

Abstract
Recent findings suggest that tau oligomers, which form before neurofibrillary tangles (NFTs), are the true neurotoxic tau entities in neurodegenerative tauopathies, including Alzheimer's disease (AD). Studies in animal models of tauopathy suggest that tau oligomers play a key role in eliciting behavioral and cognitive impairments. Here, we used a novel tau oligomer-specific monoclonal antibody (TOMA) for passive immunization in mice expressing mutant human tau. A single dose of TOMA administered either intravenously or intracerebroventricularly was sufficient to reverse both locomotor and memory deficits in a mouse model of tauopathy for 60 d, coincident with rapid reduction of tau oligomers but not phosphorylated NFTs or monomeric tau. Our data demonstrate that antibody protection is mediated by extracellular and rapid peripheral clearance. These findings provide the first direct evidence in support of a critical role for tau oligomers in disease progression and validate tau oligomers as a target for the treatment of AD and other neurodegenerative tauopathies.
Keywords: Alzheimer's disease, immunotherapy, tau oligomers, tauopathies
Introduction
The accumulation of tau protein into neurofibrillary tangles (NFTs) is a pathological hallmark of several sporadic neurodegenerative diseases termed tauopathies, which include Alzheimer's disease (AD). Although tau in its native form exists as an unfolded monomer essential for microtubule dynamics, misfolded forms of the protein are found in disease states, leading to toxicity and the formation of tau aggregates. In addition, tau mutations can cause disorders such as familial frontotemporal dementia (for review, see Ballatore et al., 2007). Although tau pathology appears to be essential for neurodegeneration (Ballatore et al., 2007; Haroutunian et al., 2007) and amyloid-β (Aβ)-mediated neurotoxicity (Rapoport et al., 2002; Roberson et al., 2007), tau-based therapeutic approaches have lagged behind those targeting Aβ (Schneider and Mandelkow, 2008; Lasagna-Reeves et al., 2011b; Ubhi and Masliah, 2011). Active vaccination using phosphorylated tau fragments (Asuni et al., 2007; Boimel et al., 2010; Bi et al., 2011), and passive vaccination using antibodies directed against NFT-associated tau phosphoepitopes (Boutajangout et al., 2011; Chai et al., 2011; Gu and Sigurdsson, 2011) have been reported in animal models. However, a growing body of evidence suggests that large, metastable tau aggregates such as NFTs are not causally linked to tauopathy phenotypes in animal models (Wittmann et al., 2001; Santacruz et al., 2005; Berger et al., 2007; Yoshiyama et al., 2007; Polydoro et al., 2009; Lasagna-Reeves et al., 2011b; Spires-Jones et al., 2011; Cowan et al., 2012). Furthermore, in human AD brain, NFT-containing neurons can survive for decades (Morsch et al., 1999) and neuronal loss precedes or is independent of NFT formation (Gómez-Isla et al., 1997; Terry, 2000; Maeda et al., 2006; van de Nes et al., 2008), suggesting that other tau species may contribute to earlier stages of disease.
Although both active immunization with tau protein and tau fragments and passive immunization with sequence-specific antibodies to tau have considerable appeal, there are concerns regarding these approaches. Because tau is an endogenous protein with normal cellular functions, such treatments carry the risk of inducing autoimmunity and/or other related complications (Rosenmann et al., 2006). To protect against negative side effects, we developed a novel anti-tau oligomer-specific mouse monoclonal antibody (anti-TOMA). This antibody recognizes tau oligomers specifically and does not recognize monomeric functional tau or mature meta-stable NFTs. We used TOMA to study tau oligomers in a well established mouse model of tauopathy, JNPL3, which expresses the mutant human tau protein (P301L), which is responsible for frontotemporal dementia (Lewis et al., 2000). TOMA proved to have ideal characteristics for immunotherapy due to its unique specificity for tau oligomers, its high affinity, and its ability to sequester tau oligomer toxicity in vitro. Furthermore, other IgG antibodies have been shown to enter the brain in P301L mice, lending evidence to the ability of TOMA to cross the blood brain barrier (BBB) if administered intravenously (Asuni et al., 2007). We treated male homozygous P301L mice with TOMA via two separate routes of administration, evaluated the ability of TOMA to modulate the pathological effects of tau oligomers in vivo, and studied its mechanism of action.
Materials and Methods
Animals.
BALB/c mice (Taconic) were used for the generation of the TOMA antibody. For analysis of immunotherapy, C57BL/6 (C57) wild-type (stock # 1638M; Taconic) and male homozygous P301L (JNPL3) mice (Taconic) were used. The JNPL3 mouse model (Lewis et al., 2000) expresses a frontotemporal dementia-associated human mutant tau transgene (P301L) and has been used previously for tau immunotherapy (Asuni et al., 2007; Chai et al., 2011; Bi et al., 2011). C57 mice are B6D2F1xSW hybrid mice, which were recommended by Taconic as nontransgenic controls for P301L mice. All animals used were male to control for changes in female hormone state that may impact cognitive data. Mice were housed at the University of Texas Medical Branch animal care facility and maintained according to US Department of Agriculture standards (12 h light/dark cycle, food and water ad libitum) in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). P301L and C57 mice of various ages were used for our experiments, including biochemical and immunohistochemical analyses and passive immunization. Tissue and blood were collected at the appropriate time points. Before euthanasia, mice were deep anesthetized and perfused transcardially with 1× PBS before decapitation. Animals were anesthetized according to Institutional Animal Care and Use Committee-approved procedures for animal euthanasia. After euthanasia, brains and spinal cords were dissected and stored at −80°C.
Generation of TOMA antigen.
To generate TOMA antigen, tau oligomers were prepared in PBS as described previously (Lasagna-Reeves et al., 2012a). Briefly, recombinant tau [tau-441 (2N4R) 45.9 kDa; Margittai and Langen, 2004; Lasagna-Reeves et al., 2012a] was treated with 8 m urea to obtain monomeric tau and then dialyzed overnight against 1× PBS buffer, pH 7.4. Samples were adjusted to 1 mg/ml with PBS and aliquots of tau monomer (in PBS) were kept at −20°C. Tau oligomers were obtained by mixing 300 μl of the tau stock (1 mg/ml) with 700 μl of 1× PBS, yielding a final concentration 0.3 mg/ml. Samples were then incubated at room temperature for 1 h on an orbital shaker (Lasagna-Reeves et al., 2010; Lasagna-Reeves et al., 2011c). The resulting tau oligomers were purified by fast protein liquid chromatography (Superdex 200 HR 10/30 column; GE Healthcare).
Immunization.
We immunized 2-month-old BALB/c mice with tau oligomers. The antigen was mixed with an equal volume of saline or Freund's complete adjuvant. The mice received an intraperitoneal injection of 100 μl of 1:1 (antigen:adjuvant) on the ventral side (20 μg/mouse). Two weeks later, a second injection of antigen with Freund's incomplete adjuvant was performed followed by boosts after 28, 47, 60, 80, and 103 d. Before fusion, mice were given daily booster injections for 4 consecutive days.
TOMA screening.
Anti-tau oligomer antibody response was determined by screening serial dilutions of animal sera using an enzyme-linked immunosorbent assay (ELISA). ELISA plates were coated with 50 or 200 ng of tau oligomers, Aβ oligomers, or α-synuclein oligomers to rule out cross-reactivity with other amyloid oligomers. Dot blot was also used to test TOMA specificity. Each strip had seven protein dots: dot #1 (tau monomer), dots #2–4 (tau oligomers from different preparations), dot #5 (Aβ oligomers), dot #6 (tau fibrils), and dot #7 (Aβ fibrils). Selected clones were tested by Western blot using in vitro prepared samples and dot blot using brain homogenates. Finally, the selected clones (TOMA) were tested using human and mouse brains. Antibody isotype and light chain composition (κ or λ) were determined using a commercially available mouse monoclonal antibody isotyping kit (Rapid Isotyping kit Plus Kappa and Lambda-mouse; Pierce).
Antibody purification and quality control.
TOMA was produced from hybridoma cells grown in X-VIVO 15 (Lonza) medium following standard conditions for cell culture. The antibody was purified from the medium by standard affinity chromatography methods followed by high-performance liquid chromatography purification (purity >95%). TOMA used in immunization studies was endotoxin free, as confirmed using a commercially available kit (Limulus amebocyte lysate, Chromogenic Endpoint Assay; Hycult Biotechnology). The endotoxin concentrations of different TOMA batches were measured concurrently with the standards and determined using a standard curve. TOMA batches with detectable endotoxin level of 3 ng/ml or higher were not used in immunotherapy studies. These batches were treated with an endotoxin removal microkit (ProteoSpin Endotoxin Removal Micro kit; Norgen Biotek) and designated for biochemical assays only. All TOMA samples were stored in appropriate endotoxin free vials at −80°C until use.
Immunotherapy with TOMA.
Eight-month-old P301L mice (n = 10 animals/group) were divided into three groups: treated mice that were injected intracerebroventricularly or intravenously with either 1 μg/per animal or 30 μg/animal of TOMA antibody and a control group that received a nonspecific IgG (rhodamine, catalog #GTX29093; Genetex). A fourth group of 8-month-old wild-type mice (stock #1638M, Taconic) received saline injection.
Intracerebroventricular injection.
Briefly, P301L and wild-type mice were anesthetized with ketamine (80–100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and placed in a stereotactic apparatus (Motorized Stereotaxic StereoDrive; Neurostar). For each mouse, the scalp was shaved, an incision was made through the midline to expose the top of the skull, and the bregma was located. A hole was drilled into the skull at −2.06 mm caudal to the bregma and 1.7 mm lateral to the midline at a depth of 2.5 mm. A 5.0 μl Hamilton syringe was used to inject approximately 1.0 μl of antibody (1 mg/ml) into both sides at a rate of 0.5 μl/min. The incision was closed using Vet-Bond and the mice were placed on an isothermal pad at 37°C and continuously monitored after surgery until recovery.
Intravenous injection.
The TOMA group was immunized with 30 μg of antibody/animal and the control group received a nonspecific IgG, rhodamine (30 μg/animal). Wild-type mice were intravenously injected with 30 μl of saline solution. Briefly, mice were placed in a restrainer (Braintree Scientific) and an inch of the tail was shaved and introduced into warm water to dilate the veins. A total of 30 μg of rhodamine was then injected into the lateral tail vein. Mice were placed in cages and kept under observation.
Intravenous injection of biotinylated TOMA.
To demonstrate that TOMA antibody enters the brain, 2 mg of TOMA antibody was biotinylated using the EZ-link Sulfo-NHS-SS Biotinylation kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Next, 30 μg of either biotinylated TOMA or nonbiotinylated TOMA (control group) was intravenously administered into the tail veins of 8-month-old P301L mice. Mice were bled preinjection as well as 30 min, 1 h, 24 h, and 1 week after injection. Mice were terminated at 2, 6, and 24 h and 1 week after injection. Euthanasia was preceded by deep anesthesia followed by transcardial perfusion with 1× PBS. Spinal cord and brain were rapidly excised and frozen. Brains were divided into two hemispheres. The left hemisphere was homogenized in PBS containing protease inhibitor and centrifuged at 10,000 rpm for 10 min at 4°C. The right hemisphere was sliced into 10 μm sagittal sections and used for histological examination. Brain sections were stained with streptavidin-horseradish peroxidase (Vectastain Elite; Vector Laboratories) and DAB.
In vivo imaging.
Eight-month-old P301L and wild-type C57 mice were intravenously injected with 30 μg of labeled TOMA into the tail vein. The TOMA was labeled with the far red probe (Kodak X-Sight 640 LSS Dye, excitation 650 nm, emission 750 nm; Carestream Molecular Imaging) according to manufacturer's instructions. Briefly, 2 mg/ml of antibody was incubated with the dye for 1 h (no light, on ice). Unlabeled antibody was removed by purifying the conjugation reaction in a column. The successful labeling of the antibody was calculated. Before injection, P301L and wild-type mice were shaved and anesthetized with a ketamine-xylazine mixture. Immediately after injection, the animals were placed in the dorsal position on animal trays and imaged using the Kodak Multispectral Image Station. Mice were imaged at 30 min and 1, 2, and 4 h after injection. After imaging, mice were transcardially perfused with 1× PBS. Brains and spinal cords were extracted from animals and imaged immediately. Whole-body optical and x-ray imaging were performed using the Kodak multispectral in vivo FX imaging system (Carestream).
Treatment of neuroblastoma cells with labeled TOMA.
SH-SY5Y human neuroblastoma cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS, glutamine (4 mm), penicillin (200 U/ml), streptomycin (200 μg/ml), and sodium pyruvate (1 μm). Cells were maintained at 37°C in 5% CO2. Cells (∼10,000/well) were cultured in 24-well plates (Corning) containing poly-L-lysine coverslips (BD) and grown overnight. TOMA (2 mg) was labeled with Alexa Fluor 594 dye (excitation/emission maxima ∼590/617 nm) using a labeling kit (Invitrogen) according to the manufacturer's instructions. Next, 1 μg of labeled TOMA was added to the cells, which were incubated for 30, 60, and 90 minutes. Media was removed and coverslips were washed twice with 1× PBS. Cells were permeabilized with cold methanol and kept at −80°C.
Detection of tau oligomer-TOMA complexes in serum.
TOMA bound to tau oligomers (complexed) in serum from 8-month-old P301L mice after intravenous immunization with TOMA and controls that received nonspecific IgG was investigated using NAb Protein-A Plus Spin (kit #89952; Thermo Fisher Scientific). Briefly, 100 μl of diluted sample were added to the protein A, incubated for 1 h at 4°C, and centrifuged. The flow-through (containing free tau oligomers) was collected and kept at −80°C for analyses. The bound antibodies (containing TOMA-tau oligomer complexes) were recovered with 100 μl of elution buffer. Oligomeric and total tau in both the antibody fractions and flow-through were measured by direct ELISA using T22 and HT7, respectively.
Tissue harvesting.
Frozen brains and spinal cord extracted from P301L and C57 mice were diced and homogenized in PBS with a protease inhibitor mixture (Roche) and 0.02% NaN3 using a 1:3 (w/v) dilution of brain: PBS. Samples were then centrifuged at 10,000 rpm for 10 min at 4°C. The supernatants were aliquoted, snap-frozen, and stored at −80°C until use (Lasagna-Reeves et al., 2011a).
Western blot analysis.
Each lane was loaded with 20–25 μg of total protein from one sample on precast NuPAGE 4–12% Bis-Tris Gels for SDS-PAGE (Invitrogen) and subsequently transferred onto nitrocellulose membranes. After blocking overnight at 4°C with 10% nonfat dried milk, membranes were probed for 1 h at room temperature with TOMA (1:100), Tau-5 (1:1000; Covance) AT8, AT100, and AT180 (1:1000; Thermo Fisher Scientific), Tau-13 (1:1000; Covance), annexin (1:500; Abcam), lamin (1:500; Abcam), actin (1:1000) and GAPDH (1:1000; Sigma) diluted in 5% nonfat dried milk. T22 immunoreactivity was detected with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:3000; GE Healthcare). TOMA, Tau-5, Tau-13, AT8, actin, annexin, GAPDH, and lamin antibody detection was done with HRP-conjugated IgG anti-mouse secondary antibody (1:3000; GE Healthcare). ECL Plus (GE Healthcare) was used for signal generation. For protein quantification, the densitometry of each band in the Western blot was normalized with actin. Analysis was performed using Labworks 4.5 software (UVP). All densitometry results represent the mean and SDs.
Dot blot analysis.
Dot blot strips were prepared on nitrocellulose membranes. Briefly, 0.5–1.2 μl of each sample was applied onto the strips and blocked with 10% nonfat dried milk in 1× TBST buffer, pH 7.4, overnight at 4°C. The strips were washed once with 1× TBST buffer and incubated with biotinylated TOMA, TOMA, Tau-5, 4D6, and 6E10 for 1 h at room temperature. The strips were washed 3 times with TBST and then incubated with streptavidin-HRP (1:3000; Southern Biotech) and HRP-conjugated IgG anti-mouse secondary antibody (1:3000; GE Healthcare), respectively, for 1 h at room temperature. Finally, the membranes were washed three times with TBST and developed using ECL plus chemiluminiscence kit (GE Healthcare). Dot quantification was performed using Labworks 4.5 software (UVP).
ELISA.
ELISA plates were coated with 15 μl of sample (PBS soluble fraction of brains, spinal cord, or diluted serum) using 0.05 m sodium bicarbonate, pH 9.6, as coating buffer and incubated overnight at 4°C. Plates were washed one time with TBST (containing 0.01% Tween 20), followed by blocking for 2 h at room temperature with 10% nonfat dry milk in TBST. The plates were then washed one time with TBST. T22 (1:250), AT8 (1:250), or HT7 (1:1000) diluted in 5% nonfat milk in TBST was then added and allowed to react for 1 h at room temperature. The plates were then washed three times with TBST. T22 immunoreactivity was detected using 100 μl of HRP-conjugated anti-rabbit IgG (GE Healthcare) and AT8 and HT7 were detected using HRP-conjugated anti-mouse IgG (GE Healthcare). The secondary antibody (diluted 1:3000 in 5% nonfat milk in TBST) was added, followed by incubation for 1 h at room temperature. Finally, plates were washed three times with TBST and incubated with 100 μl of 3,3,5,5-tetramethylbenzidine (TMB-1 component substrate; Dako) for 1 h in the dark. The reaction was stopped with 100 μl of 2 m HCl and the plates were read at 450 nm in a Polar Star Omega plate reader (BMG Labtech). To evaluate the levels of inflammation markers such as IL-1β and IL-6, we used ELISA kits (EM2IL6 and EM2ILB; Pierce) and analyzed brain homogenates from P301L mice immunized with either TOMA antibody or nonspecific IgG and wild-type mice that received saline injections. Briefly, samples were incubated on plates precoated with biotinylated antibodies against IL-1β and IL-6 for 1 h. After incubation with streptavidin-HRP, tetramethylbenzidine chromogen was applied and the manufacturer-supplied stop solution was used. The concentration of samples was determined by reading at 450 nm. To quantify the amount of oligomeric and monomeric tau from brain homogenates, spinal cord homogenates, and sera, we used recombinant tau oligomer and monomer preparations as standards for the direct ELISAs using T22 and HT7 antibodies. The concentrations of recombinant tau samples were measured by the BCA method (Pierce) using BSA as standard. Optical density values obtained from standard curves were fed into GraphPad Prism 5 software and linear regression analyses were performed. Concentrations of tau oligomers and tau monomers (pg/μl) present in the samples were obtained and plotted in the graphs.
Sandwich ELISA.
ELISA plates (NUNC MaxiSorp, 96 well; Thermo Fisher Scientific) were coated with two different concentrations of each of the capture antibodies Tau-5 or HT7 (0.5 μg and 0.25 μg, respectively) diluted in 0.05 m sodium bicarbonate buffer, pH 9.6. The plate was incubated at 4°C overnight and then washed twice with TBST and blocked with 10% nonfat dry milk solution for 2 h at room temperature. The plate was then loaded with 0.5 or 4 μg of brain homogenates in 1× PBS and incubated for 90 min at 37°C. The plate was washed and incubated with rabbit anti-tau antibody (1:1000; Abcam) for 90 min at room temperature, followed by additional washes. The plate was then incubated with secondary anti-rabbit IgG antibody (1:3000) for 1 h at room temperature. Detection was performed using TMB-substrate plus chromogen (Dako) incubated for 1 h in the dark. The reaction was stopped by adding equal volume of 2 m HCl. The plate was read at 450 nm.
Brain tissue fractionation.
Cell fractions of brain tissue from mice immunized with TOMA or nonspecific IgG were obtained using a Qproteome cell compartment kit (Qiagen). Briefly, 20 mg of mouse brain tissue was cut into 4 pieces and washed with 1 ml of ice-cold 1× PBS. Tissue was disrupted at low speed in a buffer containing protease inhibitor. Homogenate was put into a mini-column and centrifuged at 510 × g. Cell fractions were recovered by sequential addition of differential extraction buffers and centrifugation steps. Cell fractions were stored at −80°C.
Gallyas-silver staining.
Conventional Gallyas staining was performed as described previously (Gallyas, 2008) on paraffin-embedded sections. The sections were examined using a bright-field Eclipse 800 microscope equipped with a DXM1200 color CCD camera (both Nikon Instruments).
Immunofluorescence.
Paraffin sections were deparaffinized, rehydrated, and washed in 0.01 m phosphate buffer 3 times (5 min each). After blocking in normal horse serum (Invitrogen) for 1 h, sections were incubated overnight with TOMA (1:100) or T22 (1:300). The next day, the sections were washed in PBS three times (10 min each) and then incubated with goat anti-mouse IgG Alexa Fluor 488 (1:700; Invitrogen) or goat anti-rabbit IgG Alexa Fluor 568 (1:700; Invitrogen) for 1 h. The sections were then washed three times (10 min each) in PBS before incubation overnight with anti-mouse Tau-5 (1:300), Thr231 (1:100), AT8 (1:100), HT7 (1:400; Thermo Fisher Scientific), Lamp-2 (1:100; Abcam), axon marker (1:700, anti-68 kDa neurofilament antibody; Abcam), or the microglial marker Iba-1 (Abcam). The next day, the sections were washed in PBS three times for 10 min each before incubation with donkey anti-mouse IgG Alexa Fluor 488 (1:700; Invitrogen) or goat anti-rabbit IgG Alexa Fluor 568 (1:700; Invitrogen) for 1 h. Sections were washed and mounted using Fluoromount G (Southern Biotech) mounting medium with DAPI (Invitrogen). The sections were examined using an epifluorescence microscope (Eclipse 800; Nikon) equipped with a CoolSnap-FX monochrome CCD camera (Photometrics) using standard Nikon FITC and DAPI filters and images were acquired. Images were analyzed using the Metavue version 7.1 software (Molecular Devices).
Immunohistochemistry.
Immunohistochemistry was performed on frozen and paraffin-embedded sections. All sections were processed simultaneously under the same conditions. In brief, frozen sections were fixed with chilled acetone for 10 min at room temperature. Paraffin sections (5 μm) were deparaffinized and rehydrated. After blocking in normal goat serum for 1 h, sections were incubated overnight with primary antibodies: T22 (1:700), TOMA (1:300), AT100, AT180, AT8 (1:1000l Thermo Fisher Scientific), Thr231 and HT7 (1:400; Thermo Fisher Scientific). The next day, the sections were washed in PBS three times for 10 min each and then incubated with biotinylated goat anti-mouse IgG (1:2000; Jackson ImmunoResearch) or biotinylated goat anti-rabbit IgG (1:1800; Jackson ImmunoResearch) for 1 h. The sections were then washed three times for 10 min each in PBS and visualized using an ABC reagent kit (Vector Laboratories) according to the manufacturer's recommendations. Finally, sections were counterstained with hematoxylin (Vector Laboratories) for nuclear staining and mounted.
Quantitative histochemical analyses.
To examine cell morphology, we performed hematoxylin staining and acquired bright-field images using a Multizoom AZ100 microscope equipped with a DS-2M color CCD camera (both Nikon Instruments). For quantification, five sections per animal were randomly chosen and immunostained with each antibody and analyzed. ImageJ was used to quantify six visual fields in CA1, dentate gyrus, and posterior hypothalamus (PH) from P301L immunized with TOMA or nonspecific IgG. The number of NFT-positive cells per field in the PH corresponds to the average of NFTs in all six fields from five sections. The number of processes in CA1 and cell bodies in the dentate gyrus stained by T22, HT7, and Thr231 was automatically counted using the ImageJ plugin counting cell. The results are displayed as the percentage of labeled neuronal processes in CA1 divided by the total number of neuronal cells (represented by total number of neuronal nuclei stained in CA1). In the dentate gyrus, percentage represents the number of immunoreactive neuronal cell bodies in the measurement field divided by the total number of neuronal nuclei stained by hematoxylin.
Behavioral tests.
To determine the effect of immunotherapy on behavioral deficits, the spatial memory and motor function of P301L and wild-type mice were evaluated by rotarod and Y-maze tests (Holcomb et al., 1999; Morgan et al., 2008). Before the Y-maze test, mice were habituated to frequent experimenter handling. The Y-maze was performed 4 d after immunization and the rotarod test was performed 6 d after immunization, except in the case of the long-term experiment.
Y-maze task.
The Y-maze task provides a measure of spatial working memory and is based on the innate preference of mice to alternate arms when exploring a new environment. The animals were placed in a symmetrical Y-shaped maze. Arms were 40 cm long, 8 cm wide, and 12 cm high (San Diego Instruments), beige in color, nonreflective, and randomly designated A, B, or C. Each mouse was placed in an arm facing the center (arm A) and allowed to explore the maze for 8 min. The number of arms entered and the sequence of entries was recorded. A correct alternation occurred when the animal moved from the arm in which it began to the other two arms without retracing its steps (i.e., ABC or ACB). Spontaneous alternation, expressed as a percentage, was calculated by dividing the number of entries into all 3 arms on consecutive choices (correct choices) by number of arm entries subtracted by 2, then multiplying the quotient by 100. A high spontaneous alternation rate is indicative of sustained working memory because the animals must remember which arm was entered last to know not to reenter it.
Rotarod.
The rotarod test has been used to assess motor function and is based on the ability of a rodent to maintain balance on a rotating rod (diameter 3.2 cm). Mice were first habituated in one session of four trials to reach a baseline level of performance. The next day, mice were tested in one session of four trials (Rotarod meter; Stoelting). Five mice were placed onto the rod, one per testing station. The speed started at 4 rpm and accelerated at 0.1 rpm/s. The latency to fall from the rotating rod was determined and taken as a measure of motor function.
Statistics.
Means ± SEM for all data were analyzed with GraphPad Prism 5 and Excel. One-way ANOVA followed by Bonferroni's multiple-comparison test was used for the time course of tau pathology in the P301L mice, as measured by ELISA, Western, and dot blot quantifications and the analysis of the rotarod and Y-maze data. The unpaired t test was used to analyze ELISA measurements and the quantification of immunoreactivity in tissues. Two-way ANOVA followed by post hoc Bonferroni's multiple-comparison test was performed when the animals were tracked or samples were collected over sequential time points. One-tailed power analysis using Cohen's power table when α = 0.01 was completed using the means and SDs of previously completed rotarod task analysis of passively immunized mice in our laboratory. A sample size of 7 was determined to be sufficient to allow us to detect a significant difference between groups on the rotarod task 99% of the time. A similar sample size in another tau immunotherapy study was found to be suitable to detect significant differences for the Y-maze task as well (Troquier et al., 2012). Three additional animals were included in each group to protect against any unforeseen attrition.
Study approval.
Animal handling and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and according to protocols approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch
Results
TOMA specifically recognizes tau oligomers
We developed TOMA using tau oligomers derived from full-length human tau as described previously for the generation of polyclonal anti-tau oligomer antibody (T22) (Lasagna-Reeves et al., 2012a). Antibody specificity was confirmed by Western blot, ELISA, and dot blot analyses (Fig. 1), which showed that this novel antibody mainly recognizes dimers and trimers of tau protein and does not react with monomeric tau or oligomers from other amyloidogenic proteins such as Aβ recognized by 6E10 or α-synuclein recognized by 4D6 (Fig. 1A–D). TOMA is an IgG2a antibody with high affinity for tau oligomers (dissociation constant, Kd = 3.1 × 10−7 m), as determined by the dilution method using ELISA. The specificity of the antibody for tau oligomers was also confirmed by Western blot using AD human brain homogenates (Fig. 1E).
Figure 1.

Specificity of TOMA antibody. A, Western blot analysis of tau, α-synuclein, and Aβ aggregates formed in vitro (2 μg of protein/lane) probed with TOMA antibody. B, Same membranes used in A reprobed with sequence-specific antibodies. TOMA specifically recognized tau oligomers, but did not recognize oligomers from other amyloidogenic proteins or monomeric tau (red rectangle). C, Representative dot blot analysis from recombinant tau monomer, oligomers, and fibrils; recombinant α-synuclein; and Aβ42 oligomers probed with Tau-5, TOMA, 4D6, 6E10, and anti-mouse IgG, respectively. D, ELISA analysis confirms that TOMA does not show reactivity for monomeric tau or any significant reactivity with tau fibrils prepared in vitro (50 ng/well). TOMA does not recognize oligomers from other amyloidogenic proteins such as Aβ or α-synuclein even in the sensitive ELISA analysis. E, Western blot of the PBS soluble fraction from AD brain using TOMA, which is tau-oligomer-specific, and Tau-5, which recognizes all forms of tau. TOMA specifically detected bands corresponding to tau oligomers (red brace), but did not recognize the monomeric tau (black arrow) that is abundant in AD brain as shown by immunoreactivity with Tau-5 (25 μg of protein/lane).
Tau oligomers in the P301L mouse model
We used TOMA to analyze the brains of male homozygous P301L mice and observed abundant tau oligomers (Fig. 2). Western blots (Fig. 2A–D) of brain homogenate PBS soluble fractions from 5-, 6- and 7-month-old mice demonstrated that TOMA recognizes tau oligomers but not tau monomers, whereas both sequence-dependent anti-tau monoclonal antibodies (mAbs), Tau-5 (residues 210–230) and Tau-13 (residues 9–18), showed immunoreactivity with both tau monomers and oligomers (Figs. 2B,D, respectively). Conversely, 8-month-old wild-type mice did not exhibit an oligomeric band. Immunohistochemical and ELISA analyses of P301L brains (Fig. 2E–J) demonstrated that tau oligomers formed as early as 2–4 months of age and increased in an age-dependent manner, as did AT8 (Ser202/Thr205 phosphorylated tau)-immunoreactive NFTs (Fig. 2I–M). Peroxidase-stained hypothalamus from P301L brains displayed characteristic intracellular pre-NFT patterns (Fig. 2E,G), whereas immunofluorescence using TOMA showed partial overlap with the Tau-5 signal and some discrete foci of oligomer immunoreactivity (Fig. 2N–P). Tau oligomers were not found in 8-month-old wild-type controls (Fig. 2F,H). ELISA analysis showed a significant increase in tau oligomers peaking at 6 months of age, which then began to decline at 10 months, likely reflecting a shift toward oligomer fibrillization and the formation of NFTs. The differences in tau oligomer levels at 4, 6, and 8 months of age were not statistically significant. Immunofluorescence analysis using TOMA did not show colocalization with AT8, signifying a difference in oligomeric tau detected with TOMA and tau contained in NFTs (Fig. 2M). Moreover, analysis of spinal cord tissue showed that tau oligomer reactivity partially overlapped with Tau-5 and revealed distinct TOMA staining (data not shown).
Figure 2.

Tau oligomers in the P301L tau (JNPL3) mouse model. A–D, Western blot analysis of P301L brains at different ages using TOMA (A,C) and generic tau antibodies Tau-5 and Tau-13 (B, D). TOMA detected tau oligomers, but did not recognize tau monomers (red arrow), whereas both species were detected using Tau-5 and Tau-13. E–H, Photomicrographs of TOMA staining in paraffin sections using avidin-biotin complex and hematoxylin counterstaining (E, G) tau oligomers were detected by IHC in the hypothalamus of 4- and 8-month-old P301L brains using TOMA compared with wild-type C57 mice (F, H). Scale bar, 50 μm. I, J, Quantitative results of tau oligomers were determined in PBS-soluble fraction of brain homogenates from P301L mice by ELISA using TOMA. Oligomers were significantly higher at 6 months compared with 8-month-old wild-type mice and 2-month-old and 10-month-old P301L, respectively. ★★★★p < 0.0001; ★★★p < 0.0003, one-way ANOVA, Bonferroni post hoc comparison. J, Increased immunoreactivity against AT8 in 10-month-old P301L compared with wild-type mice and 2-month-old P301L and 4- to 8-month-old P301L, respectively. ★★★★p < 0.0001; ★★★p < 0.0003; ★★p < 0.001, one-way ANOVA, Bonferroni post hoc comparison. Bars represent the means and error bars the SEM. K–P, Epifluorescence images of frontal cortex from 8-month-old P301L stained using AT8 (K–M) and Tau-5 (N–P) (green), TOMA (red), and DAPI (blue). Scale bar, 25 μm.
Single intracerebroventricular injection of TOMA reverses phenotypes and specifically clears tau oligomers in P301L mice
Injection of anti-amyloid antibodies in AD animal models via intracerebroventricular injection has been performed in a few well executed studies (Chauhan and Siegel, 2003; Oddo et al., 2004; Chauhan, 2007; Thakker et al., 2009). In a similar manner using TOMA, we evaluated the effect of a single intracerebroventricular injection of 1 μg/animal of the antibody on 8-month-old P301L phenotypes 4 d after injection (Fig. 3A) using behavioral tasks and biochemical analyses. Rotarod performance of P301L mice immunized with TOMA was restored to that of age-matched nontransgenic controls, whereas no rescue was observed in control P301L mice immunized with anti-rhodamine IgG antibody (Fig. 3A). Analysis of tau species in treated animals and controls by Western blot using T22 (polyclonal anti-tau oligomer antibody) and Tau-5, which recognizes all forms of tau, revealed reduction of oligomeric but not monomeric tau (Fig. 3B,C). The reduction of tau oligomers in mice immunized with TOMA was confirmed by immunofluorescence with T22 (Fig. 4). These data show significant reduction in tau oligomers in both neuronal processes and cell bodies of TOMA-immunized animals (Fig. 4H,K). We also observed a significant reduction in phospho-Thr231 (p-Thr231)-immunoreactive tau aggregates (Fig. 4A–G,J), which is consistent with our recent finding of a strong association between p-Thr231 and tau oligomers in human brain (Lasagna-Reeves et al., 2012a). Interestingly, we found an increase in HT7, which specifically recognizes human tau that P301L mice overexpress in the neuronal processes (Fig. 4L), that may reflect an increase in the levels of functional tau monomer. Furthermore, Western blot and densitometric quantification analyses using the mAbs Tau-5 for total tau and AT100 (Ser212/Thr214) and AT180 (Thr231) for p-epitopes that are associated with early-stage tau aggregation (Augustinack et al., 2002) revealed that the decrease in tau oligomers coincided with significant reduction of soluble phospho-tau dimers in the immunized mice (Fig. 4M,N). Reduction of tau oligomers in mice immunized with TOMA was confirmed by immunostaining with polyclonal anti-tau oligomer antibody T22 and HT7 (data not shown).
Figure 3.

A single intracerebroventricular injection of TOMA reverses phenotypes and removes tau oligomers in 8-month-old P301L mice. A, Behavioral improvement of P301L mice immunized with TOMA (1 μg/animal) intracerebroventricular. Rotarod test was performed on three groups of mice (n = 10/group): (1) Wt: 8-month-old wild-type mice that received saline injection, (2) Control IgG: 8-month-old P301L mice immunized with anti-rhodamine IgG antibody (1 μg/animal), and (3) TOMA: 8-month-old P301L mice immunized with TOMA (1 μg/animal). Six days after injection, we evaluated mice by placing them onto the rod four times, increasing the speed each round. One day before the test, all mice were trained for four trial sessions. The rotarod test showed statistically significant behavioral improvement in mice immunized with TOMA (gray bar) compared with the control group (black bar). ★p < 0.04, one-way ANOVA, Bonferroni post hoc comparison. No statistically significant differences were found when the TOMA-immunized group was compared with the wild-type group. B–C, Reduction of tau oligomers in P301L mice immunized with TOMA. The levels of tau oligomers and monomer in mouse brain homogenates were assessed by Western blot using T22 and Tau-5, which recognizes all forms of tau. B, Representative Western blot of PBS homogenates from mouse immunized with the nonspecific IgG (lane 1) and a mouse immunized with TOMA (lane 2). C, The bar graph represents the percentage of band density relative to Tau-5 that corresponds to tau oligomers and monomers in mice injected with TOMA (black bars) and controls injected with nonspecific IgG (white bars). Quantitation (n = 10/group) shows a reduction of tau oligomers compared with the control group. ★★★★p < 0.0001, one-way ANOVA, Bonferroni post hoc comparison.
Figure 4.

Intracerebroventricular TOMA effects on tau pathology in P301L. Representative epifluorescence images of the CA1 region of 8-month-old P301L mice intracerebroventricularly immunized with TOMA or nonspecific IgG antibody (anti-rhodamine), immunostained with Thr231-green (A, D), T22-red (B, E), and merged + DAPI (C, F). A, Strong immunoreactivity with Thr231 in the perikaryon (solid white arrows) and neuronal processes (solid yellow arrows) of control IgG-treated mice compared with TOMA-treated mice (open white and yellow arrows; D). B, Tau oligomers as detected by T22 in somata (solid white arrows) and neuronal processes (solid yellow arrows) of IgG-treated mice. E, TOMA reduced tau oligomers in the perikaryon (open white arrows), neuronal processes (open yellow arrows), and the perinuclear area (asterisks). Scale bar, 15 μm. G–L, Quantification of changes observed in brain sections of frontal cortex from 8-month-old P301L mice immunized with TOMA and P301L mice injected with control IgG and immunostained with Thr231, T22, and HT7. G–I, Percentage of total immunoreactivity in cell bodies in the dentate gyrus. Shown is the reduction of Thr231 (G), T22 (H), and HT7 (I) immunoreactivity in cell bodies of mice immunized with TOMA compared with control mice. ★p < 0.02; ★p < 0.05; ★p < 0.02, respectively; Student's t test. J–L, Percentage of the immunoreactivity in neuronal processes in CA1 from P301L mice treated with TOMA and control mice. Reduction of Thr231 (J) and T22 (K) immunoreactivity in the TOMA group compared with the control group. ★p < 0.01; ★★p < 0.003. L, Significant increase in HT7 (specific for human tau) immunoreactivity in the TOMA-treated mice compared with control mice. ★p < 0.04. Bars represent the means and error bars the SEM, unpaired Student's t test. The levels of tau oligomers and monomers in mouse brain homogenates were assessed by Western blot using Tau-5, AT100, and AT180. M, Western blot of PBS soluble fraction from brain homogenate of mice immunized with the nonspecific IgG antibody (lanes 1–4) and mice immunized with TOMA (lanes 5–8) detected with Tau-5 antibody and reprobed with AT100 and AT180. Internal control is shown at the bottom. N, Graphs represent the relative band intensity for AT100 and AT180 (arbitrary units, AU). The differences were statistically significant. ★★p < 0.005; ★p < 0.02, Student's t test, respectively. Bars represent means and error bars SEM.
Single intravenous injection of TOMA also reverses P301L phenotypes and clears tau oligomers
After study of intracerebroventricular administration, we investigated the ability of TOMA to ameliorate tau oligomer toxicity after a single dose administered by the more translationally relevant intravenous route. Each animal was immunized with 30 μg of TOMA. Cognitive performance was evaluated 4 d after injection using the Y-maze task; motor activity was assessed 6 d after injection in the rotarod task. Intravenous TOMA treatment rescued the locomotor phenotype and cleared oligomeric tau similarly to intracerebroventricular TOMA treatment (Fig. 5). Animals immunized with TOMA exhibited improved performance in the rotarod test (Fig. 5A) and in the Y-maze task that measures working memory (Fig. 5B,C). P301L mice treated with TOMA showed no significant difference in rotarod or Y-maze performance compared with wild-type mice and performed significantly better on both tasks than P301L mice treated with anti-rhodamine IgG (Fig. 5B). These results suggest that intravenous delivery of TOMA reversed the memory deficits associated with oligomeric tau pathology in P301L mice. There were no significant differences between groups in total arm entries in the Y-maze, indicating no effect of treatment on the ability to complete the task (Fig. 5C). Moreover, we evaluated the long-term effects of TOMA on working memory and found that TOMA provided protection for at least 2 months, as demonstrated by TOMA-injected P301L mice committing significantly more correct alternations in the Y-maze task compared with control mice immunized with control antibody (data not shown). Western blot analyses using rabbit polyclonal anti-tau antibody, which recognizes all tau aggregates, revealed a reduction of tau oligomers but not monomeric tau in mice treated with TOMA compared with high levels observed in the control group (Fig. 5D,E). Quantitative ELISA analysis showed that improvements in locomotor and memory performance coincided with reduced tau oligomers (Fig. 6A,B) and total tau (Fig. 6D,E) in the brain and spinal cord. Reduction of oligomeric tau in the brain was confirmed by sandwich ELISA (Fig. 6G,H). Surprisingly, there was an increase in total and oligomeric tau in serum (Fig. 6C,F), implying peripheral involvement in the clearance of tau oligomers reminiscent of the peripheral mechanism described for Aβ clearance in animal models (DeMattos et al., 2001; Lemere et al., 2003; Levites et al., 2006; Winkler et al., 2010).
Figure 5.

A single intravenous injection of TOMA reverses phenotypes and clears tau oligomers in 8-month-old P301L mice. Three groups of mice (n = 10 /group): (1) Wt: wild-type mice that received saline injection, (2) Control IgG: P301L immunized with nonspecific IgG antibody (anti-rhodamine, 30 μg/animal), (3) TOMA: P301L immunized with TOMA (30 μg/animal). Animals were evaluated using the rotarod task for locomotor deficits and the Y-maze task for memory deficits. A, The rotarod test showed improvement of motor performance in mice immunized with TOMA (gray bar) compared with the control group (black bar). ★★★ p < 0.0003, one-way ANOVA, Bonferroni post hoc comparison. No statistically significant differences were found between the TOMA group and wild-type mice (white bar). B, The Y-maze memory test showed improved retention in TOMA-treated animals as depicted by the number of completed alternations in the Y-maze, defined as successive entry into each of the three arms of the maze without reentry into a previously visited arm. The differences were statistically significant. ★p < 0.02, one-way ANOVA, Bonferroni post hoc comparison. C, Total number of entries into all arms of the maze. D, Representative Western blot of PBS soluble fraction from brain homogenate of mice intravenous immunized with the nonspecific IgG antibody (lanes 1–6) and mice immunized with TOMA (lanes 7–12) detected with rabbit anti-tau antibody, which recognizes all tau aggregates. Internal control is shown at the bottom. E, Graphs represent the band intensity relative to rabbit anti-tau antibody (arbitrary units, AU). The differences were statistically significant. ★p < 0.005, two-way ANOVA, Bonferroni post hoc comparison. Bars represent means and error bars SEM.
Figure 6.
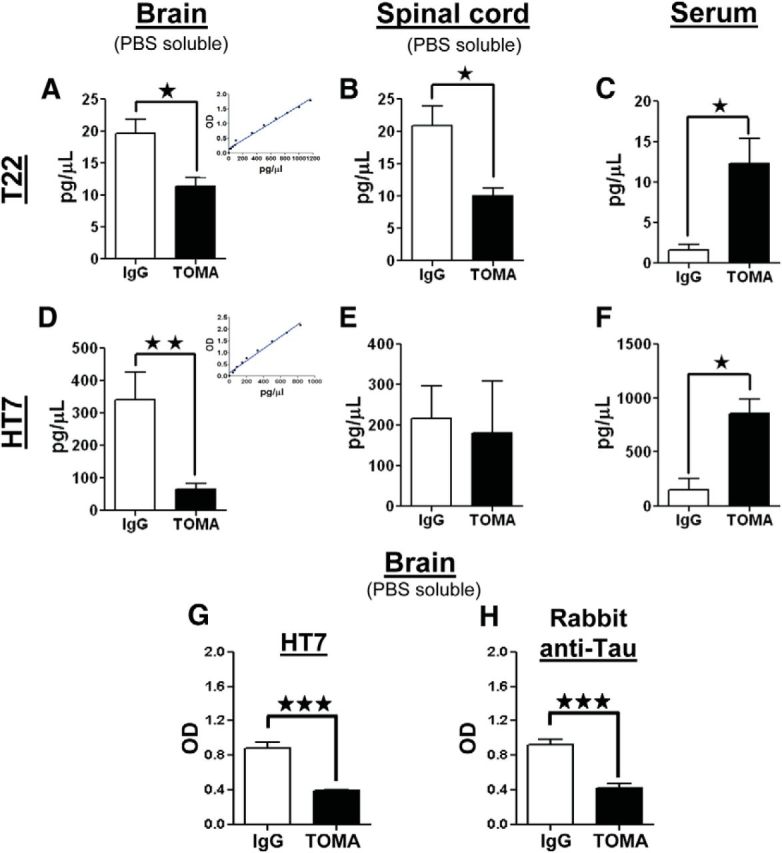
Quantitative analyses of oligomeric and total tau in brain, spinal cord, and serum. Analyses were performed by both direct and sandwich ELISA. T22 was used for tau oligomers; HT7 and polyclonal-Tau antibodies were used for total tau. Measurements were performed using brain and spinal cord homogenates and serum from 8-month-old P301L mice immunized with either TOMA or nonspecific IgG (anti-rhodamine). A–F, The graphs represent the amount (pg/μl) of oligomeric tau and total tau derived from brain (A, D) and spinal cord homogenates (B, E) and serum (C, F). Quantity of protein was calculated using standard curves (inserts in A and D). P301L animals treated with TOMA have significantly reduced oligomeric tau levels in both the brain and spinal cord and increased levels in the serum compared with the animals treated with nonspecific IgG. ★p < 0.01, Student's t test. D–F, Quantification of total tau shows significantly reduced levels in the brain and significantly increased levels in the serum of the TOMA-treated group compared with the control group. ★★p < 0.009; ★p < 0.01, Student's t test. Changes in levels of total tau in the spinal cord are not statistically significant. The reduction of total tau in the brain homogenates from mice immunized with TOMA compared with mice immunized with nonspecific IgG was confirmed by sandwich ELISA (G) using Tau-5 as capture antibody and HT7 as detection antibody (H) using Tau-5 as capture antibody and polyclonal-Tau as detection antibody. The decrease in total tau in TOMA-treated mice was statistically significant. ★★★p < 0.0002, Student's t test. Bars represent means and error bars SEM.
Immunofluorescence analysis revealed that intravenous TOMA-treated animals had reduced levels of tau oligomers in neuronal cell bodies and axons in the CA1 region (data not shown) compared with animals treated with control IgG antibody. Most importantly, as seen in intracerebroventricular TOMA-treated P301L mice, intravenous TOMA immunization did not have any effect on AT8 immunoreactive NFTs or those positively stained using the Gallyas method (Fig. 7A–I), thus confirming the specificity of TOMA for tau oligomers and further supporting the hypothesis that NFTs do not significantly contribute to tauopathy-related motor and cognitive phenotype of P301L mice. No differences were found between TOMA-immunized and control mice in levels of insoluble tau as measured by ELISA and Western blot using AT8 (Ser202/Thr205) and PHF13 (Ser396), which are both indicators of late stage aggregation and paired helical filament (PHF) formation (Augustinack et al., 2002) (Fig. 7J–M).
Figure 7.
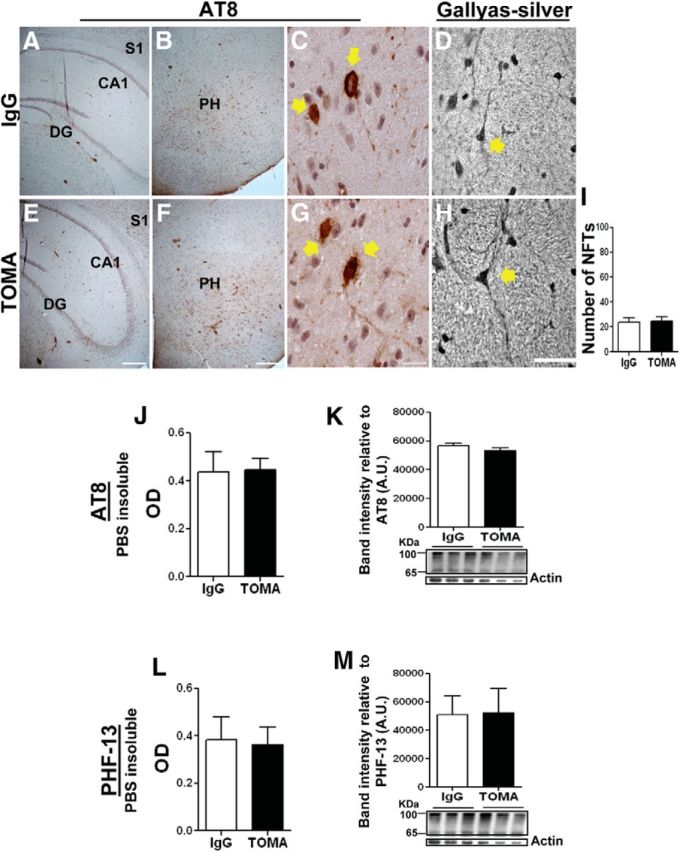
TOMA does not affect NFTs in posterior hypothalamus or cortex. Coronal sections of 8-month-old P301L brain immunized with nonspecific IgG antibody (anti-rhodamine) (A–D) and TOMA (E–H) were immunostained with AT8 (A–C and E–G) and stained with the Gallyas-silver method (D, H). A and E are hippocampus; B–D and F–H are posterior hypothalamus (PH). C–D and G–H are images at higher-magnification showing NFTs (arrows) in PH. Scale bars: A, B, E, F, 100 μm; C, G, 15 μm. Graph represents the number of NFT-positive cells per field in the PH stained with AT8. J–M, PBS-insoluble tau fraction from P301L mice immunized with nonspecific IgG and TOMA was analyzed by ELISA (J, L) and Western blot (K, M) using AT8 (J, K) and PHF-13 (L, M). The gel band intensity immunoreactive to AT8 and PHF-13 (∼56–100 kDa) was quantified and normalized using actin as an internal control. The differences were not statistically significant using Student's t test.
Insight into the mechanisms of tau oligomer clearance by intravenously administered TOMA
Previous studies have suggested that clearance of tau aggregates can occur via endosomal/lysosomal degradation after internalization of tau-antibody complexes (Krishnamurthy et al., 2011) in addition to microglia receptor-mediated clearance, autophagy-mediated clearance, and/or the peripheral sink mechanism, all of which have been described for other amyloid proteins (Masliah et al., 2011; Morgan, 2011; Ubhi and Masliah, 2011). Importantly, these mechanisms are not mutually exclusive and it has been demonstrated that different clearance mechanisms can be activated to different degrees depending on the antibody and its target (DeMattos et al., 2001; Levites et al., 2006; Sigurdsson, 2008; Golde et al., 2009; Masliah et al., 2011; Morgan, 2011; Ubhi and Masliah, 2011). We investigated the mechanism of action of intravenously injected TOMA by first using in vivo imaging to demonstrate that a fraction of TOMA injected into the tail vein was able to cross the BBB and bind to tau oligomers in the P301L brain and spinal cord (Fig. 8A,B). Binding of TOMA to tau oligomers was confirmed by imaging brain and spinal cord sections prepared from animals 4 h after TOMA treatment (Fig. 8C,D). Moreover, biotinylated TOMA was detected in the brains of intravenously injected P301L mice at 2, 6, and 24 h after injection (Fig. 8E–J).
Figure 8.

Extracellular and peripheral clearance of tau oligomers by TOMA. A–D, In vivo imaging demonstrates that a fraction of TOMA injected into the tail vein crosses the BBB and binds to tau oligomers in the 8-month-old P301L brain and spinal cord. A, TOMA at the injection site in 8-month-old wild-type-C57 and P301L mice at time 0; animals were imaged immediately after the injection. B, Animals imaged 4 h after injection showed retention of TOMA only in the brains of P301L animals, whereas TOMA was cleared from the wild-type animals. C–D, To confirm that TOMA entered the CNS, we imaged brain and spinal cord sections derived from cryotome cross-sectioning of P301L and wild-type brains and spinal cords extracted from animals killed 4 h after TOMA injection. E–J, Biotinylated TOMA antibody (30 μg/animal) was injected into the tail veins of 8-month-old P301L mice. E–H, Representative images of brain sections stained with streptavidin-peroxidase demonstrate that biotinylated TOMA antibody crosses the BBB and remains in hippocampus 2, 6, and 24 h after injection. Scale bar, 30 μm. I, Dot blot analyses of PBS soluble fractions from brain homogenates of mice injected with biotinylated TOMA confirmed the presence of the biotinylated antibody in the brain compared with the control (brain homogenate from P301L mouse injected with unlabeled TOMA). A 0.5 μl aliquot of biotinylated-TOMA was used as a positive control. J, Quantification of the dot intensities. ★★★p < 0.0005; ★★p < 0.001, one-way ANOVA, Bonferroni post hoc comparison. K–M, Detailed quantitative analysis of the short-term effects of intravenous TOMA injection on the levels of both oligomeric and total tau in the serum. K, Tau oligomers were quantified by direct ELISA using T22 before and at different time points shortly after TOMA injections in 8-month-old P301L and wild-type mice. The effects of TOMA were rapid and a significant increase in tau oligomer levels occurred in the serum, peaking at 1 h. By 24 h, levels began to decline. L, Levels of total tau measured by direct ELISA using HT7 confirmed TOMA effects and correlated with the changes observed for tau oligomers. ★★★★p < 0.0001; ★★★p < 0.001, two-way ANOVA, Bonferroni post hoc comparison. M, Tau oligomer reduction in brain homogenates measured 168 h after TOMA injection. ★★★p < 0.0003; ★★p < 0.001, one-way ANOVA, Bonferroni post hoc comparison. Bars represent means and error bars SEM.
Next, we investigated cellular uptake of intravenously injected TOMA and its ability to clear intracellular tau aggregates. TOMA did not colocalize with the endosomal/lysosomal marker Lamp-2 in either brain slices derived from animals treated with Alexa Fluor 594-labeled TOMA or in cells treated with Alexa Fluor 594-labeled TOMA (data not shown), indicating that cellular TOMA internalization is unlikely. Nonetheless, intravenously injected TOMA reduced intracellular tau in cell somata and biochemical analyses of cellular fractions from brain homogenates showed reduction of both cytosolic and membrane-associated tau oligomers in animals immunized with TOMA (data not shown). It is possible that TOMA depletion of extracellular tau may shift the equilibrium between the intracellular and extracellular tau oligomer pools, thereby leading to removal of cytosolic and membrane-associated tau oligomers, similar to what has been reported to occur for Aβ and α-synuclein after passive immunization (Oddo et al., 2004; Masliah et al., 2005).
The inflammatory response has been implicated as a mechanism or side effect of anti-amyloid immunotherapy approaches (Bard et al., 2000; Wilcock et al., 2003; Wilcock et al., 2004; Masliah et al., 2011). We therefore examined the inflammatory markers IL-6 and IL-1β using ELISA kits and Iba1 using immunofluorescence microscopy (Sy et al., 2011) on brain sections prepared from 8-month-old P301L mice immunized with either TOMA or control IgG. No significant difference in immunostaining was observed between animals immunized with TOMA and IgG controls (data not shown), suggesting that the removal of tau oligomers by TOMA passive immunotherapy is not mediated by microglia, which is consistent with previous results for active tau immunotherapy (Asuni et al., 2007). The fact that clearance of tau oligomers did not affect inflammation provides evidence for the safety of this approach relative to active immunotherapy.
To further investigate the peripheral mechanism for TOMA-mediated tau oligomer clearance, we performed detailed analyses of the short-term effects of immunotherapy on oligomeric and total tau in serum (Fig. 8K,L). TOMA effects were rapid, resulting in a significant increase in serum tau oligomer levels peaking at 1 h after administration (Fig. 8K). These results parallel total tau levels in the serum (Fig. 8L), suggesting that the rapid reversal of motor and cognitive deficits in P301L mice and the reduction of tau oligomers in the brain (Fig. 8M) occur via clearance from the brain to the periphery. In addition, we performed an ELISA analysis of oligomeric and total tau to compare free tau oligomers and TOMA-oligomer complexes (Fig. 9). The majority of tau oligomers in the serum formed complexes with TOMA antibody (Fig. 9A,B), suggesting that TOMA-tau complexes are critical for the rapid clearance of tau oligomers by intravenously administered TOMA.
Figure 9.
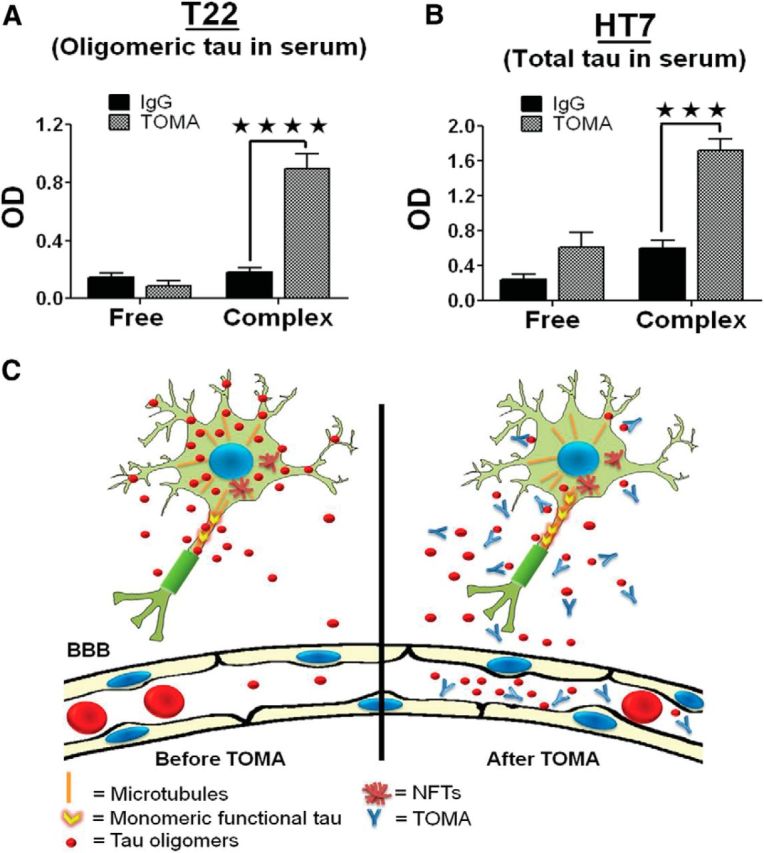
TOMA forms complexes with tau oligomers. A, B, Quantitative analyses of tau oligomers as free or in complex with TOMA assessed using ELISA. Serum samples from P301L mice immunized either with TOMA or nonspecific IgG antibody were passed through a protein-A column. Tau oligomers and total tau in flow-through (“free” tau oligomers that did not form complex with the antibody) and the eluted fraction (antibodies and TOMA-oligomer complexes) were measured by ELISA. Quantitation of tau oligomers (A) and total tau (B) in the eluted fraction were higher compared with flow-through “free oligomers” as measured by T22 and HT7, respectively. ★★★★p < 0.0001; ★★★p < 0.001, two-way ANOVA, Bonferroni post hoc comparison. Most of the tau oligomers are complexed with TOMA. C, Schematic representation of the proposed mechanism of action for intravenously injected TOMA, which specifically modulates tau oligomers. A fraction of the injected TOMA crosses the BBB and binds extracellular oligomeric tau and tau oligomer levels in the serum peak at 1 h after injection.
The mechanism of tau oligomer clearance appears to occur via a peripheral pathway similar to several previously described for the clearance of other amyloid proteins (DeMattos et al., 2001; Lemere et al., 2003; Oddo et al., 2004; Masliah et al., 2005; Winkler et al., 2010). Accordingly, we propose a model in which TOMA traps and neutralizes extracellular tau oligomers and promotes the egress of intracellular oligomers, resulting in a net decrease of CNS tau oligomers and eventual serum clearance (Fig. 9C).
Discussion
The pathological role of prefilamentous tau aggregates (i.e., tau oligomeric intermediates) in tauopathies is poorly understood; however, novel polyclonal and monoclonal antibodies that define specific tau species have enabled the study of tau oligomers in vivo (Patterson et al., 2011; Lasagna-Reeves et al., 2012a; Blair et al., 2013; Hawkins et al., 2013; Wu et al., 2013). We engineered a novel polyclonal tau oligomer-specific antibody, T22, by collecting the serum of rabbits injected with tau oligomers and demonstrated that oligomeric tau is elevated in human AD brain samples and appears to contribute to NFT formation (Lasagna-Reeves et al., 2012a). We also showed that AD-brain-derived tau oligomers isolated using T22 disrupt memory and propagate the abnormal conformation of endogenous tau in wild-type mice, implicating tau oligomers in the spread of pathology but not NFTs (Lasagna-Reeves et al., 2012b). Furthermore, widespread tau oligomers accumulate at the synapses (Henkins et al., 2012), impairing the ubiquitin-proteasome system (Tai et al., 2012) and contributing to synaptic dysfunction and loss. These findings suggest that targeting oligomeric tau species through immunotherapy may be superior to targeting NFTs across the spectrum of neurodegenerative tauopathies and, therefore, this approach has outstanding potential as a disease-modifying intervention (Lasagna-Reeves et al., 2011b; Ubhi and Masliah, 2011; Castillo-Carranza et al., 2013a; Castillo-Carranza et al., 2013b).
We demonstrate here for the first time the benefits of using an antibody against tau oligomers for passive immunotherapy in a tauopathy mouse model. We have shown that a single dose of peripherally administered TOMA entered the P301L mouse brain and specifically removed tau oligomers without affecting functional monomeric tau. The dose used in this study was 5- to 10-fold lower than those previously used to nonselectively target tau aggregates by passive immunotherapy (Boutajangout et al., 2011; Chai et al., 2011), which may also contribute to increased safety while maintaining efficacy because oligomers represent a small percentage of total tau in the brain. Moreover, our vaccination study has provided evidence that intravenous delivery of IgG antibodies can be detected within the P301L mouse brain, suggesting that the BBB may be impaired in this mouse model (Asuni et al., 2007), as in amyloid precursor protein transgenic mice (Bard et al., 2000; Yamada et al., 2009) and synuclein mice (Masliah et al., 2011). Because the BBB has also been shown to be impaired in AD patients (Bowman et al., 2007), immunization with IgG antibody may represent a good therapeutic approach in humans.
The therapeutic effects of the TOMA antibody in P301L results in a reduction of pathogenic tau oligomers coinciding with the rescue of tauopathy-related motor and cognitive phenotypes in mice, reinforcing the concept that oligomers are the pathological form of tau in animal models of neurodegenerative tauopathy (Lasagna-Reeves et al., 2011b; Patterson et al., 2011; Cowan et al., 2012; Lasagna-Reeves et al., 2012a). In support of this, we recently demonstrated that tau oligomers prepared in vitro from full-length recombinant human tau are responsible for the behavioral deficits observed in wild-type mice (Lasagna-Reeves et al., 2011c). Furthermore, it has been reported that reduction of soluble tau oligomers after treatment with curcumin rescues synaptic and behavioral deficits in aged tau transgenic mice (Ma et al., 2013). In addition, environmental enrichment reduced soluble tau and restored Arc messenger RNA levels in rTg4510-transgenic mice overexpressing P301L mutant human tau protein despite the continued presence of NFT pathology (Fox et al., 2011).
In contrast to our work, other tau immunotherapy studies have reported a reduction in NFT burden; however, in this study, insoluble hyperphosphorylated tau aggregate levels were not altered. A possible explanation for this is that previous studies (Asuni et al., 2007; Boimel et al., 2010; Boutajangout et al., 2011; Chai et al., 2011; Gu and Sigurdsson, 2011; Bi et al., 2011) specifically targeted tau pathology using phosphorylated antigens or antibodies directed against p-epitopes associated with later-stage tau pathology. Moreover, the use of conformational antibodies in immunotherapy appears to be superior at reducing tau pathology in P301L mice compared with pan-tau antibodies, which are not specific to any conformational state (d'Abramo et al., 2013). In addition, our study found that the reduction of tau oligomers and soluble p-tau epitopes that are associated with earlier stages of tau aggregation affected the levels of total tau in the brain. It is important to note that other immunotherapy studies did not find changes in the levels of total tau, which may be explained by the fact that they did not investigate the fate of these toxic tau species and that p-tau antibodies, which disassemble NFTs containing large amounts of tau, may influence the measurements of total tau in the brain. Furthermore, the striking increase of TOMA-oligomer complex in serum suggests that TOMA removed extracellular tau oligomers from the brain to the periphery, analogous to immunotherapy against Aβ (Levites et al., 2006; Yamada et al., 2009).
Finally, anti-tau oligomer immunization may prevent the spread of tau pathology by sequestering extracellular tau oligomers, possibly preventing tau toxicity and allowing neurons to recover (Gerson and Kayed, 2013). As described recently in cell culture experiments, anti-tau antibodies can trap tau aggregates in the extracellular space and thus prevent the uptake and propagation of tau in adjacent cells (Kfoury et al., 2012). Indeed, a recent passive immunization study in P301L mice failed to demonstrate that the antibody was internalized into neurons, suggesting that tau released to the extracellular space was targeted by the antibody (Yanamandra et al., 2013; d'Abramo et al., 2013). This extracellular clearance model argues that TOMA would have the additional benefit of halting the cell-to-cell spread of tau pathology (Frost and Diamond, 2010; Ubhi and Masliah, 2011; Lasagna-Reeves et al., 2012b; Wu et al., 2013). Moreover, another study demonstrated that the clearance of extracellular α-synuclein by passive immunization prevents cell-to-cell aggregate transmission and ameliorates neurodegeneration and behavioral deficits (Bae et al., 2012; Yanamandra et al., 2013).
In summary, our studies support the notion that tau oligomer immunization strategies may be beneficial for AD and other tauopathies (Guzmán-Martinez et al., 2013). These multifactorial diseases have a long asymptomatic phase and a prolonged survival period once symptoms begin. In the latter, brain degeneration and clinical manifestations arise from several different and perhaps related molecular events, during which tau oligomers develop and likely contribute to disease progression. Therefore, studies designed to evaluate immunotherapeutic approaches targeting tau aggregates are critical and should not be viewed as alternative approaches to Aβ immunotherapy, but rather as complementary strategies. Our findings indicate that TOMA antibody not only recognizes oligomers in P301L mice, but that the antibody can also detect nonmutant tau oligomers formed from recombinant tau protein and in situ species from AD brain; therefore, TOMA therapy may be able to clear tau oligomers that are specifically associated with AD and FTD pathology. Passive immunization with TOMA may also bypass potential complications by removing the true toxic entities of tau without affecting normal tau function and without affecting inflammation associated with active immunotherapy in AD. In conclusion, the data described herein lay a solid foundation for future studies to optimize the development of disease-modifying immunization strategies for AD and other neurodegenerative tauopathies.
Footnotes
This work was supported by the Alzheimer's Drug Discovery Foundation, the Cullen Family Trust for Health Care, and the Mitchell Center for Neurodegenerative Diseases. We thank Shashirekha Krishnamurthy and Malika Farhed for excellent technical assistance; the University of Texas Medical Branch Rodent In Vivo Assessment Core (directed by Kelly T. Dineley) and Caterina Hernandez for assistance with experimental design and data analysis for the animal work; and Adriana Paulucci and Yogesh Wairkar for help with microscopy and image analysis.
R.K. has patent applications on the compositions and methods related to tau oligomers and antibodies. The remaining authors declare no competing financial interests.
References
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Ittner A, Ke YD, Götz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One. 2011;6:e26860. doi: 10.1371/journal.pone.0026860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair LJ, Nordhues BA, Hill SE, Scaglione KM, O'Leary JC, 3rd, Fontaine SN, Breydo L, Zhang B, Li P, Wang L, Cotman C, Paulson HL, Muschol M, Uversky VN, Klengel T, Binder EB, Kayed R, Golde TE, Berchtold N, Dickey CA. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J Clin Invest. 2013;123:4158–4169. doi: 10.1172/JCI69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;224:472–485. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011;118:658–667. doi: 10.1111/j.1471-4159.2011.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood-brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology. 2007;68:1809–1814. doi: 10.1212/01.wnl.0000262031.18018.1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Lasagna-Reeves CA, Kayed R. Tau aggregates as immunotherapeutic targets. Front Biosci (Schol Ed) 2013a;5:426–438. doi: 10.2741/s381. [DOI] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Guerrero-Muñoz MJ, Kayed R. Immunotherapy for the treatment of Alzheimer's disease: amyloid-β or tau, which is the right target? ImmunoTargets and Therapy. 2013b;2014:19–28. doi: 10.2147/ITT.S40131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O'Neill MJ, Hutton ML, Citron M. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan NB. Intracerebroventricular passive immunization with anti-oligoAbeta antibody in TgCRND8. J Neurosci Res. 2007;85:451–463. doi: 10.1002/jnr.21110. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization with anti-Abeta antibody in Tg2576. J Neurosci Res. 2003;74:142–147. doi: 10.1002/jnr.10721. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Quraishe S, Mudher A. What is the pathological significance of tau oligomers? Biochem Soc Trans. 2012;40:693–697. doi: 10.1042/BST20120135. [DOI] [PubMed] [Google Scholar]
- d'Abramo C, Acker CM, Jimenez HT, Davies P. Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One. 2013;8:e62402. doi: 10.1371/journal.pone.0062402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LM, William CM, Adamowicz DH, Pitstick R, Carlson GA, Spires-Jones TL, Hyman BT. Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J Neuropathol Exp Neurol. 2011;70:588–595. doi: 10.1097/NEN.0b013e318220a658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11:155–159. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallyas F. Physicochemical mechanisms of histological silver staining and their utilization for rendering individual silver methods selective and reliable. Biotech Histochem. 2008;83:221–238. doi: 10.1080/10520290802538543. [DOI] [PubMed] [Google Scholar]
- Gerson JE, Kayed R. Formation and propagation of tau oligomeric seeds. Front Neurol. 2013;4:93. doi: 10.3389/fneur.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Das P, Levites Y. Quantitative and mechanistic studies of Abeta immunotherapy. CNS Neurol Disord Drug Targets. 2009;8:31–49. doi: 10.2174/187152709787601830. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gu J, Sigurdsson EM. Immunotherapy for tauopathies. J Mol Neurosci. 2011;45:690–695. doi: 10.1007/s12031-011-9576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán-Martinez L, Farías GA, Maccioni RB. Tau Oligomers as Potential Targets for Alzheimer's Diagnosis and Novel Drugs. Front Neurol. 2013;4:167. doi: 10.3389/fneur.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutunian V, Davies P, Vianna C, Buxbaum JD, Purohit DP. Tau protein abnormalities associated with the progression of alzheimer disease type dementia. Neurobiol Aging. 2007;28:1–7. doi: 10.1016/j.neurobiolaging.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, Sengupta U, Prough DS, Jackson GR, DeWitt DS, Kayed R. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. J Biol Chem. 2013;288:17042–17050. doi: 10.1074/jbc.M113.472746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkins KM, Sokolow S, Miller CA, Vinters HV, Poon WW, Cornwell LB, Saing T, Gylys KH. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer's cortical synapses. Brain Pathol. 2012;22:826–833. doi: 10.1111/j.1750-3639.2012.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/A:1021691918517. [DOI] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy PK, Deng Y, Sigurdsson EM. Mechanistic studies of antibody-mediated clearance of tau aggregates using an ex vivo brain slice model. Front Psychiatry. 2011;2:59. doi: 10.3389/fpsyt.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Glabe CG, Kayed R. Amyloid-{beta} annular protofibrils evade fibrillar fate in Alzheimer's disease brain. J Biol Chem. 2011a;286:22122–22130. doi: 10.1074/jbc.M111.236257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Jackson GR, Kayed R. Tau oligomers as potential targets for immunotherapy for Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2011b;8:659–665. doi: 10.2174/156720511796717177. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau Oligomers Impair Memory and Induce Synaptic and Mitochondrial Dysfunction in Wild-type Mice. Mol Neurodegener. 2011c;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 2012a;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, Jackson GR, Kayed R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012b;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, Matsuoka Y, Taylor JW, DeMattos RB, Holtzman DM, Clements JD, Selkoe DJ, Duff KE. Evidence for peripheral clearance of cerebral Abeta protein following chronic, active Abeta immunization in PSAPP mice. Neurobiol Dis. 2003;14:10–18. doi: 10.1016/S0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, Dakin RS, Yuan B, Sierks MR, Kim J, McGowan E, Reed DK, Rosenberry TL, Das P, Golde TE. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer's disease mouse models. FASEB J. 2006;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, Teng E, Hu S, Chen PP, Maiti P, Teter B, Cole GM, Frautschy SA. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J Biol Chem. 2013;288:4056–4065. doi: 10.1074/jbc.M112.393751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci U S A. 2004;101:10278–10283. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Morgan D. Immunotherapy for Alzheimer's disease. J Intern Med. 2011;269:54–63. doi: 10.1111/j.1365-2796.2010.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Munireddy S, Alamed J, DeLeon J, Diamond DM, Bickford P, Hutton M, Lewis J, McGowan E, Gordon MN. Apparent behavioral benefits of tau overexpression in P301L tau transgenic mice. J Alzheimers Dis. 2008;15:605–614. doi: 10.3233/jad-2008-15407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006;63:1459–1467. doi: 10.1001/archneur.63.10.1459. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Mandelkow E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 2008;5:443–457. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer's disease and related tauopathies. J Alzheimers Dis. 2008;15:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT. Are tangles as toxic as they look? J Mol Neurosci. 2011;45:438–444. doi: 10.1007/s12031-011-9566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD. Do neuronal inclusions kill the cell? J Neural Transm Suppl. 2000;59:91–93. doi: 10.1007/978-3-7091-6781-6_12. [DOI] [PubMed] [Google Scholar]
- Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF, Stewart GR, Shafer LL. Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2009;106:4501–4506. doi: 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troquier L, Caillierez R, Burnouf S, Fernandez-Gómez FJ, Grosjean ME, Zommer N, Sergeant N, Schraen-Maschke S, Blum D, Buee L. Targeting phospho-Ser422 by active Tau Immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res. 2012;9:397–405. doi: 10.2174/156720512800492503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Masliah E. Recent advances in the development of immunotherapies for tauopathies. Exp Neurol. 2011;230:157–161. doi: 10.1016/j.expneurol.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Nes JA, Nafe R, Schlote W. Non-tau based neuronal degeneration in Alzheimer's disease–an immunocytochemical and quantitative study in the supragranular layers of the middle temporal neocortex. Brain Res. 2008;1213:152–165. doi: 10.1016/j.brainres.2008.03.043. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DT, Abramowski D, Danner S, Zurini M, Paganetti P, Tolnay M, Staufenbiel M. Rapid cerebral amyloid binding by Abeta antibodies infused into beta-amyloid precursor protein transgenic mice. Biol Psychiatry. 2010;68:971–974. doi: 10.1016/j.biopsych.2010.01.030. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 2013;288:1856–1870. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Yabuki C, Seubert P, Schenk D, Hori Y, Ohtsuki S, Terasaki T, Hashimoto T, Iwatsubo T. Abeta immunotherapy: intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci. 2009;29:11393–11398. doi: 10.1523/JNEUROSCI.2021-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]