

Short abstract
Multiple cathepsins promote particle‐induced cell death independently of inflammasomes, suggests that cathepsin inhibitors may therapeutically target multiple mechanisms of particle‐induced inflammatory disease.
Keywords: inflammation, macrophage, caspase‐1, pyroptosis, peritonitis
Abstract
Sterile particles cause several chronic, inflammatory diseases, characterized by repeating cycles of particle phagocytosis and inflammatory cell death. Recent studies have proposed that these processes are driven by the NLRP3 inflammasome, a platform activated by phagocytosed particles, which controls both caspase‐1–dependent cell death (pyroptosis) and mature IL‐1β secretion. After phagocytosis, particles can disrupt lysosomes, and inhibitor studies have suggested that the resulting release of a lysosomal protease—cathepsin B—into the cytosol somehow activates NLRP3. However, using primary murine macrophages, we found that particle‐induced cell death occurs independent of NLRP3/caspase‐1 and depends instead on multiple, redundant cathepsins. In contrast, nigericin, a soluble activator of NLRP3 inflammasomes, induced cell death that was dependent on the NLRP3. Interestingly, nigericin‐induced cell death depended partly on a single cathepsin, cathepsin X. By inhibiting or silencing multiple cathepsins in macrophages, several key proinflammatory events induced by sterile particles are blocked, including cell death, pro–IL‐1β production, and IL‐1β secretion. These data suggest that cathepsins might be potential therapeutic targets in particulate‐mediated inflammatory disease. In support of this concept, we find that a broad‐spectrum cathepsin inhibitor can suppress particle‐induced IL‐1–dependent peritonitis.
Abbreviations
- ASC−/−
ASC deficient
- AIM2
absent in melanoma 2
- ASC
apoptosis‐associated spec‐like protein
- BCS
WT or BCS−/−
- BCS−/−
cathepsins B, C, and S deficient
- BCSXL
cathepsins B, C, S, X, and L
- BL
WT or B&L−/−
- B&L−/−
cathepsin B and L deficient
- BLXS
cathepsins B, L, X, and S
- BMDM
bone marrow–derived macrophage
- Casp1−/−
caspase‐1 deficient
- CC
cholesterol crystal
- dAdT
poly(deoxyadenylic‐deoxythymidylic) acid
- LLOMe
Leu‐Leu‐O‐methyl ester
- LMD
lysosomal membrane disruption
- MSU
monosodium urate
- NLRP3
NOD‐like receptor containing a pyrin domain containing 3
- NLRP3−/−
NLRP3 deficient
- PM
peritoneal macrophage
- RIP3
receptor‐interacting protein kinase 3
- RIP3−/−
RIP3 deficient
- WT
wild‐type
Introduction
Particle‐induced, sterile inflammation underlies the pathogenesis of many common and often chronic diseases [1]. Silica, MSU, and CC are some of the many exogenous or endogenous particles implicated in such diseases [2, 3, 4, 5, 6, 7–8]. When a macrophage ingests one of these particles, two events follow: cell death, and the generation of inflammatory mediators [1]. Cell death after the ingestion of a pathogen may be beneficial to the host, helping to limit infections [9, 10–11]. However, for sterile particles, which cannot replicate or be readily destroyed by immune defenses, cell death is not thought to be beneficial but, instead, results in pathology [12, 13–14]. In fact, both particles and dead cells can stimulate macrophages to produce inflammatory mediators [7, 8, 12, 15]. Therefore, a repeating cycle of particle ingestion, inflammatory cell death, and the release of particles back into the environment for reingestion by other cells is thought to drive such pathology [16].
A better understanding of the processes regulating both cell death and production of inflammatory mediators by macrophages may help identify tractable drug targets for suppressing particle‐induced sterile inflammation. Although the initial inflammatory response to particles has been shown to depend on signaling through the IL‐1 receptor, the exact mechanisms responsible for the production/release of IL‐1β and concomitant cell death are not completely understood [15, 17, 18].
In macrophages, the induction of IL‐1β secretion by sterile particles requires 2 signals. The first signal (“priming”) can be initiated through a receptor (eg, TLR4), which leads to the activation of the transcription factor NF‐κB, which then drives the transcription of pro–IL‐1β and NLRP3 [19, 20]. The second signal (“activation”) occurs after the ingestion of particles by macrophages, which stimulates the activation of the multimolecular NLRP3 inflammasome (NLRP3, the adaptor ASC caspase activation and recruitment and the effector caspases 1 or 11) [3, 6, 7, 21, 22]. Upon NLRP3 activation, caspase‐1 converts pro–IL‐1β into active IL‐1β (and pro–IL‐18 into active IL‐18) for secretion. Active caspase‐1 (and caspase‐11) can also drive a lytic form of inflammatory programmed cell death called pyroptosis [23, 24], and this has often been assumed to be the mechanism through which ingestion of particles kills primed macrophages. Therefore, NLRP3 activation can lead to both mature IL‐1β secretion and cell death.
One of the models proposed for particle‐induced NLRP3 activation and lytic cell death originated from the observation that phagocytosed particles cause LMD [3, 7, 8]. According to this model, LMD releases a lysosomal protease—cathepsin B—into the cytosol, which activates NLRP3 [3, 7, 8]. This model has been controversial. In several studies, an inhibitor that had been assumed specific for cathepsin B—Ca074Me—was shown to suppress cell death and IL‐1β secretion during NLRP3 activation induced by particulate and lysosome‐disrupting stimuli [25, 26, 27–28]. However, genetic deficiency of either cathepsins B or L have showed either a partial role for both of these proteases in NLRP3 activation [7, 29, 30] or no role for either [28, 31, 32]. The discrepancies among the findings in studies inactivating cathepsins genetically versus with inhibitors might reflect Ca074Me inhibiting several other cathepsins (like cathepsin L) at concentrations used to block NLRP3 activation in prior studies [26, 33, 34, 35, 36–37]. In addition, the loss of one cathepsin can result in a compensatory up‐regulation of other functionally redundant cathepsins [37, 38–39]. In fact, we have found that multiple redundant cathepsins, not just cathepsin B, participate in particle‐stimulated IL‐1β production, and they do so by both enhancing the priming of pro–IL‐1β synthesis and NLRP3‐dependent pro–IL‐1β cleavage [37]. Whether multiple cathepsins are also involved in particle‐induced cell death remains an open question.
In vivo, IL‐1β is a critical mediator of particle‐induced sterile inflammatory diseases [40, 41, 42–43]. Although IL‐1β responses to particles absolutely require inflammasomes (and caspase‐1) in vitro, several studies have shown that caspase‐1–deficient mice have, on average, <50% reduction in particle or dead cell–induced responses compared with WT animals [2, 6, 7, 17]; some of those studies showed no reduction at all. Therefore, in vivo, there seems to be some caspase‐1–independent mechanisms driving particle and dead cell–induced IL‐1β activation [17].
Because cell death is known to be a key parameter in particle‐induced inflammatory disease, understanding the cellular mediators of particle‐induced cell death may be mechanistically and therapeutically relevant. In the current study, we examined, with both inhibitors and genetic models, whether particle‐induced cell death during NLRP3 activation depended on inflammasomes or cathepsins. Furthermore, we examined the potential for cathepsin inhibitors to suppress IL‐1–dependent inflammatory responses in an in vivo model of particle‐induced acute peritonitis.
MATERIALS AND METHODS
Reagents and antibodies
Abs for flow cytometry were against mouse Ly‐6G (1A8; BD Biosciences, Franklin Lakes, NJ, USA), Ly‐6C (7/4; formerly AbD Serotec, now Bio‐Rad Laboratories, Hercules, CA, USA), and the dead cell marker was 7‐AAD (Thermo Fisher Scientific, Waltham, MA, USA). Antibodies for Western blots were against β‐actin (C4; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and GAPDH (6C5; EMD Millipore, Billerica, MA, USA). ELISA kits were purchased for mouse IL‐1β (BD Biosciences) and TNF‐α (eBioscience, San Diego, CA, USA). Ultrapure LPS was from Salmonella minnesota (InvivoGen, San Diego, CA, USA). dAdT acid and nigericin were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Silica crystals (MIN‐ U‐SIL 15) were obtained from U.S. Silica (Frederick, MD, USA). CCs were synthesized by acetone supersaturation and cooling [7], Alum (Imject alum adjuvant, a mixture of aluminum hydroxide and magnesium hydroxide) was from Pierce Biotechnology (Rockford, IL, USA; now, Thermo Fisher Scientific, Waltham, MA, USA); LLOMe–HCl was from Chem‐Impex International (Wood Dale, IL, USA), and MSU crystals were prepared, as previously described [44]. ZVAD and Ca074Me were from Enzo Life Sciences (Farmingdale, NY, USA), and K777 was initially gifted to us by Stephanie A. Robertson and James H. McKerrow at the University of California (San Francisco, CA, USA), and further stocks obtained through services from the National Heart, Lung, and Blood Institute (Bethesda, MD, USA) SMARTT (Science Moving Towards Research and Therapy) Program. AT406 was from Selleck Chemicals (Houston, TX, USA). Lipofectamine 2000, RNAiMax, and all siRNA smart pools were from Life Technologies (Thermo Fisher Scientific), and Endoporter was from Gene Tools (Philomath, OR, USA).
Animal and cell lines
WT C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Casp1−/− mice have been previously described [45]; NLRP3−/− mice [46] and ASC−/− mice [46] were provided by Millennium Pharmaceuticals (Cambridge, MA, USA), and RIP3−/− mice were provided by Francis K. Chan (University of Massachusetts Medical School, Worcester, MA, USA). Casp1−/− mice also lack caspase‐11 [24]. Cathepsin S‐ [47], L‐ [48], and B‐deficient [49] mice were provided by Dr. Hal Chapman (University of California, San Francisco) and Dr. Hidde Ploegh (Harvard Medical School, Boston, MA, USA), cathepsin C–deficient mice [50] were provided by Dr. Christine Pham (Washington University School of Medicine, St. Louis, WA, USA), and all mice were backcrossed to mice with a C57BL/6 background. All animal protocols were approved by the University of Massachusetts Institutional Animal Care and Use Committee.
Production and measurement of IL‐1β, TNF‐α, and cell death assayed with in vitro cultures
Collection (and/or differentiation) and plating of primary PM, BMDMs, neutrophils, and mast cells have been described previously [37]. Unless otherwise stated, the standard protocol followed herein was as follows: Priming in RPMI 1640 (or MC/9 medium for mast cells [51]) for 3 h with LPS (200 ng/ml), with or without the addition of inhibitors after 2 h of priming, followed by 6 h of stimulation. Inhibitors were added in a final concentration of ≤0.1% DMSO, which has no effect on readouts compared with media alone. Supernatants were collected, and cytokine levels were analyzed by ELISA. In the same samples assayed for IL‐1β and TNF‐α, cell death was assessed using lactate dehydrogenase assays as recommended by the manufacturer (Promega, Madison, WI, USA). In all experiments, primary cells of any particular genotype were collected and combined from at least 3 individual age‐ and sex‐matched mice. Moreover, key experiments show means ± sem (error bars) of combined data from multiple (n) independent experiments and with data normalized to fold change (Δ) compared with controls as indicated on the y axes labels of figures.
siRNA knockdowns, immunoblotting, and live‐cell intracellular cathepsin activity‐labeling with BMV109
Assays were performed as previously described [37].
K777 formulation and systemic treatment of mice by injection or Alzet pump infusion
K777‐HCl doses and formulations were as follows: DMSO (10%)/dextrose (5%)/water (85%) for i.v. (100 μl = 62.5 mg/kg for ∼20‐g mice) or s.c. (200 μl = 125 mg/kg for ∼20‐g mice), and polyethyleneglycol‐300 (25%)/Glycofurol (25%)/Cremophor ELP (25%)/ethanol (15%)/propylene glycol (10%) [52] for Alzet pumps (model 2001; Direct Corporation, Cupertino, CA, USA), which were surgically implanted s.c. on the backs of mice for 1 wk, delivering drug or excipient formulation at a rate of 1 μl/h for the indicated doses of K777. All excipient ingredients used for K777 formulation were purchased from Sigma‐Aldrich (St. Louis, MO). Before injection of PBS or silica i.p. on the seventh day of treatment, plasma samples were taken from mice, and the K777 concentration was analyzed by LC‐MS/MS on a Waters Acquity UPLC coupled to a Waters Quattro Premier XE triple quadrupole mass spectrometer.
Neutrophil and monocyte recruitment to peritoneal cavity
Quantification of recruited neutrophils and monocytes to the peritoneal cavity was described previously [15].
Generation of bone marrow chimeras
Bone marrow chimeras have been described previously [37].
RESULTS
Particle‐induced cell death is independent of both inflammasomes and RIP3
Particle‐induced cell death in LPS‐primed macrophages has often been assumed to occur via pyroptosis, which is initiated by activated caspase‐1 (or caspase‐11). Contrary to that assumption, it was recently reported that alum particles actually elicit inflammasome‐independent necrosis [26, 28]. However, it is unknown whether the cell death induced by other pathogenic particles is pyroptotic. Therefore, we examined whether cell death induced by such particles required inflammasomes.
We stimulated PMs from WT mice or mice deficient in the inflammasome components NLRP3, ASC, or caspase‐1 (and caspase‐11) with various NLRP3 activators: a lysosomotropic detergent (LLOMe, an agent that disrupts lysosomes) or particles (silica, MSU, CC) ( Fig. 1a ). Although NLRP3, ASC, and caspase‐1 knockouts show a small reduction (significant in the case of CCs with ASC and capase‐1 knockouts) in cell death compared with those from WT mice, after treatment with LLOMe, silica, MSU, or CC, most of that response was inflammasome‐independent. However, IL‐1β secretion induced by those same stimuli was completely inflammasome‐dependent. In contrast, nigericin‐induced cell death and IL‐1β production were both inflammasome‐dependent, as shown with NLRP3‐deficient PMs (Fig. 1b, right panel).
Figure 1.
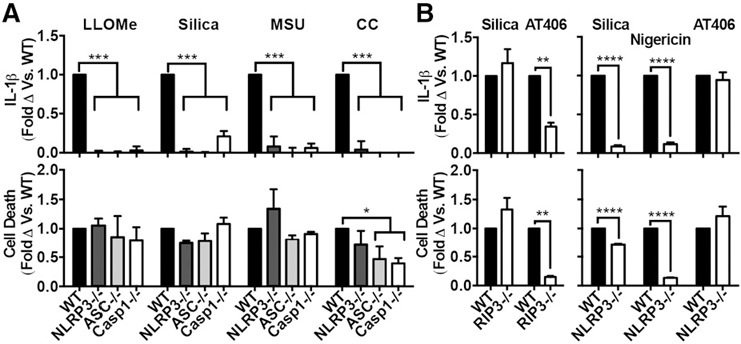
Particle‐induced cell death is independent of both inflammasomes and RIP3. In all cases, PMs were primed with LPS. (A) PMs from WT, NLRP3−/−, ASC−/−, or Casp1−/− stimulated with LLOMe (1 mM), silica (50 μg/ml), MSU (250 μg/ml), or CCs (750 μg/ml). (B, left panel) PMs from WT or RIP3−/− mice stimulated with silica (50 μg/ml) or AT406 (10 μM). (B, right panel) PMs from WT or NLRP3−/− mice were stimulated with silica (40 μg/ml), nigericin (2 μM), or AT406 (7.5 μM). Error bars represent (A) WT (LLOMe n = 6, silica n = 6, MSU n = 4, CCs n = 4), NLRP3−/− (LLOMe n = 3, silica n = 4, MSU n = 3, CCs n = 3), ASC−/− (LLOMe n = 4, silica n = 3, MSU n = 3, CCs n = 3), Casp1−/− (LLOMe n = 5, silica n = 3, MSU n = 3, CC n = 3); or (B, left panel) n = 4, (B, right panel) n = 3. Statistical analysis was performed by 2‐way ANOVA and Dunnett's multiple comparisons test (A), or 2‐tailed Student's t test (B). *P < 0.05, **P < 0.01, ***P < 0.001 ****P < 0.0001.
Because RIP3‐dependent pathways driven by Smac mimetic drugs have also been shown to induce both cell death and IL‐1β secretion independent of inflammasomes [53], it was reasonable to ask whether particles induce cell death via this RIP3‐dependent alternative pathway. Therefore, we compared the effects of silica with those of a Smac mimetic drug (AT406) [54] on PMs from both WT and RIP3−/− mice. Although both cell death and IL‐1β secretion induced by the Smac mimetic depended on RIP3, as expected, these responses to silica were completely independent of RIP3 (Fig. 1b, left panel). As expected, IL‐1β secretion and cell death induced by AT406 was NLRP3‐independent (Fig. 1b, right panel). TNF‐α was not significantly reduced under any of the above conditions (Supplemental Fig. 1). In addition to the pooled, normalized data displayed in Fig. 1, nonnormalized data from representative experiments are shown in Supplemental Fig. 1c and d. Therefore, although the pathway used by particles to stimulate IL‐1β secretion is highly dependent on the NLRP3 inflammasome, the cell death response is independent of both inflammasomes and RIP3.
Cathepsin inhibitors and not caspase inhibitors suppress both particle‐induced cell death and IL‐1β secretion
Ingested particles are known to induce LMD, which releases cathepsins into the cytosol [3, 7, 8]. Cathepsins have a role in generating signal 1 and signal 2 for inflammasome‐dependent IL‐1β production [37] and have been implicated in cell death in some other systems [55]. Therefore, we examined the role of cathepsins in particle‐induced cell death.
First, we tested the effect of a broad cathepsin inhibitor K777 on particle‐induced cell death in WT PMs or PMs deficient in the inflammasome components ASC or caspase‐1 (we have previously shown that this inhibitor blocks particle‐induced IL‐1β production [37]) ( Fig. 2a ). Again, silica‐induced IL‐1β secretion was inflammasome‐dependent, but silica‐induced cell death was largely inflammasome‐independent, showing only a small reduction in the knockouts. Importantly, K777 suppressed silica‐induced IL‐1β secretion in the WT cells, as expected, and blocked cell death in both the WT and inflammasome‐deficient cells. In contrast, both cell death and IL‐1β secretion induced by dAdT (a dsDNA stimulator of AIM2 inflammasomes) depended on inflammasome components (Fig. 2a). Unlike the K777ˈs strong inhibition of silica‐induced cell death and IL‐1β secretion, K777 treatment caused only a minor reduction in dAdT‐mediated IL‐1β secretion—likely because it can reduce the level of pro–IL‐1β available for both NLRP3 and AIM2 inflammasomes by suppressing pro–IL‐1β synthesis [37]—and no reduction in dAdT‐mediated cell death. TNF‐α secretion was not reduced by K777, demonstrating that this agent selectively reduced cell death and IL‐1β (Supplemental Fig. 2a). Therefore, even in the absence of inflammasome components, silica‐induced cell death was robust yet sensitive to cathepsin inhibition by K777. These contrasting results emphasize that inflammasomes are required for cell death in response to some stimuli, such as the soluble agents nigericin and dAdT, but not in response to sterile‐particulate stimuli.
Figure 2.
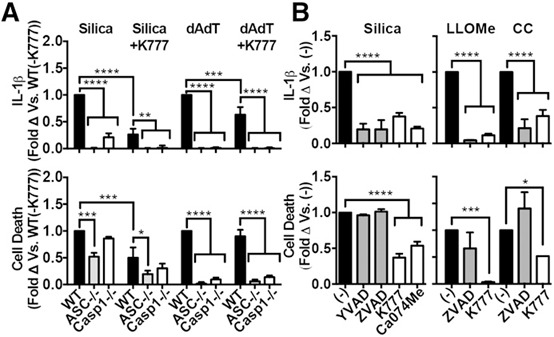
Cathepsin inhibitors and not‐caspase inhibitors suppress both particle‐induced cell death and IL‐1β secretion. In all cases, PMs were primed with LPS. (A) PMs from WT, ASC−/−, Casp‐1−/− mice treated with K777 (30 μM) before stimulation with silica (80 μg/ml) or dAdT (1 μg/ml). (B) PMs treated with media control (black bars, [−]), caspase‐1 inhibitors (gray bars, YVAD [20 μM], ZVAD [10 μM]), or cathepsin inhibitors (white bars, K777 [20 μM], Ca074Me [20 μM]) before stimulation with silica (50 μg/ml), LLOMe (1 mM), or CCs (750 μg/ml). Error bars represent (A) n = 3, or (B) silica ([−] n = 12, YVAD n = 4, ZVAD n = 6, K777 n = 8, Ca074Me n = 3), LLOMe ([−] n = 5, ZVAD n = 2, K777 n = 4), CCs ([−] n = 5, ZVAD n = 3, K777 n = 3). Statistical analysis was performed by 1‐way ANOVA and Dunnett's multiple comparisons test (A), or 2‐way ANOVA and Dunnett's multiple comparisons test (B). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Because it may be therapeutically useful for a drug to inhibit both cell death and IL‐1β secretion, and to independently verify the findings above, we also tested the effect of caspase inhibitors on particle‐induced cell death in LPS‐primed macrophages. We treated PMs with either Ca074Me (cathepsin inhibitor), K777 (cathepsin inhibitor), YVAD (caspase‐1 inhibitor), or ZVAD (pan‐caspase inhibitor), then stimulated with silica, CC, or LLOMe (Fig. 2b). Consistent with our genetic data, even though YVAD and/or ZVAD blocked IL‐1β secretion induced by silica, CC, and LLOMe, caspase inhibition did not suppress particle‐induced cell death. Although not statistically significant, ZVAD even enhanced cell death after stimulation with CC, which may be related to its inhibition of caspase‐8, leading to TNF‐α–induced necroptosis [56]. However, it is unclear why ZVAD did not also enhance cell death in PMs stimulated with either silica or LLOMe. In contrast, the cathepsin inhibitors K777 and Ca074Me (both of which inhibit multiple cathepsins at the concentrations used here, >1 μM [37]) suppressed both cell death and IL‐1β secretion. Again, TNF‐α secretion was not significantly reduced by any of the above inhibitors (Supplemental Fig. 2b). In addition to the pooled, normalized data displayed in Fig. 2, nonnormalized data from representative experiments are shown in Supplemental Fig. 2c and d. Thus, our data indicate that, at inhibitor concentrations at which IL‐1β secretions and cell death are suppressed to a similar degree, cathepsin inhibitors block particle‐induced cell death more effectively than caspase inhibitors.
Inhibition of multiple cathepsins suppresses cell death in several primary, innate, immune cell lines and in response to various lysosome‐disrupting stimuli
To extend and generalize the effects of K777 on cell death, we examined other primary cell types, various concentrations of particles/lysosome‐disrupting stimuli, and various concentrations of either K777 or Ca074Me. Again, K777 selectively suppressed particle‐induced cell death in BMDMs and mast cells, although K777 did not suppress neutrophil cell death ( Fig. 3a ). Consistent with our previous report showing that K777 selectively suppresses particle‐induced IL‐1β secretion much more effectively than that induced by nigericin or dAdT [37], K777 selectively inhibited particle‐induced cell death across a broad range of silica and alum concentrations, whereas it caused no significant reduction in cell death induced by nigericin or dAdT at any concentration tested (Fig. 3b). Thus, in addition to the K777ˈs ability to suppress IL‐1β secretion, K777 suppresses the cell death response to particles in PMs, BMDMs, and mast cells, and that effect is generalizable to all particles tested, as well as the lysosome‐disrupting agent LLOMe (see Fig. 2b). Moreover, these data suggest that K777 is a selective inhibitor of both particle‐induced IL‐1β secretion and inflammasome‐independent cell death, implicating a dualistic role for cathepsins.
Figure 3.
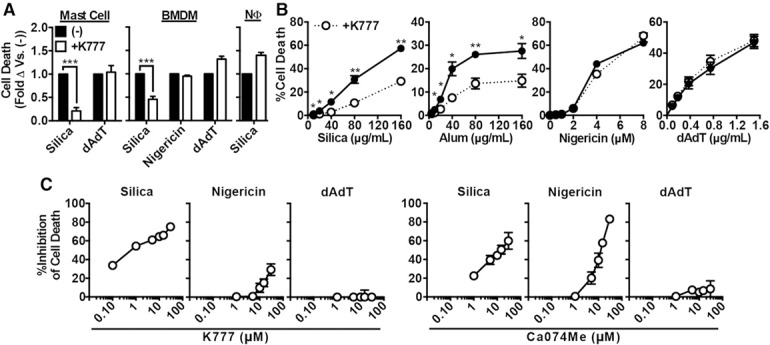
Inhibition of multiple cathepsins suppresses cell death in several primary innate immune cell lines and in response to various lysosome‐disrupting stimuli. In all cases, cells were primed with LPS. (A) BMDM, neutrophils (NΦ), or mast cells treated with media control ([−], black bars) or K777 (white bars, 15 μM), and then, stimulated with silica (40 μg/ml for BMDMs and mast cells, 100 μg/ml for NΦ), nigericin (1 μM), or dAdT (0.3 μg/ml). (B) PMs treated with media control (solid line) or K777 (dashed line, 15 μM) before stimulation with a titration of silica, alum, nigericin, or dAdT. (C) PMs were treated with media control or the indicated concentrations of K777 (left figures) or Ca074Me (right figures) and stimulated with silica (40 μg/ml), nigericin (2 μM), or dAdT (0.5 μg/ml); data shows the percentage of inhibition of cell death compared to media control. Error bars represent (A and B) sem (n = 3), (C) sem (K777 from 0.1–15 μM [n = 4], Ca074Me from 0.1–15 μM [n = 3], K777 at 30 μM [n = 3], Ca074Me at 30 μM [n = 2]). Statistical analysis was performed by 2‐tailed Student's t test (A and B). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, we determined the concentrations of Ca074Me and K777 necessary to inhibit cell death induced by silica, nigericin, or dAdT (Fig. 3c) and compared that to the cathepsin‐inhibition profile of those inhibitors that has been established in that cell type [37]. This analysis revealed that, at the concentrations of these agents needed to block cell death (>30 μM), Ca074Me and K777 inhibit multiple cathepsins; these results were similar to what we recently observed for the concentrations needed to block IL‐1β responses (>30 μM) (note: at all concentrations of K777 tested and >1 μM of Ca074Me, these inhibitors target multiple cathepsins) [37]. Interestingly, K777 inhibited particle‐induced cell death more selectively than Ca074Me. Although K777 inhibited silica‐induced cell death much more than it inhibited cell death induced by nigericin, Ca074Me strongly inhibited silica‐ and nigericin‐induced cell death with similar potency. This is likely because nigericin‐induced NLRP3 activation, which is responsible for the consequent cell‐death response, involves a nonredundant role for cathepsin X and Ca074Me is a more effective inhibitor of cathepsin X than K777 [37] (see also Fig. 4 , described below). In addition to the pooled normalized data displayed in Fig. 3, nonnormalized data from representative experiments are shown in Supplemental Fig. 3. Thus, both here and in prior studies, multiple cathepsins (not cathepsin B alone) are inhibited at the concentrations of K777 or Ca074Me required to block particle‐induced cell death during NLRP3 activation [25, 26, 27–28].
Figure 4.
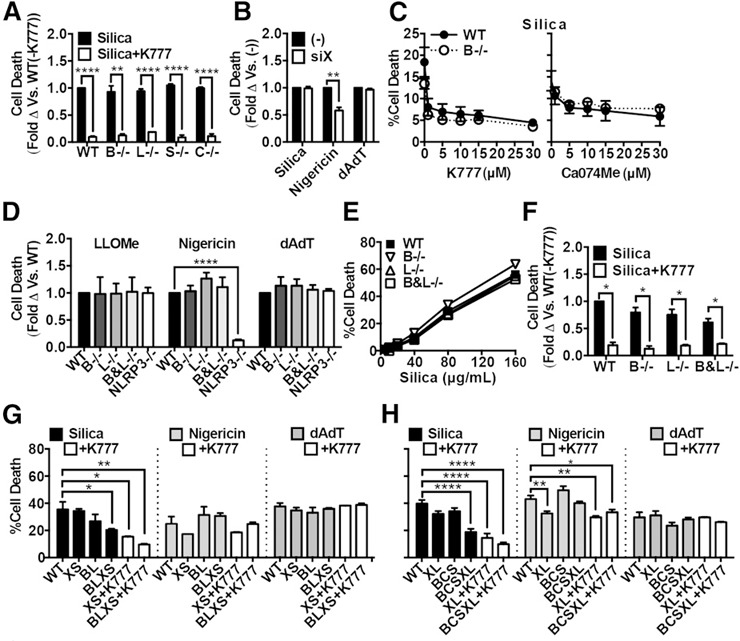
Particle‐induced cell death depends on multiple redundant cathepsins. In all cases, cells were primed with LPS. (A) WT PMs or those lacking cathepsins B (B−/−), L (L−/−), S (S−/−), or (C−/−) treated with silica (black bars, 40 μg/ml) or silica plus K777 (white bars, 15 μM). (B) PMs treated with nontargeting control siRNA (−) or siRNA targeting cathepsin X (siX) before priming with LPS and stimulating with silica (80 μg/ml), nigericin (1.5 μM), or dAdT (0.5 μg/ml). (C) WT (closed circles, solid line) or cathepsin B‐deficient (open circles, dashed line) PMs treated with a range of K777 or Ca074Me concentrations (1, 5, 10, 15, or 30 μM) before stimulation with silica (50 μg/ml). (D–F) Lethally irradiated WT mice were reconstituted with bone marrow from WT, cathepsins B (B−/−), L (−/−), or B and L (B&L−/−), or NLRP3 (NLRP3−/−)–deficient donor mice. LPS‐primed PMs elicited from those mice were stimulated with LLOMe (1 mM), nigericin (2 μM), or dAdT (0.4 μg/ml) (D), a range of silica concentrations (E), silica plus media control (black bars) or silica plus K777 (white bars, 20 μM) (F). (G and H) PMs elicited from chimeric WT or B&L−/− mice (G) described for panels D–F, or WT mice or mice deficient in the 3 cathepsins B, C, and S (BCS) (H), were treated with nontargeting siRNA (WT) or siRNA‐targeting (G) both cathepsins X and S (XS when given to WT or BLXS when given to B&L−/− [labeled as BL]), or both cathepsins X and L (XL when given to WT or BCSXL when given to BCS) (H), and subsequently primed with LPS before stimulating with silica (80 μg/ml), nigericin (1.5 μM), or dAdT (0.5 μg/ml). XL, BCSXL, BL, BLXS macrophages were also treated with K777 (white bars, 15 μM). Error bars represent (A and B) sem [n = 3], (C) sem (left panel, n = 3; right panel, n = 2), (D) sem (LLOMe [WT n = 6, B−/− n = 3, L−/− n = 3, B&L−/− n = 3, NLRP3−/− n = 4], nigericin [WT n = 7, B−/− n = 5, L−/− n = 5, B&L−/− n = 5, NLRP3−/− n = 5], dAdT [WT n = 6, B−/− n = 6, L−/− n = 6, B&L−/− n = 6, NLRP3−/− n = 3]), (E) sd of technical triplicates representing 3 independent experiments, (F) sem (WT, B−/−, L−/− [n = 6], B&L−/− [n = 3]), (G) sem (n = 2), or (H) sem (WT, XL, BCS, BCSXL [n = 5], +K777 [n = 3]). Statistical analysis was performed by 2‐tailed t test (A, B, and F) or 2‐way ANOVA with Dunnett's multiple comparisons test (D, G, and H). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Particle‐induced cell death depends on multiple redundant cathepsins
Because cathepsin inhibitors blocked particle‐induced cell death, we next sought to test whether the genetic loss of cathepsins similarly inhibited this process. Compared with WT PMs, deficiency of single cathepsins B, L, C, or S failed to significantly attenuate cell death in response to silica (Fig. 4a). In fact, K777 attenuated cell death induced by silica equally well in cells sufficient or deficient for any one of those cathepsins, indicating that K777 does not suppress this response by inhibiting any one of those cathepsins alone. In addition to the pooled, normalized data displayed in Fig. 4, nonnormalized data from representative experiments are shown in Supplemental Fig. 4. To examine the role of cathepsin X, we silenced cathepsin X in PMs with siRNA. We confirmed a 90–95% knockdown of cathepsin X mRNA by qPCR (Supplemental Fig. 5a). We further demonstrated a reduction in enzyme activity observed with the fluorescent, cathepsin activity‐based probe BMV109 [57]. Although intracellular cathepsin X activity was difficult to assess at such low levels shown in the lysates, the reduction of cathepsin X activity in the supernatants after siRNA treatment demonstrates the similar knockdown efficiency of both cathepsin X activity and expression (Supplemental Fig. 5b). After silencing cathepsin X, we stimulated those cells with silica, nigericin, or dAdT. Again, there was no significant difference between cathepsin X–sufficient and cathepsin X–deficient macrophages in the amount of cell death induced by either silica or dAdT (Fig. 4b). However, deficiency of cathepsin X significantly and selectively reduced nigericin‐induced cell death, which is similar to our previous observations for the nigericin‐induced IL‐1β response [37]. Finally, we treated both WT and cathepsin B–deficient PMs with titrations of either K777 or Ca074Me before stimulation with silica (Fig. 4c). Again, both cathepsin inhibitors suppressed cell death in WT and cathepsin B–deficient PMs across the titration range. Therefore, these data indicate that cathepsin X has a previously unrecognized, nonredundant role in nigericin‐induced, NLRP3‐dependent cell death, whereas the individual cathepsins examined (including cathepsins B, L, C, S, and X) are not essential for particle‐induced, inflammasome‐independent cell death during NLRP3 activation.
Because particle‐induced cell death does not require any one of the single cathepsins examined, and the cathepsin inhibitors block cell death at concentrations that inhibit multiple cathepsins, it seemed likely that multiple cathepsins might have redundant roles in particle‐induced cell death. To test that possibility, we first sought to analyze macrophages lacking both cathepsins B and L. Because mice deficient in both cathepsins B and L die within the first few weeks of life [38], we could not directly analyze PMs from those animals. To circumvent that limitation, we harvested bone marrow from those neonatal, double‐knockout mice and used that bone marrow to reconstitute lethally irradiated, adult WT mice. In those chimeric mice, cells of hematopoietic origin lacked cathepsin B and L (B&L−/−) activity [37]. For comparison, we also made chimeras with bone marrow from neonatal WT, cathepsin B−/−, and cathepsin L−/− mice. We elicited PMs from those various chimeric mice and stimulated cells as previously performed. Again, cathepsin B–, L–, or B and L–deficient macrophages showed no attenuation of cell death induced by LLOMe, silica titration, nigericin, or dAdT (Fig. 4d and e). Again, K777 treatment still attenuated silica‐induced cell death in the absence of those cathepsins (Fig. 4f).
Because we have shown that K777 and Ca074Me inhibit more than only cathepsins B and L at concentrations required to block particle‐induced cell death, we examined the particle‐induced response of PMs deficient in up to 4 or 5 cathepsins. To do that, we examined PMs deficient for both cathepsins B and L (described above) and also elicited PMs from WT mice or mice deficient for the 3 cathepsins B, C, and S (BCS−/−); those mice were viable with no obvious physical or behavioral pathology. In the BL macrophages, we silenced cathepsins X and S with siRNA. Similarly, in the BCS macrophages, we silenced cathepsins X and L with siRNA. In both cases, treatment with siRNA resulted in >90% reduction in the mRNA of each gene (Supplemental Fig. 5b and c). Intracellular cathepsin activity, as assayed with BMV109, was also reduced with the knockouts and knockdowns. As discussed, although the level of intracellular cathepsin X activity in unstimulated LPS‐primed PMs was close to background in those assays, the knockdown in cathepsin X activity was apparent in the cathepsin S– or L–deficient cells (in which cathepsin X activity was up‐regulated before its knockdown with siRNA) and was also apparent in the supernatants in which cathepsin X activity was great enough that it could be clearly visualized. After stimulation, compound deficiency of BLXS or BCSXL both resulted in a specific and significant attenuation of cell death induced by silica and not by nigericin or dAdT (Fig. 4g,h). Importantly, K777 was able to further attenuate particle‐induced cell death in the BLXS and BCSXL macrophages, suggesting that other and/or residual (incompletely silenced) cathepsins or noncathepsin targets may still be involved. Surprisingly, although cathepsin X&L–deficiency significantly reduced nigericin‐induced cell death, the other combinations in which cathepsin X was silenced, which all included loss of cathepsin S, did not. This may be because nigericin‐induced responses were uniquely dependent on cathepsin X activity, and cathepsin X activity was up‐regulated in cathepsin S–deficient macrophages [37]. Altogether, these data suggest that multiple cathepsins have redundant roles in particle‐induced cell death during NLRP3 activation.
Particle‐induced sterile inflammation is suppressed by cathepsin inhibition in vivo
Despite being necessary for particle‐induced IL‐1β secretion in vitro, caspase‐1 is not required for the bulk of the IL‐1–dependent response to particles in vivo [2, 6, 7, 17]. In fact, cathepsin C has been shown to have a role in this response in vivo [17], despite us finding no role for cathepsin C alone in vitro in inflammasome activation in our previous study [37] or here, in cell death. This suggests that the critical mechanism or mechanisms for particle‐induced inflammation in vivo is or are largely inflammasome‐independent. Our recent and current findings indicate that multiple cathepsins have a role in a much broader set of particle‐induced, proinflammatory events. Importantly, we have shown here that cathepsins have a role in inflammasome‐independent cell death induced by sterile particles, and sterile cell death itself is known to induce IL‐1–dependent inflammation in vivo [15, 17]. It is also thought that IL‐1 (pro–IL‐1β, IL‐1β, and IL‐1α) can be released and/or potentially activated in the extracellular milieu subsequent to cell death [17, 40, 41, 42–43]. We have shown previously that cathepsin inhibitors suppress both the priming of pro–IL‐1β synthesis and particle‐induced IL‐1β activation, suggesting that cathepsins have a role in both the inflammasome‐dependent proinflammatory events and those events that are inflammasome‐independent [37]. The contribution of cathepsins to all of these proinflammatory events raises the possibility that broadly acting cathepsin inhibitors, such as K777 (which inhibits cathepsins B, L, S, X, V, K, and C [37, 58]), might be able to suppress particle‐induced inflammation in vivo.
We examined the effect of the cathepsin inhibitor K777 in a model of IL‐1–dependent, silica‐induced acute peritonitis [17]. Pretreatment with K777, either i.v. or s.c., strongly suppressed both neutrophil and macrophage recruitment in response to silica ( Fig. 5a ). Given that cathepsin C is implicated as a component of this response [17], we performed the same experiment with cathepsin C–deficient mice and found again that K777 strongly suppressed that response (Fig. 5b). This suggests that K777 blocked the in vivo, inflammatory response by inhibiting not only cathepsin C but also additional targets, which is consistent with what we observed for IL‐1β production in vitro.
Figure 5.
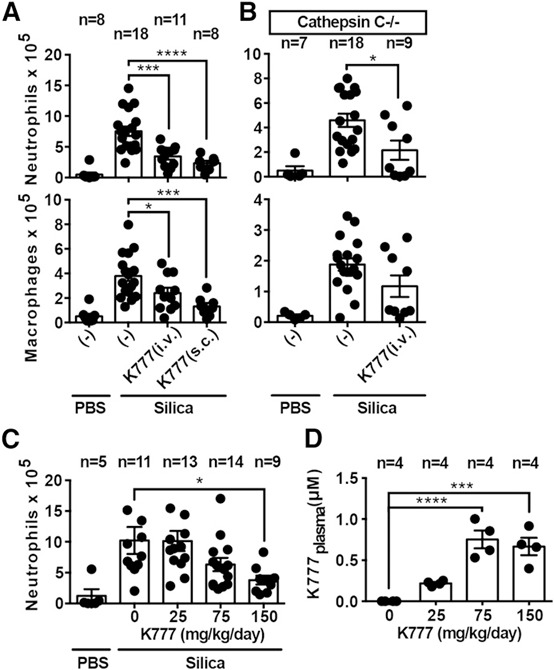
Particle‐induced sterile inflammation is suppressed by cathepsin inhibition in vivo. (A–C) Quantification of IL‐1–dependent cellular exudates by flow cytometric analysis following i.p. injection of 100 µl PBS or silica (0.2 mg) for 4 h. Effect of single‐bolus excipient (−) or K777 treatment (62.5 mg/kg i.v., 125 mg/kg s.c.) 1 h before silica injection is shown in WT mice (A) or cathepsin C−/− mice (B). (C) Effect of 1‐wk, s.c. infusion of excipient (0) or different doses of K777 (mg/kg/d) before i.p. silica injection in WT mice. (D) Concentration of K777 in the plasma after 1 wk of treatment with the doses described in panel C. Error bars represent (A–D) sem from the indicated number (n) of mice. Statistical analysis was performed by 2‐tailed Student's t test (A and B) or 1‐way ANOVA and Dunnett's multiple comparisons test (C and D). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Finally, we sought to determine the target concentration of K777 in the plasma necessary to achieve efficacy. To do that, we treated mice via s.c. infusion of K777 at different doses, delivered by osmotic Alzet pumps, which we surgically implanted in the backs of mice. After 1 wk of continuous K777 infusion, we measured steady‐state drug levels via mass spectrometry, monitoring the inflammatory response elicited by i.p. silica injection, as described above. Again, K777 markedly attenuated that response in a dose‐dependent manner (up to ∼70% reduction) (Fig. 5c). The concentration of K777 in the plasma needed to inhibit that response was ∼0.75 µM (Fig. 5d). Together, these data demonstrate, in an in vivo model, that a tolerable dose of a cathepsin inhibitor K777 can suppress the IL‐1–dependent, acute inflammatory response to sterile particles.
DISCUSSION
Inflammasome activation is known to lead to two caspase‐1–dependent events: IL‐1β secretion and pyroptotic cell death [23]. Because particles are known to activate the NLRP3 inflammasome, it has often been assumed that particles cause cell death in LPS‐primed macrophages through pyroptosis [59]. Instead, we found that particles induce inflammasome‐independent, caspase‐1–independent cell death. In LPS‐primed macrophages stimulated with particles, it seems that pyroptosis have only a minor/redundant role or that it is somehow suppressed. This is consistent with an earlier finding that alum particles induced inflammasome‐independent cell death [26, 28]. Here, our data generalized that phenomenon to multiple pathogenic particles, suggesting that, rather than an exception, this is the rule for NLRP3‐activating particles. Moreover, our extensive pharmacologic and genetic examination defines the underlying mechanism as cathepsin‐dependent.
In contrast to caspase‐1, we have found that cathepsins are important components of the particle‐induced cell death pathway in LPS‐primed macrophages. Conversely, cell death induced by soluble, microbe‐associated NLRP3 activators (like nigericin) or AIM2 activators (such as dAdT) depends on the caspase‐1 inflammasome. Cathepsin‐dependent, caspase‐independent cell death has been demonstrated in other models examining cell death unrelated to the IL‐1–dependent inflammatory response [60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71–72]. However, we have examined this phenomenon in the context of the concomitant cell death and the IL‐1β secretory response classically associated with inflammasomes. In this context, particulate/LMD‐promoting NLRP3 activators appear to be unique in their dependence on multiple cathepsins for both cell death and IL‐1β secretion. In line with those conclusions, we found that cathepsin inhibitors, and not caspase‐1 inhibitors, block cell death with the same selectivity for particulate stimuli as the respective genetic models, suggesting that cathepsins may be selective pharmacologic targets involved in particle‐induced pathology, rather than during microbial threats. Presumably, that is because both particle‐induced responses are initiated by LMD, which delivers multiple cathepsins or their products into the cytosol, whereas the soluble NLRP3 activators (nigericin and dAdT) do not cause LMD. Again, our data make the critical distinction that the events downstream of LMD are ultimately different: cathepsin‐dependent IL‐1β secretion requires inflammasomes, whereas cathepsin‐dependent cell death does not.
Although cathepsin inhibitors are effective at reducing particle‐induced cell death, there is controversy as to whether cathepsins really do promote cell death in LPS‐primed macrophages because the genetic evidence has been lacking. Many studies have shown that the supposed cathepsin B–selective inhibitor Ca074Me suppresses cell death induced by LLOMe, particles, and soluble NLRP3 activators [8, 25, 26, 27, 28, 29, 30–31, 73, 74, 75, 76, 77, 78, 79, 80–81]. Such studies have reinforced the prevailing notion that cathepsin B mediates NLRP3 activation and, therefore, pyroptotic cell death. However, several follow‐up studies have found that cathepsin B deficiency does not suppress particle‐induced NLRP3 activation in LPS‐primed macrophages, which raised doubt about earlier conclusions that cathepsins were involved in those responses [28, 31, 32]. In our current study, PMs deficient in 4 cathepsins (B, L, S, and X) or 5 cathepsins (B, C, S, L, and X) showed a large and significant reduction in cell death induced by silica. Moreover, we found that deficiency of these 4 or 5 cathepsins had no effect on cell death induced by nigericin or dAdT, demonstrating that the genetic phenotype matches the specificity of inhibitors (note: surprisingly, cathepsin X deficiency alone does reduce nigericin‐induced cell death; discussed below). Therefore, our genetic and inhibitor data demonstrate that multiple, redundant cathepsins are involved in particle‐stimulated cell death and help to reconcile earlier discordant data. The degree of redundancy in cathepsins is further emphasized by our finding that a reduction in silica‐induced cell death was not evident in single, double (B&L) or even triple (BCS), cathepsin‐deficient PMs. Moreover, even in the absence of these 5 cathepsins, there were still a few bands of residual cathepsin activity labeled by the probe BMV109. In fact, both the residual cathepsin activity and cell death were susceptible to K777‐mediated inhibition. Therefore, at the concentrations used in previous studies, it is highly likely that Ca074Me suppressed cell death by inhibiting multiple cathepsins, not just cathepsin B [26, 33, 34, 35, 36–37]. Similarly, we have shown previously that multiple redundant cathepsins are involved in particle‐induced IL‐1β secretion [37]. Thus, in addition to their role in promoting IL‐1β secretion, multiple redundant cathepsins (not inflammasomes or cathepsin B alone) promote particle‐induced cell death during NLRP3 activation.
In contrast to the redundant role of multiple cathepsins in particle‐induced cell death, we found that cathepsin X has a nonredundant role in nigericin‐induced cell death. This is consistent with our previous study, which demonstrated that cathepsin X also had a nonredundant role in nigericin‐induced NLRP3 activation [37]. Also consistent with that study, which showed that Ca074Me suppressed nigericin‐induced IL‐1β secretion more effectively than K777 did, we found here that Ca074Me was also more effective at suppressing nigericin‐induced cell death. This difference can be explained by the fact that, compared with K777, Ca074Me is a more‐potent inhibitor of cathepsin X [37]. Moreover, when compared with WT PMs, cathepsin X knockdown in cathepsin S–deficient PMs reduced nigericin‐induced cell death less effectively, likely because those cells (as well as cathepsin L–deficient PMs) up‐regulate cathepsin X activity [37]. Whether other cathepsins besides cathepsin X participate in additional steps of nigericin‐induced NLRP3 activation, like they do for particles, is not yet clear but remains a possibility.
Although our study has provided the genetic evidence that multiple cathepsins promote caspase‐1–independent, particle‐induced cell death in LPS‐primed macrophages, the biologic significance of cell death in the context of particle‐induced inflammatory disease remains unclear. This is largely because particle‐induced cell death occurs concomitant with IL‐1β secretion, making isolation of those two phenomena technically challenging in vivo. It is clear, however, that IL‐1 is necessary for particle‐induced inflammation in vivo [2, 6, 7, 17]. Moreover, we know that caspase‐1 is absolutely required for particle‐induced IL‐1β secretion in vitro [2, 6, 7]. Concomitant with IL‐1β secretion, the cell death response has often been assumed to occur via caspase‐1–mediated pyroptosis. Therefore, assuming that caspase‐1 mediates both cell death and IL‐1β secretion induced by particles, caspase‐1 appears to be an ideal therapeutic target for particle‐induced inflammatory disease. However, recent studies have shown that, despite caspase‐1 being absolutely required for particle‐induced IL‐1β secretion in vitro, it has a minimal role in the particle‐induced response in vivo [2, 6, 7, 17]. Therefore, there must be alternative mechanisms driving this IL‐1–dependent response independently of caspase‐1. In fact, two major explanatory hypotheses have been proposed for the inflammatory response to particles in vivo: 1) alternative intracellular mechanisms of caspase‐1–independent IL‐1β activation and secretion are engaged only in the in vivo environment, or 2) after cell death, pro–IL‐1β (and/or IL‐1α) is released into the extracellular milieu, where it is activated by alternative extracellular mechanisms (e.g., cathepsin‐C–dependent neutrophil proteases) [2, 6, 7, 17, 40, 41, 42–43]. Therefore, a major goal of our study was to determine whether cathepsin inhibition might suppress any of the caspase‐1–independent mechanisms that may drive the particle‐induced inflammatory response.
Our findings in this study suggest that, regardless of whether one or both of the above hypotheses turn out to be correct, cathepsin inhibitors can act at multiple levels to suppress the in vivo inflammatory response to sterile particles. Regarding hypothesis 1, we have shown previously that the cathepsin inhibitors K777 and Ca074Me can reduce the intracellular pool of pro–IL‐1β [37]. This should make less pro–IL‐1β available for intracellular and/or extracellular activation by caspase‐1–independent IL‐1β activating platforms. Regarding hypothesis 2, we have shown that, in addition to reducing pro–IL‐1β levels, cathepsin inhibitors can suppress caspase‐independent cell death induced by inflammatory particles, which may further reduce the availability of pro–IL‐1β (or release of IL‐1α) for activation in the extracellular milieu. Finally, cathepsin inhibitors, such as K777 can also directly block cathepsin C, which may prevent cathepsin C–dependent neutrophil proteases from activating IL‐1β either inside cells or in the extracellular milieu [2, 6, 7, 17, 40, 41, 42–43, 58]. Altogether, our data show that the broad cathepsin inhibitor K777 can suppress particle‐induced inflammation in vivo and suggest that it could achieve that effect through multiple modes of action. Further studies are needed to develop reliable tools that can define the relative contributions of cell death and various IL‐1β–activating mechanisms in vivo.
In summary, this study elucidates several basic aspects of particle‐induced inflammation that are conceptually and therapeutically relevant. First, we have shown that sterile particles use multiple, redundant cathepsins to promote cell death, which may release not only proinflammatory danger‐associated‐molecular patterns (DAMPs) but also pro–IL‐1β and IL‐1α. Second, we have shown that cathepsins drive the two aspects of sterile inflammation that may be critical for particulate‐mediated pathology, cell death, and pro–IL‐1β synthesis, which are not addressed by therapeutic modalities focused solely on targeting caspase‐1/inflammasomes. Therefore, our findings predict that drugs that inhibit multiple cathepsins should reduce particle‐induced, sterile inflammation. In support of that idea, we have shown that the broad cathepsin inhibitor K777 blocked both particle‐induced cell death and IL‐1β production in vitro and, most important, that it inhibited particle‐stimulated inflammatory responses in vivo. Given their newly appreciated role in both caspase‐1–dependent and –independent pathways of cell death and IL‐1β production, cathepsins may be multifaceted therapeutic targets that bring with them new possibilities for the treatment of particle‐induced sterile inflammatory diseases.
AUTHORSHIP
G.M.O. and K.L.R. performed the primary conception and design of the study and G.M.O. peformed the majority of experiments and analyses. S.S., J.D.C., M.B., S.A.R., and F.K.C. contibuted significantly to the conception, design, and analysis of the study. H.K. contributed significantly to the conception of the study, its design, and performed key experiments.
DISCLOSURES
The authors declare no conflicts of interest.
Supporting information
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health (Grants 5R01AI078287‐05 and T32AI095213‐01 to K.L.R.), services from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, and the Science Moving Towards Research Translation and Therapy program via Contract HHSN268201100015C. We thank John McCullough at the University of Massachusetts Medical School (UMMS) for advice and guidance on pharmacology and formulation, Alan Wolfe at the University of California, San Francisco, for providing the protocol for mass spectrometric analysis of K777 in plasma, and Scott Shaffer and Andre Kopoyan at the UMMS Proteomics and Mass Spectrometry Facility for standardization and performance of this analysis. In addition, we thank Havisha Karnam from the Brown and Khvorova laboratories and Myriam Aouadi from the Czech Laboratory at UMMS for help in the optimization of siRNA knockdowns.
Footnotes
SEE CORRESPONDING EDITORIAL ON PAGE 1
REFERENCES
- 1. Shen, H. , Kreisel, D. , Goldstein, D. R. (2013) Processes of sterile inflammation. J. Immunol. 191, 2857–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dostert, C. , Pétrilli, V. , Van Bruggen, R. , Steele, C. , Mossman, B. T. , Tschopp, J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hornung, V. , Bauernfeind, F. , Halle, A. , Samstad, E. O. , Kono, H. , Rock, K. L. , Fitzgerald, K. A. , Latz, E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Halle, A. , Hornung, V. , Petzold, G. C. , Stewart, C. R. , Monks, B. G. , Reinheckel, T. , Fitzgerald, K. A. , Latz, E. , Moore, K. J. , Golenbock, D. T. (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat. Immunol. 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cassel, S. L. , Eisenbarth, S. C. , Iyer, S. S. , Sadler, J. J. , Colegio, O. R. , Tephly, L. A. , Carter, A. B. , Rothman, P. B. , Flavell, R. A. , Sutterwala, F. S. (2008) The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 105, 9035–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martinon, F. , Pétrilli, V. , Mayor, A. , Tardivel, A. , Tschopp, J. (2006) Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- 7. Duewell, P. , Kono, H. , Rayner, K. J. , Sirois, C. M. , Vladimer, G. , Bauernfeind, F. G. , Abela, G. S. , Franchi, L. , Nuñez, G. , Schnurr, M. , Espevik, T. , Lien, E. , Fitzgerald, K. A. , Rock, K. L. , Moore, K. J. , Wright, S. D. , Hornung, V. , Latz, E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajamäki, K. , Lappalainen, J. , Oörni, K. , Välimäki, E. , Matikainen, S. , Kovanen, P. T. , Eklund, K. K. (2010) Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5, e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ashida, H. , Mimuro, H. , Ogawa, M. , Kobayashi, T. , Sanada, T. , Kim, M. , Sasakawa, C. (2011) Cell death and infection: a double‐edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miao, E. A. , Leaf, I. A. , Treuting, P. M. , Mao, D. P. , Dors, M. , Sarkar, A. , Warren, S. E. , Wewers, M. D. , Aderem, A. (2010) Caspase‐1‐induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brodsky, I. E. , Medzhitov, R. (2011) Pyroptosis: macrophage suicide exposes hidden invaders. Curr. Biol. 21, R72–R75. [DOI] [PubMed] [Google Scholar]
- 12. Nair, P. N. , Sjögren, U. , Sundqvist, G. (1998) Cholesterol crystals as an etiological factor in non‐resolving chronic inflammation: an experimental study in guinea pigs. Eur. J. Oral Sci. 106, 644–650. [DOI] [PubMed] [Google Scholar]
- 13. Ellson, C. D. , Dunmore, R. , Hogaboam, C. M. , Sleeman, M. A. , Murray, L. A. (2014) Danger‐associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 51, 163–168. [DOI] [PubMed] [Google Scholar]
- 14. Kono, H. , Rock, K. L. (2008) How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kono, H. , Karmarkar, D. , Iwakura, Y. , Rock, K. L. (2010) Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J. Immunol. 184, 4470–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dutta, D. , Moudgil, B. M. (2007) Crystalline silica particles mediated lung injury. Kona. 25, 76–87. [Google Scholar]
- 17. Kono, H. , Orlowski, G. M. , Patel, Z. , Rock, K. L. (2012) The IL‐1–dependent sterile inflammatory response has a substantial caspase‐1–independent component that requires cathepsin C. J. Immunol. 189, 3734–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen, C. J. , Kono, H. , Golenbock, D. , Reed, G. , Akira, S. , Rock, K. L. (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856. [DOI] [PubMed] [Google Scholar]
- 19. Bauernfeind, F. G. , Horvath, G. , Stutz, A. , Alnemri, E. S. , MacDonald, K. , Speert, D. , Fernandes‐Alnemri, T. , Wu, J. , Monks, B. G. , Fitzgerald, K. A. , Hornung, V. , Latz, E. (2009) Cutting edge: NF‐κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression.) Cutting edge: NF‐κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bauernfeind, F. , Bartok, E. , Rieger, A. , Franchi, L. , Núñez, G. , Hornung, V. (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mariathasan, S. , Newton, K. , Monack, D. M. , Vucic, D. , French, D. M. , Lee, W. P. , Roose‐Girma, M. , Erickson, S. , Dixit, V. M. (2004) Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 430, 213–218. [DOI] [PubMed] [Google Scholar]
- 22. Agostini, L. , Martinon, F. , Burns, K. , McDermott, M. F. , Hawkins, P. N. , Tschopp, J. (2004) NALP3 forms an IL‐1beta‐processing inflammasome with increased activity in Muckle‐Wells autoinflammatory disorder. Immunity 20, 319–325. [DOI] [PubMed] [Google Scholar]
- 23. Lamkanfi, M. , Dixit, V. M. (2012) Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161. [DOI] [PubMed] [Google Scholar]
- 24. Kayagaki, N. , Warming, S. , Lamkanfi, M. , Vande Walle, L. , Louie, S. , Dong, J. , Newton, K. , Qu, Y. , Liu, J. , Heldens, S. , Zhang, J. , Lee, W. P. , Roose‐Girma, M. , Dixit, V. M. (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- 25. Barlan, A. U. , Griffin, T. M. , McGuire, K. A. , Wiethoff, C. M. (2011) Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 85, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jacobson, L. S. , Lima, H., Jr. , Goldberg, M. F. , Gocheva, V. , Tsiperson, V. , Sutterwala, F. S. , Joyce, J. A. , Gapp, B. V. , Blomen, V. A. , Chandran, K. , Brummelkamp, T. R. , Diaz‐Griffero, F. , Brojatsch, J. (2013) Cathepsin‐mediated necrosis controls the adaptive immune response by Th2 (T helper type 2)‐associated adjuvants. J. Biol. Chem. 288, 7481–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brojatsch, J. , Lima, H. , Kar, A. K. , Jacobson, L. S. , Muehlbauer, S. M. , Chandran, K. , Diaz‐Griffero, F. (2014) A proteolytic cascade controls lysosome rupture and necrotic cell death mediated by lysosome‐destabilizing adjuvants. PLoS One 9, e95032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lima, H., Jr. , Jacobson, L. S. , Goldberg, M. F. , Chandran, K. , Diaz‐Griffero, F. , Lisanti, M. P. , Brojatsch, J. (2013) Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle 12, 1868–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bruchard, M. , Mignot, G. , Derangère, V. , Chalmin, F. , Chevriaux, A. , Végran, F. , Boireau, W. , Simon, B. , Ryffel, B. , Connat, J. L. , Kanellopoulos, J. , Martin, F. , Rébé, C. , Apetoh, L. , Ghiringhelli, F. (2013) Chemotherapy‐triggered cathepsin B release in myeloid‐derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64. [DOI] [PubMed] [Google Scholar]
- 30. Terada, K. , Yamada, J. , Hayashi, Y. , Wu, Z. , Uchiyama, Y. , Peters, C. , Nakanishi, H. (2010) Involvement of cathepsin B in the processing and secretion of interleukin‐1beta in chromogranin A‐stimulated microglia. Glia 58, 114–124. [DOI] [PubMed] [Google Scholar]
- 31. Newman, Z. L. , Leppla, S. H. , Moayeri, M. (2009) CA‐074Me protection against anthrax lethal toxin. Infect. Immun. 77, 4327–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dostert, C. , Guarda, G. , Romero, J. F. , Menu, P. , Gross, O. , Tardivel, A. , Suva, M. L. , Stehle, J. C. , Kopf, M. , Stamenkovic, I. , Corradin, G. , Tschopp, J. (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4, e6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Montaser, M. , Lalmanach, G. , Mach, L. (2002) CA‐074, but not its methyl ester CA‐074Me, is a selective inhibitor of cathepsin B within living cells. Biol. Chem. 383, 1305–1308. [DOI] [PubMed] [Google Scholar]
- 34. Mihalik, R. , Imre, G. , Petak, I. , Szende, B. , Kopper, L. (2004) Cathepsin B‐independent abrogation of cell death by CA‐074‐OMe upstream of lysosomal breakdown. Cell Death Differ. 11, 1357–1360. [DOI] [PubMed] [Google Scholar]
- 35. Klemenčič, I. , Carmona, A. K. , Cezari, M. H. , Juliano, M. A. , Juliano, L. , Guncar, G. , Turk, D. , Krizaj, I. , Turk, V. , Turk, B. (2000) Biochemical characterization of human cathepsin X revealed that the enzyme is an exopeptidase, acting as carboxymonopeptidase or carboxydipeptidase. Eur. J. Biochem. 267, 5404–5412. [DOI] [PubMed] [Google Scholar]
- 36. Bogyo, M. , Verhelst, S. , Bellingard‐Dubouchaud, V. , Toba, S. , Greenbaum, D. (2000) Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem. Biol. 7, 27–38. [DOI] [PubMed] [Google Scholar]
- 37. Orlowski, G. M. , Colbert, J. D. , Sharma, S. , Bogyo, M. , Robertson, S. A. , Rock, K. L. (2015) Multiple cathepsins promote pro–IL‐1β synthesis and NLRP3‐mediated IL‐1β activation. J. Immunol. 195, 1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Felbor, U. , Kessler, B. , Mothes, W. , Goebel, H. H. , Ploegh, H. L. , Bronson, R. T. , Olsen, B. R. (2002) Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc. Natl. Acad. Sci. USA 99, 7883–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vasiljeva, O. , Papazoglou, A. , Krüger, A. , Brodoefel, H. , Korovin, M. , Deussing, J. , Augustin, N. , Nielsen, B. S. , Almholt, K. , Bogyo, M. , Peters, C. , Reinheckel, T. (2006) Tumor cell‐derived and macrophage‐derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 66, 5242–5250. [DOI] [PubMed] [Google Scholar]
- 40. Stehlik, C. (2009) Multiple interleukin‐1beta‐converting enzymes contribute to inflammatory arthritis. Arthritis Rheum. 60, 3524–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joosten, L. A. , Netea, M. G. , Fantuzzi, G. , Koenders, M. I. , Helsen, M. M. , Sparrer, H. , Pham, C. T. , van der Meer, J. W. , Dinarello, C. A. , van den Berg, W. B. (2009) Inflammatory arthritis in caspase 1 gene‐deficient mice: contribution of proteinase 3 to caspase 1‐independent production of bioactive interleukin‐1β. Arthritis Rheum. 60, 3651–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fantuzzi, G. , Ku, G. , Harding, M. W. , Livingston, D. J. , Sipe, J. D. , Kuida, K. , Flavell, R. A. , Dinarello, C. A. (1997) Response to local inflammation of IL‐1 beta‐converting enzyme‐ deficient mice. J. Immunol. 158, 1818–1824. [PubMed] [Google Scholar]
- 43. Guma, M. , Ronacher, L. , Liu‐Bryan, R. , Takai, S. , Karin, M. , Corr, M. (2009) Caspase 1‐independent activation of interleukin‐1β in neutrophil‐predominant inflammation. Arthritis Rheum. 60, 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi, Y. , Evans, J. E. , Rock, K. L. (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521. [DOI] [PubMed] [Google Scholar]
- 45. Kuida, K. , Lippke, J. A. , Ku, G. , Harding, M. W. , Livingston, D. J. , Su, M. S. , Flavell, R. A. (1995) Altered cytokine export and apoptosis in mice deficient in interleukin‐1 beta converting enzyme. Science 267, 2000–2003. [DOI] [PubMed] [Google Scholar]
- 46. Kanneganti, T. D. , Ozören, N. , Body‐Malapel, M. , Amer, A. , Park, J. H. , Franchi, L. , Whitfield, J. , Barchet, W. , Colonna, M. , Vandenabeele, P. , Bertin, J. , Coyle, A. , Grant, E. P. , Akira, S. , Núñez, G. (2006) Bacterial RNA and small antiviral compounds activate caspase‐1 through cryopyrin/Nalp3. Nature 440, 233–236. [DOI] [PubMed] [Google Scholar]
- 47. Shi, G. P. , Villadangos, J. A. , Dranoff, G. , Small, C. , Gu, L. , Haley, K. J. , Riese, R. , Ploegh, H. L. , Chapman, H. A. (1999) Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity 10, 197–206. [DOI] [PubMed] [Google Scholar]
- 48. Nakagawa, T. , Roth, W. , Wong, P. , Nelson, A. , Farr, A. , Deussing, J. , Villadangos, J. A. , Ploegh, H. , Peters, C. , Rudensky, A. Y. (1998) Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science 280, 450–453. [DOI] [PubMed] [Google Scholar]
- 49. Deussing, J. , Roth, W. , Saftig, P. , Peters, C. , Ploegh, H. L. , Villadangos, J. A. (1998) Cathepsins B and D are dispensable for major histocompatibility complex class II‐mediated antigen presentation. Proc. Natl. Acad. Sci. USA 95, 4516–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pham, C. T. , Ley, T. J. (1999) Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc. Natl. Acad. Sci. USA 96, 8627–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jensen, B. M. , Swindle, E. J. , Iwaki, S. , Gilfillan, A. M. (2006). Generation, isolation, and maintenance of rodent mast cells and mast cell lines in Current Protocols Immunology John Wiley & Sons, Inc., New York, Unit 3.23. [DOI] [PubMed] [Google Scholar]
- 52. Gullapalli, R. , Wong, A. , Brigham, E. , Kwong, G. , Wadsworth, A. , Willits, C. , Quinn, K. , Goldbach, E. , Samant, B. (2012) Development of ALZET® osmotic pump compatible solvent compositions to solubilize poorly soluble compounds for preclinical studies.) Development of ALZET® osmotic pump compatible solvent compositions to solubilize poorly soluble compounds for preclinical studies. Drug Deliv. 19, 239–246. [DOI] [PubMed] [Google Scholar]
- 53. Vince, J. E. , Wong, W. W. , Gentle, I. , Lawlor, K. E. , Allam, R. , O'Reilly, L. , Mason, K. , Gross, O. , Ma, S. , Guarda, G. , Anderton, H. , Castillo, R. , Häcker, G. , Silke, J. , Tschopp, J. (2012) Inhibitor of apoptosis proteins limit RIP3 kinase‐dependent interleukin‐1 activation. Immunity 36, 215–227. [DOI] [PubMed] [Google Scholar]
- 54. Cai, Q. , Sun, H. , Peng, Y. , Lu, J. , Nikolovska‐Coleska, Z. , McEachern, D. , Liu, L. , Qiu, S. , Yang, C. Y. , Miller, R. , Yi, H. , Zhang, T. , Sun, D. , Kang, S. , Guo, M. , Leopold, L. , Yang, D. , Wang, S. (2011) A potent and orally active antagonist (SM‐406/AT‐406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J. Med. Chem. 54, 2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Conus, S. , Simon, H. U. (2008) Cathepsins: key modulators of cell death and inflammatory responses. Biochem. Pharmacol. 76, 1374–1382. [DOI] [PubMed] [Google Scholar]
- 56. Kaiser, W. J. , Upton, J. W. , Long, A. B. , Livingston‐Rosanoff, D. , Daley‐Bauer, L. P. , Hakem, R. , Caspary, T. , Mocarski, E. S. (2011) RIP3 mediates the embryonic lethality of caspase‐8‐deficient mice. Nature 471, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Verdoes, M. , Oresic Bender, K. , Segal, E. , van der Linden, W. A. , Syed, S. , Withana, N. P. , Sanman, L. E. , Bogyo, M. (2013) Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. J. Am. Chem. Soc. 135, 14726–14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen, Y. T. , Brinen, L. S. , Kerr, I. D. , Hansell, E. , Doyle, P. S. , McKerrow, J. H. , Roush, W. R. (2010) In vitro and in vivo studies of the trypanocidal properties of WRR‐483 against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 4, e825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Muñoz‐Planillo, R. , Kuffa, P. , Martínez‐Colón, G. , Smith, B. L. , Rajendiran, T. M. , Núñez, G. (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Oberle, C. , Huai, J. , Reinheckel, T. , Tacke, M. , Rassner, M. , Ekert, P. G. , Buellesbach, J. , Borner, C. (2010) Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak‐dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 17, 1167–1178. [DOI] [PubMed] [Google Scholar]
- 61. Fehrenbacher, N. , Gyrd‐Hansen, M. , Poulsen, B. , Felbor, U. , Kallunki, T. , Boes, M. , Weber, E. , Leist, M. , Jäättelä, M. (2004) Sensitization to the lysosomal cell death pathway upon immortalization and transformation. Cancer Res. 64, 5301–5310. [DOI] [PubMed] [Google Scholar]
- 62. Baskin‐Bey, E. S. , Canbay, A. , Bronk, S. F. , Werneburg, N. , Guicciardi, M. E. , Nyberg, S. L. , Gores, G. J. (2005) Cathepsin B inactivation attenuates hepatocyte apoptosis and liver damage in steatotic livers after cold ischemia‐warm reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G396–G402. [DOI] [PubMed] [Google Scholar]
- 63. Guicciardi, M. E. , Deussing, J. , Miyoshi, H. , Bronk, S. F. , Svingen, P. A. , Peters, C. , Kaufmann, S. H. , Gores, G. J. (2000) Cathepsin B contributes to TNF‐α‐mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 106, 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guicciardi, M. E. , Bronk, S. F. , Werneburg, N. W. , Gores, G. J. (2007) cFLIPL prevents TRAIL‐induced apoptosis of hepatocellular carcinoma cells by inhibiting the lysosomal pathway of apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1337–G1346. [DOI] [PubMed] [Google Scholar]
- 65. Foghsgaard, L. , Wissing, D. , Mauch, D. , Lademann, U. , Bastholm, L. , Boes, M. , Elling, F. , Leist, M. , Jäättelä, M. (2001) Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Di Piazza, M. , Mader, C. , Geletneky, K. , Herrero Y Calle, M. , Weber, E. , Schlehofer, J. , Deleu, L. , Rommelaere, J. (2007) Cytosolic activation of cathepsins mediates parvovirus H‐1‐induced killing of cisplatin and TRAIL‐resistant glioma cells. J. Virol. 81, 4186–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang, H. , Zhong, C. , Shi, L. , Guo, Y. , Fan, Z. (2009) Granulysin induces cathepsin B release from lysosomes of target tumor cells to attack mitochondria through processing of bid leading to Necroptosis. J. Immunol. 182, 6993–7000. [DOI] [PubMed] [Google Scholar]
- 68. Zhou, Z. D. , Yap, B. P. , Gung, A. Y. , Leong, S. M. , Ang, S. T. , Lim, T. M. (2006) Dopamine‐related and caspase‐independent apoptosis in dopaminergic neurons induced by overexpression of human wild type or mutant alpha‐synuclein. Exp. Cell Res. 312, 156–170. [DOI] [PubMed] [Google Scholar]
- 69. Michallet, M. C. , Saltel, F. , Flacher, M. , Revillard, J. P. , Genestier, L. (2004) Cathepsin‐dependent apoptosis triggered by supraoptimal activation of T lymphocytes: a possible mechanism of high dose tolerance. J. Immunol. 172, 5405–5414. [DOI] [PubMed] [Google Scholar]
- 70. Nylandsted, J. , Rohde, M. , Brand, K. , Bastholm, L. , Elling, F. , Jäättelä, M. (2000) Selective depletion of heat shock protein 70 (Hsp70) activates a tumor‐specific death program that is independent of caspases and bypasses Bcl‐2. Proc. Natl. Acad. Sci. USA 97, 7871–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ostenfeld, M. S. , Fehrenbacher, N. , Høyer‐Hansen, M. , Thomsen, C. , Farkas, T. , Jäättelä, M. (2005) Effective tumor cell death by sigma‐2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 65, 8975–8983. [DOI] [PubMed] [Google Scholar]
- 72. Nylandsted, J. , Gyrd‐Hansen, M. , Danielewicz, A. , Fehrenbacher, N. , Lademann, U. , Høyer‐Hansen, M. , Weber, E. , Multhoff, G. , Rohde, M. , Jäättelä, M. (2004) Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 200, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Niemi, K. , Teirilä, L. , Lappalainen, J. , Rajamäki, K. , Baumann, M. H. , Öörni, K. , Wolff, H. , Kovanen, P. T. , Matikainen, S. , Eklund, K. K. (2011) Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B‐sensitive pathway. J. Immunol. 186, 6119–6128. [DOI] [PubMed] [Google Scholar]
- 74. Meixenberger, K. , Pache, F. , Eitel, J. , Schmeck, B. , Hippenstiel, S. , Slevogt, H. , N'Guessan, P. , Witzenrath, M. , Netea, M. G. , Chakraborty, T. , Suttorp, N. , Opitz, B. (2010) Listeria monocytogenes‐infected human peripheral blood mononuclear cells produce IL‐1beta, depending on listeriolysin O and NLRP3. J. Immunol. 184, 922–930. [DOI] [PubMed] [Google Scholar]
- 75. Masters, S. L. , Dunne, A. , Subramanian, S. L. , Hull, R. L. , Tannahill, G. M. , Sharp, F. A. , Becker, C. , Franchi, L. , Yoshihara, E. , Chen, Z. , Mullooly, N. , Mielke, L. A. , Harris, J. , Coll, R. C. , Mills, K. H. , Mok, K. H. , Newsholme, P. , Nuñez, G. , Yodoi, J. , Kahn, S. E. , Lavelle, E. C. , O'Neill, L. A. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL‐1β in type 2 diabetes. Nat. Immunol. 11, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hentze, H. , Lin, X. Y. , Choi, M. S. , Porter, A. G. (2003) Critical role for cathepsin B in mediating caspase‐1‐dependent interleukin‐18 maturation and caspase‐1‐independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ. 10, 956–968. [DOI] [PubMed] [Google Scholar]
- 77. Heid, M. E. , Keyel, P. A. , Kamga, C. , Shiva, S. , Watkins, S. C. , Salter, R. D. (2013) Mitochondrial reactive oxygen species induces NLRP3‐dependent lysosomal damage and inflammasome activation. J. Immunol. 191, 5230–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rintahaka, J. , Lietzén, N. , Öhman, T. , Nyman, T. A. , Matikainen, S. (2011) Recognition of cytoplasmic RNA results in cathepsin‐dependent inflammasome activation and apoptosis in human macrophages. J. Immunol. 186, 3085–3092. [DOI] [PubMed] [Google Scholar]
- 79. Saito, M. , Nishikomori, R. , Kambe, N. , Fujisawa, A. , Tanizaki, H. , Takeichi, K. , Imagawa, T. , Iehara, T. , Takada, H. , Matsubayashi, T. , Tanaka, H. , Kawashima, H. , Kawakami, K. , Kagami, S. , Okafuji, I. , Yoshioka, T. , Adachi, S. , Heike, T. , Miyachi, Y. , Nakahata, T. (2008) Disease‐associated CIAS1 mutations induce monocyte death, revealing low‐level mosaicism in mutation‐negative cryopyrin‐associated periodic syndrome patients. Blood 111, 2132–2141. [DOI] [PubMed] [Google Scholar]
- 80. Fujisawa, A. , Kambe, N. , Saito, M. , Nishikomori, R. , Tanizaki, H. , Kanazawa, N. , Adachi, S. , Heike, T. , Sagara, J. , Suda, T. , Nakahata, T. , Miyachi, Y. (2007) Disease‐associated mutations in CIAS1 induce cathepsin B‐dependent rapid cell death of human THP‐1 monocytic cells. Blood 109, 2903–2911. [DOI] [PubMed] [Google Scholar]
- 81. Moore, S. F. , MacKenzie, A. B. (2009) NADPH oxidase NOX2 mediates rapid cellular oxidation following ATP stimulation of endotoxin‐primed macrophages. J. Immunol. 183, 3302–3308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files