

Abstract
Extinction therapy has been suggested to suppress the conditioned motivational effect of drug cues to prevent relapse. However, extinction forms a new inhibiting memory rather than erasing the original memory trace and drug memories invariably return. Perineuronal nets (PNNs) are a specialized extracellular matrix around interneurons in the brain that have been suggested to be a permissive factor that allows synaptic plasticity in the adolescent brain. The degradation of PNNs caused by chondroitinase ABC (ChABC) may generate induced juvenile-like plasticity (iPlasticity) and promote experience-dependent plasticity in the adult brain. In the present study, we investigated the effect of removing PNNs in the amygdala of rat on the extinction of drug memories. We found that extinction combined with intra-amygdala injections of ChABC (0.01 U/side) prevented the subsequent priming-induced reinstatement of morphine-induced and cocaine-induced, but not food -induced, conditioned place preference (CPP). Intra-amygdala injections of ChABC alone had no effect on the retention, retrieval, or relearning of morphine-induced CPP and storage of acquired food-induced CPP. Moreover, we found that the procedure facilitated the extinction of heroin- and cocaine-seeking behavior and prevented the spontaneous recovery and drug-induced reinstatement of heroin- and cocaine-seeking behavior. We also found that the effect of PNNs degradation combined with extinction may be mediated by the potentiation of several plasticity-related proteins in the amygdala. Altogether, our findings demonstrate that a combination of extinction training with PNNs degradation in the amygdala erases drug memories and suggest that ChABC may be an attractive candidate for the prevention of relapse.
Keywords: addiction, amygdala, extinction, memory, perineuronal nets
Introduction
Persistent drug memories that are formed between drug-paired cues and drug-induced effects play a critical role in drug use and relapse (Hyman, 2005; Milton and Everitt, 2012). Extinction therapy has been developed to inhibit the motivational effect of drug cues to prevent relapse, in which addicts are repeatedly exposed to drug-associated cues without given drugs of abuse (Marlatt, 1990; O'Brien et al., 1993). However, substantial evidence from rodent and human studies has demonstrated that the effectiveness of extinction training is limited and drug craving and drug-seeking behavior often return with the passage of time (spontaneous recovery) or after acute exposure to drugs of abuse (de Wit and Stewart, 1983; Tobeña et al., 1993; Shaham et al., 2003; reinstatement).
Extinction is an active process that forms a new inhibitory memory to temporally suppress previously conditioned responding rather than erasing acquired memory traces (Bouton, 1993). Numerous studies have focused on the circuitry and cellular mechanisms that underlie the extinction of fear and appetitive memories, demonstrating the involvement of glutamatergic, dopaminergic, and noradrenergic plasticity in the infralimbic cortex, amygdala, hippocampus, and nucleus accumbens (Quirk and Mueller, 2008; Peters et al., 2009; Herry et al., 2010; Millan et al., 2011). Manipulating extinction-induced plasticity by upregulating brain-derived neurotropic factor (BDNF) and AMPA receptors has been shown to enhance extinction to prevent drug-seeking behavior (Sutton et al., 2003; Briand et al., 2012; Wang et al., 2012).
Perineuronal nets (PNNs) are specialized extracellular matrix structures around interneurons in the brain and spinal cord (Celio and Blümcke, 1994; Celio et al., 1998). Chondroitin sulfate proteoglycans (CSPGs) are crucial components of PNNs (Kwok et al., 2008). PNNs have been suggested to be a permissive factor that allows synaptic plasticity in the adolescent CNS (Bavelier et al., 2010; Gundelfinger et al., 2010). Studies by Castrén and other researchers found that the degradation of PNNs induced by chondroitinase ABC (ChABC) converts the plasticity of the adult CNS to an adolescent phenotype; the promotion of experience-dependent plasticity in the adult CNS has been termed iPlasticity (Castrén et al., 2012; Castrén, 2013). For example, the degradation of chondroitin sulfate chains from CSPGs with ChABC promoted axon regeneration and the functional recovery of spinal cord injury (Bradbury et al., 2002; Barritt et al., 2006; García-Alías et al., 2009) and reactivated ocular dominance plasticity (Pizzorusso et al., 2002) through actions on plasticity-related proteins, including BDNF (Jain et al., 2011). Recently, Gogolla et al. (2009) found that the degradation of PNNs in adult rats reenabled the erasure of fear memories by extinction that occurred in juveniles. Therefore, we hypothesized that the elevation of extinction-related proteins together with PNNs degradation may be useful for erasing drug memories.
In the present study, we investigated whether iPlasticity with PNNs degradation enhances extinction to erase drug memories. We studied drug memories using conditioned place preference (CPP) and self-administration models in which the erasure of drug memories was assessed in a spontaneous recovery test with the passage of time and in a reinstatement test after a priming injection of drug (Li et al., 2011b; Xue et al., 2012). We examined the effect of PNNs degradation in the amygdala, a brain center that modulates the of emotional memories. The amygdala encompasses several structures with distinct connectional and functional characteristics. The basolateral amygdala (BLA) and central amygdala (CeA) are two important subregions that are involved in the drug reward memories (Milton et al., 2008; Li et al., 2010; He et al., 2011). We also explored extinction-related plasticity-related proteins that may underlie the effect of PNNs depletion.
Materials and Methods
Subjects.
Male Sprague Dawley rats, weighing 220–240 g upon arrival, were obtained from the Laboratory Animal Center of Peking University Health Science Center. They were housed in groups of 5 in a temperature- (23 ± 2°C) and humidity (50 ± 5%)-controlled animal facility with food and water freely available. The animals were maintained on a reverse 12 h/12 h light/dark cycle (lights on at 8:00 A.M.). All of the treatments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the experiments were approved by the University Animal Use Committee. A total of 414 were used in the study, from which 27 were excluded because of loss of the head cup or cannula misplacement and 28 were excluded because they exhibited strong unconditioned side preference (> 540 s) during the drug preconditioning phase (see Drug-induced CPP and Food-induced CPP, below).
Drugs.
Morphine sulfate and cocaine HCl (both from Qinghai Pharmaceutical) were dissolved in 0.9% physiological saline and injected in a volume of 10 ml/kg. Morphine was injected subcutaneously and cocaine was injected intraperitoneally. ChABC (#C3667; Sigma-Aldrich) and penicillinase (#P0389; Sigma-Aldrich) were dissolved in 0.1 m PBS (vehicle) and prepared in stock solutions of 0.02 U/μl. The control group received corresponding vehicle injections.
Surgery.
The rats weighed 280–300 g when surgery began and were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). Guide cannulae (23 gauge; Plastics One) were bilaterally implanted 1 mm above the BLA, 1 mm above the central nucleus of the amygdala (CeA), or 2 mm dorsal to the amygdala (anatomic control; Debiec et al., 2010). The stereotaxic coordinates for the BLA, CeA, and dorsal amygdala were as follows: BLA: anterior/posterior, −2.9 mm from bregma, medial/lateral, ±5.0 mm, dorsal/ventral, −7.8 mm from the skull surface; CeA: anterior/posterior, −2.9 mm, medial/lateral, ±4.2 mm, dorsal/lateral, −7.0 mm; and dorsal amygdala: anterior/posterior, −2.9 mm, medial/lateral, ±5.0 mm, dorsal/lateral, −5.8 mm (Paxinos and Watson, 2005; see Fig. 1E). A stainless steel stylet blocker was inserted into each cannula to maintain patency and to prevent infection. The rats were allowed to recover from surgery for 5–7 d.
Figure 1.
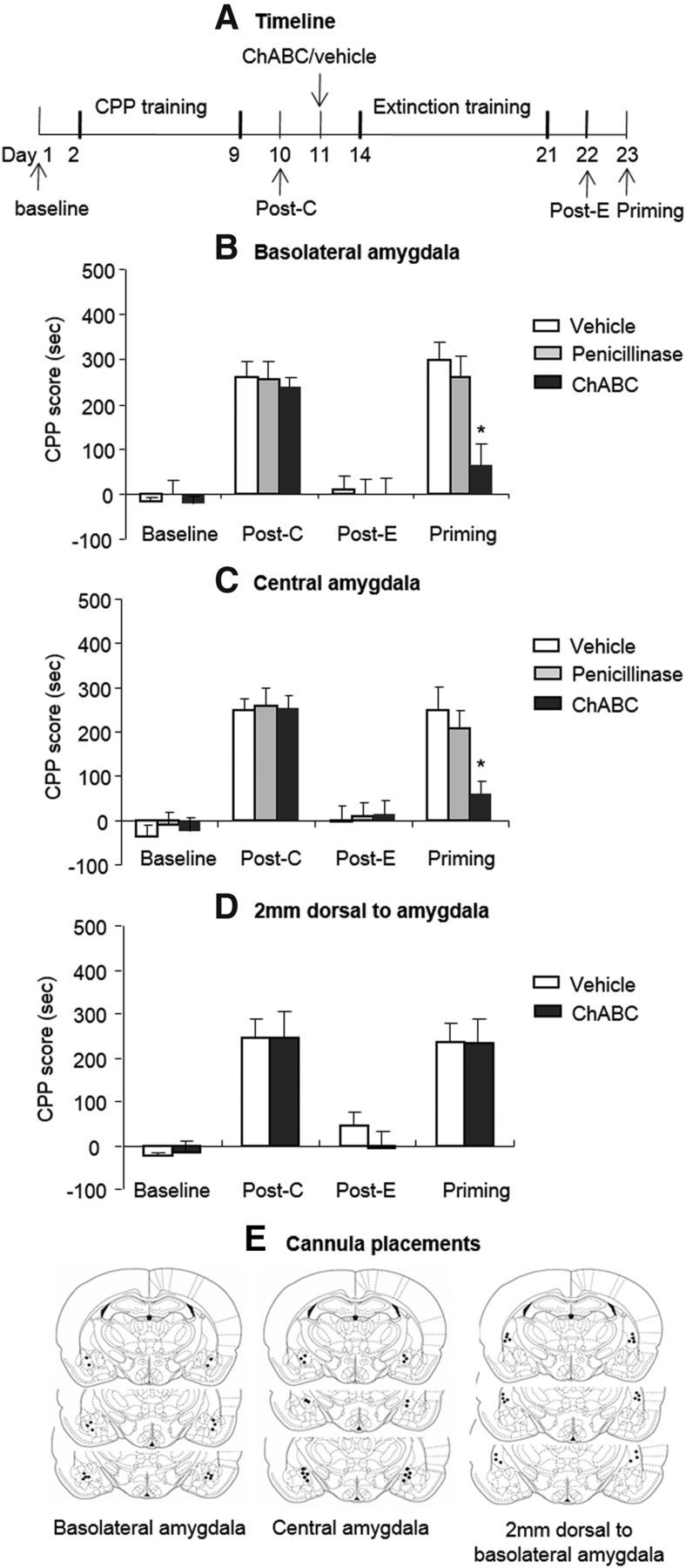
Effects of the combination of extinction training with ChABC injections into the amygdala on the subsequent reinstatement of morphine-induced CPP. A, Timeline of experimental procedure (see Materials and Methods for the experimental conditions of the groups). B, C, The combination of extinction training with microinfusions of chondroitinase ABC (0.01 U/side) and penicillinase (0.01 U/side) into the BLA (B) or CeA (C) but not 2 mm dorsal to the amygdala (D) prevented the subsequent drug-induced reinstatement of morphine-induced CPP. A significant difference was found in the morphine-induced priming test between the ChABC group and other two groups. E, Approximate placements of the intracranial injectors. The data are expressed as the mean ± SEM preference scores in seconds (time spent in the morphine-paired chamber minus time spent in the saline-paired chamber) during the CPP tests. *p < 0.05 vs vehicle group (n = 8–10 per group). Post-C, Postconditioning test; Post-E, postextinction test.
Intracranial injections.
ChABC (0.01 U/μg/side/0.5 μl), penicillinase (0.01 U/6 ng/side/0.5 μl), and vehicle were injected bilaterally into the BLA, CeA, or dorsal amygdala using 30 gauge injectors (Plastics One). The dose of ChABC was based on a previous study (Barritt et al., 2006). A total volume of 0.5 μl was injected bilaterally over 1 min and the injector was kept in place for another 1 min to allow diffusion. At the end of the experiments, the rats were anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and transcardially perfused. Cannula placements were assessed using Nissl staining with a section thickness of 40 μm under light microscopy.
Drug-induced CPP.
The CPP procedure was performed using an unbiased, counterbalanced protocol (Li et al., 2010; Li et al., 2011b; Ren et al., 2013). The CPP apparatus consisted of 10 identical three-chamber polyvinyl chloride (PVC) boxes. Two large side chambers (27.9 cm length × 21.0 cm width × 20.9 cm height) were separated by a smaller chamber (12.1 cm length × 21.0 cm width × 20.9 cm height, with a smooth PVC floor). The two larger chambers differed in their floor texture (bar or grid, respectively) and provided distinct contexts that were paired with morphine, cocaine, food, and saline injections. The three distinct chambers were separated by manual guillotine doors.
Baseline preference was assessed by placing the rats in the center compartment of the CPP apparatus, allowing them free access to all of the compartments for 15 min. On subsequent conditioning days, the rats were trained for 8 consecutive days with alternating injections of morphine (10 mg/kg, s.c.) or saline (1 ml/kg, s.c.) in the designated compartments. After each injection, the rats were confined to the corresponding conditioning chambers for 45 min and then returned to their home cages. Tests for the expression of drug-induced CPP in a drug-free state (15 min duration) were performed on different days after training (see Results, below). The procedure during testing was the same as during the initial baseline preference assessment. The CPP score was defined as the time (in seconds) spent in the reward-paired chamber minus the time spent in the saline-paired chamber during the CPP test. The training and testing procedures for cocaine-induced CPP were identical, with the exception that cocaine (10 mg/kg, i.p) was injected instead of morphine.
Food-induced CPP.
The CPP procedure for food was based on our previous work (Li et al., 2011b) and included three phases, similar to those used in the drug-induced CPP procedures. Baseline preference was assessed by placing the rats in the center compartment of the CPP apparatus, allowing them free access to all of the compartments for 15 min. The 8 d training phase included four 2 d cycles, in which the rats received a 5 g aliquot of food in one chamber on 1 day and no food in the other chamber on the other day. The duration of the daily training sessions was 45 min. The 15 min tests for the expression of CPP were conducted in the absence of food on different days after CPP training. During testing, the rats were allowed to freely explore the chambers. The CPP score is defined as the time (in seconds) spent in the food-paired chamber minus the time spent in the non-food-paired chamber.
CPP extinction training.
CPP extinction training was performed on 8 consecutive days and the procedure was similar to the establishment of CPP, with the exception that the drugs were replaced by saline (He et al., 2011). The CPP test was performed on different days after the manipulations (see Results, below).
Drug-priming-induced CPP.
The drug-priming-induced reinstatement of drug-induced CPP was based on previous studies (Wang et al., 2008; Xue et al., 2012). Five minutes before the CPP reinstatement test, the rats received a drug injection (cocaine, 5 mg/kg, i.p., or morphine, 3 mg/kg, s.c.). The conditions during the reinstatement test were the same as those for the baseline preference test.
Intravenous drug self-administration.
The drug self-administration training procedures were based on procedures we had established in our previous studies (Lu et al., 2009; Li et al., 2011a; Xue et al., 2012). Rats, weighing 300–320 g when surgery began, were anesthetized with sodium pentobarbital (60 mg/kg, i.p.). Catheters were inserted into the right jugular vein with the tip terminating at the opening of the right atrium. All of the rats were allowed to recover for 5–7 d after surgery. The operant chambers (AniLab Software and Instruments) were equipped with two nose poke operandi (AniLab Software and Instruments) located 9 cm above the chamber floor. Nose pokes in the active operandum led to cocaine or heroin infusions that were accompanied by a 5 s tone-light cue. Nose pokes in the inactive operandum were also recorded but had no programmed consequences. The modified cannula on the rat's skull was connected to a liquid swivel with polyethylene-50 tubing that was protected by a metal spring and led to a 10 ml syringe infusion pump. The rats were trained to self-administer heroin (0.05 mg/kg/infusion) or cocaine (0.75 mg/kg/infusion) during 3 1 h daily sessions separated by 5 min over 10 d. The sessions began at the onset of the dark cycle. A fixed-ratio one reinforcement schedule was used, with a 40 s timeout period after each infusion. Each session began with illumination of a house light that remained on for the entire session. The number of drug infusions was limited to 20/h. At the end of the training phase, the groups in the different experimental conditions were matched for drug intake during training.
Extinction of drug-reinforced responding.
During extinction, the conditions were the same as those during training, with the exception that drug was no longer available. The rats underwent extinction training until responding at the active nose poke operandum decreased to <20% of mean responding during the last 3 d of heroin or cocaine self-administration for at least 2 consecutive days (Xue et al., 2012).
Test for drug seeking.
The tests for drug-seeking behavior after spontaneous recovery and drug-induced reinstatement were based on previous studies from our group and others (Leri et al., 2002; Lu et al., 2004; Xue et al., 2012). The spontaneous recovery test was performed 4 weeks after extinction training. The reinstatement test was performed 24 h after the rats met the extinction criterion and all of the rats received a noncontingent heroin injection (0.25 mg/kg, s.c.) or cocaine injection (5 mg/kg, i.p.) 5 min before the test sessions. The testing conditions were the same as those during training, with the exception that active nose pokes were not reinforced by drug. Each session began with illumination of the house light, which remained on for the entire session. Nose poke responding during the test sessions resulted in contingent presentations of the tone-light cue that had previously been paired with drug infusions.
Immunofluorescence.
The rats were perfused with 4% paraformaldehyde, and the brains were removed, postfixed for 24 h, and placed in 30% sucrose in phosphate buffer. The brains were then sectioned coronally with a microtome into 15-μm-thick sections. Every third serial section was collected on gelatin-coated microscope slides. Free-floating sections were placed in a freshly prepared methanol-H2O2 solution for 10 min to block endogenous peroxidase activity. All of the sections were then treated with a biotin blocking kit and incubated for 30 min in a blocking solution composed of 3% bovine serum albumin (BSA), 20 mm lysine, and 0.2% Triton X-100 in PBS, pH 7.4. The sections were incubated overnight at 4°C with monoclonal antibodies in a solution of biotin-conjugated lectin wisteria floribunda (WFA, 10 μg/ml, #L1516; Sigma-Aldrich). All of the sections were then washed 3 times in PBS and stained with 1 h incubation in FITC-conjugated streptavidin (10 μg/ml, #S3762; Sigma-Aldrich). The method of counting the number of WFA-positive cells in these brain regions was moderately modified based on previous reports (Nixon and Crews, 2004; Gogolla et al., 2009; Shi et al., 2012), in which two or three sections from each brain region for each rat were selected. The cell numbers on either side of the specific brain region were averaged and taken as the positive immunoreactive cell number for each rat. The number of WFA-labeled cells was measured using a fluorescence microscope (Olympus) with an image-analysis program (MetaMorph, version 4.65). One in every 15 sections was sampled to avoid multiple counting of the same cell (each section was spaced 225 μm apart). This profile counting method provided results that are similar to those obtained with stereological cell counting methods in some studies (Crews et al., 2004; Nixon and Crews, 2004; Lussier et al., 2009).
Tissue sample preparation and Western blot assay.
The assay procedures were based on those used in our previous studies (Li et al., 2010; Li et al., 2011b). Twenty-four hours after the end of the last extinction session, the rats were decapitated. Their brains were quickly extracted, frozen in −60°C N-hexane, and transferred to a −80°C freezer. Using a freezing cryostat (−20°C; Reichert-Jung 2800 Frigocut E), bilateral tissue punches (12 gauge) of the BLA and CeA were taken from 1 mm thick coronal sections ∼2.9 mm from bregma. The tissue punches were homogenized (10–15 s × 3, 5 s interval) with an electrical disperser (Wiggenhauser, Sdn Bhd) after 30 min with RIPA lysis buffer (Beyotime Biotechnology, Jiangsu, China; 20 mm Tris, pH 7.5, 150 mm NaCl, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm ethylenediamine tetraacetic acid, 1% Na3VO4, 0.5 μg/ml leupeptin, and 1 mm phenylmethanesulfonyl fluoride). The homogenate was then subjected to 10,000 × g centrifugation for 10 min at 4°C. All of the above procedures were performed at 0–4°C. Protein concentrations in all of the samples were determined using the bicinchoninic acid assay (Beyotime Biotechnology). The samples were further diluted in RIPA lysis buffer to equalize the protein concentrations.
Four times loading buffer (16% glycerol, 20% mercaptoethanol, 2% SDS, and 0.05% bromophenol blue) was added to each sample (3:1 sample to loading buffer) before boiling for 3 min. The samples were cooled and subjected to SDS-PAGE (10% acrylamide/0.27% N,N′-methylenebisacrylamide resolving gel) for ∼40 min at 80 V in stacking gel and ∼1 h at 130 V in resolving gel. Proteins were transferred electrophoretically to Immobilon P transfer membranes (Millipore) at 0.25 A for 2.5 h. The membranes were washed with Tris-buffered saline plus 0.05% Tween 20 (TBST), pH 7.4, and then dipped in blocking buffer (5% BSA in TBST) overnight at 4°C. The next day, the membranes were incubated for 1 h at room temperature on an orbital shaker with anti-BDNF antibody (1:200, #sc-546; Santa Cruz Biotechnology), anti-GluR1 antibody (1:200, #sc-13152; Santa Cruz Biotechnology), anti-GluR2 antibody (1:200, #sc-7611; Santa Cruz Biotechnology), anti-GluR3 antibody (1:1000, #sc-34066; Santa Cruz Biotechnology), or β-actin (1:1000, sc-47778; Santa Cruz Biotechnology) in TBST plus 5% BSA and 0.05% sodium azide. After 3 5 min washes (3×) in TBST buffer, the blots were incubated for 45 min at room temperature on a shaker with horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit IgG, goat anti-mouse IgG, or rabbit anti-goat IgG; Santa Cruz Biotechnology) diluted 1:5000 in blocking buffer. The blots were then washed 3 times for 5 min each in TBST and incubated with a layer of Super Signal Enhanced Chemiluminescence Substrate (Detection Reagents 1 and 2, 1:1 ratio; Applygen Technologies) for 1 min at room temperature. Excess mixture was dripped off before the blots were wrapped with a clean piece of plastic wrap (with no bubbles between blot and wrap) and the blots were then exposed to x-ray film (Eastman Kodak) for 5–60 s. Band intensities were quantified using Quantity One software (version 4.4.0; Bio-Rad). Band intensities from each test sample were compared with the band intensities from the standard curves. The amount of the protein of interest in each sample was interpolated from the standard curve. The standard curve runs in all of the Western blots in the present study showed that the band intensities for each of our test samples were within the linear range of detection.
Statistical analyses.
The data are expressed as mean ± SEM. In the CPP experiment, the statistical analysis was performed using mixed ANOVA, with treatment (ChABC, penicillinase, and vehicle) as the between-subjects factor and test phase (baseline, postconditioning test, postextinction test, priming test, and posttreatment test) as the within-subjects factor. In the self-administration experiment, the statistical analysis was performed with treatment (ChABC and vehicle) as the between-subjects factor and test phase (sessions 1–11 for extinction data; last extinction session and priming test/spontaneous recovery test for reinstatement/spontaneous recovery experiment) as the within-subjects factor. Because our multifactorial ANOVAs yielded multiple main effects and interaction effects, we only report significant effects in the Results section that are critical for data interpretation. Post hoc analyses of significant effects in the ANOVA were performed using the Tukey test. Values of p < 0.05 were considered statistically significant.
Results
Experiment 1: Extinction training combined with intra-amygdala injections of ChABC prevented the subsequent reinstatement of morphine-induced CPP
In Experiment 1, we used 8 groups of rats (n = 8–10 per group) to determine the effects of ChABC in different subregions of the amygdala on the extinction and reinstatement of morphine-induced CPP (Fig. 1A). The rats were trained for morphine-induced CPP for 8 d and tested for the expression of CPP on day 10 without any injections (postconditioning test). On day 11, the rats were injected with ChABC/penicillinase/vehicle into the BLA, CeA, and a brain area 2 mm dorsal to amygdala. One day after a single injection of ChABC, the structure of PNNs is completely disrupted and this effect can last at least 4 weeks (Brückner et al., 1998). Three days after the injection, the rats underwent four consecutive blocks of extinction training. On day 22, the rats underwent the extinction test to confirm that CPP was extinguished (postextinction test). On day 23, the rats were injected with a low dose of morphine (3 mg/kg, s.c.), and the expression of CPP was subsequently retested (priming test).
The analysis revealed significant effects of test phase (F(3,66) = 61.35, p < 0.01 for BLA; F(3,72) = 49.85, p < 0.01 for CeA) and a treatment × test phase interaction (F(6,66) = 4.07, p < 0.01 for BLA; F(6,72) = 2.50, p < 0.05 for CeA; Fig. 1B,C). For injections 2 mm dorsal to the amygdala, the analysis revealed only a significant effect of test phase (F(3,45) = 20.86, p < 0.01; Fig. 1D). The post hoc analysis revealed that the morphine-priming-induced reinstatement of morphine-induced CPP was significantly decreased in the groups that received ChABC injections into the BLA and CeA (all p < 0.01). These results indicate that intra-amygdala injections of ChABC combined with extinction training erased morphine-induced CPP memory.
Experiment 2: Intra-amygdala injections of ChABC had no effect on the retention, retrieval, or reconsolidation of morphine-induced CPP
To exclude the possibility that ChABC injections into the amygdala alone impaired the retention of morphine-induced CPP (Fig. 2A), 4 groups of rats (n = 7 per group) were first trained for morphine-induced CPP and then tested for the expression of CPP on day 10 (postconditioning test). The rats were injected with ChABC or vehicle into the BLA or CeA. Ten days after the injections, these rats were tested again (posttreatment test) for morphine-induced CPP. The analysis revealed only a significant effect of test phase (F(2,24) = 20.53, p < 0.01 for BLA; F(2,24) = 22.51, p < 0.01 for CeA; Fig. 2B). These results indicate that intra-amygdala injections of ChABC alone had no effect on the retention of morphine-induced CPP.
Figure 2.
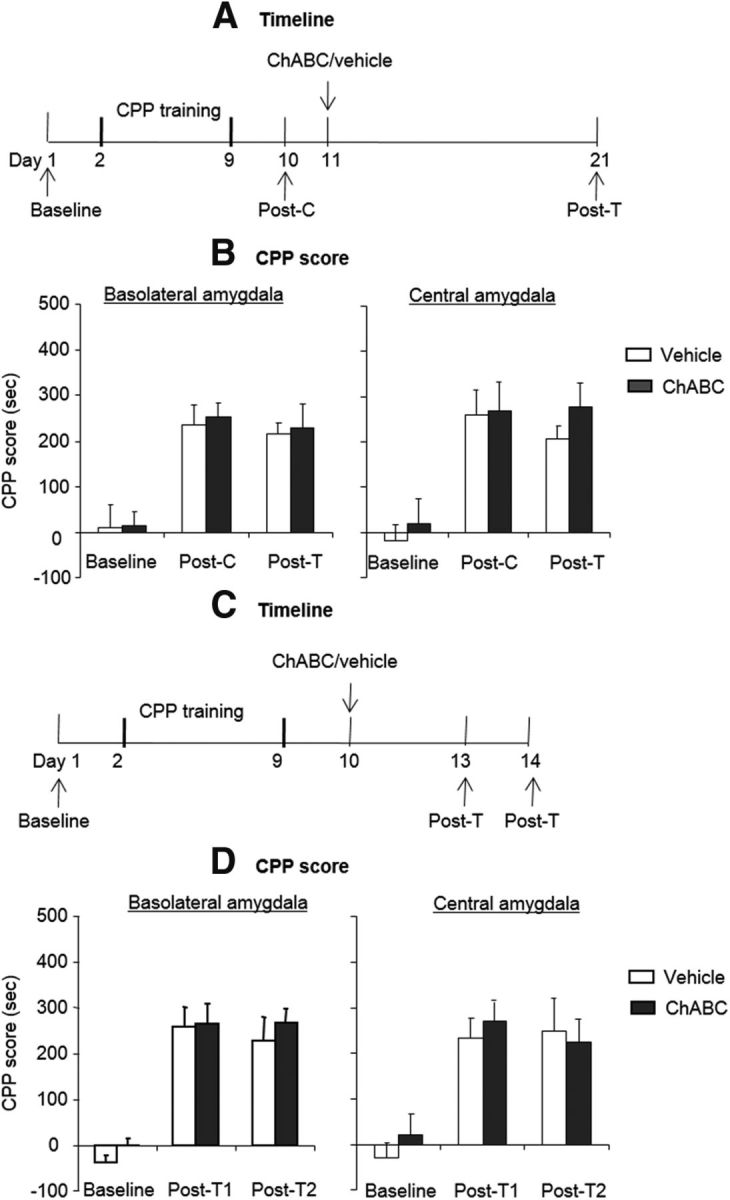
Effects of ChABC injections into the amygdala on the retrieval, reconsolidation, and retention of morphine-induced CPP. A, C, Timeline of experimental procedure. B, D, ChABC (0.01 U/side) microinfusions into the BLA or CeA 1 d after the postconditioning test had no effect on the retention (B), retrieval, or reconsolidation (D) of morphine-induced CPP. No significant difference was found in the retention, retrieval, or reconsolidation test between the ChABC group and vehicle group. The data are expressed as the mean ± SEM preference scores in seconds (time spent in the morphine-paired chamber minus time spent in the saline-paired chamber) during the CPP tests (n = 7–8 per group). Post-C, Postconditioning test; Post-T1, postextinction test 1; Post-T2, posttreatment test 2.
Memory retrieval may induce two processes, reconsolidation and extinction (Eisenberg et al., 2003; Suzuki et al., 2004). An experiment was designed to exclude the possibility that intra-amygdala injections of ChABC in conjunction with extinction produce deficits in retrieval or reconsolidation (Fig. 2C). The rats were trained for morphine-induced CPP and were then randomly assigned to 4 groups (n = 7–8 per group) that were injected with ChABC or vehicle into the BLA or CeA. Three days after the injection, these rats underwent a morphine-induced CPP retrieval test (posttreatment test 1). Twenty-four hours later, morphine-induced CPP was retested (posttreatment test 2). Posttreatment test 1 assessed the potential influence of ChABC on memory retrieval and posttreatment test 2 assessed whether memory reconsolidation induced by posttreatment test 1 was influenced by PNNs degradation. The analysis revealed only a significant effect of test phase (F(2,28) = 48.06, p < 0.01 for BLA; F(2,24) = 15.98, p < 0.01 for CeA; Fig. 2D). These results indicate that, after the intra-amygdala injection, the preference for the environment previously paired with morphine was unaffected after the CPP retrieval test and subsequent reconsolidation phase. Therefore, the erasure of morphine-induced CPP by ChABC in conjunction with extinction was not attributable to disruption of the reconsolidation of CPP.
Experiment 3: Intra-amygdala injections of ChABC had no effect on the relearning of morphine-induced CPP and maintenance of acquired food-induced CPP
Extinction training combined with ChABC injections might not produce the specific memory erasure of morphine-induced CPP, but rather may lead to amygdala dysfunction such as a deficit in reward processing. To exclude this possibility, we investigated whether the combination of an intra-amygdala infusion of ChABC and extinction training affected the relearning of morphine-induced CPP (Fig. 3A). The rats (n = 7–8 per group) were trained for morphine-induced CPP for 8 d and tested for the expression of CPP on day 10 without any injections (postconditioning test 1). On day 11, the rats were injected with ChABC or vehicle into the BLA or CeA. Twenty-four hours after the injection, the rats underwent 8 consecutive days of extinction training. On day 20, the rats underwent the CPP test to confirm that CPP was extinguished (postextinction test). Then the rats were trained for morphine-induced CPP again and tested for the expression of CPP on day 32 (postconditioning test 2). We compared the preference baseline and postconditioning CPP score between rats that experienced the manipulation and naive rats. The analysis revealed only a significant effect of test phase (F(1,13) = 30.33, p < 0.01 for BLA, Fig. 3B, right column; F(1,13) = 28.57, p < 0.01 for CeA, Fig. 4C, right column). These results indicate that the rats were capable of relearning morphine-induced CPP after the injection of ChABC in conjunction with extinction. Therefore, this manipulation did not induce a deficit in responsiveness to the rewarding stimulus.
Figure 3.

Effects of combination of intra-amygdala infusions of ChABC and extinction training on the subsequent learning of new morphine-induced CPP. A, Timeline of experimental procedure. B, C, ChABC (0.01 U/side) microinfusions into the BLA or CeA 1 d after the postconditioning test in conjunction with extinction training had no effect on the subsequent relearning of morphine-induced CPP. No significant difference was found in the new morphine-induced CPP test between the ChABC group and the naive group. The data are expressed as the mean ± SEM preference scores in seconds (time spent in the morphine-paired chamber minus time spent in the saline-paired chamber) during the CPP tests (n = 7–8 per group). Post-C1, Postconditioning test 1; Post-E, postextinction test; Post-C2, postconditioning test 2.
Figure 4.

Effects of combination of intra-amygdala infusions of ChABC and extinction training on the maintenance of acquired food-induced CPP. A, Timeline of experimental procedure. B, C, ChABC (0.01 U/side) microinfusions into the BLA or CeA 1 d after the postconditioning test in conjunction with extinction training had no effect on the maintenance of acquired food-induced CPP. No significant difference was found in the food-induced CPP test between the ChABC group and vehicle group. The data are expressed as the mean ± SEM preference scores in seconds (time spent in the food-paired chamber minus time spent in the non-food-paired chamber) during the CPP tests (n = 7–8 per group). Post-C, Postconditioning test; Post-E, postextinction test; Post-T, posttreatment test.
We then investigated whether the degradation of PNNs in the amygdala combined with the extinction of morphine memory affects the retention of acquired natural reward memory (Fig. 4A). Four groups of rats (n = 7–8 per group) were trained for morphine-induced CPP for 8 d in context A and tested for the expression of CPP on day 10 without any injections (morphine postconditioning test). All of the rats were then trained for food-induced CPP for 8 d in a new context (context B) and tested for the expression of CPP (food postconditioning test). Twenty-four hours later, all of the rats were injected with ChABC/vehicle into the BLA/CeA. Three days after the injection, the rats underwent four consecutive blocks of morphine-induced CPP extinction training and an extinction test to confirm that morphine-induced CPP was extinguished (morphine postextinction test). One day later, the rats were injected with a lower dose of morphine (3 mg/kg, s.c.) and the expression of CPP was subsequently retested (priming test). Twenty-four hours later, the rats were tested for food-induced CPP in context B (food retention test). The statistical analyses were performed separately for morphine- and food-induced CPP. For the morphine-induced CPP data, the analysis revealed significant main effects of test phase (F(3,39) = 14.31, p < 0.01 for BLA, Fig. 4B, left column; F(3,39) = 12.76, p < 0.01 for CeA, Fig. 4C, left column) and a treatment × test phase interaction (F(3,39) = 6.81, p < 0.01 for BLA; F(3,39) = 5.28, p < 0.01 for CeA). The post hoc analysis revealed that the morphine-priming-induced reinstatement of morphine-induced CPP was significantly decreased in the groups that received ChABC injections into the BLA and CeA (all p < 0.01). For the food-induced CPP data, only a significant main effect of test phase was revealed (F(2,26) = 28.35, p < 0.01 for BLA, Fig. 4B, right column; F(2,26) = 24.74, p < 0.01 for CeA, Fig. 4C, right column). These results indicate that the rats were still capable of maintaining acquired food-induced CPP after ChABC injections in conjunction with extinction. Therefore, the manipulation did not affect the acquired natural reward memory.
Experiment 4: Extinction training combined with intra-amygdala injections of ChABC prevented the subsequent reinstatement of cocaine-induced CPP, but not food-induced CPP
Previous studies demonstrated behavioral and neurobiological differences between opioid and stimulant addiction (Badiani et al., 2011). We sought to determine whether the combination of extinction training and intra-amygdala injection of ChABC has a similar effect on the memories of stimulant drugs (Fig. 5A). Four groups of rats (n = 8–10 per group) were trained for cocaine-induced CPP for 8 d and tested for the expression of CPP on day 10 without any injections (postconditioning test). On day 11, the rats were injected with ChABC/vehicle into the BLA/CeA. Three days after the injection, the rats underwent four consecutive blocks of extinction training. On day 22, the rats underwent the extinction test to confirm that CPP was extinguished (postextinction test). On day 23, the rats were injected with a lower dose of cocaine (5 mg/kg, i.p.) and the expression of CPP was subsequently retested (priming test). The analysis revealed a significant effect of test phase (F(3,45) = 28.89, p < 0.01 for BLA; F(3,51) = 20.09, p < 0.01 for CeA) and a treatment × test phase interaction (F(3,45) = 3.83, p < 0.05 for BLA, Fig. 5B; F(3,51) = 3.01, p < 0.05 for CeA, Fig. 5C). The post hoc analysis revealed that the cocaine-priming-induced reinstatement of cocaine-induced CPP was significantly decreased in the groups that received ChABC injections into the BLA and CeA (all p < 0.01). These results indicate that intra-amygdala injections of ChABC combined with extinction training also erased cocaine-induced CPP memory.
Figure 5.
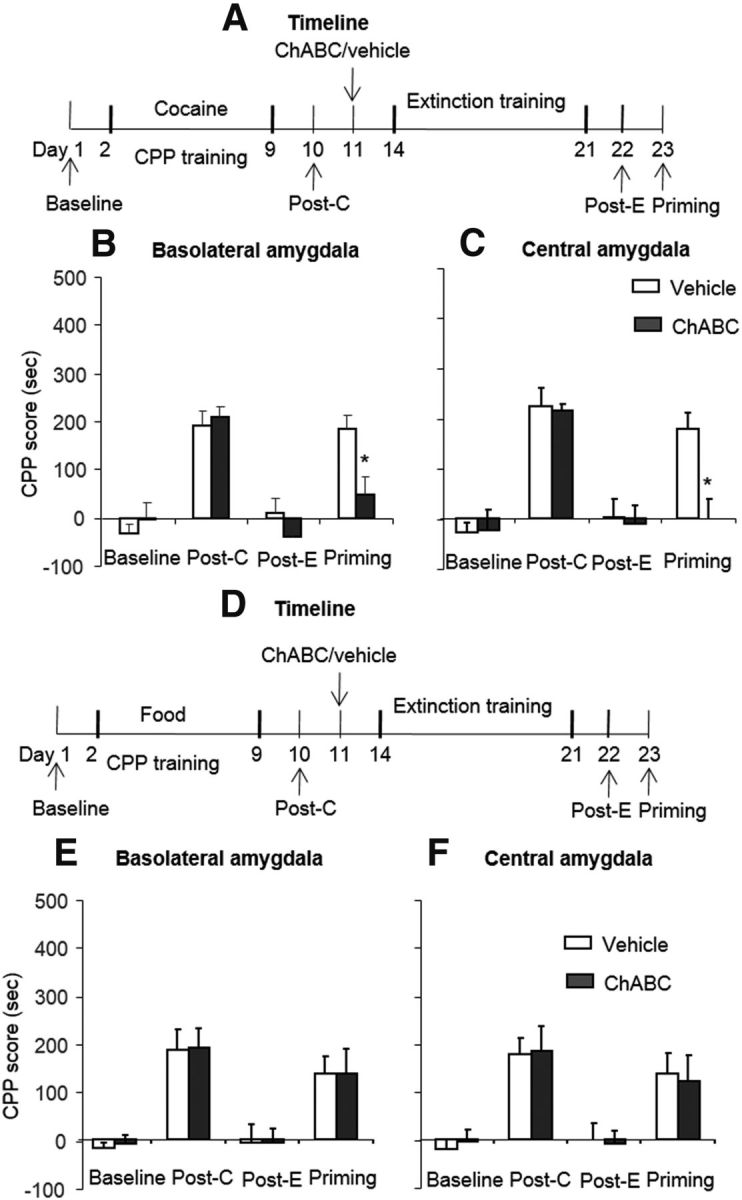
Effects of combination of extinction training with ChABC injection into the amygdala on the subsequent reinstatement of cocaine-induced CPP. A, D, Timeline of experimental procedure. B, C, Combination of extinction training with microinfusions of chondroitinase ABC (0.01 U/side) into the BLA (B) or CeA (C) 1 d after the postconditioning test prevented the subsequent cocaine-priming-induced reinstatement of cocaine-induced CPP. A significant difference was found in cocaine-priming test between the ChABC group and the vehicle group. E, F, Combination of extinction training with microinfusions of chondroitinase ABC (0.01 U/side) into the BLA (E) or CeA (F) 1 d after the postconditioning test had no effect on subsequent-priming-induced reinstatement of food-induced CPP. The data are expressed as the mean ± SEM preference scores in seconds (time spent in the cocaine- or food-paired chamber minus time spent in the saline-paired chamber) during the CPP tests. *p < 0.05 vs vehicle group (n = 7–10 per group). Post-C, Postconditioning test; Post-E, postextinction test.
We used 4 groups of rats (n = 7–8 per group) to determine whether the combination of extinction training and intra-amygdala injections of ChABC affect the memories of food-induced CPP (Fig. 5D). The procedure was similar to the cocaine-induced CPP experiment. The analysis revealed only a significant effect of test phase (F(3,39) = 16.50, p < 0.01 for BLA, Fig. 5E; F(3,39) = 13.33, p < 0.01 for CeA, Fig. 5F). These results indicate that intra-amygdala injections of ChABC combined with extinction training did not erase food-induced CPP memory.
Experiment 5: Extinction training combined with intra-amygdala injections of ChABC prevented the subsequent spontaneous recovery and reinstatement of heroin- and cocaine-seeking behavior
To further confirm the effect of extinction training in conjunction with ChABC injections in the amygdala on opioid memories, we explored whether this procedure also prevented the reinstatement and spontaneous recovery of heroin-seeking behaviors. For the reinstatement experiment, 2 groups of rats (n = 7–8 per group) were trained to self-administer heroin for 10 d. On day 11, the rats were injected with ChABC or vehicle into the BLA or CeA. Three days after the injection, the rats underwent 10 or 12 d of extinction training until they met the extinction criterion. Twenty-four hours later, all of the rats underwent a priming test that was initiated by a noncontingent injection of heroin (0.25 mg/kg, s.c.) 5 min before the test sessions (priming). The statistical analyses were performed separately for self-administration training, extinction training, drug-priming-induced reinstatement. All of the rats acquired similar heroin self-administration (Fig. 6B). For extinction responding, the analysis revealed a significant effect of extinction session (F(9,126) = 29.78, p < 0.01 for BLA; F(9,153) = 32.73, p < 0.01 for CeA) and a treatment × extinction session interaction (F(9,126) = 2.35, p < 0.05 for BLA, Fig. 6C, left column; F(9,153) = 2.81, p < 0.01 for CeA, Fig. 6D, left column). For drug-induced reinstatement, the statistical analysis revealed a main effect of reinstatement condition (F(1,14) = 35.58, p < 0.01 for BLA; F(1,17) = 17.80, p < 0.01 for CeA) and a treatment × reinstatement condition interaction (F(1,14) = 7.83, p < 0.05 for BLA, Fig. 6C, right column; F(1,17) = 9.81, p < 0.01 for CeA, Fig. 6D, right column). In the spontaneous recovery experiment, another 2 groups of rats (n = 8–11 per group) were used and the protocol for the spontaneous recovery experiment was similar to the reinstatement experiment, with the exception that the reinstatement test was replaced by the spontaneous recovery test 4 weeks after extinction training (Fig. 6E). The data for the spontaneous recovery of heroin-seeking behavior revealed a main effect of test phase (F(1,17) = 52.43, p < 0.01 for BLA; F(1,14) = 17.87, p < 0.01 for CeA) and a treatment × test phase interaction (F(1,17) = 7.73, p < 0.05 for BLA, Fig. 6F, left column; F(1,14) = 5.7, p < 0.05 for CeA, Fig. 6G, right column). The post hoc analysis revealed that the spontaneous recovery of drug seeking were significantly decreased in the groups that received ChABC injections into the BLA and CeA (all p < 0.05). These results indicate that intra-amygdala injections of ChABC facilitated extinction and prevented the subsequent spontaneous recovery and heroin-priming-induced reinstatement of drug-seeking behavior.
Figure 6.

Effects of combination of extinction training with ChABC injections into the amygdala on the subsequent spontaneous recovery and reinstatement of heroin-seeking behavior. A, E, Timeline of experimental procedure. B–D, All rats acquired similar heroin self-administration behavior (B), and a combination of extinction training with microinfusions of chondroitinase ABC (0.01 U/side) into the BLA (C) or CeA (D) 1 d after heroin self-administration training facilitated subsequent extinction training and prevented the reinstatement of heroin-seeking behavior. F, G, Combination of extinction training with microinfusions of chondroitinase ABC (0.01 U/side) into the BLA (F) or CeA (G) 1 d after heroin self-administration training prevented the spontaneous recovery of heroin-seeking behavior. Significant differences were found in the rates of extinction training and spontaneous recovery and in the heroin-priming test between the ChABC group and vehicle group. The data are expressed as the mean ± SEM number of responses on the active and inactive nose poke devices during the extinction sessions and heroin-priming test. *p < 0.05 vs vehicle group (n = 8–11 per group).
We also investigated whether this procedure exerts a memory erasure effect in the cocaine self-administration model (Fig. 7A). The procedure was similar to the heroin self-administration experiment. All of the rats had similar cocaine self-administration behavior (Fig. 7B). For extinction responding, the analysis revealed a main effect of extinction session (F(11,143) = 20.64, p < 0.01 for BLA; F(11,143) = 26.47, p < 0.01 for CeA) and a treatment × extinction session interaction (F(11,143) = 2.01, p < 0.05 for BLA, Fig. 7C, left column; F(11,143) = 2.43, p < 0.01 for CeA, Fig. 7D, left column). For drug-induced reinstatement, the statistical analysis revealed a main effect of reinstatement condition (F(1,13) = 40.96, p < 0.01 for BLA; F(1,13) = 20.94, p < 0.01 for CeA) and a treatment × reinstatement condition interaction (F(1,13) = 13.00, p < 0.05 for BLA, Fig. 7C, right column; F(1,13) = 11.87, p < 0.01 for CeA, Fig. 7D, right column). The post hoc analysis revealed that the cocaine-priming-induced reinstatement of drug seeking was significantly decreased in the groups that received ChABC injections into the BLA and CeA (all p < 0.05). Altogether, these results indicate that intra-amygdala injections of ChABC facilitated extinction and prevented the subsequent cocaine-priming-induced reinstatement of drug-seeking behavior.
Figure 7.

Effects of combination of extinction training with ChABC injection into the amygdala on the subsequent reinstatement of cocaine-seeking behavior. A, Timeline of experimental procedure. B–D, All rats acquired similar heroin self-administration behavior (B), and a combination of extinction training with microinfusions of chondroitinase ABC (0.01 U/side) into the BLA (C) or CeA (D) 1 d after cocaine self-administration training facilitated subsequent extinction training and prevented the reinstatement of cocaine-seeking behavior. A significant difference was found in the rate of extinction training and in the cocaine-induced-priming test between the ChABC group and the vehicle group. The data are expressed as the mean ± SEM number of responses on the active and inactive nose poke devices during the extinction sessions and cocaine-priming test. *p < 0.05 vs vehicle group (n = 7–8 per group).
Experiment 6: PNNs depletion potentiated the extinction-induced alterations of plasticity-related proteins in the amygdala
Extinction training has been suggested to produce neuroplastic alterations, such as the upregulation of AMPA receptors and neurotropic factors in the mesolimbic system that control drug-seeking behavior (Sutton et al., 2003; Briand et al., 2012; Wang et al., 2012). Therefore, Experiment 6 was designed to explore the plasticity-related proteins that participate in the effect of PNNs degradation (Fig. 8A). PNNs levels were detected by WFA staining as described previously (Härtig et al., 1992; Gogolla et al., 2009). The rats were trained for morphine-induced CPP for 8 d and tested for the expression of CPP on day 10 without any injections (postconditioning test). On day 11, the rats were injected with ChABC/vehicle into the BLA. Three days after the injection, the rats underwent four consecutive blocks of extinction training. On day 22, the rats were perfused with 4% paraformaldehyde, and the brains were removed for the immunofluorescent detection of WFA-positive cells. Another four groups of rats were killed 24 h after extinction training and the amygdala was removed to assess BDNF and glutamate receptor 1 (GluR1), GluR2, and GluR3 protein levels. A two-way ANOVA of WFA-positive cells was performed, with treatment (ChABC and vehicle) and extinction as the between-subjects factors. The analysis revealed a significant main effect of treatment (F(1,15) = 145.53, p < 0.01; Fig. 5C), but no significant effect of extinction and no treatment × extinction interaction (p > 0.05). The post hoc analysis revealed that WFA-positive cells were significantly decreased in the ChABC + no extinction and ChABC + extinction groups compared with the other two groups in the BLA (p < 0.01), but not in the CeA (p > 0.05). These results confirmed that PNNs in the amygdala were significantly decreased after ChABC injections and that extinction training alone had no effect on PNNs levels.
Figure 8.
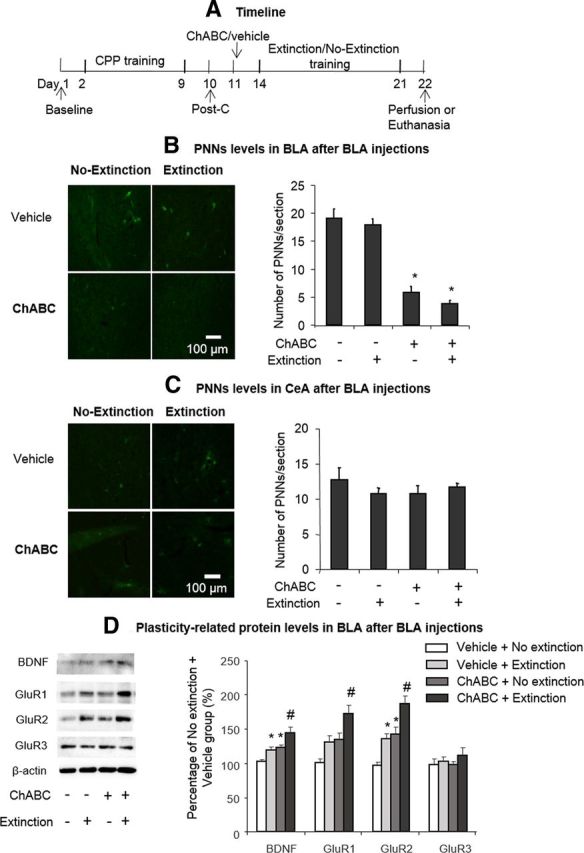
Effects of intra-BLA ChABC injections and extinction training on PNNs and plasticity-related protein levels. A, Timeline of experimental procedure. B, C, Representative WFA-positive PNN immunostaining in the BLA and CeA under different experimental conditions (left column). Scale bar, 100 μm. ChABC infusions into the BLA significantly decreased WFA-positive PNNs in the BLA but not CeA (right column). D, Effect of the different manipulations on plasticity-related protein levels in the BLA. The data are expressed as mean ± SEM percentage differences from the vehicle + no extinction group. *p < 0.05 vs vehicle group; #p < 0.05 vs vehicle + extinction and ChABC + no extinction groups (n = 4–7 per group).
For the Western bolt analysis, a two-way ANOVA was conducted, with treatment (ChABC and vehicle) and extinction as the between-subjects factor. ChABC injections into the BLA potentiated the effects of extinction training alone on BDNF (treatment: F(1,24) = 14.01, p < 0.01; extinction: F(1,24) = 9.63, p < 0.01; Fig. 8C), GluR1 (treatment: F(1,24) = 11.91, p < 0.01; extinction: F(1,24) = 9.34, p < 0.01; Fig. 8C), and GluR2 (treatment: F(1,24) = 22.63, p < 0.01; extinction: F(1,24) = 17.10, p < 0.01; Fig. 8C) levels in the BLA. These results indicate that PNNs may not be involved in the extinction of morphine-induced CPP, but may provide metaplastic conditions that facilitate extinction training.
Discussion
In the present study, we investigated whether extinction training combined with ChABC-induced PNNs degradation erases drug memories. We found that extinction training combined with intra-BLA and intra-CeA injections of ChABC (but not injections 2 mm dorsal to the amygdala) prevented the subsequent reinstatement of morphine-induced CPP. Intra-amygdala injections of ChABC alone had no effect on the retention, retrieval, reconsolidation, or relearning of morphine-induced CPP or storage of acquired food memory. In addition, the action of ChABC on drug memory occurred across drug types. Intra-amygdala injections of ChABC combined with extinction also prevented the subsequent reinstatement of cocaine-induced, but not food-induced, CPP. Furthermore, we confirmed the effects of the procedure in the operant self-administration model and found that a combination of extinction training and ChABC injections into the amygdala prevented the spontaneous recovery and reinstatement of heroin- and cocaine-seeking behavior. Last, we explored the potential mechanisms that underlie the effect of PNNs depletion on drug memories and found that it may potentiate the extinction-related levels of plasticity-related proteins in the amygdala, including BDNF, GluR1, and GluR2. Altogether, our findings indicate that intra-amygdala injections of ChABC combined with extinction training erase drug memories and prevent the return of drug-seeking behavior.
Some issues should be considered with regard to our results. First, the effect of PNNs degradation in the amygdala on extinction training was region specific in that we found that microinfusions of ChABC into a brain area 2 mm dorsal to the basolateral amygdala did not have a synergistic effect with extinction on morphine-induced CPP. Second, the effects of ChABC injections into the amygdala on extinction training cannot be explained by immunologic reactivity induced by foreign proteins. We used penicillinase as a control (Pizzorusso et al., 2002) and found that intra-amygdala injections of penicillinase had no such effect. Third, the effect of intra-amygdala injections of ChABC on morphine memory was not attributable to functional deficits in memory maintenance or reconsolidation. Two weeks of abstinence or a retrieval trial after intra-amygdala ChABC injections, rather than extinction training after intra-amygdala ChABC injections, had no effect on morphine-induced CPP, which is consistent with a previous study (Gogolla et al., 2009). In addition, the effect of intra-amygdala injections of ChABC on morphine memory was not attributable to amygdala dysfunction. We found that the rats were able to learn new morphine-induced CPP after the combined ChABC amygdala injections and extinction training and maintained intact acquired food-induced CPP memory. Last, although some studies indicated some behavioral and neurobiological differences between psychostimulant and opioid addiction (Badiani et al., 2011), we found that the present protocol affected both opioid memories (i.e., morphine-induced CPP memory and operant heroin self-administration memory) and cocaine-induced CPP memory and operant cocaine self-administration memory. Moreover, we found that the procedure did not erase food CPP memory. These results indicate a general effect of the procedure on preventing the return of drug memories.
Although numerous studies have elucidated the mechanisms that underlie the extinction of drug memories (Quirk and Mueller, 2008; Peters et al., 2009; Parsons and Ressler, 2013), only a few manipulations, such as GluR2 overexpression (Sutton et al., 2003), N-methyl-d-aspartate receptor partial agonist d-Cycloserine administration (Torregrossa et al., 2010), Homer1 overexpression (Knackstedt et al., 2010), and chromatin modulation (Malvaez et al., 2010; Malvaez et al., 2013), effectively promote extinction training to prevent relapse. Furthermore, all of these manipulations target neuronal signal transduction, but it is unknown whether modulation of the extracellular matrix affects extinction and original drug memories. Our study extends these findings, in which the degradation of PNNs in the amygdala, a specialized extracellular matrix, enhanced extinction efficacy and erased drug memories. Furthermore, we discovered that extinction training had no effect on PNNs levels, indicating that PNNs may not be involved in extinction training. However, PNNs degradation is sufficient to increase BDNF, GluR1, and GluR2 and to potentiate the extinction training on their protein levels. Considering the important roles of BDNF and AMPA receptors in the extinction of drug memories (Sutton et al., 2003; Briand et al., 2012; Wang et al., 2012), we speculate that PNN depletion may provide metaplastic conditions that enhance extinction training to prevent the relapse of drug-seeking behavior. Our results also support the notion that extinction training can ameliorate or reverse the neuroplasticity produced by previous acquired memory (Sutton et al., 2003; Lai et al., 2012; Ren et al., 2013). Enhancing extinction-related neuroadaptations within extinction circuits may erase the original memory trace (Xu et al., 2009). Notably, we used an extinction procedure to extinguish responses to both contextual cues and discrete cues (light and sound) and we did not consider the effect of extinguishing the memory of one cue (such as a contextual cue or discrete cue) combined with PNNs removal on the memories of other cues. Further studies are needed to explore the effect of PNNs removal on the generalization of extinction training.
Previous studies demonstrated that the degradation of PNNs may induce juvenile-like plasticity and remove the inhibitory factor that constrains experience-dependent plasticity. CSPGs are components of the extracellular matrix in the CNS that inhibit axonal sprouting and growth (Fitch and Silver, 1997; Fawcett and Asher, 1999). During late development, CSPGs condense around the soma and dendrites of GABA neurons in the form of PNNs (Celio, 1993; Celio and Blümcke, 1994). The degradation of PNNs and CSPGs by ChABC results in the functional recovery of cortical plasticity (Pizzorusso et al., 2002) and promotes the functional recovery of nervous system injury (Bradbury et al., 2002). The degradation of PNNs and CSPGs with ChABC reenabled neuronal plasticity and traumatic memories that are susceptible to erasure by extinction (Gogolla et al., 2009). Our findings are consistent with these studies showing that the pharmacological induction of plasticity with ChABC may act synergistically with extinction training and erase the original drug memory trace. In addition, previous studies showed that some other components of the extracellular matrix play roles in drug-seeking behavior (Nagai et al., 2004; Brown et al., 2007; Van den Oever et al., 2010; Wiggins et al., 2011). Intracerebroventricular or intra-mPFC injections of a metalloproteinase inhibitor disrupted the acquisition and reconsolidation of cocaine-induced CPP (Brown et al., 2007) and cue-induced heroin-seeking behavior (Van den Oever et al., 2010; Wiggins et al., 2011). Our results appear to be inconsistent with these findings. We found that PNNs did not participate in the learning, retrieval, or reconsolidation of drug memories, suggesting that different components of the extracellular matrix may play differential roles in drug-associated behaviors.
Interestingly, Gogolla et al. (2009) found that pretraining, but not posttraining, destruction of amygdala PNNs prevented the recovery of fear memories after extinction, whereas we observed the effect of PNNs removal after memory training in the present study. We speculate that this may be attributable to specific neuroplastic changes in the extracellular matrix during the formation of drug-associated memories. Indeed, different neural mechanisms may be involved in natural reward memory and drug memory (Wang et al., 2010; Young et al., 2014). Consistent with this, we found that PNNs removal combined with extinction training did not erase food-induced CPP memory. The differences in other conditions, such as the training sessions and extinction sessions (i.e., one session vs multiple sessions) may have also contributed to this discrepancy. In addition, in this study, we found that the degradation of PNNs in two subregions of the amygdala, the BLA and CeA, had the same effect on drug memories. This seems to be incompatible with some studies reporting that the BLA, but not the CeA, is critical for the consolidation, retrieval, reconsolidation, and storage of drug memories (Milton et al., 2008; Li et al., 2010; He et al., 2011). This discrepancy may be attributable to a specific memory process (i.e., memory extinction) that we focused on in this study. Our findings are consistent with other studies finding that the CeA is involved in extinction processes of aversive and appetitive memories (Paré et al., 2004; Amano et al., 2010; Duvarci et al., 2011; Cai et al., 2013). Although the observed effect of PNNs removal was amygdala dependent, we cannot conclusively state that the BLA and CeA play an exclusive role in drug reward memory. Other factors may also be relevant, such as minor drug diffusion into the BLA after CeA injections and possible interactions between the BLA and CeA, in which PNNs removal in the CeA may affect the function of the BLA. Although our molecular data in the BLA demonstrated that several plasticity-related proteins may mediate the effect of PNNs removal on the erasure of drug reward memories, the precise mechanism is still unknown. More research is needed to explore the common and different molecular mechanisms that underlie the effects of PNNs removal in the BLA and CeA on drug memories and the interaction between these two subregions.
In summary, we found that PNNs degradation in the amygdala combined with extinction training erased drug reward memory. The present results corroborate the growing evidence that ChABC is a powerful pharmacological iPlastic agent that modulates neuroplasticity in the adult CNS (Castrén et al., 2012). This increased efficiency of extinction training on the prevention of relapse to drug use makes ChABC an attractive candidate as a novel pharmacological adjunct that may improve existing treatments for drug abuse and relapse.
Footnotes
This work was supported in part by the Natural Science Foundation of China (Grants 81271489, 31230033, 31300930, and 91132716) and the Specialized Research Fund for the Doctoral Program of Higher Education (Grant 20130001120135). We thank Yavin Shaham for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Amano T, Unal CT, Paré D. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci. 2010;13:489–494. doi: 10.1038/nn.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Belin D, Epstein D, Calu D, Shaham Y. Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci. 2011;12:685–700. doi: 10.1038/nrn3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barritt AW, Davies M, Marchand F, Hartley R, Grist J, Yip P, McMahon SB, Bradbury EJ. Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. J Neurosci. 2006;26:10856–10867. doi: 10.1523/JNEUROSCI.2980-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavelier D, Levi DM, Li RW, Dan Y, Hensch TK. Removing brakes on adult brain plasticity: from molecular to behavioral interventions. J Neurosci. 2010;30:14964–14971. doi: 10.1523/JNEUROSCI.4812-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME. Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull. 1993;114:80–99. doi: 10.1037/0033-2909.114.1.80. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Briand LA, Lee FS, Blendy JA, Pierce RC. Enhanced extinction of cocaine seeking in brain-derived neurotrophic factor Val66Met knock-in mice. Eur J Neurosci. 2012;35:932–939. doi: 10.1111/j.1460-9568.2012.08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TE, Forquer MR, Cocking DL, Jansen HT, Harding JW, Sorg BA. Role of matrix metalloproteinases in the acquisition and reconsolidation of cocaine-induced conditioned place preference. Learn Mem. 2007;14:214–223. doi: 10.1101/lm.476207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner G, Bringmann A, Härtig W, Köppe G, Delpech B, Brauer K. Acute and long-lasting changes in extracellular-matrix chondroitin-sulphate proteoglycans induced by injection of chondroitinase ABC in the adult rat brain. Exp Brain Res. 1998;121:300–310. doi: 10.1007/s002210050463. [DOI] [PubMed] [Google Scholar]
- Cai YQ, Wang W, Hou YY, Zhang Z, Xie J, Pan ZZ. Central amygdala GluA1 facilitates associative learning of opioid reward. J Neurosci. 2013;33:1577–1588. doi: 10.1523/JNEUROSCI.1749-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry. 2013;70:983–989. doi: 10.1001/jamapsychiatry.2013.1. [DOI] [PubMed] [Google Scholar]
- Castrén E, Elgersma Y, Maffei L, Hagerman R. Treatment of neurodevelopmental disorders in adulthood. J Neurosci. 2012;32:14074–14079. doi: 10.1523/JNEUROSCI.3287-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio MR. Perineuronal nets of extracellular matrix around parvalbumin-containing neurons of the hippocampus. Hippocampus. 1993;3:55–60. [PubMed] [Google Scholar]
- Celio MR, Blümcke I. Perineuronal nets–a specialized form of extracellular matrix in the adult nervous system. Brain Res Brain Res Rev. 1994;19:128–145. doi: 10.1016/0165-0173(94)90006-X. [DOI] [PubMed] [Google Scholar]
- Celio MR, Spreafico R, De Biasi S, Vitellaro-Zuccarello L. Perineuronal nets: past and present. Trends Neurosci. 1998;21:510–515. doi: 10.1016/S0166-2236(98)01298-3. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Wilkie ME. Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol. 2004;33:63–71. doi: 10.1016/j.alcohol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Debiec J, Díaz-Mataix L, Bush DE, Doyère V, Ledoux JE. The amygdala encodes specific sensory features of an aversive reinforcer. Nat Neurosci. 2010;13:536–537. doi: 10.1038/nn.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Stewart J. Drug reinstatement of heroin-reinforced responding in the rat. Psychopharmacology (Berl) 1983;79:29–31. doi: 10.1007/BF00433012. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Popa D, Paré D. Central amygdala activity during fear conditioning. J Neurosci. 2011;31:289–294. doi: 10.1523/JNEUROSCI.4985-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg M, Kobilo T, Berman DE, Dudai Y. Stability of retrieved memory: inverse correlation with trace dominance. Science. 2003;301:1102–1104. doi: 10.1126/science.1086881. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/S0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Glial cell extracellular matrix: boundaries for axon growth in development and regeneration. Cell Tissue Res. 1997;290:379–384. doi: 10.1007/s004410050944. [DOI] [PubMed] [Google Scholar]
- García-Alías G, Barkhuysen S, Buckle M, Fawcett JW. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat Neurosci. 2009;12:1145–1151. doi: 10.1038/nn.2377. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Lüthi A, Herry C. Perineuronal nets protect fear memories from erasure. Science. 2009;325:1258–1261. doi: 10.1126/science.1174146. [DOI] [PubMed] [Google Scholar]
- Gundelfinger ED, Frischknecht R, Choquet D, Heine M. Converting juvenile into adult plasticity: a role for the brain's extracellular matrix. Eur J Neurosci. 2010;31:2156–2165. doi: 10.1111/j.1460-9568.2010.07253.x. [DOI] [PubMed] [Google Scholar]
- Härtig W, Brauer K, Brückner G. Wisteria floribunda agglutinin-labelled nets surround parvalbumin-containing neurons. Neuroreport. 1992;3:869–872. doi: 10.1097/00001756-199210000-00012. [DOI] [PubMed] [Google Scholar]
- He YY, Xue YX, Wang JS, Fang Q, Liu JF, Xue LF, Lu L. PKMzeta maintains drug reward and aversion memory in the basolateral amygdala and extinction memory in the infralimbic cortex. Neuropsychopharmacology. 2011;36:1972–1981. doi: 10.1038/npp.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Lüthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- Jain A, McKeon RJ, Brady-Kalnay SM, Bellamkonda RV. Sustained delivery of activated Rho GTPases and BDNF promotes axon growth in CSPG-rich regions following spinal cord injury. PLoS One. 2011;6:e16135. doi: 10.1371/journal.pone.0016135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci. 2010;30:7984–7992. doi: 10.1523/JNEUROSCI.1244-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JC, Afshari F, García-Alías G, Fawcett JW. Proteoglycans in the central nervous system: plasticity, regeneration and their stimulation with chondroitinase ABC. Restor Neurol Neurosci. 2008;26:131–145. [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- Leri F, Flores J, Rodaros D, Stewart J. Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci. 2002;22:5713–5718. doi: 10.1523/JNEUROSCI.22-13-05713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Xue YX, Wang JS, Fang Q, Li YQ, Zhu WL, He YY, Liu JF, Xue LF, Shaham Y, Lu L. Basolateral amygdala cdk5 activity mediates consolidation and reconsolidation of memories for cocaine cues. J Neurosci. 2010;30:10351–10359. doi: 10.1523/JNEUROSCI.2112-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QQ, Luo YX, Sun CY, Xue YX, Zhu WL, Shi HS, Zhai HF, Shi J, Lu L. A morphine/heroin vaccine with new hapten design attenuates behavioral effects in rats. J Neurochem. 2011a;119:1271–1281. doi: 10.1111/j.1471-4159.2011.07502.x. [DOI] [PubMed] [Google Scholar]
- Li YQ, Xue YX, He YY, Li FQ, Xue LF, Xu CM, Sacktor TC, Shaham Y, Lu L. Inhibition of PKMzeta in nucleus accumbens core abolishes long-term drug reward memory. J Neurosci. 2011b;31:5436–5446. doi: 10.1523/JNEUROSCI.5884-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Dempsey J, Shaham Y. Cocaine seeking over extended withdrawal periods in rats: different time courses of responding induced by cocaine cues versus cocaine priming over the first 6 months. Psychopharmacology. 2004;176:101–108. doi: 10.1007/s00213-004-1860-4. [DOI] [PubMed] [Google Scholar]
- Lu L, Wang X, Wu P, Xu C, Zhao M, Morales M, Harvey BK, Hoffer BJ, Shaham Y. Role of ventral tegmental area glial cell line-derived neurotrophic factor in incubation of cocaine craving. Biol Psychiatry. 2009;66:137–145. doi: 10.1016/j.biopsych.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier AL, Caruncho HJ, Kalynchuk LE. Repeated exposure to corticosterone, but not restraint, decreases the number of reelin-positive cells in the adult rat hippocampus. Neurosci Lett. 2009;460:170–174. doi: 10.1016/j.neulet.2009.05.050. [DOI] [PubMed] [Google Scholar]
- Malvaez M, Sanchis-Segura C, Vo D, Lattal KM, Wood MA. Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol Psychiatry. 2010;67:36–43. doi: 10.1016/j.biopsych.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlatt GA. Cue exposure and relapse prevention in the treatment of addictive behaviors. Addict Behav. 1990;15:395–399. doi: 10.1016/0306-4603(90)90048-3. [DOI] [PubMed] [Google Scholar]
- Millan EZ, Marchant NJ, McNally GP. Extinction of drug seeking. Behav Brain Res. 2011;217:454–462. doi: 10.1016/j.bbr.2010.10.037. [DOI] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ. The persistence of maladaptive memory: addiction, drug memories and anti-relapse treatments. Neurosci Biobehav Rev. 2012;36:1119–1139. doi: 10.1016/j.neubiorev.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Milton AL, Lee JL, Butler VJ, Gardner R, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. J Neurosci. 2008;28:8230–8237. doi: 10.1523/JNEUROSCI.1723-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Yoshimura M, Ishikawa K, Miyamoto Y, Hashimoto K, Noda Y, Nitta A, Nabeshima T. The tissue plasminogen activator-plasmin system participates in the rewarding effect of morphine by regulating dopamine release. Proc Natl Acad Sci U S A. 2004;101:3650–3655. doi: 10.1073/pnas.0306587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon K, Crews FT. Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J Neurosci. 2004;24:9714–9722. doi: 10.1523/JNEUROSCI.3063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CP, Childress AR, McLellan AT, Ehrman R. Developing treatments that address classical conditioning. NIDA Res Monogr. 1993;135:71–91. [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Parsons RG, Ressler KJ. Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci. 2013;16:146–153. doi: 10.1038/nn.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Amsterdam: Elsevier Academic; 2005. [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren ZY, Liu MM, Xue YX, Ding ZB, Xue LF, Zhai SD, Lu L. A critical role for protein degradation in the nucleus accumbens core in cocaine reward memory. Neuropsychopharmacology. 2013;38:778–790. doi: 10.1038/npp.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- Shi HS, Zhu WL, Liu JF, Luo YX, Si JJ, Wang SJ, Xue YX, Ding ZB, Shi J, Lu L. PI3K/Akt signaling pathway in the basolateral amygdala mediates the rapid antidepressant-like effects of trefoil factor 3. Neuropsychopharmacology. 2012;37:2671–2683. doi: 10.1038/npp.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D, Karanian DA, Monteggia LM, Neve RL, Self DW. Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature. 2003;421:70–75. doi: 10.1038/nature01249. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobeña A, Fernández-Teruel A, Escorihuela RM, Núñez JF, Zapata A, Ferré P, Sánchez R. Limits of habituation and extinction: implications for relapse prevention programs in addictions. Drug Alcohol Depend. 1993;32:209–217. doi: 10.1016/0376-8716(93)90085-5. [DOI] [PubMed] [Google Scholar]
- Torregrossa MM, Sanchez H, Taylor JR. D-cycloserine reduces the context specificity of Pavlovian extinction of cocaine cues through actions in the nucleus accumbens. J Neurosci. 2010;30:10526–10533. doi: 10.1523/JNEUROSCI.2523-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Oever MC, Lubbers BR, Goriounova NA, Li KW, Van der Schors RC, Loos M, Riga D, Wiskerke J, Binnekade R, Stegeman M, Schoffelmeer AN, Mansvelder HD, Smit AB, De Vries TJ, Spijker S. Extracellular matrix plasticity and GABAergic inhibition of prefrontal cortex pyramidal cells facilitates relapse to heroin seeking. Neuropsychopharmacology. 2010;35:2120–2133. doi: 10.1038/npp.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WS, Kang S, Liu WT, Li M, Liu Y, Yu C, Chen J, Chi ZQ, He L, Liu JG. Extinction of aversive memories associated with morphine withdrawal requires ERK-mediated epigenetic regulation of brain-derived neurotrophic factor transcription in the rat ventromedial prefrontal cortex. J Neurosci. 2012;32:13763–13775. doi: 10.1523/JNEUROSCI.1991-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XY, Zhao M, Ghitza UE, Li YQ, Lu L. Stress impairs reconsolidation of drug memory via glucocorticoid receptors in the basolateral amygdala. J Neurosci. 2008;28:5602–5610. doi: 10.1523/JNEUROSCI.0750-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Luo YX, He YY, Li FQ, Shi HS, Xue LF, Xue YX, Lu L. Nucleus accumbens core mammalian target of rapamycin signaling pathway is critical for cue-induced reinstatement of cocaine seeking in rats. J Neurosci. 2010;30:12632–12641. doi: 10.1523/JNEUROSCI.1264-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins A, Smith RJ, Shen HW, Kalivas PW. Integrins modulate relapse to cocaine-seeking. J Neurosci. 2011;31:16177–16184. doi: 10.1523/JNEUROSCI.3816-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu CM, Wang J, Wu P, Zhu WL, Li QQ, Xue YX, Zhai HF, Shi J, Lu L. Glycogen synthase kinase 3beta in the nucleus accumbens core mediates cocaine-induced behavioral sensitization. J Neurochem. 2009;111:1357–1368. doi: 10.1111/j.1471-4159.2009.06414.x. [DOI] [PubMed] [Google Scholar]
- Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, Zhu WL, Ding ZB, Bao YP, Shi J, Epstein DH, Shaham Y, Lu L. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science. 2012;336:241–245. doi: 10.1126/science.1215070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EJ, Aceti M, Griggs EM, Fuchs RA, Zigmond Z, Rumbaugh G, Miller CA. Selective, retrieval-independent disruption of methamphetamine-associated memory by actin depolymerization. Biol Psychiatry. 2014;75:96–104. doi: 10.1016/j.biopsych.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]