

Short abstract
Applications of available multidimensional flow cytometry software are compared to determine which are most appropriate to portray complex datasets using T cell polyfunctionality as a model.
Keywords: polychromatic flow cytometry, computational biology/bioinformatics, ACCENSE, Pestle, SPICE
Abstract
Advancement in flow cytometry reagents and instrumentation has allowed for simultaneous analysis of large numbers of lineage/functional immune cell markers. Highly complex datasets generated by polychromatic flow cytometry require proper analytical software to answer investigators’ questions. A problem among many investigators and flow cytometry Shared Resource Laboratories (SRLs), including our own, is a lack of access to a flow cytometry‐knowledgeable bioinformatics team, making it difficult to learn and choose appropriate analysis tool(s). Here, we comparatively assess various multidimensional flow cytometry software packages for their ability to answer a specific biologic question and provide graphical representation output suitable for publication, as well as their ease of use and cost. We assessed polyfunctional potential of TCR‐transduced T cells, serving as a model evaluation, using multidimensional flow cytometry to analyze 6 intracellular cytokines and degranulation on a per‐cell basis. Analysis of 7 parameters resulted in 128 possible combinations of positivity/negativity, far too complex for basic flow cytometry software to analyze fully. Various software packages were used, analysis methods used in each described, and representative output displayed. Of the tools investigated, automated classification of cellular expression by nonlinear stochastic embedding (ACCENSE) and coupled analysis in Pestle/simplified presentation of incredibly complex evaluations (SPICE) provided the most user‐friendly manipulations and readable output, evaluating effects of altered antigen‐specific stimulation on T cell polyfunctionality. This detailed approach may serve as a model for other investigators/SRLs in selecting the most appropriate software to analyze complex flow cytometry datasets. Further development and awareness of available tools will help guide proper data analysis to answer difficult biologic questions arising from incredibly complex datasets.
Abbreviations
- 2D
2‐dimensional
- ACCENSE
automated classification of cellular expression by nonlinear stochastic embedding
- AF
Alexa Fluor
- BV
brilliant violet
- DBSCAN
density‐based spatial clustering of applications with noise
- FLOCK
FLOw clustering without K
- FlowSOM
self‐organizing maps of visualizing and interpretation of cytometry data
- HCV
hepatitis C virus
- NIAID
National Institute of Allergy and Infectious Diseases
- pMHC
peptide MHC
- PSM
probability state modeling
- SPADE
spanning‐tree progression analysis of density normalized events
- SPICE
simplified presentation of incredibly complex evaluations
- SRL
Shared Resource Laboratory
- t‐SNE
t‐distributed stochastic neighbor embedding
- Tyro
tyrosinase
- WT
wild‐type
Introduction
Advancement in flow cytometry instrumentation and reagent development has lifted the ceiling for the analysis of large numbers of cellular parameters. These improvements have allowed for synchronized identification of numerous immune system components, including lineage and functional markers of leukocytes, which have significant implications in understanding basic biologic behavior, immune monitoring techniques, and immunotherapy design. However, the use of polychromatic flow cytometry requires proper analytical software to analyze these highly complex datasets. Furthermore, different research questions may require individual tools to analyze the same dataset.
Currently, many investigators use a flow cytometry SRL at their institution for assistance in acquiring and analyzing flow cytometry data. However, there is a significant need for both investigators and SRLs to be knowledgeable of, acquire, and implement software that can adequately analyze highly complex datasets specific to individual needs. Furthermore, investigators and SRLs lacking access to bioinformatics cores struggle with the ability to choose and understand these complex software, which range greatly in their accessibility, user‐friendliness, and readability of graphical output. In our setting, there is no readily available bioinformatics team to assist with this complex data analysis, so it can become a daunting task to use these tools.
Here, we use a complex dataset case study as a model to choose an appropriate multidimensional software tool. Specifically, we are interested in the polyfunctional potential of TCR gene‐modified T cells and how alterations in pMHC ligands influence polyfunctional profiles, which could have significant implications in the design, efficacy, and safety of gene‐modified T cells. We have extensively studied a cross‐reactive TCR (HCV1406 TCR) that can transfer its reactivity to PBLs, recognizing naturally occurring mutant HCV epitopes and HCV+ hepatocellular carcinoma cells [1, 2, 3–4]. Whereas these studies have primarily relied on evaluation of IFN‐γ release to characterize changes in reactivity, we wanted to expand our functional profile by simultaneously evaluating IFN‐γ, TNF‐α, IL‐2, IL‐4, IL‐17A, IL‐22, and the lytic marker CD107a. We wanted to determine if polyfunctional phenotypes changed across mutant peptide and tumor stimulation conditions and/or between CD4+ and CD8+ T cell subsets.
The simultaneous analysis of 7 functional parameters resulted in 128 possible combinations of positivity/negativity for these functional markers. The evaluation of multiple peptide or tumor stimulation conditions and 2 T cell subsets yielded a grossly complex dataset that would be difficult to analyze with basic flow cytometry software. As such, the goals of this investigation were the following: to 1) identify which tools could appropriately perform this high‐dimensional analysis without the aid of a bioinformatics core that has expertise in flow cytometry data, an issue many SRLs, including ours, face and 2) generate graphical output to visually display T cell polyfunctionality simple enough to make comparisons across multiple treatment groups. Some of the requirements of tools evaluated were to represent rare and high‐frequency populations, visualize the data at a single‐cell level, preserve the relationships and geometry of the data, and provide an interpretable view of the data for publication or presentation. Programs currently available use manual and/or unsupervised manipulations for gating, and some use algorithms containing clustering, dimension reduction, hierarchy extraction, and/or t‐SNE. Of the various approaches investigated, coupled analysis in Pestle and SPICE provided the most user‐friendly manipulations and simple, comprehensible graphical output. ACCENSE also provides a meaningful interpretation while removing subjectivity by automated analysis, measuring intensity rather than Boolean gates and preserving single‐cell discrimination. Throughout, we use the same experimental dataset for each tool's evaluation. For simplicity, we demonstrate the ability of these software packages to illustrate polyfunctionality of CD8+ T cells against WT NS3 HCV:1406–1415 peptide stimulation. We comment on how various graphical representations may or may not be suitable for making comparisons across multiple treatment groups within this dataset. Increased awareness and demonstration of these and other tools will help guide investigators’ and SRLs’ decisions and proper implementation of sophisticated multidimensional analysis software.
MATERIALS AND METHODS
Cell lines, media, and reagents
293GP, PG13, and T2 cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). All media were obtained from Corning Life Sciences (Corning, NY, USA) unless otherwise noted. 293 GP cell lines were maintained in DMEM, supplemented with 10% FBS (Tissue Culture Biologicals, Long Beach, CA, USA). PG13 cells were maintained in IMDM + 10% FBS. The T2 cell line was maintained in RPMI‐1640 medium + 10% FBS. HCV NS3:1406–1415 (KLVALGINAV) and Tyro:368–376 (TMDGTMSQV) peptides were obtained at 95% purity from Synthetic Biomolecules (San Diego, CA, USA) and stored at −80°C in 100% DMSO (Sigma‐Aldrich, St. Louis, MO, USA).
T cells
PBMCs were isolated from apheresis products of normal donors purchased from Key Biologics (Memphis, TN, USA) using Ficoll‐Hypaque (Sigma‐Aldrich) density gradient centrifugation. PBMCs were stimulated with 50 ng/ml anti‐CD3 mAb (Miltenyi Biotec, Bergisch Gladbach, Germany), 3 d before retroviral transduction. T Cells were maintained in AIM V Medium (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 5% heat‐inactivated, pooled human AB serum (Valley Biomedical Products & Services, Winchester, VA, USA), 300 IU/ml recombinant human IL‐2 (Novartis Pharmaceuticals, East Hanover, NJ, USA), and 100 ng/ml recombinant human IL‐15 (NIH, Biological Resources Branch, Bethesda, MD, USA) at 37°C in a humidified 5% CO2 incubator.
Retroviral transduction
Retroviral supernatants were prepared using a stable retroviral producer cell line PG13 expressing HCV1406 TCR with a truncated CD34 expression marker in a modified SAMEN retroviral described previously [5]. Generation of producer cell lines, retrovirus harvest, and transduction by spinoculation have been described elsewhere [3, 4]. Transduced T cells were enriched for high/uniform transgene expression by positive selection with anti‐CD34‐labeled immunomagnetic beads (Miltenyi Biotec) [3, 6].
Antigen stimulation coculture
T2 cells were loaded with 10 μg/ml peptide for 2 h T2 cells (3 × 105) and 3 × 105 T cells were cocultured in a 1:1 ratio in 96‐well U‐bottom tissue‐culture plates with 2.5 μg/ml anti‐CD107a mAb, 5 ng/ml brefeldin‐A, and 2 nM monensin (all BioLegend, San Diego, CA, USA). Cocultures were incubated at 37°C/5% CO2 for 5 h. Cells were harvested, stained for cell‐surface antigens, fixed, permeabilized, and counterstained for intracellular cytokines, according to the manufacturer's protocols (BioLegend). Samples were analyzed by flow cytometry (see below).
Immunofluorescence staining panel
The staining panel consisted of the following mAb: anti‐CD3‐allophycocyanin/Cy7, anti‐CD4‐PE/Cy7, anti‐CD8‐FITC, anti‐CD34‐AF700, anti‐CD107a‐BV510, anti‐IFN‐γ‐BV421, anti‐TNF‐α‐BV711, anti‐IL‐2‐PerCP/Cy5.5, anti‐IL‐4‐AF647, anti‐IL‐17A‐BV570, and anti‐IL‐22‐PE. All fluorochrome‐conjugated antibodies were purchased from BioLegend.
Compensation and gating strategy
After antigen stimulation and immunofluorescence staining, cells were analyzed using a BD LSRFortessa instrument (BD Biosciences, San Jose, CA, USA). Compensation was performed using fluorochrome‐stained beads (BD Biosciences) and calculated using FACSDiva software (BD Biosciences). With the use of FlowJo X software (Tree Star, Ashland, OR, USA), lymphocyte populations were identified by forward‐scatter versus side‐scatter comparison, and events were gated on CD3 expression and subsequently gated as CD4+CD8− or CD4−CD8+ populations. CD4+ or CD8+ T cells were then gated on CD34+ expression to define our transduced cell populations. These CD3+CD4+CD8−CD34+ or CD3+CD4−CD8+CD34+ were then used as starting points for subsequent analysis of functional markers using software strategies described below.
Data analysis
To evaluate the different analysis methods uniformly, the same collected dataset was analyzed in each approach. Strategies used include FlowJo X (Tree Star), GemStone (Verity Software House, Topsham, ME, USA) [7], SPADE (Cytobank, Santa Clara, CA, USA) [8], FlowSOM (Bioconductor, http://www.bioconductor.org/) [9], viSNE (Cytobank) [10], FLOCK (ImmPort, https://immport.niaid.nih.gov/home) [11], ACCENSE (http://www.cellaccense.com/) [12], Pestle (http://www.drmr.com/pestle.zip), and SPICE (https://exon.niaid.nih.gov/spice) [13]. A detailed description of analysis methods is included for each approach within Results.
RESULTS
We analyzed complex multidimensional datasets to evaluate diverse polyfunctional behavior of TCR‐transduced T cell populations and how changes in pMHC ligands influence polyfunctional phenotypes. Our systematic approach to identify available software to analyze these datasets appropriately may help serve as a model in choosing the appropriate software, as the field is increasingly generating highly complex flow cytometry data. To evaluate uniformly the different analysis methods, we chose a single experimental dataset to analyze with the different outlined strategies. Here, we present a detailed description of the analysis methods and output generated by these approaches. For simplicity, we showed only comparisons of negatively (Tyro:368–376) or positively (WT HCV NS3:1406–1415) peptide‐stimulated HCV TCR‐transduced CD8+ T cells. It is pointed out where additional comparisons between CD8+ and CD4+ T cells, as well across variant peptide or tumor stimulation conditions, would be easily interpreted in subsequent analytical approaches or would make the data more complicated to discern a biologic message. It was imperative to generate graphical output that simply and clearly translates a message fit for publication or oral presentation.
Multidimensional analysis within FlowJo can be limited
FlowJo is a popular, effective, and user‐friendly tool for flow cytometry data analysis. Whereas it is the workhorse for flow cytometry data analysis in our laboratory, unidimensional histograms and biaxial dotplots limit its ability to display analysis graphically in >3 dimensions. After gating on our TCR‐transduced T cell populations, we can deduce which cytokines are present after antigen‐specific stimulation, but this is generally limited to coexpression of 1 or 2 functional markers at a time. However, simple and meaningful observations can still be made. For example, when comparing IFN‐γ production and lytic activity by CD107a expression, HCV NS3:1406–1415 peptide stimulation induced T cells to produce IFN‐γ and/or degranulate. (Supplemental Fig. 1A). This analysis also suggests that if a CD8+ T cell produces IL‐2, then it will almost always also produce TNF‐α (Supplemental Fig. 1A). A full matrix of pairwise comparisons allows for a simple interpretation of the relationship between 2 functional parameters (Supplemental Fig. 1B). Boolean gating within FlowJo also generated gates for each of the 128 possible phenotypes (2n, where n = number of parameters) given 7 parameters, but the graphical output in FlowJo does not easily accommodate these multiparameter comparisons simultaneously. However, population frequencies of each combination could be summed and binned into categories of 0–7 functional parameters (Supplemental Fig. 1C). This sequential gating method also provided the basis for other methods discussed below. Overall, whereas a very effective tool for low‐dimensional datasets, FlowJo is limited in its ability to graphically display comparisons easily for multidimensional data. The newest versions of FlowJo contain Plugins, applications that allow FlowJo to perform both data dimension reduction using t‐SNE and cluster analysis using SPADE. Use of these Plugins does require, however, installation of R and writing some R script. FCS Express (De Novo Software, Glendale, CA, USA), another flow cytometry analysis program, is now available with transformation modules for k‐means clustering analysis, Principal Component Analysis, and t‐SNE data dimension reduction. As these tools become commonly used, the interface with commercially available software should become more user friendly.
GemStone‐generated TriCOMs are effective for comparing a limited number of cytokines
GemStone is a commercially available product from Verity House Software. It uses PSM, which represents a set of cellular progressions or “cell types,” and was originally engineered to analyze and visualize multidimensional cellular populations [7]. The basis of PSM is a user‐defined model consisting of different cell types. With the use of the information available for each of the cell types, parameter profiles are created, and these cell types are used as progression steps in the model. Relationships among parameters may be discovered, and transitional cell types may also be defined. As a model is built, complexity may be added and then tested with the data to be analyzed. TriCOM is a visualization tool contained within the GemStone package that shows different phenotypes and activation states. TriCOM analysis displays concentric pie charts with multiple levels of positive coexpression ( Fig. 1A ). These pie charts can also be scaled to relative frequency within the levels of staining in a single sample across sample groups (Fig. 1B). Highlighted in red at the top of each TriCOM are the number of events or percentage of cells positive, respectively, and listed against each colored wedge of each TriCOM is a lettered code corresponding to the functional markers.
Figure 1.
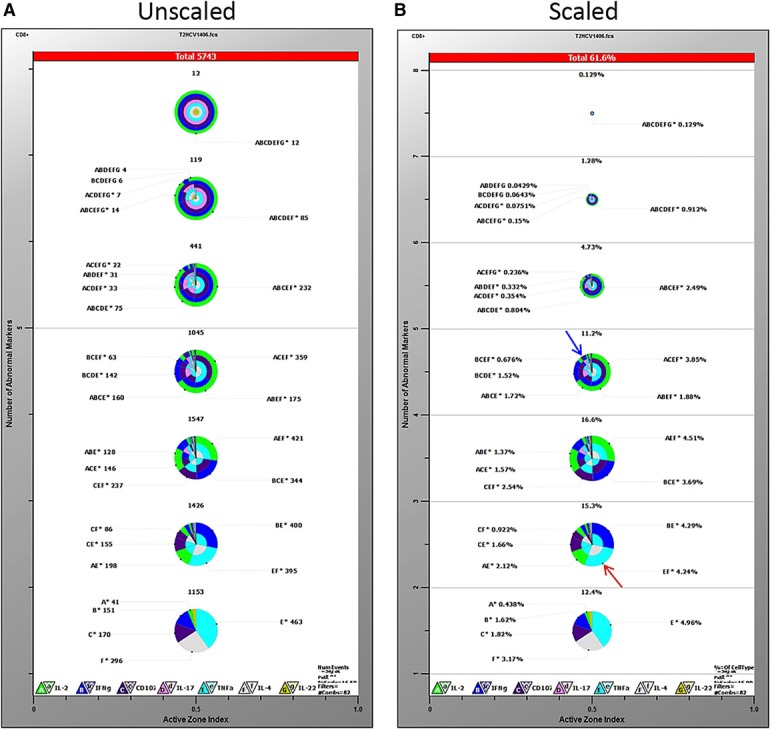
Gemstone TriCOM analysis. TriCOMs display percentages of cells positive for each functional marker in concentric pie charts in increasing orders of staining. The bottom pie chart represents cells positive for only 1 marker, and each pie chart above it represents an additional level of staining, up to 7 simultaneous markers (top pie chart). Higher order combinations represent single wedges with multiple colors. Overall percent reactive cells is shown at the top of each chart. Frequencies of various combinations are listed next to each wedge and are both letter and color coded: A/green = IL‐2, B/blue = IFN‐γ, C/purple = CD107a, D/pink = IL‐17A, E/turquoise = TNF‐α, F/gray = IL‐4, G/yellow = IL‐22. Pie charts can be represented as equal size (A) or scaled to the relative frequency within the levels of staining in a single sample (B). Percentages reflect that of CD8+ HCV TCR‐transduced T cells cocultured with the HCV NS3:1406–1415 peptide.
Major benefits of PSM analysis include accounting for population overlap, simple clustering routines, and identifying populations without giving biologic interrelationships. Additionally, gates are not drawn using GemStone, so there is neither subjectivity nor operator variability. Whereas gating defines positive and negative, GemStone allows for transitional populations and can allow for up‐ and down‐regulation of markers. One disadvantage, however, is that design of templates used for analysis requires knowledge of some biology of the experimental system. Verity House Software does provide significant help in generating such models.
For our purposes, the TriCOMs showed a distinct visualization of cytokines present at each order of staining, but the concentric pie charts became much harder to read when analyzing higher orders of staining (≥3 parameters). For example, percentages of cells expressing only 1 cytokine are clearly shown (TNF‐α = 4.96%, IL‐4 = 3.17%; bottom pie chart Fig. 1B). It is also still legible to discern that 4.24% of the transduced cells produce a combination of both TNF‐α and IL‐4 but no other functional parameters measured (Fig. 1B, highlighted by red arrow). It becomes less clear, however, when discerning populations >3 parameters or looking at even 2 parameter‐positive cells that occur in low frequencies (Fig. 1B, highlighted by blue arrow). This design makes it substantially harder to resolve potentially rare or infrequent functional populations. Overall, whereas TriCOMs are useful tools that clearly visualize phenotypes, they can became difficult to read at >3 parameters or for low‐frequency populations. GemStone also requires a large number of TriCOMs to make comparisons across treatment groups with such a large number of evaluated parameters.
Tools using clustering algorithms are not optimal if phenotypes measured fail to cluster
To analyze the potential of available clustering tools, we compared SPADE and FlowSOM's ability to analyze and present our T cell polyfunctionality data.
SPADE was created to aid in visualization and analysis of multiparameter data. This algorithm, originally available through Cytobank and now available in FlowJo and FCS Express, is designed to extract a hierarchy from high‐dimensional cytometry data in an unsupervised manner, which enables multiple cell types to be visualized in a branched tree structure [8]. SPADE contains 4 modules: 1) Density‐dependent down‐sampling allows rare, as well as abundant, populations to be represented equally. 2) Agglomerative clustering separates the down‐sampled data into groups containing cells with similar phenotypes. Since rare cells are represented equally, a node for these cells can be created. 3) A minimum spanning tree is created that connects all of the clusters with a minimal edge length. 4) The upsampling of data map cells to the appropriate cluster. The clusters are then organized and displayed as a 2D tree.
The major benefits of SPADE are that the user does not need to know a hierarchical order before analysis and that rare cell types are identifiable. It is also scalable with increasing numbers of parameters. As such, investigators can infer likely cellular processes and hierarchies without needing predetermined hierarchies. The spanning tree is only limited by choice of markers used in an experiment and the subsets used for building a tree. The major limitation of SPADE, however, is that data are represented as clusters rather than individual cells, so single cell resolution is lost.
SPADE has been most useful for studies looking at cell populations with mixed lineages or tracking distinct cellular progressions [8, 14, 15–16]. Clusters form nicely when markers are mutually exclusive from others or if there is a clear progression of cellular phenotypes. After spanning trees are generated, markers can be analyzed for their intensity within these nodes but only one at a time. In our analysis, a SPADE‐derived spanning tree is not optimal in determining a cellular progression of cytokine production, as one does seem to exist in our dataset, nor are certain cytokines mutually exclusive with others (consistent with FlowJo and GemStone‐generated output). Thus, instead of a tree, a web‐like pattern is generated. It is clear, however, that there are visual differences between spanning trees of HCV and Tyro stimulated transduced T cells ( Fig. 2A ). For our purposes, SPADE did not visually demonstrate well enough specific differences among populations for us to make meaningful conclusions. Perhaps additional parameters that would cluster naive, memory, activating, or inhibitory T cell markers would create more nodal or mutually exclusive populations, lending itself to SPADE analysis; however, such additions would restrict the number of functional markers evaluated with current instrument limitations.
Figure 2.
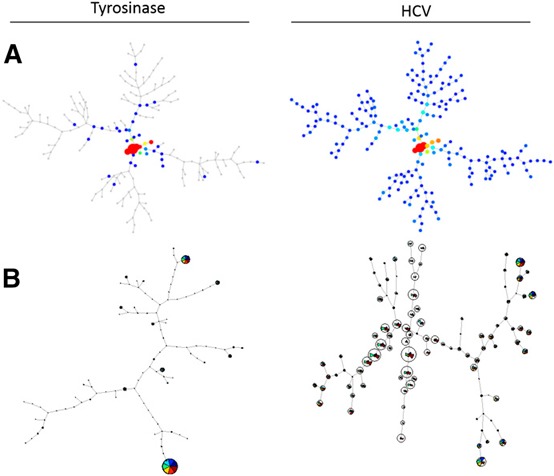
Graphical output provided by clustering programs SPADE and FlowSOM. All plots represent CD8+ HCV TCR‐transduced T cells after coculture stimulation with Tyro:368–376 or HCV NS3:1406–1415. (A) Spanning trees generated in SPADE. (B) FlowSOM‐created minimal spanning trees.
FlowSOM also focuses on clustering, as well as being a visualization aid [9]. As an R‐based program, available through Bioconductor, this algorithm consists of 4 steps. First, the data are read. Data can be compensated, transformed, and gated and then exported by FlowJo, or these functions may be performed using FlowSOM itself within R. Second, a self‐organizing map is built, which is an artificial neural network containing a grid of nodes. These nodes represent a single point in multidimensional space. Cells are then classified to the nearest node, and the grid places nodes that closely resemble each other spatially and proportionally. Third, a minimal spanning tree is built connecting the nodes that are most similar to each other in minimal branches. Lastly, meta‐clustering calculates the expected number of nodes if there is a much larger amount of clusters than the expected number of cell types. After this process, the data may be visualized either as a minimal spanning tree (similar to SPADE) or as a grid. Each node is coded as a pie chart with information about the phenotype of cells in that node.
The benefits of using FlowSOM are that even though this is an R‐based program, the use of the provided documentation and only a minimal understanding of R will allow one to use this program successfully. The program also does not tax the memory use of a desktop computer, and the output is very detailed. Each node of the minimal spanning tree contains colored wedges corresponding to the measured functional parameters (Fig. 2B). Similar to SPADE, there are clear visual differences between HCV peptide stimulation compared with negative control Tyro but for our purposes, was difficult to interpret. Overall, clustering tools can be very useful when tracking distinct cellular progressions or measuring changes in mixed cell populations with distinct lineages. However, when looking at a subset of T cells without mutually exclusive or markers present, clustering tools are not optimal to make these multidimensional analyses.
viSNE preserves cells in space but is difficult to compare multiple cytokines in a single plot
viSNE is also available through Cytobank, but FlowJo and FCS Express contain modules for a similar analysis. viSNE is a tool for the visualization of high‐dimensional, single‐cell data that place a cell in a 2D map but preserve the separation among types. This mapping takes advantage of the inherent structure of the data where different types are in separate regions in high‐dimensional space. viSNE uses a nonlinear dimensionality reduction algorithm that is based on t‐SNE [10]. Basically, t‐SNE calculates a distance matrix in a high‐dimensional space, which is transformed into a similarity matrix. Low‐dimensional similarities are calculated using Student's t distribution. viSNE generates a representation of this data that are similar to a biaxial plot and retains the geometry of the populations. The data are represented as cells in high‐dimensional data space and do so without relying on traditional gating strategies. viSNE can also discretely and automatically separate cells based on subtype, provided they exist. The cyt feature allows for coloring of cells based on selected expression markers. The data appear as a cloud biaxial plot with a specific geometry. Differences in populations can be seen as changes in the geometry, and events may be colored to determine which parameter or parameters have changed.
A major benefit with using viSNE is that comparisons are made in a high‐dimensional space. Populations that do not resolve in a 2D space can be resolved using viSNE. viSNE is also unsupervised and does not require in‐depth knowledge of the system being investigated. A limitation of viSNE is that low‐dimensional mapping cannot represent all of the information in high‐dimensional space, as viSNE only captures the most dominant structures. Additionally, plots become too crowded when >30,000 cells are shown.
For our purposes, viSNE was able to detect distinct changes in our populations when examining cytokines or CD107a singly ( Fig. 3 ). Whereas cyt plots provide clear distribution of individual markers, identification of multifunctional populations requires cumbersome, mental overlap of individual plots. The formation of comparisons across different T cell subsets or peptide stimulation conditions would even be more difficult to convey. Whereas others have shown cyt plots with color gradients coding for the number of cytokines present using t‐SNE within R [17], viSNE is currently unable to compare multiple cytokines spatially in the same plot and is not optimal for our analysis.
Figure 3.
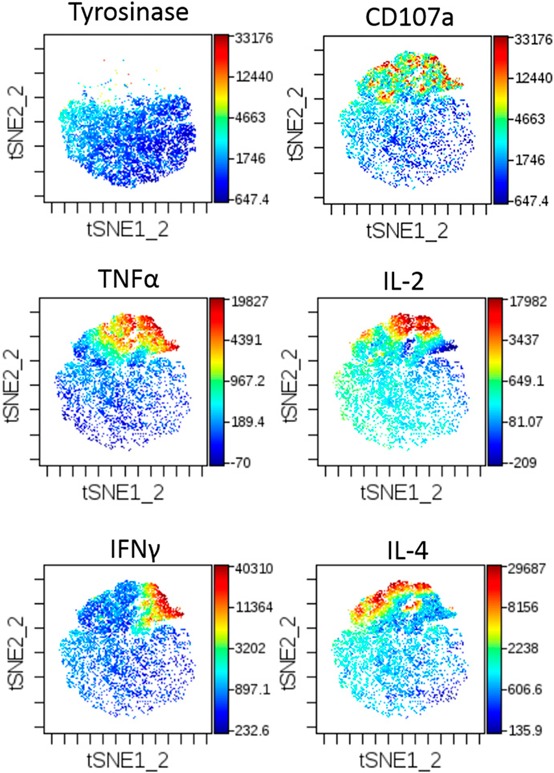
viSNE‐generated cyt maps. cyt maps display collected events in a Tyro peptide stimulation (top left) or HCV peptide stimulation (remaining panels) for expression of CD107a, TNF‐α, IL‐2, IFN‐γ, and IL‐4.
FLOCK allows for a quantitative analysis but with complicated presentation
As an unsupervised analysis using FLOCK, this algorithm is sponsored by NIAID for open use by the immunology research community and is accessible at https://immportgalaxy.org. FLOCK uses a rapid binning algorithm to identify unique populations in high‐dimension data. It can identify and compare populations from different samples and determine which parameters are most informative without subjective gating bias [11]. FLOCK also produces color‐coded populations within a matrix of biaxial plots, enhancing traditional 2D representation. FLOCK also has the ability to calculate many useful summary statistics through cross‐sample analysis to compare differences across treatment groups and is a model‐independent approach to multiparameter analysis.
In brief, there are 4 steps in this analysis algorithm. 1) Data preprocessing: compensated FCS files are converted to tab‐delimited text files, or “.fcs” files can be uploaded and converted to text files through FLOCK. 2) Grid‐based density clustering: data are separated into equally sized bins, and partitioning is applied to all dimensions in the dataset simultaneously. Each dimension is portioned into the same number of bins, resulting in partitions of n‐dimensional space called hyper‐regions. Each hyper‐region is assessed to determine the number of events in the region. If events exceed a specific threshold, then the hyper‐region is called dense. 3) Centroid generation: hyper‐regions are grouped together, and if they are adjacent to each other in n‐dimensional space, then they are called a dense hyper‐region group. The centroids representing all events in each dense hyper‐region group are then determined. 4) Event assignment: after centroids are determined, each event is assigned to its closest centroid. The centroids are recalculated, and cluster membership is computed again with the new centroids. This procedure, after a few iterations, is regarded as a modified k‐means determination. FLOCK quickly determines a stable centroid recalculation, and unique populations are identified. The visualization module of FLOCK contains 2D dot plots with each clustered population colored uniquely. Expression profiles of a population are presented to indicate the approximate expression level of each marker of a population as negative, low, or high.
Major advantages of FLOCK are that any up‐front gating can be performed using commercially available software and FLOCK's ease of use. Data can be easily uploaded and after answering a few questions, an analysis is requested. Users then receive notification after analysis has been completed. In our analysis, FLOCK is able to generate distinct populations based on intensity of staining for each parameter combination ( Fig. 4 ). For example, the red population (Pop. 1) corresponds to the third‐most prevalent population consisting of IL‐2 low, IFN‐γ negative, CD107a low, IL‐17A low, TNF‐α negative, IL‐4 low, and IL‐22 low expressing CD8+ T cells. The major drawback for our use of FLOCK, however, is the difficulty to demonstrate simply the differences among populations or across multiple treatment conditions given the large number of plots generated for a single sample. However, the ability to visualize each phenotypic cluster with corresponding quantitative data is a useful aspect of FLOCK and can resolve rare or unexpected phenotypes.
Figure 4.
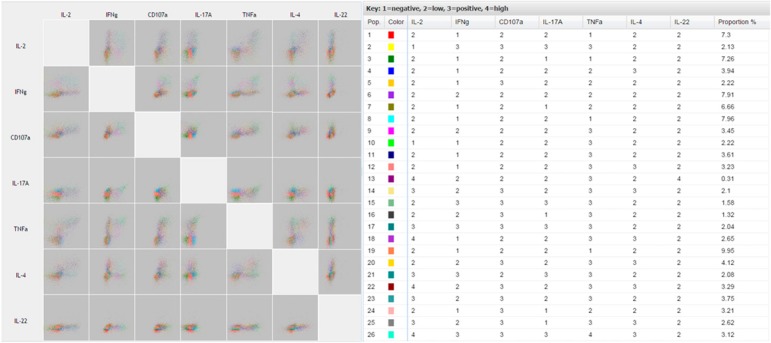
Quantitative multidimensional dotplots generated in FLOCK. FLOCK analysis shows pairwise comparisons using a matrix of dotplots to display the relationship of expression among the 7 functional parameters. Color‐coded populations for various combinations of markers are accompanied with frequency percentages and description of staining intensity: 1 = negative fluorescence; 2 = low fluorescence; 3 = positive fluorescence; 4 = high fluorescence.
Tandem analysis in Pestle/SPICE is user friendly; provides simple, pleasing graphical output; but relies on manual gating
SPICE uses large FlowJo data sets to normalize data graphically. The preprocessing of the data is performed using both FlowJo and Pestle. After sequential gating in FlowJo, Pestle offers data formatting and background subtraction of multivariate data sets, which are imported into SPICE for graphical and statistical analyses. Whereas background subtraction can result in below‐0 values, SPICE has a threshold approach that will minimize systematic bias and can maximize the amount of information that can be gained from positive measurements [13]. SPICE also offers the ability to compare the distributions statistically for all parameters. A χ2 measurement uses a nonparametric partial permutation (Monte Carlo simulation) to determine the differences among samples. Visualization of data includes pie charts, bar graphs, and cool plots—a type of heat map.
The major benefits of using Pestle/SPICE are the ease of use, readily available software, and its range of simple visualization of data. A major drawback of SPICE analysis, however, is that the data are initially manipulated in FlowJo, which means a significant amount of subjectivity is present. Another potential drawback is that the software is currently only offered for Apple Mac use.
For our purposes, tandem analysis in Pestle/SPICE offers graphical output that cannot only resolve and visually represent individual polyfunctional populations but also is useful for making comparisons across a large number of treatment groups. Figure 5 demonstrates the multiple ways that SPICE can graphically represent our polyfunctional data. Figure 5A displays frequencies of any of the 128 possible combinations of functional expression markers in our positive and negative stimulations (Tyro vs. HCV). A feature allows for overlaying arcs for up to 6 markers (Fig. 5B) to indicate which markers are present in each wedge. Still, the attempt to evaluate this many markers in a single pie chart does not easily, visually resolve dominant populations. Functional phenotypes can also be represented in a traditional bar chart format but require 128 different groups along the x‐axis to show the frequency of each phenotype in our dataset (not shown). A unique heat‐map feature, called a cool plot, is better suited for identifying polyfunctional populations present above an indicated threshold frequency, and it is the only tool evaluated thus far that can be used to display easily differences in phenotypes across multiple treatment groups. Figure 5C shows a representative cool plot, truncated to only show frequencies above background, displaying polyfunctional phenotypes by HCV TCR‐transduced CD8+ T cells stimulated with the WT and several mutant HCV peptides. This approach allows for a condensed display of combinatorial cytokine‐producing populations present in a given stimulation (read along the x‐axis), whereas perturbations in these individual population frequencies (shades of blue) by differential ligand stimulations are resolved in the y‐axis direction. From this representation, we can clearly define populous phenotypes, including IL‐2+CD107a+TNF‐α+IL‐4+ T cells, and that their frequency is consistent between WT and mutant peptides 1 and 3 but decreased in the presence of mutants 2 and 4. Overall, this approach is easy to use with familiar data processing techniques and offers a wide variety of simple graphical representations capable of making sophisticated comparisons, despite user subjectivity generated by preprocessing in FlowJo.
Figure 5.
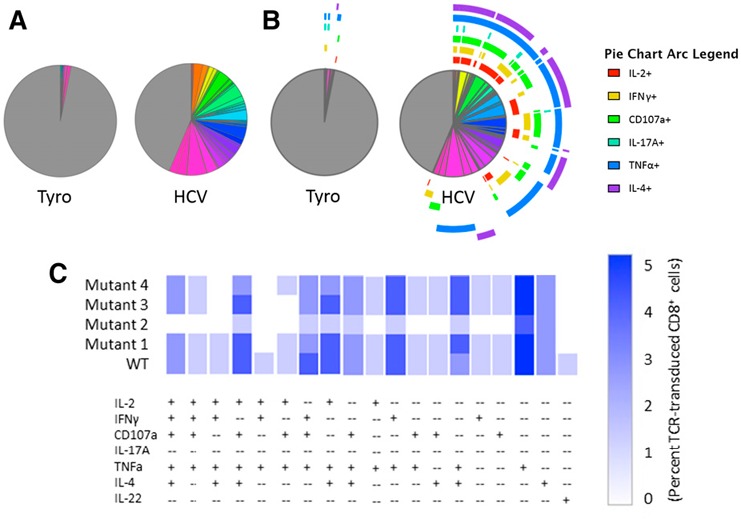
SPICE‐generated graphical output. Boolean‐gated frequencies within FlowJo were formatted within Pestle and exported into SPICE. Various graphical options include pie charts without (A) or with (B) overlaying phenotype arcs, bar graphs (data not shown), or cool plot (C). A representative cool plot condensed to show only functional phenotypes present above background compares phenotypes for WT and several mutant HCV peptide‐stimulated HCV TCR‐transduced CD8+ T cells.
ACCENSE uses stochastic neighbor embedding to calculate a 2D distribution and clusters the data into phenotypic subpopulations
ACCENSE data preprocessing is performed using commercially available software, if necessary. When the populations to be analyzed have been identified and gated, the data are exported as either a “.cvs” or “.fcs” file. This file is directly imported into ACCENSE. t‐SNE is used for data dimension reduction. Two varieties of t‐SNE are available: standard t‐SNE is used for small data sets, and the Barnes‐Hut‐SNE is for large data sets (>5000). The cell clustering can be performed using either k‐means or DBSCAN. k‐means clusters cells into groups with similar protein expression, and DBSCAN finds clusters when the population is not normally distributed.
To run ACCENSE, data are imported, variables are defined, and the data dimension reduction proceeds. After completion, the data can be visualized, and phenotype clusters are displayed. An output file, in “.cvs” form, is created, which contains all of the previous data, as well as t‐SNE and cell classification for each event. This data can then be analyzed further using a variety of programs. The data can be visually demonstrated using a sample clustered plot, and the exported “.cvs” file can be analyzed in any variety of programs to demonstrate differences in populations.
ACCENSE is available at http://www.cellaccense.com/ in both Mac and personal computer versions. This is freeware and can be run on a desktop computer. The program is easy to use and can be performed on a local computer. Although it is difficult for some users to understand dimension reduction, the differences between Tyro and HCV peptide stimulations from this type of analysis are evident ( Fig. 6 ). As cluster analysis is also performed, we can determine which populations have been changed among multiple samples. This type of analysis has been demonstrated to reveal heterogeneity in populations that previously had not been known as heterogeneous [12]. The color coding of subpopulations also makes it easier to visualize differences rather than viSNE plots alone. However, comparisons across altered pMHC ligand stimulations and between CD4+ and CD8+ T cell subsets would require a large number of cluster plots. Moreover, the identity of discrete phenotypes is not immediately evident from this type of display. The most significant advantages in using ACCENSE are that the analysis is not subjective, cells are grouped in clusters that are similar, and a single plot is able to demonstrate all of the dimensions of the data. The disadvantage in using ACCENSE or any other data dimension reduction analysis, however, is in communicating to users, reviewers, and audiences a way to trust and interpret this data.
Figure 6.
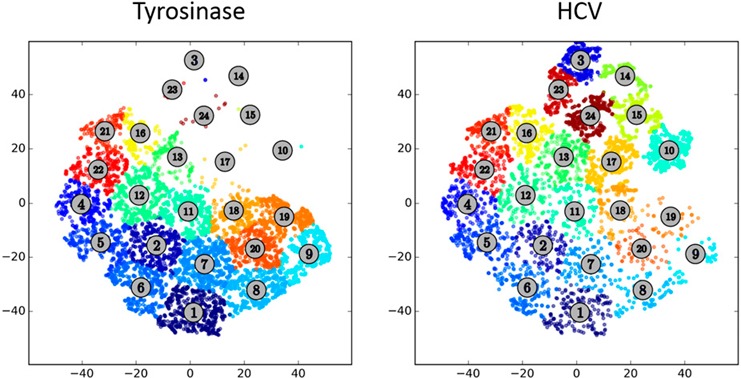
ACCENSE‐generated cluster pots. Clustered populations in ACCENSE reveal heterogeneity of populations between Tyro (left) or HCV (right) peptide‐stimulated, TCR‐transduced CD8+ T cells.
DISCUSSION
As vast developments in available reagents and instrumentation have provided investigators seemingly limitless ways to target large numbers of cellular markers of leukocytes and other immune cells, the ability to analyze complex datasets is lagging behind the ability to perform the experiments. Whereas useful software is still being developed and optimized, access to and knowledge of the currently available tools to answer biologic questions appropriately are not necessarily well known. Additionally, the absence of a readily available bioinformatics team to assist with complex data analysis can grossly impede the choice and/or use of appropriate software.
As such, we reported on several available software packages’ abilities to assess the polyfunctionality of HCV TCR‐transduced T cells after antigen‐specific stimulation to serve as a model approach in identifying appropriate tools to analyze multidimensional flow cytometry datasets. Our comparative analysis highlighted strengths and weaknesses in the methods of each strategy, as well as the appropriateness for using them for our studies. User‐friendliness, graphical output, and simple interpretation of the data were certainly not equal among the software types, but each offers its own advantages and optimal applications. A summary of these findings can be found in Table 1 . For our purposes, the ability to visualize polyfunctional potential by HCV TCR‐transduced T cells was best achieved using either ACCENSE or tandem analysis in Pestle/SPICE, providing a global few of polyfunctional phenotypes, the ability to resolve rare and unexpected populations, and the capability of displaying simple graphical output for large treatment group comparisons. ACCENSE removes subjectivity, visualizes populations that cannot be resolved in 1 or 2 dimensions in identifiable clusters, and does not rely on a binary classification of positive or negative phenotypes by using centroid values. SPICE, however, was more user friendly, interfacing with the widely popular FlowJo, and offered the simplest graphical visualization to make large comparisons when scaling analyses to include comparisons across numerous stimulation or treatment conditions. Although with the reliance on manual gating and discrete positive or negative phenotyping, this type of analysis is more comfortable. However, approaches similar to ACCENSE for a more unbiased analysis of complex datasets should be encouraged. Questions still unanswered in this data set are which clusters of cells change upon stimulation and how to compare and quantify those changes.
Table 1.
Summary of approaches used to analyze antigen‐specific polyfunctional T cell responses
| Software | Analysis strategy | Advantages | Disadvantages | Graphical display | Availability |
|---|---|---|---|---|---|
| FlowJo | Basic flow cytometry software package; manual, sequential gating | Popular, widely used software; output can be imported into other multidimensional software tools | Limited in scope for analyzing >2 dimensions; time consuming and subjective gating | Histograms and dot plots | Tree Star (P) |
| GemStone | PSM; template‐driven analysis | Accounts for population overlap and simple clustering routines; lack of gating eliminates subjectivity and operator variability | Templates require knowledge of some biology of the system; TriCOMs visually hard to interpret and compare ≥3 parameters | TriCOMs | Verity Software House (P) |
| SPADE | Unsupervised clustering extracts cellular hierarchy | No prior knowledge of hierarchical order needed; scalable; better for mutually exclusive markers and mixed lineage populations | Represents data as clusters rather than individual cells; does not appropriately analyze if data do not lend themselves to clustering | Spanning trees | Cytobank (F/P) |
| FlowSOM | R‐based clustering tool | Similar to SPADE; although R based, only requires minimal understanding of R to use effectively | Similar to SPADE; detailed data challenging to compare across multiple treatment groups | Minimal spanning trees or grids | Bioconductor (F) |
| viSNE | Nonlinear dimensionality reduction algorithm based on t‐SNE | Unsupervised and does not require in‐depth knowledge of experimental system; preserves cell separation and retains prior gating information | Low‐dimensional mapping cannot represent all of the information in a high‐dimensional space; a large number of cyt maps require visual overlay to make multidimensional comparisons | cyt maps | Cytobank (F/P) |
| FLOCK | Unsupervised rapid binning | Up‐front gating uses familiar FlowJo; delineates population‐based intensity of expression profiles; cross‐sample statistical analysis | Difficult to demonstrate differences among populations with very complex matrices | Color‐coded dot plots | ImmPort (F) |
| ACCENSE | Combines nonlinear dimensionality reduction with k‐means clustering | Automated cell classification while retaining single‐cell resolution; color codes identified populations; facilitates downstream statistical analysis with tabular data output | This type of analysis is more challenging to communicate to users; individual plot per sample makes comparison across multiple treatment groups more difficult | Color‐coded, multi‐dimensional cluster plots | ACCENSE (F) |
| SPICE | Quantitatively compares discrete phenotypic profiles in a mixture; uses FlowJo output with intermediary formatting tool Pestle | Ease of use and clear visualization of complex datasets; offers background subtraction and permutation statistical analysis; permits comparison across large numbers of treatment groups | Manual gating within FlowJo affords significant amount of subjectivity; software only works on Macintosh operating systems | Pie charts, bar graphs, cool plots | NIAID (F) |
Certainly the list of approaches used in this study is not exhaustive, and there exist a variety of other software types designed for multidimensional analysis, including Flame [18], Wanderlust [19], Citrus [20], and a host of R‐based tools, such as RchyOptimyx [21]. But even with numerous available options, not all are well known and easily accessible, provide interpretable output, or are appropriate for answering certain questions. However, we believe that our approach outlined in this report may serve as a guide for laboratories with limited access to bioinformatics teams in choosing appropriate tools to aid investigators in complex data analysis. It is imperative that communication of available tools and their use become more widespread. As this happens, laboratory personnel will be better able to help identify, acquire, and implement the appropriate software to analyze adequately the growing number of complex datasets in flow cytometry.
AUTHORSHIP
T.T.S., M.I.N., and P.E.S. provided conception/design, acquisition/analysis/interpretation, drafting/revising, and final approval of manuscript.
DISCLOSURES
The authors have reviewed the Conflict of Interest Disclosure policy and declare no conflicts of interest.
Supporting information
Supplementary Material Files
ACKNOWLEDGMENTS
Support for this work was provided by the U.S. National Institutes of Health, National Cancer Institute: P01 CA154778 (to M.I.N.), R01 CA104947 (to M.I.N.), R01 CA104947‐S1 (to M.I.N.), R01 CA90873 (to M.I.N.), R01 CA102280 (to M.I.N.), and F30 CA180731 (to T.T.S.). The authors thank Beth Hill of Verity Software House for providing assistance in using GemStone templates and generation of TriCOMs and Rochelle Diamond for critical review of this manuscript.
Footnotes
P, Software for purchase; F, freeware; F/P, freeware with additional purchasable options.
REFERENCES
- 1. Callender, G. G. , Rosen, H. R. , Roszkowski, J. J. , Lyons, G. E. , Li, M. , Moore, T. , Brasic, N. , McKee, M. D. , Nishimura, M. I. (2006) Identification of a hepatitis C virus‐reactive T cell receptor that does not require CD8 for target cell recognition. Hepatology 43, 973–981. [DOI] [PubMed] [Google Scholar]
- 2. Rosen, H. R. , Hinrichs, D. J. , Leistikow, R. L. , Callender, G. , Wertheimer, A. M. , Nishimura, M. I. , Lewinsohn, D. M. (2004) Cutting edge: identification of hepatitis C virus‐specific CD8+ T cells restricted by donor HLA alleles following liver transplantation. J. Immunol. 173, 5355–5359. [DOI] [PubMed] [Google Scholar]
- 3. Spear, T. T. , Callender, G. G. , Roszkowski, J. J. , Moxley, K. M. , Simms, P. E. , Foley, K. C. , Murray, D. C. , Scurti, G. M. , Li, M. , Thomas, J. T. , Langerman, A. , Garrett‐Mayer, E. , Zhang, Y. , Nishimura, M. I. (2016) TCR gene‐modified T cells can efficiently treat established hepatitis C‐associated hepatocellular carcinoma tumors. Cancer Immunol. Immunother. 65, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spear, T. T. , Riley, T. P. , Lyons, G. E. , Callender, G. G. , Roszkowski, J. J. , Wang, Y. , Simms, P. E. , Scurti, G. M. , Foley, K. C. , Murray, D. C. , Hellman, L. M. , McMahan, R. H. , Iwashima, M. , Garrett‐Mayer, E. , Rosen, H. R. , Baker, B. M. , Nishimura, M. I. (2016) Hepatitis C virus‐cross‐reactive TCR gene‐modified T cells: a model for immunotherapy against diseases with genomic instability. J. Leukoc. Biol. 100, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spear, T. T. , Callender, G. G. , Roszkowski, J. J. , Moxley, K. M. , Simms, P. E. , Foley, K. C. , Murray, D. C. , Scurti, G. M. , Li, M. , Thomas, J. T. , Langerman, A. , Garrett‐Mayer, E. , Zhang, Y. , Nishimura, M. I. (2016) TCR gene‐modified T cells can efficiently treat established hepatitis C‐associated hepatocellular carcinoma tumors. Cancer Immunol. Immunother. 65, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Norell, H. , Zhang, Y. , McCracken, J. , Martins da Palma, T. , Lesher, A. , Liu, Y. , Roszkowski, J. J. , Temple, A. , Callender, G. G. , Clay, T. , Orentas, R. , Guevara‐Patiño, J. , Nishimura, M. I. (2010) CD34‐based enrichment of genetically engineered human T cells for clinical use results in dramatically enhanced tumor targeting. Cancer Immunol. Immunother. 59, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bagwell, C. B. , Hunsberger, B. C. , Herbert, D. J. , Munson, M. E. , Hill, B. L. , Bray, C. M. , Preffer, F. I. (2015) Probability state modeling theory. Cytometry A 87, 646–660. [DOI] [PubMed] [Google Scholar]
- 8. Qiu, P. , Simonds, E. F. , Bendall, S. C. , Gibbs, K. D. Jr. , Bruggner, R. V. , Linderman, M. D. , Sachs, K. , Nolan, G. P. , Plevritis, S. K. (2011) Extracting a cellular hierarchy from high‐dimensional cytometry data with SPADE. Nat. Biotechnol. 29, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Gassen, S. , Callebaut, B. , Van Helden, M. J. , Lambrecht, B. N. , Demeester, P. , Dhaene, T. , Saeys, Y. (2015) FlowSOM: using self‐organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- 10. Amir, el. A.D. , Davis, K. L. , Tadmor, M. D. , Simonds, E. F. , Levine, J. H. , Bendall, S. C. , Shenfeld, D. K. , Krishnaswamy, S. , Nolan, G. P. , Pe'er, D. (2013) viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qian, Y. , Wei, C. , Eun‐Hyung Lee, F. , Campbell, J. , Halliley, J. , Lee, J. A. , Cai, J. , Kong, Y. M. , Sadat, E. , Thomson, E. , Dunn, P. , Seegmiller, A. C. , Karandikar, N. J. , Tipton, C. M. , Mosmann, T. , Sanz, I. , Scheuermann, R. H. (2010) Elucidation of seventeen human peripheral blood B‐cell subsets and quantification of the tetanus response using a density‐based method for the automated identification of cell populations in multidimensional flow cytometry data. Cytometry B Clin. Cytom. 78 (Suppl 1), S69–S82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shekhar, K. , Brodin, P. , Davis, M. M. , Chakraborty, A. K. (2014) Automatic classification of cellular expression by nonlinear stochastic embedding (ACCENSE). Proc. Natl. Acad. Sci. USA 111, 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roederer, M. , Nozzi, J. L. , Nason, M. C. (2011) SPICE: exploration and analysis of post‐cytometric complex multivariate datasets. Cytometry A 79, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diggins, K. E. , Ferrell, Jr., P. B. , Irish, J. M. (2015) Methods for discovery and characterization of cell subsets in high dimensional mass cytometry data. Methods 82, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jobin, C. , Cloutier, M. , Simard, C. , Neron, S. (2015) Heterogeneity of in vitro‐cultured CD34+ cells isolated from peripheral blood. Cytotherapy 17, 1472–1484. [DOI] [PubMed] [Google Scholar]
- 16. Qiu, P. (2012) Inferring phenotypic properties from single‐cell characteristics. PLoS One 7, e37038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin, L. , Frelinger, J. , Jiang, W. , Finak, G. , Seshadri, C. , Bart, P. A. , Pantaleo, G. , McElrath, J. , DeRosa, S. , Gottardo, R. (2015) Identification and visualization of multidimensional antigen‐specific T‐cell populations in polychromatic cytometry data. Cytometry A 87, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pyne, S. , Hu, X. , Wang, K. , Rossin, E. , Lin, T. I. , Maier, L. M. , Baecher‐Allan, C. , McLachlan, G. J. , Tamayo, P. , Hafler, D. A. , De Jager, P. L. , Mesirov, J. P. (2009) Automated high‐dimensional flow cytometric data analysis. Proc. Natl. Acad. Sci. USA 106, 8519–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bendall, S. C. , Davis, K. L. , Amir, el‐A. D. , Tadmor, M. D. , Simonds, E. F. , Chen, T. J. , Shenfeld, D. K. , Nolan, G. P. , Pe'er, D. (2014) Single‐cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruggner, R. V. , Bodenmiller, B. , Dill, D. L. , Tibshirani, R. J. , Nolan, G. P. (2014) Automated identification of stratifying signatures in cellular subpopulations. Proc. Natl. Acad. Sci. USA 111, E2770–E2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aghaeepour, N. , Jalali, A. , O'Neill, K. , Chattopadhyay, P. K , Roederer, M. , Hoos, H. H. , Brinkman, R. R. (2012) RchyOptimyx: cellular hierarchy optimization for flow cytometry. Cytometry A 81, 1022–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files