36–37], as well as to a recent neutrophil‐depletion model [22]. However, also, this issue needs to be elucidated further, as evidence that neutrophils promote IMQ‐induced psoriasis also exists [38].
In sum, the findings reported in this study add new insights into the specific MyD88‐dependent and ‐independent contribution of skin‐resident stromal vs. hematopoietic cells to disease initiation and progression in the IMQ‐induced mouse model of psoriasis. In particular, this study uncovers a potential role of monocytes/Mϕ as additional crucial cells promoting γδ T cell effector functions and disease development in this experimental model. In view of the little knowledge currently existing on the role of monocytes/Mϕ in psoriatic patients, future studies should be aimed to clarify better whether these cells or their functions could represent additional therapeutic targets for psoriasis treatment.
AUTHORSHIP
S.C., G.G., and P.S. designed the research study and performed data analysis. S.C., O.M., D.B., S.L., W.V., P.R., A.P., and F.T. performed experiments. A.L.D. and B.H. provided intellectual guidance. C.A.L., M.A.C., G.G., and P.S. wrote the paper.
DISCLOSURES
The authors declare no conflicts of interest.
Supporting information
Supplementary Material Files
ACKNOWLEDGMENTS
This work was supported by Ministero dell´Istruzione dell´Università e della Ricerca (RBFR12I3UB_003; to P.S.) and Associazione Italiana per la Ricerca sul Cancro (AIRC; IG‐15454; to M.A.C.). S.L. is supported by Fondazione Beretta (Brescia, Italy). We thank S. Ugel (University of Verona) and M. Eberl (University of Cardiff) for critical suggestions and helpful comments. We thank F. Granucci (University of Milano‐Bicocca) for providing Myd88 −/− mice.
REFERENCES
- 1. Deng, Y. , Chang, C. , Lu, Q. (2016) The inflammatory response in psoriasis: a comprehensive review. Clin. Rev. Allergy Immunol. 50, 377–389. [DOI] [PubMed] [Google Scholar]
- 2. Boehncke, W. H. , Schön, M. P. (2015) Psoriasis. Lancet 386, 983–994. [DOI] [PubMed] [Google Scholar]
- 3. Harden, J. L. , Krueger, J. G. , Bowcock, A. M. (2015) The immunogenetics of psoriasis: a comprehensive review. J. Autoimmun. 64, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jariwala, S. P. (2007) The role of dendritic cells in the immunopathogenesis of psoriasis. Arch. Dermatol. Res. 299, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diani, M. , Altomare, G. , Reali, E. (2015) T cell responses in psoriasis and psoriatic arthritis. Autoimmun. Rev. 14, 286–292. [DOI] [PubMed] [Google Scholar]
- 6. Perera, G. K. , Di Meglio, P. , Nestle, F. O. (2012) Psoriasis. Annu. Rev. Pathol. 7, 385–422. [DOI] [PubMed] [Google Scholar]
- 7. Coimbra, S. , Figueiredo, A. , Castro, E. , Rocha‐Pereira, P. , Santos‐Silva, A. (2012) The roles of cells and cytokines in the pathogenesis of psoriasis. Int. J. Dermatol. 51, 389–395; quiz 395–388. [DOI] [PubMed] [Google Scholar]
- 8. Baliwag, J. , Barnes, D. H. , Johnston, A. (2015) Cytokines in psoriasis. Cytokine 73, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Terui, T. , Ozawa, M. , Tagami, H. (2000) Role of neutrophils in induction of acute inflammation in T‐cell‐mediated immune dermatosis, psoriasis: a neutrophil‐associated inflammation‐boosting loop. Exp. Dermatol. 9, 1–10. [DOI] [PubMed] [Google Scholar]
- 10. Van der Fits, L. , Mourits, S. , Voerman, J. S. , Kant, M. , Boon, L. , Laman, J. D. , Cornelissen, F. , Mus, A. M. , Florencia, E. , Prens, E. P. , Lubberts, E. (2009) Imiquimod‐induced psoriasis‐like skin inflammation in mice is mediated via the IL‐23/IL‐17 axis. J. Immunol. 182, 5836–5845. [DOI] [PubMed] [Google Scholar]
- 11. Flutter, B. , Nestle, F. O. (2013) TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. Eur. J. Immunol. 43, 3138–3146. [DOI] [PubMed] [Google Scholar]
- 12. Hawkes, J. E. , Gudjonsson, J. E. , Ward, N. L. (2017) The snowballing literature on imiquimod‐induced skin inflammation in mice: a critical appraisal. J. Invest. Dermatol. 137, 546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cai, Y. , Shen, X. , Ding, C. , Qi, C. , Li, K. , Li, X. , Jala, V. R. , Zhang, H. G. , Wang, T. , Zheng, J. , Yan, J. (2011) Pivotal role of dermal IL‐17‐producing γδ T cells in skin inflammation. Immunity 35, 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uribe‐Herranz, M. , Lian, L. H. , Hooper, K. M. , Milora, K. A. , Jensen, L. E. (2013) IL‐1R1 signaling facilitates Munro's microabscess formation in psoriasiform imiquimod‐induced skin inflammation. J. Invest. Dermatol. 133, 1541–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rabeony, H. , Pohin, M. , Vasseur, P. , Petit‐Paris, I. , Jégou, J. F. , Favot, L. , Frouin, E. , Boutet, M. A. , Blanchard, F. , Togbe, D. , Ryffel, B. , Bernard, F. X. , Lecron, J. C. , Morel, F. (2015) IMQ‐induced skin inflammation in mice is dependent on IL‐1R1 and MyD88 signaling but independent of the NLRP3 inflammasome. Eur. J. Immunol. 45, 2847–2857. [DOI] [PubMed] [Google Scholar]
- 16. Tortola, L. , Rosenwald, E. , Abel, B. , Blumberg, H. , Schäfer, M. , Coyle, A. J. , Renauld, J. C. , Werner, S. , Kisielow, J. , Kopf, M. (2012) Psoriasiform dermatitis is driven by IL‐36‐mediated DC‐keratinocyte crosstalk. J. Clin. Invest. 122, 3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Milora, K. A. , Fu, H. , Dubaz, O. , Jensen, L. E. (2015) Unprocessed interleukin‐36α regulates psoriasis‐like skin inflammation in cooperation with interleukin‐1. J. Invest. Dermatol. 135, 2992–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Belle, A. B. , de Heusch, M. , Lemaire, M. M. , Hendrickx, E. , Warnier, G. , Dunussi‐Joannopoulos, K. , Fouser, L. A. , Renauld, J. C. , Dumoutier, L. (2012) IL‐22 is required for imiquimod‐induced psoriasiform skin inflammation in mice. J. Immunol. 188, 462–469. [DOI] [PubMed] [Google Scholar]
- 19. Pantelyushin, S. , Haak, S. , Ingold, B. , Kulig, P. , Heppner, F. L. , Navarini, A. A. , Becher, B. (2012) Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J. Clin. Invest. 122, 2252–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wohn, C. , Ober‐Blöbaum, J. L. , Haak, S. , Pantelyushin, S. , Cheong, C. , Zahner, S. P. , Onderwater, S. , Kant, M. , Weighardt, H. , Holzmann, B. , Reizis, B. , Becher, B. , Prens, E. P. , Clausen, B. E. (2013) Langerin(neg) conventional dendritic cells produce IL‐23 to drive psoriatic plaque formation in mice. Proc. Natl. Acad. Sci. USA 110, 10723–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshiki, R. , Kabashima, K. , Honda, T. , Nakamizo, S. , Sawada, Y. , Sugita, K. , Yoshioka, H. , Ohmori, S. , Malissen, B. , Tokura, Y. , Nakamura, M. (2014) IL‐23 from Langerhans cells is required for the development of imiquimod‐induced psoriasis‐like dermatitis by induction of IL‐17A‐producing γδ T cells. J. Invest. Dermatol. 134, 1912–1921. [DOI] [PubMed] [Google Scholar]
- 22. Singh, T. P. , Zhang, H. H. , Borek, I. , Wolf, P. , Hedrick, M. N. , Singh, S. P. , Kelsall, B. L. , Clausen, B. E. , Farber, J. M. (2016) Monocyte‐derived inflammatory Langerhans cells and dermal dendritic cells mediate psoriasis‐like inflammation. Nat. Commun. 7, 13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alrefai, H. , Muhammad, K. , Rudolf, R. , Pham, D. A. , Klein‐Hessling, S. , Patra, A. K. , Avots, A. , Bukur, V. , Sahin, U. , Tenzer, S. , Goebeler, M. , Kerstan, A. , Serfling, E. (2016) NFATc1 supports imiquimod‐induced skin inflammation by suppressing IL‐10 synthesis in B cells. Nat. Commun. 7, 11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yanaba, K. , Kamata, M. , Ishiura, N. , Shibata, S. , Asano, Y. , Tada, Y. , Sugaya, M. , Kadono, T. , Tedder, T. F. , Sato, S. (2013) Regulatory B cells suppress imiquimod‐induced, psoriasis‐like skin inflammation. J. Leukoc. Biol. 94, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walter, A. , Schäfer, M. , Cecconi, V. , Matter, C. , Urosevic‐Maiwald, M. , Belloni, B. , Schönewolf, N. , Dummer, R. , Bloch, W. , Werner, S. , Beer, H. D. , Knuth, A. , van den Broek, M. (2013) Aldara activates TLR7‐independent immune defence. Nat. Commun. 4, 1560. [DOI] [PubMed] [Google Scholar]
- 26. Callahan, J. A. , Hammer, G. E. , Agelides, A. , Duong, B. H. , Oshima, S. , North, J. , Advincula, R. , Shifrin, N. , Truong, H. A. , Paw, J. , Barrera, J. , DeFranco, A. , Rosenblum, M. D. , Malynn, B. A. , Ma, A. (2013) Cutting edge: ABIN‐1 protects against psoriasis by restricting MyD88 signals in dendritic cells. J. Immunol. 191, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hou, B. , Reizis, B. , DeFranco, A. L. (2008) Toll‐like receptors activate innate and adaptive immunity by using dendritic cell‐intrinsic and ‐extrinsic mechanisms. Immunity 29, 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abram, C. L. , Roberge, G. L. , Hu, Y. , Lowell, C. A. (2014) Comparative analysis of the efficiency and specificity of myeloid‐Cre deleting strains using ROSA‐EYFP reporter mice. J. Immunol. Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hou, B. , Benson, A. , Kuzmich, L. , DeFranco, A. L. , Yarovinsky, F. (2011) Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll‐like receptors. Proc. Natl. Acad. Sci. USA 108, 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elliott, E. R. , Van Ziffle, J. A. , Scapini, P. , Sullivan, B. M. , Locksley, R. M. , Lowell, C. A. (2011) Deletion of Syk in neutrophils prevents immune complex arthritis. J. Immunol. 187, 4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Ziffle, J. A. , Lowell, C. A. (2009) Neutrophil‐specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood 114, 4871–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wohn, C. T. , Pantelyushin, S. , Ober‐Blöbaum, J. L. , Clausen, B. E. (2014) Aldara‐induced psoriasis‐like skin inflammation: isolation and characterization of cutaneous dendritic cells and innate lymphocytes. Methods Mol. Biol. 1193, 171–185. [DOI] [PubMed] [Google Scholar]
- 33. Batycka‐Baran, A. , Maj, J. , Wolf, R. , Szepietowski, J. C. (2014) The new insight into the role of antimicrobial proteins‐alarmins in the immunopathogenesis of psoriasis. J. Immunol. Res. 2014, 628289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clausen, B. E. , Burkhardt, C. , Reith, W. , Renkawitz, R. , Förster, I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277. [DOI] [PubMed] [Google Scholar]
- 35. Tsuda, Y. , Takahashi, H. , Kobayashi, M. , Hanafusa, T. , Herndon, D. N. , Suzuki, F. (2004) Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin‐resistant Staphylococcus aureus. Immunity 21, 215–226. [DOI] [PubMed] [Google Scholar]
- 36. Zhang, X. , Majlessi, L. , Deriaud, E. , Leclerc, C. , Lo‐Man, R. (2009) Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity 31, 761–771. [DOI] [PubMed] [Google Scholar]
- 37. Smuda, C. , Wechsler, J. B. , Bryce, P. J. (2011) TLR‐induced activation of neutrophils promotes histamine production via a PI3 kinase dependent mechanism. Immunol. Lett. 141, 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sumida, H. , Yanagida, K. , Kita, Y. , Abe, J. , Matsushima, K. , Nakamura, M. , Ishii, S. , Sato, S. , Shimizu, T. (2014) Interplay between CXCR2 and BLT1 facilitates neutrophil infiltration and resultant keratinocyte activation in a murine model of imiquimod‐induced psoriasis. J. Immunol. 192, 4361–4369. [DOI] [PubMed] [Google Scholar]
- 39. Lowes, M. A. , Suárez‐Fariñas, M. , Krueger, J. G. (2014) Immunology of psoriasis. Annu. Rev. Immunol. 32, 227–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ueyama, A. , Yamamoto, M. , Tsujii, K. , Furue, Y. , Imura, C. , Shichijo, M. , Yasui, K. (2014) Mechanism of pathogenesis of imiquimod‐induced skin inflammation in the mouse: a role for interferon‐alpha in dendritic cell activation by imiquimod. J. Dermatol. 41, 135–143. [DOI] [PubMed] [Google Scholar]
- 41. Schön, M. P. , Schön, M. , Klotz, K. N. (2006) The small antitumoral immune response modifier imiquimod interacts with adenosine receptor signaling in a TLR7‐ and TLR8‐independent fashion. J. Invest. Dermatol. 126, 1338–1347. [DOI] [PubMed] [Google Scholar]
- 42. Kono, T. , Kondo, S. , Pastore, S. , Shivji, G. M. , Tomai, M. A. , McKenzie, R. C. , Sauder, D. N. (1994) Effects of a novel topical immunomodulator, imiquimod, on keratinocyte cytokine gene expression. Lymphokine Cytokine Res. 13, 71–76. [PubMed] [Google Scholar]
- 43. Carrier, Y. , Ma, H. L. , Ramon, H. E. , Napierata, L. , Small, C. , O'Toole, M. , Young, D. A. , Fouser, L. A. , Nickerson‐Nutter, C. , Collins, M. , Dunussi‐Joannopoulos, K. , Medley, Q. G. (2011) Inter‐regulation of Th17 cytokines and the IL‐36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J. Invest. Dermatol. 131, 2428–2437. [DOI] [PubMed] [Google Scholar]
- 44. Stratis, A. , Pasparakis, M. , Rupec, R. A. , Markur, D. , Hartmann, K. , Scharffetter‐Kochanek, K. , Peters, T. , van Rooijen, N. , Krieg, T. , Haase, I. (2006) Pathogenic role for skin macrophages in a mouse model of keratinocyte‐induced psoriasis‐like skin inflammation. J. Clin. Invest. 116, 2094–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang, H. , Peters, T. , Kess, D. , Sindrilaru, A. , Oreshkova, T. , Van Rooijen, N. , Stratis, A. , Renkl, A. C. , Sunderkötter, C. , Wlaschek, M. , Haase, I. , Scharffetter‐Kochanek, K. (2006) Activated macrophages are essential in a murine model for T cell‐mediated chronic psoriasiform skin inflammation. J. Clin. Invest. 116, 2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morimura, S. , Oka, T. , Sugaya, M. , Sato, S. (2016) CX3CR1 deficiency attenuates imiquimod‐induced psoriasis‐like skin inflammation with decreased M1 macrophages. J. Dermatol. Sci. 82, 175–188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files