

Abstract
The three-layered piriform cortex, an integral part of the olfactory system, processes odor information relayed by olfactory bulb mitral cells. Specifically, mitral cell axons form the lateral olfactory tract (LOT) by targeting lateral olfactory tract (lot) guidepost cells in the piriform cortex. While lot cells and other piriform cortical neurons share a pallial origin, the factors that specify their precise phenotypes are poorly understood. Here we show that in mouse, the proneural genes Neurog1 and Neurog2 are coexpressed in the ventral pallium, a progenitor pool that first gives rise to Cajal-Retzius (CR) cells, which populate layer I of all cortical domains, and later to layer II/III neurons of the piriform cortex. Using loss-of-function and gain-of-function approaches, we find that Neurog1 has a unique early role in reducing CR cell neurogenesis by tempering Neurog2's proneural activity. In addition, Neurog1 and Neurog2 have redundant functions in the ventral pallium, acting in two phases to first specify a CR cell fate and later to specify layer II/III piriform cortex neuronal identities. In the early phase, Neurog1 and Neurog2 are also required for lot cell differentiation, which we reveal are a subset of CR neurons, the loss of which prevents mitral cell axon innervation and LOT formation. Consequently, mutation of Trp73, a CR-specific cortical gene, results in lot cell and LOT axon displacement. Neurog1 and Neurog2 thus have unique and redundant functions in the piriform cortex, controlling the timing of differentiation of early-born CR/lot cells and specifying the identities of later-born layer II/III neurons.
Keywords: Cajal-Retzius neurons, lateral olfactory tract guidepost cells, Neurog1 and Neurog2, piriform cortex, proneural genes, ventral pallium
Introduction
The cerebral cortex, which includes the archicortex, neocortex, and piriform cortex, is derived from the dorsal telencephalon, or pallium. Based on gene expression, the pallium is subdivided into medial, dorsal, lateral, and ventral domains, each giving rise to distinct cortical territories (Puelles et al., 2000; Yun et al., 2001). Cajal-Retzius (CR) cells are an early-born cortical lineage, differentiating between embryonic day (E) 10.5 and E12.5 in mouse (Smart and Smart, 1977; Wood et al., 1992; Marin-Padilla, 1998; Supèr et al., 1998). CR cells arise from three sites in the pallial margins: (1) medially, the cortical hem/choroid plexus; (2) rostrally, the pallial septum/rostrobulbar area; and (3) laterally, the ventral pallium (Takiguchi-Hayashi et al., 2004; Bielle et al., 2005; Yoshida et al., 2006; Zhao et al., 2006; García-Moreno et al., 2007; Imayoshi et al., 2008). Given their pallial origins, most CR cells are labeled in an Emx1-lineage trace (Gorski et al., 2002) and express Tbr1 (Hevner et al., 2003), both cortical-specific transcription factors. Many CR cells also express Reelin (Alcántara et al., 1998), a secreted glycoprotein that guides radial migration of neocortical neurons (Caviness, 1982; Howell et al., 1997).
While neocortical development has been well studied, much less is known about the three-layered piriform cortex. The piriform cortex is a central component of the olfactory system, which together with the olfactory epithelium (OE) and olfactory bulb (OB) is responsible for detecting and processing odors. Superficial layer II and deep layer III of the piriform cortex contain glutamatergic projection neurons and interspersed GABAergic interneurons (Sarma et al., 2011), while layer I is a cell-sparse zone containing CR neurons and lateral olfactory tract (lot) guidepost cells. Lot cells guide innervation of the anterior piriform cortex by OB mitral cells, the axons of which form the LOT (note that lot guidepost cells are distinguished from LOT axons by small and capital letters, respectively; Sato et al., 1998). We noted several striking similarities between lot cells (Sato et al., 1998; Tomioka et al., 2000) and CR neurons (Wood et al., 1992; Hevner et al., 2003; Takiguchi-Hayashi et al., 2004): both act as cellular guideposts for axonal tract formation, have a pallial origin, are among the earliest born cortical neurons (lot cells also differentiate between E9.5 and E11), and migrate tangentially from their pallial sites of origin to populate the piriform cortex (Sato et al., 1998; Tomioka et al., 2000). However, while CR cell differentiation is well understood, the factors that specify a lot cell identity, and more globally control neuronal fate specification in the piriform cortex, remain poorly characterized.
Here we demonstrate that the proneural basic helix-loop-helix (bHLH) transcription factors Neurog1 and Neurog2, which specify a neocortical projection neuron identity (Fode et al., 2000; Schuurmans et al., 2004), also specify piriform cortical neuronal identities. Specifically, Neurog1 and Neurog2 are required in two differentiation waves—first acting in opposition to control lot cell differentiation, which we reveal are a subpopulation of CR neurons, the localization of which depends on Trp73, and later controlling the differentiation of layer II/III piriform cortical neurons.
Materials and Methods
Animals and genotyping.
Animal procedures followed guidelines of the Canadian Council of Animal Care and were approved by the University of Calgary Animal Care Committee (Protocol AC11-0053), the Institutional Animal Care and Use Committee of State University of New York Upstate Medical University, and the Hospital for Sick Children Animal Care Committee. Embryos of either sex were used throughout. Neurog1 (Ma et al., 1998) and Neurog2GFPKI (Britz et al., 2006) mutant alleles were maintained on a CD1 background (Charles River) and genotyped as described (Ma et al., 1998; Britz et al., 2006). Lef/Tcf-lacZ transgenics were provided by Valerie Wallace and Daniel Dufort (Mohamed et al., 2004) and genotyped using forward (CCATCCAGAGACAAGCGAAGAC) and reverse (TTGAGGGGACGACGACAGT ATC) primers (35 cycles of 95°C/1′, 58°C/1′, 72°C/1.5′, then final extension 72°C/10′). TAp73 mutants were genotyped using the following primers: TAp73WT:CTGGTCCAGGAGGTGAGACTGAGGC; TAp73 Common: CTGGCCCTCTCAGCTTGTGCCACTTC, and TAp73Neo: GTGGGGGTGGGATTAGATAAATGCCTG (TAp73WT and TAp73 Common for wild-type allele, 1 kb; and TAp73Neo and TAp73 Common for mutant allele, 1.2 kb; 35 cycles of 94°C/45 s, 65°C/30 s, 72°C/1.5 min). Trp73 (p73) mutants were genotyped using three primer PCRs: p73-1:GGGCCATGCCTGTCTACAAGAA; p73-2: CCTTCTACACGGATGAGGTG; p73-3:GAAAGCGAAGGAGCAAAGCTG (wild type, 550 bp; mutant, 400 bp; 40 cycles of 94°C/30 s, 64°C/30 s, 72°C/40 s). Reeler mutants (B6C3Fe a/a-Relnrl/+) were obtained from Jackson Laboratories and genotyped as described previously (D'Arcangelo et al., 1996).
Tissue processing, histology, and β-galactosidase histochemical staining.
For histology, whole E18.5 heads were fixed in Bouin's solution, embedded in paraffin, and cut into 7 μm sections, which were stained with hematoxylin and eosin as previously described (Fode et al., 1998). For all other tissue studies, embryos were fixed overnight in 4% paraformaldehyde (PFA) in 1× PBS, pH 7.4, at 4°C, washed in 1× PBS, and cryoprotected in 20% sucrose in 1× PBS overnight at 4°C before embedding and freezing in Tissue Tek OCT (Sakura Finetek). Ten-micrometer cryosections were collected on Superfrost plus slides (Fisher Scientific). X-gal histochemical staining was performed on cryostat sections as follows. Sections were first washed 3 × 10 min in lacZ wash buffer (2 mm MgCl2/0.01% sodium deoxycholate/0.02% Nonidet-P40/100 mm sodium phosphate, pH 7.3) before staining overnight at 37°C in wash buffer containing 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, and 0.67 mg/ml X-gal (Invitrogen). Slides were then washed in PBS, rinsed in water, and allowed to dry before mounting in Permount (Fisher Scientific).
RNA in situ hybridization.
RNA in situ hybridization was performed as previously described (Alam et al., 2005) using digoxygenin-labeled riboprobes that were generated using a 10× labeling mix according to the manufacturer's instructions (Roche Diagnostics). Riboprobes were generated from linearized plasmid templates as follows: Reln (EcoRI/T3), Trp73 (SalI/T3), Etv1 (SpeI/T7), Ctip2 (IMAGE 4457123; SalI/T7), Id2 (HindIII/T3), Dlx1 (XbaI/T3), Wnt7b (NcoI/SP6), mGluR1 (IMAGE 30536724; EcoRI/T3), and Dbx1 (IMAGE 5718470; EcoRI/T3).
Immunostaining and imaging.
Immunostaining was performed on 10 μm cryostat sections that were processed and collected as described above. Cryosections were blocked either in 10% normal goat or donkey serum in 0.1% Triton X-100 in 1 × PBS or in 1 × Tris-buffered saline (25 mm Tris-HCl, pH 7.4, 0.14 m NaCl). Primary antibodies included: rabbit anti-calretinin (1:500; Swant), mouse anti-Ascl1 (1:200; BD Biosciences), mouse anti-Neurog2 (1:20; gift from David Anderson), rabbit anti-Neurog2 (1:500; gift from Masato Nakafuku), rabbit anti-Neurog1 (1:500; gift from Jane Johnson), rabbit anti-GFP (1:500; Millipore Bioscience Research Reagents), sheep anti-GFP (1:750; Biogenesis), rabbit anti-Tbr1 (1:3000, Millipore Bioscience Research Reagents), mouse anti-Reelin (1:500; Millipore Bioscience Research Reagents), rabbit anti-activated caspase 3 (1:100; Promega), mouse anti-MAP2 (1:500; Sigma-Aldrich), rabbit anti-Pax6 (1:500; Covance), rabbit-anti-Trp73 (1:500; Bethyl Laboratories), and rat anti-lot1 (1:200; gift from Tatsumi Hirata). Species-specific secondary antibodies were conjugated to Alexa488 (1:500; Invitrogen), Cy3 (1:500; Jackson Immunoresearch), or horseradish peroxidase (HRP). Sections were counterstained with DAPI (4,6-diamidino-2-phenylindole, 1:10,000; Sigma-Aldrich) and mounted in AquaPolymount (Polysciences). DAB staining of HRP-conjugated antibodies was performed using the Vectastain ABC kit according to the manufacturer's instructions (Vector Laboratories).
DiI tracing.
E18.5 brains were fixed for 2 d in 4% PFA in 1× PBS at 4°C. Carbocyanin DiI crystals (Invitrogen) were introduced into the OB, and the brains were incubated at 37°C in 4% PFA in 1× PBS to allow dye diffusion for 2–3 weeks, followed by imaging.
In vitro electroporation and quantitation.
The pCIG2-Neurog2 expression vector was previously described (Mattar et al., 2008). The Neurog1 cDNA was similarly PCR amplified and subcloned into pCIG2. Ex vivo electroporation and culture of E10.5 embryos were performed as previously described (Zimmer et al., 2010). Cell counts were performed on ≥3 independent embryos and on three sections per embryo. Error bars reflect SEM. Student's t tests were performed with p values denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.005.
Results
Neurog1 and Neurog2 are coexpressed in ventral pallial progenitors and derivative lineages in both the neocortex and piriform cortex
Neocortical progenitors undergo temporal identity transitions (Pearson and Doe, 2004), first giving rise to CR neurons, then sequentially generating glutamatergic pyramidal neurons in neocortical layers VI, V, IV, and finally II/III (fused in mouse; Takahashi et al., 1999). Neurog2 functions iteratively in this process, first promoting the differentiation of CR neurons (Imayoshi et al., 2008) and then functioning with Neurog1, a related proneural gene, to specify the glutamatergic identities of layer V/VI neurons (Fode et al., 2000; Schuurmans et al., 2004). Currently, it is not known whether Neurog1 also functions in CR development, nor is it known whether Neurog1 and Neurog2 specify neuronal identities in the three-layered piriform cortex, which is an evolutionarily more ancient structure. We addressed these two questions herein.
The piriform cortex is derived from the ventral pallium (Puelles et al., 2000; Hirata et al., 2002), which also serves as one of three CR cell progenitor domains (in addition to the pallial septum and cortical hem; Fig. 1A). Thus, first asking whether Neurog1 and Neurog2 were expressed in ventral pallial progenitors, we examined cortices between E10.5 and E12.5, the period when CR cells (Wood et al., 1992; Hevner et al., 2003; Takiguchi-Hayashi et al., 2004) and layer III piriform cortex neurons arise (Hirata et al., 2002). At E10.5 (data not shown) and E11.5, Neurog1 and Neurog2 transcripts (Fig. 1B,C) and protein (data not shown) were detected in scattered pallial progenitors in a high-lateral to low-medial gradient, with elevated expression levels particularly evident in the ventral pallium, cortical hem, and pallial septum.
Figure 1.
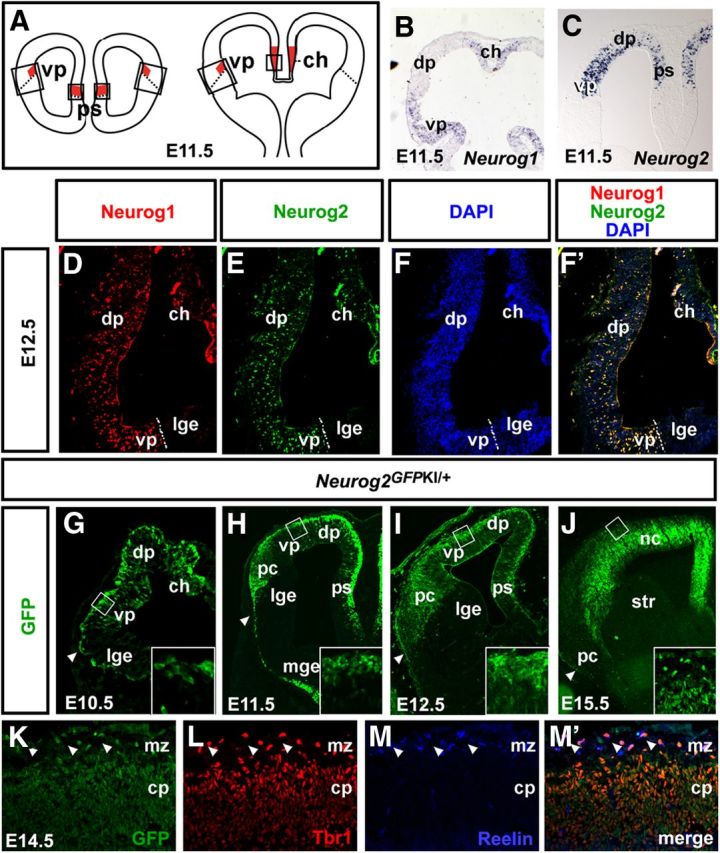
Neurog1 and Neurog2 are coexpressed in CR progenitors. A, Schematic representation of the three main sites of CR cell production. B, C, Distribution of Neurog1 (B) and Neurog2 (C) transcripts in the E11.5 telencephalon. D–F, F', Expression of Neurog1 (D, F', red), Neurog2 (E, F', green), and DAPI counterstain (F, F', blue) in E12.5 telencephalon. The dotted lines mark the pallial–subpallial border. G–J, Expression of GFP in E10.5 (G), E11.5 (H), E12.5 (I), and E15.5 (J) Neurog2GFPKI/+ cortices. Insets are high-magnification images of the boxed areas in the preplate (E10.5–E12.5) and marginal zone (E15.5). Arrowhead in mark the pallial-subpallial border. K–M, M', Coexpression of GFP (K, M', green), Tbr1 (L, M', red), and Reelin (M, M') in E15.5 Neurog2GFPKI/+ neocortex. Arrowheads mark CR neurons coexpressing all three markers in the marginal zone. ch, Cortical hem; cp, cortical plate; dp, dorsal pallium; lge, lateral ganglionic eminence; mge, medial ganglionic eminence; mz, marginal zone; nc, neocortex; ps, pallial septum; str, striatum; vp, ventral pallium.
By E12.5, Neurog1 and Neurog2 were more broadly expressed in scattered progenitors throughout the pallial ventricular zone (VZ), including in the three sites of CR cell production (Fig. 1D–F'; data not shown). Quantification of proneural expression in E12.5 CR progenitor domains revealed that Neurog1 (ventral pallium; 11.3 ± 1.1% of DAPI+ cells, n = 3, 1372 cells; cortical hem: 6.9 ± 0.2% of DAPI+ cells; n = 3, 2980 cells) was expressed in fewer CR progenitors than Neurog2 (ventral pallium; 24.4 ± 6.1% of DAPI+ cells, n = 3, 1372 cells; cortical hem: 11.6 ± 0.5% of DAPI+ cells; n = 3, 2980 cells). Nevertheless, the vast majority of Neurog1+ progenitors coexpressed Neurog2 (ventral pallium: 97.8 ± 1.5%, n = 3, 1372 cells; cortical hem: 89.1 ± 4.3%, n = 3, 2980 cells), indicating that these proneural genes are coexpressed in a common lineage.
Given that Neurog2+ pallial progenitors largely encompass the Neurog1+ population, we performed short-term lineage tracing in Neurog2GFPKI/+ cortices to capture both lineages, taking advantage of GFP perdurance in derivative neurons. In E10.5–E15.5 Neurog2GFPKI/+ cortices, GFP+ cells were detected in the neocortical and piriform cortical marginal zones (Fig. 1G–J), where CR cells reside. GFP expression was also detected in the germinal zones and mantle layers of the neocortex and piriform cortex (Fig. 1G–J,K,M'). Within the E14.5 Neurog2GFPKI/+ marginal zone, the vast majority of GFP+ cells coexpressed Reelin, a CR cell marker (Alcántara et al., 1998), and Tbr1, a cortical-specific T-box protein (Hevner et al., 2003; Fig. 1K–M'). This is consistent with the pallial identity of CR neurons (Hevner et al., 2003) and with our previous short-term lineage-tracing experiments in E11.5 Neurog2GFPKI/+ neocortices (Dixit et al., 2011b).
Thus, Neurog2+ pallial progenitors, which largely encompass the Neurog1+ pallial progenitor pool, give rise to CR cells that populate the marginal zones of both the piriform cortex and neocortex as well as the mantle zones of both structures.
Neurog2 single and Neurog1 and Neurog2 double mutants display early and spatially distinct deficiencies in CR cells
It was previously reported that fewer CR cells are generated in E12.5 and E14.5 Neurog2−/− neocortices (Imayoshi et al., 2008). However, this study did not address Neurog2 function in CR cells in the piriform cortex, nor did it address whether Neurog1 also contributes to CR cell development. We therefore compared CR cell numbers in Neurog1 and Neurog2 single mutants as well as Neurog1 and Neurog2 double mutants (hereafter Neurog1/2) at E12.5, when CR differentiation is mostly complete. We first analyzed the neocortical preplate for Reln expression. Compared with E12.5 wild-type neocortices (111.4 ± 7.0 Reln+ cells/field; n = 3; Fig. 2A,A',M), the number of Reln+ cells was reduced to a similar extent (1.3-fold and 1.4-fold, respectively) in Neurog2−/− (88.6 ± 5.0 Reln+ cells/field; n = 3; p < 0.05; Fig. 2C,C',M) and Neurog1−/−;Neurog2−/− (hereafter referred to as Neurog1/2−/−) neocortices (75.4 ± 5.9 Reln+ cells/field; n = 2; p < 0.01; Fig. 2D,D',M). In contrast, 1.3-fold more Reln+ cells were detected in the Neurog1−/− neocortical preplate (143.3 ± 6.0 Reln+ cells/field; n = 3; p < 0.01; Fig. 2B,B',M). Notably, the increase in Reln+ CR cells in Neurog1−/− neocortices is consistent with our previous demonstration that E10.5–E12.5 Neurog1−/− neocortices undergo precocious neurogenesis, although these ectopic neurons were not previously identified as CR cells (Fode et al., 2000; Mattar et al., 2004). Strikingly, precocious neurogenesis is also observed in Neurog1/2−/− neocortices (Fode et al., 2000; Mattar et al., 2004), yet these neurons do not acquire a CR cell fate (Fig. 2D,D',M). We interpret these results in the following way. While either Neurog1 or Neurog2 can specify a CR cell identity, Neurog1 has an added role, which is to slow down the rate of early cortical neurogenesis. Consistent with this interpretation, Neurog2−/− CR cell loss is restricted to dorsomedial domains (Fig. 2C), where Neurog1 expression is lost (Fode et al., 2000), such that this region is equivalent to a Neurog1/2 double mutant.
Figure 2.
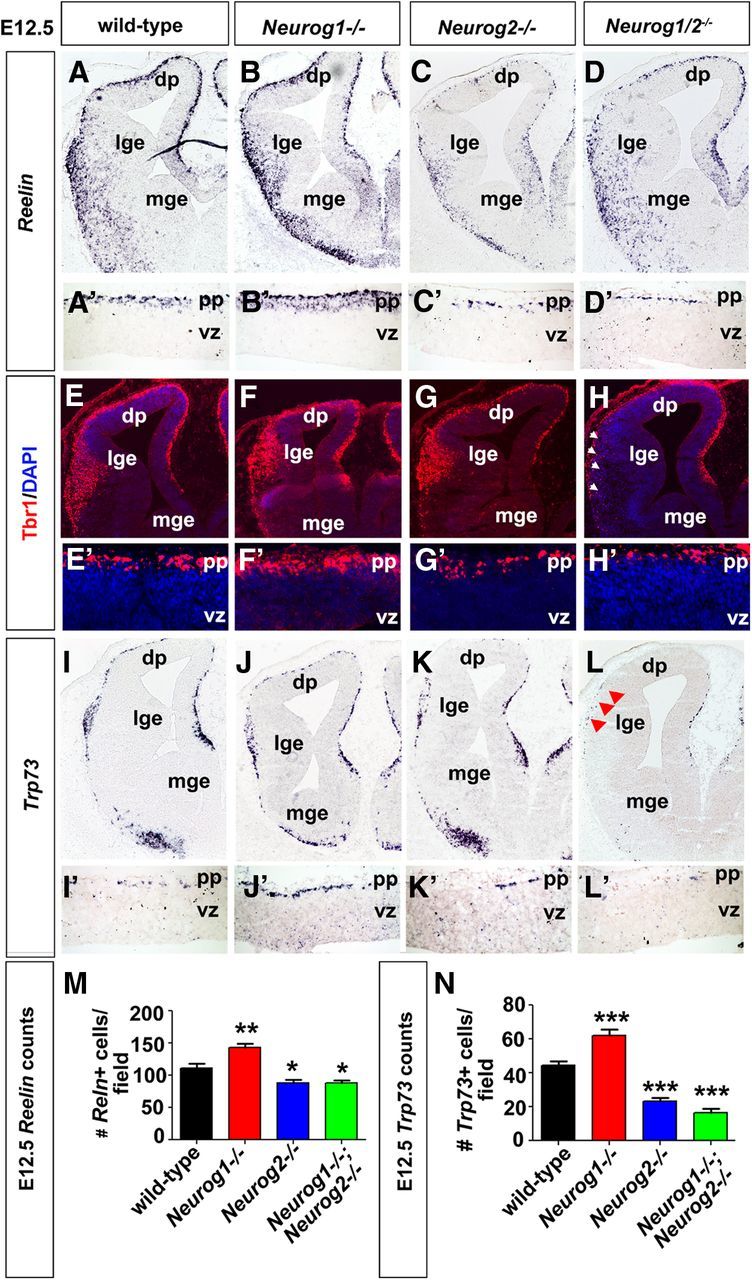
Reduced CR cell differentiation in E12.5 Neurog2−/− and Neurog1/2−/− neocortices and Neurog1/2−/− piriform cortex. A–D', Expression of Reelin in E12.5 wild-type (A, A'), Neurog1−/− (B, B'), Neurog2−/− (C, C'), and Neurog1/2−/− (D, D') cortices. A'–D' are high-magnification images of A–D in the neocortical preplate. E–H', Expression of Tbr1 in E12.5 wild-type (E, E'), Neurog1−/− (F, F'), Neurog2−/− (G, G'), and Neurog1/2−/− (H, H') cortices. E'–H' are high-magnification images of neocortical preplate in E–H. Arrowheads in H mark the loss of Tbr1+ CR cells in the presumptive piriform cortex. I–L', Expression of Trp73 in E12.5 wild-type (I, I'), Neurog1−/− (J, J'), Neurog2−/− (K, K'), and Neurog1/2−/− (L, L') cortices. I'–L' are high-magnification images of neocortical preplate in I–L. Arrowheads in L mark the loss of Trp73+ CR cells in the presumptive piriform cortex. M, N, Quantitation of the number of Reelin+ (M) and Trp73+ (N) cells in the neocortex of embryos for each genotype. Error bars in M and N represent SEM. Student's t tests were performed comparing all genotypes individually to wild type with p values denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.005. dp, Dorsal pallium; lge, lateral ganglionic eminence; mge, medial ganglionic eminence; pp, preplate; vz, ventricular zone.
To further substantiate the roles of Neurog1 and Neurog2 in CR cell genesis, we also analyzed Tbr1 expression, which labels CR neurons as well as deep-layer cortical neurons (Hevner et al., 2003). In E12.5 embryos of all genotypes, Tbr1 was expressed in the neocortical and piriform cortical preplates, albeit at higher levels in Neurog1−/− cortices, which produce more CR neurons, and at lower levels in Neurog2−/− and Neurog1/2−/− cortices, where CR cell numbers are reduced (Fig. 2E–H'). In addition, Tbr1 was expressed in a wedge of cells adjacent to the lateral ganglionic eminence (LGE), demarcating the nascent piriform cortex, in all genotypes, except Neurog1/2−/− double mutants (Fig. 2E–H). Similar results were obtained when monitoring Trp73 expression, which labels CR cells derived from the pallial septum and cortical hem (Meyer et al., 2002, 2004): 1.4-fold more Trp73+ cells in the Neurog1−/− preplate (62.2 ± 3.6 Trp73+ cells/field; n = 3; p < 0.0001; Fig. 2J,J',N) compared with wild type (44.5 ± 2.2 Trp73+ cells/field; n = 3; Fig. 2I,I',N). Conversely, Trp73+ cell numbers were reduced 1.9-fold and 2.7-fold in Neurog2−/− (23.2 ± 2.0 Trp73+ cells/field; n = 3; p < 0.0001; Fig. 2K,K',N) and Neurog1/2−/− (16.6 ± 2.1 Trp73+ cells/field; n = 2; p < 0.0001; Fig. 2L,L',N) neocortices, respectively. Moreover, in the nascent piriform cortex, where Trp73+ cells normally accumulate in a lateral wedge, a striking loss of Trp73+ CR cells was observed specifically within Neurog1/2−/− embryos (Fig. 2L).
Thus, CR cell differentiation in both the neocortex and piriform cortex requires either Neurog1 or Neurog2. In addition, Neurog1 has an additional role, which is to slow down the genesis of early-born cortical neurons, including CR cells, ensuring that appropriate numbers of these early-born neurons are generated.
Defects in CR cell differentiation persist in later stage Neurog2−/− and Neurog1/2−/− neocortices and piriform cortices
The presence of fewer CR cells in E12.5 Neurog2−/− and Neurog1/2−/− cortices could be due to a delay rather than a reduction in differentiation. To address this possibility, we examined Neurog1 and Neurog2 single and Neurog1/2 double mutants at E15.5, 3 d after CR cell differentiation is normally complete. At E15.5, Reln was expressed in a relatively continuous layer in the neocortical marginal zones of wild-type and Neurog1−/− neocortices, whereas distinct gaps in Reln expression were detected in Neurog2−/− and Neurog1/2−/− mutants (Fig. 3A–D'). Accordingly, quantitation of Reln+ CR cells in the marginal zone revealed 1.3-fold and 1.5-fold reductions, respectively, in Neurog2−/− (88.6 ± 3.6 Reln+ cells/field; n = 3; p = 0.0002) and Neurog1/2−/− (78.1 ± 3.8 Reln+ cells/field; n = 2; p < 0.0001) neocortical marginal zones compared with wild-type (116.2 ± 5.4 Reln+ cells/field; n = 3; Fig. 3I). Neurog1−/− CR numbers had normalized by E15.5 and were not significantly different than in wild-type neocortices (114.1 ± 5.8 Reln+ cells/field; n = 3; p = 0.79; Fig. 3I). A similar result was observed when analyzing Trp73+ CR cell numbers, which were reduced 3.4-fold and 3.8-fold, respectively, in Neurog2−/− (20.8 ± 1.3 Trp73+ cells/field; n = 3; p < 0.0001; Fig. 3G,G',J) and Neurog1/2−/− (18.8 ± 1.5 Trp73+ cells/field; n = 2; p < 0.0001; Fig. 3H,H',J) neocortical marginal zones compared with wild-type embryos (72.6 ± 3.3 Trp73+ cells/field; n = 3; Fig. 3E,E',J), while Neurog1−/− embryos had normal numbers of Trp73+ cells (63.1 ± 3.9 Trp73+ cells/field; n = 3; p = 0.07; Fig. 3F,F',J). Moreover, similar to observations made at E12.5, almost no CR cells were detected in the presumptive piriform cortex of Neurog1/2−/− embryos (Fig. 3D”,H”), whereas CR cells lined the marginal zone of the piriform cortex in all other genotypes (Fig. 3A”–C”,E”–G”).
Figure 3.

Defects in CR cell differentiation in the E15.5 Neurog2−/− neocortex and Neurog1/2−/− neocortex and piriform cortex. A–D”, Expression of Reelin in E15.5 wild-type (A–A”), Neurog1−/− (B–B”), Neurog2−/− (C–C”), and Neurog1/2−/− (D–D”) cortices. A'–D' are high-magnification images of boxed neocortical regions in A–D. A”–D” are high-magnification images of boxed piriform cortex regions in A–D. E–H”, Expression of Trp73 in E15.5 wild-type (E–E”), Neurog1−/− (F–F”), Neurog2−/− (G–G”), and Neurog1/2−/− (H–H”) cortices. E'–H' are high-magnification images of boxed neocortical regions in E–H. E”–H” are high-magnification images of boxed piriform cortex regions in E–H. I, J, Quantitation of the number of Reelin+ (I) and Trp73+ (J) cells per field in the marginal zone (i.e., CR cells). Error bars represent SEM. Student's t tests were performed comparing all genotypes individually to wild type with p values denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.005. mz, Marginal zone; nc, neocortex; pc, piriform cortex; str, striatum.
The reduction in CR cell number in Neurog2−/− and Neurog1/2−/− cortices thus persists at E15.5, and hence is unlikely to be due to a delay in CR cell genesis. Moreover, the original increase in CR cell number observed in E12.5 Neurog1−/− cortices is rectified by E15.5, indicating that Neurog1 is only required to delay cortical neurogenesis at early developmental stages.
Neurog2 is sufficient to induce CR cell differentiation while Neurog1 limits the formation of neocortical CR cells
The loss of CR cells in Neurog2−/− and Neurog1/2−/− cortices could arise if the medial signaling domains from which some CR cells arise are defective (Hanashima et al., 2007; Griveau et al., 2010; Zimmer et al., 2010), or if Neurog1 and Neurog2 function as instructive determinants of a CR cell fate. To distinguish between these possibilities, we first examined marker expression in the Fgf-rich pallial septum (Bielle et al., 2005; Zimmer et al., 2010), Wnt/Bmp-rich cortical hem (Takiguchi-Hayashi et al., 2004; Yoshida et al., 2006; García-Moreno et al., 2007; Imayoshi et al., 2008), and Egf-rich ventral pallium (Assimacopoulos et al., 2003). In E12.5 Neurog1 and Neurog2 single and Neurog1/2 double mutant cortices, we observed normal patterns of expression of Fgf8/17 in the pallial septum (data not shown), Wnt3a in the cortical hem (Fig. 4A–D; Imayoshi et al., 2008), and Dbx1 in the ventral pallium (Fig. 4E–H). CR cell progenitor domains are thus properly established in Neurog2−/− and Neurog1/2−/− cortices.
Figure 4.
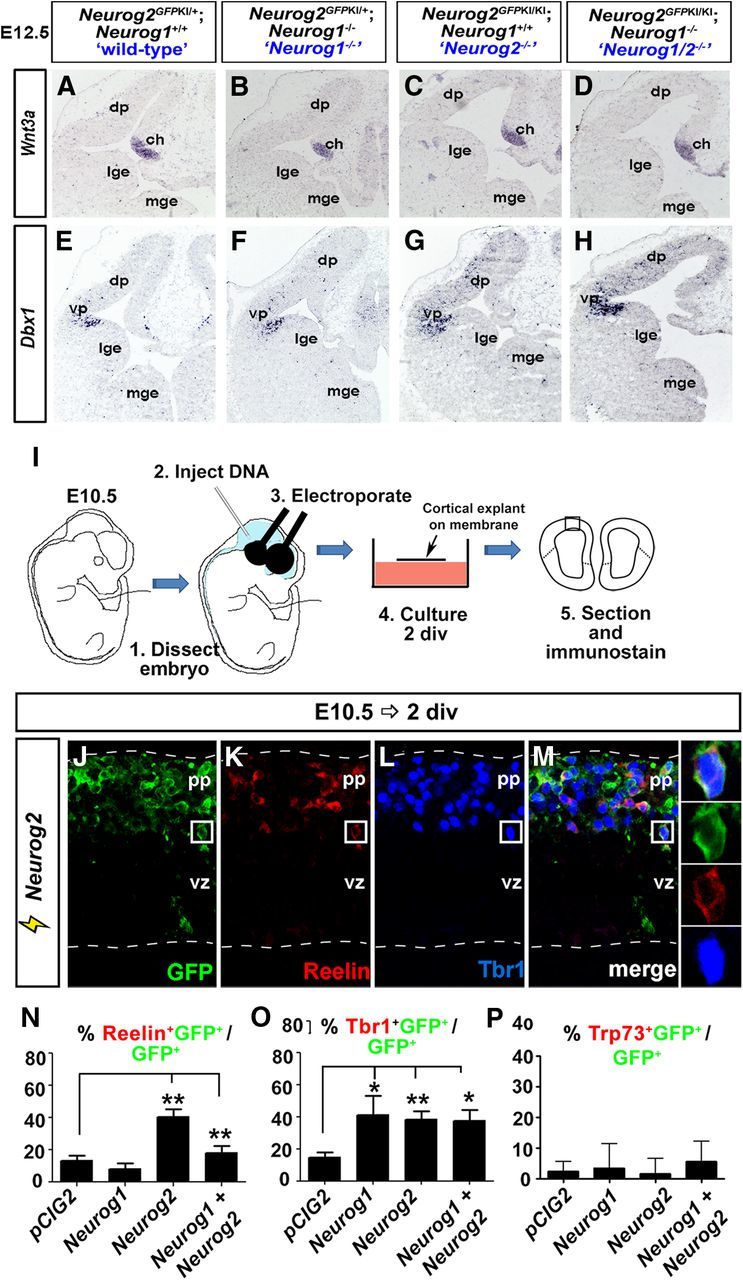
Neurog1 and Neurog2 are sufficient to induce the expression of a subset of CR markers. A–H, Expression of Wnt3a (A–D) and Dbx1 (E–H) in wild-type (A, E), Neurog1−/− (B, F), Neurog2−/− (C, G), and Neurog1/2−/− (D, H) E12.5 cortices. I, Schematic representation of in vitro electroporation protocol: expression constructs were injected into the E10.5 mouse telencephalon, followed by dissection of the dorsal telencephalon, embryo culture for 2 DIV, and analysis. J–M, Misexpression of Neurog2-IRES-GFP in E10.5 cortices, followed after 2 DIV by immunolabeling to visualize expression of GFP (J, M, green), Reelin (K, M, red), and Tbr1 (L, M, blue). Boxes to the right of M are high-magnification images of the boxed areas in J–M. N–P, Quantitation of the percentage of GFP+ electroporated cells expressing Reelin (N), Tbr1 (O), or Trp73 (P). Error bars represent SEM. Student's t tests were performed comparing all genotypes individually to wild type with p values denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.005. ch, Cortical hem; div, days in vitro; dp, dorsal pallium; lge, lateral ganglionic eminence; mge, medial ganglionic eminence; pp, preplate; vz, ventricular zone; vp, ventral pallium.
We next asked whether Neurog1 and Neurog2 could function as instructive determinants of a CR cell fate. Consistent with this idea, Neurog2 was previously shown to promote ectopic Reelin expression when misexpressed in the E9.5 choroid plexus (Imayoshi et al., 2008). Here we asked whether Neurog1 and Neurog2 were sufficient to promote some or all aspects of a CR cell identity when misexpressed in E10.5 pallial progenitors. Expression vectors for Neurog1 or Neurog2 and a pCIG2 vector control, all expressing GFP, were introduced into the E10.5 pallium via in utero electroporation (Dixit et al., 2011a). After 2 d of in vitro explant culture, cortices were harvested and assessed for the induction of CR marker expression (Fig. 4I–M). Consistent with the Imayoshi et al. (2008) study, Neurog2 was sufficient to induce ectopic Reelin expression in the E10.5 pallium (3.0-fold increase; control: 13.4 ± 2.9% Reelin+GFP+/GFP+ cells; n = 4; 567 cells; Neurog2: 40.5 ± 4.8% Reelin+GFP+/GFP+; n = 3; 1469 cells; p < 0.005; Fig. 4I–M,N). In contrast, Neurog1 did not induce ectopic Reelin expression and, moreover, Neurog1 inhibited the ability of Neurog2 to induce a CR fate when the two genes were coelectroporated (2.3-fold decrease; Neurog1: 8.1 ± 3.7% Reelin+ GFP+/GFP+ cells; n = 3; 79 cells; Neurog1 plus Neurog2: 18.0 ± 4.6% Reelin+GFP+/GFP+; n = 3; 757 cells; Fig. 4N). In contrast, neither Neurog1 nor Neurog2 were sufficient to induce the expression of Trp73 (Fig. 4P), even though loss-of-function studies indicate that these proneural genes are required for the generation of Trp73+ CR neurons (Figs. 2, 3).
The inability of Neurog1 to promote a CR cell fate is not at first glance consistent with our loss-of-function studies, which suggest that Neurog1 can promote a CR cell fate in the absence of Neurog2. One possibility is that Neurog1 can induce some aspects of a CR cell fate, such as a cortical-specific neuronal identity, and not subtype-specific CR pathways, a mode of action that would be consistent with Neurog1's partial fate specification properties in the OE (Cau et al., 2002). To test this model, we asked whether Neurog1 and Neurog2 could induce the expression of Tbr1, a cortical identity marker. In striking contrast to Reelin, both Neurog1 and Neurog2 induced ectopic Tbr1 expression whether expressed together or apart (2.8-fold, 2.6-fold, and 2.5-fold increases respectively; control vector: 14.8 ± 3.0% Tbr1+GFP+/GFP+; n = 4; 251 cells; Neurog1: 41.3 ± 12.0% Tbr1+GFP+/GFP+; n = 3; 140 cells; p < 0.05 vs pCIG2; Neurog2: 38.6 ± 4.9% Tbr1+GFP+/GFP+; n = 3; 923 cells; p < 0.005 vs pCIG2; Neurog1 plus Neurog2: 37.7 ± 6.6% Tbr1+GFP+/GFP+; n = 3; 806 cells; p < 0.05 vs pCIG2; Fig. 4O).
Thus, while Neurog2 is sufficient to promote the expression of cortical and some CR cell identity markers in early pallial progenitors, as previously shown in the choroid plexus (Imayoshi et al., 2008), Neurog1 is only sufficient to induce a cortical identity, at least in the cellular context and time frame tested. Moreover, Neurog1 impedes Neurog2's ability to induce CR marker expression, consistent with a role for this factor in controlling the rate of early CR cell production.
Early defects in cell fate specification and not migration in the Neurog1/2−/− piriform cortex
Neurog2 has sequential functions in the neocortex: first promoting a CR identity and later specifying deep-layer neocortical cell fates. We thus asked whether Neurog1 and Neurog2 have similar sequential functions in the ventral pallium/piriform cortex. After forming CR cells, ventral pallial progenitors give rise to neurons in the piriform cortex, with layer I lot cells differentiating between E10 and E11, followed by deep-layer III neurons at E12, and layer II neurons arising between E13 and E14 (de Carlos et al., 1996; Tomioka et al., 2000; Hirata et al., 2002; Vyas et al., 2003). To first determine whether Neurog2-expressing progenitors in the ventral pallium populate the piriform cortex, we examined GFP expression in E12.5 “wild-type” (i.e., Neurog2GFPKI/+ heterozygous) and Neurog1 and Neurog2 single and double mutant embryos carrying a Neurog2GFPKI allele (Fig. 5A–D). In E12.5 Neurog2GFPKI+/− heterozygotes, GFP+ cells emanated from the ventral pallium, accumulating in a ventrolateral wedge adjacent to the LGE in the nascent piriform cortex (Fig. 5A). The accumulation of GFP+ cells in a wedge shape in the piriform cortex closely mimicked the spatial pattern of Tbr1+ and Trp73+ cells observed above (Fig. 2E,I). A similar ventrolateral stream of GFP+ cells was also observed in Neurog1−/− mutants carrying one copy of the Neurog2GFPKI allele, in Neurog2GFPKI/KI single mutants, and in Neurog1−/−;Neurog2GFPKI/KI double mutants (Fig. 5B–D). Thus, the tangential migration of neurons derived from the ventral pallium into the piriform cortex is not grossly perturbed by the loss of Neurog1 and Neurog2. However, immunostaining with anti-activated caspase 3 revealed that there is elevated levels of apoptosis in the Neurog2−/− and Neurog1/2−/− ventral pallium at E12.5, after CR cell production has ended and the genesis of piriform cortical neurons has begun (Fig. 5E–H). To test whether cells with a piriform cortex identity were selectively lost in Neurog2−/− and Neurog1/2−/− cortices, we examined the expression of Lhx2, a cortical selector gene that regulates a key decision between making neocortical versus piriform cortical structures (Chou et al., 2009). In E12.5 wild-type and Neurog1−/− cortices (Fig. 5I,J), a stream of Lhx2+ cells emanated ventrally from the ventral pallium, marking the presumptive piriform cortex. In contrast, the Lhx2+ presumptive piriform cortical neurons were disorganized in Neurog2−/− embryos and, strikingly, were lost in Neurog1/2−/− cortices (Fig. 5K,L).
Figure 5.
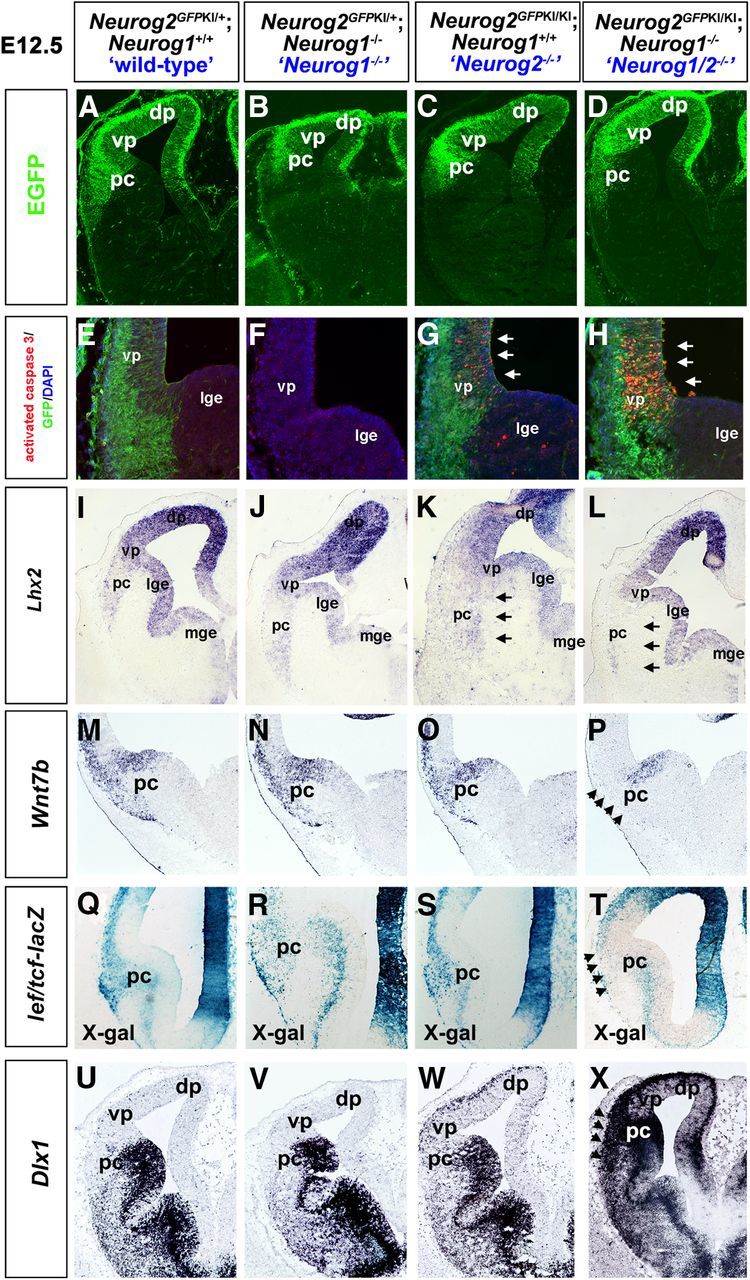
Early defects in cell fate specification in the ventral pallium and presumptive piriform cortex in Neurog1/2−/− embryos. A–H, Expression of GFP (A–D) and activated caspase 3 (E–H, red) in E12.5 Neurog2GFPKI+/− heterozygotes (“wild-type”; A, E), Neurog1−/− mutants carrying one copy of the Neurog2GFPKI allele (B, F), Neurog2GFPKI/KI mutants (C, G), and Neurog1−/−;Neurog2GFPKI/KI double mutants (Neurog1/2−/−; D, H). Arrowheads in G and H mark elevated levels of apoptosis in the Neurog2−/− and Neurog1/2−/− ventral pallium. I–P, Expression of Lhx2 (I–L) and Wnt7b (M–P) in E12.5 wild-type (I, M), Neurog1−/− (J, N), Neurog2−/− (K, O), and Neurog1/2−/− (L, P) cortices. Arrowheads in L and P mark reduced expression of Lhx2 and Wnt7b in the double-mutant ventral pallium. Q–T, X-gal staining (blue) of E12.5 wild-type (Q), Neurog1−/− (R), Neurog2−/− (S), and Neurog1/2−/− (T) cortices, all from embryos carrying a lef/tcf-lacZ transgene. Arrowhead in T marks reduced X-gal staining. U–X, Expression of Dlx1 in E12.5 wild-type (U), Neurog1−/− (V), Neurog2−/− (W), and Neurog1/2−/− (X) cortices. Arrowhead in X marks ectopic Dlx1 expression in double mutants. dp, Dorsal pallium; lge, lateral ganglionic eminence; pc, piriform cortex; vp, ventral pallium.
To further assess piriform cortex development in E12.5 Neurog1 and Neurog2 single and double mutants, we monitored canonical Wnt signaling, which specifies a dorsal telencephalic cell fate (Gunhaga et al., 2003; Hirabayashi et al., 2004; Israsena et al., 2004; Backman et al., 2005; Machon et al., 2005; Watanabe et al., 2005). Wnt7b, which can initiate canonical signaling (Wang et al., 2005), was expressed in neurons migrating into the outer layers of the developing piriform cortex in E12.5 wild-type, Neurog1−/−, and Neurog2−/− embryos (Fig. 5M–P). In contrast, Wnt7b expression was strikingly absent in the presumptive Neurog1/2−/− piriform cortex, although transcripts were still detected in the dorsal LGE (Fig. 5P). To further test whether canonical Wnt signaling was indeed disrupted, we monitored β-galactosidase activity in a transgenic line carrying a lef/tcf-lacZ reporter (Mohamed et al., 2004). In E12.5 wild-type, Neurog1−/−, and Neurog2−/− embryos, lef/tcf-lacZ reporter activity was detected throughout the wedge of cells corresponding to the presumptive piriform cortex (Fig. 5Q–T). In contrast, the presumptive piriform cortex in E12.5 Neurog1/2−/− embryos was devoid of lef/tcf-lacZ reporter activity, indicating that canonical Wnt signaling was strongly reduced in the Neurog1/2−/− piriform cortex (Fig. 5T). Finally, to test whether loss of a dorsal telencephalic identity was due to a transition to a ventral fate, we examined the expression of Dlx1. Dlx1 was ectopically expressed throughout the Neurog1/2−/− piriform cortex (Fig. 5U–X), as previously shown in adjacent neocortical territories (Fode et al., 2000; Schuurmans et al., 2004).
Thus, the loss of both Neurog1 and Neurog2 does not prevent the migration of neurons derived from the ventral pallium into the presumptive piriform cortex, but rather results in a ventralization of these cells, which may contribute to their subsequent death.
Lamination and cell fate specification defects in Neurog1/2−/− piriform cortex
To further characterize defects in piriform cortex development in Neurog1/2−/− embryos, we first examined histological sections at E18.5, when neurogenesis and lamination are essentially complete. In E18.5 wild-type, Neurog1−/−, and Neurog2−/− brains, the transition from a six-layered neocortex to three-layered piriform cortex was evident as a medial inflection and narrowing of the densely stained neuronal layers (Fig. 6A–C). In contrast, in E18.5 Neurog1/2−/− brains, the presumptive piriform cortex was thinner, hypocellular, and lacked densely stained neuronal layers (Fig. 6D).
Figure 6.
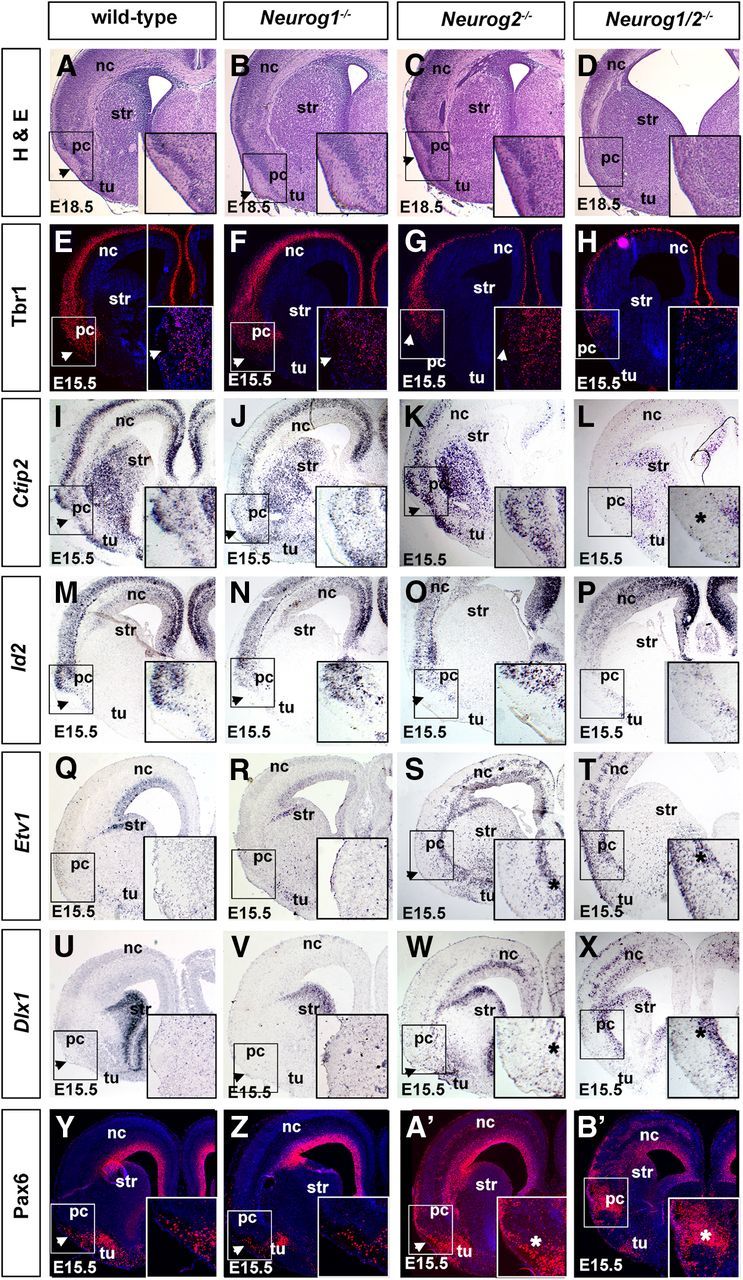
Defects in lamination and cell fate specification in the Neurog1/2−/− piriform cortex. A–D, Hematoxylin and eosin staining of E18.5 wild-type (A), Neurog1−/− (B), Neurog2−/− (C), and Neurog1/2−/− (D) cortices. Arrowheads in A–C mark site of LOT formation. E–Z, A', B', Expression of Tbr1 (E–H), Ctip2 (I–L), Id2 (M–P), Etv1 (Q–T), Dlx1 (U–X), and Pax6 (Y, Z, A', B') in E15.5 wild-type (E, I, M, Q, U, Y), Neurog1−/− (F, J, N, R, V, Z), Neurog2−/− (G, K, O, S, W, A'), and Neurog1/2−/− (H, L, P, T, X, B') cortices. Insets in I–P are high-magnification images of the piriform cortex. Asterisks in L and P show reduced expression of Ctip2 and Id2 in the Neurog1/2−/− piriform cortex. Arrowheads in S, T, W, X, A', and B' mark the ectopic expression of Etv1, Dlx1, and Pax6 in Neurog2−/− and Neurog1/2−/− piriform cortices. nc, Neocortex; pc, piriform cortex; str, striatum; tu, olfactory tubercle.
To examine the basis of these histological defects, we used markers to label the different layers of the piriform cortex. In E15.5 wild-type, Neurog1−/−, and Neurog2−/− embryos, Tbr1, which marks neocortical and piriform cortical neurons (Hevner et al., 2001), was expressed in all three layers of the piriform cortex (Fig. 6E–G). In contrast, many fewer neurons expressed Tbr1 in the E15.5 Neurog1/2−/− piriform cortex (Fig. 6H), which is consistent with a similar reduction observed at E12.5 (Fig. 2H). Similarly, the expression of Ctip2, which was expressed in layer II of the E15.5 wild-type, Neurog1−/−, and Neurog2−/− piriform cortex, as well as Id2, which was expressed in layers II and III, were both strongly reduced in the Neurog1/2−/− piriform cortex (Fig. 6I–P). The loss of piriform cortex marker expression in Neurog1/2−/− embryos was not due to a transformation from a piriform to neocortical identity, as Fezf1 and Tle4, which mark upper and deep layer neocortical neurons, respectively, and which are not expressed in the piriform cortex, were not ectopically expressed in the presumptive piriform cortex of Neurog1/2−/− embryos (data not shown).
While the piriform cortex has a ventral location, it has a dorsal origin, arising from lateral and ventral pallial progenitors. We thus reasoned that the loss of “dorsal” (cortical)-specific marker expression in the Neurog1/2−/− piriform cortex could be due to a dorsal-to-ventral fate transition, resulting in the generation of ectopic interneurons, as previously shown in Neurog2−/− and Neurog1/2−/− neocortices (Fode et al., 2000; Schuurmans et al., 2004) and OBs (Shaker et al., 2012). We thus examined the expression of Etv1 and Pax6, which label dopaminergic periglomerular interneurons in the OB (Stenman et al., 2003; Kohwi et al., 2005; Saino-Saito et al., 2007), and Dlx1, which labels GABAergic interneurons in both the cortex and OB (Anderson et al., 1997; Long et al., 2007). All three interneuron markers were ectopically expressed in E15.5 Neurog2−/− and Neurog1/2−/− piriform cortices, whereas only scattered piriform cortex interneurons expressed these markers in E15.5 wild-type and Neurog1−/− embryos (Fig. 6Q–Z,A',B'). However, neuronal mis-specification was much more severe in the double-mutant piriform cortex, with ectopic Etv1 and Pax6 expression detected in all three layers (Fig. 6T,B'), while ectopic Dlx1 expression was restricted to deep layer III (Fig. 6X).
Together, these results suggest that the Neurog1/2−/− piriform cortex is comprised of neurons with aberrant molecular identities. Strikingly, the “ventralized” neurons have a similar molecular signature as OB interneurons, which are derived from the dorsal LGE, a subpallial progenitor domain that lies immediately adjacent to the ventral pallium (Vergaño-Vera et al., 2006; Flames and Hobert, 2009).
LOT formation is perturbed in the Neurog1/2−/− double-mutant piriform cortex
The piriform cortex is part of a three-part neural network that also includes the OE and OB, which together mediate the primary sense of olfaction. We previously showed that Neurog1/2−/− double mutants only form a remnant of an OB-like structure (OBLS) in an aberrant ventral position (Shaker et al., 2012). Nevertheless, despite this abnormal architecture, some glutamatergic mitral cells, which are the output neurons of the OB that target the piriform cortex, differentiate in the Neurog1/2−/− OBLS (Shaker et al., 2012). We therefore questioned whether Neurog1/2−/− mitral cell axons are capable of forming a LOT that innervates the piriform cortex.
To trace LOT axons, a DiI crystal was inserted into the OB of E18.5 wild-type and Neurog1 and Neurog2 single-mutant and double-mutant embryos (Fig. 7A–D). In E18.5 wild-type embryos, DiI-labeled LOT axons formed a narrow axon bundle that extended along the lateral surface of the telencephalon before turning in a ventral direction to innervate the piriform cortex (Fig. 7A). DiI crystals inserted into the Neurog1−/− OB also anterogradely labeled the LOT (Fig. 7B), even though the Neurog1−/− OB is smaller and abnormally laminated (Shaker et al., 2012). Likewise, Neurog2−/− mitral cells, which differentiate normally (Shaker et al., 2012), also formed a LOT (Fig. 7C). In contrast, in E18.5 Neurog1/2−/− brains, DiI crystals placed in the rostroventral telencephalon did not label a distinct LOT, suggesting that the ectopically localized mitral cells in the Neurog1/2−/− OBLS failed to extend axons to innervate the piriform cortex (Fig. 7D).
Figure 7.
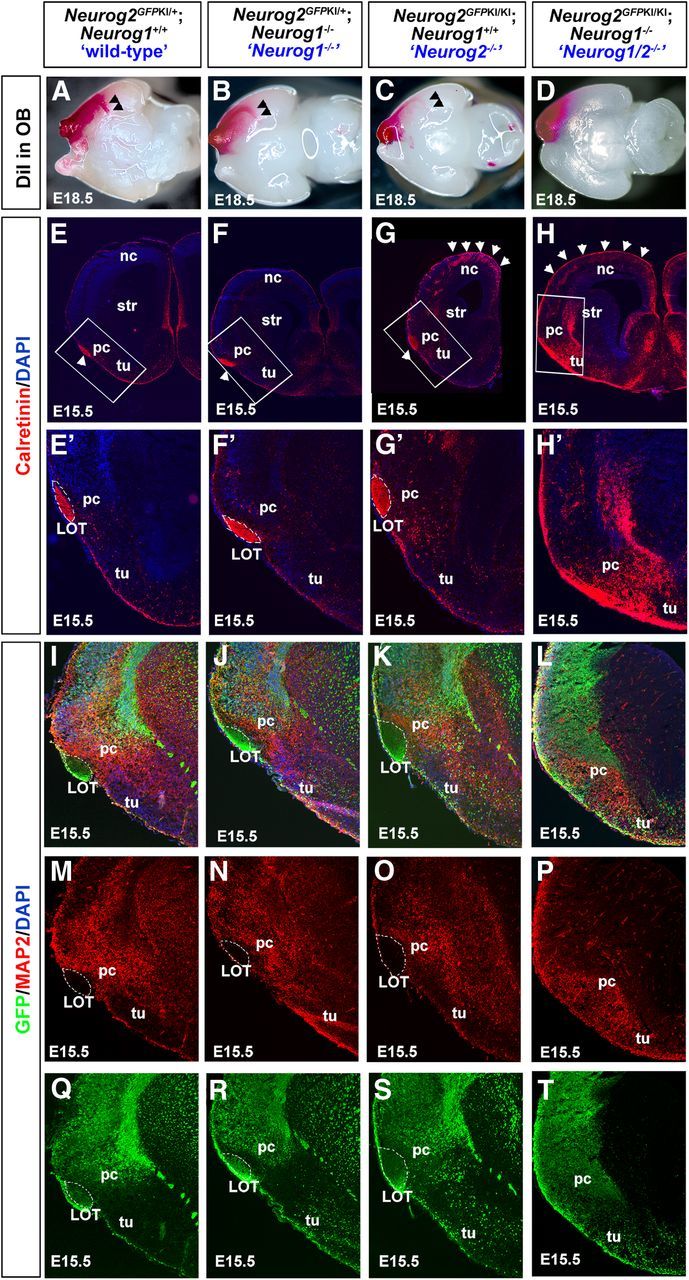
LOT formation is perturbed in the Neurog1/2−/− piriform cortex. A–D, LOT axons were traced by inserting a DiI crystal into E18.5 wild-type (A), Neurog1−/− (B), Neurog2−/− (C), and Neurog1/2−/− (D) OBs. Arrowheads mark the ventromedial turn of the forming LOT, which fails to form in Neurog1/2−/− embryos. E–H', Labeling of OB mitral cell axons in the LOT with anti-calretinin in E15.5 wild-type (E, E'), Neurog1−/− (F, F'), Neurog2−/− (G, G'), and Neurog1/2−/− (H, H') piriform cortices. Blue is a DAPI counterstain. E'–H' are higher-magnification images of boxed areas in E–H. Arrowheads in G and H mark supernumerary calretinin+ interneurons in Neurog2−/− and Neurog1/2−/− neocortices, respectively. I–T, Expression of GFP (green) and MAP2 (red) in E15.5 Neurog2GFPKI+/− heterozygotes (“wild-type”; I, M, Q), Neurog1−/− mutants carrying one copy of the Neurog2GFPKI allele (J, N, R), Neurog2GFPKI/KI mutants (K, O, S), and Neurog1−/−;Neurog2GFPKI/KI double mutants (Neurog1/2−/−; L, P, T). Blue is a DAPI counterstain. The dotted white line outlines the LOT. nc, Neocortex; pc, piriform cortex; str, striatum; tu, olfactory tubercle.
To confirm that LOT formation did not occur in Neurog1/2−/− embryos, we examined calretinin expression. Calretinin immunolabeling was detected in mitral cell axon bundles innervating the piriform cortex in E15.5 wild-type, Neurog1−/−, and Neurog2−/− brains (Fig. 7E–H'). In contrast, a calretinin+ axonal bundle was not observed in the presumptive piriform cortex in Neurog1/2−/− mutants (Fig. 7H,H'). To further examine LOT formation, we examined embryos carrying a Neurog2GFPKI allele, which serves as a short-term lineage trace of glutamatergic projection neurons in both the neocortex and OB (Britz et al., 2006; Shaker et al., 2012). A distinct GFP+ LOT was observed in the piriform cortex of Neurog2GFPKI+/− heterozygotes (Fig. 7I,M,Q), as well as in Neurog1−/− mutants carrying one copy of the Neurog2GFPKI allele (Fig. 7J,N,R) and Neurog2GFPKI/KI single mutants (Fig. 7K,O,S). In contrast, a GFP+ LOT was not evident in the Neurog1−/−;Neurog2GFPKI/KI piriform cortex (Fig. 7L,P,T). Notably, the LOT, which is comprised of mitral cell axons, was also distinguishable in wild-type, Neurog1−/−, and Neurog2−/− piriform cortex by the absence of MAP2 expression, which is a dendritic marker (Fig. 7M–O), whereas a MAP2-free zone was not observed in the presumptive Neurog1/2−/− piriform cortex (Fig. 7P). Thus, although some mitral cells differentiate in the Neurog1/2−/− OBLS, they fail to innervate the piriform cortex.
Defects in the differentiation of lot1 guidepost neurons in the Neurog1/2−/− piriform cortex
LOT formation depends on lot guidepost cells, which have a dorsal pallial origin (Sato et al., 1998; Tomioka et al., 2000). We speculated that the failure of LOT formation in the Neurog1/2−/− piriform cortex could be due to defects in the differentiation of lot cells, which could be derived from a Neurog2-lineage. To monitor lot cell formation, we took advantage of two markers: a lot-cell-specific monoclonal antibody called lot1 (Sato et al., 1998), which recently was shown to recognize metabotropic glutamate receptor subtype-1 (mGluR1), and an mGluR1 riboprobe (Hirata et al., 2012). To first determine whether lot cells were derived from the Neurog2 lineage, we performed lot1 immunolabeling on embryos carrying a Neurog2GFPKI allele. In E12.5 Neurog2GFPKI+/− heterozygotes (wild type), Neurog1−/− mutants carrying one copy of the Neurog2GFPKI allele, Neurog2GFPKI/KI single mutants, and Neurog2GFPKI/KI; Neurog1−/− double mutants, lot1 and GFP were coexpressed (Fig. 8A–C). However, in Neurog1/2−/− embryos, many fewer lot1+ cells migrated into the presumptive piriform cortex compared with all other genotypes (Fig. 8D). Similarly, mGluR1 was expressed in the presumptive piriform cortex in E12.5 wild-type, Neurog1−/−, and Neurog2−/− piriform cortex, whereas many fewer mGluR1+ lot cells were detected in Neurog1/2−/− embryos (Fig. 8E–H). Expression of lot1 persisted in the piriform cortex at E15.5, with notable defects only observed in Neurog1/2−/− piriform cortex (Fig. 8I–L'). The differentiation of lot cells is thus strikingly reduced in Neurog1/2−/− embryos.
Figure 8.

Lot cells, which are reduced in number in the Neurog1/2−/− piriform cortex, are a subpopulation of CR cells. A–H, Labeling of E12.5 telencephalons from Neurog2GFPKI+/− heterozygotes (“wild-type”; A, E), Neurog1−/− mutants carrying one copy of the Neurog2GFPKI allele (B, F), Neurog2GFPKI/KI mutants (C, G), and Neurog1−/−;Neurog2GFPKI/KI double mutants (Neurog1/2−/−; D, H) with anti-lot1 (A–D, red), anti-GFP (A–D, green), and a mGluR1 riboprobe (E–H). Insets in A–D are lot1 immunostaining of the lot. Arrows in D and H mark a loss of lot cells in the Neurog1/2−/− piriform cortex. (I–L') Expression of lot1 in E15.5 wild-type (I, I'), Neurog1−/− (J, J'), Neurog2−/− (K, K'), and Neurog1/2−/− (L, L') cortices. The images in I'–L' are high-magnification images of the boxed areas in I–L. M–P, Coexpression of lot1 (M, N′, red) and Reelin (N, N′, green), and of lot1 (O, P', red) and Trp73 (P, P') in the lot. Blue is DAPI counterstain in N′ and P'. Insets in N′ and P' are higher-magnification images of the lot. Q, Quantitation of the coexpression of lot1 with Reelin and Trp73. Cells that expressed high levels of lot1, which surround the LOT, were counted separately from those displaced from the lot (high and low lot+ cells, respectively). R, Schematic representation of E12.5 telencephalon, depicting the suggested dorsal pallial source of lot cells (red), and the cortical hem and ventral pallium as the sites of CR cell genesis (blue). S, Summary of our findings, demonstrating that Neurog1 and Neurog2 are coexpressed in dorsal and ventral pallial progenitors, which give rise to lot cells, a subset of Reelin+/Trp73+ CR cells. ch, Cortical hem; dp, dorsal pallium; lge, lateral ganglionic eminence; lot, lot guidepost cells; vp, ventral pallium.
We were struck by the similarities in the E12.5 expression profiles of lot1 (Fig. 8A–D) and Trp73 (Fig. 2I–L), which labels a subset of CR cells (Meyer et al., 2002, 2004). Given their similarities, we speculated that lot guidepost cells could be a subpopulation of CR neurons. While previous studies have suggested that there is limited overlap in the expression of lot1 and Reelin (Sato et al., 1998), in our studies, which were conducted on sections instead of in whole-mount, we found that a subset of E12.5 lot1+ cells indeed coexpressed Reelin (Fig. 8M,N,N′) and Trp73, the full-length Trp73 isoform (Fig. 8O,P,P'). Moreover, quantitation of coexpression rates revealed that the high lot1+ cells that surround the LOT frequently coexpressed Reelin (94.5 ± 0.8%, n = 3, 697 cells) and Trp73 (96.0 ± 1.2%, n = 3, 1068 cells), while the lower-expressing lot1+ cells that are farther away from the LOT displayed reduced levels of Reelin (39.6 ± 4.0%, n = 3, 2228 cells) and Trp73 (43.5 ± 4.2%, n = 3, 2218 cells) coexpression (Fig. 8Q).
We thus conclude that lot guidepost cells are a subset of CR neurons, revealing that these cells have a pallial origin, and depend on both Neurog1 and Neurog2 for their differentiation (Fig. 8R,S).
Trp73 is required for LOT formation and lot cell positioning
We reasoned that if lot cells were indeed a subpopulation of Reelin+/Trp73+ CR neurons, that the mutation of Reelin and/or Trp73 could in turn influence lot cell formation and/or function. Indeed, Reelin is required for cellular orientation of neocortical layer VI neurons (O'Dell et al., 2012), while Trp73 is required for CR cell genesis (Meyer et al., 2004). Notably, Trp73 has two transcriptional start sites, and it is only the larger TAp73 protein, which is generated via transcription from an upstream P1 promoter, that is a functional transcription factor (Conforti et al., 2012). Moreover, the TAp73 isoform regulates the expression of Hey2, a repressor of bHLH proteins, such as Neurog1 and Neurog2 (Sakamoto et al., 2003; Fujitani et al., 2010). Thus, to test whether genes involved in CR genesis and/or function also influenced lot formation, we analyzed E15.5 Reeler, Trp73−/− (p73−/−), and TAp73−/− cortices.
We first examined whether CR cell production was affected in these mutants. Interestingly, while the expression of Trp73 in neocortical and cortical CR cells appeared normal in E15.5 Reeler mutants (Fig. 9A,B), the expression of Reelin was strikingly reduced in the p73−/− and TAp73−/− piriform cortical and neocortical marginal zones (Fig. 9C–E), which is consistent with previous studies (Meyer et al., 2002, 2004). Thus, if CR cells remain in p73−/− and TAp73−/− cortices, they have an abnormal molecular identity.
Figure 9.

Defects in LOT formation and lot cell positioning in p73 and TAp73 mutants. A–J, Expression of Trp73 (A, B), Reelin (C, D), and calretinin (F–J) in E15.5 wild-type (A, C, F, H), Reeler−/− (B, G), p73−/− (D, I), and TAp73−/− (E, J) cortices. K–T, DAPI staining (K–N; P–S, blue counterstain) and lot1 immunolabeling (P–S, red label) of E15.5 wild-type (K, P), Reeler−/− (L, Q), p73−/− (M, R), and TAp73−/− (N, S) piriform cortices. The LOT is outlined by a dashed white line, following the contours of the area of tight cellular packing. Quantitation of relative area measurements of the LOT in each genotype (O). Quantitation of the number of DAPI+ cells displaced within the LOT (T). nc, Neocortex; pc, piriform cortex; str, striatum; tu, olfactory tubercle.
Next we examined whether LOT formation, which depends on lot cells (Sato et al., 1998), was disrupted in the absence of Reelin and p73. Strikingly, calretinin immunolabeling marked a larger and less clearly defined LOT in both p73−/− and TAp73−/− piriform cortices (Fig. 9H–J,M–O), whereas LOT formation was normal in Reeler mutants (Fig. 9F,G,K,L,O). In addition, the borders of the LOT were not well circumscribed in p73−/− and TAp73−/− piriform cortices. Instead, DAPI+ cells were found within the interior of the LOT (Fig. 9K–N,T). Furthermore, immunostaining with lot1 revealed that lot guidepost cells were in some instances aberrantly localized within the LOT in p73−/− and TAp73−/− mutants (Fig. 9M,N,R–T), whereas lot1+ cells were rarely observed in the LOT in wild-type or Reeler mutant cortices (Fig. 9K,L,P,Q,T).
Thus, not only is Trp73 expressed in lot cells, which are a subpopulation of CR neurons, but p73 is also required for the proper organization of lot cells, and for the subsequent innervation of the piriform cortex by LOT axons.
Discussion
The cerebral cortex is comprised of the archicortex, neocortex, and piriform cortex, each derived from distinct pallial progenitor domains during development. Within each pallial territory, progenitor cells undergo temporal identity transitions, so that the diverse neuronal populations that make up each cortical structure are generated at their correct times and in proper numbers. Here we demonstrate that the proneural genes Neurog1 and Neurog2 form a regulatory loop to temporally control successive cell fate decisions in the ventral pallium. At early stages, either Neurog1 or Neurog2 are required to specify a CR cell identity. In addition, Neurog1 has an added early role—preventing precocious neurogenesis by limiting Neurog2's ability to specify a CR cell fate. Hence, in the absence of Neurog1, more CR cells are generated. Furthermore, we reveal that Neurog1 and Neurog2 are required to specify the identity of a subset of CR neurons in the piriform cortex: lot guidepost cells. Accordingly, we demonstrate that Trp73, which is expressed in CR cells (Meyer et al., 2002, 2004) and lot cells (this study), is required for normal LOT axon innervation of the piriform cortex. Finally, we demonstrate that at later developmental stages, Neurog1 and Neurog2 change their fate specification properties in the ventral pallium, guiding the differentiation of layer II/III piriform cortical neurons.
Neurog1 and Neurog2 have overlapping and nonoverlapping functions in CR progenitors
Neurog1 and Neurog2 are expressed in CR cell lineages derived from all three progenitor domains (this study; Dixit et al., 2011b), including the rostrobulbar area/pallial septum, cortical hem/choroid plexus, and ventral pallium (Takiguchi-Hayashi et al., 2004; Bielle et al., 2005; Yoshida et al., 2006; Zhao et al., 2006; García-Moreno et al., 2007; Imayoshi et al., 2008). Neurog2-derived CR cells are widespread, populating the piriform cortex, neocortex, and hippocampus (this study; Dixit et al., 2011b). It is thus not surprising that we observed a global reduction in CR cell numbers in all cortical domains in Neurog1/2−/− embryos. What was unexpected was the increase in CR numbers in Neurog1−/− embryos, and the ability of Neurog1 to limit Neurog2's ability to specify a Reelin+ CR cell identity in gain-of-function assays. We suggest that the transition from generating CR neurons to deep-layer cortical neurons in the neocortex and piriform cortex depends on Neurog1's ability to curtail Neurog2's early proneural and cell fate specification properties. While future studies are required to address mechanism, it may be that Neurog1-Neurog2 heterodimers might have a reduced capacity to transactivate target genes compared with Neurog2-Neurog2 homodimers. Indeed, in a previous study we revealed that Neurog2's proneural activities are temporally regulated by their dimerization partner, and that Neurog2-Neurog2 homodimers are more active than Neurog2-E47 heterodimers (Li et al., 2012).
Unexpected was the striking and specific loss of Trp73+ CR cells in a V-shaped wedge in the Neurog1/2−/− piriform cortex. Trp73+ CR cells are thought to arise from the cortical hem and pallial septum (Meyer et al., 2002, 2004; Hanashima et al., 2007). However, in a Dbx1-lineage trace (Bielle et al., 2005), which labels CR cells derived from the ventral pallium and pallial septum, lacZ+ cells accumulate in the piriform cortex in a pattern that closely resembles the wedge-shaped distribution of Trp73+ cells (this study). While it has been suggested that pallial septum-derived CR cells in the Dbx1 lineage are Trp73+ (Griveau et al., 2010), our data support the idea that some of these Trp73+ CR cells may also be derived from the ventral pallium. Interestingly, the pattern of Trp73 expression in the wild-type piriform cortex is the same as lot1/mGluR1, which label lot guidepost cells (Sato et al., 1998; Hirata et al., 2012). Moreover, lot1/mGluR1 expression was strikingly reduced in the Neurog1/2−/− piriform cortex. These data, combined with our coexpression data, are good evidence that lot guidepost cells are a subpopulation of CR cells that depend on Neurog1 and Neurog2 for their differentiation.
Neurog1 and Neurog2 specify the identities of piriform cortical neurons
Recent studies have suggested that CR cells influence regional identities and boundary formation in the underlying cortical neuroepithelium (Griveau et al., 2010). Specifically, the loss of Dbx1, which is expressed in CR progenitors in the ventral pallium and pallial septum, alters the distribution of the remaining CR cells (Bielle et al., 2005), thereby shifting the regional borders that prefigure cortical areas (Griveau et al., 2010). Similar compensatory changes likely influence the distribution of remaining CR cells in Neurog2−/− and Neurog1/2−/− cortices. This raises the question of whether the patterning defects in Neurog2−/− and Neurog1/2−/− cortices are related in any way to CR cell changes. It is difficult to reach a definitive answer to this question, since the mis-specification of Neurog2−/− and Neurog1/2−/− cortical progenitors may instead be linked to a requirement to suppress expression of Ascl1, another proneural gene that specifies a ventral telencephalic phenotype (Fode et al., 2000; Schuurmans et al., 2004). Nevertheless, it is remarkable that the phenotype of the ectopic interneurons in Neurog2−/− and Neurog1/2−/− cortices most closely resemble OB interneurons (i.e., Etv1+, Pax6+, Dlx1+, calretinin+; Yun et al., 2001, 2003), an interneuron population that is normally derived from the dorsal LGE (Long et al., 2003, 2007; Stenman et al., 2003; Yun et al., 2003; Kohwi et al., 2005). Interestingly, the dorsal LGE is a subpallial progenitor domain that immediately abuts the ventral pallium (Yun et al., 2001). It is thus tempting to speculate that the mis-specification of Neurog2−/− and Neurog1/2−/− cortical territories may be due to a shift in the pallial–subpallial border, and that this shift may be related to alterations in the distribution and identity of CR cells. In contrast, the border between the neocortex and piriform cortex does not appear to be shifted in Neurog1/2−/− embryos, as the loss of piriform cortex markers does not correlate with an expansion of neocortical markers Fezf1 and Tle4. In this regard, Neurog1/2−/− mutants differ from Tbr1 mutants, which ectopically express neocortical markers in the piriform cortex (Hevner et al., 2001).
Based on marker expression (Hirata et al., 2002), as well as short-term lineage tracing using Neurog2GFPKI mice (this study) and long-term lineage tracing using Dbx1-creKI mice (Bielle et al., 2005), neurons in the piriform cortex are generated from progenitor cells in the ventral pallium. Hypocellularity of the Neurog2−/− and Neurog1/2−/− piriform cortices is likely related to increased apoptosis in the ventral pallium. Notably, in Pax6 mutants, neuronal mis-specification leads to elevated apoptosis due to altered expression of the neurotrophin receptors TrkB and p75NTR (Nikoletopoulou et al., 2007). Interestingly, Dbx1 expression, which marks the ventral pallium, is lost in Pax6 mutants (Yun et al., 2001), whereas in Neurog2−/− and Neurog1/2−/− cortices, Dbx1 expression is retained (this study). Moreover, TrkB expression persists in the Neurog2−/− and Neurog1/2−/− ventral pallium (data not shown), which undergoes massive cell death, suggesting that the expression of this neurotrophin receptor alone may not be sufficient to prevent apoptosis in mis-specified pallial progenitors.
CR cells function in axon guidance
The piriform cortex is part of the olfactory system, which also includes the OE and OB. While the OE, OB, and piriform cortex are separate functional entities, they develop as an interconnected unit (de Castro, 2009). Interestingly, Neurog1 and/or Neurog2 are expressed in progenitor cells for each of these components of the olfactory system, including the OE (Cau et al., 2002), OB (Winpenny et al., 2011; Shaker et al., 2012), and piriform cortex (this study). While the differentiation of olfactory sensory neurons is perturbed in Neurog1−/− single mutants, disruptions in OB (Shaker et al., 2012) and piriform cortex (this study) development are most severe in Neurog1/2−/− double mutants. Neurog1/2−/− embryos develop a hypocellular OBLS in an aberrant ventrolateral position (Shaker et al., 2012). However, even though some mitral cells differentiate in the Neurog1/2−/− OBLS (Shaker et al., 2012), we show here that these OB projection neurons fail to innervate the piriform cortex (i.e., form a LOT) due to the loss of lot guidepost cells (Sato et al., 1998). Notably, a similar OBLS phenotype is observed in Pax6−/− (Hirata et al., 2002), Lhx2−/− (Saha et al., 2007), and Gli3−/− (Balmer and LaMantia, 2004) mutants, yet only Pax6−/− mitral cells extend their axons out of the OB to form the LOT and innervate the piriform cortex (Hirata et al., 2002). In contrast, in Gli3−/− mutants, in which lot cells fail to migrate into the piriform cortex and instead accumulate in the neocortex (Tomioka et al., 2000; Balmer and LaMantia, 2004), and in Lhx2−/− cortices, where lot cells fail to differentiate (Saha et al., 2007), LOT formation is perturbed. CR cells thus have multiple roles in building the architecture of the cortex: not only in patterning the cortical VZ (Griveau et al., 2010), but also in axonal guidance of olfactory tracts (this study).
Through these findings, we provide new insights into the control of temporal identity transitions and neuronal fate specification in the piriform cortex, a poorly understood brain region. Moreover, we identify a novel population of CR neurons (lot cells), and ascribe a new function to Trp73, which is expressed in these lot/CR cells, in LOT formation.
Footnotes
This work was supported by a Canadian Institutes of Health Research (CIHR; MOP-44094) Operating Grant to C.S., C.S. is an Alberta Innovates Health Solutions Senior Scholar. R.D. was supported by a CIHR Canada Hope Scholarship and G.W. and S.L. were supported by a CIHR Training Grant in Genetics, Child Development and Health. We thank François Guillemot, David Anderson, Tak Mak, Tatsumi Hirata, Daniel Dufort, Masato Nakafuku, and Valerie Wallace for generously providing reagents or mice. We also thank Nicole Gruenig, Dawn Zinyk, Pierre Mattar, and Natasha Klenin for technical support.
The authors declare no competing financial interests.
References
- Alam S, Zinyk D, Ma L, Schuurmans C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev Dyn. 2005;234:772–782. doi: 10.1002/dvdy.20577. [DOI] [PubMed] [Google Scholar]
- Alcántara S, Ruiz M, D'Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL. Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron. 1997;19:27–37. doi: 10.1016/S0896-6273(00)80345-1. [DOI] [PubMed] [Google Scholar]
- Assimacopoulos S, Grove EA, Ragsdale CW. Identification of a Pax6-dependent epidermal growth factor family signaling source at the lateral edge of the embryonic cerebral cortex. J Neurosci. 2003;23:6399–6403. doi: 10.1523/JNEUROSCI.23-16-06399.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, Krauss S. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol. 2005;279:155–168. doi: 10.1016/j.ydbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Balmer CW, LaMantia AS. Loss of Gli3 and Shh function disrupts olfactory axon trajectories. J Comp Neurol. 2004;472:292–307. doi: 10.1002/cne.20053. [DOI] [PubMed] [Google Scholar]
- Bielle F, Griveau A, Narboux-Nême N, Vigneau S, Sigrist M, Arber S, Wassef M, Pierani A. Multiple origins of Cajal-Retzius cells at the borders of the developing pallium. Nat Neurosci. 2005;8:1002–1012. doi: 10.1038/nn1511. [DOI] [PubMed] [Google Scholar]
- Britz O, Mattar P, Nguyen L, Langevin LM, Zimmer C, Alam S, Guillemot F, Schuurmans C. A role for proneural genes in the maturation of cortical progenitor cells. Cereb Cortex. 2006;16(Suppl 1):i138–i151. doi: 10.1093/cercor/bhj168. [DOI] [PubMed] [Google Scholar]
- Cau E, Casarosa S, Guillemot F. Mash1 and Ngn1 control distinct steps of determination and differentiation in the olfactory sensory neuron lineage. Development. 2002;129:1871–1880. doi: 10.1242/dev.129.8.1871. [DOI] [PubMed] [Google Scholar]
- Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Chou SJ, Perez-Garcia CG, Kroll TT, O'Leary DD. Lhx2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat Neurosci. 2009;12:1381–1389. doi: 10.1038/nn.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti F, Sayan AE, Sreekumar R, Sayan BS. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012;3:e285. doi: 10.1038/cddis.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Curran T. Detection of the reelin breakpoint in reeler mice. Brain Res Mol Brain Res. 1996;39:234–236. doi: 10.1016/0169-328X(96)00046-0. [DOI] [PubMed] [Google Scholar]
- de Castro F. Wiring olfaction: the cellular and molecular mechanisms that guide the development of synaptic connections from the nose to the cortex. Front Neurosci. 2009;3:52. doi: 10.3389/neuro.22.004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carlos JA, López-Mascaraque L, Valverde F. Dynamics of cell migration from the lateral ganglionic eminence in the rat. J Neurosci. 1996;16:6146–6156. doi: 10.1523/JNEUROSCI.16-19-06146.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Lu F, Cantrup R, Gruenig N, Langevin LM, Kurrasch DM, Schuurmans C. Efficient gene delivery into multiple CNS territories using in utero electroporation. J Vis Exp. 2011a;pii:2957. doi: 10.3791/2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Zimmer C, Waclaw RR, Mattar P, Shaker T, Kovach C, Logan C, Campbell K, Guillemot F, Schuurmans C. Ascl1 participates in Cajal-Retzius cell development in the neocortex. Cereb Cortex. 2011b;21:2599–2611. doi: 10.1093/cercor/bhr046. [DOI] [PubMed] [Google Scholar]
- Flames N, Hobert O. Gene regulatory logic of dopamine neuron differentiation. Nature. 2009;458:885–889. doi: 10.1038/nature07929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. The bHLH protein NEUROGENIN 2 is a determination factor for epibranchial placode-derived sensory neurons. Neuron. 1998;20:483–494. doi: 10.1016/S0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- Fode C, Ma Q, Casarosa S, Ang SL, Anderson DJ, Guillemot F. A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes Dev. 2000;14:67–80. [PMC free article] [PubMed] [Google Scholar]
- Fujitani M, Cancino GI, Dugani CB, Weaver IC, Gauthier-Fisher A, Paquin A, Mak TW, Wojtowicz MJ, Miller FD, Kaplan DR. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr Biol. 2010;20:2058–2065. doi: 10.1016/j.cub.2010.10.029. [DOI] [PubMed] [Google Scholar]
- García-Moreno F, López-Mascaraque L, De Carlos JA. Origins and migratory routes of murine Cajal-Retzius cells. J Comp Neurol. 2007;500:419–432. doi: 10.1002/cne.21128. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griveau A, Borello U, Causeret F, Tissir F, Boggetto N, Karaz S, Pierani A. A novel role for Dbx1-derived Cajal-Retzius cells in early regionalization of the cerebral cortical neuroepithelium. PLoS Biol. 2010;8:e1000440. doi: 10.1371/journal.pbio.1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunhaga L, Marklund M, Sjödal M, Hsieh JC, Jessell TM, Edlund T. Specification of dorsal telencephalic character by sequential Wnt and FGF signaling. Nat Neurosci. 2003;6:701–707. doi: 10.1038/nn1068. [DOI] [PubMed] [Google Scholar]
- Hanashima C, Fernandes M, Hebert JM, Fishell G. The role of Foxg1 and dorsal midline signaling in the generation of Cajal-Retzius subtypes. J Neurosci. 2007;27:11103–11111. doi: 10.1523/JNEUROSCI.1066-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/S0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Neogi T, Englund C, Daza RA, Fink A. Cajal-Retzius cells in the mouse: transcription factors, neurotransmitters, and birthdays suggest a pallial origin. Brain Res Dev Brain Res. 2003;141:39–53. doi: 10.1016/S0165-3806(02)00641-7. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development. 2004;131:2791–2801. doi: 10.1242/dev.01165. [DOI] [PubMed] [Google Scholar]
- Hirata T, Nomura T, Takagi Y, Sato Y, Tomioka N, Fujisawa H, Osumi N. Mosaic development of the olfactory cortex with Pax6-dependent and -independent components. Brain Res Dev Brain Res. 2002;136:17–26. doi: 10.1016/S0165-3806(02)00304-8. [DOI] [PubMed] [Google Scholar]
- Hirata T, Kumada T, Kawasaki T, Furukawa T, Aiba A, Conquet F, Saga Y, Fukuda A. Guidepost neurons for the lateral olfactory tract: expression of metabotropic glutamate receptor 1 and innervation by glutamatergic olfactory bulb axons. Dev Neurobiol. 2012;72:1559–1576. doi: 10.1002/dneu.22030. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Shimogori T, Ohtsuka T, Kageyama R. Hes genes and neurogenin regulate nonneural versus neural fate specification in the dorsal telencephalic midline. Development. 2008;135:2531–2541. doi: 10.1242/dev.021535. [DOI] [PubMed] [Google Scholar]
- Israsena N, Hu M, Fu W, Kan L, Kessler JA. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol. 2004;268:220–231. doi: 10.1016/j.ydbio.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Kohwi M, Osumi N, Rubenstein JL, Alvarez-Buylla A. Pax6 is required for making specific subpopulations of granule and periglomerular neurons in the olfactory bulb. J Neurosci. 2005;25:6997–7003. doi: 10.1523/JNEUROSCI.1435-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mattar P, Zinyk D, Singh K, Chaturvedi CP, Kovach C, Dixit R, Kurrasch DM, Ma YC, Chan JA, Wallace V, Dilworth FJ, Brand M, Schuurmans C. GSK3 temporally regulates neurogenin 2 proneural activity in the neocortex. J Neurosci. 2012;32:7791–7805. doi: 10.1523/JNEUROSCI.1309-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JE, Garel S, Depew MJ, Tobet S, Rubenstein JL. DLX5 regulates development of peripheral and central components of the olfactory system. J Neurosci. 2003;23:568–578. doi: 10.1523/JNEUROSCI.23-02-00568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JE, Garel S, Alvarez-Dolado M, Yoshikawa K, Osumi N, Alvarez-Buylla A, Rubenstein JL. Dlx-dependent and -independent regulation of olfactory bulb interneuron differentiation. J Neurosci. 2007;27:3230–3243. doi: 10.1523/JNEUROSCI.5265-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ. Neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469–482. doi: 10.1016/S0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- Machon O, Backman M, Krauss S, Kozmik Z. The cellular fate of cortical progenitors is not maintained in neurosphere cultures. Mol Cell Neurosci. 2005;30:388–397. doi: 10.1016/j.mcn.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Marín-Padilla M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci. 1998;21:64–71. doi: 10.1016/S0166-2236(97)01164-8. [DOI] [PubMed] [Google Scholar]
- Mattar P, Britz O, Johannes C, Nieto M, Ma L, Rebeyka A, Klenin N, Polleux F, Guillemot F, Schuurmans C. A screen for downstream effectors of Neurogenin2 in the embryonic neocortex. Dev Biol. 2004;273:373–389. doi: 10.1016/j.ydbio.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Mattar P, Langevin LM, Markham K, Klenin N, Shivji S, Zinyk D, Schuurmans C. Basic helix-loop-helix transcription factors cooperate to specify a cortical projection neuron identity. Mol Cell Biol. 2008;28:1456–1469. doi: 10.1128/MCB.01510-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Perez-Garcia CG, Abraham H, Caput D. Expression of p73 and Reelin in the developing human cortex. J Neurosci. 2002;22:4973–4986. doi: 10.1523/JNEUROSCI.22-12-04973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Cabrera Socorro A, Perez Garcia CG, Martinez Millan L, Walker N, Caput D. Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J Neurosci. 2004;24:9878–9887. doi: 10.1523/JNEUROSCI.3060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed OA, Clarke HJ, Dufort D. Beta-catenin signaling marks the prospective site of primitive streak formation in the mouse embryo. Dev Dyn. 2004;231:416–424. doi: 10.1002/dvdy.20135. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, Plachta N, Allen ND, Pinto L, Götz M, Barde YA. Neurotrophin receptor-mediated death of misspecified neurons generated from embryonic stem cells lacking Pax6. Cell Stem Cell. 2007;1:529–540. doi: 10.1016/j.stem.2007.08.011. [DOI] [PubMed] [Google Scholar]
- O'Dell RS, Ustine CJ, Cameron DA, Lawless SM, Williams RM, Zipfel WR, Olson EC. Layer 6 cortical neurons require Reelin-Dab1 signaling for cellular orientation, Golgi deployment, and directed neurite growth into the marginal zone. Neural Dev. 2012;7:25. doi: 10.1186/1749-8104-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BJ, Doe CQ. Specification of temporal identity in the developing nervous system. Annu Rev Cell Dev Biol. 2004;20:619–647. doi: 10.1146/annurev.cellbio.19.111301.115142. [DOI] [PubMed] [Google Scholar]
- Puelles L, Kuwana E, Puelles E, Bulfone A, Shimamura K, Keleher J, Smiga S, Rubenstein JL. Pallial and subpallial derivatives in the embryonic chick and mouse telencephalon, traced by the expression of the genes Dlx-2, Emx-1, Nkx-2.1, Pax-6, and Tbr-1. J Comp Neurol. 2000;424:409–438. doi: 10.1002/1096-9861(20000828)424:3<409::AID-CNE3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Saha B, Hari P, Huilgol D, Tole S. Dual role for LIM-homeodomain gene Lhx2 in the formation of the lateral olfactory tract. J Neurosci. 2007;27:2290–2297. doi: 10.1523/JNEUROSCI.5571-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saino-Saito S, Cave JW, Akiba Y, Sasaki H, Goto K, Kobayashi K, Berlin R, Baker H. ER81 and CaMKIV identify anatomically and phenotypically defined subsets of mouse olfactory bulb interneurons. J Comp Neurol. 2007;502:485–496. doi: 10.1002/cne.21293. [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Hirata H, Ohtsuka T, Bessho Y, Kageyama R. The basic helix-loop-helix genes Hesr1/Hey1 and Hesr2/Hey2 regulate maintenance of neural precursor cells in the brain. J Biol Chem. 2003;278:44808–44815. doi: 10.1074/jbc.M300448200. [DOI] [PubMed] [Google Scholar]
- Sarma AA, Richard MB, Greer CA. Developmental dynamics of piriform cortex. Cereb Cortex. 2011;21:1231–1245. doi: 10.1093/cercor/bhq199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Hirata T, Ogawa M, Fujisawa H. Requirement for early-generated neurons recognized by monoclonal antibody lot1 in the formation of lateral olfactory tract. J Neurosci. 1998;18:7800–7810. doi: 10.1523/JNEUROSCI.18-19-07800.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H, Cunningham JM, Dyck R, Walsh C, Campbell K, Polleux F, Guillemot F. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. EMBO J. 2004;23:2892–2902. doi: 10.1038/sj.emboj.7600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker T, Dennis D, Kurrasch DM, Schuurmans C. Neurog1 and Neurog2 coordinately regulate development of the olfactory system. Neural Dev. 2012;7:28. doi: 10.1186/1749-8104-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart IH, Smart M. The location of nuclei of different labelling intensities in autoradiographs of the anterior forebrain of postnatial mice injected with [3H]thymidine on the eleventh and twelfth days post-conception. J Anat. 1977;123:515–525. [PMC free article] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supèr H, Soriano E, Uylings HB. The functions of the preplate in development and evolution of the neocortex and hippocampus. Brain Res Brain Res Rev. 1998;27:40–64. doi: 10.1016/S0165-0173(98)00005-8. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS., Jr Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiguchi-Hayashi K, Sekiguchi M, Ashigaki S, Takamatsu M, Hasegawa H, Suzuki-Migishima R, Yokoyama M, Nakanishi S, Tanabe Y. Generation of reelin-positive marginal zone cells from the caudomedial wall of telencephalic vesicles. J Neurosci. 2004;24:2286–2295. doi: 10.1523/JNEUROSCI.4671-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomioka N, Osumi N, Sato Y, Inoue T, Nakamura S, Fujisawa H, Hirata T. Neocortical origin and tangential migration of guidepost neurons in the lateral olfactory tract. J Neurosci. 2000;20:5802–5812. doi: 10.1523/JNEUROSCI.20-15-05802.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergaño-Vera E, Yusta-Boyo MJ, de Castro F, Bernad A, de Pablo F, Vicario-Abejón C. Generation of GABAergic and dopaminergic interneurons from endogenous embryonic olfactory bulb precursor cells. Development. 2006;133:4367–4379. doi: 10.1242/dev.02601. [DOI] [PubMed] [Google Scholar]
- Vyas A, Saha B, Lai E, Tole S. Paleocortex is specified in mice in which dorsal telencephalic patterning is severely disrupted. J Comp Neurol. 2003;466:545–553. doi: 10.1002/cne.10900. [DOI] [PubMed] [Google Scholar]
- Wang Z, Shu W, Lu MM, Morrisey EE. Wnt7b activates canonical signaling in epithelial and vascular smooth muscle cells through interactions with Fzd1, Fzd10, and LRP5. Mol Cell Biol. 2005;25:5022–5030. doi: 10.1128/MCB.25.12.5022-5030.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- Winpenny E, Lebel-Potter M, Fernandez ME, Brill MS, Götz M, Guillemot F, Raineteau O. Sequential generation of olfactory bulb glutamatergic neurons by Neurog2-expressing precursor cells. Neural Dev. 2011;6:12. doi: 10.1186/1749-8104-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Martin S, Price DJ. Evidence that the earliest generated cells of the murine cerebral cortex form a transient population in the subplate and marginal zone. Brain Res Dev Brain Res. 1992;66:137–140. doi: 10.1016/0165-3806(92)90150-U. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133:537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- Yun K, Potter S, Rubenstein JL. Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development. 2001;128:193–205. doi: 10.1242/dev.128.2.193. [DOI] [PubMed] [Google Scholar]
- Yun K, Garel S, Fischman S, Rubenstein JL. Patterning of the lateral ganglionic eminence by the Gsh1 and Gsh2 homeobox genes regulates striatal and olfactory bulb histogenesis and the growth of axons through the basal ganglia. J Comp Neurol. 2003;461:151–165. doi: 10.1002/cne.10685. [DOI] [PubMed] [Google Scholar]
- Zhao C, Guan W, Pleasure SJ. A transgenic marker mouse line labels Cajal-Retzius cells from the cortical hem and thalamocortical axons. Brain Res. 2006;1077:48–53. doi: 10.1016/j.brainres.2006.01.042. [DOI] [PubMed] [Google Scholar]
- Zimmer C, Lee J, Griveau A, Arber S, Pierani A, Garel S, Guillemot F. Role of Fgf8 signalling in the specification of rostral Cajal-Retzius cells. Development. 2010;137:293–302. doi: 10.1242/dev.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]