

Short abstract
Review on novel concepts in wound angiogenesis; although more capillaries are generally considered better, new studies suggest otherwise.
Keywords: wound healing, capillary, VEGF, scar, fibrosis, inflammation
Abstract
All animals heal, and the ability to heal is requisite for human health. One aspect of repair that has always been considered to be essential for adequate healing is the creation of a new vasculature via angiogenesis. As adult skin wounds heal, a period of rapid and robust capillary growth creates a vascular bed that has many fold more capillaries than does normal tissue. Over time, most of the newly formed capillaries regress, resulting in a final vascular density similar to that of normal skin. Certainly, new capillaries are necessary to bring nutrients, immune cells, and oxygen to healing wounds. Yet, the presumed functional importance of an overabundance of capillaries has recently been challenged, creating questions about whether excess capillary growth is truly necessary for healing. In particular, studies of wounds that heal exceptionally quickly and with less scar formation, such as those in fetal skin and oral mucosa, show that these tissues heal with a reduced angiogenic burst composed of more mature vessels that provide better oxygenation. The level of angiogenesis in wounds often correlates with the inflammatory response, largely because inflammatory cells produce an abundance of proangiogenic mediators. Both the selective reduction of inflammation and the selective reduction of angiogenesis have now been suggested as ways to improve scarring. These concepts link excessive inflammation and the production of a dense but poorly perfused capillary bed to inferior healing outcomes.
Introduction
Wound healing is a complex process that involves a large number of cell types that act in a specific sequence to repair tissue [1, 2]. One highly observable part of normal healing is the creation of a new capillary bed via angiogenesis. In skin wounds, angiogenesis proceeds by the creation of a dense but poorly organized capillary bed that is eventually trimmed back to normal density and capillary architecture [3]. It has long been assumed that a high level of capillary growth is essential for optimal healing, but a recent study has challenged that notion [4]. The emerging data suggest that normally healing wounds exhibit an overly robust and largely dysfunctional angiogenic response that may have detrimental effects on repair outcomes.
THE PROCESS OF WOUND ANGIOGENESIS
The normally healing skin wound provides an outstanding model of both robust capillary growth and controlled capillary regression [3, 5]. In healing wounds, new capillaries grow into the wound at a ferocious rate, producing an abundant network of new blood vessels that is 2, 3, or even up to 10 times more dense than normal tissue ( Fig. 1 ). The onset of angiogenesis is positively regulated by several soluble factors, most conspicuously VEGF‐A [6]. In mammals, the VEGF family includes 5 members (VEGF‐A, ‐B, ‐C, and ‐D and placenta growth factor ). Several studies have shown that VEGF‐A, which is produced in response to hypoxia, is the most dominant proangiogenic factor in healing wounds [6, 7]. VEGF‐A acts as a potent proangiogenic mediator, but also increases vascular permeability, contributing to wound edema [8, 9]. In addition to VEGF‐A, fibroblast growth factor ‐2, platelet‐derived growth factor, members of the TGF‐β family, and other factors, such as cardiac ankyrin repeat protein, also promote wound angiogenesis [10, 11, 12, 13–14]. After an injury, levels of proangiogenic factors increase, reaching a peak slightly before maximum capillary content occurs, and then subside to nearly undetectable levels.
Figure 1.
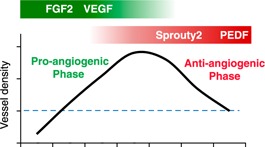
Regulation of wound angiogenesis in skin. During the healing of adult skin wounds, the number of capillaries increases dramatically to a level much greater than that in normal tissue. During the antiangiogenic phase, most of these newly formed vessels are pruned, creating a final vessel density that is similar to that of normal skin. Bars at the top of the graph show some critical proangiogenic (green) and antiangiogenic (red) signals. The blue dashed line indicates vessel density in normal tissue.
After the growth of blood vessels into the wounds, a period of vascular pruning occurs. Over time, most of the newly formed vessels regress until eventually the density of blood vessels returns to that of normal, uninjured skin. The regression process is carefully regulated and includes the selective apoptosis of many of the recently formed capillaries, followed by maturation of the remaining ones [15]. A loss of the proangiogenic stimulus appears to be only part of the reason for capillary regression in wounds, and several negative regulators of angiogenesis are produced in the resolving wound, including Sprouty2, pigment epithelium‐derived factor, and CXCR3 ligands such, as IFN‐γ‐inducible protein‐10 (CXCL10) [16, 17–18]. At the molecular level, microRNA 200b has been proposed to regulate the switch from pro‐ to antiangiogenic phenotype [19]. The selective coverage of capillaries by pericytes plays an important role in capillary maturation. Pericytes are multifunctional cells that can stabilize capillaries, and their presence has been suggested to protect capillaries from negative signals [20]. In wounds, pericytes cover only a portion of the newly formed vessels, probably rendering the remainder susceptible to antiangiogenic stimuli [21, 22]. Pruning of the vessels concludes as the levels of antiangiogenic factors in the wound subside [17].
REQUIREMENTS FOR ANGIOGENESIS IN WOUNDS
One widely accepted view is that wound healing requires a vigorous and dynamic angiogenic response [3, 23]. Indeed, the cellular proliferation, migration, and metabolic activities in wounds create a demand for oxygen and nutrients. Yet, at least 8 separate studies have suggested that skin wound closure is perfectly normal when angiogenesis is reduced. In these studies, many methods have been used to reduce wound angiogenesis, such as treatment with antibodies to VEGF, the application of antiangiogenic agents, and blockade of integrin signaling [17, 24, 25, 26, 27, 28, 29–30]. Together, the findings support the idea that the high level of angiogenesis that occurs in skin wounds is excessive and perhaps unnecessary. Additional support for this idea comes from studies of wounds that heal exceptionally well. In adult skin, any wound that is more than very superficial always heals imperfectly and with at least a small scar. Scars represent less than ideal outcomes, because scar tissue has substantially less strength than normal tissue, has less elasticity, and can create cosmetic and psychosocial problems [1]. In contrast to skin, several types of wounds have the ability to heal very quickly and with no or imperceptible scars. The first of these are wounds produced on the fetal skin in utero, at times before the end of the second trimester [31, 32]. The scarless healing phenotype of early fetal skin has been well described and includes both reduced inflammation and reduced fibrosis [33]. A second type of wound that heals quickly with little scar formation occurs inside the mouth, on the oral mucosa [34], a location well known to be a privileged site of repair and, similar to fetal skin, to exhibit reduced inflammation [34]. Moreover, hypertrophic scars are rare in the oral cavity. Perhaps surprisingly, these wounds that heal exceptionally well—fetal skin and oral mucosal wounds—have much less angiogenesis than normally healing adult skin wounds ( Fig. 2 ) [30, 35]. In short, wounds that heal faster and with less scar formation exhibit reduced inflammation and capillary growth and a more rapidly maturing capillary network.
Figure 2.
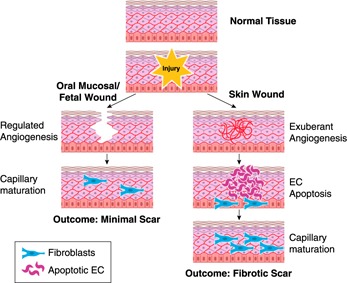
Comparison of angiogenesis in skin, oral mucosal, and fetal wounds. In skin, after an injury to the dermis and normal capillary bed occurs, a vigorous angiogenic response occurs (right). Eventually most of the new capillaries undergo apoptosis and capillary maturation results. In contrast, the angiogenic process in wounds of the oral mucosa and fetal skin (left) is limited, leading to more rapid capillary maturation. The differences in the angiogenic response have been linked to the final amount of scar formation. EC, endothelial cell.
The explanation of how a rapidly healing wound in the fetus or the oral mucosa can survive with fewer capillaries probably involves capillary architecture and performance [36]. Many of the capillaries that are formed in skin wounds are not highly functional [37, 38]. Most of the new vasculature in the healing skin wound is not effectively perfused, and many of the capillaries in the early wound are immature and highly permeable [9, 17]. Together, these studies suggest that the characteristic pattern of skin wound angiogenesis includes a dense bed of capillaries that are not very effective in delivering oxygen and nutrients. Wounds that heal well seem to circumvent the development of excess vasculature, proceeding directly to a well‐formed vascular network (Fig. 2).
THE INTERSECTION OF INFLAMMATION AND ANGIOGENESIS
Two processes that appear likely to be intricately connected in wounds are the inflammatory response to injury and subsequent capillary growth. Tissue injury leads to a rapid acute inflammatory response designed to clear microbes, dying cells, and debris. As a part of this response, stimulated macrophages and keratinocytes produce high levels of proangiogenic factors such as VEGF, and at least a part of the wound angiogenic response lies downstream of inflammation ( Fig. 3 ) [7,39,40]. In skin, multiple sophisticated depletion studies have indicated that macrophages are an important source of the overall proangiogenic stimulus [41, 42–43]. Thus, inflammation and the ultimate angiogenic response seem to be linked in the healing wound. In support of this concept, the level of inflammation is low in both fetal and oral mucosal wounds, correlating with a decreased angiogenic response at these sites ( Table 1 ). Therefore, both decreased inflammation and decreased angiogenesis are features of optimal healing and reduced scar formation.
Figure 3.
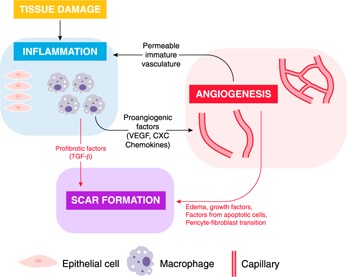
Intersection of wound inflammation and angiogenesis with scar formation. Both inflammation and angiogenesis can directly influence scar formation (red arrows). During the acute inflammatory response, inflammatory cells may directly release profibrotic factors, notably TGF‐β. Independent of inflammation, the presence of a growing yet malformed vasculature that must eventually be removed can directly influence fibroblasts and scar outcomes, perhaps via edema, apoptosis, or the transition of recruited pericytes to an active fibroblast phenotype. Inflammation and angiogenesis are also intertwined as each process influences the other (black arrows). Macrophages, activated epithelial cells, and other inflammatory cells can release proangiogenic mediators such as VEGF and CXC chemokines, supporting robust capillary growth. In turn, the creation of the highly permeable temporary vasculature supports continued inflammation. Overall, inflammation and the immature capillary network can intersect to modulate scar formation. Each process can modulate the other, and each can independently influence scar formation in wounds. Dampening of either process, even independent of the other, has been shown to reduce scar formation.
Table 1.
Levels of inflammation, angiogenesis, and scar formation in different types of wounds
| Wound location | Inflammation | Angiogenesis | Scar formation |
| Adult skin | High | Robust | Moderate |
| Fetal skin | Low–absent | Refined | None |
| Adult oral mucosa | Moderate | Refined | Minimal |
Because inflammation is decreased in fetal and oral mucosal wounds, the role of inflammation itself in dictating scar formation in wounds has received a great deal of experimental attention [44]. Many studies have suggested that a reduction in inflammation can improve skin wound healing outcomes and reduces scar formation. Modulation of specific inflammatory mediators or inflammatory cells, including macrophage‐derived mediators, epithelial mediators, and mast cells, has been shown to yield reduced scar formation and improved healing [44, 45, 46, 47, 48, 49–50]. The correlation of inflammation, angiogenesis, and scar formation seems to indicate that both inflammation and angiogenesis directly affect the final scar outcome (Fig. 3).
When considering wound angiogenesis, an important distinction is the separation of situations where angiogenesis may be excessive, such as normal skin healing, and situations in which angiogenesis is wholly deficient, such as in diabetic wound healing [51, 52]. Diabetes is associated with poorly healing outcomes, and both immunologic aberrancies and a deficit of angiogenesis have been shown to play a role in the pathobiology. It is not surprising, then, that treatment of diabetic wounds with proangiogenic factors or cells to stimulate angiogenesis has been shown to improve diabetic wound healing, at least in animal models [53].
REDUCING WOUND ANGIOGENESIS TO REDUCE SCAR FORMATION
The association of robust angiogenesis with scar formation has led to the suggestion that antiangiogenic therapy could be used to reduce scar formation [30, 54, 55–56]. This idea is supported by clinical studies that have demonstrated that hypertrophic scar formation is linked to increased microvascular content [57, 58]. In addition, a high level of angiogenesis has been described in the formation of keloids, a particularly vigorous type of skin scarring [56]. In tissues other than skin, angiogenesis has been linked to fibrotic outcomes, such as liver and lung fibrosis [59, 60].
Although antiangiogenic therapies targeting improved wound healing have yet to be attempted in human subjects, studies in animal models have already demonstrated that pharmacologic inhibition of angiogenesis can improve healing outcomes. In adult skin wounds in mice, neutralization of VEGF via antibody treatment causes an ∼50% reduction in peak wound vascularity and leads to a significant reduction in wound scar width [30]. Similar results have been obtained when the wound angiogenic response is blunted by the addition of antiangiogenic factors, such as pigment epithelial derived factor [17]. This pharmacologic inhibition of wound angiogenesis leads to a more rapid maturation of the vasculature, with improved pericyte coverage and decreased edema. In a rabbit model of hypertrophic scar formation, systemic administration of the angiogenic inhibitor endostatin was shown to reduce scar formation [55]. In sum, these results suggest that a partial inhibition of the angiogenic response could reduce scar formation or minimize the likelihood of severe scarring. In some ways, these results may seem surprising, as an inhibition of angiogenesis may be expected to impair healing. However, as discussed above, many studies now suggest that a partial reduction in wound angiogenesis does not inhibit wound closure, so long as the reduced number of capillaries provides adequate perfusion [17, 24, 25, 26, 27, 28, 29–30].
HOW DOES THE ANGIOGENIC RESPONSE SPUR SCARRING?
The mechanism by which excessive angiogenesis influences fibrosis and fibroblasts is currently unknown (Fig. 3). However, the effect seems to include some direct effects that are independent of inflammation, given that treatment with antiangiogenic agents has been shown to diminish fibrosis without diminishing inflammation. For example, when acute wounds in mice were treated with anti‐VEGF, a notable decrease in scar formation was seen. Concerning inflammation, though, a quantitative assessment found no differences in acute macrophage and neutrophil levels in control and anti‐VEGF‐treated wounds [30]. Therefore, inflammation can spur both scar formation and angiogenesis, but angiogenesis itself seems also to directly influence scar formation.
There are several possible mechanisms by which excessive angiogenesis could directly stimulate scar formation (Fig. 3). One way by which a reduction in angiogenesis may directly influence healing is via a decrease in edema or in levels of growth factors derived from endothelial cells. Pericyte content and function may also be involved, as the influence of pericytes as mediators of the fibrotic response has also been suggested. Pericytes have been proposed to transition to myofibroblasts, a contractile fibroblast phenotype that supports fibrosis [21, 61]. In a situation of abundant angiogenesis, increased pericyte recruitment to wounds may provide a large population that can adopt a myofibroblast phenotype. Finally, apoptotic cells have been shown to influence fibrosis in a variety of positive ways [62, 63–64]. Thus, the high number of apoptotic endothelial cells that are formed as dense capillary networks regress may play a role in scar formation. Whatever the mechanism of the capillary–fibrosis relationship, a major challenge to any antiangiogenic approach in wounds is fine‐tuning the response. An effective treatment must assure that capillary growth is not too severely thwarted, but instead reaches normal density, stops, and then quickly matures.
In approaching the possible clinical use of antiangiogenic therapy to modify wound scar formation, an important positive factor is the availability of FDA‐approved antiangiogenic therapeutics. Antiangiogenic treatments, such as anti‐VEGF antibodies, are currently approved for therapeutic use in certain cancers and in age‐related macular degeneration, both conditions that are associated with excessive angiogenesis [65, 66]. These available therapeutics may be adapted to and tested for efficacy in scar‐forming wounds.
OTHER CONSIDERATIONS IN SCAR FORMATION
Beyond the roles of inflammation and angiogenesis in wound scar formation, many other factors have been proposed to influence scar formation in healing wounds. In particular, other site‐specific tissue characteristics may contribute to the differences in healing found in skin vs. oral mucosal and fetal wounds.
A major difference between skin and oral mucosa is the epithelial response to injury. Some evidence has suggested that the rate and quality of epithelial restoration greatly influence scar formation in healing wounds [67]. After an injury, oral epithelial cells exhibit more rapid proliferation and migration, characteristics that would support rapid healing [68]. This rapid renewal of the epithelium restores tissue hydration, as water is easily lost when the epithelial barrier is perturbed. The restitution of an effective barrier has been shown to support limited scar formation by crosstalk with the underlying connective tissue. Recent studies suggest that the crosstalk involves detection of the hydration status of the tissue and subsequent detection of increased sodium concentrations in epithelium at injured sites [69]. Oral mucosal and fetal wounds both close rapidly and occur in a liquid environment, features that would support a rapid return to appropriate hydration status and nonscarring outcomes. Another epithelial consideration that could distinguish skin and oral mucosal healing is the presence and availability of local stem cells and progenitor cells. Oral epithelium contains a large and complex assortment of stem cells that may support rapid closure [70].
Finally, a very obvious difference between oral mucosal, fetal, and skin wounds is the local microflora [71]. Although the role of the microbiome in the differential healing in these tissues has yet to be rigorously investigated, several studies have demonstrated that microflora have a substantial influence on healing outcomes [72, 73–74]. Skin wound healing in germ‐free mice has been shown to exhibit greatly reduced scar formation, a finding that could be directly applicable to the relatively germ‐free environment of fetal wound repair [74]. Conversely, perturbations in the normal skin microbiome have been shown to associate with delayed skin wound healing [72, 73]. Further study is needed to determine whether normally occurring microbial differences explain the site‐specific changes in skin, oral mucosal, and fetal wound‐healing outcomes.
In sum, both structural and functional aspects of the tissue are likely to be involved in differential scar formation between skin and oral and fetal mucosa. Although the level of the inflammation and the angiogenic response are surely part of the picture, other distinguishing elements also play a role in the distinctive healing outcomes of certain tissues.
CONCLUDING REMARKS
The dissection of the role of inflammation, angiogenesis, and scar formation has led to many surprising ideas about the roles and intersections of these aspects of the wound‐healing process. More information is needed to fully understand the mechanisms of scar formation vs. regenerative healing, as this distinction certainly involves the complicated interacting systems of inflammation and angiogenesis. Clinical studies should be conducted to investigate whether therapeutics that partially block wound capillary growth will indeed yield an abridged yet efficient vasculature that provides excellent perfusion, thus improving healing. Given the link between excess capillary growth and scar formation, the mechanisms by which a reduction in capillaries improves healing should be investigated further.
AUTHORSHIP
The article was written by Luisa A. DiPietro.
DISCLOSURE
The author declares no conflicts of interest.
ACKNOWLEDGMENTS
This paper was adapted from a presentation at the Society for Leukocyte Biology Annual Meeting, September 2015. Research in the author's laboratory is supported by the U.S. National Institutes of Health (NIH), National Institute of General Medicine Grant R01‐GM50875 and National Institute of Dental Research Grant R21‐DE025926. The content is solely the responsibility of the author and does not necessarily represent the official views of the NIH.
References
- 1. Gurtner, G. C. , Werner, S. , Barrandon, Y. , Longaker, M. T. (2008) Wound repair and regeneration. Nature 453, 314–321. [DOI] [PubMed] [Google Scholar]
- 2. Eming, S. A. , Martin, P. , Tomic‐Canic, M. (2014) Wound repair and regeneration: mechanisms, signaling, and translation. Sci. Transl. Med. 6, 265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eming, S. A. , Brachvogel, B. , Odorisio, T. , Koch, M. (2007) Regulation of angiogenesis: wound healing as a model. Prog. Histochem. Cytochem. 42, 115–170. [DOI] [PubMed] [Google Scholar]
- 4. DiPietro, L. A. (2013) Angiogenesis and scar formation in healing wounds. Curr. Opin. Rheumatol. 25, 87–91. [DOI] [PubMed] [Google Scholar]
- 5. Banda, M. J. , Knighton, D. R. , Hunt, T. K. , Werb, Z. (1982) Isolation of a nonmitogenic angiogenesis factor from wound fluid. Proc. Natl. Acad. Sci. USA 79, 7773–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nissen, N. N. , Polverini, P. J. , Koch, A. E. , Volin, M. V. , Gamelli, R. L. , DiPietro, L. A. (1998) Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am. J. Pathol. 152, 1445–1452. [PMC free article] [PubMed] [Google Scholar]
- 7. Brown, L. F. , Yeo, K. T. , Berse, B. , Yeo, T. K. , Senger, D. R. , Dvorak, H. F. , van de Water, L. (1992) Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med. 176, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dvorak, H. F. , Detmar, M. , Claffey, K. P. , Nagy, J. A. , van de Water, L. , Senger, D. R. (1995) Vascular permeability factor/vascular endothelial growth factor: an important mediator of angiogenesis in malignancy and inflammation. Int. Arch. Allergy Immunol. 107, 233–235. [DOI] [PubMed] [Google Scholar]
- 9. Nagy, J. A. , Benjamin, L. , Zeng, H. , Dvorak, A. M. , Dvorak, H. F. (2008) Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 11, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nissen, N. N. , Polverini, P. J. , Gamelli, R. L. , DiPietro, L. A. (1996) Basic fibroblast growth factor mediates angiogenic activity in early surgical wounds. Surgery 119, 457–465. [DOI] [PubMed] [Google Scholar]
- 11. Shi, Y. , Reitmaier, B. , Regenbogen, J. , Slowey, R. M. , Opalenik, S. R. , Wolf, E. , Goppelt, A. , Davidson, J. M. (2005) CARP, a cardiac ankyrin repeat protein, is up‐regulated during wound healing and induces angiogenesis in experimental granulation tissue. Am. J. Pathol. 166, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grotendorst, G. R. , Grotendorst, C. A. , Gilman, T. (1988) Production of growth factors (PDGF & TGF‐beta) at the site of tissue repair. Prog. Clin. Biol. Res. 266, 47–54. [PubMed] [Google Scholar]
- 13. Uutela, M. , Wirzenius, M. , Paavonen, K. , Rajantie, I. , He, Y. , Karpanen, T. , Lohela, M. , Wiig, H. , Salven, P. , Pajusola, K. , Eriksson, U. , Alitalo, K. (2004) PDGF‐D induces macrophage recruitment, increased interstitial pressure, and blood vessel maturation during angiogenesis. Blood 104, 3198–3204. [DOI] [PubMed] [Google Scholar]
- 14. Pardali, E. , Goumans, M. J. , ten Dijke, P. (2010) Signaling by members of the TGF‐beta family in vascular morphogenesis and disease. Trends Cell Biol. 20, 556–567. [DOI] [PubMed] [Google Scholar]
- 15. Iruela‐Arispe, M. L. , Dvorak, H. F. (1997) Angiogenesis: a dynamic balance of stimulators and inhibitors. Thromb. Haemost. 78, 672–677. [PubMed] [Google Scholar]
- 16. Wietecha, M. S. , Chen, L. , Ranzer, M. J. , Anderson, K. , Ying, C. , Patel, T. B. , DiPietro, L. A. (2011) Sprouty2 downregulates angiogenesis during mouse skin wound healing. Am. J. Physiol. Heart Circ. Physiol. 300, H459–H467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wietecha, M. S. , Król, M. J. , Michalczyk, E. R. , Chen, L. , Gettins, P. G. , DiPietro, L. A. (2015) Pigment epithelium‐derived factor as a multifunctional regulator of wound healing. Am. J. Physiol. Heart Circ. Physiol. 309, H812–H826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bodnar, R. J. , Yates, C. C. , Rodgers, M. E. , Du, X. , Wells, A. (2009) IP‐10 induces dissociation of newly formed blood vessels. J. Cell Sci. 122, 2064–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sinha, M. , Ghatak, S. , Roy, S. , Sen, C. K. (2015) microRNA‐200b as a switch for inducible adult angiogenesis. Antioxid. Redox Signal. 22, 1257–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kelly‐Goss, M. R. , Sweat, R. S. , Stapor, P. C. , Peirce, S. M. , Murfee, W. L. (2014) Targeting pericytes for angiogenic therapies. Microcirculation 21, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dulmovits, B. M. , Herman, I. M. (2012) Microvascular remodeling and wound healing: a role for pericytes. Int. J. Biochem. Cell Biol. 44, 1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crocker, D. J. , Murad, T. M. , Geer, J. C. (1970) Role of the pericyte in wound healing: an ultrastructural study. Exp. Mol. Pathol. 13, 51–65. [DOI] [PubMed] [Google Scholar]
- 23. Tonnesen, M. G. , Feng, X. , Clark, R. A. (2000) Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 5, 40–46. [DOI] [PubMed] [Google Scholar]
- 24. Lange‐Asschenfeldt, B. , Velasco, P. , Streit, M. , Hawighorst, T. , Pike, S. E. , Tosato, G. , Detmar, M. (2001) The angiogenesis inhibitor vasostatin does not impair wound healing at tumor‐inhibiting doses. J. Invest. Dermatol. 117, 1036–1041. [DOI] [PubMed] [Google Scholar]
- 25. Berger, A. C. , Feldman, A. L. , Gnant, M. F. , Kruger, E. A. , Sim, B. K. , Hewitt, S. , Figg, W. D. , Alexander, H. R. , Libutti, S. K. (2000) The angiogenesis inhibitor, endostatin, does not affect murine cutaneous wound healing. J. Surg. Res. 91, 26–31. [DOI] [PubMed] [Google Scholar]
- 26. Roman, C. D. , Choy, H. , Nanney, L. , Riordan, C. , Parman, K. , Johnson, D. , Beauchamp, R. D. (2002) Vascular endothelial growth factor‐mediated angiogenesis inhibition and postoperative wound healing in rats. J. Surg. Res. 105, 43–47. [DOI] [PubMed] [Google Scholar]
- 27. Klein, S. A. , Bond, S. J. , Gupta, S. C. , Yacoub, O. A. , Anderson, G. L. (1999) Angiogenesis inhibitor TNP‐470 inhibits murine cutaneous wound healing. J. Surg. Res. 82, 268–274. [DOI] [PubMed] [Google Scholar]
- 28. Bloch, W. , Huggel, K. , Sasaki, T. , Grose, R. , Bugnon, P. , Addicks, K. , Timpl, R. , Werner, S. (2000) The angiogenesis inhibitor endostatin impairs blood vessel maturation during wound healing. FASEB J. 14, 2373–2376. [DOI] [PubMed] [Google Scholar]
- 29. Jang, Y. C. , Arumugam, S. , Gibran, N. S. , Isik, F. F. (1999) Role of alpha(v) integrins and angiogenesis during wound repair. Wound Repair Regen. 7, 375–380. [DOI] [PubMed] [Google Scholar]
- 30. Wilgus, T. A. , Ferreira, A. M. , Oberyszyn, T. M. , Bergdall, V. K. , Dipietro, L. A. (2008) Regulation of scar formation by vascular endothelial growth factor. Lab. Invest. 88, 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Longaker, M. T. , Whitby, D. J. , Adzick, N. S. , Crombleholme, T. M. , Langer, J. C. , Duncan, B. W. , Bradley, S. M. , Stern, R. , Ferguson, M. W. , Harrison, M. R. (1990) Studies in fetal wound healing, VI, second and early third trimester fetal wounds demonstrate rapid collagen deposition without scar formation. J. Pediatr. Surg. 25, 63–68, discussion 68–69. [DOI] [PubMed] [Google Scholar]
- 32. Leung, A. , Crombleholme, T. M. , Keswani, S. G. (2012) Fetal wound healing: implications for minimal scar formation. Curr. Opin. Pediatr. 24, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilgus, T. A. (2007) Regenerative healing in fetal skin: a review of the literature. Ostomy Wound Manage. 53, 16–31; quiz 32–33. [PubMed] [Google Scholar]
- 34. Szpaderska, A. M. , Zuckerman, J. D. , DiPietro, L. A. (2003) Differential injury responses in oral mucosal and cutaneous wounds. J. Dent. Res. 82, 621–626. [DOI] [PubMed] [Google Scholar]
- 35. Szpaderska, A. M. , Walsh, C. G. , Steinberg, M. J. , DiPietro, L. A. (2005) Distinct patterns of angiogenesis in oral and skin wounds. J. Dent. Res. 84, 309–314. [DOI] [PubMed] [Google Scholar]
- 36. Dvorak, H. F. (2015) Tumors: wounds that do not heal‐redux. Cancer Immunol. Res. 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bluff, J. E. , O'Ceallaigh, S. , O'Kane, S. , Ferguson, M. W. , Ireland, G. (2006) The microcirculation in acute murine cutaneous incisional wounds shows a spatial and temporal variation in the functionality of vessels. Wound Repair Regen. 14, 434–442. [DOI] [PubMed] [Google Scholar]
- 38. Erba, P. , Ogawa, R. , Ackermann, M. , Adini, A. , Miele, L. F. , Dastouri, P. , Helm, D. , Mentzer, S. J. , D'Amato, R. J. , Murphy, G. F. , Konerding, M. A. , Orgill, D. P. (2011) Angiogenesis in wounds treated by microdeformational wound therapy. Ann. Surg. 253, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thakral, K. K. , Goodson, W. H. , III, Hunt, T. K. (1979) Stimulation of wound blood vessel growth by wound macrophages. J. Surg. Res. 26, 430–436. [DOI] [PubMed] [Google Scholar]
- 40. Spiller, K. L. , Anfang, R. R. , Spiller, K. J. , Ng, J. , Nakazawa, K. R. , Daulton, J. W. , Vunjak‐Novakovic, G. (2014) The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 35, 4477–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goren, I. , Allmann, N. , Yogev, N. , Schürmann, C. , Linke, A. , Holdener, M. , Waisman, A. , Pfeilschifter, J. , Frank, S. (2009) A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin‐driven lysozyme M‐specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am. J. Pathol. 175, 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koh, T. J. , DiPietro, L. A. (2011) Inflammation and wound healing: the role of the macrophage. Expert Rev. Mol. Med. 13, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lucas, T. , Waisman, A. , Ranjan, R. , Roes, J. , Krieg, T. , Müller, W. , Roers, A. , Eming, S. A. (2010) Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 184, 3964–3977. [DOI] [PubMed] [Google Scholar]
- 44. Stramer, B. M. , Mori, R. , Martin, P. (2007) The inflammation‐fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J. Invest. Dermatol. 127, 1009–1017. [DOI] [PubMed] [Google Scholar]
- 45. Peranteau, W. H. , Zhang, L. , Muvarak, N. , Badillo, A. T. , Radu, A. , Zoltick, P. W. , Liechty, K. W. (2008) IL‐10 overexpression decreases inflammatory mediators and promotes regenerative healing in an adult model of scar formation. J. Invest. Dermatol. 128, 1852–1860. [DOI] [PubMed] [Google Scholar]
- 46. Mori, R. , Shaw, T. J. , Martin, P. (2008) Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J. Exp. Med. 205, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zgheib, C. , Xu, J. , Liechty, K. W. (2014) Targeting inflammatory cytokines and extracellular matrix composition to promote wound regeneration. Adv. Wound Care (New Rochelle) 3, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gallant‐Behm, C. L. , Du, P. , Lin, S. M. , Marucha, P. T. , DiPietro, L. A. , Mustoe, T. A. (2011) Epithelial regulation of mesenchymal tissue behavior. J. Invest. Dermatol. 131, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gallant‐Behm, C. L. , Hildebrand, K. A. , Hart, D. A. (2008) The mast cell stabilizer ketotifen prevents development of excessive skin wound contraction and fibrosis in red Duroc pigs. Wound Repair Regen. 16, 226–233. [DOI] [PubMed] [Google Scholar]
- 50. Chen, L. , Schrementi, M. E. , Ranzer, M. J. , Wilgus, T. A. , DiPietro, L. A. (2014) Blockade of mast cell activation reduces cutaneous scar formation. PLoS One 9, e85226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Costa, P. Z. , Soares, R. (2013) Neovascularization in diabetes and its complications: unraveling the angiogenic paradox. Life Sci. 92, 1037–1045. [DOI] [PubMed] [Google Scholar]
- 52. Falanga, V. (2005) Wound healing and its impairment in the diabetic foot. Lancet 366, 1736–1743. [DOI] [PubMed] [Google Scholar]
- 53. Demidova‐Rice, T. N. , Durham, J. T. , Herman, I. M. (2012) Wound healing angiogenesis: innovations and challenges in acute and chronic wound healing. Adv. Wound Care (New Rochelle) 1, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Diao, J. S. , Xia, W. S. , Guo, S. Z. (2010) Bevacizumab: a potential agent for prevention and treatment of hypertrophic scar. Burns 36, 1136–1137. [DOI] [PubMed] [Google Scholar]
- 55. Ren, H. T. , Hu, H. , Li, Y. , Jiang, H. F. , Hu, X. L. , Han, C. M. (2013) Endostatin inhibits hypertrophic scarring in a rabbit ear model. J. Zhejiang Univ. Sci. B 14, 224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gira, A. K. , Brown, L. F. , Washington, C. V. , Cohen, C. , Arbiser, J. L. (2004) Keloids demonstrate high‐level epidermal expression of vascular endothelial growth factor. J. Am. Acad. Dermatol. 50, 850–853. [DOI] [PubMed] [Google Scholar]
- 57. Amadeu, T. , Braune, A. , Mandarim‐de‐Lacerda, C. , Porto, L. C. , Desmoulière, A. , Costa, A. (2003) Vascularization pattern in hypertrophic scars and keloids: a stereological analysis. Pathol. Res. Pract. 199, 469–473. [DOI] [PubMed] [Google Scholar]
- 58. Van der Veer, W. M. , Niessen, F. B. , Ferreira, J. A. , Zwiers, P. J. , de Jong, E. H. , Middelkoop, E. , Molema, G. (2011) Time course of the angiogenic response during normotrophic and hypertrophic scar formation in humans. Wound Repair Regen. 19, 292–301. [DOI] [PubMed] [Google Scholar]
- 59. Farkas, L. , Gauldie, J. , Voelkel, N. F. , Kolb, M. (2011) Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am. J. Respir. Cell Mol. Biol. 45, 1–15. [DOI] [PubMed] [Google Scholar]
- 60. Elpek, G. O. (2015) Angiogenesis and liver fibrosis. World J. Hepatol. 7, 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Greenhalgh, S. N. , Conroy, K. P. , Henderson, N. C. (2015) Healing scars: targeting pericytes to treat fibrosis. QJM 108, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Laplante, P. , Sirois, I. , Raymond, M. A. , Kokta, V. , Béliveau, A. , Prat, A. , Pshezhetsky, A. V. , Hébert, M. J. (2010) Caspase‐3‐mediated secretion of connective tissue growth factor by apoptotic endothelial cells promotes fibrosis. Cell Death Differ. 17, 291–303. [DOI] [PubMed] [Google Scholar]
- 63. Wang, P. , Koyama, Y. , Liu, X. , Xu, J. , Ma, H. Y. , Liang, S. , Kim, I. H. , Brenner, D. A. , Kisseleva, T. (2016) Promising therapy candidates for liver fibrosis. Front. Physiol. 7, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Johnson, A. , DiPietro, L. A. (2013) Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J. 27, 3893–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Santarelli, M. , Diplotti, L. , Samassa, F. , Veritti, D. , Kuppermann, B. D. , Lanzetta, P. (2015) Advances in pharmacotherapy for wet age‐related macular degeneration. Expert Opin. Pharmacother. 16, 1769–1781. [DOI] [PubMed] [Google Scholar]
- 66. Jayson, G. C. , Kerbel, R. , Ellis, L. M. , Harris, A. L. (2016) Antiangiogenic therapy in oncology: current status and future directions. Lancet. S0140‐6736(15)01088‐0 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 67. Mustoe, T. A. , Gurjala, A. (2011) The role of the epidermis and the mechanism of action of occlusive dressings in scarring. Wound Repair Regen. 19 (Suppl 1), s16–s21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Turabelidze, A. , Guo, S. , Chung, A. Y. , Chen, L. , Dai, Y. , Marucha, P. T. , DiPietro, L. A. (2014) Intrinsic differences between oral and skin keratinocytes. PLoS One 9, e101480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu, W. , Hong, S. J. , Zhong, A. , Xie, P. , Jia, S. , Xie, Z. , Zeitchek, M. , Niknam‐Bienia, S. , Zhao, J. , Porterfield, D. M. , Surmeier, D. J. , Leung, K. P. , Galiano, R. D. , Mustoe, T. A. (2015) Sodium channel Nax is a regulator in epithelial sodium homeostasis. Sci. Transl. Med. 7, 312ra177. [DOI] [PubMed] [Google Scholar]
- 70. Jones, K. B. , Klein, O. D. (2013) Oral epithelial stem cells in tissue maintenance and disease: the first steps in a long journey. Int. J. Oral Sci. 5, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Costalonga, M. , Herzberg, M. C. (2014) The oral microbiome and the immunobiology of periodontal disease and caries. Immunol. Lett. 162, 22–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Misic, A. M. , Gardner, S. E. , Grice, E. A. (2014) The wound microbiome: modern approaches to examining the role of microorganisms in impaired chronic wound healing. Adv. Wound Care (New Rochelle) 3, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Holmes, C. J. , Plichta, J. K. , Gamelli, R. L. , Radek, K. A. (2015) Dynamic role of host stress responses in modulating the cutaneous microbiome: implications for wound healing and infection. Adv. Wound Care (New Rochelle) 4, 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Canesso, M. C. , Vieira, A. T. , Castro, T. B. , Schirmer, B. G. , Cisalpino, D. , Martins, F. S. , Rachid, M. A. , Nicoli, J. R. , Teixeira, M. M. , Barcelos, L. S. (2014) Skin wound healing is accelerated and scarless in the absence of commensal microbiota. J. Immunol. 193, 5171–5180. [DOI] [PubMed] [Google Scholar]