

Short abstract
Review on emerging combinations of immunotherapeutic agents, and treatment regimens which combine immunotherapy with drugs targeting cancer cells.
Keywords: immune checkpoints, immunogenic cell death, senescence, melanoma
Abstract
Immune‐checkpoint blockade therapy with antibodies targeting CTLA‐4 and PD‐1 has revolutionized melanoma treatment by eliciting responses that can be remarkably durable and is now advancing to other malignancies. However, not all patients respond to immune‐checkpoint inhibitors. Extensive preclinical evidence suggests that combining immune‐checkpoint inhibitors with other anti‐cancer treatments can greatly improve the therapeutic benefit. The first clinical success of the combinatorial approach to cancer immunotherapy was demonstrated using a dual‐checkpoint blockade with CTLA‐4 and PD‐1 inhibitors, which resulted in accelerated FDA approval of this therapeutic regimen. In this review, we discuss the combinations of current and emerging immunotherapeutic agents in clinical and preclinical development and summarize the insights into potential mechanisms of synergistic anti‐tumor activity gained from animal studies. These promising combinatorial partners for the immune‐checkpoint blockade include therapeutics targeting additional inhibitory receptors of T cells, such as TIM‐3, LAG‐3, TIGIT, and BTLA, and agonists of T cell costimulatory receptors 4‐1BB, OX40, and GITR, as well as agents that promote cancer cell recognition by the immune system, such as tumor vaccines, IDO inhibitors, and agonists of the CD40 receptor of APCs. We also review the therapeutic potential of regimens combining the immune‐checkpoint blockade with therapeutic interventions that have been shown to enhance immunogenicity of cancer cells, including oncolytic viruses, RT, epigenetic therapy, and senescence‐inducing therapy.
Abbreviations
- 5‐AZA‐CdR
= 5‐aza‐2′‐deoxycytidine
- AML
= acute myelogenous leukemia
- ASCO
= American Society of Clinical Oncology
- BTLA
= B and T lymphocyte attenuator
- CD40L
= cluster of differentiation 40 ligand
- DAMP
= damage‐associated molecular pattern
- DNMT
= DNA methyltransferase
- FDA
= U.S. Food and Drug Administration
- Gal‐9
= Galectin‐9
- GITR
= glucocorticoid‐induced TNFR‐related protein
- HDAC
= histone deacetylase
- HMGB1
= high‐mobility group box 1
- ICD
= immunogenic cell death
- LAG‐3
= lymphocyte‐activated gene 3
- MDSC
= myeloid‐derived suppressor cell
- MHC I/II
= MHC class I/II
- MICA/B
= MHC class I‐related chain A/B
- NKG2D
= NK group 2, member D
- NSCLC
= nonsmall cell lung carcinoma
- ORR
= objective response rate
- OS
= overall survival
- PD‐1
= programmed cell death 1
- PD‐L1/2
= programmed cell death ligand 1/2
- PFS
= progression‐free survival
- RCC
= renal cell carcinoma
- RT
= radiotherapy
- SASP
= senescence‐associated secretory phenotype
- T‐VEC
= talimogene laherperpvec
- TA
= tumor antigen
- TIGIT
= T cell Ig and ITIM domain
- TIL
= tumor‐infiltrating lymphocyte
- TIM‐3
= T cell Ig‐ and mucin domain‐containing molecule 3
- TME
= tumor microenvironment
- Treg
= regulatory T cell
- ULBP
= UL16‐binding protein
Introduction
The ultimate goal of immunotherapeutic cancer treatment is to make the immune system more efficient in killing tumor cells. Early approaches to cancer immunotherapy included peptide vaccination and high‐dose cytokines and had modest clinical activity overall. The breakthrough in the field of cancer immunotherapy came from the basic immunology research investigating fundamental mechanisms of T cell function. These studies have led to the clinical development and licensing of therapeutics targeting 2 distinct inhibitory receptors of T cells, CTLA‐4 and PD‐1, which induce durable responses in a small but significant proportion of patients. These agents have transformed the therapy of metastatic melanoma, a disease notorious for promptly developing resistance to traditional systemic treatments. Inspired by this success, a major focus of current translational cancer research is exploring the clinical use of manipulating other molecules involved in T cell regulation, as well as finding other ways of rendering tumors responsive to immunotherapies.
IMMUNE‐CHECKPOINT INHIBITORS APPROVED FOR TREATMENT OF CANCER
Ipilimumab, a mAb targeting the coinhibitory receptor CTLA‐4 (CD152), was the first immune‐checkpoint inhibitor to be approved for treatment of human cancer. CTLA‐4 plays a critical role in negative regulation of T cell function. It attenuates T cell responses by interfering with their costimulation via costimulatory receptor CD28 [1]. CD28 has 2 ligands, B7‐1 (CD80) and B7‐2 (CD86), which are expressed on APCs. When a T cell recognizes an MHC‐bound peptide antigen via the TCR, CD28 provides the second signal critical for activation of naïve T cells, which results in their proliferation, production of cytokines, and survival [1]. CTLA‐4 shares structural similarity with CD28 and can bind to the ligands of CD28, B7‐1 and B7‐2, albeit with much higher affinity [2, 3]. Furthermore, CTLA‐4 is known to be critical for inhibitory function of Tregs [4]. The blocking of interactions between CTLA‐4 and the B7 molecules leads to persistent T cell responses and tumor eradication [5, 6]. The anti‐tumor effect of anti‐CTLA‐4 therapy is attributed to the enhancement of T cell priming, which is associated with generation of new tumor‐responsive T cells [7], as well as the Fc‐dependent depletion of Tregs in the TME [8].
Anti‐CTLA‐4 therapy with ipilimumab significantly improved OS for patients with metastatic melanoma in clinical trials [9, 10] and was approved by the FDA for treatment of this disease in 2011. One large Phase 3 study of 676 patients receiving ipilimumab and/or a peptide vaccine reported a significant benefit in OS for ipilimumab‐treated cohorts, with a median survival of 10.0 mo for ipilimumab plus vaccine versus 10.1 mo for ipilimumab alone versus 6.4 mo for peptide vaccine alone [9]. Furthermore, in a Phase 3 trial of tremelimumab—another anti‐CTLA‐4 antibody in development—the response duration was longer for patients who received tremelimumab compared with the chemotherapy‐treated cohort (35.8 vs. 13.7 mo), although the difference in OS between treatment groups did not meet statistical significance [11]. Despite the demonstrated OS advantage, the ORRs in multiple clinical trials of anti‐CTLA‐4 antibodies are typically only ∼10% [12]. Some patients may experience prolonged stable disease or atypical immune‐related responses (including tumor growth, followed by shrinkage).
On the downside, the treatment with anti‐CTLA‐4 antibodies is often associated with significant side‐effects related to aberrant immune activation in multiple organ systems, including skin (dermatitis, pruritus), gastrointestinal tract (diarrhea, colitis), liver (hepatitis), and endocrine organs (hypophysitis, thyroiditis, hypothyroidism, adrenal insufficiency) [9]. Common treatment‐related toxicities together with modest response rates prompted an extensive search for biomarkers to guide the selection of patients who are likely to respond to therapy without developing serious complications. Snyder et al. [13] sequenced exomes in 64 melanoma tumors and showed that a high level of mutations in tumors correlates with a sustained clinical benefit from the CTLA‐4 blockade. However, high mutational load alone is not sufficient to predict clinical benefit, as not all of the tumors with a high mutational burden responded to therapy. The authors then identified candidate tumor neoantigens that arose from the somatic tumor mutations using genome‐wide somatic neoepitope analysis and found that these predicted neoantigens activated T cells from the patients treated with ipilimumab. Likewise, Van Allen et al. [14] showed that the number of nonsynonymous mutations per tumor correlated with clinical responses in ipilimumab‐treated melanoma patients in their whole exome analysis of 110 pretreatment melanoma tumor biopsies and matching germline tissue samples. However, no recurrent neoantigen peptide sequences that can predict patients’ benefit to the CTLA‐4 blockade were found. Among other potential biomarkers, absolute lymphocyte counts and increased frequency of ICOS+ CD4 T cells after the first 2 doses of ipilimumab therapy have been associated with therapeutic benefit, whereas baseline serum concentrations of soluble CD25 predicted resistance to therapy [15, 16–17].
Superior clinical outcomes in a wider range of cancer types have been demonstrated by the antibodies that target PD‐1, another immune‐checkpoint receptor that inhibits responses of activated T cells [18]. The ligands of PD‐1, PD‐L1 and PD‐L2, are expressed on many cell types, including T cells, epithelial cells, endothelial cells, and tumor cells. These ligands are induced in response to IFN‐γ, which is produced by activated T cells [19]. Therefore, unlike CTLA‐4 that functions early in T cell activation to block costimulation, the PD‐1/PD‐L1 pathway inhibits T cells that have already been activated, thus protecting healthy host cells from T cell attack. Tumors often use the PD‐1/PD‐L1 immune checkpoint to escape immune surveillance by overexpressing PD‐L1 (B7‐H1) in the TME [20]. Consequently, the blocking of the PD‐1/PD‐L1 axis in such tumors is expected to “release the breaks” on the anti‐tumor immune response.
Several PD‐1 antagonists, such as nivolumab and pembrolizumab, demonstrate excellent activity against melanoma, RCC, and NSCLC, leading to significant improvement of patients’ OS. PD‐1‐ and PD‐L1‐blocking antibodies are currently being tested against a spectrum of other solid and hematologic malignancies, including bladder, prostate, head and neck, breast cancer, and Hodgkin lymphoma [21]. In a Phase 3 study involving 418 previously untreated patients with metastatic melanoma without a BRAF mutation, nivolumab outperformed chemotherapy with a 1 yr OS rate of 72.9% and an ORR of 40% (compared with 42.1% 1 yr OS and 13.9 ORR in dacarbazine group) [22]. Likewise, pembrolizumab showed a significant improvement of response rates compared with ipilimumab (33.7 vs. 11.9%), as well as improved estimated 12 mo survival rates (74.1 vs. 58.2%) [23]. Furthermore, anti‐PD‐1 therapy with pembrolizumab or nivolumab is effective after ipilimumab failure [24, 25]. Notably, compared with CTLA‐4, substantially fewer serious immune‐related adverse events were reported with anti‐PD‐1 therapy. Anti‐PD‐1 immunotherapy has been cleared for clinical use in the United States, as well as in several other developed countries, including Europe, Australia, and Japan. Given the efficacy and relative lack of toxicities of anti‐PD‐1 therapy, the rise of its global use is exciting. However, the high cost of this therapy will be a barrier for many patients and will need to decrease substantially before it is accessible to low‐income countries. Identification of biomarkers that accurately pinpoint potential responders would certainly help to facilitate the deployment of anti‐PD‐1/PD‐L1 therapy in low‐income settings.
There are several potential biomarkers being evaluated for their ability to predict clinical response to PD‐1/PD‐L1 therapy. Among those, the level of PD‐L1 expression in the tumor is, perhaps, the potential biomarker of most debate. The analysis of tumors from patients on an anti‐PD‐1 clinical trial identified absence of tumor PD‐L1 expression as a potential predictor of poor therapeutic response. In this study, 9 out of 25 patients with PD‐L1‐positive tumors responded to the PD‐1 blockade, whereas none of the 17 patients with undetectable tumor PD‐L1 expression responded [26]. Likewise, in a clinical study of anti‐PD‐L1, therapeutic response was associated with high levels of PD‐L1 in tumors, especially when PD‐L1 was expressed by tumor‐infiltrating immune cells [27]. However, whereas many other trials confirmed that PD‐L1‐positive tumors tend to respond better to the PD‐1/PD‐L1 blockade, collective evidence demonstrates that PD‐L1 status alone is not always an accurate predictor of response. A recent meta analysis of 20 distinct trials of anti‐PD‐1 and anti‐PD‐L1 agents, involving 1475 patients collectively, reported a statistically significant association between the tumor PD‐L1 expression and the response to PD‐1 therapy; however, there were responders in both PD‐L1‐positive and ‐negative groups. Specifically, an ORR of 34.1% was found in patients with PD‐L1‐positive and 19.9% in patients with PD‐L1‐negative tumors [28]. Moreover, recently reported results from randomized Phase 3 clinical trials of PD‐1 antagonists in melanoma, RCC, and NSCLC demonstrated efficacy in patients with either PD‐L1‐positive or ‐negative tumors [29, 30–31]. Based on these facts, PD‐L1 should not be used as a prerequisite for prescribing anti‐PD‐1/PD‐L1 therapy. Clearly, additional predictive biomarkers are needed to guide anti‐PD‐1 treatment in adherence with the principals of precision medicine. A study of 46 metastatic melanoma tumors biopsied before and during anti‐PD‐1 therapy demonstrated that responsive patients have higher numbers of CD8‐, PD‐1‐, and PD‐L1‐expressing cells at the invasive margin and inside the tumor and increased clonality in antigen specificity of T cells within the tumor [32]. Furthermore, we have recently demonstrated that expression of MHC II on melanoma cells can predict response to anti‐PD‐1/PD‐L1 therapy in patients. Collectively, these findings point out that the therapeutic benefit from the PD‐1/PD‐L1 blockade requires hindered pre‐existing immunity that gets reinvigorated by the treatment.
COMBINATIONS OF IMMUNOTHERAPEUTIC AGENTS
Dual CTLA‐4 and PD‐1 blockade
In view of the success of anti‐PD‐1 and anti‐CTLA‐4 as single agents, combining these agents was considered as a potential strategy. Indeed, early preclinical studies supported this concept [33] and led to several clinical trials. In the first clinical trial of combined immunotherapy with CTLA‐4 and PD‐1 antagonists involving 53 patients with advanced melanoma, an ORR of 53% was achieved with acceptable level of adverse events [34]. Likewise, another Phase 2 study of 142 patients with previously untreated advanced melanoma reported improved ORR and the PFS with combined nivolumab/ipilimumab compared with ipilimumab monotherapy. A subsequent large Phase 3 study involving 945 previously untreated patients with unresectable Stage 3 or 4 melanoma revealed a median PFS of 11.5 mo with nivolumab plus ipilimumab compared with 2.9 mo with ipilimumab and 6.9 mo with nivolumab [35]. As a result, the FDA has granted accelerated approval to the combination of the PD‐1 inhibitor nivolumab and the CTLA‐4 inhibitor ipilimumab to treat advanced melanoma [36]. Based on these exciting findings in melanoma, nivolumab/ipilimumab is now undergoing clinical trials in many other human malignancies (see Table 1 for examples). The synergy of ipilimumab and nivolumab is likely a result of the collaboration of 2 nonredundant mechanisms that boost anti‐tumor immunity via the amplification of anti‐tumor T cells and broadening of their repertoire in response to CTLA‐4 targeting and through overcoming immunosuppression induced in the TME by blocking PD‐1/PD‐L1 interaction. Notably, combination therapy demonstrated increased immune‐related adverse events compared with either agent alone [37]. Overall, however, toxicities were similar in nature and were well‐managed with standard treatment algorithms. In addition to the obvious benefit of the combined CTLA‐4/PD‐1 blockade, there are several potential drawbacks, including added toxicities and the high cost of therapy. Therefore, the detection of ways to identify patients who will gain a long‐term benefit from single‐agent treatment alone remains a priority of current translational melanoma research. Furthermore, a number of additional immunostimulatory approaches are being tested in combination with the CTLA‐4 or PD‐1 immune‐checkpoint blockade in an effort to achieve high rates of durable responses while increasing the “benefit/risk” ratio.
Table 1.
Selected clinical studies of therapeutic combinations with an immune‐checkpoint blockade
| Method | Combination therapy and drug used | Immunotherapy target and drug used | Trial phase | Tumor types | Trial reference |
| Blockade of T cell inhibition | Anti‐CTLA‐4 | Anti‐PD‐1 | |||
| Ipilimimab | Nivolumab | 1–3 | Melanoma, uveal melanoma, melanoma brain metastasis, NSCLC, small cell lung cancer, colon cancer, liver cancer, breast cancer, RCC, lymphoma, multiple myeloma, glioblastoma, gliosarcoma | NCT01844505 NCT02477826 NCT02538666 NCT02374242 NCT02060188 NCT01658878 NCT02453620 NCT01592370 NCT02210117 NCT01585194 NCT02311920 | |
| Anti‐LAG‐3 | Anti‐PD‐1 | ||||
| LAG525 | PDR001 | 1/2 | Advanced cancer | NCT02460224 | |
| BMS‐986016 | Nivolumab | 1 | Advanced solid tumors | NCT01968109 | |
| Anti‐TIM‐3 | Anti‐PD‐1 | ||||
| MBG453 | PDR001 | 1/2 | Advanced cancer | NCT02608268 | |
| T cell costimulation | Anti‐4‐1BB | Anti‐PD‐1 | |||
| Urelumab | Nivolumab | 1/2 | Advanced solid tumors, B cell non‐Hodgkin lymphoma | NCT02253992 | |
| PF‐05082566 | MK3475 | 1 | Advanced solid tumors | NCT02179918 | |
| Anti‐PD‐L1 | |||||
| PF‐05082566 | Avelumab | 2 | Melanoma, lung, head and neck cancer | NCT02554812 | |
| Anti‐OX40 | Anti‐CTLA‐4 | ||||
| MEDI6469 | Tremelimumab | 1/2 | Advanced solid tumors | NCT02205333 | |
| Anti‐PD‐L1 | |||||
| MEDI6383 | MEDI4736 | 1 | Advanced solid tumors | NCT02221960 | |
| Anti‐CD27 | Anti‐CTLA‐4 | ||||
| Varlilumab | Ipilimumab | 1/2 | Melanoma | NCT02413827 | |
| Anti‐PD‐1 | |||||
| Varlilumab | Nivolumab | 1/2 | Advanced solid tumors | NCT02335918 | |
| Anti‐PD‐L1 | |||||
| Varlilumab | Atezolizumab | 1/2 | Advanced tumors, RCC | NCT02543645 | |
| Therapeutic cancer vaccines | Peptide vaccines | Anti‐CTLA‐4 | |||
| 6MHP | Ipilimumab | 1/2 | Melanoma | NCT02385669 | |
| Anti‐PD‐1 | |||||
| 6MHP | Pembrolizumab | 1/2 | Melanoma | NCT02515227 | |
| Tumor cell vaccine | Anti‐CTLA‐4 | ||||
| GVAX | Ipilimumab | 2 | Pancreatic cancer | NCT01896869 | |
| Anti‐PD‐1 | |||||
| GVAX | Nivolumab | 2 | Pancreatic cancer | NCT02243371 | |
| GM.CD40L | Nivolumab | 1/2 | Lung cancer | NCT02466568 | |
| Viagenpumatucel‐L | Nivolumab | 1 | NSCLC | NCT02439450 | |
| Vigil | Pembrolizumab | 1 | Melanoma | NCT02574533 | |
| Tuberculosis vaccine | Anti‐CTLA‐4 | ||||
| BCG | Ipilimumab | 1 | Melanoma | NCT01838200 | |
| Dendritic cell vaccine | Anti‐PD‐1 | ||||
| Sipuleucel‐T | Pidilizumab | 2 | Prostate cancer | NCT01420965 | |
| AML fusion vaccine | Pidilizumab | 2 | AML | NCT01096602 | |
| DNA vaccine | Anti‐PD‐1 | ||||
| pTVG‐HP plasmid | Pembrolizumab | 1/2 | Prostate cancer | NCT02499835 | |
| Viro therapy | Oncolytic virus | Anti‐CTLA‐4 | |||
| T‐VEC | Ipilimumab | 1b/2 | Melanoma | NCT01740297 | |
| Anti‐PD‐1 | |||||
| T‐VEC | Pembrolizumab | 1b/3 | Melanoma | NCT02263508 | |
| IDO inhibitors | IDO1i | Anti‐PD‐1 | |||
| Epacadostat | Pembrolizumab | 1/2 | Advanced solid tumors | NCT02178722 | |
| Anti‐PD‐L1 | |||||
| Epacadostat | Durvalumab | 1/2 | Advanced solid tumors | NCT02318277 | |
| Epacadostat | Atezolizumab | 1 | NSCLC | NCT02298153 | |
| Anti‐CTLA‐4 | |||||
| Epacadostat | Ipilimumab | 1/2 | Melanoma | NCT01604889 | |
| Targeted therapy | BRAFi + MEKi | Anti‐PD‐1 | |||
| Dabrafenib + trametinib | Pembrolizumab | 1/2 | Melanoma | NCT02130466 | |
| Anti‐PD‐L1 | |||||
| Dabrafenib + trametinib | Durvalumab | 1/2 | Melanoma | NCT02027961 | |
| Dabrafenib + trametinib | Atezolizumab | 1 | Melanoma | NCT01656642 | |
| EGFRi | Anti‐PD‐1 | ||||
| Gefitinib or erlotinib | Pembrolizumab | 1/2 | NSCLC | NCT02039674 | |
| Gefitinib | Tremelimumab | 1 | NSCLC | NCT02040064 | |
| Anti‐CTLA‐4 | |||||
| Cetuximab + radiation | Ipilimumab | 1 | Head and neck cancer | NCT01935921 | |
| RT and chemotherapy | Stereotactic body RT | Anti‐CTLA‐4 | |||
| Ipilimumab | 2 | Melanoma, liver, lung cancer | NCT01970527 NCT02107755 NCT02239900 | ||
| Tremelimumab | 1 | Unresectable pancreatic cancer | NCT02311361 | ||
| Anti‐PD‐1 | |||||
| Nivolumab | 2–3 | Glioblastoma, triple‐negative breast cancer | NCT02617589 NCT02499367 | ||
| Chemotherapy | Anti‐PD‐1 | ||||
| Temsirolimus, irinotecan, capecitabin | Nivolumab | 1/2 | Advanced tumors | NCT02423954 | |
| Nab‐paclitaxel, gemcitabine, carboplatin | Nivolumab | 1 | Pancreatic, breast, NSCLC | NCT02309177 | |
| Paclitaxel, carboplatin, pemetrexed | Pembrolizumab | 1/2 | Lung cancer | NCT02039674 | |
| Anti‐CTLA‐4 | |||||
| Gemcitabine | Ipilimumab | 1 | Pancreatic cancer | NCT01473940 | |
| Epigenetic therapy | DNMTi | Anti‐CTLA‐4 | |||
| SGI‐110 | Ipilimumab | 1 | Melanoma | NCT02608437 | |
| HDACi | Anti‐PD‐1 | ||||
| Vorinostat | Pembrolizumab | 1/1b | RCC | NCT02619253 | |
| Entinostat | Pembrolizumab | 1b/2 | NSCLC, melanoma | NCT02437136 | |
| Entinostat | Anti‐PD‐1/CTLA‐4 nivolumab + ipilimum. | Breast cancer | NCT02453620 | ||
| DNMT + HDACi | Anti‐PD‐1 | ||||
| Azacitidine + entinostat | Nivolumab | 2 | NSCLC | NCT01928576 |
GM.CD40L, GM‐CSF‐producing and CD40L‐expressing bystander cell line; BCG, bacillus Calmette‐Guérin; IDOi, BRAFi, MEKi, EGFRi, DNMTi, and HDACi, IDO, BRAF, MEK, epidermal growth factor, DNMT, and HDAC inhibitors.
Emerging combinations of immune‐checkpoint inhibitors
Besides CTLA‐4 and PD‐1, TILs express a diverse array of additional inhibitory coreceptors that function as immune‐checkpoint regulators and can be targeted to boost tumor immunity. Many promising emerging immunotherapeutic modalities are based on targeting these molecules in the TME ( Fig. 1 ).
Figure 1.
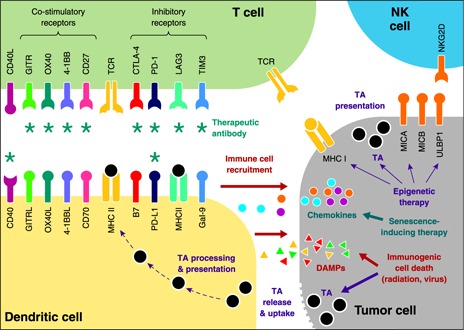
Current and emerging targets for therapeutic modulation of anti‐tumor immune response. Inhibition of immune checkpoints with antibodies targeting PD‐1 and CTLA‐4 inhibitory receptors of T cells produces durable responses in patients with many deadly malignancies. Several strategies are use to improve further the success rate of immunotherapies. These strategies include the following: 1) combining PD‐1 and CTLA‐4 blockers with each other or with antagonists of other inhibitory receptors on T cells, such as TIM‐3, LAG‐3, TIGIT, and BTLA; 2) combining the immune‐checkpoint blockade with agonists of costimulatory receptors of T cells, including CD27, 4‐1BB, OX40, and GITR; and 3) blocking immune checkpoints in conjunction with stimulation of TA recognition using vaccines and dendritic cell activation by CD40 agonists. An alternative approach involves combining the immune‐checkpoint blockade with therapy that enhances immunogenicity of tumors as a result of ICD (radiation, oncolytic viruses). Immunogenic death of tumor cells promotes immune cell recruitment and presentation of TAs. Enhancement of anti‐tumor immune responses can also be achieved by epigenetic therapies that restore silenced expression of molecules targeted by NK cells, TAs, and antigen‐presenting molecules (MHC I). In addition, therapy‐induced senescence can promote recruitment of anti‐tumor immune cells via the increased production of chemokines. *Molecules that can be modulated by therapeutic antibodies currently used in clinics or undergoing clinical development. DAMPs can include calreticulin, heat‐shock proteins, type I IFNs, ATP, and HMGB1. GITRL, OX40L, and 4‐1BBL, GITR, OX40, and 4‐1BB ligand.
TIM‐3.
TIM‐3 is a coinhibitory receptor known to negatively regulate T cell responses. It was initially identified as a specific marker of fully differentiated IFN‐γ‐producing CD4 Th1 [38] but later has been shown to play a significant biologic role in other cells, including CD8 T cells, Tregs, dendritic cells, macrophages, and MDSCs[39, 40]. TIM‐3 is activated by binding its ligand Gal‐9 that is ubiquitously expressed in a variety of tissues. This binding induces aggregation and death of TIM‐3+ Th1 cells [41]. The blocking of the TIM‐3/Gal‐9 interaction induces hyperproliferation of effector T cells and increases Th1 cytokine production and CD8 T cell cytotoxicity [41, 42]. Interestingly, Gal‐9‐independent activation of TIM‐3 was also reported. Chiba et al. [43] showed that TIM‐3 expressed on tumor‐infiltrating dendritic cells binds HMGB1, thus preventing HMGB1 from binding the DNA released from dying tumor cells and delivering it to the innate cells. The loss of HMGB1 binding to DNA ultimately dampens the innate immune response in tumor tissue. Not surprisingly, the targeting of TIM‐3 is currently being developed as a promising, emerging modality of cancer immunotherapy [44]. There is experimental evidence that TIM‐3 antagonists will be an excellent therapeutic partner for PD‐1/PD‐L1‐blocking antibodies. Coexpression of TIM‐3 and PD‐1 marks exhausted T cells [45, 46], and this coexpression links to loss of function of TA‐specific CD8+ T cells in melanoma patients [47]. In addition, intratumoral forkhead box P3+ Tregs that coexpress TIM‐3 and PD‐1 are highly suppressive [48]. The cotargeting of TIM‐3 and PD‐1 pathways can reverse T cell exhaustion and restore anti‐tumor immunity in experimental and carcinogen‐induced mouse models of melanoma, sarcoma, and colon cancer [49, 50]. Furthermore, the combination of the blockade of the TIM‐3/PD‐1 pathways extended survival in mice with AML [46]. Early‐phase clinical trials of this combination are ongoing for patients with advanced malignancies (Table 1).
LAG‐3.
Another targetable inhibitory receptor that is expressed on activated CD4 and CD8 T cells is the LAG‐3 (CD223) [41]. This surface molecule is highly homologous to CD4 in structure and like CD4, can bind to MHC II molecules, but the binding affinity for LAG‐3 is much higher than for CD4 [51, 52]. It negatively regulates T cell proliferation, homeostasis, and function via the inhibition of TCR signaling following antigen activation [53, 54]. LAG‐3 is also necessary for the optimal function of immunosuppressive Tregs, and its expression on conventional T cells makes them more susceptible to Treg‐based suppression [55, 56]. Interestingly, LAG‐3 and PD‐1 were found to be coexpressed on TILs of ovarian cancer patients [57], as well as on TILs derived from mouse melanoma, fibrosarcoma, and colon adenocarcinoma tumors [58]. Furthermore, whereas single PD‐1 and LAG‐3 knockout had minimal immunopathological consequences in C57BL/6 mice, dual knockout mice abrogated self‐tolerance, resulting in autoimmune infiltrates in multiple organs and lethality [58].These findings suggest that LAG‐3 and PD‐1 act synergistically to control immune homeostasis and mediate tumor‐induced tolerance. Notably, combined anti‐LAG‐3/anti‐PD‐1 antibody treatment cured most mice of established fibrosarcoma and colon adenocarcinoma tumors that were largely resistant to single‐antibody therapy [58]. Likewise, in a mouse model of recurrent melanoma, the simultaneous immune‐checkpoint blockade with combination therapy with PD‐L1 and LAG‐3 antagonists caused regression of aggressive B16F10 tumors [59].
TIGIT.
TIGIT is an emerging tumor immunotherapy target in preclinical development. It is an inhibitory receptor of the Ig superfamily expressed on subsets of activated T cells and NK cells [60]. One study showed that TIGIT is up‐regulated on TA‐specific CD8+ T cells and CD8+ TILs from patients with melanoma and cooperates with PD‐1 to impair anti‐tumor activity of these cells [61]. In contrast to these findings, another recent study reported that TIGIT primarily suppresses anti‐tumor immunity via its ability to stimulate Tregs rather than by inhibiting CD8+ T cells [62]. A recent report demonstrated that cotargeting TIGIT and PD‐L1 results in complete regression of subcutaneous tumors formed by mouse colorectal carcinoma cells [63], highlighting the potential of this treatment strategy.
BTLA.
BTLA is another inhibitory receptor found on B and T cells. BTLA may also be targeted for cancer therapy. Up‐regulation of BTLA and PD‐1 in the TME was shown to inhibit the function of TA‐specific CD8+ T cells, and dual antagonism of these receptors ex vivo enhanced the expansion, proliferation, and cytokine production in these cells [64]. However, a more recent study reported that the role of BTLA in anti‐tumor response may be complex, as it can act as a dual signaling molecule, inhibiting the activation of T cells and at the same time, enhancing the survival of CD8+ T cells [65].
Combinations of immune‐checkpoint inhibitors with agonists of costimulatory receptors
An alternative to the checkpoint blockade approach for reactivation of anti‐tumor immune responses is to activate costimulatory receptors of T cells that in conjunction with TCR signaling, promote their proliferation, as well as proinflammatory and cytotoxic activity [66]. Costimulatory receptors are often constitutively expressed on the surface of T cells, including resting antigen‐naïve T cells, or become transiently available in response to antigen recognition. A classic example of costimulatory receptors is CD28, which plays a crucial role in T cell activation [67]. Antibody agonists of CD28 are used routinely in laboratory practice for stimulation and expansion of T cells in vitro. Unfortunately, these agents may be too potent to be used as therapeutics, as they can induce a very severe systemic inflammatory syndrome driven by a cytokine storm with multiple organ failure, even when administered at extremely low doses [68].
CD27.
More clinical potential has been demonstrated for another constitutive costimulatory receptor expressed on T and plasma cells, CD27. Activation of this receptor via the ligation with its ligand CD70 promotes cell survival, expansion, and effector functions of T and B cells [69]. Costimulation of T cells with the CD27 agonist antibody in vitro can activate memory CD4+ and CD8+ T cells but not purified Tregs [70]. Furthermore, costimulation with CD27 and OX40 was shown to synergize with the anti‐PD‐L1 blockade by forcing exhausted CD8+ T cells to exit quiescence [71]. A fully humanized antibody agonist of CD27, varlilumab, has completed a Phase 1 dose‐escalation study. The manufacturer of the drug (Celldex Therapeutics, Hampton, NJ, USA) reported potent immunologic and anti‐tumor activity in patients with advanced, refractory disease. Furthermore, minimal toxicity was observed, and the maximal tolerated dose was not reached (http://www.celldex.com/science/publications.php#cdx1127). Furthermore, the company reports improved survival after combining varlilumab with immune‐checkpoint inhibitors in animal models, and clinical trials of varlilumab in combination with the CTLA‐4‐, PD‐1‐, or PD‐L1‐targeting antibody are ongoing (Table 1). However, another study has shown that in some tumors, the CD27–CD70 interaction may promote tumor growth and inhibit, rather than activate, anti‐tumor immune response [72]. Currently, there are several antagonists of CD70 undergoing clinical development for cancer treatment [73]. Therefore, clinical development of therapeutics acting on the CD27–CD70 pathway may require particular precision to achieve potent anti‐tumor activity without exaggerating tumor‐immune escape.
Several agonists are being developed for therapeutic targeting of costimulatory receptors specific for activated T cells, such as 4‐1BB, OX40, GITR, and others (Fig. 1). These receptors, in contrast to ubiquitous CD28 and CD27, are exclusively expressed or substantially up‐regulated on a surface of T cells within a few days following TCR engagement.
4‐1BB.
Lymphocyte costimulatory receptor 4‐1BB (CD137/TNF superfamily 9) possesses an unequaled capacity to promote survival, expansion, and enhanced effector function of activated T cells [74]. It is expressed on many subtypes of immune cells and plays a critical role in sustaining effective T cell immune responses and generating immunologic memory [75]. The anti‐tumor potential of 4‐1BB activation was first shown in 1997 by Melero and colleagues [76], who reported that antibodies against 4‐1BB eradicate established tumors in a murine model of mastocytoma and sarcoma. Therapeutic stimulation of 4‐1BB was also effective in mouse model of squamous cell cancer [77], lymphoma [78], hepatocellular carcinoma [79], melanoma [80], and colon cancer [81]. A subsequent Phase 1 study of urelumab (BMS‐663513), a fully human anti‐CD137 agonist mAb, in patients with advanced cancer (NCT0030902), reported evidence of clinical activity, as well as generally manageable adverse effects, including fatigue and neutropenia [82]. However, a follow‐up Phase 2 (NCT00612664) had to be terminated as a result of unusually high incidence of Grade 4 hepatitis. Consequently, a number of other trials of CD137 agonists were terminated or withdrawn (reviewed in ref. [83]). Studies in mice confirmed hepatotoxicity, resulting from 4‐1BB activation that was associated with the influx of inflammatory cells into the liver of the treated animals [84, 85]. It is plausible that using a low dose of urelumab in combination with other immunotherapies could potentially augment anti‐tumor immune response while limiting the severity of therapy‐associated adverse events. Recently, a number of trials were initiated using a putatively safer low dose of urelumab in combination with other therapeutics (Table 1). There is another CD137 agonist PF‐05082566 currently undergoing clinical development, which in contrast to urelumab, has been reported to be well tolerated [86].
Several animal studies have demonstrated a promising therapeutic benefit of combining 4‐1BB agonists with the PD‐1 and CTLA‐4 blockade. For instance, the combination of 4‐1BB agonist and PD‐1 antagonist promoted anti‐tumor effector/memory CD8 T cells in a poorly immunogenic, aggressive B16F10 murine melanoma model, resulting in tumor inhibition that was dependent on IFN‐γ and CD8+ T cells [87]. Likewise, tumor rejection and enhanced anti‐tumor immunity after combined anti‐PD‐1 and anti‐4‐1BB therapy were seen in mouse models of colon and ovarian cancer [88, 89]. Anti‐4‐1BB antibody treatment also augmented the therapeutic response to CTLA‐4 antagonist in MC38 colon cancer tumors but not in B16 melanoma tumors [90]. Another study found that the combination of CTLA‐4 and 4‐1BB agonists had a synergistic effect on tumor rejection in the context of a Flt3 ligand‐based B16 melanoma vaccine [91].
OX40.
OX40 (CD134) is another receptor that serves costimulatory functions. It is expressed on activated CD4+ (including both Th1 and Th2 cells) and CD8+ T cells within 24–96 h following TCR engagement [92, 93]. OX40 is activated by its ligand OX40L (CD252) that is expressed on APCs; this binding augments proliferation and activity of activated T cells [93]. This receptor has been detected on tumor‐infiltrating leukocytes and in tumor‐draining lymph nodes in patients with melanoma, head and neck, breast, and colorectal cancer [94, 95, 96–97]. Moreover, in colorectal and melanoma tumors, high expression of OX40 on tumor‐infiltrating leukocytes was associated with decreased metastasis and longer survival [96, 97]. Interestingly, OX40 has also been implicated in inhibiting activity of Tregs [98, 99]. Agonists of OX40 have demonstrated promising anti‐tumor activity in preclinical development [100]. A Phase 1 clinical trial of the OX40 agonist antibody demonstrated increased anti‐tumor activity of T and B cells in melanoma patients. This study also reported tumor regression in 12 of 30 patients and an acceptable level of toxicity [101]. Preclinical evidence also suggests that addition of OX40 agonists may enhance the clinical benefit of an immune‐checkpoint blockade. For instance, antibodies specific for OX40 combined with the CTLA‐4 blockade demonstrated synergistic anti‐tumor activity in mouse lymphoma, leukemia, breast, and sarcoma tumors [98, 102, 103]. Furthermore, OX40 activation combined with the PD‐1 blockade was effective against murine ovarian cancer [104]. Phase 1/2 clinical studies, combining the OX40 agonist humanized antibody MEDI6383 with anti‐PD‐L1 and anti‐CTLA‐4 antagonists, are ongoing (Table 1).
GITR.
Another targetable costimulatory receptor of T cells is GITR. It is expressed at a low level on naïve cells and is induced after TCR engagement in CD4+ and CD8+ subsets. Activation of GITR by its ligand (GITRL), which is highly expressed on activated APCs and endothelial cells, promotes survival, proliferation, and effector function of T cells [93, 105, 106]. GITR is constitutively expressed on Tregs and blocks their inhibitory function [107, 108]. In mice, GITR agonists showed activity against CT26 colon tumors and small B16 melanoma tumors [109, 110]. An agonist of GITR synergized with the CTLA‐4 blockade to inhibit the growth of sarcoma and colon tumors [111]. Likewise, the combination of the PD‐1 blockade with the GITR agonist induced potent anti‐tumor immunity in mice with peritoneal ovarian cancer tumors [112]. Humanized antibody agonists of GITR are undergoing clinical evaluation.
Combination of immune‐checkpoint inhibitors with therapies that promote tumor cell recognition by T cells
Stimulation of APCs.
APCs play a critical role in the establishment of effective anti‐tumor immunity and are attractive therapeutic targets. For instance, CD40 is a costimulatory molecule essential for activation of APCs, such as dendritic cells, B cells, and macrophages [113, 114]. CD40 is activated when ligated to CD40L (expressed on CD4+ T cells), which substantially increases antigen presentation and costimulatory capacity of APCs. Notably, a synergistic response was observed after combined treatment with the CD40 agonist and PD‐1 antagonist, inducing rejection of colon and breast tumor implants in 50% of the tumor‐bearing mice [115]. This synergistic response suggests that CD40 may be used potentially as a therapeutic partner for immune‐checkpoint inhibitors.
Tumor vaccines.
In light of the exceptional success of preventative vaccination against infectious diseases, including polio, tetanus, measles, and many others, the vaccination of patients against antigens expressed by tumors to boost anti‐tumor immunity seems like an attractive therapeutic approach. Somatic mutations in tumors can give rise to neoantigens. Interestingly, Snyder et al. [13] reported significant similarities of tumor neoantigens to certain viral and bacterial antigens with tetrapeptide sequences of tumor neoantigens matching those in known antigenic peptides of pathogens [116]. Response to neoantigens is especially relevant in malignancies with high‐mutation burden, such as melanoma. A large number of therapeutic vaccination studies in patients with nonviral cancers have been conducted. As summarized in recent reviews, many Phase 1/2 cancer vaccine studies showed modest clinical benefit, and objective durable tumor regressions were rarely seen [117, 118]. Use of appropriate cotreatments, for instance, immune‐checkpoint inhibitors, can potentially alleviate immunosuppressive mechanisms in the TME and boost vaccine performance, and there are numerous clinical studies currently testing checkpoint blockade‐vaccine combinations in a variety of tumor types. However, despite the promising reports from mouse model studies, no strong evidence of therapeutic potential for these approaches has been demonstrated in the clinic so far (reviewed recently in refs. [119, 120]). For instance, no evidence of clinical synergy from vaccine plus ipilimumab treatment was shown in a clinical trial, where melanoma patients received CTLA‐4 antagonist ipilimumab plus gp100 peptide vaccine, ipilimumab alone, or gp100 alone. In fact, survival was similar for the ipilimumab, with or without the GP100 vaccine, and exceeded survival for the GP100 alone [9]. Furthermore, peptide vaccination did not add to the clinical activity of the anti‐PD‐1 therapy in a clinical study of the PD‐1 antagonist nivolumab in combination with a multipeptide vaccine in melanoma patients [121].
IDO inhibitors.
IDO1 is a critical enzyme in the metabolic pathway that converts l‐tryptophan to l‐kynurenine [122]. It plays an important role in the establishment of local immunosuppression and is responsible for protection of allogeneic fetuses from the attack of maternal T cells [123]. IDO1 is expressed by distinct subsets of macrophages, monocytes, and dendritic cells with potent immunosuppressive activity [124, 125–126] and is often found in tumors, where it is implicated in enabling immunologic tolerance and immune escape; however, the exact mechanism of these effects is still a subject of debate (reviewed in ref. [127]). Inhibition of IDO1 demonstrated anti‐tumor activity in a mouse model, which was associated with the induction of an anti‐tumor immune response [128], and enhanced the efficacy of chemotherapy [129]. In clinical studies, IDO inhibitors epacadostat (INCB024360) and indoximod (NLG‐8189) were well tolerated by the majority of enrolled patients. Disease stabilization was often reported; however, tumor eradication from stand‐alone IDO targeting was not achieved [130, 131]. More exciting results were obtained in studies combining IDO inhibitors with the immune‐checkpoint blockade. For instance, epacadostat showed promising clinical activity in combination with ipilimumab with ORR of 31% (10 out of 32 immunotherapy‐naive patients) and was well tolerated [132]. Furthermore, a number of clinical trials of epacadostat, in combination with PD‐1/PD‐L1 targeting agents, are ongoing (Table 1). The level of optimism for these approaches is high, based on preliminary results from a Phase 1/2 study of an epacadostat and pembrolizumab (anti‐PD‐1) combination, reported at the 2015 Society for Immunotherapy of Cancer Annual Meeting, which demonstrated an ORR of 53% (10 out of 19 patients) and a disease control rate of 74% (15 out of 19 patients) across multiple malignancies, with an even more profound effect in patients with melanoma [133].
Combination of immune‐checkpoint inhibitors with therapies that enhance tumor immunogenicity
Inducers of ICD.
It is well established that a certain type of cell death, ICD, induced by some anti‐tumor agents, carries immunostimulatory potential. ICD results from ordered activation of stress‐response pathways associated with the emission of danger signals by dying cancer cells, called DAMPs, which promote recognition of dying tumor cells by the innate and adaptive immune system, ultimately eliciting tumor‐targeting immune responses [134, 135–136]. Among the most studied of these are the following: membrane exposure of the endoplasmic reticulum chaperone calreticulin and various heat‐shock proteins, production of type I IFNs, secretion of ATP, and the release of the nuclear protein HMGB1 into the extracellular space (reviewed in refs. [136, 137–138]). Notably, outside of its cancer‐related effect, DAMP release plays a key role on the induction of a systemic inflammatory response syndrome in response to injury [139]. Among the most studied inducers of ICD are several chemotherapeutic drugs and RT. Furthermore, ICD has been reported in response to treatment with oncolytic viruses and drugs that target HDAC [137]. Notably, a recent review of the clinical data on cancer patients, with a variety of different malignancies, showed the prognostic and predictive value of ICD markers, DAMPs and DAMP‐associated stress responses [140].
It is plausible that the immunogenic potential of therapies that promote DAMPs can be exploited in combination with immune‐checkpoint blockade drugs to stimulate further immune‐mediated tumor destruction (Fig. 1). The synergistic relationship between RT and the CTLA‐4 blockade was shown in a preclinical model of breast cancer [141, 142], with the PD‐1 blockade in melanoma, breast, and glioma models [143, 144], as well as with anti‐PD‐L1 in melanoma, breast, and colon cancer [145, 146]. In 2012, several anecdotal cases have reported tumor regression of multiple distant, unirradiated metastases (abscopal effect) in patients treated with radiation and anti‐CTLA‐4 [147, 148]. Later, a small study of 21 melanoma patients, who received RT after progressing on ipilimumab, suggested that RT after ipilimumab leads to abscopal responses in some patients, which correlates with prolonged OS [149]. Another study of 47 patients with metastatic melanoma, treated with ipilimumab and RT, showed an abscopal effect in 68% of cases [150]. Recently, Twyman‐Saint Victorand colleagues [151] reported major tumor regression in a subset of patients with metastatic melanoma treated with an anti‐CTLA‐4 antibody and radiation. However, the majority of patients was resistant to this treatment. The authors recapitulated these results in a mouse model and showed that resistance was a result of up‐regulation of PD‐L1 on melanoma cells and was associated with T cell exhaustion. Notably, improved responses in the mouse model were seen when RT and anti‐CTLA‐4 cotreatment was combined with the anti‐PD‐L1/PD‐1 blockade [151]. Multiple clinical trials of immunotherapy/RT regimens for treating patients with melanoma and other cancers are ongoing (examples in Table 1).
In contrast to immune‐stimulating effects of modern RT that target tumors with precision, systemic chemotherapy is generally associated with leukopenia and immunosuppression, as it negatively impacts normal proliferation of cells. Nevertheless, a number of clinical studies that test combinations of immunotherapy with chemotherapeutic drugs are ongoing, with a yet‐unknown outcome (Table 1). In particular, low‐dose chemotherapy (e.g., cyclophosphamide) has demonstrated a proimmune effect by depleting Tregs and other immune‐suppressive cellular subsets [152].
Oncolytic viruses.
Oncolytic viruses can be engineered so that they preferentially replicate in, and kill, tumor cells. Compelling evidence has demonstrated that oncolytic viruses can stimulate anti‐tumor responses. In 1999, the induction of systemic anti‐tumor immunity by replication‐competent, attenuated HSV was shown in a syngeneic murine model of neuroblastoma. In this study, experimental mice were protected from tumor rechallenge and had elevated anti‐tumor activity of the specific CTL [153]. Furthermore, oncolytic viral therapy is capable of producing regression, not only in injected but also in distant, uninjected tumor lesions. In a murine model of glioma, tumor cells infected with oncolytic rodent parvovirus activated APCs, such as dendritic cells and microglia, resulting in anti‐tumor immunity, demonstrated in rechallenge experiments using uninfected tumor cells, and this effect of oncoviral therapy was abrogated in immunodeficient mice [154]. Another study using viral therapy with Newcastle disease virus showed long‐term survival and the establishment of the anti‐tumor immune memory in a murine glioma model [155]. Furthermore, Zamarin et al. [156] found that injection of oncolytic Newcastle disease virus into subcutaneous melanoma, colon, and prostate tumors in immunocompetent mouse models augments the response to the CTLA‐4 blockade via the induction of tumor inflammation. Notably, virotherapy promoted lymphocyte recruitment and tumor regression not only in the tumor that was injected with the virus but also in the uninjected tumor that was grown on the opposite flank of the mouse.
A modified herpes virus T‐VEC was the first oncolytic virus approved in the United States for therapy of cancer. T‐VEC is designed to replicate exclusively in the tumor cells and produces GM‐CSF to enhance anti‐tumor immune responses. This virus is injected directly into the tumor; therefore, only superficial cutaneous or nodal lesions, but not the distant metastatic lesions, could be treated with this therapy. In a Phase 3 clinical trial, T‐VEC demonstrated durable responses lasting >6 mo in 16% of advanced melanoma patients with an ORR of 26% [157]. Combinations of CTLA‐4 and PD‐1 antagonist antibodies, T‐VEC, and other injectable oncolytic viruses are currently in clinical development (Table 1). Preliminary results of the Phase 1b trial of T‐VEC plus ipilimumab, reported at the 2015 ASCO Annual Meeting, were very promising, with an ORR of 56%, including 33% complete responses in 18 patients, receiving this combination [158].The early report from the T‐VEC‐plus‐pembrolizumab study highlights the relatively low toxicity of this therapeutic combination, and although the efficacy has not been officially published as of May 2016, a Phase 3 arm of this trial is already recruiting participants [159].
Epigenetic therapy.
Epigenetic silencing is a common strategy used by cancer cells to escape immune surveillance. It leads to down‐regulation of tumor‐associated antigens or molecules that are required for processing and presentation of these antigens and thereby, interferes with recognition of neoplastic cells by the immune system. Maintenance of gene silencing requires continued activity of DNMT and HDAC; therefore, “epigenetic drugs” that target these enzymes may restore/improve immunologic recognition of cancer cells [160, 161]. Indeed, DNA hypomethylating agent 5‐aza‐deoxycytidine up‐regulates expression of tumor‐associated antigens [162], as well as HLA class I molecules [163, 164], in various models. HDAC inhibitors can also restore expression of HLA molecules in cancer cells [165, 166]. Furthermore, HDAC inhibitors have been shown to promote recognition and lysis of cancer cells by NK cells by inducing neoplastic cell expression of MICA, MICB, and/or ULBP1–3, the ligands for the NK cell receptor NKG2D [167, 168]. West et al. [169] demonstrated that an intact immune system was required for the robust anticancer effects of the HDAC inhibitors, vorinostat and panobinostat, against a colon adenocarcinoma and leukemia/lymphoma. In this study, the authors reported signs of ICD, including surface calreticulin exposure, ATP, and HMGB1 release in vorinostat‐treated cells. Likewise, calreticulin on the cellular membrane was expressed in cells from childhood brain tumors after exposure to HDAC inhibitors [170].
Several studies have reported synergistic anti‐tumor activity of combined epigenetic and immune‐checkpoint blockade therapies. For instance, in mouse tumor models, treatment with 5‐azacytidine (DNMT inhibitor) and entinostat (HDAC inhibitor), combined with anti‐PD‐1‐ and anti‐CTLA‐4‐targeted antibodies, was able to produce a significantly improved outcome [171]. Intriguingly, the primary targets of the epigenetic modulators were MDSCs and not the tumor cells. Furthermore, mice bearing B16F10 tumors that received combination therapy with HDAC inhibitors and the PD‐1 blockade exhibited slower progression of tumors and increased survival compared with control and single‐agent treatments [172]. Recently, Covre et al. [173] demonstrated significant immune‐related anti‐tumor activity of 5‐aza‐deoxycytidine combined with the anti‐CTLA‐4 therapy in a mouse model of mammary carcinoma and mesothelioma. Several ongoing human studies of immunotherapy/epigenetic therapy are shown in Table 1.
Senescence‐inducing therapy.
Senescent cells are characterized by metabolic activity in “permanent” cell‐cycle arrest. Senescence is induced by aberrant oncogene activation and other cellular stressors. Senescent cells are characterized by enhanced secretion of many proinflammatory cytokines and chemokines, described as the SASP [174, 175], which has been shown to attract cells of the innate and adaptive immune system that kill and clear senescent cells [176, 177]. Based on these findings, it was proposed that senescence and SASP are essential steps in the process of immunosurveillance and elimination of premalignant cells in the body [178]. Notably, senescence can be induced in established tumors by overexpression of p53 or in response to certain therapies. It has been shown that senescence subjects tumors to immune targeting [176, 179, 180]. We have reported that senescence induced in melanoma tumors by treatment with the inhibitors of cell‐cycle kinases aurora kinase A and cyclin‐dependent kinase 4/6 promotes secretion of chemokines that recruit T cells and APCs into the tumor [181, 182]. This activity is potentiated further in cells treated with a combination of senescence‐inducing drugs and p53‐activating therapy. Furthermore, senescent melanoma cells overexpress TNF family death receptors that make them vulnerable to cell death induced by immune cells secreting death receptor ligands [183]. Importantly, we have recently demonstrated that senescence‐inducing therapy can synergize with the agonist antibody to T cell costimulatory receptor 4‐1BB (CD137), leading to complete tumor regression in a significant proportion of melanoma‐bearing mice [182]. Whereas senescence‐inducing and immune therapy combinations have not yet entered clinical development, this approach has proved promising in preclinical studies.
Targeted therapy
Targeted therapy is designed to disrupt specific pathways critical for survival and proliferation of cancer cells. The inhibition of oncogenic kinase BRAF (BRAFV600E), activated in ∼½ of melanoma cases, has been among the most successful targeted approaches to date. Specific small‐molecule inhibitors of BRAF, combined with inhibitors of its downstream signaling mediator MEK, are widely used for patients with BRAFV600E mutations (see ref. [184] for review). Whereas response rates from these drugs are high, prompt acquisition of resistance is a major drawback [35, 185]. It is suggested that combining targeted therapy with immunotherapy may induce durable responses in a large subset of patients [186]. Recent reviews on the subject highlight the immunostimulatory effects of several targeted therapeutics and compile impressive evidence for the potent anti‐tumor activity of targeted/immunotherapy combinations [187, 188]. However, higher than expected incidences of toxicity, observed with targeted/immunotherapy combinations, have been hindering the clinical development of these strategies. For instance, severe liver toxicity was responsible for terminating a clinical trial combining the BRAF inhibitor vemurafenib with ipilimumab [189]. In addition, several episodes of severe colitis with bowel perforation in response to the BRAF inhibitor dabrafenib, the MEK inhibitor trametinib, and ipilimumab led to another study closure [190]. Notably, a 2015 ASCO report from an ongoing Phase 1 study that tested combinations of BRAF and MEK inhibitors with anti‐PD‐L1 therapy in melanoma patients showed strong evidence of clinical activity with a manageable safety profile [191]. These exciting results suggest that PD‐L1 blockers may be better combinatorial partners for BRAF/MEK‐targeted therapy compared with ipilimumab. Sequential administration is another solution proposed to address toxicity issues with targeted/immunotherapy combinations (see ref. [187] for review).
CONCLUDING REMARKS
Just a few decades ago, aggressive malignancies, such as metastatic melanoma and NSCLC, were terminal diseases with very low life expectancy. The development of immune‐checkpoint agents, such as CTLA‐4‐ and PD‐1‐targeting antibodies, has allowed patients to achieve a durable therapeutic response and in some cases, even complete regression of advanced disease. Furthermore, the knowledge gained in cancer immunotherapy research has led to significant advances in the treatment of nonmalignant diseases, including getting closer to a cure of historically devastating, but currently manageable, HIV‐1 infections [192]. One current goal of translational cancer research is to capitalize on the success of novel immunotherapeutics to improve further the incidence and durability of patients’ responses. As we have described here, a large number of promising immunotherapeutic combinations are transitioning into clinical practice, including 1 approved combination (dual‐checkpoint inhibition with anti‐CTLA‐4 and anti‐PD‐1). Furthermore, numerous emerging drug combinations are being investigated in clinical trials for treatment of aggressive malignancies, such as melanoma, lung, breast cancer, liver, renal, brain, and pancreatic cancer. The immune‐checkpoint blockade is commonly combined with other antagonists of inhibitory receptors of T cells or with agonists of their costimulatory receptors. Other promising therapeutic partners of the checkpoint blockade are treatments that promote presentation and recognition of TAs by effector T cells and therapies that increase antigenicity of tumor cells. Early clinical and preclinical studies have highlighted the major challenges of combining immunotherapies with other immune‐stimulatory agents or targeted therapies, including unexpected toxicities and the importance of treatment dose and schedule. In summary, whereas current immunotherapeutic agents have already demonstrated their impressive potential in immunogenic tumors, such as melanoma, rational combinations with these agents hold promise to improve further clinical response rates and overall patient survival. We envision an extended use of immunotherapy for the treatment of a wide spectrum of malignancies in the immediate future.
AUTHORSHIP
A.E.V., D.B.J., and A.R. wrote the paper.
DISCLOSURES
D.B.J. is on the Advisory Boards for Bristol‐Myers Squibb and Genoptix.
ACKNOWLEDGMENTS
This work was supported by the Department of Veterans Affairs (MERIT Grant Number 5101BX000196‐04 and a Senior Research Career Scientist award; to A.R.), Harry J. Lloyd Charitable Trust award (to A.E.V.), and National Cancer Institute at the U.S. National Institutes of Health (Grant Numbers CA116021, CA90625, and CA68485). The authors thank C. Andrew Johnston for help in manuscript preparation.
References
- 1. Rudd, C. E. , Taylor, A. , Schneider, H. (2009) CD28 and CTLA‐4 coreceptor expression and signal transduction. Immunol. Rev. 229, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stamper, C. C. , Zhang, Y. , Tobin, J. F. , Erbe, D. V. , Ikemizu, S. , Davis, S. J. , Stahl, M. L. , Seehra, J. , Somers, W. S. , Mosyak, L. (2001) Crystal structure of the B7‐1/CTLA‐4 complex that inhibits human immune responses. Nature 410, 608–611. [DOI] [PubMed] [Google Scholar]
- 3. Pentcheva‐Hoang, T. , Egen, J. G. , Wojnoonski, K. , Allison, J. P. (2004) B7‐1 and B7‐2 selectively recruit CTLA‐4 and CD28 to the immunological synapse. Immunity 21, 401–413. [DOI] [PubMed] [Google Scholar]
- 4. Wing, K. , Onishi, Y. , Prieto‐Martin, P. , Yamaguchi, T. , Miyara, M. , Fehervari, Z. , Nomura, T. , Sakaguchi, S. (2008) CTLA‐4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. [DOI] [PubMed] [Google Scholar]
- 5. Leach, D. R. , Krummel, M. F. , Allison, J. P. (1996) Enhancement of antitumor immunity by CTLA‐4 blockade. Science 271, 1734–1736. [DOI] [PubMed] [Google Scholar]
- 6. Sharma, P. , Allison, J. P. (2015) The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- 7. Kvistborg, P. , Philips, D. , Kelderman, S. , Hageman, L. , Ottensmeier, C. , Joseph‐Pietras, D. , Welters, M. J. , van der Burg, S. , Kapiteijn, E. , Michielin, O. , Romano, E. , Linnemann, C. , Speiser, D. , Blank, C. , Haanen, J. B. , Schumacher, T. N. (2014) Anti‐CTLA‐4 therapy broadens the melanoma‐reactive CD8+ T cell response. Sci. Transl. Med. 6, 254ra128. [DOI] [PubMed] [Google Scholar]
- 8. Simpson, T. R. , Li, F. , Montalvo‐Ortiz, W. , Sepulveda, M. A. , Bergerhoff, K. , Arce, F. , Roddie, C. , Henry, J. Y. , Yagita, H. , Wolchok, J. D. , Peggs, K. S. , Ravetch, J. V. , Allison, J. P. , Quezada, S. A. (2013) Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti‐CTLA‐4 therapy against melanoma. J. Exp. Med. 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hodi, F. S. , O'Day, S. J. , McDermott, D. F. , Weber, R. W. , Sosman, J. A. , Haanen, J. B. , Gonzalez, R. , Robert, C. , Schadendorf, D. , Hassel, J. C. , Akerley, W. , van den Eertwegh, A. J. , Lutzky, J. , Lorigan, P. , Vaubel, J. M. , Linette, G. P. , Hogg, D. , Ottensmeier, C. H. , Lebbé, C. , Peschel, C. , Quirt, I. , Clark, J. I. , Wolchok, J. D. , Weber, J. S. , Tian, J. , Yellin, M. J. , Nichol, G. M. , Hoos, A. , Urba, W. J. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robert, C. , Thomas, L. , Bondarenko, I. , O'Day, S. , Weber, J. , Garbe, C. , Lebbe, C. , Baurain, J. F. , Testori, A. , Grob, J. J. , Davidson, N. , Richards, J. , Maio, M. , Hauschild, A. , Miller, W. H. , Jr, Gascon, P. , Lotem, M. , Harmankaya, K. , Ibrahim, R. , Francis, S. , Chen, T. T. , Humphrey, R. , Hoos, A. , Wolchok, J. D. (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526. [DOI] [PubMed] [Google Scholar]
- 11. Ribas, A. , Kefford, R. , Marshall, M. A. , Punt, C. J. , Haanen, J. B. , Marmol, M. , Garbe, C. , Gogas, H. , Schachter, J. , Linette, G. , Lorigan, P. , Kendra, K. L. , Maio, M. , Trefzer, U. , Smylie, M. , McArthur, G. A. , Dreno, B. , Nathan, P. D. , Mackiewicz, J. , Kirkwood, J. M. , Gomez‐Navarro, J. , Huang, B. , Pavlov, D. , Hauschild, A. (2013) Phase III randomized clinical trial comparing tremelimumab with standard‐of‐care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 31, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Callahan, M. K. , Wolchok, J. D. (2015) Clinical activity, toxicity, biomarkers, and future development of CTLA‐4 checkpoint antagonists. Semin. Oncol. 42, 573–586. [DOI] [PubMed] [Google Scholar]
- 13. Snyder, A. , Makarov, V. , Merghoub, T. , Yuan, J. , Zaretsky, J. M. , Desrichard, A. , Walsh, L. A. , Postow, M. A. , Wong, P. , Ho, T. S. , Hollmann, T. J. , Bruggeman, C. , Kannan, K. , Li, Y. , Elipenahli, C. , Liu, C. , Harbison, C. T. , Wang, L. , Ribas, A. , Wolchok, J. D. , Chan, T. A. (2014) Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Allen, E. M. , Miao, D. , Schilling, B. , Shukla, S. A. , Blank, C. , Zimmer, L. , Sucker, A. , Hillen, U. , Geukes Foppen, M. H. , Goldinger, S. M. , Utikal, J. , Hassel, J. C. , Weide, B. , Kaehler, K. C. , Loquai, C. , Mohr, P. , Gutzmer, R. , Dummer, R. , Gabriel, S. , Wu, C. J. , Schadendorf, D. , Garraway, L. A. (2015) Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ku, G. Y. , Yuan, J. , Page, D. B. , Schroeder, S. E. , Panageas, K. S. , Carvajal, R. D. , Chapman, P. B. , Schwartz, G. K. , Allison, J. P. , Wolchok, J. D. (2010) Single‐institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer 116, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ng Tang, D. , Shen, Y. , Sun, J. , Wen, S. , Wolchok, J. D. , Yuan, J. , Allison, J. P. , Sharma, P. (2013) Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti‐CTLA‐4 therapy. Cancer Immunol. Res. 1, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hannani, D. , Vétizou, M. , Enot, D. , Rusakiewicz, S. , Chaput, N. , Klatzmann, D. , Desbois, M. , Jacquelot, N. , Vimond, N. , Chouaib, S. , Mateus, C. , Allison, J. P. , Ribas, A. , Wolchok, J. D. , Yuan, J. , Wong, P. , Postow, M. , Mackiewicz, A. , Mackiewicz, J. , Schadendorff, D. , Jaeger, D. , Zörnig, I. , Hassel, J. , Korman, A. J. , Bahjat, K. , Maio, M. , Calabro, L. , Teng, M. W. , Smyth, M. J. , Eggermont, A. , Robert, C. , Kroemer, G. , Zitvogel, L. (2015) Anticancer immunotherapy by CTLA‐4 blockade: obligatory contribution of IL‐2 receptors and negative prognostic impact of soluble CD25. Cell Res. 25, 208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Freeman, G. J. , Long, A. J. , Iwai, Y. , Bourque, K. , Chernova, T. , Nishimura, H. , Fitz, L. J. , Malenkovich, N. , Okazaki, T. , Byrne, M. C. , Horton, H. F. , Fouser, L. , Carter, L. , Ling, V. , Bowman, M. R. , Carreno, B. M. , Collins, M. , Wood, C. R. , Honjo, T. (2000) Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong, H. , Strome, S. E. , Salomao, D. R. , Tamura, H. , Hirano, F. , Flies, D. B. , Roche, P. C. , Lu, J. , Zhu, G. , Tamada, K. , Lennon, V. A. , Celis, E. , Chen, L. (2002) Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800. [DOI] [PubMed] [Google Scholar]
- 20. Teng, M. W. , Ngiow, S. F. , Ribas, A. , Smyth, M. J. (2015) Classifying cancers based on T‐cell infiltration and PD‐L1. Cancer Res. 75, 2139–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lipson, E. J. , Forde, P. M. , Hammers, H. J. , Emens, L. A. , Taube, J. M. , Topalian, S. L. (2015) Antagonists of PD‐1 and PD‐L1 in cancer treatment. Semin. Oncol. 42, 587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robert, C. , Long, G. V. , Brady, B. , Dutriaux, C. , Maio, M. , Mortier, L. , Hassel, J. C. , Rutkowski, P. , McNeil, C. , Kalinka‐Warzocha, E. , Savage, K. J. , Hernberg, M. M. , Lebbé, C. , Charles, J. , Mihalcioiu, C. , Chiarion‐Sileni, V. , Mauch, C. , Cognetti, F. , Arance, A. , Schmidt, H. , Schadendorf, D. , Gogas, H. , Lundgren‐Eriksson, L. , Horak, C. , Sharkey, B. , Waxman, I. M. , Atkinson, V. , Ascierto, P. A. (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330. [DOI] [PubMed] [Google Scholar]
- 23. KEYNOTE‐006 Investigators . (2015) Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 372, 2521–2532. [DOI] [PubMed] [Google Scholar]
- 24. Ribas, A. , Puzanov, I. , Dummer, R. , Schadendorf, D. , Hamid, O. , Robert, C. , Hodi, F. S. , Schachter, J. , Pavlick, A. C. , Lewis, K. D. , Cranmer, L. D. , Blank, C. U. , O'Day, S. J. , Ascierto, P. A. , Salama, A. K. , Margolin, K. A. , Loquai, C. , Eigentler, T. K. , Gangadhar, T. C. , Carlino, M. S. , Agarwala, S. S. , Moschos, S. J. , Sosman, J. A. , Goldinger, S. M. , Shapira‐Frommer, R. , Gonzalez, R. , Kirkwood, J. M. , Wolchok, J. D. , Eggermont, A. , Li, X. N. , Zhou, W. , Zernhelt, A. M. , Lis, J. , Ebbinghaus, S. , Kang, S. P. , Daud, A. (2015) Pembrolizumab versus investigator‐choice chemotherapy for ipilimumab‐refractory melanoma (KEYNOTE‐002): a randomised, controlled, phase 2 trial. Lancet Oncol. 16, 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber, J. S. , D'Angelo, S. P. , Minor, D. , Hodi, F. S. , Gutzmer, R. , Neyns, B. , Hoeller, C. , Khushalani, N. I. , Miller, W. H. , Jr, Lao, C. D. , Linette, G. P. , Thomas, L. , Lorigan, P. , Grossmann, K. F. , Hassel, J. C. , Maio, M. , Sznol, M. , Ascierto, P. A. , Mohr, P. , Chmielowski, B. , Bryce, A. , Svane, I. M. , Grob, J. J. , Krackhardt, A. M. , Horak, C. , Lambert, A. , Yang, A. S. , Larkin, J. (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti‐CTLA‐4 treatment (CheckMate 037): a randomised, controlled, open‐label, phase 3 trial. Lancet Oncol. 16, 375–384. [DOI] [PubMed] [Google Scholar]
- 26. Topalian, S. L. , Hodi, F. S. , Brahmer, J. R. , Gettinger, S. N. , Smith, D. C. , McDermott, D. F. , Powderly, J. D. , Carvajal, R. D. , Sosman, J. A. , Atkins, M. B. , Leming, P. D. , Spigel, D. R. , Antonia, S. J. , Horn, L. , Drake, C. G. , Pardoll, D. M. , Chen, L. , Sharfman, W. H. , Anders, R. A. , Taube, J. M. , McMiller, T. L. , Xu, H. , Korman, A. J. , Jure‐Kunkel, M. , Agrawal, S. , McDonald, D. , Kollia, G. D. , Gupta, A. , Wigginton, J. M. , Sznol, M. (2012) Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herbst, R. S. , Soria, J. C. , Kowanetz, M. , Fine, G. D. , Hamid, O. , Gordon, M. S. , Sosman, J. A. , McDermott, D. F. , Powderly, J. D. , Gettinger, S. N. , Kohrt, H. E. , Horn, L. , Lawrence, D. P. , Rost, S. , Leabman, M. , Xiao, Y. , Mokatrin, A. , Koeppen, H. , Hegde, P. S. , Mellman, I. , Chen, D. S. , Hodi, F. S. (2014) Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carbognin, L. , Pilotto, S. , Milella, M. , Vaccaro, V. , Brunelli, M. , Caliò, A. , Cuppone, F. , Sperduti, I. , Giannarelli, D. , Chilosi, M. , Bronte, V. , Scarpa, A. , Bria, E. , Tortora, G. (2015) Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death‐ligand‐1 (PD‐L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS One 10, e0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brahmer, J. , Reckamp, K. L. , Baas, P. , Crinò, L. , Eberhardt, W. E. E. , Poddubskaya, E. , Antonia, S. , Pluzanski, A. , Vokes, E. E. , Holgado, E. , Waterhouse, D. , Ready, N. , Gainor, J. , Arén Frontera, O. , Havel, L. , Steins, M. , Garassino, M. C. , Aerts, J. G. , Domine, M. , Paz‐Ares, L. , Reck, M. , Baudelet, C. , Harbison, C. T. , Lestini, B. , Spigel, D. R. (2015) Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N. Engl. J. Med. 373, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolchok, J. D. , Chiarion‐Sileni, V. , Gonzalez, R. , Rutkowski, P. , Grob, J. J. , Cowey, C. L. , Lao, C. D. , Schadendorf, D. , Ferrucci, P. F. , Smylie, M. , Dummer, R. , Hill, A. G. , Haanen, J. B. A. G. , Maio, M. , McArthur, G. A. , Yang, A. , Rollin, L. , Horak, C. E. , Larkin, J. M. G. , Hodi, F. S. (2015) Efficacy and safety results from a phase III trial of nivolumab (NIVO) alone or combined with ipilimumab (IPI) versus IPI alone in treatment‐naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). J. Clin. Oncol. 33 (Suppl; abs. LBA1). [Google Scholar]
- 31. CheckMate 025 Investigators . (2015) Nivolumab versus Everolimus in Advanced Renal‐Cell Carcinoma. N. Engl. J. Med. 373, 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tumeh, P. C. , Harview, C. L. , Yearley, J. H. , Shintaku, I. P. , Taylor, E. J. , Robert, L. , Chmielowski, B. , Spasic, M. , Henry, G. , Ciobanu, V. , West, A. N. , Carmona, M. , Kivork, C. , Seja, E. , Cherry, G. , Gutierrez, A. J. , Grogan, T. R. , Mateus, C. , Tomasic, G. , Glaspy, J. A. , Emerson, R. O. , Robins, H. , Pierce, R. H. , Elashoff, D. A. , Robert, C. , Ribas, A. (2014) PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Curran, M. A. , Montalvo, W. , Yagita, H. , Allison, J. P. (2010) PD‐1 and CTLA‐4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 107, 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wolchok, J. D. , Kluger, H. , Callahan, M. K. , Postow, M. A. , Rizvi, N. A. , Lesokhin, A. M. , Segal, N. H. , Ariyan, C. E. , Gordon, R. A. , Reed, K. , Burke, M. M. , Caldwell, A. , Kronenberg, S. A. , Agunwamba, B. U. , Zhang, X. , Lowy, I. , Inzunza, H. D. , Feely, W. , Horak, C. E. , Hong, Q. , Korman, A. J. , Wigginton, J. M. , Gupta, A. , Sznol, M. (2013) Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larkin, J. , Chiarion‐Sileni, V. , Gonzalez, R. , Grob, J. J. , Cowey, C. L. , Lao, C. D. , Schadendorf, D. , Dummer, R. , Smylie, M. , Rutkowski, P. , Ferrucci, P. F. , Hill, A. , Wagstaff, J. , Carlino, M. S. , Haanen, J. B. , Maio, M. , Marquez‐Rodas, I. , McArthur, G. A. , Ascierto, P. A. , Long, G. V. , Callahan, M. K. , Postow, M. A. , Grossmann, K. , Sznol, M. , Dreno, B. , Bastholt, L. , Yang, A. , Rollin, L. M. , Horak, C. , Hodi, F. S. , Wolchok, J. D. (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larkin, J. , Chiarion‐Sileni, V. , Gonzalez, R. , Grob, J. J. , Cowey, C. L. , Lao, C. D. , Schadendorf, D. , Dummer, R. , Smylie, M. , Rutkowski, P. , Ferrucci, P. F. , Hill, A. , Wagstaff, J. , Carlino, M. S. , Haanen, J. B. , Maio, M. , Marquez‐Rodas, I. , McArthur, G. A. , Ascierto, P. A. , Long, G. V. , Callahan, M. K. , Postow, M. A. , Grossmann, K. , Sznol, M. , Dreno, B. , Bastholt, L. , Yang, A. , Rollin, L. M. , Horak, C. , Hodi, F. S. , Wolchok, J. D. (2015) First immunotherapy combo approved for cancer. Cancer Discov. 5, 1228–1229.26487304 [Google Scholar]
- 37. Postow, M. A. , Chesney, J. , Pavlick, A. C. , Robert, C. , Grossmann, K. , McDermott, D. , Linette, G. P. , Meyer, N. , Giguere, J. K. , Agarwala, S. S. , Shaheen, M. , Ernstoff, M. S. , Minor, D. , Salama, A. K. , Taylor, M. , Ott, P. A. , Rollin, L. M. , Horak, C. , Gagnier, P. , Wolchok, J. D. , Hodi, F. S. (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 372, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monney, L. , Sabatos, C. A. , Gaglia, J. L. , Ryu, A. , Waldner, H. , Chernova, T. , Manning, S. , Greenfield, E. A. , Coyle, A. J. , Sobel, R. A. , Freeman, G. J. , Kuchroo, V. K. (2002) Th1‐specific cell surface protein Tim‐3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541. [DOI] [PubMed] [Google Scholar]
- 39. Li, X. , Hu, W. , Zheng, X. , Zhang, C. , Du, P. , Zheng, Z. , Yang, Y. , Wu, J. , Ji, M. , Jiang, J. , Wu, C. (2015) Emerging immune checkpoints for cancer therapy. Acta Oncol. 54, 1706–1713. [DOI] [PubMed] [Google Scholar]
- 40. Le Mercier, I. , Lines, J. L. , Noelle, R. J. (2015) Beyond CTLA‐4 and PD‐1, the generation Z of negative checkpoint regulators. Front. Immunol. 6, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu, C. , Anderson, A. C. , Schubart, A. , Xiong, H. , Imitola, J. , Khoury, S. J. , Zheng, X. X. , Strom, T. B. , Kuchroo, V. K. (2005) The Tim‐3 ligand galectin‐9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252. [DOI] [PubMed] [Google Scholar]
- 42. Boenisch, O. , D'Addio, F. , Watanabe, T. , Elyaman, W. , Magee, C. N. , Yeung, M. Y. , Padera, R. F. , Rodig, S. J. , Murayama, T. , Tanaka, K. , Yuan, X. , Ueno, T. , Jurisch, A. , Mfarrej, B. , Akiba, H. , Yagita, H. , Najafian, N. (2010) TIM‐3: a novel regulatory molecule of alloimmune activation. J. Immunol. 185, 5806–5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chiba, S. , Baghdadi, M. , Akiba, H. , Yoshiyama, H. , Kinoshita, I. , Dosaka‐Akita, H. , Fujioka, Y. , Ohba, Y. , Gorman, J. V. , Colgan, J. D. , Hirashima, M. , Uede, T. , Takaoka, A. , Yagita, H. , Jinushi, M. (2012) Tumor‐infiltrating DCs suppress nucleic acid‐mediated innate immune responses through interactions between the receptor TIM‐3 and the alarmin HMGB1. Nat. Immunol. 13, 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anderson, A. C. (2014) Tim‐3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol. Res. 2, 393–398. [DOI] [PubMed] [Google Scholar]
- 45. Jin, H. T. , Anderson, A. C. , Tan, W. G. , West, E. E. , Ha, S. J. , Araki, K. , Freeman, G. J. , Kuchroo, V. K. , Ahmed, R. (2010) Cooperation of Tim‐3 and PD‐1 in CD8 T‐cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 107, 14733–14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou, Q. , Munger, M. E. , Veenstra, R. G. , Weigel, B. J. , Hirashima, M. , Munn, D. H. , Murphy, W. J. , Azuma, M. , Anderson, A. C. , Kuchroo, V. K. , Blazar, B. R. (2011) Coexpression of Tim‐3 and PD‐1 identifies a CD8+ T‐cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fourcade, J. , Sun, Z. , Benallaoua, M. , Guillaume, P. , Luescher, I. F. , Sander, C. , Kirkwood, J. M. , Kuchroo, V. , Zarour, H. M. (2010) Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sakuishi, K. , Ngiow, S. F. , Sullivan, J. M. , Teng, M. W. , Kuchroo, V. K. , Smyth, M. J. , Anderson, A. C. (2013) TIM3(+)FOXP3(+) regulatory T cells are tissue‐specific promoters of T‐cell dysfunction in cancer. OncoImmunology 2, e23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakuishi, K. , Apetoh, L. , Sullivan, J. M. , Blazar, B. R. , Kuchroo, V. K. , Anderson, A. C. (2010) Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J. Exp. Med. 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ngiow, S. F. , von Scheidt, B. , Akiba, H. , Yagita, H. , Teng, M. W. , Smyth, M. J. (2011) Anti‐TIM3 antibody promotes T cell IFN‐γ‐mediated antitumor immunity and suppresses established tumors. Cancer Res. 71, 3540–3551. [DOI] [PubMed] [Google Scholar]
- 51. Triebel, F. , Jitsukawa, S. , Baixeras, E. , Roman‐Roman, S. , Genevee, C. , Viegas‐Pequignot, E. , Hercend, T. (1990) LAG‐3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huard, B. , Prigent, P. , Tournier, M. , Bruniquel, D. , Triebel, F. (1995) CD4/major histocompatibility complex class II interaction analyzed with CD4‐ and lymphocyte activation gene‐3 (LAG‐3)‐Ig fusion proteins. Eur. J. Immunol. 25, 2718–2721. [DOI] [PubMed] [Google Scholar]
- 53. Workman, C. J. , Cauley, L. S. , Kim, I. J. , Blackman, M. A. , Woodland, D. L. , Vignali, D. A. A. (2004) Lymphocyte activation gene‐3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 172, 5450–5455. [DOI] [PubMed] [Google Scholar]
- 54. Gandhi, M. K. , Lambley, E. , Duraiswamy, J. , Dua, U. , Smith, C. , Elliott, S. , Gill, D. , Marlton, P. , Seymour, J. , Khanna, R. (2006) Expression of LAG‐3 by tumor‐infiltrating lymphocytes is coincident with the suppression of latent membrane antigen‐specific CD8+ T‐cell function in Hodgkin lymphoma patients. Blood 108, 2280–2289. [DOI] [PubMed] [Google Scholar]
- 55. Durham, N. M. , Nirschl, C. J. , Jackson, C. M. , Elias, J. , Kochel, C. M. , Anders, R. A. , Drake, C. G. (2014) Lymphocyte activation gene 3 (LAG‐3) modulates the ability of CD4 T‐cells to be suppressed in vivo. PLoS One 9, e109080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang, C. T. , Workman, C. J. , Flies, D. , Pan, X. , Marson, A. L. , Zhou, G. , Hipkiss, E. L. , Ravi, S. , Kowalski, J. , Levitsky, H. I. , Powell, J. D. , Pardoll, D. M. , Drake, C. G. , Vignali, D. A. A. (2004) Role of LAG‐3 in regulatory T cells. Immunity 21, 503–513. [DOI] [PubMed] [Google Scholar]
- 57. Matsuzaki, J. , Gnjatic, S. , Mhawech‐Fauceglia, P. , Beck, A. , Miller, A. , Tsuji, T. , Eppolito, C. , Qian, F. , Lele, S. , Shrikant, P. , Old, L. J. , Odunsi, K. (2010) Tumor‐infiltrating NY‐ESO‐1‐specific CD8+ T cells are negatively regulated by LAG‐3 and PD‐1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 107, 7875–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Woo, S. R. , Turnis, M. E. , Goldberg, M. V. , Bankoti, J. , Selby, M. , Nirschl, C. J. , Bettini, M. L. , Gravano, D. M. , Vogel, P. , Liu, C. L. , Tangsombatvisit, S. , Grosso, J. F. , Netto, G. , Smeltzer, M. P. , Chaux, A. , Utz, P. J. , Workman, C. J. , Pardoll, D. M. , Korman, A. J. , Drake, C. G. , Vignali, D. A. (2012) Immune inhibitory molecules LAG‐3 and PD‐1 synergistically regulate T‐cell function to promote tumoral immune escape. Cancer Res. 72, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goding, S. R. , Wilson, K. A. , Xie, Y. , Harris, K. M. , Baxi, A. , Akpinarli, A. , Fulton, A. , Tamada, K. , Strome, S. E. , Antony, P. A. (2013) Restoring immune function of tumor‐specific CD4+ T cells during recurrence of melanoma. J. Immunol. 190, 4899–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu, X. , Harden, K. , Gonzalez, L. C. , Francesco, M. , Chiang, E. , Irving, B. , Tom, I. , Ivelja, S. , Refino, C. J. , Clark, H. , Eaton, D. , Grogan, J. L. (2009) The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 10, 48–57. [DOI] [PubMed] [Google Scholar]
- 61. Chauvin, J. M. , Pagliano, O. , Fourcade, J. , Sun, Z. , Wang, H. , Sander, C. , Kirkwood, J. M. , Chen, T. H. , Maurer, M. , Korman, A. J. , Zarour, H. M. (2015) TIGIT and PD‐1 impair tumor antigen‐specific CD8+ T cells in melanoma patients. J. Clin. Invest. 125, 2046–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kurtulus, S. , Sakuishi, K. , Ngiow, S. F. , Joller, N. , Tan, D. J. , Teng, M. W. , Smyth, M. J. , Kuchroo, V. K. , Anderson, A. C. (2015) TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest. 125, 4053–4062. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Johnston, R. J. , Comps‐Agrar, L. , Hackney, J. , Yu, X. , Huseni, M. , Yang, Y. , Park, S. , Javinal, V. , Chiu, H. , Irving, B. , Eaton, D. L. , Grogan, J. L. (2014) The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937. [DOI] [PubMed] [Google Scholar]
- 64. Fourcade, J. , Sun, Z. , Pagliano, O. , Guillaume, P. , Luescher, I. F. , Sander, C. , Kirkwood, J. M. , Olive, D. , Kuchroo, V. , Zarour, H. M. (2012) CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD‐1. Cancer Res. 72, 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Haymaker, C. L. , Wu, R. C. , Ritthipichai, K. , Bernatchez, C. , Forget, M. A. , Chen, J. Q. , Liu, H. , Wang, E. , Marincola, F. , Hwu, P. , Radvanyi, L. G. (2015) BTLA marks a less‐differentiated tumor‐infiltrating lymphocyte subset in melanoma with enhanced survival properties. OncoImmunology 4, e1014246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen, L. P. , Flies, D. B. (2013) Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat. Rev. Immunol. 13, 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Alegre, M. L. , Frauwirth, K. A. , Thompson, C. B. (2001) T‐Cell regulation by CD28 and CTLA‐4. Nat. Rev. Immunol. 1, 220–228. [DOI] [PubMed] [Google Scholar]
- 68. Suntharalingam, G. , Perry, M. R. , Ward, S. , Brett, S. J. , Castello‐Cortes, A. , Brunner, M. D. , Panoskaltsis, N. (2006) Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355, 1018–1028. [DOI] [PubMed] [Google Scholar]
- 69. Akiba, H. , Nakano, H. , Nishinaka, S. , Shindo, M. , Kobata, T. , Atsuta, M. , Morimoto, C. , Ware, C. F. , Malinin, N. L. , Wallach, D. , Yagita, H. , Okumura, K. (1998) CD27, a member of the tumor necrosis factor receptor superfamily, activates NF‐kappaB and stress‐activated protein kinase/c‐Jun N‐terminal kinase via TRAF2, TRAF5, and NF‐kappaB‐inducing kinase. J. Biol. Chem. 273, 13353–13358. [DOI] [PubMed] [Google Scholar]
- 70. Ramakrishna, V. , Sundarapandiyan, K. , Zhao, B. , Bylesjo, M. , Marsh, H. C. , Keler, T. (2015) Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J. Immunother. Cancer 3, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buchan, S. L. , Manzo, T. , Flutter, B. , Rogel, A. , Edwards, N. , Zhang, L. , Sivakumaran, S. , Ghorashian, S. , Carpenter, B. , Bennett, C. L. , Freeman, G. J. , Sykes, M. , Croft, M. , Al‐Shamkhani, A. , Chakraverty, R. (2015) OX40‐ and CD27‐mediated costimulation synergizes with anti‐PD‐L1 blockade by forcing exhausted CD8+ T cells to exit quiescence. J. Immunol. 194, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Claus, C. , Riether, C. , Schürch, C. , Matter, M. S. , Hilmenyuk, T. , Ochsenbein, A. F. (2012) CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 72, 3664–3676. [DOI] [PubMed] [Google Scholar]
- 73. Jacobs, J. , Deschoolmeester, V. , Zwaenepoel, K. , Rolfo, C. , Silence, K. , Rottey, S. , Lardon, F. , Smits, E. , Pauwels, P. (2015) CD70: an emerging target in cancer immunotherapy. Pharmacol. Ther. 155, 1–10. [DOI] [PubMed] [Google Scholar]
- 74. Bartkowiak, T. , Curran, M. A. (2015) 4‐1BB agonists: multi‐potent potentiators of tumor immunity. Front. Oncol. 5, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vinay, D. S. , Kwon, B. S. (2011) 4‐1BB signaling beyond T cells. Cell. Mol. Immunol. 8, 281–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Melero, I. , Shuford, W. W. , Newby, S. A. , Aruffo, A. , Ledbetter, J. A. , Hellström, K. E. , Mittler, R. S. , Chen, L. (1997) Monoclonal antibodies against the 4‐1BB T‐cell activation molecule eradicate established tumors. Nat. Med. 3, 682–685. [DOI] [PubMed] [Google Scholar]
- 77. Adappa, N. D. , Sung, C. K. , Choi, B. , Huang, T. G. , Genden, E. M. , Shin, E. J. (2008) The administration of IL‐12/GM‐CSF and Ig‐4‐1BB ligand markedly decreases murine floor of mouth squamous cell cancer. Otolaryngol. Head Neck Surg. 139, 442–448. [DOI] [PubMed] [Google Scholar]
- 78. Houot, R. , Goldstein, M. J. , Kohrt, H. E. , Myklebust, J. H. , Alizadeh, A. A. , Lin, J. T. , Irish, J. M. , Torchia, J. A. , Kolstad, A. , Chen, L. , Levy, R. (2009) Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood 114, 3431–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gauttier, V. , Judor, J. P. , Le Guen, V. , Cany, J. , Ferry, N. , Conchon, S. (2014) Agonistic anti‐CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int. J. Cancer 135, 2857–2867. [DOI] [PubMed] [Google Scholar]
- 80. Li, B. , Lin, J. , Vanroey, M. , Jure‐Kunkel, M. , Jooss, K. (2007) Established B16 tumors are rejected following treatment with GM‐CSF‐secreting tumor cell immunotherapy in combination with anti‐4‐1BB mAb. Clin. Immunol. 125, 76–87. [DOI] [PubMed] [Google Scholar]
- 81. Tirapu, I. , Arina, A. , Mazzolini, G. , Duarte, M. , Alfaro, C. , Feijoo, E. , Qian, C. , Chen, L. , Prieto, J. , Melero, I. (2004) Improving efficacy of interleukin‐12‐transfected dendritic cells injected into murine colon cancer with anti‐CD137 monoclonal antibodies and alloantigens. Int. J. Cancer 110, 51–60. [DOI] [PubMed] [Google Scholar]
- 82. Sznol, M. , Hodi, F. S. , Margolin, K. , McDermott, D. F. , Ernstoff, M. S. , Kirkwood, J. M. , Wojtaszek, C. , Feltquate, D. , Logan, T. (2008) Phase I study of BMS‐663513, a fully human anti‐CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). J. Clin. Oncol. 26 (Suppl 3007; abs.). [Google Scholar]
- 83. Ascierto, P. A. , Simeone, E. , Sznol, M. , Fu, Y. X. , Melero, I. (2010) Clinical experiences with anti‐CD137 and anti‐PD1 therapeutic antibodies. Semin. Oncol. 37, 508–516. [DOI] [PubMed] [Google Scholar]
- 84. Niu, L. , Strahotin, S. , Hewes, B. , Zhang, B. , Zhang, Y. , Archer, D. , Spencer, T. , Dillehay, D. , Kwon, B. , Chen, L. , Vella, A. T. , Mittler, R. S. (2007) Cytokine‐mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti‐CD137‐treated mice. J. Immunol. 178, 4194–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dubrot, J. , Milheiro, F. , Alfaro, C. , Palazón, A. , Martinez‐Forero, I. , Perez‐Gracia, J. L. , Morales‐Kastresana, A. , Romero‐Trevejo, J. L. , Ochoa, M. C. , Hervás‐Stubbs, S. , Prieto, J. , Jure‐Kunkel, M. , Chen, L. , Melero, I. (2010) Treatment with anti‐CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol. Immunother. 59, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Segal, N. H. , Gopal, A. K. , Bhatia, S. , Kohrt, H. E. , Levy, R. , Pishvalan, M. J. , Houot, R. , Bartlett, N. , Nghlem, P. , Kronenberg, S. A. , Thali, A. D. , Mugundu, G. , Huang, B. , Davis, C. (2014) A phase 1 study of PF‐05082566 (auti‐4‐1BB) in patients with advanced cancer. J. Clin. Oncol. 32 (Suppl; abs. 3007). [Google Scholar]
- 87. Chen, S. , Lee, L. F. , Fisher, T. S. , Jessen, B. , Elliott, M. , Evering, W. , Logronio, K. , Tu, G. H. , Tsaparikos, K. , Li, X. , Wang, H. , Ying, C. , Xiong, M. , VanArsdale, T. , Lin, J. C. (2015) Combination of 4‐1BB agonist and PD‐1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol. Res. 3, 149–160. [DOI] [PubMed] [Google Scholar]
- 88. Shindo, Y. , Yoshimura, K. , Kuramasu, A. , Watanabe, Y. , Ito, H. , Kondo, T. , Oga, A. , Ito, H. , Yoshino, S. , Hazama, S. , Tamada, K. , Yagita, H. , Oka, M. (2015) Combination immunotherapy with 4‐1BB activation and PD‐1 blockade enhances antitumor efficacy in a mouse model of subcutaneous tumor. Anticancer Res. 35, 129–136. [PubMed] [Google Scholar]
- 89. Duraiswamy, J. , Freeman, G. J. , Coukos, G. (2013) Therapeutic PD‐1 pathway blockade augments with other modalities of immunotherapy T‐cell function to prevent immune decline in ovarian cancer. Cancer Res. 73, 6900–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kocak, E. , Lute, K. , Chang, X. , May, K. F. , Jr, Exten, K. R. , Zhang, H. , Abdessalam, S. F. , Lehman, A. M. , Jarjoura, D. , Zheng, P. , Liu, Y. (2006) Combination therapy with anti‐CTL antigen‐4 and anti‐4‐1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 66, 7276–7284. [DOI] [PubMed] [Google Scholar]
- 91. Curran, M. A. , Kim, M. , Montalvo, W. , Al‐Shamkhani, A. , Allison, J. P. (2011) Combination CTLA‐4 blockade and 4‐1BB activation enhances tumor rejection by increasing T‐cell infiltration, proliferation, and cytokine production. PLoS One 6, e19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lane, P. (2000) Role of OX40 signals in coordinating CD4 T cell selection, migration, and cytokine differentiation in T helper (Th)1 and Th2 cells. J. Exp. Med. 191, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Croft, M. (2010) Control of immunity by the TNFR‐related molecule OX40 (CD134). Annu. Rev. Immunol. 28, 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vetto, J. T. , Lum, S. , Morris, A. , Sicotte, M. , Davis, J. , Lemon, M. , Weinberg, A. (1997) Presence of the T‐cell activation marker OX‐40 on tumor infiltrating lymphocytes and draining lymph node cells from patients with melanoma and head and neck cancers. Am. J. Surg. 174, 258–265. [DOI] [PubMed] [Google Scholar]
- 95. Ramstad, T. , Lawnicki, L. , Vetto, J. , Weinberg, A. (2000) Immunohistochemical analysis of primary breast tumors and tumor‐draining lymph nodes by means of the T‐cell costimulatory molecule OX‐40. Am. J. Surg. 179, 400–406. [DOI] [PubMed] [Google Scholar]
- 96. Petty, J. K. , He, K. , Corless, C. L. , Vetto, J. T. , Weinberg, A. D. (2002) Survival in human colorectal cancer correlates with expression of the T‐cell costimulatory molecule OX‐40 (CD134). Am. J. Surg. 183, 512–518. [DOI] [PubMed] [Google Scholar]
- 97. Ladányi, A. , Somlai, B. , Gilde, K. , Fejös, Z. , Gaudi, I. , Tímár, J. (2004) T‐Cell activation marker expression on tumor‐infiltrating lymphocytes as prognostic factor in cutaneous malignant melanoma. Clin. Cancer Res. 10, 521–530. [DOI] [PubMed] [Google Scholar]
- 98. Marabelle, A. , Kohrt, H. , Sagiv‐Barfi, I. , Ajami, B. , Axtell, R. C. , Zhou, G. , Rajapaksa, R. , Green, M. R. , Torchia, J. , Brody, J. , Luong, R. , Rosenblum, M. D. , Steinman, L. , Levitsky, H. I. , Tse, V. , Levy, R. (2013) Depleting tumor‐specific Tregs at a single site eradicates disseminated tumors. J. Clin. Invest. 123, 2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Piconese, S. , Valzasina, B. , Colombo, M. P. (2008) OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 205, 825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Aspeslagh, S. , Postel‐Vinay, S. , Rusakiewicz, S. , Soria, J. C. , Zitvogel, L. , Marabelle, A. (2016) Rationale for anti‐OX40 cancer immunotherapy. Eur. J. Cancer 52, 50–66. [DOI] [PubMed] [Google Scholar]
- 101. Curti, B. D. , Kovacsovics‐Bankowski, M. , Morris, N. , Walker, E. , Chisholm, L. , Floyd, K. , Walker, J. , Gonzalez, I. , Meeuwsen, T. , Fox, B. A. , Moudgil, T. , Miller, W. , Haley, D. , Coffey, T. , Fisher, B. , Delanty‐Miller, L. , Rymarchyk, N. , Kelly, T. , Crocenzi, T. , Bernstein, E. , Sanborn, R. , Urba, W. J. , Weinberg, A. D. (2013) OX40 is a potent immune‐stimulating target in late‐stage cancer patients. Cancer Res. 73, 7189–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Redmond, W. L. , Linch, S. N. , Kasiewicz, M. J. (2014) Combined targeting of costimulatory (OX40) and coinhibitory (CTLA‐4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol. Res. 2, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Houot, R. , Levy, R. (2009) T‐Cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood 113, 3546–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Guo, Z. , Wang, X. , Cheng, D. , Xia, Z. , Luan, M. , Zhang, S. (2014) PD‐1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS One 9, e89350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nocentini, G. , Giunchi, L. , Ronchetti, S. , Krausz, L. T. , Bartoli, A. , Moraca, R. , Migliorati, G. , Riccardi, C. (1997) A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor‐induced apoptosis. Proc. Natl. Acad. Sci. USA 94, 6216–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tone, M. , Tone, Y. , Adams, E. , Yates, S. F. , Frewin, M. R. , Cobbold, S. P. , Waldmann, H. (2003) Mouse glucocorticoid‐induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA 100, 15059–15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. McHugh, R. S. , Whitters, M. J. , Piccirillo, C. A. , Young, D. A. , Shevach, E. M. , Collins, M. , Byrne, M. C. (2002) CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid‐induced TNF receptor. Immunity 16, 311–323. [DOI] [PubMed] [Google Scholar]
- 108. Shimizu, J. , Yamazaki, S. , Takahashi, T. , Ishida, Y. , Sakaguchi, S. (2002) Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self‐tolerance. Nat. Immunol. 3, 135–142. [DOI] [PubMed] [Google Scholar]
- 109. Zhou, P. , L'italien, L. , Hodges, D. , Schebye, X. M. (2007) Pivotal roles of CD4+ effector T cells in mediating agonistic anti‐GITR mAb‐induced‐immune activation and tumor immunity in CT26 tumors. J. Immunol. 179, 7365–7375. [DOI] [PubMed] [Google Scholar]
- 110. Cohen, A. D. , Schaer, D. A. , Liu, C. , Li, Y. , Hirschhorn‐Cymmerman, D. , Kim, S. C. , Diab, A. , Rizzuto, G. , Duan, F. , Perales, M. A. , Merghoub, T. , Houghton, A. N. , Wolchok, J. D. (2010) Agonist anti‐GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra‐tumor accumulation. PLoS One 5, e10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mitsui, J. , Nishikawa, H. , Muraoka, D. , Wang, L. , Noguchi, T. , Sato, E. , Kondo, S. , Allison, J. P. , Sakaguchi, S. , Old, L. J. , Kato, T. , Shiku, H. (2010) Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin. Cancer Res. 16, 2781–2791. [DOI] [PubMed] [Google Scholar]
- 112. Lu, L. , Xu, X. , Zhang, B. , Zhang, R. , Ji, H. , Wang, X. (2014) Combined PD‐1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Caux, C. , Massacrier, C. , Vanbervliet, B. , Dubois, B. , Van Kooten, C. , Durand, I. , Banchereau, J. (1994) Activation of human dendritic cells through CD40 cross‐linking. J. Exp. Med. 180, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cella, M. , Scheidegger, D. , Palmer‐Lehmann, K. , Lane, P. , Lanzavecchia, A. , Alber, G. (1996) Ligation of CD40 on dendritic cells triggers production of high levels of interleukin‐12 and enhances T cell stimulatory capacity: T‐T help via APC activation. J. Exp. Med. 184, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zippelius, A. , Schreiner, J. , Herzig, P. , Müller, P. (2015) Induced PD‐L1 expression mediates acquired resistance to agonistic anti‐CD40 treatment. Cancer Immunol. Res. 3, 236–244. [DOI] [PubMed] [Google Scholar]
- 116. Melero, I. , Lasarte, J. J. (2015) Genetic basis for clinical response to CTLA‐4 blockade. N. Engl. J. Med. 372, 783. [DOI] [PubMed] [Google Scholar]
- 117. Melero, I. , Gaudernack, G. , Gerritsen, W. , Huber, C. , Parmiani, G. , Scholl, S. , Thatcher, N. , Wagstaff, J. , Zielinski, C. , Faulkner, I. , Mellstedt, H. (2014) Therapeutic vaccines for cancer: an overview of clinical trials. Nat. Rev. Clin. Oncol. 11, 509–524. [DOI] [PubMed] [Google Scholar]
- 118. Melief, C. J. , van Hall, T. , Arens, R. , Ossendorp, F. , van der Burg, S. H. (2015) Therapeutic cancer vaccines. J. Clin. Invest. 125, 3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kleponis, J. , Skelton, R. , Zheng, L. (2015) Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol. Med. 12, 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Morse, M. A. , Lyerly, H. K. (2015) Checkpoint blockade in combination with cancer vaccines. Vaccine 33, 7377–7385. [DOI] [PubMed] [Google Scholar]
- 121. Weber, J. S. , Kudchadkar, R. R. , Yu, B. , Gallenstein, D. , Horak, C. E. , Inzunza, H. D. , Zhao, X. , Martinez, A. J. , Wang, W. , Gibney, G. , Kroeger, J. , Eysmans, C. , Sarnaik, A. A. , Chen, Y. A. (2013) Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab‐refractory or ‐naive melanoma. J. Clin. Oncol. 31, 4311–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Löb, S. , Königsrainer, A. , Rammensee, H. G. , Opelz, G. , Terness, P. (2009) Inhibitors of indoleamine‐2,3‐dioxygenase for cancer therapy: can we see the wood for the trees? Nat. Rev. Cancer 9, 445–452. [DOI] [PubMed] [Google Scholar]
- 123. Munn, D. H. , Zhou, M. , Attwood, J. T. , Bondarev, I. , Conway, S. J. , Marshall, B. , Brown, C. , Mellor, A. L. (1998) Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 281, 1191–1193. [DOI] [PubMed] [Google Scholar]
- 124. Hwu, P. , Du, M. X. , Lapointe, R. , Do, M. , Taylor, M. W. , Young, H. A. (2000) Indoleamine 2,3‐dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 164, 3596–3599. [DOI] [PubMed] [Google Scholar]
- 125. Mellor, A. L. , Baban, B. , Chandler, P. , Marshall, B. , Jhaver, K. , Hansen, A. , Koni, P. A. , Iwashima, M. , Munn, D. H. (2003) Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 171, 1652–1655. [DOI] [PubMed] [Google Scholar]
- 126. Yu, J. , Wang, Y. , Yan, F. , Zhang, P. , Li, H. , Zhao, H. , Yan, C. , Yan, F. , Ren, X. (2014) Noncanonical NF‐kPB activation mediates STAT3‐stimulated IDO upregulation in myeloid‐derived suppressor cells in breast cancer. J. Immunol. 193, 2574–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Vacchelli, E. , Aranda, F. , Eggermont, A. , Sautès‐Fridman, C. , Tartour, E. , Kennedy, E. P. , Platten, M. , Zitvogel, L. , Kroemer, G. , Galluzzi, L. (2014) Trial watch: IDO inhibitors in cancer therapy. OncoImmunology 3, e957994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Uyttenhove, C. , Pilotte, L. , Théate, I. , Stroobant, V. , Colau, D. , Parmentier, N. , Boon, T. , Van den Eynde, B. J. (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3‐dioxygenase. Nat. Med. 9, 1269–1274. [DOI] [PubMed] [Google Scholar]
- 129. Muller, A. J. , DuHadaway, J. B. , Donover, P. S. , Sutanto‐Ward, E. , Prendergast, G. C. (2005) Inhibition of indoleamine 2,3‐dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 11, 312–319. [DOI] [PubMed] [Google Scholar]
- 130. Beatty, G. L. , O'Dwyer, P. J. , Clark, J. , Shi, J. G. , Newton, R. C. , Schaub, R. , Maleski, J. , Leopold, L. , Gajewski, T. (2013) Phase I study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of the oral inhibitor of indoleamine 2,3‐dioxygenase (IDO1) INCB024360 in patients (pts) with advanced malignancies. J. Clin. Oncol. 31 (Suppl; abs. 3025). [Google Scholar]
- 131. Soliman, H. H. , Antonia, S. , Sullivan, D. , Vanahanian, N. , Link, C. (2009) Overcoming tumor antigen anergy in human malignancies using the novel indeolamine 2,3‐dioxygenase (IDO) enzyme inhibitor, 1‐methyl‐D‐tryptophan (1MT). J. Clin. Oncol. 27 (Suppl; abs. 3004). [Google Scholar]
- 132. Gibney, G. , Hamid, O. , Lutzky, J. , Olszanski, A. , Gangadhar, T. , Gajewski, T. , Chmielowski, B. , Hanks, B. , Boasberg, P. , Zhao, Y. , Newton, R. , Bowman, J. , Maleski, J. , Leopold, L. , Weber, J. S. (2015) Updated results from a phase 1/2 study of epacadostat (INCB024360) in combination with ipilimumab in patients with metastatic melanoma. Eur. J. Cancer 51 (Suppl 3), S106–S107. [Google Scholar]
- 133. Gangadhar, T. C. , Hamid, O. , Smith, D. C. , Bauer, T. M. , Wasser, J. S. , Luke, J. J. , Balmanoukian, A. S. , Kaufman, D. R. , Zhao, Y. , Maleski, J. , Leopold, L. , Gajewski, T. F. (2015) Preliminary results from a phase I/II study of epacadostat (incb024360) in combination with pembrolizumab in patients with selected advanced cancers. J. Immunother. Cancer 3 (Suppl 2), O7. [Google Scholar]
- 134. Kroemer, G. , Galluzzi, L. , Kepp, O. , Zitvogel, L. (2013) Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72. [DOI] [PubMed] [Google Scholar]
- 135. Garg, A. D. , Martin, S. , Golab, J. , Agostinis, P. (2014) Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 21, 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Krysko, D. V. , Garg, A. D. , Kaczmarek, A. , Krysko, O. , Agostinis, P. , Vandenabeele, P. (2012) Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875. [DOI] [PubMed] [Google Scholar]
- 137. Garg, A. D. , Galluzzi, L. , Apetoh, L. , Baert, T. , Birge, R. B. , Bravo‐San Pedro, J. M. , Breckpot, K. , Brough, D. , Chaurio, R. , Cirone, M. , Coosemans, A. , Coulie, P. G. , De Ruysscher, D. , Dini, L. , de Witte, P. , Dudek‐Peric, A. M. , Faggioni, A. , Fucikova, J. , Gaipl, U. S. , Golab, J. , Gougeon, M. L. , Hamblin, M. R. , Hemminki, A. , Herrmann, M. , Hodge, J. W. , Kepp, O. , Kroemer, G. , Krysko, D. V. , Land, W. G. , Madeo, F. , Manfredi, A. A. , Mattarollo, S. R. , Maueroder, C. , Merendino, N. , Multhoff, G. , Pabst, T. , Ricci, J. E. , Riganti, C. , Romano, E. , Rufo, N. , Smyth, M. J. , Sonnemann, J. , Spisek, R. , Stagg, J. , Vacchelli, E. , Vandenabeele, P. , Vandenberk, L. , Van den Eynde, B. J. , Van Gool, S. , Velotti, F. , Zitvogel, L. , Agostinis, P. (2015) Molecular and translational classifications of DAMPs in immunogenic cell death. Front. Immunol. 6, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bezu, L. , Gomes‐de‐Silva, L. C. , Dewitte, H. , Breckpot, K. , Fucikova, J. , Spisek, R. , Galluzzi, L. , Kepp, O. , Kroemer, G. (2015) Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 6, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhang, Q. , Raoof, M. , Chen, Y. , Sumi, Y. , Sursal, T. , Junger, W. , Brohi, K. , Itagaki, K. , Hauser, C. J. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fucikova, J. , Moserova, I. , Urbanova, L. , Bezu, L. , Kepp, O. , Cremer, I. , Salek, C. , Strnad, P. , Kroemer, G. , Galluzzi, L. , Spisek, R. (2015) Prognostic and predictive value of DAMPs and DAMP‐associated processes in cancer. Front. Immunol. 6, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Dewan, M. Z. , Galloway, A. E. , Kawashima, N. , Dewyngaert, J. K. , Babb, J. S. , Formenti, S. C. , Demaria, S. (2009) Fractionated but not single‐dose radiotherapy induces an immune‐mediated abscopal effect when combined with anti‐CTLA‐4 antibody. Clin. Cancer Res. 15, 5379–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Demaria, S. , Kawashima, N. , Yang, A. M. , Devitt, M. L. , Babb, J. S. , Allison, J. P. , Formenti, S. C. (2005) Immune‐mediated inhibition of metastases after treatment with local radiation and CTLA‐4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 11, 728–734. [PubMed] [Google Scholar]
- 143. Sharabi, A. B. , Nirschl, C. J. , Kochel, C. M. , Nirschl, T. R. , Francica, B. J. , Velarde, E. , Deweese, T. L. , Drake, C. G. (2015) Stereotactic radiation therapy augments antigen‐specific PD‐1‐mediated antitumor immune responses via cross‐presentation of tumor antigen. Cancer Immunol. Res. 3, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zeng, J. , See, A. P. , Phallen, J. , Jackson, C. M. , Belcaid, Z. , Ruzevick, J. , Durham, N. , Meyer, C. , Harris, T. J. , Albesiano, E. , Pradilla, G. , Ford, E. , Wong, J. , Hammers, H. J. , Mathios, D. , Tyler, B. , Brem, H. , Tran, P. T. , Pardoll, D. , Drake, C. G. , Lim, M. (2013) Anti‐PD‐1 blockade and stereotactic radiation produce long‐term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 86, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Deng, L. , Liang, H. , Burnette, B. , Beckett, M. , Darga, T. , Weichselbaum, R. R. , Fu, Y. X. (2014) Irradiation and anti‐PD‐L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Invest. 124, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Dovedi, S. J. , Adlard, A. L. , Lipowska‐Bhalla, G. , McKenna, C. , Jones, S. , Cheadle, E. J. , Stratford, I. J. , Poon, E. , Morrow, M. , Stewart, R. , Jones, H. , Wilkinson, R. W. , Honeychurch, J. , Illidge, T. M. (2014) Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD‐L1 blockade. Cancer Res. 74, 5458–5468. [DOI] [PubMed] [Google Scholar]
- 147. Hiniker, S. M. , Chen, D. S. , Knox, S. J. (2012) Abscopal effect in a patient with melanoma. N. Engl. J. Med. 366, 2035; author reply 2035–2036. [DOI] [PubMed] [Google Scholar]
- 148. Postow, M. A. , Callahan, M. K. , Barker, C. A. , Yamada, Y. , Yuan, J. , Kitano, S. , Mu, Z. , Rasalan, T. , Adamow, M. , Ritter, E. , Sedrak, C. , Jungbluth, A. A. , Chua, R. , Yang, A. S. , Roman, R. A. , Rosner, S. , Benson, B. , Allison, J. P. , Lesokhin, A. M. , Gnjatic, S. , Wolchok, J. D. (2012) Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 366, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Grimaldi, A. M. , Simeone, E. , Giannarelli, D. , Muto, P. , Falivene, S. , Borzillo, V. , Giugliano, F. M. , Sandomenico, F. , Petrillo, A. , Curvietto, M. , Esposito, A. , Paone, M. , Palla, M. , Palmieri, G. , Caracò, C. , Ciliberto, G. , Mozzillo, N. , Ascierto, P. A. (2014) Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. OncoImmunology 3, e28780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chandra, R. A. , Wilhite, T. J. , Balboni, T. A. , Alexander, B. M. , Spektor, A. , Ott, P. A. , Ng, A. K. , Hodi, F. S. , Schoenfeld, J. D. (2015) A systematic evaluation of abscopal responses following radiotherapy in patients with metastatic melanoma treated with ipilimumab. OncoImmunology 4, e1046028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Twyman‐Saint Victor, C. , Rech, A. J. , Maity, A. , Rengan, R. , Pauken, K. E. , Stelekati, E. , Benci, J. L. , Xu, B. , Dada, H. , Odorizzi, P. M. , Herati, R. S. , Mansfield, K. D. , Patsch, D. , Amaravadi, R. K. , Schuchter, L. M. , Ishwaran, H. , Mick, R. , Pryma, D. A. , Xu, X. , Feldman, M. D. , Gangadhar, T. C. , Hahn, S. M. , Wherry, E. J. , Vonderheide, R. H. , Minn, A. J. (2015) Radiation and dual checkpoint blockade activate non‐redundant immune mechanisms in cancer. Nature 520, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Madondo, M. T. , Quinn, M. , Plebanski, M. (2016) Low dose cyclophosphamide: mechanisms of T cell modulation. Cancer Treat. Rev. 42, 3–9. [DOI] [PubMed] [Google Scholar]
- 153. Todo, T. , Rabkin, S. D. , Sundaresan, P. , Wu, A. , Meehan, K. R. , Herscowitz, H. B. , Martuza, R. L. (1999) Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication‐competent herpes simplex virus. Hum. Gene Ther. 10, 2741–2755. [DOI] [PubMed] [Google Scholar]
- 154. Grekova, S. P. , Raykov, Z. , Zawatzky, R. , Rommelaere, J. , Koch, U. (2012) Activation of a glioma‐specific immune response by oncolytic parvovirus Minute Virus of Mice infection. Cancer Gene Ther. 19, 468–475. [DOI] [PubMed] [Google Scholar]
- 155. Koks, C. A. , Garg, A. D. , Ehrhardt, M. , Riva, M. , Vandenberk, L. , Boon, L. , De Vleeschouwer, S. , Agostinis, P. , Graf, N. , Van Gool, S. W. (2015) Newcastle disease virotherapy induces long‐term survival and tumor‐specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 136, E313–E325. [DOI] [PubMed] [Google Scholar]
- 156. Zamarin, D. , Holmgaard, R. B. , Subudhi, S. K. , Park, J. S. , Mansour, M. , Palese, P. , Merghoub, T. , Wolchok, J. D. , Allison, J. P. (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 6, 226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Andtbacka, R. H. , Kaufman, H. L. , Collichio, F. , Amatruda, T. , Senzer, N. , Chesney, J. , Delman, K. A. , Spitler, L. E. , Puzanov, I. , Agarwala, S. S. , Milhem, M. , Cranmer, L. , Curti, B. , Lewis, K. , Ross, M. , Guthrie, T. , Linette, G. P. , Daniels, G. A. , Harrington, K. , Middleton, M. R. , Miller, W. H. , Jr, Zager, J. S. , Ye, Y. , Yao, B. , Li, A. , Doleman, S. , VanderWalde, A. , Gansert, J. , Coffin, R. S. (2015) Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, 2780–2788. [DOI] [PubMed] [Google Scholar]
- 158. Puzanov, I. , Milhem, M. M. , Andtbacka, R. H. I. , Minor, D. R. , Hamid, O. , Li, A. , Chou, J. , Kaufman, H. (2015) Survival, safety, and response patterns in a phase 1b multicenter trial of talimogene laherparepvec (T‐VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB‐IV melanoma. J. Clin. Oncol. 33 (Suppl; abs. 9063). [Google Scholar]
- 159. Long, G. V. , Dummer, R. , Ribas, A. , Puzanov, I. , Michielin, O. , Vanderwalde, A. , Andtbacka, R. H. I. , Cebon, J. , Fernandez, E. , Malvehy, J. , Olszanski, A. J. , Gajewski, T. F. , Kirkwood, J. M. , Gause, C. , Chen, L. , Kaufman, D. R. , Chou, J. , Hodi, F. S. (2015) 24LBA safety data from the phase 1b part of the MASTERKEY‐265 study combining talimogene laherparepvec (T‐VEC) and pembrolizumab for unresectable stage IIIB‐IV melanoma. Eur. J. Cancer 51 (Suppl 3), S722. [Google Scholar]
- 160. Covre, A. , Coral, S. , Di Giacomo, A. M. , Taverna, P. , Azab, M. , Maio, M. (2015) Epigenetics meets immune checkpoints. Semin. Oncol. 42, 506–513. [DOI] [PubMed] [Google Scholar]
- 161. Sigalotti, L. , Fratta, E. , Coral, S. , Maio, M. (2014) Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol. Ther. 142, 339–350. [DOI] [PubMed] [Google Scholar]
- 162. Coral, S. , Sigalotti, L. , Altomonte, M. , Engelsberg, A. , Colizzi, F. , Cattarossi, I. , Maraskovsky, E. , Jager, E. , Seliger, B. , Maio, M. (2002) 5‐aza‐2'‐Deoxycytidine‐induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin. Cancer Res. 8, 2690–2695. [PubMed] [Google Scholar]
- 163. Fonsatti, E. , Nicolay, H. J. , Sigalotti, L. , Calabrò, L. , Pezzani, L. , Colizzi, F. , Altomonte, M. , Guidoboni, M. , Marincola, F. M. , Maio, M. (2007) Functional up‐regulation of human leukocyte antigen class I antigens expression by 5‐aza‐2'‐deoxycytidine in cutaneous melanoma: immunotherapeutic implications. Clin. Cancer Res. 13, 3333–3338. [DOI] [PubMed] [Google Scholar]
- 164. Adair, S. J. , Hogan, K. T. (2009) Treatment of ovarian cancer cell lines with 5‐aza‐2'‐deoxycytidine upregulates the expression of cancer‐testis antigens and class I major histocompatibility complex‐encoded molecules. Cancer Immunol. Immunother. 58, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Magner, W. J. , Kazim, A. L. , Stewart, C. , Romano, M. A. , Catalano, G. , Grande, C. , Keiser, N. , Santaniello, F. , Tomasi, T. B. (2000) Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 165, 7017–7024. [DOI] [PubMed] [Google Scholar]
- 166. Mora‐Garcia, M. D. , Duenas‐Gonzalez, A. , Hernandez‐Montes, J. , De la Cruz‐Hernandez, E. , Perez‐Cardenas, E. , Weiss‐Steider, B. , Santiago‐Osorio, E. , Ortiz‐Navarrete, V. F. , Rosales, V. H. , Cantu, D. , Lizano‐Soberon, M. , Rojo‐Aguilar, M. P. , Monroy‐Garcia, A. (2006) Up‐regulation of HLA class‐I antigen expression and antigen‐specific CTL response in cervical cancer cells by the demethylating agent hydralazine and the histone deacetylase inhibitor valproic acid. J. Transl. Med. 4, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Armeanu, S. , Bitzer, M. , Lauer, U. M. , Venturelli, S. , Pathil, A. , Krusch, M. , Kaiser, S. , Jobst, J. , Smirnow, I. , Wagner, A. , Steinle, A. , Salih, H. R. (2005) Natural killer cell‐mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 65, 6321–6329. [DOI] [PubMed] [Google Scholar]
- 168. Skov, S. , Pedersen, M. T. , Andresen, L. , Straten, P. T. , Woetmann, A. , Odum, N. (2005) Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase‐3‐dependent expression of MHC class I‐related chain A and B. Cancer Res. 65, 11136–11145. [DOI] [PubMed] [Google Scholar]
- 169. West, A. C. , Mattarollo, S. R. , Shortt, J. , Cluse, L. A. , Christiansen, A. J. , Smyth, M. J. , Johnstone, R. W. (2013) An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 73, 7265–7276. [DOI] [PubMed] [Google Scholar]
- 170. Sonnemann, J. , Gressmann, S. , Becker, S. , Wittig, S. , Schmudde, M. , Beck, J. F. (2010) The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother. Pharmacol. 66, 611–616. [DOI] [PubMed] [Google Scholar]
- 171. Kim, K. , Skora, A. D. , Li, Z. , Liu, Q. , Tam, A. J. , Blosser, R. L. , Diaz, L. A. , Jr, Papadopoulos, N. , Kinzler, K. W. , Vogelstein, B. , Zhou, S. (2014) Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid‐derived cells. Proc. Natl. Acad. Sci. USA 111, 11774–11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Woods, D. M. , Sodre, A. L. , Villagra, A. , Sarnaik, A. , Sotomayor, E. M. , Weber, J. (2015) HDAC inhibition upregulates PD‐1 ligands in melanoma and augments immunotherapy with PD‐1 blockade. Cancer Immunol. Res. 3, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Covre, A. , Coral, S. , Nicolay, H. , Parisi, G. , Fazio, C. , Colizzi, F. , Fratta, E. , Di Giacomo, A. M. , Sigalotti, L. , Natali, P. G. , Maio, M. (2015) Antitumor activity of epigenetic immunomodulation combined with CTLA‐4 blockade in syngeneic mouse models. OncoImmunology 4, e1019978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kuilman, T. , Peeper, D. S. (2009) Senescence‐messaging secretome: SMS‐ing cellular stress. Nat. Rev. Cancer 9, 81–94. [DOI] [PubMed] [Google Scholar]
- 175. Coppé, J. P. , Desprez, P. Y. , Krtolica, A. , Campisi, J. (2010) The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Xue, W. , Zender, L. , Miething, C. , Dickins, R. A. , Hernando, E. , Krizhanovsky, V. , Cordon‐Cardo, C. , Lowe, S. W. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kang, T. W. , Yevsa, T. , Woller, N. , Hoenicke, L. , Wuestefeld, T. , Dauch, D. , Hohmeyer, A. , Gereke, M. , Rudalska, R. , Potapova, A. , Iken, M. , Vucur, M. , Weiss, S. , Heikenwalder, M. , Khan, S. , Gil, J. , Bruder, D. , Manns, M. , Schirmacher, P. , Tacke, F. , Ott, M. , Luedde, T. , Longerich, T. , Kubicka, S. , Zender, L. (2011) Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature 479, 547–551. [DOI] [PubMed] [Google Scholar]
- 178. Hoenicke, L. , Zender, L. (2012) Immune surveillance of senescent cells—biological significance in cancer‐ and non‐cancer pathologies. Carcinogenesis 33, 1123–1126. [DOI] [PubMed] [Google Scholar]
- 179. Vilgelm, A. , Richmond, A. (2015) Combined therapies that induce senescence and stabilize p53 block melanoma growth and prompt antitumor immune responses. OncoImmunology 4, e1009299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Ewald, J. A. , Desotelle, J. A. , Wilding, G. , Jarrard, D. F. (2010) Therapy‐induced senescence in cancer. J. Natl. Cancer Inst. 102, 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Liu, Y. , Hawkins, O. E. , Su, Y. , Vilgelm, A. E. , Sobolik, T. , Thu, Y. M. , Kantrow, S. , Splittgerber, R. C. , Short, S. , Amiri, K. I. , Ecsedy, J. A. , Sosman, J. A. , Kelley, M. C. , Richmond, A. (2013) Targeting aurora kinases limits tumour growth through DNA damage‐mediated senescence and blockade of NF‐kPB impairs this drug‐induced senescence. EMBO Mol. Med. 5, 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Vilgelm, A. E. , Johnson, C. A. , Prasad, N. , Yang, J. , Chen, S.‐C. , Ayers, G. D. , Pawlikowski, J. S. , Raman, D. , Sosman, J. A. , Kelley, M. , Ecsedy, J. A. , Shyr, Y. , Levy, S. E. , Richmond, A. (2015) Connecting the dots: therapy‐induced senescence and a tumor‐suppressive immune microenvironment. J. Natl. Cancer Inst. 108, pii: djv406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Liu, Y. , Hawkins, O. E. , Vilgelm, A. E. , Pawlikowski, J. S. , Ecsedy, J. A. , Sosman, J. A. , Kelley, M. C. , Richmond, A. (2015) Combining an aurora kinase inhibitor and a death receptor ligand/agonist antibody triggers apoptosis in melanoma cells and prevents tumor growth in preclinical mouse models. Clin. Cancer Res. 21, 5338–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Johnson, D. B. , Sosman, J. A. (2015) Therapeutic advances and treatment options in metastatic melanoma. JAMA Oncol. 1, 380–386. [DOI] [PubMed] [Google Scholar]
- 185. Long, G. V. , Stroyakovskiy, D. , Gogas, H. , Levchenko, E. , de Braud, F. , Larkin, J. , Garbe, C. , Jouary, T. , Hauschild, A. , Grob, J. J. , Chiarion Sileni, V. , Lebbe, C. , Mandalà, M. , Millward, M. , Arance, A. , Bondarenko, I. , Haanen, J. B. , Hansson, J. , Utikal, J. , Ferraresi, V. , Kovalenko, N. , Mohr, P. , Probachai, V. , Schadendorf, D. , Nathan, P. , Robert, C. , Ribas, A. , DeMarini, D. J. , Irani, J. G. , Casey, M. , Ouellet, D. , Martin, A. M. , Le, N. , Patel, K. , Flaherty, K. (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 371, 1877–1888. [DOI] [PubMed] [Google Scholar]
- 186. Vanneman, M. , Dranoff, G. (2012) Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 12, 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Xu, M. M. , Pu, Y. , Zhang, Y. , Fu, Y. X. (2016) The role of adaptive immunity in the efficacy of targeted cancer therapies. Trends Immunol. 37, 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Wargo, J. A. , Cooper, Z. A. , Flaherty, K. T. (2014) Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer Discov. 4, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ribas, A. , Hodi, F. S. , Callahan, M. , Konto, C. , Wolchok, J. (2013) Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 368, 1365–1366. [DOI] [PubMed] [Google Scholar]
- 190. Puzanov, I. , Callahan, M. K. , Linette, G. P. , Patel, S. P. , Luke, J. J. , Sosman, J. A. , Wolchok, J. D. , Hamid, O. , Minor, D. R. , Orford, K. W. , Hug, B. A. , Ma, B. , Matthys, G. M. , Hoos, A. (2014) Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation positive unresectable or metastatic melanoma (MM). J. Clin. Oncol. 32 (Suppl; abs. 2511). [Google Scholar]
- 191. Ribas, A. , Butler, M. , Lutzky, J. , Lawrence, D. P. , Robert, C. , Miller, W. , Linette, G. P. , Ascierto, P. A. , Kuzel, T. , Algazi, A. P. , Postow, M. A. , Nathan, P. D. , Curti, B. D. , Robbins, P. B. , Li, X. , Blake‐Haskins, J. A. , Gordon, M. S. (2015) Phase I study combining anti‐PD‐L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J. Clin. Oncol. 33 (Suppl; abs. 3003). [Google Scholar]
- 192. Smith, K. N. , Housseau, F. (2015) An unexpected journey: how cancer immunotherapy has paved the way for an HIV‐1 cure. Discov. Med. 19, 229–238. [PubMed] [Google Scholar]