

Short abstract
Peripheral blood neutrophils exhibit delayed spontaneous apoptosis in subjects with type 2 diabetes through an AGE/RAGE ligand/receptor‐mediated interaction.
Keywords: leukocyte, cell death, chronic inflammation
Abstract
The purpose of this study was to test the hypothesis that peripheral blood neutrophils (PMN) exhibit delayed spontaneous apoptosis in individuals with diabetes mellitus type 2 (T2DM) and that the delay is exacerbated further among people who coexpress chronic periodontitis (CP). Seventy‐three individuals were enrolled, including those with T2DM (n = 16), CP (n = 15), T2DM + CP (n = 21), and healthy volunteers (n = 21). PMN apoptosis was determined by flow cytometry using TUNEL and Annexin V assays. The activity of caspase‐3, ‐8, and ‐9 was measured by colorimetric assay. PMN surface death receptor quantification was performed by flow cytometry staining with fluorescence‐conjugated anti‐CD120a (TNFR1) and anti‐CD95 [Fas receptor (FasR)] antibody. Analysis of inflammatory markers in serum samples was performed using multiplexed sandwich immunoassays. In healthy volunteers and individuals with T2DM, CP, and T2DM + CP, spontaneous PMN apoptosis observed at 12 h reached 85.3 ± 3.1, 67.3 ± 3.9, 62.9 ± 3.5 and 62.5 ± 5.4%, respectively (P < 0.05). Caspase‐3 activity was significantly reduced in individuals with T2DM and T2DM + CP (P < 0.05) when compared with healthy volunteers. Caspase‐8 activity was also significantly decreased in CP and T2DM + CP (P < 0.05), associated with reduced cell‐surface FasR, TNFRs, and Fas ligand (FasL) serum levels. Glucose alone was not observed to impact PMN apoptosis; simultaneous incubation with the receptor for advanced glycation endproducts (RAGE) agonist S100B induced significant PMN apoptosis (P < 0.05). These data support the premise that the inhibition of PMN apoptosis in individuals with T2DM occurs through an advanced glycation endproducts/RAGE ligand/receptor‐mediated interaction.
Abbreviations
- APC
allophycocyanin
- BMI
body mass index
- CP
chronic periodontitis
- DM
diabetes mellitus
- FasL
Fas ligand
- FasR
Fas receptor
- HbA1c
hemoglobin A1c
- HG
hyperglycemia
- MOI
multiplicity of infection
- NG
normoglycemic
- PI
propidium iodide
- PMN
peripheral blood neutrophil
- pNA
para‐nitroaniline
- RAGE
receptor for advanced glycation endproducts
- sFasL
soluble Fas ligand
- T2DM
diabetes mellitus type 2
Introduction
DM is characterized by HG, resulting from defective insulin secretion, insulin action, or their combination [1, 2]. There are 2 forms of DM: type 1 and type 2, of which type 2 is the most common (>90% of cases are T2DM). Prolonged exposure to HG is the primary causal factor in diabetic complications [1].
CP is the most common periodontal disease caused by the extension of gingival inflammation to supporting periodontal tissues in response to bacteria colonizing the teeth [3]. The incidence and severity of CP in people with diabetes are influenced by the individual’s degree of diabetic control [4, 5]. The existence of severe generalized periodontitis in a person with DM may adversely affect glycemic control, serving as a source of chronic inflammation [6]. Mounting evidence suggests that poorly controlled DM correlates with a higher prevalence, severity, and progression rate of periodontal disease when compared with healthy individuals or those with controlled DM [7, 9, –, 11].
PMNs, or neutrophils, are the first line of host defense in the innate immune system. When infection or injury occurs, PMNs are mobilized to localize invading microorganisms and clear dead host cells and debris. Phagocyte function is altered in individuals with DM [12]; PMN phagocytosis, adherence, and chemotaxis are impaired, limiting the clearance of the pathogenic microbes in periodontal tissues and increasing tissue destruction, which further amplifies the systemic inflammatory load. These changes worsen glycemic control in patients with DM [13, 14]. PMNs are cleared from an inflammatory lesion through the initiation of spontaneous apoptosis (also called constitutive apoptosis) [15], which subsequently mediates the clearance of the apoptotic PMN by macrophages [15, –, 17]. The resolution of inflammation and restoration of homeostasis are dependent on efficient clearance of the PMN during the final phase [18]. Two major signaling pathways are involved in PMN apoptosis: the death receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway [17]. The intrinsic pathway regulates spontaneous apoptosis, whereas extrinsic apoptosis can be activated as a part of the inflammatory process [19] and is mediated by TNF‐α and FasL [20, 21].
A considerable amount of research has already been conducted that explores the alteration of PMN apoptosis in subjects with T2DM [22, 24, –, 26]; however, whether peripheral blood PMN from people with T2DM demonstrates defects in spontaneous apoptosis remains controversial. In this study, we test the hypothesis that spontaneous PMN apoptosis is delayed in T2DM and that coexpression with localized inflammatory conditions, such as CP, exacerbates delayed spontaneous PMN apoptosis.
MATERIALS AND METHODS
Reagents
S100B was purchased from Millipore Sigma (Temecula, CA, USA). RPMI‐1640 medium and FBS were obtained from American Type Culture Collection (Manassas, VA, USA). All other materials were obtained from Sigma Chemical (St. Louis, MO, USA).
Subject recruitment
In total, 73 individuals were recruited for this study. Subjects were grouped as follows: T2DM (n = 16), moderate to severe CP (n = 15), T2DM with moderate to severe CP (T2DM + CP; n = 21), and healthy volunteers (H; n = 21). All individuals were recruited from the Clinical Research Center at The Forsyth Institute. Subjects were qualified as healthy if they possessed NG status and had no systemic or local infections (e.g., periodontitis). All subjects provided informed consent, and the study was approved by The Forsyth Institute Institutional Review Board. Subjects with T2DM were selected according to the criteria of the American Diabetes Association [1] with no diabetic complications, and T2DM subjects with CP were selected according to the criteria of the American Academy of Periodontology [9]. Demographic data for all subject groups, including diabetes status, age, gender, race, smoking status, and BMI, were recorded ( Table 1 ).
Table 1.
Demographic characteristics of the study population
| Study group | ||||
|---|---|---|---|---|
| Characteristic | H | CP | T2DM | T2DM + CP |
| Sample size, n | 21 | 15 | 16 | 21 |
| Age | ||||
| means in yr ± sd | 38 ± 11 | 47 ± 9 | 57 ± 7 | 57 ± 11 |
| median | 37 | 48 | 59 | 59 |
| Gender | ||||
| Male | 11 | 9 | 7 | 15 |
| Female | 10 | 6 | 9 | 6 |
| Ethnicity | ||||
| African‐American | 5 | 6 | 5 | 13 |
| Caucasian | 14 | 8 | 9 | 5 |
| Asian | 2 | 1 | 3 | |
| Hispanic | 1 | 1 | ||
| BMI | ||||
| means ± sd | 27.0 ± 4.7 | 27.3 ± 4.5 | 32.5 ± 5.9 | 32.8 ± 6.9 |
| median | 27.7 | 27.4 | 32.2 | 31.5 |
| HbA1c | ||||
| means in % ± sd | 7.3 ± 1.4 | 7.4 ± 1.2 | ||
| median | 6.8 | 7.15 | ||
| Fasting blood glucose | ||||
| means in mg/dl ± sd | 204.1 ± 93.6 | 178.0 ± 53.4 | ||
| median | 189.5 | 160 | ||
| Duration of T2DM | ||||
| means in yr ± sd | 9.1 ± 5.9 | 7.3 ± 4.4 | ||
| median | 8 | 7 | ||
PMN isolation
Peripheral venous blood was collected into vacutainer tubes containing 10 U/ml heparin. PMNs were isolated using a discontinuous gradient system, as previously reported [27]. MonoPoly 1119 (3 ml) and 2 ml Histopaque 1077 were layered in 15 ml polystyrene culture tubes. Peripheral blood (4.5 ml) was layered on the separating medium, and the tubes were centrifuged at 1000 g for 15 min. The PMN‐enriched layers were collected, and contaminating RBCs were lysed with hypotonic NH4Cl buffer (155 mM NH4Cl, 10 mM KHCO3, 120 mM EDTA, pH 7.4). The isolated cells were washed twice with PBS. Cell preparations were routinely 99% PMN with ≥95% viability, as determined by Trypan blue exclusion.
Quantitative TUNEL assay
To analyze DNA fragmentation as a biochemical hallmark of apoptotic death in PMN, the TUNEL assay was performed. In brief, PMNs (1 × 106) were fixed with 1% (w/v) paraformaldehyde for 1 h on ice, washed in PBS, and postfixed in 70% (v/v) ethanol for 30 min on ice. Cell apoptosis was assessed by TUNEL assay. TUNEL staining of cells was performed with the APO‐DIRECT flow cytometry kit for apoptosis (Cat. No. APT110; Thermo Fisher Scientific, Waltham, MA, USA). Cells were resuspended with 1.0 ml wash buffer, twice. Cells were centrifuged, and supernatants were aspirated. Cells were incubated in DNA labeling solution for 60 min at 37°C. After incubation, 1.0 ml rinse buffer was added to each tube and centrifuged. Finally, the cell pellets were resuspended in 0.3 ml PI/RNase staining buffer and incubated without light for 30 min at room temperature. Flow cytometric analysis was performed to quantify the percentage of cells undergoing apoptosis on a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA) using CellQuest software (BD Biosciences).
Annexin V expression on PMN
An Annexin V‐FITC/PI apoptosis assay kit (BD PharMingen; BD Biosciences) and flow cytometry were used to measure Annexin V expression by PMN. The experiment was performed following the manufacturer’s instructions, with minor changes. In brief, after isolation or incubation with cell‐culture media, PMNs (1 × 106) were washed twice with ice‐cold PBS and then resuspended in binding buffer. Annexin V‐FITC and PI were added to the culture tube and incubated for 15 min without light. PMNs were analyzed by flow cytometry within 1 h of Annexin V‐PI labeling. Viable PMNs were defined as negative for Annexin V‐FITC and PI staining; apoptotic PMNs were defined as positive for Annexin V‐FITC staining but negative for PI staining. Cells positive for both Annexin V and PI staining were considered necrotic cells. Cell survival/apoptosis was expressed as a percentage of PMN relative to the total number of counted PMN.
Caspase activity in PMN
Caspase‐3, ‐8, and ‐9 protease activity was measured using the ApoTarget caspase‐3, ‐8, and ‐9 protease assay kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. The kit detects caspase proteolytic activity in lysates of mammalian cells. The caspase enzyme activity is recognized by amino acid sequences, including Asp‐Glu‐Val‐Asp (caspase‐3), Ile‐Glu‐thr‐Asp (caspase‐8), and Leu‐Glu‐His‐Asp (caspase‐9). The individual substrates provided for measuring the activity of these caspases are synthetic peptides, labeled at their C‐termini with pNA. Upon cleavage of the substrates by caspases, absorption of light by free pNA was quantified using a spectrophotometer. PMNs (1 × 106) were collected, washed with PBS, and suspended in 50 μl lysis buffer. Lysates were then centrifuged for 1 min at 10,000 g. The supernatant was incubated with substrate pNA and reaction buffer for 2 h at 37°C. Levels of the chromophore pNA released by caspase activity were spectrophotometrically quantified at 405 nm using a microplate reader (SpectraMax 340; Molecular Devices, Sunnyvale, CA, USA). The data were normalized for protein concentration through the bicinchoninic acid protein assay kit (Thermo Fisher Scientific).
PMN surface death receptor quantification
PMNs (1 × 106) were fixed with 1% paraformaldehyde at 4°C for 20 min, then resuspended in PBS, and kept at 4°C until used. Cells were incubated for 30 min at 4°C with 1% BSA in PBS to reduce nonspecific binding of antibodies and fluorochrome reagents. Subsequently, cells were stained with FITC‐conjugated mouse anti‐human CD120a (TNFR1) antibody (Thermo Fisher Scientific) and APC‐conjugated mouse anti‐human CD95 (FasR) antibody (BioLegend, San Diego, CA, USA) for 30 min without light, as recommended by the manufacturer. All samples were analyzed using a flow cytometer (FACScan) with CellQuest software.
Multiplex cytokine and inflammatory biomarker analysis
Whole blood (10 ml) was collected for serum separation in a nonheparinized tube. The sample was centrifuged at 1500 g for 15 min at 4°C, and the serum layer was aliquoted and frozen at −80°C. Multiplexed sandwich immunoassays, based on flow metric xMAP technology (Luminex, Austin, TX, USA), were conducted. Assays were carried out on a Bio‐Plex 200 platform (Bio‐Rad Laboratories, Hercules, CA, USA). Immediately before the initiation of study measurements, the Bio‐Plex platform underwent a complete on‐site maintenance cycle and operational calibration. Daily and weekly performance calibration was continuously verified. Assay kits were purchased from Thermo Fisher Scientific (Ultrasensitive kit).
Mimicking T2DM with CP delayed PMN apoptosis in healthy donors
To investigate further the mechanism of PMN apoptosis, diabetes was mimicked following the protocol of Omori et al. [28] with modifications. Healthy subject PMNs were cultured in diabetic conditions consisting of high glucose, with or without S100B, a ligand for RAGE. PMNs (1 × 106/ml) were cultured for 8 h in RPMI, supplemented with 5.5 mM glucose (NG) or 25 mM glucose (HG), with or without RAGE ligand S100B (50 µg/ml).
To induce an inflammatory response similar to one seen in an individual with T2DM + CP, Porphyromonas gingivalis (MOI 20) was coincubated with the PMN (1 × 106/ml) for 8 h in RPMI, supplemented with 25 mM glucose and S100B. P. gingivalis strain A7436 was cultured, as was described previously [29]. After 48 h of anaerobic growth in Wilkins‐Chalgren broth in an anaerobic chamber (85% N2, 5% H2, and 10% CO2), bacteria were harvested by centrifugation, washed 3 times with sterile pyrogen‐free saline, and opsonized with normal human serum for 30 min before coincubation with PMN. After 8 h, PMN apoptosis was analyzed by flow cytometry with an Annexin V‐FITC staining kit.
Statistical analysis
All data presented represent the average of at least 3 experiments with sem, except for demographic data, which used sd. For 2 group comparisons, Student’s t test was applied for significance; for multiple condition experiments, ANOVA was used with Bonferroni’s post hoc analysis.
RESULTS
Spontaneous PMN apoptosis in T2DM subjects
To measure spontaneous PMN apoptosis, we first compared the TUNEL and Annexin V methods. At 6 h, the Annexin V data demonstrated a 42.2% reduction in apoptosis in T2DM compared with the healthy group, and TUNEL showed a 55.3% reduction. This difference was also similar at 24 h; Annexin V demonstrated an 8.4% reduction in T2DM compared with the healthy group, and TUNEL showed a 17.2% reduction ( Fig. 1A ). Based on the similarities between methods, we chose TUNEL assay, as a result of the convenience of cell fixation, which does not require immediate analysis by FACS. This approach allowed us to measure apoptosis over the entire 24 h experimental period with 2 h intervals, without being dependent on the availability of the FACS device. In healthy subjects, spontaneous PMN apoptosis reached 50% in 7.4 h. By 12 h, PMN apoptosis had reached 85.3 ± 3.1%. In individuals with T2DM, spontaneous PMN apoptosis reached 50% in 8.5 h. By 12 h, PMN apoptosis had reached 67.3 ± 3.9%. Compared with PMN from healthy volunteers, circulating PMN from the T2DM group showed a significant delay of apoptosis in vitro culture after the 12 h mark (P = 0.031; Fig. 1B). Through this method, we were able to identify the shift at intermediate time points and assessed the time‐dependent change in apoptosis of cells.
Figure 1.
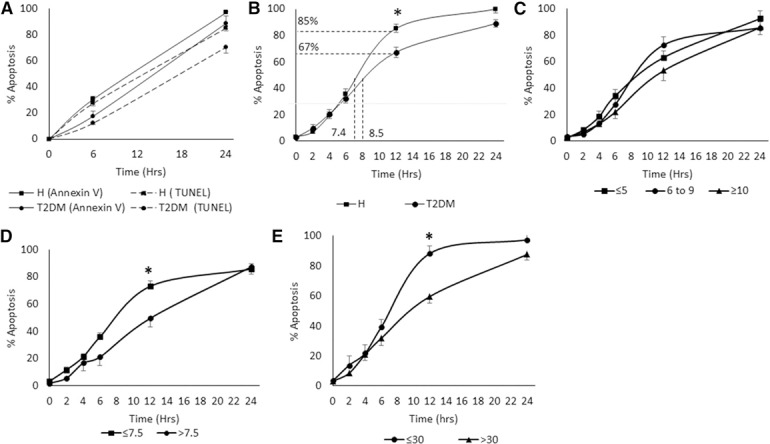
Comparison of spontaneous PMN apoptosis between healthy (H) and T2DM subjects.
PMNs (1 × 106 cells/ml) were resuspended and cultured in RPMI 1640, supplemented with 10% FBS at 37°C in a 5% CO2 atmosphere. (A) Comparison between TUNEL assay and Annexin V in measurement of PMN apoptosis. The results showed a similar trend between both methods. PMNs were collected at time points 0, 2, 4, 6, 12, and 24 h and were analyzed by TUNEL assay. (B) Spontaneous PMN apoptosis of healthy and T2DM subjects. A significant delay is seen in the T2DM group compared with the healthy group at the 12 h time point. (C) Effect of duration of diabetes on the spontaneous PMN apoptosis. Trends suggest a delay caused by increased disease duration but no significance. (D) Spontaneous PMN apoptosis in T2DM groups based on glycemic control. A significant delay is seen in the group with HbA1c >7.5 (n = 9) compared with the ≤7.5 group (n = 7) at the 12 h time point. (E) Effect of BMI on spontaneous PMN apoptosis in the T2DM group. A significant delay is observed in the group with BMI >30 (n = 6) compared with the ≤30 group (n = 10) at the 12 h time point. Error bars represent se. *P < 0.05 at the 12 h time point by Student’s t test.
Correlations between PMN apoptosis and characteristics of T2DM, such as glycemic control, duration of diabetes, and obesity control, revealed that in T2DM groups differentiated by the duration of diabetes (≤5, 6–9, and ≥10 yr), increased disease duration may cause a delay in PMN apoptosis, but findings were not significant (Fig. 1C). In T2DM groups differentiated by glycemic control (HbA1c ≤7.5%: good‐fair control; HbA1c >7.5%: poor control [30]), the poor control group exhibited a significant delay in PMN apoptosis when compared with the good‐fair control group following the 12 h mark (P = 0.041; Fig. 1D). When subjects were grouped by obesity control, where a BMI ≤30 includes ideal and overweight, and >30 is obese [31], a significant delay in spontaneous PMN apoptosis was seen in the obese group compared with the ideal‐overweight group at the 12 h mark (P = 0.006; Fig. 1E).
Spontaneous PMN apoptosis in patients with T2DM and CP
Several studies have previously reported delayed neutrophil apoptosis in chronic periodontal disease [32, 33]. Based on our results, which show a delay in spontaneous apoptosis of PMN in T2DM, we performed further investigations to determine if local infection and inflammation play a role in altering the PMN apoptosis profile in the presence of both T2DM and CP. The spontaneous PMN apoptosis profile for each group was measured by TUNEL assay ( Fig. 2 ). Individuals with T2DM + CP show a significant delay in spontaneous PMN apoptosis when compared with those in the healthy group. Spontaneous PMN apoptosis in the CP and T2DM + CP groups reached 50% by the 9.4 h mark, whereas PMN apoptosis in the healthy group reached 50% in 7.4 h. After 12 h, PMN apoptosis was 62.9 ± 3.5% in the CP group and 62.5 ± 5.4% in the T2DM + CP group, significantly lower than what was observed in the healthy group, where PMN apoptosis reached 85.3 ± 3.1% (P = 0.009 and P = 0.003, respectively). There was no difference among the CP, T2DM, and T2DM + CP groups, suggesting that CP and T2DM did not have additive effects on PMN apoptosis.
Figure 2.

Comparison of spontaneous PMN apoptosis among healthy (H), T2DM, CP, and T2DM + CP subjects.
PMNs (1 × 106 cells/ml) were resuspended and cultured in RPMI 1640, supplemented with 10% FBS at 37°C in a 5% CO2 atmosphere and were collected at time points 0, 2, 4, 6, 12, and 24 h. The collected PMNs were analyzed with TUNEL assay. A significant delay in apoptosis is observed in the CP and T2DM + CP compared with healthy groups at the 12 h time point. Healthy PMN reached 85.3 ± 3.1% apoptosis after 12 h. Meanwhile, CP and T2DM + CP PMN reached only 62.9 ± 3.5% and 62.5 ± 5.4% apoptosis, respectively, after 12 h. Error bars represent se. *P < 0.05 vs. healthy PMN by 1‐way ANOVA.
Caspase activity in patients with T2DM and CP
Our results demonstrated that spontaneous PMN apoptosis was delayed in patients with T2DM and CP. We further measured caspase enzyme activity, an essential downstream enzyme regulating the apoptosis cascade, with a caspase colorimetric assay. A time‐dependent analysis of caspase‐3 activity was performed at 2, 4, 6, 12, and 24 h. At a 12 h time point, caspase‐3 activity in patients with T2DM and T2DM + CP was decreased (P = 0.018 and P = 0.031, respectively; Fig. 3A ).
Figure 3.
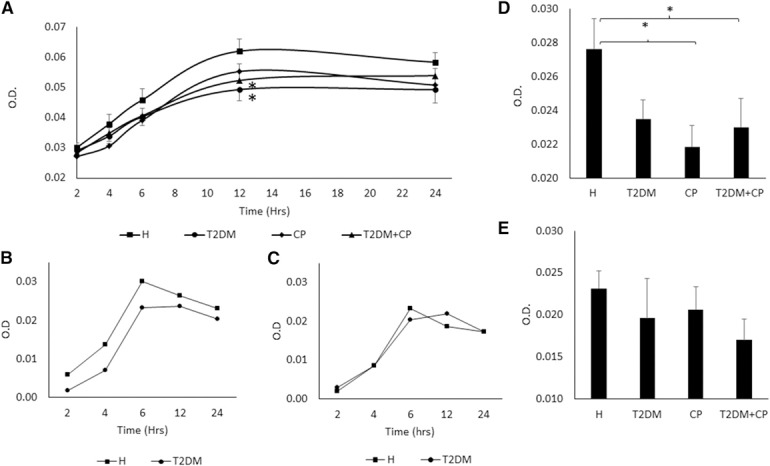
Comparison of caspase‐3, ‐8, and ‐9 activities among healthy (H), T2DM, CP, and T2DM + CP subjects.
PMNs (1 × 106 cells/ml) were resuspended and cultured in RPMI 1640, supplemented with 10% FBS at 37°C in a 5% CO2 atmosphere and were analyzed with a caspase protease assay kit. (A) Comparison of caspase‐3 activity among groups after PMN incubation for 0, 2, 4, 6, 12, and 24 h (n = 12). Healthy PMNs show significantly higher levels of caspase‐3 activity than the T2DM and T2DM + CP groups in 12 h. (B and C) Comparison of caspase‐8 and ‐9 activity between H and T2DM after PMN incubation for 0, 2, 4, 6, 12, and 24 h. The highest levels of caspase activity were observed in 6 h and gradually decreased over 24 h in both caspase‐8 and ‐9. (D) Caspase‐8 activity between healthy and diseased groups after 6 h (n = 5). A significant decrease in caspase‐8 activity is seen in CP and T2DM + CP compared with the H group. A decrease is also seen in the T2DM group but is not significant. (E) Comparison of caspase‐9 activity among groups after 6 h incubation (n = 5). No significant differences between healthy and diseased groups. Error bars represent se. *P < 0.05 vs. healthy PMN by 1‐way ANOVA.
We then determined whether the reduced caspase‐3 activity resulted from an upstream decrease in caspase‐9 activity, the initiator caspase enzyme representing the intrinsic (mitochondrial) pathway; an upstream decrease of caspase‐8 activity, representing the extrinsic (death receptor) pathway; or both. Firstly, we started by conducting a time‐point experiment to find the best time point to analyze the data. Fig. 3B and C showed that caspase‐8 and ‐9 activities, respectively, reached their maximum by 6 h and remained stable for the rest of the 24 h assay in both healthy and T2DM groups. A 6 h time point was then chosen for caspase‐8 and ‐9 investigation between H and all diseased groups. PMN caspase‐8 enzyme activity from the CP and T2DM + CP groups was significantly decreased compared with healthy subjects (P = 0.009 and P = 0.046, respectively; Fig. 3D). A decrease in caspase‐8 activity was also observed in the T2DM group. However, the result was not significant (P = 0.059). When compared with the healthy group, PMN from the T2DM, CP, and T2DM + CP groups was not observed to undergo a profound change in caspase‐9 activity (Fig. 3E).
Expression of cell‐surface death receptors and their ligands in PMN from subjects with T2DM and CP
To characterize further the mechanism of decrease in caspase‐8 in individuals from the T2DM + CP group, the expression of cell‐surface death receptors was measured. FasR (CD95) and TNFR1 (CD120a) activate caspase‐8 through cooperation with their adapter proteins. A drastic reduction of the cell‐surface FasR in PMN was observed from T2DM (P = 0.001) and T2DM + CP (P = 0.016) subjects when compared with healthy controls ( Fig. 4A ). The T2DM group also showed a significant decrease in cell‐surface TNFRs compared with healthy subjects (P = 0.005). Interestingly, in individuals with both T2DM and CP, the amount of cell‐surface TNFR expression was significantly restored compared with those solely afflicted with T2DM (P = 0.049; Fig. 4B).
Figure 4.

Cell surface death receptors expression and their ligands in T2DM and CP.
PMNs (1 × 106 cells) were fixed with 1% paraformaldehyde at 4°C. Cells were incubated with PBS with 1% BSA. Cells were then stained with FITC‐conjugated anti‐human CD120a (TNFR1) antibody and APC‐conjugated anti‐human CD95 (FasR). All samples were analyzed using a flow cytometer. (A) Cell‐surface FasR expression in healthy subjects (H), T2DM, CP, and T2DM + CP. A significant decrease in serum FasR expression is observed in T2DM and T2DM + CP compared with H (n = 6). (B) Cell‐surface TNFR expression in H, T2DM, CP, and T2DM + CP. A significant decrease in serum TNFR expression is observed in T2DM compared with H and T2DM + CP subjects (n = 6). Whole blood was collected for serum separation, and multiplexed sandwich immunoassays were conducted. (C) sFasL serum levels in H, T2DM, CP, and T2DM + CP. A significant decrease in serum sFasL levels is seen in T2DM + CP compared with H serum (n = 10). (D) TNF‐α serum levels in H, T2DM, CP, and T2DM + CP (n = 10). No difference is seen among groups. Error bars represent se. *P < 0.05 vs. healthy control by 1‐way ANOVA.
We also measured the levels of FasL and TNF‐α in serum. Results revealed a decrease in FasL in all T2DM groups compared with those in the healthy group. This finding was more pronounced in the T2DM + CP group, which presented a significantly lower level than the healthy group (P = 0.035; Fig. 4C). However, there was no significant difference in TNF‐α level between diseased and healthy subjects (Fig. 4D).
Mimicking T2DM with CP delayed PMN apoptosis in healthy donors
To investigate further the mechanism of PMN apoptosis, we adapted a model that we developed before. As described in Omori et al. [28], we cultured PMNs from healthy subjects in diabetic conditions in high glucose, supplemented with or without S100B. The results revealed that high glucose alone did not affect PMN apoptosis, but when incubated with high glucose and S100B, PMN apoptosis was delayed in a concentration‐dependent manner starting from 0, 5, 10, 20, and 50 μg/ml. The difference was significant when the concentration of S100B was 50 μg/ml (P = 0.007; Fig. 5A ). To simulate the CP and T2DM + CP in vitro, P. gingivalis was coincubated with PMN with high glucose and S100B (50 μg/ml) at MOI = 20 (Fig. 5B). Results from Annexin V staining indicated a significant delay in PMN apoptosis in response to P. gingivalis in NG, HG, and HG + S100B compared with normal glucose control (P = 0.002, P = 0.002, and P = 0.003, respectively).
Figure 5.

Mimicking T2DM and CP delays PMN apoptosis in healthy donors.
PMNs (1 × 106/ml) from healthy (H) subjects (n = 3) were cultured for 8 h in RPMI with 25 mM glucose (HG), with or without S100B (50 µg/ml) and with or without P. gingivalis. (A) Dose response of high glucose and S100B on spontaneous PMN apoptosis, analyzed by an Annexin V staining kit. A concentration of 50 µg/ml produced a significant decrease in the percentage of apoptosis. (B) Effect of HG, S100B, and P. gingivalis (P.g.; MOI 20) conditions on spontaneous PMN apoptosis, analyzed by the Annexin V staining kit. The combination of HG + S100B produced a significant decrease in spontaneous PMN apoptosis compared with the NG control. The addition of P. gingivalis to the NG, HG, and HG + S100B condition produced a dramatic decrease in PMN apoptosis compared with NG control. (C and D) PMNs were fixed with 1% paraformaldehyde at 4°C. Cells were incubated with PBS with 1% BSA. Cells were then stained with FITC‐conjugated anti‐human CD120a (TNFR1) antibody and APC‐conjugated anti‐human CD95 (FasR). All samples were analyzed using a flow cytometer. (C) Cell‐surface FasR expression. The combination of HG + S100B produced a significant decrease in cell‐surface FasR expression compared with NG and HG groups. The addition of P. gingivalis to NG, HG, and HG + S100B conditions produced a dramatic decrease in PMN surface FasRs compared with NG control. (D) Cell‐surface TNFR expression. By adding P. gingivalis to NG, HG, and HG + S100B conditions, significant decreases in PMN surface TNFRs compared with NG control were observed. Error bars represent se. *P < 0.05 vs. healthy control by 1‐way ANOVA.
Since our results showed a decrease in cell‐surface FasR and TNFR on PMN in the presence of T2DM, we then measured the impact of HG, S100B, and P. gingivalis on the expression of cell‐surface death receptors on PMN obtained from healthy donors. There was a significant reduction of the cell‐surface FasR in PMN incubated under HG when compared with normal glucose control (P = 0.025). After P. gingivalis was coincubated in NG, HG, and HG + S100B, there was a significant reduction in FasR expression when compared with normal glucose control (P = 0.025, P = 0.003, and P = 0.007, respectively; Fig. 5C). Cell‐surface TNFR in PMN, incubated under NG, HG, and HG with S100B, showed no significant differences. However, after P. gingivalis was coincubated in NG, HG, and HG + S100B, there was a substantial reduction in TNFR expression when compared with normal glucose control (P = 0.05, P = 0.031, and P = 0.01, respectively; Fig. 5D).
DISCUSSION
DM is a group of diseases characterized by chronic HG and other metabolic abnormalities. The majority of diabetic complications, including coronary artery disease, cerebrovascular and peripheral vascular disease, inhibited wound healing, and periodontitis, are now recognized as primarily stemming from prolonged exposure to HG [1, 12]. Dysfunctional PMNs with abnormal chemotaxis, adherence, and phagocytosis have been linked to T2DM as a potential mechanism for impaired host resistance to infection [34]. Functional abnormalities of PMN apoptosis would cause persistence at the site of infection, exacerbating a proinflammatory potential, furthering host tissue destruction, and ultimately causing delayed wound healing [35]. To date, research indicating that PMN apoptosis plays a crucial role in the resolution of inflammation in patients who have diabetes is still controversial. In this study, we measured the spontaneous PMN apoptotic response in patients with T2DM and found a significant delay in apoptosis compared with the healthy controls. The results of our research support the findings of Sudo et al. [22], who demonstrated a delayed PMN apoptosis in diabetic subjects compared with healthy controls through the measurement of the active caspase‐3‐positive rate. Higher amounts of anti‐apoptosis cytokines were shown to be produced from PMN in diabetic patients [23]. Other studies, performed in mice and rats, suggested that there was no significant difference between spontaneous PMN apoptosis in diabetic and nondiabetic groups [24, –, 26].
Based on our demographic data, there was an age difference among our study groups, but this factor has no apparent impact on PMN apoptosis, as demonstrated by Fülöp et al. [36], who compared PMN apoptosis of healthy, young with elderly subjects. They concluded that under unstimulated conditions, there were no significant differences between age groups. We also evaluated the correlations between PMN apoptosis and characteristics of T2DM. HbA1c, which is the gold‐standard measurement for the assessment of long‐term glycemic control, was used to evaluate the relationship between PMN apoptosis and diabetic control. Our results revealed that poor glycemic control greatly delayed PMN apoptosis. On the contrary, the duration of diabetes did not play a significant role in altering PMN apoptosis. Interestingly, when PMN apoptosis in T2DM subjects was related to the obesity index, higher BMI significantly delayed PMN apoptosis, whereas obese individuals showed reduced PMN apoptosis, regardless of their diabetic control. This result contradicted the previous literature, which reported no significant difference between half‐life of resting and unstimulated PMN from obese and lean subjects [37]. Our finding further emphasized the impact of T2DM in delaying PMN apoptosis, in which the combination of T2DM with obesity had a different impact on PMN compared with obesity alone. Further investigation is needed to explore the relationship between these two diseases.
PMN apoptosis has been shown to be delayed in chronic periodontal disease. Gamonal et al. [32] demonstrated that GM‐CSF and TNF‐α, responsible for inhibition of PMN apoptosis, were present in the gingival crevicular fluid collected from patients exhibiting CP. Likewise, other studies have observed a decrease in PMN apoptosis in subjects with periodontitis using a variety of methods, including measurement of in situ DNA breaks, electron microscopy, caspase‐3 measurement, and in vitro coculture of HL‐60‐derived PMN with P. gingivalis, a well‐known periodontal pathogen [33, 38, 39]. As CP was observed to increase anti‐apoptotic cytokines and delay PMN apoptosis at the local tissue level, we wanted to investigate further the impact that CP has on spontaneous PMN apoptosis at a systemic level, which is also complicated by chronic HG. Our results show a significant delay in spontaneous PMN apoptosis from subjects in the T2DM + CP group compared with healthy individuals; the effect was observed to be additive but not synergistic. This demonstrates that periodontal disease affects PMN apoptosis at the local site of periodontal infection but also systemically influences the resolution of inflammation and PMN clearance, which may intensify other systemic inflammatory conditions.
Our results demonstrate that spontaneous PMN apoptosis was delayed in subjects exhibiting T2DM and CP. These results were confirmed by measuring the caspase‐3 enzyme, a well‐known and reliable method for measuring apoptosis [40]. Caspase‐3 is a member of the cysteine‐aspartic acid protease family produced in the cascade pathway; it acts on protein substrates and plays a vital role in the execution phase of cell death by apoptosis. This process occurs before DNA fragmentation, a characteristic of apoptosis, which can be detected by the TUNEL assay, used in this study. As expected, our results show that healthy subjects exhibit the highest level of caspase‐3 activity when compared with diseased subjects. This reveals that delays in spontaneous PMN apoptosis in T2DM and CP occur via a caspase‐dependent pathway. In CP, no significant difference was found in caspase‐3 activity compared with the healthy group, although an analysis by the TUNEL assay showed a robust inhibition of apoptosis. This might be a result of a complex pathogenesis of periodontitis, which involves both a hyperinflammatory phenotype of PMN and bacterial infection. One study suggested an alternative and caspase‐independent, oxidant‐dependent apoptosis pathway in PMN in which excessive NADPH oxidase stimulation can generate copious amounts of superoxide and inhibit apoptosis. This mechanism could play a role in inhibiting PMN apoptosis in patients with CP and thus, lead to excessive production of reactive oxygen species [41]. Taken together with our previous findings [42], the data suggest that a delayed PMN apoptosis, associated with an increased superoxide formation in T2DM and CP, contributed to this pathology.
As a significant decrease in diseased subjects’ caspase‐3 activity was observed compared with healthy individuals, further investigation was undertaken to determine if the process of caspase‐3 activation was the result of an upstream increase in initiator caspase‐9 activity, representing the intrinsic pathway; an increase in caspase‐8 activity, representing the extrinsic pathway; or both. The findings demonstrate a significant increase in caspase‐8 activity in the healthy group compared with diseased groups. Caspase‐9 activity was observed to exhibit the same trend, but findings were not statistically significant. Taken together, these results demonstrate that delayed spontaneous PMN apoptosis in T2DM + CP is triggered by an inhibition of the caspase‐dependent pathway via down‐regulation of caspase activation by the death receptor pathway. The extracellular signals can be triggered by engaging the cell‐surface death receptor with their specific death ligands, which play a central role in apoptosis.
To investigate if a discrepancy exists between diseased and nondiseased groups in FasR and TNFR levels, cell‐surface death receptor expression was measured. The findings indicate that both TNFR and FasR are decreased in the T2DM group, but TNFR levels were restored in the T2DM + CP group. The levels of sFasL and TNF‐α in serum, separated from the subjects’ whole blood, were also assessed. Our results show no significant difference in the TNF‐α level in all groups, supporting the findings of Makino et al. [43], who observed no significant difference in serum TNF‐α levels between uncomplicated diabetic patients and healthy normal participants. A significant decrease in sFasL was noted in the T2DM + CP group when compared with the healthy controls. This finding is in line with existing literature, which has concluded that the sFasL is decreased or equivalent in T2DM subjects [44]. However, Guillot et al. [44] have reported an increase in the sFasL in diabetic patients, especially in patients with diabetic neuropathy.
To investigate further the mechanism of PMN apoptosis, a model mimicking T2DM was created by culturing healthy subject PMNs in diabetic conditions, modified from Omori et al. [28]. Our results demonstrate that high glucose levels did not affect spontaneous PMN apoptosis, similar to what has been reported in previous literature [45]. This indicates that HG may not be the main cause of delayed spontaneous apoptosis of PMN in T2DM subjects. However, our study also observed a significant delay in spontaneous PMN apoptosis in poor glycemic control T2DM subjects (HbA1c >7.5), and it is known that poor glycemic control can increase RAGE activation [46]. Therefore, it is possible that RAGE may play a role in delaying spontaneous PMN apoptosis in individuals afflicted with T2DM. There is strong existing evidence of RAGE involvement in delayed apoptosis in monocytes, which aggravates inflammation and alters PMN functions in diabetics [47, 48]. Therefore, we assessed if RAGE could be a potential factor in delaying PMN apoptosis by incubating healthy PMN with high glucose and the RAGE agonist S100B. The results of this investigation show that there is a delay in spontaneous PMN apoptosis in a concentration‐dependent manner in the presence of both high glucose levels and S100B. The results also show that when incubating healthy PMN with high glucose and S100B, cell‐surface FasR expression was decreased compared with normal glucose control. This may explain our results that show a decrease in the FasR on PMNs from T2DM subjects. To emulate the conditions of coexisting T2DM and CP on delayed PMN apoptosis in healthy donors, P. gingivalis was included with PMN in high glucose and S100B. The findings from this experiment demonstrate that apoptosis was delayed significantly compared with control, an impact similar to the result obtained in the T2DM + CP group. Our results support a previous paper by Preshaw et al. [49], who reported that the components of P. gingivalis, such as LPS and lipid A, delayed PMN apoptosis. In the PMN–P. gingivalis coincubation experiment, we were not able to obtain readable data using the TUNEL assay with FACS. Because as shown in Fig. 1A, Annexin V provides comparable outcomes, we had to choose the Annexin V assay over TUNEL. It is interesting that Annexin V has been used by other investigators studying the apoptosis in PMN and bacteria coculture experiments [50, 51].
In conclusion, we have shown that peripheral blood spontaneous PMN apoptosis was impaired in subjects with T2DM, and CP acts as a confounding factor. These findings provide a better understanding of a systemically impeded resolution of inflammation in both chronic inflammatory conditions investigated. Possible explanations for this occurrence include a lack of sFasL and its receptor expression on the PMN cell surface, and in patients with T2DM, that RAGE may play a crucial role in diminishing spontaneous PMN apoptosis.
AUTHORSHIP
A.M. designed and conducted the experiments, and wrote the draft of the paper. A.K. designed, wrote, and revised the paper. H.H. contributed in designing the paper and revised the text. D.S. worked with A.M. in executing the assays and revised the text. T.V.D. contributed in designing the paper and revised the text.
DISCLOSURES
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health, National Institute of Dental and Craniofacial Research Grants DE025020 and DE025383.
Contributor Information
Aggasit Manosudprasit, Email: akantarci@forsyth.org.
Alpdogan Kantarci, Email: akantarci@forsyth.org.
REFERENCES
- 1. American Diabetes Association . (2012) Diagnosis and classification of diabetes mellitus. Diabetes Care 35 (Suppl 1), S64–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kahn, B. B. , Flier, J. S. (2000) Obesity and insulin resistance. J. Clin. Invest. 106, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armitage, G. C. (1999) Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 4, 1–6. [DOI] [PubMed] [Google Scholar]
- 4. Nishimura, F. , Takahashi, K. , Kurihara, M. , Takashiba, S. , Murayama, Y. (1998) Periodontal disease as a complication of diabetes mellitus. Ann. Periodontol. 3, 20–9. [DOI] [PubMed] [Google Scholar]
- 5. Yalda, B. , Offenbacher, S. , Collins, J. G. (1994) Diabetes as a modifier of periodontal disease expression. Periodontol. 2000 6, 37–49. [DOI] [PubMed] [Google Scholar]
- 6. Grossi, S. G. , Genco, R. J. (1998) Periodontal disease and diabetes mellitus: a two‐way relationship. Ann. Periodontol. 3, 51–61. [DOI] [PubMed] [Google Scholar]
- 7. Demmer, R. T. , Desvarieux, M. , Holtfreter, B. , Jacobs, D. R., Jr. , Wallaschofski, H. , Nauck, M. , VÖlzke, H. , Kocher, T. (2010) Periodontal status and A1C change: longitudinal results from the Sstudy of Health in Pomerania (SHIP). Diabetes Care 33, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mealey, B. L. , Ocampo, G. L. (2007) Diabetes mellitus and periodontal disease. Periodontol. 2000 44, 127–153. [DOI] [PubMed] [Google Scholar]
- 9. Soskolne, W. A. , Klinger, A. (2001) The relationship between periodontal diseases and diabetes: an overview. Ann. Periodontol. 6, 91–98. [DOI] [PubMed] [Google Scholar]
- 10. Taylor, G. W. (2001) Bidirectional interrelationships between diabetes and periodontal diseases: an epidemiologic perspective. Ann. Periodontol. 6, 99–112. [DOI] [PubMed] [Google Scholar]
- 11. Taylor, G. W. , Borgnakke, W. S. (2008) Periodontal disease: associations with diabetes, glycemic control and complications. Oral Dis. 14, 191–203. [DOI] [PubMed] [Google Scholar]
- 12. Mealey, B. (1999) Diabetes and periodontal diseases. J. Periodontol. 70, 935–949. [DOI] [PubMed] [Google Scholar]
- 13. Manouchehr‐Pour, M. , Spagnuolo, P. J. , Rodman, H. M. , Bissada, N. F. (1981) Comparison of neutrophil chemotactic response in diabetic patients with mild and severe periodontal disease. J. Periodontol. 52, 410–415. [DOI] [PubMed] [Google Scholar]
- 14. McMullen, J. A. , Van Dyke, T. E. , Horoszewicz, H. U. , Genco, R. J. (1981) Neutrophil chemotaxis in individuals with advanced periodontal disease and a genetic predisposition to diabetes mellitus. J. Periodontol. 52, 167–173. [DOI] [PubMed] [Google Scholar]
- 15. Geering, B. , Simon, H. U. (2011) Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 18, 1457–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lockshin, R. A. , Williams, C. M. (1965) Programmed cell death‐I. Cytology of degeneration in the intersegmental muscles of the pernyi silkmoth. J. Insect Physiol. 11, 123–133. [DOI] [PubMed] [Google Scholar]
- 17. Peter, M. E. (2011) Programmed cell death: apoptosis meets necrosis. Nature 471, 310–312. [DOI] [PubMed] [Google Scholar]
- 18. Dibbert, B. , Weber, M. , Nikolaizik, W. H. , Vogt, P. , SchÖni, M. H. , Blaser, K. , Simon, H. U. (1999) Cytokine‐mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc. Natl. Acad. Sci. USA 96, 13330–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Savill, J. S. , Wyllie, A. H. , Henson, J. E. , Walport, M. J. , Henson, P. M. , Haslett, C. (1989) Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 83, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Renshaw, S. A. , Timmons, S. J. , Eaton, V. , Usher, L. R. , Akil, M. , Bingle, C. D. , Whyte, M. K. (2000) Inflammatory neutrophils retain susceptibility to apoptosis mediated via the Fas death receptor. J. Leukoc. Biol. 67, 662–668. [DOI] [PubMed] [Google Scholar]
- 21. Salamone, G. , Giordano, M. , Trevani, A. S. , Gamberale, R. , Vermeulen, M. , Schettinni, J. , Geffner, J. R. (2001) Promotion of neutrophil apoptosis by TNF‐alpha. J. Immunol. 166, 3476–3483. [DOI] [PubMed] [Google Scholar]
- 22. Sudo, C. , Ogawara, H. , Saleh, A. W. , Nishimoto, N. , Utsugi, T. , Ooyama, Y. , Fukumura, Y. , Murakami, M. , Handa, H. , Tomono, S. , Murakami, H. (2007) Clinical significance of neutrophil apoptosis in peripheral blood of patients with type 2 diabetes mellitus. Lab. Hematol. 13, 108–112. [PubMed] [Google Scholar]
- 23. Hatanaka, E. , Monteagudo, P. T. , Marrocos, M. S. , Campa, A. (2006) Neutrophils and monocytes as potentially important sources of proinflammatory cytokines in diabetes. Clin. Exp. Immunol. 146, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alba‐Loureiro, T. C. , de Lima, T. M. , Lagranha, C. J. , Mendonca, J. R. , Pithon‐Curi, T. C. , Curi, R. (2007) Apoptosis of neutrophils from diabetic rats. FASEB J. 21, A1346. [Google Scholar]
- 25. Tennenberg, S. D. , Finkenauer, R. , Dwivedi, A. (1999) Absence of lipopolysaccharide‐induced inhibition of neutrophil apoptosis in patients with diabetes. Arch. Surg. 134, 1229–1233, discussion 1233–1234. [DOI] [PubMed] [Google Scholar]
- 26. Hanses, F. , Park, S. , Rich, J. , Lee, J. C. (2011) Reduced neutrophil apoptosis in diabetic mice during staphylococcal infection leads to prolonged Tnfα production and reduced neutrophil clearance. PLoS One 6, e23633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujita, T. , Zawawi, K. H. , Kurihara, H. , Van Dyke, T. E. (2005) CD38 cleavage in fMLP‐ and IL‐8‐induced chemotaxis is dependent on p38 MAP kinase but independent of p44/42 MAP kinase. Cell. Signal. 17, 167–175. [DOI] [PubMed] [Google Scholar]
- 28. Omori, K. , Ohira, T. , Uchida, Y. , Ayilavarapu, S. , Batista, E. L., Jr. , Yagi, M. , Iwata, T. , Liu, H. , Hasturk, H. , Kantarci, A. , Van Dyke, T. E. (2008) Priming of neutrophil oxidative burst in diabetes requires preassembly of the NADPH oxidase. J. Leukoc. Biol. 84, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Genco, C. A. , Cutler, C. W. , Kapczynski, D. , Maloney, K. , Arnold, R. R. (1991) A novel mouse model to study the virulence of and host response to Porphyromonas (bacteroides) gingivalis. Infect. Immun. 59, 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. American Diabetes Association . (2014) Standards of medical care in diabetes—2014. Diabetes Care 37 (Suppl 1), S14–S80. [DOI] [PubMed] [Google Scholar]
- 31. Kuczmarski, R. J. , Flegal, K. M. (2000) Criteria for definition of overweight in transition: background and recommendations for the United States. Am. J. Clin. Nutr. 72, 1074–1081. [DOI] [PubMed] [Google Scholar]
- 32. Gamonal, J. , Sanz, M. , O'Connor, A. , Acevedo, A. , Suarez, I. , Sanz, A. , Martínez, B. , Silva, A. (2003) Delayed neutrophil apoptosis in chronic periodontitis patients. J. Clin. Periodontol. 30, 616–623. [DOI] [PubMed] [Google Scholar]
- 33. Murray, D. A. , Wilton, J. M. (2003) Lipopolysaccharide from the periodontal pathogen Porphyromonas gingivalis prevents apoptosis of HL60‐derived neutrophils in vitro. Infect. Immun. 71, 7232–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geerlings, S. E. , Hoepelman, A. I. (1999) Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol. Med. Microbiol. 26, 259–265. [DOI] [PubMed] [Google Scholar]
- 35. Kim, M. H. , Liu, W. , Borjesson, D. L. , Curry, F. R. , Miller, L. S. , Cheung, A. L. , Liu, F. T. , Isseroff, R. R. , Simon, S. I. (2008) Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J. Invest. Dermatol. 128, 1812–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. FülÖp, T., Jr. , Fouquet, C. , Allaire, P. , Perrin, N. , Lacombe, G. , Stankova, J. , Rola‐Pleszczynski, M. , Gagné, D. , Wagner, J. R. , Khalil, A. , Dupuis, G. (1997) Changes in apoptosis of human polymorphonuclear granulocytes with aging. Mech. Ageing Dev. 96, 15–34. [DOI] [PubMed] [Google Scholar]
- 37. Trottier, M. D. , Naaz, A. , Kacynski, K. , Yenumula, P. R. , Fraker, P. J. (2012) Functional capacity of neutrophils from class III obese patients. Obesity (Silver Spring) 20, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 38. Gamonal, J. , Bascones, A. , Acevedo, A. , Blanco, E. , Silva, A. (2001) Apoptosis in chronic adult periodontitis analyzed by in situ DNA breaks, electron microscopy, and immunohistochemistry. J. Periodontol. 72, 517–525. [DOI] [PubMed] [Google Scholar]
- 39. Bulut, S. , Uslu, H. , Ozdemir, B. H. , Bulut, O. E. (2006) Expression of caspase‐3, p53 and Bcl‐2 in generalized aggressive periodontitis. Head Face Med. 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Danial, N. N. , Korsmeyer, S. J. (2004) Cell death: critical control points. Cell 116, 205–219. [DOI] [PubMed] [Google Scholar]
- 41. Fadeel, B. , Ahlin, A. , Henter, J. I. , Orrenius, S. , Hampton, M. B. (1998) Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood 92, 4808–4818. [PubMed] [Google Scholar]
- 42. Karima, M. , Kantarci, A. , Ohira, T. , Hasturk, H. , Jones, V. L. , Nam, B. H. , Malabanan, A. , Trackman, P. C. , Badwey, J. A. , Van Dyke, T. E. (2005) Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J. Leukoc. Biol. 78, 862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Makino, N. , Maeda, T. , Sugano, M. , Satoh, S. , Watanabe, R. , Abe, N. (2005) High serum TNF‐alpha level in type 2 diabetic patients with microangiopathy is associated with eNOS down‐regulation and apoptosis in endothelial cells. J. Diabetes Complications 19, 347–355. [DOI] [PubMed] [Google Scholar]
- 44. Guillot, R. , Bringuier, A. F. , Porokhov, B. , Guillausseau, P. J. , Feldmann, G. (2001) Increased levels of soluble Fas in serum from diabetic patients with neuropathy. Diabetes Metab. 27, 315–321. [PubMed] [Google Scholar]
- 45. Turina, M. , Miller, F. N. , Tucker, C. , Polk, H. C. (2006) Effects of hyperglycemia, hyperinsulinemia, and hyperosmolarity on neutrophil apoptosis. Surg. Infect. (Larchmt) 7, 111–121. [DOI] [PubMed] [Google Scholar]
- 46. Motawi, T. M. , Abou‐Seif, M. A. , Bader, A. M. , Mahmoud, M. O. (2013) Effect of glycemic control on soluble RAGE and oxidative stress in type 2 diabetic patients. BMC Endocr. Disord. 13, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Collison, K. S. , Parhar, R. S. , Saleh, S. S. , Meyer, B. F. , Kwaasi, A. A. , Hammami, M. M. , Schmidt, A. M. , Stern, D. M. , Al‐Mohanna, F. A. (2002) RAGE‐mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs). J. Leukoc. Biol. 71, 433–444. [PubMed] [Google Scholar]
- 48. Hou, F. F. , Miyata, T. , Boyce, J. , Yuan, Q. , Chertow, G. M. , Kay, J. , Schmidt, A. M. , Owen, W. F. (2001) beta(2)‐Microglobulin modified with advanced glycation end products delays monocyte apoptosis. Kidney Int. 59, 990–1002. [DOI] [PubMed] [Google Scholar]
- 49. Preshaw, P. M. , Schifferle, R. E. , Walters, J. D. (1999) Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J. Periodontal Res. 34, 197–202. [DOI] [PubMed] [Google Scholar]
- 50. Perskvist, N. , Long, M. , Stendahl, O. , Zheng, L. (2002) Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase‐3 and altering expression of Bax/Bcl‐xL via an oxygen‐dependent pathway. J. Immunol. 168, 6358–6365. [DOI] [PubMed] [Google Scholar]
- 51. Brest, P. , Bétis, F. , Cuburu, N. , Selva, E. , Herrant, M. , Servin, A. , Auberger, P. , Hofman, P. (2004) Increased rate of apoptosis and diminished phagocytic ability of human neutrophils infected with Afa/Dr diffusely adhering Escherichia coli strains. Infect. Immun. 72, 5741–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]