

Short abstract
Review on gut microbiota regulation of development and function of myeloid cells at distant sites and the impact on response to infection and cancer therapy.
Keywords: systemic immunity, host–microbial interactions, cancer, infection, mononuclear phagocytes
Abstract
The gut microbiota is a complex and dynamic microbial ecosystem that plays a fundamental role in host physiology. Locally, the gut commensal microbes/host symbiotic relationship is vital for barrier fortification, nutrient absorption, resistance against intestinal pathogens, and the development and maintenance of the mucosal immune system. It is now clear that the effects of the indigenous intestinal flora extend beyond the gut, ranging from shaping systemic immune responses to metabolic and behavioral functions. However, the underlying mechanisms of the gut microbiota/systemic immune system interactions remain largely unknown. Myeloid cells respond to microbial signals, including those derived from commensals, and initiate innate and adaptive immune responses. In this review, we focus on the impact of the gut microbiota on myeloid cells at extraintestinal sites. In particular, we discuss how commensal‐derived signals affect steady‐state myelopoiesis and cellular function and how that influences the response to infection and cancer therapy.
Abbreviations
- Abx
= antibiotics
- BM
= bone marrow
- CDP
= common dendritic cell precursor
- CMP
= common myeloid progenitor
- CpG‐ODN
= CpG‐oligodeoxynucleotide
- CT
= cholera toxin
- CTX
= cyclophosphamide
- DC
= dendritic cell
- GF
= germ‐free
- GMP
= granulocyte‐macrophage progenitor
- GPR
= G‐protein‐coupled receptor
- HSC
= hematopoietic stem cell
- HSPC
= hematopoietic stem and progenitor cell
- IBD
= inflammatory bowel disease
- Jax
= The Jackson Laboratory
- LDC
= lung dendritic cell
- LOS
= late‐onset sepsis
- MP
= mononuclear phagocyte
- MPP
= multipotent progenitor
- Nod1/2
= nucleotide‐binding oligomerization domain‐containing protein 1/2
- PAMP
= pathogen‐associated molecular pattern
- PD‐L1
= programmed death ligand 1
- PRR
= pattern recognition receptor
- pTh17
= pathogenic effector Th17 cells
- RNS
= reactive nitrogen species
- ROS
= reactive oxygen species
- SAA
= serum amyloid A
- SCFA
= short‐chain fatty acid
- SPF
= specific pathogen‐free
- Tac
= Taconic Farms
- TIV
= trivalent‐inactivated vaccine
Introduction
The human microbiota refers to the complex communities of bacteria, fungi, archea, and viruses that inhabit the human body. This dynamic microbial ecosystem forms a symbiotic relationship with the host and shapes its physiology [1, 2]. An estimated 100 trillion microbial cells colonize all surfaces of the body that are exposed to the environment, such as the skin, oral cavity, genitourinary, gastrointestinal, and respiratory tracts, outnumbering the host cells by at least 3–10 times [3, 4, 5–6]. The collective microbial genomes, referred to as the microbiome, are estimated to contain 100 times more genes than the human genome, representing an integral part of our genetic landscape [6, 7].
Our partnership with this diverse microbial community has been appreciated for almost as long as the discovery of bacteria themselves, dating back to the pioneer work of Antonie van Leeuwenhoek in the 1680s. In his letters to the Royal Society of London, van Leeuwenhoek described his observations about little animals—“animalcules”—present on tooth‐surface scrapings and other parts of the body from himself and other individuals of different age and health status [8]. Ever since, scientists have been interested in the consequences of the microbial/host relationship, and the development of GF animals—born and raised free of microorganisms—during the first half of the 20th century has allowed investigators to uncover many of them [9]. However, the renewed interest in the microbiome research field ignited by recent technological advances and lower costs of sequencing techniques, together with the development of bioinformatics tools, has furthered our understanding of the role of the microbiota in health and disease [10, 11, 12, 13–14].
The vast majority of commensal microbes includes bacterial cells residing in the gut and dominated by members of the phyla Bacteroidetes and Firmicutes [15]. Recent studies suggested that the human gut microbiota can be classified in 3 enterotypes, mostly based on species composition [16]. Nevertheless, much like fingerprints, each individual's microbiota is unique, and its composition is determined by a plethora of factors, including the mode of delivery, diet, lifestyle, use of Abx, and host genetic and environmental factors [17, 18, 19, 20, 21–22].
Whereas the role of the gut microbiota on nutrient absorption has long been recognized, new studies reveal a much farther reach of the gut microbiota on the host's physiology, ranging from metabolic to behavioral and cognitive functions, and a critical role in the development of the immune system [23, 24–25]. In fact, for over millions of years of coevolution, the microbiota and host immune system developed an interdependent relationship, where the innate and adaptive arms of the immune system rely on their microbial partners for proper maturation and function, while the immune system stabilizes and contains the microbial ecosystem [24, 26, 27–28]. Changes in the microbiota composition—commonly referred to as dysbiosis—have been linked to numerous disease states, many of which are mediated by altered host immune and inflammatory responses [2, 29]. The role of commensal microbes in shaping the immune response within the intestinal microenvironment has been studied extensively [30]; however, the mechanism by which gut commensals regulate systemic immunity is only beginning to be uncovered. Myeloid cells are equipped to sense microbial‐derived products, they play an instrumental role in host defense, and they are a major component of the tumor microenvironment, thereby performing a key role in the host/commensals dialogue. In this review, we will discuss the distant effects of the gut microbiota on myeloid cell development and function during steady state and response to infection and cancer therapy.
MPs
MPs, e.g., monocytes, macrophages, and DCs, are major effector cells in inflammatory and immune responses [31]. These myeloid cells derive from committed precursors in the BM and populate virtually all lymphoid and nonlymphoid tissues, where they play crucial roles in the maintenance of homeostasis, the initiation of innate and adaptive immunity, the resolution of inflammation, and tissue repair. Although MPs and neutrophils share several phenotypic and functional characteristics (e.g., exceptional phagocytic activity and microbicidal mechanisms), even within the individual subpopulations, they are heterogeneous and developmentally and functionally distinct [32, 33, 34–35]. Rather than redundant, they act in concert to eliminate potential insults and restore tissue integrity while minimizing collateral damage to self [36]: tissue resident macrophages conduct local surveillance and maintain homeostasis, whereas neutrophils and monocytes are rapidly recruited to inflamed tissues and perform effector functions in pathologic settings [31, 33]; DCs are unique in their ability to prime naïve antigen‐specific T cells and guide their effector choice [32]. Neutrophils, macrophages, DCs and monocytes can also exert immunosuppressive functions [32, 33, 37].
For the most part, MPs are short lived and are constantly replenished by BM‐derived precursors. However, the traditional view in which all tissue resident MPs originate from circulating monocytes and therefore, depend on input from BM stem and progenitor cells, has recently changed. Studies in mice showed that Langerhans cells in the skin and tissue resident macrophage populations (e.g., in spleen, liver, lung, kidney, and brain) derive from embryonic precursors, are long lived, and capable of self‐renewal throughout the adult life [38, 39, 40–41]. Although certain resident populations are exclusively of embryonic origin (e.g., microglia in the brain), tissues such as the intestinal lamina propria and the dermis, rely entirely on monocyte replacement, whereas others show macrophages of mixed origin. In addition, circulating monocytes can contribute to the resident pools following pathologic conditions.
MYELOPOIESIS
In an oversimplified view, MPs develop within the classic hierarchical hematopoiesis tree in the following sequence of events: long‐lived, self‐renewing HSCs give rise to short‐lived HSCs and MPPs and together, constitute the HSPC population; MPPs differentiate into common lymphoid progenitors that would give rise to lymphoid progeny, and into CMPs. CMPs give rise to GMPs, megakaryocyte‐erythrocyte progenitors, macrophage and DC progenitors, and CDPs. GMPs give origin to neutrophils and other granulocyte populations and to monocytes. Circulating monocytes can further differentiate into macrophages and monocyte‐derived DCs in different tissues, particularly under inflammatory circumstances [42]. In addition to the BM, increased numbers of circulating HSPCs during inflammation can generate extramedullary myelopoiesis in peripheral tissues, such as liver, lung, and spleen [43, 41, 42, 43, 44, 45–46].
Myelopoiesis is tightly regulated by transcription factors and extrinsic signals to ensure homeostasis maintenance during steady state and to provide a rapid cell output to meet the increased demand of pathologic conditions, such as infection, inflammation, and cancer [42, 43, 47, 48–49]. The latter is commonly referred to as “demand adapted” or “emergency” hematopoiesis [42]. However, in addition to cytokine, chemokine, and myelopoietic growth factor receptors, both mouse and human HSPCs express PRRs, such as TLRs [42, 50, 51, 52, 53–54], and can directly sense pathogens via binding of PAMPs. Therefore, rather than a compensatory mechanism, dysregulated myelopoiesis may represent the initial step of inflammatory and stress responses [43, 45].
MICROBIOTA REGULATION OF MYELOPOIESIS
Several lines of evidence demonstrate that the hematopoietic compartment directly participates in the response to infection and that different infectious stimuli shape the appropriate myelopoietic response needed to fight the specific pathogens [46, 52, 55, 56–57]. Thus, it is plausible that the same cells may sense signals derived from the distant gut microbiota. Systemic administration of LPS has been shown to induce HSPC mobilization and homing to the spleen [58], as well as proliferation and enhanced self‐renewal capacity of HSCs in the BM [59], suggesting that commensal‐derived products can reach the BM and modulate myelopoiesis.
In the 1970s and early 1980s, several studies in dogs and mice, using Abx and GF animals in the case of rodents, implicated gut commensal microbes in the regulation of BM granulopoiesis, with a particular emphasis on the role of gram‐negative bacteria and endotoxin levels [60, 61, 62, 63–64]. These observations were later confirmed by Tada et al. [65], who observed decreased numbers of granulocytes in the BM and blood of kanamycin‐treated mice, whereas lymphocyte subsets were not affected. In the same study, the authors reported an overall reduction in the number of mononuclear cells and in particular, granulocytes, in the BM of GF mice. Goris et al. [63] observed decreased levels of splenic and BM progenitor cells forming GM‐CFU colonies in polymyxin‐treated and GF mice compared with SPF animals. However, in a different study, GF animals displayed normal levels of M‐CSF‐responding progenitors in the BM but reduced levels in the spleen, as evidenced by similar or lower numbers of monocyte/macrophage colonies (M‐CFU), respectively [66]. In the latter study, association of GF mice with a complete flora or with Escherichia coli alone was sufficient to correct the defect observed in GF mice, pointing to the role of gram‐negative bacteria in regulating myelopoiesis.
In the current microbiome era, studies with newly available and more sophisticated tools highlight again the preponderant role of the gut microbiota in regulating myelopoiesis. In line with earlier observations, Khosravi et al. [67] have recently reported that GF mice show a global defect in MPs and neutrophils in the BM, spleen, and liver. Splenic resident F4/80hi macrophages are, for the most part, derived from embryonic precursors, whereas F4/80lo populations derive from BM HSCs [38, 68]. Interestingly, in the spleen of GF mice, both populations were reduced in number, suggesting that the commensal microbiota affects myeloid cell progenitors of both embryonic and BM origin [67]. The proportion and differentiation potential of HSCs, MPPs, and CMPs were not affected in GF mice compared with SPF animals; however, the proportion of GMPs was significantly reduced in BM from GF mice ( Fig. 1 ). In addition, GMPs from GF mice gave rise to lower numbers of granulocyte and monocyte/macrophage colonies in response to G‐CSF or M‐CSF, respectively. Similar to BM GMPs, splenic precursors from GF animals yielded fewer neutrophils and monocytes. Further analysis of the commensal bacteria‐derived signals involved in the regulation of myelopoiesis revealed that oral treatment of GF mice with SCFAs—end‐products of bacterial fermentation of dietary fibers that have been shown to be immune modulators [69]—did not change the frequencies of neutrophils or monocytes in the BM or of F4/80hi resident splenic macrophages. However, oral administration of PAMPs in the form of heat‐killed E. coli or autoclaved cecal contents from SPF mice was sufficient to recover the granulocytic and monocytic progeny of BM GMPs from GF mice. Nevertheless, a live complex flora was required to restore myelopoiesis completely, including the expansion of embryonic‐derived F4/80hi cells [67].
Figure 1.
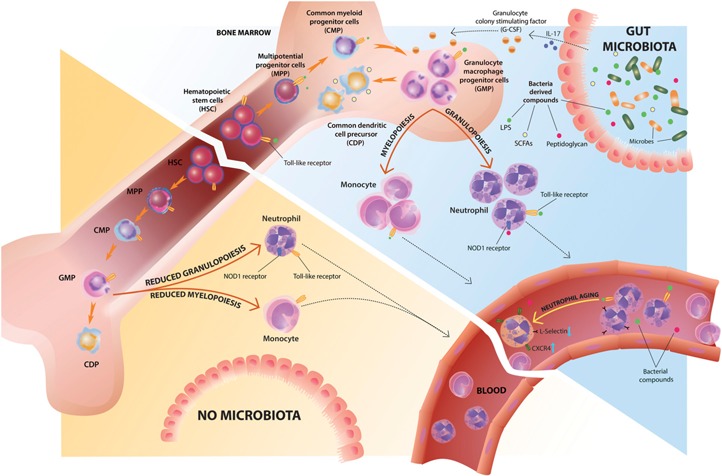
Microbiota regulation of steady‐state myelopoiesis and neutrophil homeostasis. HSPCs and myeloid progenitors express PRRs that enable them to sense bacteria‐derived products directly. Commensal‐derived signals modulate myelopoiesis, mostly by affecting GMP frequencies and differentiation potential. The gut microbiota promotes granulopoiesis, at least in part, through the local induction of IL‐17 production and increased plasma levels of G‐CSF. The absence of gut commensal microbiota leads to reduced numbers and differentiation potential of GMPs, leading to reduced neutrophil, monocyte, and macrophage numbers in the BM and peripheral tissues. Bacterial metabolites, such as SCFAs, modulate DC differentiation by increasing the numbers of CDPs. Commensal microbes also regulate neutrophil function in the BM through direct recognition of microbial‐derived peptidoglycan via the Nod1 receptor. Circulating neutrophil aging is driven by microbiota‐derived signals via TLR/MyD88 signaling, and absence of commensals leads to reduced numbers of CXCR4hi/L‐Selectinlow‐aged neutrophils in circulation.
A previous study showed that in steady‐state conditions, MyD88 (a key adaptor molecule for the recognition of PAMPs through TLR signaling) deficiency also leads to decreased numbers of neutrophils, monocytes, and macrophages in the spleen and a reduced myeloid compartment in the BM, mostly as a result of a reduction in granulocytes [70]. As observed by Khosravi et al. [67], GMPs obtained from MyD88‐deficient BM yielded fewer granulocytic and monocytic colonies. Although changes in HSPC proportions in GF mice were not apparent in the study by Khosravi et al. [67], MyD88‐deficient mice showed reduced numbers and a decreased proliferation rate of HSCs in the BM [70]. In agreement with the latter observation, reduced absolute numbers of HSPCs in BM from GF animals were also observed in a recent study [71]. Thus, MyD88‐dependent TLR signaling, induced by commensal microbes or possibly endogenous ligands as well, can impact early hematopoietic development and terminal myeloid differentiation potential (Fig. 1).
In their study, Balmer et al. [71] showed that incrementing the microbiota degree of complexity leads to consistently increased numbers of granulocytes, monocytes, and GMPs in the BM under noninflammatory conditions. However, this effect reverted after the mice regained GF status, suggesting that steady‐state myelopoiesis is dynamically regulated by the current levels of microbiota exposure [71]. Transfer of sterile‐filtered, boiled serum from SPF but not GF mice was sufficient to expand the BM myeloid cell pool of GF animals in an MyD88/Toll‐IL‐1 receptor‐containing adaptor molecule‐dependent manner, indicating that heat‐stable, circulating, commensal‐derived products may be responsible for maintaining steady‐state myelopoiesis levels [71].
A “one fits all” model does not seem to apply when it comes to microbiota control of myelopoiesis. Whereas SCFAs did not affect the generation of BM monocytes and neutrophils, they influenced DC hematopoiesis by increasing the number of macrophage and DC precursors and CDPs in the BM [72] (Fig. 1).
THE GUT MICROBIOTA MODULATES NEUTROPHIL HOMEOSTASIS
In addition to granulopoiesis, neutrophil homeostasis in neonates is dependent on the presence of gut commensals [73, 74, 75–76]. Reduced and altered microbiota in offspring of Abx‐treated dams was associated with decreased numbers of circulating and BM neutrophils and lower GMPs in the BM. Microbiota‐derived components, such as LPS via TLR4/MyD88 signaling, induced IL‐17 production mainly by group 3 innate lymphoid cells in the intestine and increased plasma levels of G‐CSF leading to granulocytosis [73]. Neonatal gut mucosal colonization also enhanced recruitment and reprogramming of B cell helper neutrophils to the splenic perimarginal zone, where they elicited class‐switching and antibody production against highly conserved microbial T cell‐independent antigens [75]. An altered and less‐diverse composition of the gut microbiota enriched in Staphylococcus in neonatal nonobese diabetic mice, correlated with increased immature granulocytes in the liver and spleen, also implicating commensal microbes in neutrophil maturation [76].
Sterile neutropenia induces feedback granulopoiesis characterized by expansion of BM HSPCs and GMPs, along with increased plasma concentration of G‐CSF, IL‐17, and IL‐23—cytokines known to be involved in granulopoiesis [77]. Although steady‐state granulopoiesis is dependent on the presence of commensal microbes, as discussed above, Bugl et al. [77] showed that feedback granulopoiesis in GF mice was not affected. However, in the same study, TLR4‐mediated signaling was shown to be required for feedback granulopoiesis in SPF animals. A caveat of this study, however, is that to prevent infection as a result of their neutropenic status, all mice received amoxicillin in the drinking water, which would induce changes in the microbiota composition of SPF animals. Further studies are needed to distinguish between a possible masking effect of Abx and the relative contribution of endogenous versus microbial ligands triggering TLR4 signaling. Contamination of the “sterile” food of GF mice with bacteria‐derived products (e.g., LPS) may also be a confounding factor [78].
Although the impact of the microbiota on myelopoiesis has been clearly established in the studies described thus far, they are all based on ex vivo analysis of BM or peripheral populations, which may not necessarily fully recapitulate the physiologic conditions of a live organism. The zebrafish offers a unique model to examine the interaction of commensal microbes with immune cells in real time in a living host. With the use of this model, direct in vivo visualization showed that the commensal microbiota establishes and maintains neutrophil homeostasis in a process involving MyD88 [79] and that it regulates neutrophil localization and recruitment in response to injury at extraintestinal sites [80]. The latter effect was mediated by microbiota‐induced, host‐derived SAA through the NF‐κB signaling axis. Colonization of mice with segmented filamentous bacteria induces SAA production in the terminal ileum, which in turn, promotes Th17 differentiation and local IL‐17A production [81] [82]. SAA has also been shown to induce G‐CSF expression and neutrophilia [83], possibly through the induction of IL‐17. Altogether, these findings indicate that SAA is an important microbial‐induced host factor modulating neutrophil development and function.
In a recent elegant study using gnotobiotic zebrafish colonized with zebrafish‐derived bacterial isolates, Rolig et al. [84] showed that individual commensal species within a community have a wide range of per‐capita, neutrophil‐stimulatory potential that does not necessarily correlate with the relative abundance. In their association studies, a low‐abundance member dominated the host immune phenotype via a bacterial‐secreted factor. Importantly, in this system, bacteria–bacteria interactions determined relative abundances in a given community. Thus, microbiota complexity can be an important factor controlling myelopoiesis, and microbial function, rather than abundance, may be of critical relevance.
PHYSIOLOGIC CONSEQUENCES OF THE MICROBIOTA–MYELOID CELL CROSSTALK
The microbiota‐dependent changes in myelopoiesis and granulocyte homeostasis have an impact in the host response to inflammatory and infectious insults. In their study, Balmer et al. [71] found a reduced BM pool of neutrophils and myeloid cell precursors and a decreased turnover of granulocytes in the blood; however, they did not observe steady‐state kinetic differences in myelopoiesis. This would implicate that the microbiota controls the initial size of the BM myeloid cell pool soon after colonization. The reduced initial myelopoietic pool results in a lower number of cells readily available to fight an invading pathogen, as evidenced by delayed clearance of bacteria following systemic administration [71]. In line with this, reduced steady‐state numbers of tissue resident and BM‐derived phagocytes rendered GF and Abx‐treated mice susceptible to acute systemic Lysteria monocytogenes infection, whereas their ability to mount adaptive immunity following secondary infection is unaffected or even enhanced, indicating a defect in the early innate rather than adaptive response [67, 85].
Likewise, impaired neutrophil homeostasis in neonates is associated with LOS. The neutropenia in neonates caused by Abx exposure through their dams resulted in heightened susceptibility to E. coli serotype K1 and Klebsiella pneumoniae—2 pathogens commonly associated with LOS and infection in human neonates [73]. Dysregulated neonatal neutrophil homeostasis has also been linked to increased risk of type 1 diabetes [76].
Among the many consequences of altered microbiota composition on host physiology is the increased risk of developing allergic diseases [2]. Two recent studies indicated that this is mediated, at least in part, through alterations in steady‐state hematopoiesis. With the use of the model of house dust mite extract‐induced allergic airway inflammation, Trompette et al. [72] showed that SCFAs enhanced DC precursor hematopoiesis in the BM and altered lung DC functionality, rendering them impaired in their ability to induce Th2 cell effector functions. The SCFAs protective effect was mediated via binding to GPR41 but not GPR43, and it was characterized by the rapid resolution of the inflammatory response. SCFAs were shown to modulate human monocyte‐derived DCs as well, although the extent of their effect varies among individual SCFAs [86]. In a report by Maslowski et al. [87], SCFAs were also necessary to prevent exacerbated inflammatory responses and for the timely resolution of inflammation in mouse models of inflammatory arthritis and OVA‐induced airway inflammation. In this case, SCFA binding to GPR43 was required, and although the precise mechanism underlying its protective role in allergic airway inflammation was not elucidated, altered neutrophil function was the suggested cause for the exacerbated inflammatory arthritis. Another study showed that microbiota‐derived signals influenced Th2 cell‐dependent allergic inflammation through the modulation of basophil hematopoiesis by limiting the proliferation of basophil precursors in the BM [88].
Altered myelopoiesis as a result of the lack of commensals can be beneficial in certain circumstances. For instance, circulating neutrophil aging is driven by microbiota‐derived signals in a cell‐intrinsic, TLR/MyD88‐dependent manner. Reduced numbers of circulating overly activated aged neutrophils, in the absence of commensal microbes, ameliorates inflammation‐induced tissue damage in murine models of endotoxin‐driven septic shock and sickle‐cell disease (Fig. 1) [89].
It was also suggested that fluctuations in the amount of circulating bacteria‐derived products absorbed from the gut could influence the emigration of inflammatory monocytes from the BM. Sensing low levels of circulating endotoxin (i.e., LPS) by BM mesenchymal stem and progenitor cells resulted in their expression of MCP1 and the release of inflammatory monocytes into the bloodstream [90]. Therefore, this provides another mechanism by which commensal microbes may fine tune the systemic innate immune tone enhancing antimicrobial defenses but not without potentially detrimental consequences, such as worsening noninfectious inflammatory conditions.
The microbiota seems to exert its effect on hematopoiesis, mostly at the level of GMPs in the BM and at sites of extramedullary hematopoiesis, affecting the monocytic and granulocytic progenies with a pronounced defect on neutrophil homeostasis (Fig. 1). The lymphocytic compartment, on the other hand, seems to be unaffected. However, how the host–microbe crosstalk occurs remains ill defined. Nevertheless, a large body of evidence suggests that circulating microbiota‐derived products or bacterial metabolites may reach the BM or extramedullar sites, where they can be directly sensed by HSPCs and committed myeloid progenitors [67, 71, 72–73, 75, 91, 92]. Alternatively, circulating HSCs can encounter bacteria or their products in the periphery before re‐entering the BM [93, 94].
MICROBIOTA MODULATION OF MP FUNCTION: IMPACT ON THE RESPONSE TO INFECTION
The clear role of the microbiota on steady‐state myelopoiesis and the heightened susceptibility of microbiota‐deprived animals to acute systemic infections [9] begged the question of whether commensal microbes could also regulate cellular function. Indeed, during early studies with GF animals, several groups reported reduced chemotaxis, phagocytosis, lysosomal content, microbicidal activity, and production of ROS and RNS in neutrophils and macrophages from GF animals [95, 96, 97, 98, 99, 100–101].
Recent studies expanded on these initial observations and uncovered some of the mechanistic pathways. For instance, Abrams and Bishop [96] observed a marked reduction in infiltrating neutrophils in the peritoneal cavity of GF mice following the induction of sterile peritonitis. An effect that could not be attributed to the lower number of steady‐state circulating neutrophils, as in their model, inflammation elicited similar numbers of neutrophils in circulation. Follow‐up studies using GF rats showed that whereas the influx of neutrophils to the site of inflammation was significantly impaired, the in vitro chemotactic response was not affected. Karmarkar and Rock [102] have recently suggested a plausible explanation to this phenomenon, so as to stem from defective neutrophil extravasation from the bloodstream into the inflamed tissue. They observed that GF mice displayed a marked reduction in the neutrophil recruitment to the peritoneal cavity in response to diverse stimuli, including microbial components and sterile ligands. The number of circulating neutrophils following zymosan‐induced inflammation was even increased in flora‐deficient animals, prompting the authors to speculate that an impaired ability of neutrophils to extravasate into the inflamed tissue was responsible for the reduced influx ( Fig. 2 ). With the use of conditional MyD88 knockout animals to delete MyD88 expression at the time of the inflammatory challenge, they showed that microbiota‐derived signals via a MyD88‐dependent pathway are required to precondition the neutrophil inflammatory response to zymosan‐induced peritonitis, whereas MyD88 signaling was dispensable during the actual challenge. Addition of LPS to the drinking water was sufficient to restore the neutrophil response. Although the precise mechanism of the defective neutrophil extravasation was not elucidated, these results are consistent with the notion that microbiota priming is required for myeloid cell response to further inflammatory stimuli.
Figure 2.
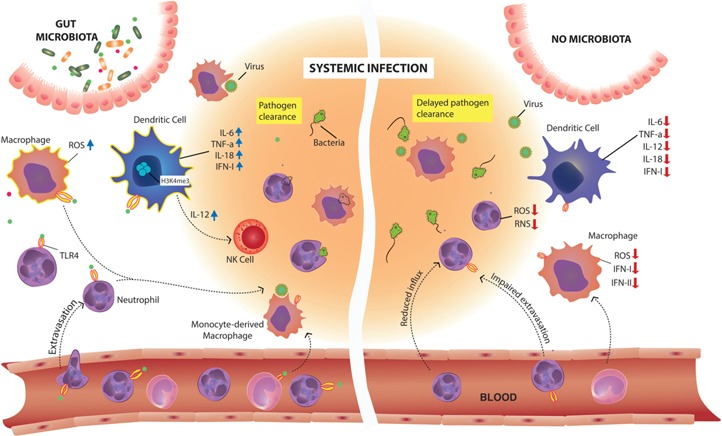
Microbiota modulation of myeloid cell function at extraintestinal sites: impact on response to invading pathogens. The preconditioning of neutrophils by microbiota‐derived signals is required for their extravasation from the bloodstream into the inflamed tissue and for their optimal production of ROS and RNS and bactericidal activity. The microbiota provides tonic signals needed for macrophage bacterial killing capacity. Gut commensal microbes also calibrate macrophage and DC responsiveness to viral infections through chromatin remodeling of antiviral genes that constitutively express histone marks of transcriptionally activated genes (H3K4me3). IL‐12, produced by the microbiota‐calibrated DCs, leads to NK cell activation. Migratory and antigen‐presenting capabilities of lung DCs are modulated by the microbiota as well. In the absence of commensal microbes, there is a reduced influx of neutrophils, macrophages, and DCs to sites of inflammation and infection, and the cells display impaired functions, leading to delayed pathogen clearance.
The antimicrobial capacity of neutrophils was shown to be dependent on recognition of microbiota‐derived peptidoglycan from Gram‐negative bacteria via the PRR Nod1 but not Nod2 or TLR4 [92] (Fig. 2). In this study, Clarke et al. [92] found that during colonization, peptidoglycan translocates from the gut, increases in circulation, and accumulates in the BM, where it primes neutrophils for optimal oxidative and nonoxidative bacterial killing mechanisms. Ex vivo killing of Staphylococcus aureus and Streptococcus pneumoniae was impaired when BM neutrophils were obtained from GF, Abx‐treated, or Nod1‐deficient animals. Systemic administration of a Nod1 ligand was sufficient to restore the ability of BM neutrophils to kill S. pneumoniae. Consistent with impaired innate priming, Nod1‐deficient mice were unable to control S. pneumoniae early sepsis. The ability of Nod1 ligand to promote neutrophil microbicidal activity directly was also confirmed on human blood neutrophils [92]. In line with these findings, GF mice also display increased bacterial burden following systemic challenge with S. aureus [67].
Early innate resistance to lung infection with the bacterial pathogen K. pneumoniae was also impaired in microbiota‐depleted mice [103]. In this case, the defect was ascribed to reduced ROS‐mediated bacterial killing by alveolar macrophages and required Nod1‐ and Nod2‐ but not TLR‐dependent signaling. Intragastric administration of Nod1/Nod2 ligands to Abx‐treated mice could restore their ability to control early bacterial burdens; however, the same ligands administered intranasally had no effect. This suggests a role for the indigenous intestinal, rather than upper‐airway, microbiota and further supports that peptidoglycan translocated from the gut can modulate systemic innate immunity. Although this study showed impaired ex vivo bacterial killing capacity of alveolar macrophages, in vitro assays showed no difference in the ability of IFN‐γ‐primed peritoneal macrophages from GF mice to kill L. monocytogenes [67]. It is possible that not all macrophage populations are functionally regulated by the microbiota to the same extent or that some but not all bactericidal mechanisms are affected. Furthermore, different experimental conditions (e.g., in vitro IFN‐γ priming of macrophages) may account for the differences observed in those studies.
Other examples of the systemic effect of the gut microbiota on MP function during infection have been provided by studies showing the increased susceptibility of GF and Abx‐treated animals to respiratory and systemic viral infections [9, 104, 105–106] (Fig. 2). Disruption of the microbiota composition by means of Abx treatment led to impaired early innate immunity to influenza A virus infection, which in turn, resulted in diminished virus‐specific adaptive‐immune responses, subsequent delayed viral clearance, and increased host mortality [104, 106]. The impaired antiviral response in Abx‐treated mice was shown to be a result of reduced constitutive expression of pro‐IL‐1β and pro‐IL‐18 in the lung, along with decreased numbers and migratory and antigen‐presenting capabilities of lung CD103+ DCs [104]. Weakened host‐protective immunity to influenza virus infection was also linked to reduced expression of antiviral‐associated response genes, including Ifnb, IFN regulatory factor 7, Myxovirus (Influenza virus) Resistance 1 (Mx1), and Stat1, in lung macrophages from infected, Abx‐treated mice [107]. Intranasal and intrarectal administration of TLR ligands was sufficient to reestablish the anti‐viral immunity in Abx‐treated mice [104], and loss of commensal bacteria in the upper respiratory tract, as well as in the gut, was observed following Abx treatment [107]. Therefore, the relative contribution of the local microbiota versus the distal effect of intestinal commensals remains to be determined.
A broad reduction in the expression of IFN response‐associated genes was observed in naïve peritoneal macrophages from Abx mice, and the same cells showed an impaired in vitro response to type I and type II IFN, as well as to infection with influenza virus. Hence, it is possible that preconditioning by microbiota‐derived signals regulates systemic antiviral innate responses [107]. In accordance with these findings, splenic DCs purified from GF animals failed to produce IFN‐I and to up‐regulate the expression of genes encoding proinflammatory cytokines, such as Il12, Il6, Il18, and Tnf, in response to TL3 and TLR4 stimulation [105]. In vivo, the diminished inflammatory cytokine production led to impaired NK cell activation and enhanced susceptibility to systemic viral infection. The proposed underlying mechanism for these defects is epigenetic chromatin modifications, as splenic DCs from GF mice showed reduced activating histone marks (i.e., H3K4me3) at the promoters of the proinflammatory cytokine genes [105]. Thus, in addition to defective inflammasome activation and lung DC function, impaired antiviral innate responses result from lack of systemic microbiota‐derived tonic signaling of MPs (Fig. 2).
IMPACT OF THE MICROBIOTA ON VACCINE EFFICACY
A major repercussion of the microbiota influence on innate and adaptive immunity is its potential impact on vaccine efficacy. This is exemplified by the poor response to oral immunizations in developing countries, where malnutrition and early‐life exposure to fecal‐oral contamination—which leads to altered microbiota composition—are prevalent [108]. Two recent studies in mice implicated the gut microbiota, through the modulation of APC function, in the blunted vaccine response following systemic [109] and intranasal immunizations [110]. In a systems biology analysis of immunity induced by the unadjuvanted TIV in humans, Bali Pulendran and colleagues [111] found that TLR5 expression strongly correlates with the magnitude of TIV‐specific antibody responses. This interesting observation prompted them to ask whether there was a causal link, and if so, what was the role of commensal microbes? In their recent study, the authors observed a markedly diminished TIV‐induced humoral response in TLR5 deficient, Abx‐treated, and GF mice compared with conventional wild‐type animals following subcutaneous immunization [109]. Similar to the immune response to influenza infection [104], targeting the gut‐resident microbial community with an Abx that is poorly absorbed in the gastrointestinal tract was sufficient to impair the vaccine‐induced response. The allowance of the natural reconstitution of the indigenous flora in GF animals, the oral gavage of flagellated E. coli, or the coinjection of the TLR5 ligand flagellin with TIV in Abx‐treated mice led to increased TIV‐specific IgG antibody responses, therefore, suggesting a role for the gut microbiota‐derived flagellin in modulating vaccine efficacy [109]. Whereas TLR5/MyD88‐mediated signaling on DCs was dispensable for the induction of TIV‐specific antibodies, deletion of MyD88 in macrophages or their depletion abrogated the vaccine‐induced response. The requirement for TLR5 was also observed for the efficacy of subcutaneous immunization with another purified viral subunit vaccine but not with adjuvanted or live‐attenuated vaccines, indicating that gut‐resident commensal microbes act as natural adjuvants, priming APCs for optimal subcutaneous vaccine effectiveness.
In a different report, Ruane et al. [110] showed that lung DC populations induce IgA class‐switch and up‐regulation of gut‐homing molecules on B cells and induce their migration to the gastrointestinal tract. CD103+ and CD24+CD11b+ DCs (LDCs), but not macrophages, purified from the lungs of conventional mice, were able to induce IgA class‐switch, whereas the same DC populations obtained from GF or Abx‐treated animals failed to do so. The effect was mediated by TGF‐β produced by LDCs in a MyD88‐dependent fashion. Oral LPS administration or reconstitution of GF mice with gut microbial flora from conventional animals was able to restore the ability of LDCs to induce IgA class‐switch. After intranasal immunization with inactive CT, lung IgA+ B cells up‐regulated the expression of the gut‐homing molecules α4β7 and CCR9. The in vivo vaccine efficacy was tested in a model of CT‐induced diarrhea that relies on secretory IgA in the intestinal lumen for protection. Intranasal immunization of conventional but not GF animals induced similar levels of IgA as the oral route of immunization [110]. This study suggests a crosstalk between the lung and gut immunity in which signals derived from commensal microbes regulate the ability of LDCs to induce mucosal IgA class‐switch, modulating the efficacy of intranasal vaccination. Nevertheless, although the results presented by Ruane et al. [110] point to a distant effect of the enteric microbiota, the contribution of signals originating from the upper‐airway tract resident microbes remains to be determined.
GUT MICROBIOTA INFLUENCE ON CANCER THERAPY
Cancer has historically been viewed as a disease determined by genetic and environmental factors; however, it is now clear that inflammation affects tumor initiation, progression, and dissemination [112]. Approximately 18% of human malignancies can be attributed to known infectious agents [113]. Recently, several studies have uncovered a role for commensal microbes in carcinogenesis, both locally and at distant, nonmucosal sites [114, 115]. How the microbiota favors cancer development is the focus of intense research, and a variety of mechanisms has been proposed [114, 116], including the mobilization and recruitment of myeloid cell populations that can promote tumor progression. Fusobacterium nucleatum, which is associated with colorectal carcinoma in humans, has been shown to increase tumor multiplicity through the selective recruitment of tumor‐infiltrating myeloid cells in the adenomatous polyposis coli gene/multiple intestinal neoplasia mouse model of intestinal tumorigenesis [117]. At extraintestinal sites, gut microbes promote the development of mammary carcinomas in genetically prone mice via a neutrophil‐mediated mechanism [118], and microbiota‐driven mobilization of myeloid‐derived suppressor cells via TLR5‐dependent signaling favors malignant progression of tumors with ablated p53 and activated K‐ras [119].
As in other facets of the intricate microbiota–host relationship, there is also a beneficial side when it comes to cancer: intestinal microbes, through the modulation of myeloid cell function, seem to be required for the optimal response to cancer chemo‐ and immunotherapy [120]. New experimental findings revealed 2 aspects of the microbiota contribution to therapy efficacy: an adjuvant effect of translocated bacteria following therapy‐induced intestinal epithelial barrier damage and a priming effect provided by the gut indigenous microbial flora before therapy ( Fig. 3 ). Initial observations of an adjuvant effect of the gut microbiota during cancer immunotherapy were made in a mouse model of adoptive T cell transfer therapy [121]. In that study, the authors observed that following the total body irradiation preconditioning regime, intestinal mucosal damage allowed the translocation of bacteria into the mesenteric lymph nodes and increased levels of circulating bacterial products, which in turn, resulted in activation of DCs and increased activity of the transferred T cells. Abx‐treated or TLR4‐deficient mice showed reduced response to therapy and administration of serum from irradiated mice or ultra‐pure LPS‐improved therapy efficacy [121].
Figure 3.
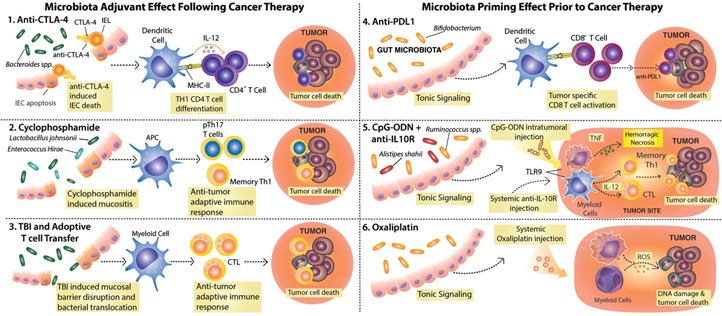
Commensal intestinal microbes modulate the response to various cancer therapies and promote spontaneous anti‐tumor immunity. (1–3) Therapies that cause damage to the intestinal epithelial barrier allow for changes in microbial composition and/or bacterial translocation and benefit from the adjuvant effect of commensal microbes. (1) Anti‐CTLA‐4 treatment leads to intraepithelial cell (IEC) apoptosis and disrupts the microbiota–host equilibrium, leading to enrichment in Bacteroides spp., which in turn, activate DCs for IL‐12 production and priming of anti‐bacteria CD4+ T cells that contribute to therapy efficacy. IEL, Intraepithelial lymphocyte; MHC‐II, MHC class II. (2) CTX induces mucositis, followed by dysbiosis and translocation of Gram‐positive bacteria that promote the induction of pTh17 and Th1 memory cells. (3) Total body irradiation (TBI), a preconditioning regime used before adoptive T cell transfer therapy, leads to mucosal barrier disruption and translocation of bacteria and bacterial products that lead to DC activation and promote the transferred cell anti‐tumor activity. (4–6) Microbiota‐derived signals prime myeloid cells for effective response to therapy. (4) Members of the Bifidobacterium genus constitutively present in the gut microbiota activate DCs, resulting in enhanced, spontaneous anti‐tumor CD8+ T cell responses. The increased frequencies of anti‐tumor CD8+ T cells in mice harboring Bifidobacterium spp. facilitate the response to anti‐PD‐L1 treatment. (5) Signals from the microbiota and in particular, Alistipes shaii and Ruminococcus spp. enable tumor‐infiltrating myeloid cells to respond to CpG‐ODN following TLR9 ligation and to produce proinflammatory cytokines (e.g., TNF, IL‐12). The CpG‐ODN activity, in combination with IL‐10R blockade (anti‐IL‐10R), leads to hemorrhagic necrosis and priming of Th1 and CTL anti‐tumor responses needed for therapy efficacy. (6) Microbiota tonic signaling conditions tumor‐infiltrating myeloid cells to produce ROS in response to oxaliplatin. ROS production by myeloid cells is required for oxaliplatin‐induced DNA damage, thus contributing to the early genotoxic effect of the drug.
Two recent studies in animal models showed that changes in microbial composition and/or systemic translocation of gut commensal microbes following therapy‐induced intestinal mucosal barrier damage are also needed for the therapeutic efficacy of CTX, an alkylating anti‐cancer drug commonly used in the clinic [122], and checkpoint blockade immunotherapy with a mAb against CTLA‐4 [123]. CTX is among the chemotherapeutic drugs that induce the so‐called “immunogenic cell death,” leading to the activation of APCs and induction of anti‐tumor adaptive immune responses that contribute to its efficacy [124]. Viaud et al. [122] showed that translocation of selective gram‐positive bacteria (e.g., Lactobacillus johnsonii and Enterococcus hirae) following CTX‐induced mucositis was required for the induction of pTh17 cells and memory Th1 cells. The anti‐tumor effect of CTX on established MCA205 sarcomas was largely reduced in GF animals or mice pre‐exposed to Abx that specifically targeted gram‐positive but not gram‐negative bacteria. This blunted response correlated with decreased accumulation of pTh17 in the spleen and reduced frequencies of intratumoral Th1 cells.
Among the side‐effects of immunotherapy with the anti‐CTLA‐4 mAb Ipilimumab, approved for the treatment of patients with metastatic melanoma, are the immune‐related events commonly occurring at sites in direct contact with commensal microbes, such as the intestine [125]. This observation inspired Laurence Zitvogel's group [123] to assess whether gut‐commensal microorganisms also have an impact on the anti‐cancer therapeutic efficacy of the CTLA‐4 blockade. Whereas SPF mice treated with anti‐CTLA‐4 were able to control tumor progression, therapy efficacy was severely compromised in GF or Abx‐treated animals [123]. When the authors studied the underlying mechanism, they found that anti‐CTLA‐4 led to a rapid T cell‐dependent increase in intestinal epithelial cell death and changes in the microbial composition. Bacteroides spp., in particular, Bacteroides thetaiotaomicron and Bacteroides fragilis, and the proteobacteria Burkholderia cepacia were capable of restoring the efficacy of anti‐CTLA‐4 in GF or Abx‐treated mice. These commensal microbes modulated the anti‐CTLA‐4 efficacy through the maturation of DCs and their IL‐12 production, which in turn, elicited anti‐bacterial Th1 T cell responses that contributed to tumor eradication. Changes in the relative abundance of Bacteroides spp. were also found in patients following anti‐CTLA‐4 treatment, and GF mice reconstituted with the Bacteroides spp.‐enriched microbiota of these patients responded to the CTLA‐4 blockade [123].
Without affecting the integrity of the intestinal mucosa, treatment of subcutaneously implanted tumors with an intratumoral injection of CpG‐ODN combined with systemic blockade of IL‐10 (CpG/anti‐IL‐10R) leads to tumor eradication in conventional animals; however, Abx‐treated or GF mice fail to respond [126]. Iida et al. [126] showed that the impaired response in the microbe‐deprived animals was a result of the inability of tumor‐infiltrating myeloid cell populations to respond to CpG and to produce proinflammatory cytokines, such as TNF and IL‐12, necessary to elicit an effective anti‐tumor response. CpG/anti‐IL‐10R treatment was inefficient in TLR4‐deficient animals, and the blunted response in Abx‐treated mice could be partially restored by oral administration of LPS. Animals that produce high levels of TNF in response to CpG/anti‐IL‐10R treatment could be segregated from those that were poor responders based on their fecal microbial composition. Further microbiota analysis revealed several bacterial species that positively or negatively correlated with the TNF response, and in vivo association experiments confirmed these findings. For example, gavage administration of the gram‐negative A. shaii to Abx‐treated mice restored the ability of tumor‐infiltrating myeloid cells to produce TNF, whereas oral administration of Lactobacillus fermentum to intact animals was sufficient to dampen the response [126].
The same study showed that microbiota tonic signaling was also required for the response to chemotherapy with platinum compounds [126]. GF or Abx‐treated animals bearing subcutaneously transplanted EL‐4 lymphomas did not respond to cisplatin or oxaliplatin to the same extent as conventional mice. The lack of response was attributed to the failure of tumor‐infiltrating, monocyte‐derived macrophages and neutrophils to produce ROS in response to the platinum salt and a reduced, early genotoxic effect of the drug. MyD88‐deficient mice and animals lacking the myeloid NADPH oxidase 2, which is needed for ROS production, also failed to respond to oxaliplatin, therefore, suggesting that microbial signals modulate the efficacy of oxaliplatin therapy by systemically priming myeloid cells to produce the ROS required for the early genotoxicity of the drug.
A role for the microbiota in spontaneous anti‐tumor responses was also recognized recently [127]. Sivan et al. [127] took advantage of the known differences in the microbiota composition of animals obtained from different vendors, namely Jax (Bar Harbor, ME, USA) and Tac (Germantown, NY, USA), to study whether the presence or absence of certain microbes affected anti‐tumor immunity. Indeed, they observed that B16 melanoma tumors progressed more aggressively in Tac mice compared with animals obtained from Jax. The slower tumor growth in Jax animals correlated with enhanced activation of DCs and increased frequency of tumor‐specific CD8+ T cells. The differences could be erased by allowing Jax and Tac mice to exchange their microbes during cohousing experiments or by performing fecal transplant. Analysis of the microbiota composition and its correlation with anti‐tumor immunity revealed Bifidobacterium as the only genus present in the microbiota of Jax mice showing a significant and positive association with the anti‐tumor response. This association was confirmed by gavage of Bifidobacterium spp. to Tac animals, which was sufficient to improve their spontaneous tumor control. The enhanced response of Jax mice, Bifidobacterium fed, and microbiota‐transplanted Tac animals to anti‐PD‐L1 treatment further emphasized the importance of the commensal‐dependent heightened spontaneous anti‐tumor CD8+ T cell response [127]. Although the precise mechanism still needs to be elucidated, gut‐resident microbes, through the activation of APCs, can lead to increased frequencies of tumor‐specific CD8+ T cells and consequently, improved anti‐PD‐L1 efficacy.
The studies mentioned above have focused on the impact of gut‐resident microbes; however, the influence of the microbiota at other anatomic sites (e.g., skin, lung, oral cavity) remains to be determined.
CONCLUDING REMARKS
The work discussed here offers compelling evidence that gut commensal microbes regulate myeloid cell homeostatic levels, provide tonic signaling, and serve as natural adjuvants, greatly influencing the host's ability to respond to infection, tumors, and cancer therapy ( Fig. 4 ). Nevertheless, the extent to which these experimental findings translate to humans remains to be determined, and several critical issues need to be addressed before we can design and perform preclinical and clinical intervention studies.
Figure 4.

The gut microbiota modulates the response to systemic infection and cancer therapy via the regulation of myeloid cell development and function. The gut microbiota regulates myelopoiesis in the BM and at extramedullary sites (liver and spleen). Both infection and cancer induce dysregulated myelopoiesis. Factors produced at the inflamed sites trigger the mobilization, proliferation, and differentiation of HSCs and the recruitment of neutrophils and inflammatory monocytes to the infected tissues and tumor microenvironment. At the site of infection, inflammatory monocytes differentiate into inflammatory DCs and macrophages that promote Th1 responses and pathogen eradication. On the other hand, in the suppressive tumor microenvironment, these cells favor tumor growth. Following chemo‐ and immunotherapy, myeloid cells can become proinflammatory and lead to tumor destruction, whereas at the site of infection, immunosuppressive myeloid cell function contributes to resolution of inflammation. Microbiota tonic signaling allows for the proper differentiation and function of myeloid cells, facilitating pathogen killing and resolution of inflammation to avoid collateral damage to self, and the efficient response to cancer therapy.
Although investigators have started to uncover the molecular basis of the commensal microbes/systemic immunity crosstalk, a large proportion of the microbiome research is still only based on correlation and association of specific bacterial species with a given immune function. Those associations imply that the particular bacteria species (or their products) can contribute to that said immune function but are not necessarily the main or solely responsible for it. Functional studies are restrained by our current limited ability to isolate and culture species that may be of interest. Furthermore, with the consideration of the uniqueness of an individual's microbiota and that bacteria–bacteria interactions modulate each other's metabolic pathways and membership in the overall microbial ecosystem [128], the functional impact of a particular microbe may not be the same in the context of 2 distinct microbiota. Thus, this underscores the importance of microbial transcriptomic analysis to understand the function of the microbial community as a whole.
Transplant of clinical microbial isolates into mice is a valuable tool to validate correlation studies. Nevertheless, the bidirectional nature of the microbiota–host interaction, in which host genetic and environmental factors play an import role, should be taken into account. The same consideration should be made when thinking about fecal transplants in patients, as what constitutes a healthy microbiota in one individual may be inflammatory in another. Nonetheless, this approach has proven successful in the treatment of Abx‐associated diarrhea caused by Clostridium difficile infection [129], where it was shown to work, at least in part, through the ability of specific bacterial species to synthesize C. difficile inhibiting metabolites [130]. The effectiveness of fecal transplant to treat other conditions, such as IBD, is less consistent [131], and the identification of bacterial functional pathways to be targeted may prove more efficient in this case, as well as in other complex inflammatory disorders.
The current knowledge on the role of the gut microbiota in cancer therapy is mostly based on experimental animal studies [121, 122–123, 126, 127], and the influence of the microbiota at other anatomic sites (e.g., skin, lung, oral cavity) remains to be determined. The gut microbiota‐mediated effect on the response to different therapeutic approaches occurred via the systemic modulation of myeloid cell function; albeit, distinct bacterial species were capable of either promoting or suppressing the anti‐cancer response. Although the findings need to be validated in humans, the results are quite striking and may help explain the variable response observed in patients. Hence, the gut microbiota may be a plausible target for adjuvant cancer therapy.
Much of the current knowledge on the role of commensal microbes in modulating immune responses comes from studies looking at the bacterial component of the intestinal microbiota. However, emerging evidence underscores the contribution of other gut‐resident microbes (e.g., viruses, fungi, and parasites), as well as the interaction among microbial communities. Interactions among microbes are of particular relevance, as they may modulate each other's metabolic pathways and membership in the overall microbial ecosystem. Researchers normally work under the assumption that Abx treatment only disturbs bacterial cells; however, they may indirectly impact other microorganisms. For instance, overgrowth of the commensal fungal Candida species is not uncommon after Abx treatment and in mice, resulted in elevated plasma concentrations of PGE2 and M2 macrophage polarization in the lung promoting allergic airway inflammation [132]. The enteric murine norovirus offers an example of the relevance of the mammalian virome; it affects intestinal homeostasis and mucosal immunity directly [133], and its persistence is influenced by the bacterial microbiota [134]. Another example of trans‐kingdom interaction was recently provided by a study showing that helminth‐induced type 2 intestinal responses modulate the bacterial microbiota composition, resulting in a protective phenotype in IBD‐susceptible mice [135]. Mice also harbor distinct symbiotic protozoa that can directly regulate intestinal type 2 immunity [136]. Further studies are needed to determine if and how these other microbial communities and their interactions affect systemic inflammatory and immune responses.
We have come a long way since van Leeuwenhoek's time or the early GF animal studies of the 20th century. Yet, despite the extraordinary progress made in recent years, we are still in the early stages toward deciphering the mechanisms underlying the microbiota–host crosstalk. Nevertheless, the field is moving at a very fast pace, and the prospects for developing microbiota‐targeted interventions for disease prevention and cure hold much promise.
AUTHORSHIP
S.G. researched the literature, wrote the first draft of the article, and did the illustrations. R.G. performed the literature review, structured and wrote the article, and reviewed and edited the figures.
DISCLOSURES
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the U.S. National Institutes of Health National Cancer Institute. The authors are grateful to Dr. Amiran Dzutsev for helpful discussion and critical reading of the manuscript. The authors regret omission of important articles as a result of scope and space limitations.
References
- 1. McFall‐Ngai, M. , Hadfield, M. G. , Bosch, T. C. , Carey, H. V. , Domazet‐Losno, T. , Douglas, A. E. , Dubilier, N. , Eberl, G. , Fukami, T. , Gilbert, S. F. , Hentschel, U. , King, N. , Kjelleberg, S. , Knoll, A. H. , Kremer, N. , Mazmanian, S. K. , Metcalf, J. L. , Nealson, K. , Pierce, N. E. , Rawls, J. F. , Reid, A. , Ruby, E. G. , Rumpho, M. , Sanders, J. G. , Tautz, D. , Wernegreen, J. J. (2013) Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 110, 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clemente, J. C. , Ursell, L. K. , Parfrey, L. W. , Knight, R. (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. NISC Comparative Sequencing Program . (2009) Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Costello, E. K. , Lauber, C. L. , Hamady, M. , Fierer, N. , Gordon, J. I. , Knight, R. (2009) Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nasidze, I. , Li, J. , Quinque, D. , Tang, K. , Stoneking, M. (2009) Global diversity in the human salivary microbiome. Genome Res. 19, 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ley, R. E. , Peterson, D. A. , Gordon, J. I. (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. [DOI] [PubMed] [Google Scholar]
- 7. MetaHIT Consortium . (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leewenhoeck, A. (1684) An abstract of a letter from Mr. Anthony Leewenhoeck at Delft, dated Sep. 17. 1683. containing some microscopical observations, about animals in the scurf of the teeth, the substance call'd worms in the nose, the cuticula consisting of scales. Philos. Trans. R. Soc. Lond. 14, 568–574. [Google Scholar]
- 9. Smith, K. , McCoy, K. D. , Macpherson, A. J. (2007) Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 19, 59–69. [DOI] [PubMed] [Google Scholar]
- 10. Shendure, J. A. , Porreca, G. J. , Church, G. M. , Gardner, A. F. , Hendrickson, C. L. , Kieleczawa, J. , Slatko, B. E. (2011) Overview of DNA sequencing strategies. Curr. Protoc. Mol. Biol. Chapter 7, Unit 7 1. doi: 10.1002/0471142727.mb0701s96. [DOI] [PubMed] [Google Scholar]
- 11. Vincent, A. T. , Derome, N. , Boyle, B. , Culley, A. I. , Charette, S. J. (2016) Next‐generation sequencing (NGS) in the microbiological world: how to make the most of your money. J. Microbiol. Methods. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 12. Ursell, L. K. , Metcalf, J. L. , Parfrey, L. W. , Knight, R. (2012) Defining the human microbiome. Nutr. Rev. 70 (Suppl 1), S38–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamady, M. , Knight, R. (2009) Microbial comtunity profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 19, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eckburg, P. B. , Bik, E. M. , Bernstein, C. N. , Purdom, E. , Dethlefsen, L. , Sargent, M. , Gill, S. R. , Nelson, K. E. , Relman, D. A. (2005) Diversity of the human intestinal microbial flora. Science 308, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turnbaugh, P. J. , Ley, R. E. , Mahowald, M. A. , Magrini, V. , Mardis, E. R. , Gordon, J. I. (2006) An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. [DOI] [PubMed] [Google Scholar]
- 16. MetaHIT Consortium . (2011) Enterotypes of the human gut microbiome. Nature 473, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu, G. D. , Chen, J. , Hoffmann, C. , Bittinger, K. , Chen, Y. Y. , Keilbaugh, S. A. , Bewtra, M. , Knights, D. , Walters, W. A. , Knight, R. , Sinha, R. , Gilroy, E. , Gupta, K. , Baldassano, R. , Nessel, L. , Li, H. , Bushman, F. D. , Lewis, J. D. (2011) Linking long‐term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benson, A. K. , Kelly, S. A. , Legge, R. , Ma, F. , Low, S. J. , Kim, J. , Zhang, M. , Oh, P. L. , Nehrenberg, D. , Hua, K. , Kachman, S. D. , Moriyama, E. N. , Walter, J. , Peterson, D. A. , Pomp, D. (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 107, 18933–18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spor, A. , Koren, O. , Ley, R. (2011) Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 9, 279–290. [DOI] [PubMed] [Google Scholar]
- 20. Dominguez‐Bello, M. G. , Costello, E. K. , Contreras, M. , Magris, M. , Hidalgo, G. , Fierer, N. , Knight, R. (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 107, 11971–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jernberg, C. , Löfmark, S. , Edlund, C. , Jansson, J. K. (2007) Long‐term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 1, 56–66. [DOI] [PubMed] [Google Scholar]
- 22. Dethlefsen, L. , Huse, S. , Sogin, M. L. , Relman, D. A. (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6, e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gill, S. R. , Pop, M. , Deboy, R. T. , Eckburg, P. B. , Turnbaugh, P. J. , Samuel, B. S. , Gordon, J. I. , Relman, D. A. , Fraser‐Liggett, C. M. , Nelson, K. E. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palm, N. W. , de Zoete, M. R. , Flavell, R. A. (2015) Immune‐microbiota interactions in health and disease. Clin. Immunol. 159, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burokas, A. , Moloney, R. D. , Dinan, T. G. , Cryan, J. F. (2015) Microbiota regulation of the mammalian gut‐brain axis. Adv. Appl. Microbiol. 91, 1–62. [DOI] [PubMed] [Google Scholar]
- 26. Bouskra, D. , Brézillon, C. , Bérard, M. , Werts, C. , Varona, R. , Boneca, I. G. , Eberl, G. (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510. [DOI] [PubMed] [Google Scholar]
- 27. Macpherson, A. J. , Slack, E. , Geuking, M. B. , McCoy, K. D. (2009) The mucosal firewalls against commensal intestinal microbes. Semin. Immunopathol. 31, 145–149. [DOI] [PubMed] [Google Scholar]
- 28. Kuhn, K. A. , Stappenbeck, T. S. (2013) Peripheral education of the immune system by the colonic microbiota. Semin. Immunol. 25, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldszmid, R. S. , Trinchieri, G. (2012) The price of immunity. Nat. Immunol. 13, 932–938. [DOI] [PubMed] [Google Scholar]
- 30. Belkaid, Y. , Hand, T. W. (2014) Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koenderman, L. , Buurman, W. , Daha, M. R. (2014) The innate immune response. Immunol. Lett. 162 (2 Pt B), 95–102. [DOI] [PubMed] [Google Scholar]
- 32. Merad, M. , Sathe, P. , Helft, J. , Miller, J. , Mortha, A. (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wynn, T. A. , Chawla, A. , Pollard, J. W. (2013) Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hettinger, J. , Richards, D. M. , Hansson, J. , Barra, M. M. , Joschko, A. C. , Krijgsveld, J. , Feuerer, M. (2013) Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 14, 821–830. [DOI] [PubMed] [Google Scholar]
- 35. Wong, K. L. , Yeap, W. H. , Tai, J. J. , Ong, S. M. , Dang, T. M. , Wong, S. C. (2012) The three human monocyte subsets: implications for health and disease. Immunol. Res. 53, 41–57. [DOI] [PubMed] [Google Scholar]
- 36. Soehnlein, O. , Lindbom, L. (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 10, 427–439. [DOI] [PubMed] [Google Scholar]
- 37. Youn, J. I. , Gabrilovich, D. I. (2010) The biology of myeloid‐derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 40, 2969–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schulz, C. , Gomez Perdiguero, E. , Chorro, L. , Szabo‐Rogers, H. , Cagnard, N. , Kierdorf, K. , Prinz, M. , Wu, B. , Jacobsen, S. E. , Pollard, J. W. , Frampton, J. , Liu, K. J. , Geissmann, F. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. [DOI] [PubMed] [Google Scholar]
- 39. Ginhoux, F. , Greter, M. , Leboeuf, M. , Nandi, S. , See, P. , Gokhan, S. , Mehler, M. F. , Conway, S. J. , Ng, L. G. , Stanley, E. R. , Samokhvalov, I. M. , Merad, M. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoeffel, G. , Wang, Y. , Greter, M. , See, P. , Teo, P. , Malleret, B. , Leboeuf, M. , Low, D. , Oller, G. , Almeida, F. , Choy, S. H. , Grisotto, M. , Renia, L. , Conway, S. J. , Stanley, E. R. , Chan, J. K. , Ng, L. G. , Samokhvalov, I. M. , Merad, M. , Ginhoux, F. (2012) Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac‐derived macrophages. J. Exp. Med. 209, 1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hashimoto, D. , Chow, A. , Noizat, C. , Teo, P. , Beasley, M. B. , Leboeuf, M. , Becker, C. D. , See, P. , Price, J. , Lucas, D. , Greter, M. , Mortha, A. , Boyer, S. W. , Forsberg, E. C. , Tanaka, M. , van Rooijen, N. , García‐Sastre, A. , Stanley, E. R. , Ginhoux, F. , Frenette, P. S. , Merad, M. (2013) Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takizawa, H. , Boettcher, S. , Manz, M. G. (2012) Demand‐adapted regulation of early hematopoiesis in infection and inflammation. Blood 119, 2991–3002. [DOI] [PubMed] [Google Scholar]
- 43. King, K. Y. , Goodell, M. A. (2011) Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 11, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ueha, S. , Shand, F. H. , Matsushima, K. (2011) Myeloid cell population dynamics in healthy and tumor‐bearing mice. Int. Immunopharmacol. 11, 783–788. [DOI] [PubMed] [Google Scholar]
- 45. Goldszmid, R. S. , Dzutsev, A. , Trinchieri, G. (2014) Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe 15, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burberry, A. , Zeng, M. Y. , Ding, L. , Wicks, I. , Inohara, N. , Morrison, S. J. , NúnTez, G. (2014) Infection mobilizes hematopoietic stem cells through cooperative NOD‐like receptor and Toll‐like receptor signaling. Cell Host Microbe 15, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Orkin, S. H. , Zon, L. I. (2008) Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paul, F. , Arkin, Y. , Giladi, A. , Jaitin, D. A. , Kenigsberg, E. , Keren‐Shaul, H. , Winter, D. , Lara‐Astiaso, D. , Gury, M. , Weiner, A. , David, E. , Cohen, N. , Lauridsen, F. K. , Haas, S. , Schlitzer, A. , Mildner, A. , Ginhoux, F. , Jung, S. , Trumpp, A. , Porse, B. T. , Tanay, A. , Amit, I. (2015) Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- 49. Metcalf, D. (2008) Hematopoietic cytokines. Blood 111, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nagai, Y. , Garrett, K. P. , Ohta, S. , Bahrun, U. , Kouro, T. , Akira, S. , Takatsu, K. , Kincade, P. W. (2006) Toll‐like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmid, M. A. , Takizawa, H. , Baumjohann, D. R. , Saito, Y. , Manz, M. G. (2011) Bone marrow dendritic cell progenitors sense pathogens via Toll‐like receptors and subsequently migrate to inflamed lymph nodes. Blood 118, 4829–4840. [DOI] [PubMed] [Google Scholar]
- 52. YánTez, A. , Goodridge, H. S. , Gozalbo, D. , Gil, M. L. (2013) TLRs control hematopoiesis during infection. Eur. J. Immunol. 43, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Luca, K. , Frances‐Duvert, V. , Asensio, M. J. , Ihsani, R. , Debien, E. , Taillardet, M. , Verhoeyen, E. , Bella, C. , Lantheaume, S. , Genestier, L. , Defrance, T. (2009) The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia 23, 2063–2074. [DOI] [PubMed] [Google Scholar]
- 54. Zhao, J. L. , Ma, C. , O'Connell, R. M. , Mehta, A. , DiLoreto, R. , Heath, J. R. , Baltimore, D. (2014) Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress‐induced hematopoiesis. Cell Stem Cell 14, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh, P. , Yao, Y. , Weliver, A. , Broxmeyer, H. E. , Hong, S. C. , Chang, C. H. (2008) Vaccinia virus infection modulates the hematopoietic cell compartments in the bone marrow. Stem Cells 26, 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boettcher, S. , Manz, M. G. (2016) Sensing and translation of pathogen signals into demand‐adapted myelopoiesis. Curr. Opin. Hematol. 23, 5–10. [DOI] [PubMed] [Google Scholar]
- 57. Baldridge, M. T. , King, K. Y. , Boles, N. C. , Weksberg, D. C. , Goodell, M. A. (2010) Quiescent haematopoietic stem cells are activated by IFN‐gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vos, O. , Buurman, W. A. , Ploemacher, R. E. (1972) Mobilization of haemopoietic stem cells (CFU) into the peripheral blood of the mouse; effects of endotoxin and other compounds. Cell Tissue Kinet. 5, 467–479. [DOI] [PubMed] [Google Scholar]
- 59. Takizawa, H. , Regoes, R. R. , Boddupalli, C. S. , Bonhoeffer, S. , Manz, M. G. (2011) Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J. Exp. Med. 208, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. MacVittie, T. J. , Walker, R. I. (1978) Canine granulopoiesis: alterations induced by suppression of gram‐negative flora. Exp. Hematol. 6, 639–647. [PubMed] [Google Scholar]
- 61. Chang, C. F. , Pollard, M. (1973) Effects of microbial flora on levels of colonay stimulating factor in serums of irradiated CFW mice. Proc. Soc. Exp. Biol. Med. 144, 177–180. [DOI] [PubMed] [Google Scholar]
- 62. Joshi, J. H. , Entringer, M. A. , Robinson, W. A. (1979) Bacterial stimulation of serum colony‐stimulating activity and neutrophil production in germ‐free mice. Proc. Soc. Exp. Biol. Med. 162, 44–47. [DOI] [PubMed] [Google Scholar]
- 63. Goris, H. , de Boer, F. , van der Waaij, D. (1985) Myelopoiesis in experimentally contaminated specific‐pathogen‐free and germfree mice during oral administration of polymyxin. Infect. Immun. 50, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Staber, F. G. , Tarcsay, L. , Dukor, P. (1978) Modulations of myelopoiesis in vivo by chemically pure preparations of cell wall components from gram‐negative bacteria: effects at different stages. Infect. Immun. 20, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tada, T. , Yamamura, S. , Kuwano, Y. , Abo, T. (1996) Level of myelopoiesis in the bone marrow is influenced by intestinal flora. Cell. Immunol. 173, 155–161. [DOI] [PubMed] [Google Scholar]
- 66. Nicaise, P. , Gleizes, A. , Sandre, C. , Forestier, F. , Kergot, R. , Quero, A. M. , Labarre, C. (1998) Influence of intestinal microflora on murine bone marrow and spleen macrophage precursors. Scand. J. Immunol. 48, 585–591. [DOI] [PubMed] [Google Scholar]
- 67. Khosravi, A. , YánTez, A. , Price, J. G. , Chow, A. , Merad, M. , Goodridge, H. S. , Mazmanian, S. K. (2014) Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 15, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoeffel, G. , Ginhoux, F. (2015) Ontogeny of tissue‐resident macrophages. Front. Immunol. 6, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Arpaia, N. , Rudensky, A. Y. (2014) Microbial metabolites control gut inflammatory responses. Proc. Natl. Acad. Sci. USA 111, 2058–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fiedler, K. , Kokai, E. , Bresch, S. , Brunner, C. (2013) MyD88 is involved in myeloid as well as lymphoid hematopoiesis independent of the presence of a pathogen. Am. J. Blood Res. 3, 124–140. [PMC free article] [PubMed] [Google Scholar]
- 71. Balmer, M. L. , Schürch, C. M. , Saito, Y. , Geuking, M. B. , Li, H. , Cuenca, M. , Kovtonyuk, L. V. , McCoy, K. D. , Hapfelmeier, S. , Ochsenbein, A. F. , Manz, M. G. , Slack, E. , Macpherson, A. J. (2014) Microbiota‐derived compounds drive steady‐state granulopoiesis via MyD88/TICAM signaling. J. Immunol. 193, 5273–5283. [DOI] [PubMed] [Google Scholar]
- 72. Trompette, A. , Gollwitzer, E. S. , Yadava, K. , Sichelstiel, A. K. , Sprenger, N. , Ngom‐Bru, C. , Blanchard, C. , Junt, T. , Nicod, L. P. , Harris, N. L. , Marsland, B. J. (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 20, 159–166. [DOI] [PubMed] [Google Scholar]
- 73. Deshmukh, H. S. , Liu, Y. , Menkiti, O. R. , Mei, J. , Dai, N. , O'Leary, C. E. , Oliver, P. M. , Kolls, J. K. , Weiser, J. N. , Worthen, G. S. (2014) The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 20, 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kristensen, M. B. , Metzdorff, S. B. , Bergström, A. , Damlund, D. S. , Fink, L. N. , Licht, T. R. , Frøkiær, H. (2015) Neonatal microbial colonization in mice promotes prolonged dominance of CD11b(+)Gr‐1(+) cells and accelerated establishment of the CD4(+) T cell population in the spleen. Immun. Inflamm. Dis. 3, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Puga, I. , Cols, M. , Barra, C. M. , He, B. , Cassis, L. , Gentile, M. , Comerma, L. , Chorny, A. , Shan, M. , Xu, W. , Magri, G. , Knowles, D. M. , Tam, W. , Chiu, A. , Bussel, J. B. , Serrano, S. , Lorente, J. A. , Bellosillo, B. , Lloreta, J. , Juanpere, N. , Alameda, F. , Baró, T. , de Heredia, C. D. , Torán, N. , Català, A. , Torrebadell, M. , Fortuny, C. , Cusí, V. , Carreras, C. , Diaz, G. A. , Blander, J. M. , Farber, C. M. , Silvestri, G. , Cunningham‐Rundles, C. , Calvillo, M. , Dufour, C. , Notarangelo, L. D. , Lougaris, V. , Plebani, A. , Casanova, J. L. , Ganal, S. C. , Diefenbach, A. , Aróstegui, J. I. , Juan, M. , Yagüe, J. , Mahlaoui, N. , Donadieu, J. , Chen, K. , Cerutti, A. (2011) B Cell‐helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 13, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Damlund, D. S. , Metzdorff, S. B. , Hasselby, J. P. , Wiese, M. , Lundsager, M. , Nielsen, D. S. , Buschard, K. S. , Hansen, A. K. , Frøkiær, H. (2016) Postnatal hematopoiesis and gut microbiota in NOD mice deviate from C57BL/6 mice. J. Diabetes Res. 2016, 6321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bugl, S. , Wirths, S. , Radsak, M. P. , Schild, H. , Stein, P. , André, M. C. , Müller, M. R. , Malenke, E. , Wiesner, T. , Märklin, M. , Frick, J. S. , Handgretinger, R. , Rammensee, H. G. , Kanz, L. , Kopp, H. G. (2013) Steady‐state neutrophil homeostasis is dependent on TLR4/TRIF signaling. Blood 121, 723–733. [DOI] [PubMed] [Google Scholar]
- 78. Hrncir, T. , Stepankova, R. , Kozakova, H. , Hudcovic, T. , Tlaskalova‐Hogenova, H. (2008) Gut microbiota and lipopolysaccharide content of the diet influence development of regulatory T cells: studies in germ‐free mice. BMC Immunol. 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bates, J. M. , Akerlund, J. , Mittge, E. , Guillemin, K. (2007) Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kanther, M. , Tomkovich, S. , Xiaolun, S. , Grosser, M. R. , Koo, J. , Flynn, E. J. , III, Jobin, C. , Rawls, J. F. (2014) Commensal microbiota stimulate systemic neutrophil migration through induction of serum amyloid A. Cell. Microbiol. 16, 1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ivanov, I. I. , Atarashi, K. , Manel, N. , Brodie, E. L. , Shima, T. , Karaoz, U. , Wei, D. , Goldfarb, K. C. , Santee, C. A. , Lynch, S. V. , Tanoue, T. , Imaoka, A. , Itoh, K. , Takeda, K. , Umesaki, Y. , Honda, K. , Littman, D. R. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sano, T. , Huang, W. , Hall, J. A. , Yang, Y. , Chen, A. , Gavzy, S. J. , Lee, J. Y. , Ziel, J. W. , Miraldi, E. R. , Domingos, A. I. , Bonneau, R. , Littman, D. R. (2015) An IL‐23R/IL‐22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. He, R. L. , Zhou, J. , Hanson, C. Z. , Chen, J. , Cheng, N. , Ye, R. D. (2009) Serum amyloid A induces G‐CSF expression and neutrophilia via Toll‐like receptor 2. Blood 113, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rolig, A. S. , Parthasarathy, R. , Burns, A. R. , Bohannan, B. J. , Guillemin, K. (2015) Individual members of the microbiota disproportionately modulate host innate immune responses. Cell Host Microbe 18, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mittrücker, H. W. , Seidel, D. , Bland, P. W. , Zarzycka, A. , Kaufmann, S. H. , Visekruna, A. , Steinhoff, U. (2014) Lack of microbiota reduces innate responses and enhances adaptive immunity against Listeria monocytogenes infection. Eur. J. Immunol. 44, 1710–1715. [DOI] [PubMed] [Google Scholar]
- 86. Nastasi, C. , Candela, M. , Bonefeld, C. M. , Geisler, C. , Hansen, M. , Krejsgaard, T. , Biagi, E. , Andersen, M. H. , Brigidi, P. , Ødum, N. , Litman, T. , Woetmann, A. (2015) The effect of short‐chain fatty acids on human monocyte‐derived dendritic cells. Sci. Rep. 5, 16148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Maslowski, K. M. , Vieira, A. T. , Ng, A. , Kranich, J. , Sierro, F. , Yu, D. , Schilter, H. C. , Rolph, M. S. , Mackay, F. , Artis, D. , Xavier, R. J. , Teixeira, M. M. , Mackay, C. R. (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hill, D. A. , Siracusa, M. C. , Abt, M. C. , Kim, B. S. , Kobuley, D. , Kubo, M. , Kambayashi, T. , Larosa, D. F. , Renner, E. D. , Orange, J. S. , Bushman, F. D. , Artis, D. (2012) Commensal bacteria‐derived signals regulate basophil hematopoiesis and allergic inflammation. Nat. Med. 18, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang, D. , Chen, G. , Manwani, D. , Mortha, A. , Xu, C. , Faith, J. J. , Burk, R. D. , Kunisaki, Y. , Jang, J. E. , Scheiermann, C. , Merad, M. , Frenette, P. S. (2015) Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shi, C. , Jia, T. , Mendez‐Ferrer, S. , Hohl, T. M. , Serbina, N. V. , Lipuma, L. , Leiner, I. , Li, M. O. , Frenette, P. S. , Pamer, E. G. (2011) Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll‐like receptor ligands. Immunity 34, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zeng, M. Y. , Cisalpino, D. , Varadarajan, S. , Hellman, J. , Warren, H. S. , Cascalho, M. , Inohara, N. , NúnTez, G. (2016) Gut microbiota‐induced immunoglobulin G controls systemic infection by symbiotic bacteria and pathogens. Immunity 44, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Clarke, T. B. , Davis, K. M. , Lysenko, E. S. , Zhou, A. Y. , Yu, Y. , Weiser, J. N. (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 16, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wright, D. E. , Wagers, A. J. , Gulati, A. P. , Johnson, F. L. , Weissman, I. L. (2001) Physiological migration of hematopoietic stem and progenitor cells. Science 294, 1933–1936. [DOI] [PubMed] [Google Scholar]
- 94. Massberg, S. , Schaerli, P. , Knezevic‐Maramica, I. , Köllnberger, M. , Tubo, N. , Moseman, E. A. , Huff, I. V. , Junt, T. , Wagers, A. J. , Mazo, I. B. , von Andrian, U. H. (2007) Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 131, 994–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bauer, H. , Paronetto, F. , Burns, W. A. , Einheber, A. (1966) The enhancing effect of the microbial flora on macrophage function and the immune response. A study in germfree mice. J. Exp. Med. 123, 1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Abrams, G. D. , Bishop, J. E. (1965) Normal flora and leukocyte mobilization. Arch. Pathol. 79, 213–217. [PubMed] [Google Scholar]
- 97. Jungi, T. W. , McGregor, D. D. (1978) Impaired chemotactic responsiveness of macrophages from gnotobiotic rats. Infect. Immun. 19, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Trippestad, A. , Midtvedt, T. (1971) Chemotaxis of polymorphonuclear leucocytes from germ‐free rats and generation of chemotactic activity in germ‐free rat sera. Clin. Exp. Immunol. 8, 639–646. [PMC free article] [PubMed] [Google Scholar]
- 99. Mitsuyama, M. , Ohara, R. , Amako, K. , Nomoto, K. , Yokokura, T. , Nomoto, K. (1986) Ontogeny of macrophage function to release superoxide anion in conventional and germfree mice. Infect. Immun. 52, 236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mørland, B. , Midtvedt, T. (1984) Phagocytosis, peritoneal influx, and enzyme activities in peritoneal macrophages from germfree, conventional, and ex‐germfree mice. Infect. Immun. 44, 750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ohkubo, T. , Tsuda, M. , Suzuki, S. , El Borai, N. , Yamamura, M. (1999) Peripheral blood neutrophils of germ‐free rats modified by in vivo granulocyte‐colony‐stimulating factor and exposure to natural environment. Scand. J. Immunol. 49, 73–77. [DOI] [PubMed] [Google Scholar]
- 102. Karmarkar, D. , Rock, K. L. (2013) Microbiota signalling through MyD88 is necessary for a systemic neutrophilic inflammatory response. Immunology 140, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Clarke, T. B. (2014) Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via nod‐like receptor ligands. Infect. Immun. 82, 4596–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ichinohe, T. , Pang, I. K. , Kumamoto, Y. , Peaper, D. R. , Ho, J. H. , Murray, T. S. , Iwasaki, A. (2011) Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 108, 5354–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ganal, S. C. , Sanos, S. L. , Kallfass, C. , Oberle, K. , Johner, C. , Kirschning, C. , Lienenklaus, S. , Weiss, S. , Staeheli, P. , Aichele, P. , Diefenbach, A. (2012) Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37, 171–186. [DOI] [PubMed] [Google Scholar]
- 106. Abt, M. C. , Osborne, L. C. , Monticelli, L. A. , Doering, T. A. , Alenghat, T. , Sonnenberg, G. F. , Paley, M. A. , Antenus, M. , Williams, K. L. , Erikson, J. , Wherry, E. J. , Artis, D. (2012) Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Abt, M. C. , Artis, D. (2013) The dynamic influence of commensal bacteria on the immune response to pathogens. Curr. Opin. Microbiol. 16, 4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Valdez, Y. , Brown, E. M. , Finlay, B. B. (2014) Influence of the microbiota on vaccine effectiveness. Trends Immunol. 35, 526–537. [DOI] [PubMed] [Google Scholar]
- 109. Oh, J. Z. , Ravindran, R. , Chassaing, B. , Carvalho, F. A. , Maddur, M. S. , Bower, M. , Hakimpour, P. , Gill, K. P. , Nakaya, H. I. , Yarovinsky, F. , Sartor, R. B. , Gewirtz, A. T. , Pulendran, B. (2014) TLR5‐mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 41, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ruane, D. , Chorny, A. , Lee, H. , Faith, J. , Pandey, G. , Shan, M. , Simchoni, N. , Rahman, A. , Garg, A. , Weinstein, E. G. , Oropallo, M. , Gaylord, M. , Ungaro, R. , Cunningham‐Rundles, C. , Alexandropoulos, K. , Mucida, D. , Merad, M. , Cerutti, A. , Mehandru, S. (2016) Microbiota regulate the ability of lung dendritic cells to induce IgA class‐switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 213, 53–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Nakaya, H. I. , Wrammert, J. , Lee, E. K. , Racioppi, L. , Marie‐Kunze, S. , Haining, W. N. , Means, A. R. , Kasturi, S. P. , Khan, N. , Li, G. M. , McCausland, M. , Kanchan, V. , Kokko, K. E. , Li, S. , Elbein, R. , Mehta, A. K. , Aderem, A. , Subbarao, K. , Ahmed, R. , Pulendran, B. (2011) Systems biology of vaccination for seasonal influenza in humans. Nat. Immunol. 12, 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Trinchieri, G. (2012) Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 30, 677–706. [DOI] [PubMed] [Google Scholar]
- 113. de Martel, C. , Ferlay, J. , Franceschi, S. , Vignat, J. , Bray, F. , Forman, D. , Plummer, M. (2012) Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 13, 607–615. [DOI] [PubMed] [Google Scholar]
- 114. Schwabe, R. F. , Jobin, C. (2013) The microbiome and cancer. Nat. Rev. Cancer 13, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dzutsev, A. , Goldszmid, R. S. , Viaud, S. , Zitvogel, L. , Trinchieri, G. (2015) The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur. J. Immunol. 45, 17–31. [DOI] [PubMed] [Google Scholar]
- 116. Sears, C. L. , Garrett, W. S. (2014) Microbes, microbiota, and colon cancer. Cell Host Microbe 15, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kostic, A. D. , Chun, E. , Robertson, L. , Glickman, J. N. , Gallini, C. A. , Michaud, M. , Clancy, T. E. , Chung, D. C. , Lochhead, P. , Hold, G. L. , El‐Omar, E. M. , Brenner, D. , Fuchs, C. S. , Meyerson, M. , Garrett, W. S. (2013) Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor‐immune microenvironment. Cell Host Microbe 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lakritz, J. R. , Poutahidis, T. , Mirabal, S. , Varian, B. J. , Levkovich, T. , Ibrahim, Y. M. , Ward, J. M. , Teng, E. C. , Fisher, B. , Parry, N. , Lesage, S. , Alberg, N. , Gourishetti, S. , Fox, J. G. , Ge, Z. , Erdman, S. E. (2015) Gut bacteria require neutrophils to promote mammary tumorigenesis. Oncotarget 6, 9387–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Rutkowski, M. R. , Stephen, T. L. , Svoronos, N. , Allegrezza, M. J. , Tesone, A. J. , Perales‐Puchalt, A. , Brencicova, E. , Escovar‐Fadul, X. , Nguyen, J. M. , Cadungog, M. G. , Zhang, R. , Salatino, M. , Tchou, J. , Rabinovich, G. A. , Conejo‐Garcia, J. R. (2015) Microbially driven TLR5‐dependent signaling governs distal malignant progression through tumor‐promoting inflammation. Cancer Cell 27, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Goldszmid, R. S. , Dzutsev, A. , Viaud, S. , Zitvogel, L. , Restifo, N. P. , Trinchieri, G. (2015) Microbiota modulation of myeloid cells in cancer therapy. Cancer Immunol. Res. 3, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Paulos, C. M. , Wrzesinski, C. , Kaiser, A. , Hinrichs, C. S. , Chieppa, M. , Cassard, L. , Palmer, D. C. , Boni, A. , Muranski, P. , Yu, Z. , Gattinoni, L. , Antony, P. A. , Rosenberg, S. A. , Restifo, N. P. (2007) Microbial translocation augments the function of adoptively transferred self/tumor‐specific CD8+ T cells via TLR4 signaling. J. Clin. Invest. 117, 2197–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Viaud, S. , Saccheri, F. , Mignot, G. , Yamazaki, T. , Daillère, R. , Hannani, D. , Enot, D. P. , Pfirschke, C. , Engblom, C. , Pittet, M. J. , Schlitzer, A. , Ginhoux, F. , Apetoh, L. , Chachaty, E. , Woerther, P. L. , Eberl, G. , Bérard, M. , Ecobichon, C. , Clermont, D. , Bizet, C. , Gaboriau‐Routhiau, V. , Cerf‐Bensussan, N. , Opolon, P. , Yessaad, N. , Vivier, E. , Ryffel, B. , Elson, C. O. , Doré, J. , Kroemer, G. , Lepage, P. , Boneca, I. G. , Ghiringhelli, F. , Zitvogel, L. (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vétizou, M. , Pitt, J. M. , Daillère, R. , Lepage, P. , Waldschmitt, N. , Flament, C. , Rusakiewicz, S. , Routy, B. , Roberti, M. P. , Duong, C. P. , Poirier‐Colame, V. , Roux, A. , Becharef, S. , Formenti, S. , Golden, E. , Cording, S. , Eberl, G. , Schlitzer, A. , Ginhoux, F. , Mani, S. , Yamazaki, T. , Jacquelot, N. , Enot, D. P. , Bérard, M. , Nigou, J. , Opolon, P. , Eggermont, A. , Woerther, P. L. , Chachaty, E. , Chaput, N. , Robert, C. , Mateus, C. , Kroemer, G. , Raoult, D. , Boneca, I. G. , Carbonnel, F. , Chamaillard, M. , Zitvogel, L. (2015) Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science 350, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kroemer, G. , Galluzzi, L. , Kepp, O. , Zitvogel, L. (2013) Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72. [DOI] [PubMed] [Google Scholar]
- 125. Hodi, F. S. , O'Day, S. J. , McDermott, D. F. , Weber, R. W. , Sosman, J. A. , Haanen, J. B. , Gonzalez, R. , Robert, C. , Schadendorf, D. , Hassel, J. C. , Akerley, W. , van den Eertwegh, A. J. , Lutzky, J. , Lorigan, P. , Vaubel, J. M. , Linette, G. P. , Hogg, D. , Ottensmeier, C. H. , Lebbé, C. , Peschel, C. , Quirt, I. , Clark, J. I. , Wolchok, J. D. , Weber, J. S. , Tian, J. , Yellin, M. J. , Nichol, G. M. , Hoos, A. , Urba, W. J. (2010) Improved survival with Ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Iida, N. , Dzutsev, A. , Stewart, C. A. , Smith, L. , Bouladoux, N. , Weingarten, R. A. , Molina, D. A. , Salcedo, R. , Back, T. , Cramer, S. , Dai, R. M. , Kiu, H. , Cardone, M. , Naik, S. , Patri, A. K. , Wang, E. , Marincola, F. M. , Frank, K. M. , Belkaid, Y. , Trinchieri, G. , Goldszmid, R. S. (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sivan, A. , Corrales, L. , Hubert, N. , Williams, J. B. , Aquino‐Michaels, K. , Earley, Z. M. , Benyamin, F. W. , Lei, Y. M. , Jabri, B. , Alegre, M. L. , Chang, E. B. , Gajewski, T. F. (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Flint, H. J. , Duncan, S. H. , Scott, K. P. , Louis, P. (2007) Interactions and competition within the microbial community of the human colon: links between diet and health. Environ. Microbiol. 9, 1101–1111. [DOI] [PubMed] [Google Scholar]
- 129. van Nood, E. , Vrieze, A. , Nieuwdorp, M. , Fuentes, S. , Zoetendal, E. G. , de Vos, W. M. , Visser, C. E. , Kuijper, E. J. , Bartelsman, J. F. , Tijssen, J. G. , Speelman, P. , Dijkgraaf, M. G. , Keller, J. J. (2013) Duodenal infusion of donor feces for recurrent Clostridium difficile . N. Engl. J. Med. 368, 407–415. [DOI] [PubMed] [Google Scholar]
- 130. Buffie, C. G. , Bucci, V. , Stein, R. R. , McKenney, P. T. , Ling, L. , Gobourne, A. , No, D. , Liu, H. , Kinnebrew, M. , Viale, A. , Littmann, E. , van den Brink, M. R. , Jenq, R. R. , Taur, Y. , Sander, C. , Cross, J. R. , Toussaint, N. C. , Xavier, J. B. , Pamer, E. G. (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile . Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Colman, R. J. , Rubin, D. T. (2014) Fecal microbiota transplantation as therapy for inflammatory bowel disease: a systematic review and meta‐analysis. J. Crohn's Colitis 8, 1569–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kim, Y. G. , Udayanga, K. G. , Totsuka, N. , Weinberg, J. B. , NúnTez, G. , Shibuya, A. (2014) Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi‐induced PGE2 . Cell Host Microbe 15, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kernbauer, E. , Ding, Y. , Cadwell, K. (2014) An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Baldridge, M. T. , Nice, T. J. , McCune, B. T. , Yokoyama, C. C. , Kambal, A. , Wheadon, M. , Diamond, M. S. , Ivanova, Y. , Artyomov, M. , Virgin, H. W. (2015) Commensal microbes and interferon‐λ determine persistence of enteric murine norovirus infection. Science 347, 266–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ramanan, D. , Bowcutt, R. , Lee, S. C. , Tang, M. S. , Kurtz, Z. D. , Ding, Y. , Honda, K. , Gause, W. C. , Blaser, M. J. , Bonneau, R. A. , Lim, Y. A. , Loke, P. , Cadwell, K. (2016) Helminth infection promotes colonization resistance via type 2 immunity. Science 352, 608–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Howitt, M. R. , Lavoie, S. , Michaud, M. , Blum, A. M. , Tran, S. V. , Weinstock, J. V. , Gallini, C. A. , Redding, K. , Margolskee, R. F. , Osborne, L. C. , Artis, D. , Garrett, W. S. (2016) Tuft cells, taste‐chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]