

Abstract
The dramatic increase in the prevalence of obesity reflects a lack of progress in combating one of the most serious health problems of this century. Recent studies have improved our understanding of the appetitive network by focusing on the paraventricular hypothalamus (PVH), a key region responsible for the homeostatic balance of food intake. Here we show that mice with PVH-specific ablation of LIM domain only 4 (Lmo4) become rapidly obese when fed regular chow due to hyperphagia rather than to reduced energy expenditure. Brain slice recording of LMO4-deficient PVH neurons showed reduced basal cellular excitability together with reduced voltage-activated Ca2+ currents. Real-time PCR quantification revealed that LMO4 regulates the expression of Ca2+ channels (Cacna1h, Cacna1e) that underlie neuronal excitability. By increasing neuronal activity using designer receptors exclusively activated by designer drugs technology, we could suppress food intake of PVH-specific LMO4-deficient mice. Together, these results demonstrate that reduced neural activity in LMO4-deficient PVH neurons accounts for hyperphagia. Thus, maintaining PVH activity is important to prevent hyperphagia-induced obesity.
Introduction
Hyperphagia (overeating) is a leading cause of diet-induced obesity. The hypothalamus is the key region controlling feeding behavior. Metabolic signals are sensed by interoceptive neurons of the arcuate (ARC) and ventromedial (VMH) nuclei of the hypothalamus that send synaptic projections to the paraventricular hypothalamic (PVH) nucleus where metabolic signals are integrated to control feeding behavior. PVH neurons are excitatory glutamatergic neurons (Ziegler et al., 2002; Rosin et al., 2003; Hrabovszky et al., 2005; Hur and Zaborszky, 2005; Stocker et al., 2006). Inhibitory inputs to the PVH evoke eating even when animals are well fed (Atasoy et al., 2012; Sternson, 2013). Thus, reduced activity of PVH neurons likely underlies hyperphagia-induced obesity, although current understanding of the mechanisms that control the firing pattern of PVH neurons remains incomplete.
Our previous studies revealed that mice with glutamatergic neuron-specific ablation of LIM domain only 4 (Lmo4), including all neurons of the PVH, the majority of neurons in the VMH, and some neurons in the dorsomedial hypothalamus (DMH), displayed metabolic defects, including diabetes and obesity (Zhou et al., 2012; Pandey et al., 2013). However, given the diverse functions ascribed to each of these hypothalamic nuclei and the complexity of the metabolic phenotype of these LMO4-deficient (CamK2αCre/Lmo4flox/flox) mice, LMO4 may affect multiple modalities of metabolic homeostasis.
LMO4 is a transcription cofactor, essential for CNS development (Chen et al., 2002; Hahm et al., 2004; Tse et al., 2004; Joshi et al., 2009; Duquette et al., 2010; Cederquist et al., 2013). LMO4 mediates calcium-dependent transcription in cortical neurons (Aizawa et al., 2004; Kashani et al., 2006). Our studies further showed that LMO4 regulates calcium-induced calcium release and synaptic plasticity in neurons of the hippocampus (Qin et al., 2012). Thus, several lines of evidence indicate that LMO4 is tied to the regulation of neuronal activity through calcium signaling.
To determine what LMO4 does specifically in the PVH to affect metabolic homeostasis, we ablated Lmo4 selectively in the PVH through Cre recombinase-dependent excision driven by the single-minded 1 (Sim1) promoter (Balthasar et al., 2005). Compared with the CamK2αCre/Lmo4flox/flox mice, Sim1Cre/Lmo4flox/flox mice exhibit early-onset hyperphagia that progresses rapidly to obesity and insulin resistance. In contrast to the CamK2αCre/Lmo4flox/flox mice, insulin resistance in Sim1Cre/Lmo4flox/flox mice is prevented by calorie restriction, demonstrating that the metabolic phenotype is entirely driven by hyperphagia. Electrophysiological studies showed that LMO4 is required to maintain the expression of voltage-activated Ca2+ channels to set the activity of parvocellular PVH neurons. Hyperphagia was suppressed using a pharmacogenetic approach to activate PVH neurons in Sim1Cre/Lmo4flox/flox mice. Collectively, our data show that by setting the basal neural activity of PVH neurons, LMO4 is critically important to control proper food intake.
Materials and Methods
All experimental protocols were approved by the University of Ottawa Animal Care and Veterinary Service and were in accordance with the institutional guidelines.
Mice.
A triple transgenic Sim1Cre/Lmo4flox/flox/ROSA26-EGFP mouse was obtained by breeding Lmo4flox mice (Schock et al., 2008; Zhou et al., 2012; Pandey et al., 2013) with the Sim1Cre (Balthasar et al., 2005) and the ROSA26-EGFP (Mao et al., 2001) mouse lines on the C57BL/6J genetic background for >6 generations. GFP-positive Sim1Cre cells in the PVH were visualized by immunofluorescence for patch-clamp experiments. Genotypes were determined by PCR analysis as described previously (Mao et al., 2001; Balthasar et al., 2005; Schock et al., 2008). The mice were housed at room temperature (23 ± 2°C), fed ad libitum, and were maintained under a 12 h light/dark cycle (lights on at 0700 h). In most cases, experiments were performed with male mice unless stated otherwise. For all experiments, the examiners were blinded to the genotype of the mice.
Brain slice preparation.
Three- to 5-week-old Lmo4 knock-out (KO) and littermate control mice were anesthetized with isoflurane (Sigma). Caudal PVH (bregma −1.30 mm to −2.0 mm) brain slices of 250 μm thickness were sectioned in the coronal plane, in ice-cold carbogen-equilibrated solution containing the following (in mm): 2.5 KCl, 10 MgS04, 1.25 NaH2PO4, 24 NaHCO3, 0.5 CaCl2-2H2O, 11 glucose, and 234 sucrose. The slices were incubated at room temperature (20–24°C) in solution containing the following (in mm): 124 NaCl, 3 KCl, 3 MgS04, 1.25 NaH2PO4, 26 NaHCO3, 1 CaCl2-2H2O, and 10 glucose.
Electrophysiology.
Electrophysiological experiments were performed at room temperature (20–24°C), and the techniques were similar to those reported previously (Zaman et al., 2011).
Intrinsic firing properties were recorded in coronal sections containing the caudal region of the PVH in recording solution (in mm) as follows: 124 NaCl, 3 KCl, 3 MgS04, 1.25 NaH2PO4, 26 NaHCO3, 2.4 CaCl2-2H2O, and 10 glucose bubbled with 95% O2/5% CO2. The Sim1 neurons expressing Cre-dependent GFP marker were visualized through an upright epifluorescence microscope (Nikon FN). Recording electrodes were pulled on Narishige PC-10 from fabricated borosilicate glass capillaries (G150F-4, OD; 1.50 mm, ID; 0.86 mm, Warner Instruments) and had 4–6 mΩ tip resistance when filled with an intracellular solution containing the following (in mm): 140 K-gluconate, 10 KCl, 1 MgCl2, 10 HEPES, 0.02 EGTA, 3 Mg-ATP, and 0.5 Na2-GTP.
Recordings for Ca2+ currents were performed in an extracellular solution as described previously (Sun et al., 2001; Zaman et al., 2011) consisting of the following (in mm): 100 NaCl, 25 tetraethyl ammonium (TEA)-Cl, 5 CaCl2, 20 HEPES, 2 MgCl2, 5 4-amino pyridine (4-AP), 10 glucose, and 0.001 TTX. Recording pipettes were filled with cesium-based internal solution containing 130 mm CsCl, 10 mm HEPES, 5 mm TEA-Cl, 10 mm EGTA, 4 mm MgCl2, 4 mm Mg-ATP, and 0.3 Na2-GTP. The currents were corrected for capacitive and leak currents.
The pH was adjusted to 7.35 with KOH and CsOH for current- and voltage-clamp experiments, respectively. The osmolarity was maintained to 290–300 mosmol/L with sucrose. Signals were amplified with a Multiclamp700-B amplifier (Molecular Devices) and analyzed using pClamp10 and Mini Analysis Program (Synaptosoft).
Stereotaxic adeno-associated viruses (AAV)-designer receptors exclusively activated by designer drugs (DREADD) injections.
Caudal PVH coordinates were as follows: bregma −1.50 mm; midline ±0.25 mm; dorsoventral −4.90 mm; infusion speed = 50 nl/min. Tissue adhesive (3M Vetbond) was applied to promote natural healing. Postoperative analgesia was provided (Tylenol, 1 mg/kg). The 0.3 μl of AAV virus containing hM3Dq or hM4Di was injected bilaterally into caudal PVH of 6-week-old Sim1Cre/Lmo4flox/wt and Sim1Cre/Lmo4flox/flox mice. After surgery, mice were housed at standard temperature, light/dark cycle, chow and water ad libitum, and allowed 3 weeks for recovery and transgene expression.
Food intake studies in AAV-DREADD-injected mice.
Mice were habituated to handling and the metabolic cages. Food intake studies were performed on regular chow. All mice were injected intraperitoneally with saline on the first day and with clozapine-N-oxide (CNO) on the following day. For mice infected with hM3Dq, CNO was administered at 18:00 h (1 mg/kg) and food intake was assessed for 2 h. For mice infected with hM4Di, CNO was administered at 12:00 h (5 mg/kg) (Atasoy et al., 2012) and food intake was assessed for 3 h.
Immunohistochemistry.
Mice were anesthetized with 100 μl (i.p.) of a ketamine/xylazine/acepromazine (100/20/10 mg/ml) mixture. Mice were immediately perfused intracardially with PBS solution followed by 4% PFA, pH 7.4. Brains were fixed in 4% PFA, treated with 20% sucrose, and quick-frozen in isopentane at −35°C. The 30 μm sections containing ARC and caudal PVH were made using Leica cryotome. Slides were dried at 37°C for 30 min and then blocked at room temperature for 1 h in 5% donkey serum containing 0.1% Triton X100. Slides were then incubated for 24 h at 4°C with primary antibodies in 1% donkey serum. Slides were then washed 5 times for 5 min each in PBS before incubation for 1 h at room temperature with secondary antibodies. The antibodies used include goat anti-LMO4 (C-15) (Santa Cruz Biotechnology; 1:200), rabbit anti-c-fos (Santa Cruz Biotechnology; 1:400), Cy2 and Cy5 (Jackson ImmunoResearch Laboratories; 1:800) secondary antibodies. Negative controls omitting the primary antibody were imaged using identical settings (data not shown).
Real-time qRT-PCR.
Total RNA from the caudal hypothalamus containing the PVH was extracted using TRIzol reagent (Invitrogen) followed by ethanol purification, reverse-transcribed to cDNA with random decamers, and reverse-transcriptase (Ambion). An aliquot of cDNA was used for qPCR with specific primers or actin primers together with TaqDNA polymerase/SYBR Green PCR mix (New England Biolabs) with the Rotor-Gene 3000 System (Corbett Life Science). All mRNA levels were normalized to CycloA as described previously (Qin et al., 2012). Gene-specific qPCR primers used included the following: CycloA: forward, 5-GGC CGA TGA CGA GCC C-3; reverse, 5-TGT CTT TGG AAC TTT GTC TGC AAA T-3; Lmo4: forward, 5-GGA CCG CTT TCT GCT CTA TG-3; reverse, 5-AAG CAC CGC TAT TCC CAA AT-3; Cacna1e: forward, 5-CCG ATG ATG ATG AGA GGG AT-3; reverse, 5-TGC TGA CTG TCT TCC AAT GC-3; Cacna1 g: forward, 5-GTA GAC GAG CAG CTT CAG CA-3; reverse, 5-GGT CAA TAC CCT CAG CAT GG-3; Cacna1h: forward, 5-CTC GGT CAT GGT GGC AGA-3; reverse, 5-CCG AGG AGG CGA TAC TGG-3; Cacna1i: forward, 5-CAT GAA GAC CAT GGA CAA CG-3; reverse, 5-TGT CCA TTG GGT GTC ATG G-3; Mc3R: forward, 5-GCCTGCTTATTGGCTTTGTA-3; reverse, 5-TGTAAGTTCTGGAAGGGAGC-3; Mc4R: forward, 5-GGTCGGAAACCATCGTCA-3; reverse, 5-GGAAAGCAGGCTGCAAAT-3; Mc5R: forward, 5-GGAGCAGAGCAGAATGGT-3; reverse, 5-ATGGGTGAGTGCAGGTTT-3.
Glucose and insulin tolerance test.
Mice were fasted overnight (∼16 h) in fresh cages with free access to water. Tests were performed at 10:00 h. Basal blood glucose was measured before mice receiving 20% d-glucose (2 g/kg body weight, i.p.). At 15, 30, 60, and 120 min, blood glucose was sampled from the saphenous vein using a standard glucometer (Pandey et al., 2013). Mice were fasted for 4 h before the insulin tolerance test. The test was performed between 14:00 and 17:00 h. Human recombinant insulin (Sigma, catalog #91077C) diluted in sterile saline was administered by intraperitoneal injection at 0.75 U/kg. Blood glucose levels were monitored in the same manner as described for the glucose tolerance test protocol at 4 different time points: before (T0), and 15, 30, and 60 min after insulin injection. Data were presented as percentage T0 blood glucose versus time.
Paired feeding.
Mice were housed individually throughout the study (4–17 weeks). Food intake was measured in littermate control mice for 1 week as described previously (Zhou et al., 2012). In the paired-feeding experiment, Lmo4 KO mice and their littermate controls were provided an average amount of food consumed by age-matched control mice. Mice were fed twice daily, one-third of food was given at 9 A.M. and two-thirds at 5 P.M. to ensure that mice never underwent long periods of fasting. Body weight was measured twice a week over the pair-feeding period, after which mice were subjected to glucose tolerance or insulin tolerance tests.
Indirect calorimetry.
Mice were single-housed in metabolic chambers for 24 h and oxygen consumption (VO2) and carbon dioxide production (VCO2) were measured using an Oxymax System with automatic temperature and light controls (Columbus Instruments). Temperature was maintained at 24°C, and lighting was on a normal 12 h light/dark cycle. System settings included a flow rate of 0.5 L/min, a sample line-purge time of 2 min, and a measurement period of 60 s every 12 min.
Protein-phosphatase 1B (PTP1B) phosphatase activity assay.
PTP1B phosphatase activity was measured with the PhosphoSeek PTP1B Assay Kit (BioVision) in extracts from the dorsal hypothalamus (mainly PVH and DMH) wedges according to the manufacturer's instructions with PTP1B enzyme and phosphatase inhibitor as positive and negative controls, respectively (Pandey et al., 2013).
Statistical analysis.
Data acquisition, analyses, and presentation were performed using a combination of pClamp10 (Molecular Devices), the Statistical Package for the Social Sciences version 14 (SPSS), and the Sigma Plot 11 (Jandel Scientific). All data are presented as mean ± SEM unless stated otherwise. p values of < 0.05 were considered statistically significant. Two-way ANOVA and two-tailed t tests were used for comparisons, where appropriate.
Results
Ablation of Lmo4 in Sim1 PVH neurons causes hyperphagia and obesity
In the hypothalamus, Sim1 is expressed in neurons of the PVH nucleus and Sim1Cre transgenic mice expressing Cre recombinase under the control of the Sim1 promoter enable PVH-selective ablation of floxed alleles (Balthasar et al., 2005). qRT-PCR analysis showed that Lmo4 mRNA levels were reduced in the dorsal hypothalamus of Sim1Cre/Lmo4flox/flox KO mice compared with their littermate controls (Sim1Cre/Lmo4flox/wt, WT) (Fig. 1A). Absence of LMO4 in the PVH was confirmed by immunofluorescence (Fig. 1B, top). Sim1 is also expressed in the supraoptic nucleus, the nucleus of the lateral olfactory tract, and the medial amygdala (Balthasar et al., 2005), but we did not detect LMO4 expression in these regions (data not shown). Ablation of LMO4 did not reduce the number of Sim1-positive PVH neurons (Fig. 1B, bottom). Sim1Cre/Lmo4flox/flox mice are morbidly obese even when fed regular chow (Fig. 1C). The early-onset obesity started at 5 weeks (2 weeks after weaning) and was observed in both male and female mice (Fig. 1D). By 3 months of age, mice had impaired glucose homeostasis (Fig. 1E) and were insulin resistant (Fig. 1F). No change in locomotor activity (Fig. 2A) or energy expenditure (Fig. 2B) was observed at 5 weeks of age (the onset of weight gain). Instead, increased body weight was associated with hyperphagia detected as early as 5 weeks of age (Fig. 2C). To test whether increased food consumption is the result of increased hedonic behavior (i.e., the pleasure of eating), we conducted a sugar preference test. Lmo4 KO mice consumed as much sugar water as littermate controls (Fig. 2D), suggesting that aberrant hedonic behavior is unlikely to be the cause of hyperphagia. It is also worth noting that qPCR analysis showed no change in the hypothalamic mRNA levels of neuropeptides, including Agouti-related protein, neuropeptide Y, pro-opiomelanocortin, oxytocin, vasopressin, and thyrotropin releasing hormone in the Lmo4 KO PVH (Fig. 2E). Also of note, mRNA levels of Sim1 were not affected by Lmo4 ablation (Fig. 2E). Similarly, mRNA levels for melanocortin receptors that mediate the satiety response to αMSH were not different in the Lmo4 KO PVH (Fig. 2F).
Figure 1.
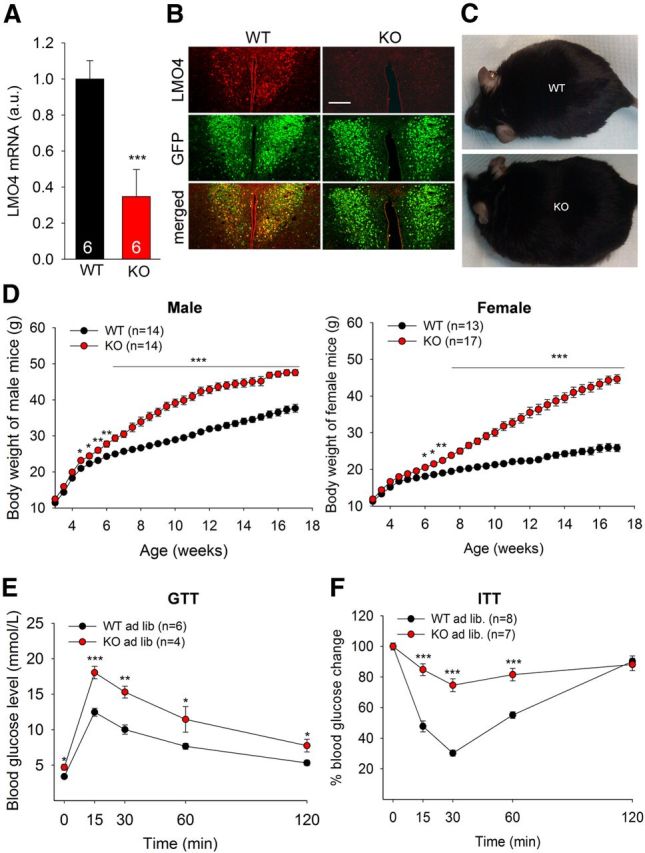
Lmo4 ablation in the PVH causes obesity and insulin resistance. A, Real-time qRT-PCR (n = 6 per genotype) showed reduced LMO4 mRNA from hypothalamic extracts. B, Immunofluorescent staining showed that LMO4 (in red) is expressed in Cre-expressing GFP-positive cells (in green) of the PVH of Sim1-Cre/Lmo4flox/wt/ROSA26-EGFP (WT) mice and confirmed loss of LMO4 in Cre-expressing GFP-positive cells in Sim1-Cre/Lmo4flox/flox /ROSA26-EGFP mice (KO). Scale bar, 150 μm. C, Lmo4 KO mice are morbidly obese at 3 months. D, Body weight progression in mice fed regular chow ad libitum revealed significant weight gain in Lmo4 KO mice (male: F(1,28) = 996.714; female: F(1,28) = 1401.379 by two-way ANOVA). *p < 0.05. **p < 0.01. ***p < 0.001. Glucose tolerance test (E) and insulin tolerance test (F) at 3 months. Data are mean ± SEM. *p < 0.05 (Student's two-tailed t test). **p < 0.01 (Student's two-tailed t test). ***p < 0.001 (Student's two-tailed t test).
Figure 2.

Hyperphagia accounts for metabolic phenotypes in mice with Lmo4 ablation in the PVH. A, Beam-break counts for locomotor activity. B, Energy expenditure measured by oxygen consumption and CO2 production (n = 8 WT and 6 KO male mice). C, Food intake >5 d. D, Sucrose preference test. Mice were offered the choice between water and water with 1% sucrose for the test. qRT-PCR of hypothalamic mRNAs shows no difference in the expression levels of (E) Sim1 and various neurotransmitters and (F) melanocortin receptors. n = 6 male KO and 6 male littermate control (WT) mice. Body weight progression (G), glucose tolerance test (H), and insulin tolerance test (I) under calorie-restricted paired-feeding in female mice. All the measurements were done at 5 weeks of age, except for paired-feeding. Data are mean ± SEM. *p < 0.05 (Student's two-tailed t test). POMC, Pro-opiomelanocortin; AgRP, Agouti-related protein; NPY, neuropeptide Y; OT, oxytocin; AVP, arginine vasopressin; TRH, thyrotropin releasing hormone.
To determine whether obesity and diabetes are the result of increased food intake, we matched the caloric intake of Lmo4 KO mice with littermate controls. Paired-feeding normalized body weight progression (Fig. 2G) and prevented glucose intolerance (Fig. 2H) and insulin resistance (Fig. 2I), confirming that the lack of LMO4 in the PVH caused obesity and diabetes entirely through hyperphagia.
Lmo4-ablated Sim1 PVH neurons have reduced activity
Using patch-clamp recording, we compared neuronal activity in Sim1 neurons of the caudal PVH in Lmo4 KO and littermate control mice. To visualize Sim1 neurons, Sim1Cre/Lmo4flox/flox mice were bred with ROSA26-EGFP mice allowing enhanced GFP expression in Sim1 neurons that express Cre-recombinase (Fig. 3A). Spontaneous firing was much reduced in Lmo4 KO mice compared with littermate controls (Fig. 3B,C), despite similar resting membrane potentials (Table 1). Reduced spontaneous firing could result from increased inhibitory inputs, reduced excitatory inputs, or a cell-intrinsic reduction in excitability resulting from altered expression of membrane channels. Membrane resistance was significantly elevated (by 50%) in Lmo4-ablated neurons (Table 1), indicating that fewer membrane channels are present in Lmo4 KO PVH neurons. Of note, membrane capacitance (a measure of cell size) was not different between Lmo4 KO and littermate control PVH neurons.
Figure 3.
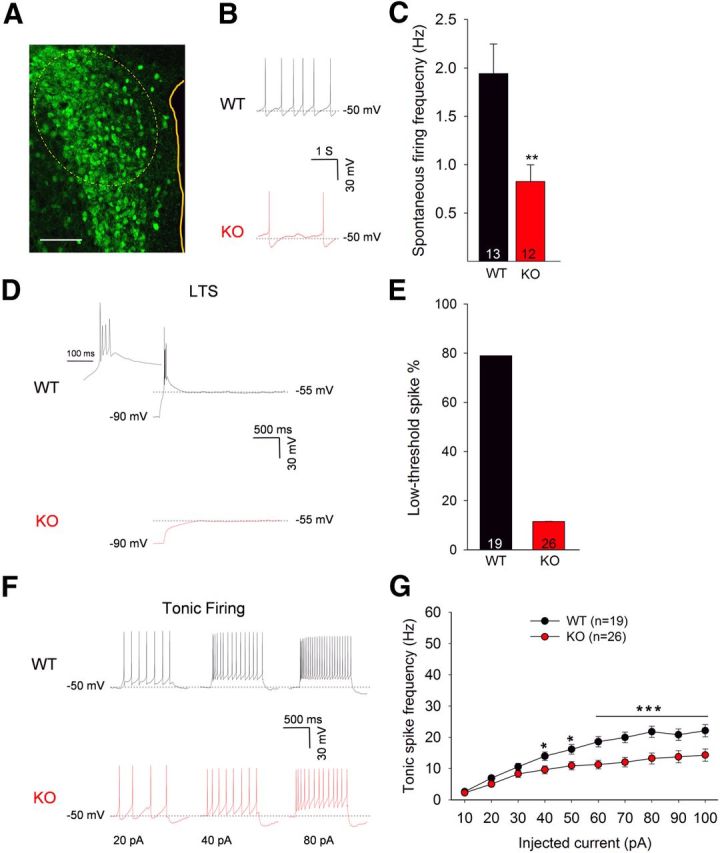
Neuronal activity of LMO4-deficient Sim1 neurons is reduced. A, GFP-positive neurons of the PVH (dashed oval, area recorded). Scale bar, 100 μm. B, Representative traces showing spontaneous firing. C, Lmo4 KO neurons have a lower spontaneous firing frequency than their littermate controls (WT). D, Representative traces show low-threshold spiking (LTS) in Lmo4 KO and littermate control (WT) neurons of the caudal PVH. LTS, containing 2–4 action potentials at ∼60 Hz, were elicited by injecting negative current (1 s) to hyperpolarize the membrane from −50 mV (holding potential) to −90 mV. Enlarged (200 ms) traces (inset) are shown. E, Percentage of the Sim1 neurons (WT, n = 19 from 7 mice; KO, n = 26 from 9 mice) showing LTS. F, Representative traces showing neuronal activity in response to depolarizing currents (20, 40, and 60 pA) from −50 mV of holding potential. G, Tonic spike frequency in response to various depolarizing currents presented as mean ± SEM, F(1,9) = 64.90; two-way ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001.
Table 1.
Membrane properties of Sim1 GFP-positive parvocellular neurons of the PVH
| Genotype of neurons | Cm (pF) | Rm (mΩ) | RMP (mV) |
|---|---|---|---|
| WT (N = 18)a | 35.25 ± 1.97 | 491.57 ± 48.61 | −54.22 ± 1.34 |
| Lmo4 KO (N = 23)b | 30.91 ± 1.22 | 735.62 ± 83.88 | −54.22 ± 1.04 |
| p value | 0.058 | 0.025 | 0.976 |
Cm, Membrane capacitance; Rm, membrane resistance; RMP, resting membrane potential.
aCells from *7 littermate control (WT) mice.
bCells from 9 Lmo4 KO mice.
To determine whether calcium currents are affected in Lmo4 KO PVH neurons and might account for the reduced excitability, we examined low-threshold spiking (LTS) and tonic firing, two properties closely tied to the low-voltage-activated (LVA) and high-voltage-activated (HVA) calcium channels, respectively. LTS was evoked by a hyperpolarizing current injection (Sun et al., 2001; Zaman et al., 2011). Under current-clamp mode, releasing a hyperpolarizing current generates LTS by allowing LVA Ca2+ channels to recover from inactivation (Suzuki and Rogawski, 1989). LTS was detected in 78.5% of littermate control PVH neurons (n = 15 of 19), whereas ∼21% (n = 4 of 19) of the cells exhibited no LTS (Fig. 3D,E), as reported previously for parvocellular neurons of the caudal PVH (Luther et al., 2002). In contrast, only 12% of GFP-positive LMO4-deficient neurons (n = 3 of 26) exhibited LTS (Fig. 3D,E).
Tonic firing was induced by injecting depolarizing currents in 10 pA stepwise increments (10 steps, 1 s duration) from a holding potential of −50 mV. Increased tonic spike frequency was observed with each increment of injected current. However, a much lower response was observed in Lmo4 KO neurons (Fig. 3F,G). Thus, ablation of Lmo4 in PVH neurons likely decreases the expression of both low- and high-voltage-gated calcium channels, and these would reduce spontaneous firing and cell excitability as we observed in Lmo4-ablated PVH neurons.
Reduced low- and high-voltage-gated Ca2+ currents in Lmo4 KO PVH neurons
Ca2+ channels play a critical role in neuronal excitability (Perez-Reyes, 2003). The markedly reduced LTS we observed in LMO4-deficient PVH neurons (Fig. 3D,E) led us to examine the status of LVA Ca2+ currents. We measured LVA currents in Cs+-based whole-cell patch clamping as described previously (Sun et al., 2001). Neurons were held at −100 mV (1 s) and depolarized to −40 mV, below the activation threshold for HVA Ca2+ channels. This depolarization step induces a fast-inactivating T-type Ca2+ current (Fox et al., 1987). We observed a significant reduction in peak current density of Ca2+ in LMO4-deficient PVH neurons (−1.60 ± 0.45 pApF−1 [n = 12] in KO vs −7.28 ± 2.01 pApF−1 [n = 18] in WT; p > 0.05; Fig. 4A,B).
Figure 4.

Total calcium current is reduced in LMO4-deficient Sim1 neurons of the PVH. A, Left, Representative traces of LVA Ca2+ currents elicited by depolarizing voltage steps from −100 mV to a test potential of −40 mV for 1 s in littermate control (WT) and Lmo4 KO PVH neurons. Right, HVA Ca2+ currents elicited by depolarizing steps from a holding potential of −50 mV to a test potential of 0 mV for 1 s. B, Normalized LVA Ca2+current at the test potential −40 mV. C, I-V curves for HVA Ca2+currents measured from the peak amplitudes (WT: n = 22 cells, 7 mice; KO: n = 19 cells, 6 mice). Data are mean ± SEM. F(1,8) = 80.38 by two-way ANOVA: *p < 0.05; ***p < 0.001.
In addition, the reduced tonic firing frequency we observed in response to depolarizing inputs (Fig. 4F,G) suggests that HVA Ca2+ channels may also be altered. To test this possibility, an inward current was evoked by depolarizing voltage steps from a holding potential of −50 mV to test potentials between −50 to 30 mV (Sun et al., 2001; Zaman et al., 2011). Most LVA Ca2+ channels should stay inactivated under these conditions. The peak current density in Lmo4 KO (−8.33 ± 2.13, −13.25 ± 1.57, and −14.97 ± 1.21 pApF−1; n = 19) compared with WT (−19.28 ± 2.29, − 22.34 ± 1.54, and −22.53 ± 1.61 pApF−1; n = 22) Sim1 neurons was significantly reduced at test potentials of −10 mV (p < 0.001), 0 mV (p < 0.001), and 10 mV (p < 0.001), respectively (Figure 3A,C).
To further examine which component of HVA Ca2+ current was affected in these neurons, we isolated nifedipine-sensitive L-type and SNX-482-sensitive R-type (α1E) Ca2+ components, currents that contribute the larger fraction of extracellular Ca2+ influx (Tsien, 1983; Perez-Reyes, 2003). Comparative analysis showed that the SNX-482-sensitive Ca2+ component was significantly reduced in LMO4-deficient PVH neurons at a test potential of 0 mV (WT, −7.043 ± 1.82 pApF−1 vs KO, −0.67 ± 0.29 pApF−1; p < 0.01; Fig. 5A,B).
Figure 5.

R-type component of HVA Ca2+ currents is significantly reduced in LMO4-deficient PVH neurons. A, Representative traces showing HVA Ca2+ currents elicited by depolarizing voltage steps from −50 mV to a test potential of 0 mV for 1 s. B, Peak current density of littermate control (WT) and Lmo4 KO PVH neurons under different experimental conditions: Ctr., Control; Nif., nifedipine 10 μm; +SNX, SNX-482. Data are mean ± SEM. **p < 0.01 (Student's two-tailed t test). C, Real-time qRT-PCR of mRNAs for different α-subunits of voltage-activated Ca2+ channels in GFP-positive PVH neurons from 5-week-old mice. Data are mean ± SEM. *p < 0.05 (Student's two-tailed t test).
Our recent findings suggest that LMO4 is a metabolic responsive inhibitor of PTP1B (Pandey et al., 2013). In light of this finding, we asked whether ablation of Lmo4 alters PTP1B activity in the PVH and thereby affects Ca2+ currents. Surprisingly, no significant change in PTP1B activity was detected in Lmo4 KO PVH (data not shown) and pharmacological blockade of PTP1B by trodusquemine (10 μm) (Lantz et al., 2010) had no effect on Ca2+ currents in LMO4-deficient neurons (data not shown), suggesting that LMO4 likely regulates calcium channels at the transcriptional rather than the post-transcriptional level. This result also suggests that PTP1B does not affect the activity of these Ca2+ channels.
Given the role of LMO4 as a transcriptional cofactor (Kashani et al., 2006), we examined whether the expression of genes encoding the Ca2+ channel α subunit is affected by ablation of Lmo4 by real-time qRT-PCR using mRNA purified from the cadual PVH. We found that mRNA of Cacna1e and Cacna1h, α subunits of R-type and T-type low-voltage calcium channels, respectively, were significantly reduced in Lmo4 KO PVH neurons (Fig. 5C), confirming that LMO4 is required for the normal expression of voltage-activated Ca2+ channels to maintain the cellular excitability of PVH neurons.
Pharmacogenetic activation or suppression of PVH neuronal activity oppositely affects food intake
Increased food intake associated with reduced basal cellular excitability in PVH neurons of KO mice led us to postulate whether directly modulating PVH activity in vivo might alter feeding behavior. This was achieved using a pharmacogenetic approach: DREADD (Alexander et al., 2009). AAV expressing either the silencer Gαi protein-coupled receptor (hM4Di) or the activator Gαq protein-coupled receptor (hM3Dq) were injected stereotactically and bilaterally into the caudal PVH of 6-week-old mice (Fig. 6A) and subsequently activated by the designer drug (i.e., the pharmacologically inert ligand) CNO.
Figure 6.

Pharmacogenetic silencing and activation of Sim1 neurons modulate food intake in opposite directions. A, Schematic diagram indicating stereotaxic injection of Cre-inducible AAV-hM3Dq-mCherry (activator) and AAV-hM4Di-mCherry (inhibitor) into the PVH of littermate control (WT) and Lmo4 KO mice (left) and fluorescent image showing the expression of mCherry from injected viruses (right). Scale bar, 100 μm. B, Food intake is increased 3 h after CNO intraperitoneal administration in AAV-hM4Di-mCherry-injected littermate control (WT, Sim1Cre/Lmo4flox/wt/ROSA-EGFP, n = 6). C, Food intake was suppressed 2 h after CNO intraperitoneal administration in AAV-hM3Dq-mCherry-injected littermate control (WT, n = 6) and Sim1Cre/Lmo4flox/flox/ROSA26-EGFP (Lmo4 KO, n = 6). Data are mean ± SEM. **p < 0.01 (Student's two-tailed t test). ***p < 0.001 (Student's two-tailed t test). D, Fluorescent staining showed that nearly all (98%) Sim1-positive neurons (green) expressing Cre-recombinase activate the expression of the injected AAV-hM3Dq-mCherry viral transgene (magenta) and appear white in the merged image. Scale bar, top: 50 μm. CNO treatment activates c-fos expression (82%, in AAV-hM3Dq-mCherry-injected PVH neurons) in Sim1Cre/ROSA26-EGFP mice. Scale bar, bottom: 75 μm.
Reduced PVH activity in WT mice (Sim1-Cre/Lmo4flox/wt) with the silencer hM4Di significantly increased food intake 3 h after CNO administration (5 mg/kg i.p.) (Atasoy et al., 2012) compared with saline treatment (0.22 ± 0.04 g in saline vs 0.58 ± 0.03 g in CNO; n = 6; p < 0.0001; Fig. 6B). The increase in food intake was observed during the light period, when mice normally refrain from eating. Conversely, increasing PVH activity with the activator hM3Dq suppressed food intake in WT mice over a 2 h period after CNO administration (1 mg/kg i.p.) (0.72 ± 0.03 g in saline vs 0.38 ± 0.08 g in CNO; n = 6; p = 0.005; Fig. 6C). The reduction in food intake was observed during the dark period, when most food intake normally occurs.
Last, we tested whether pharmacogenetic activation of the PVH could override the hyperphagic effect of Lmo4 deletion. CNO-induced depolarization is thought to occur through its inhibition of M current carried by PIP2-gated KCNQ channels (Biervert et al., 1998; Wang et al., 1998), a slowly inactivating, outwardly rectifying potassium current important for depression of neuronal excitability (Brown and Yu, 2000). Indeed, the activator hM3Dq suppressed food intake in Lmo4 KO mice to the same degree as was observed in littermate control mice (Fig. 6C). Immunostaining for the immediate early gene c-fos confirmed the precision of virus injection and that CNO administration specifically activated PVH neurons (Fig. 6D). Thus, these results show that maintaining neuronal excitability in the PVH is required to suppress food intake. In summary, our findings indicate that LMO4 is critical for the normal expression of voltage-activated Ca2+ channels required for cellular excitability underlying homeostatic regulation of appetite.
Discussion
In this study, we have identified an essential role for LMO4 in maintaining neuronal activity of the PVH to control feeding behavior. We have shown that the lack of LMO4 in PVH neurons impairs normal activity and the normal expression of voltage-activated Ca2+ channels required for neuronal activity. Consequently, reduced activity of the PVH profoundly increases body weight resulting from hyperphagia. In addition, pharmacogenetic inactivation of PVH neurons induced hyperphagia in WT mice, whereas their pharmacogenetic activation blocked the hyperphagic phenotype of Lmo4 KO mice.
A recent study elegantly demonstrated that inhibitory input from orexigenic AgRP neurons to the PVH neurons, particularly parvocellular oxytocin neurons, increases acute feeding behavior (Atasoy et al., 2012). This study also showed that suppressing the activity of PVH Sim1 neurons using the same pharmacogenetic approach (Sim1Cre and hMD4i) that we used here could effectively trigger acute feeding behavior. It is important to note that we observed no change in the mRNA levels of hypothalamic neuropeptides, including, AgRP, NPY, POMC, oxytocin, vasopressin, and TRH. Thus, the chronic hyperphagia we observe in Sim1Cre/Lmo4flox/flox mice cannot be explained from altered upstream signaling from the ARC. In addition, because Sim1 is not expressed in other brain regions that have been implicated in feeding behavior, like the VMH and DMH nuclei where LMO4 expression remains unaffected in Sim1-Cre mice (data not shown), the selective loss of Lmo4 in Sim1 neurons of the PVH is highly likely to account for the observed phenotype of hyperphagia. Both spontaneous activity and excitability (LTS) of the Sim1 PVH neurons were significantly reduced in Lmo4 KO mice as early as 3 weeks. Consistent with this notion, pharmacogenetic activation of Sim1 neurons with AAV-hMD3q acutely suppressed food intake in WT Sim1Cre mice during the dark cycle, when mice are hungry and AgRP neurons are normally activated. The fact that we were able to suppress food intake in adult Lmo4 KO (Sim1Cre/Lmo4flox/flox) mice at 9 weeks of age to the same extent as in WT Sim1Cre mice further strengthens our conclusion that hyperphagia in Lmo4 KO mice results from loss of Sim1 neuron excitability. This is not the result of reduced Sim1 expression, as revealed by qPCR, or loss of Sim1-positive neurons, as revealed by GFP immunofluorescence, or cellular atrophy because a similar membrane capacitance (an index of cell size) was observed in Lmo4 KO PVH neurons. In addition, that feeding suppression was restored by activating Sim1 neurons argues that Sim1 neurons of the PVH and their projections remain intact despite the loss of LMO4. These observations strongly indicate a role for LMO4 in maintaining the activity of Sim1 PVH neurons to prevent excess food intake.
Reduced excitability in Lmo4 KO PVH neurons was associated with reduced expression of R-type and T-type channels. Parvocellular nonsecretory neurons of the posterior PVH respond to release from a hyperpolarizing current-clamp with LTS through T-type calcium channels (Luther et al., 2002). Among low-voltage-activated T-type Ca2+ channels, Cacna1i was barely detectable, Cacna1g expression was unchanged, and only Cacna1h was significantly reduced by Lmo4 ablation. In addition, expression of the intermediate-voltage-activated R-type channel Cacna1e mRNA was also reduced with Lmo4 ablation. Little is known about the transcriptional regulation of Cacna1e. Cacna1h can be induced by the transcription factor Egr1 (van Loo et al., 2012). It remains to be seen whether LMO4 directly modulates Egr1-dependent activation of Cacna1h.
The phenotype of Sim1Cre/Lmo4flox/flox mice is less complex than that of CamK2αCre/Lmo4flox/flox mice. The early-onset obesity observed in Sim1Cre/Lmo4flox/flox mice contrasts with the late-onset obesity we observed in CamK2αCre/Lmo4flox/flox mice (Zhou et al., 2012). In the latter model, Lmo4 was ablated in glutamatergic neurons that make up all of the neurons of the PVH, most of the VMH, and some of the DMH (Bailey et al., 2003; Xu and Tong, 2011). Thus, we were surprised by the early voracious appetite of Sim1Cre/Lmo4flox/flox mice because no other hypothalamic nuclei other than the PVH are affected in these mice. The obesity and diabetes phenotypes could be prevented by calorie restriction through paired feeding, indicating that the metabolic phenotype was entirely the result of overeating. In contrast, paired feeding only prevented obesity but did not rescue the diabetes phenotype of CamK2αCre/Lmo4flox/flox mice (Pandey et al., 2013). CamK2αCre/Lmo4flox/flox mice are defective in central leptin signaling in the VMH and DMH and consequently have impaired glucose homeostasis and reduced sympathetic outflow to peripheral tissues, respectively. Reduced sympathetic outflow was found to affect peripheral insulin sensitivity, insulin secretion from the pancreas, lipid metabolism, and blood pressure (Zhou et al., 2012; Pandey et al., 2013). Whereas CamK2αCre/Lmo4flox/flox mice had reduced systolic and diastolic blood pressures (Pandey et al., 2013), Sim1Cre/Lmo4flox/flox mice had normal blood pressures, even when they were morbidly obese (data not shown).
It is noteworthy that the response of PVH neurons to leptin signaling (i.e., a suppression of spontaneous firing frequency that was reported previously; Ghamari-Langroudi et al., 2011) was not affected by ablation of Lmo4 in Sim1Cre/Lmo4flox mice (data not shown). This result contrasts with the loss of leptin signaling we noted in the VMH and DMH of CamK2αCre/Lmo4flox/flox mice (Zhou et al., 2012; Pandey et al., 2013) and indicates that the effect of leptin to reduce spontaneous firing in PVH neurons is not affected by LMO4 deficiency and may involve a Jak/Stat3-independent signaling mechanism.
If Lmo4 was ablated in the PVH of CamK2αCre/Lmo4flox/flox mice, then why do they not develop hyperphagia early on as do the Sim1Cre/Lmo4flox/flox mice? One possibility may relate to an attenuating effect of the ablation of Lmo4 in the glutamatergic neurons of the DMH in CamK2αCre/Lmo4flox/flox mice. DMH neurons project to orexin neurons that increase feeding and energy expenditure (Stotz-Potter et al., 1996; Sakurai et al., 2005). Thus, ablation of Lmo4 in the glutamatergic population of the DMH might reduce feeding and counteract the hyperphagic effect of Lmo4 ablation in the PVH.
A mutation that disrupts Sim1 causes hyperphagia and obesity in humans (Holder et al., 2000), and a study of mice with Sim1 haploinsufficiency revealed that loss of oxytocin signaling from the PVH accounted for the severe hyperphagia (Kublaoui et al., 2008). Loss of function of oxytocin neurons in the PVH has been linked to Prader-Willi syndrome, a neurological disorder associated with voracious appetite and obesity (Swaab et al., 1995). It will be interesting to see whether the Prader-Willi syndrome affects LMO4 expression in PVH neurons. Our study reveals that LMO4 plays a central role in establishing caudal Sim1 PVH neuron excitability through modulation of voltage-gated Ca2+ channels and that Sim1 neuron-specific ablation of Lmo4 during development causes hyperphagia. It remains to be seen whether ablation of Lmo4 after birth or in adulthood would have a similar effect on feeding behavior. A recent study showed that activation of oxytocin neurons, a subset of Sim1 neurons of the caudal PVH, strongly suppresses feeding behavior (Atasoy et al., 2012). We suspect that the main hyperphagic effect of Lmo4 ablation occurs by disrupting excitability of oxytocin neurons. However, a recent study noted that ablated oxytocin neurons in adult mice did not cause hyperphagia but rather affected energy expenditure (Wu et al., 2012). In addition to oxytocin neurons, other PVH neurons like those expressing arginine vasopressin might be responsible for the control of feeding behavior (Aoyagi et al., 2009). Future studies using an oxytocin or arginine vasopressin promoter-Cre transgenic mouse will be required to address whether LMO4 is required for oxytocin or arginine vasopressin neuron-dependent control of feeding behavior.
Footnotes
This work was supported by the Canadian Diabetes Association (OG-3-11-3520-HC), Ontario Research Fund, Canada Foundation for Innovation, Heart and Stroke Foundation of Canada (Grants-in-Aid NA6301 and G-13-0002596), Canadian Institutes of Health Research (MOP-86745 and MOP-130567) and HSFO Centre for Stroke Recovery Catalyst Grants to H.-H.C., H.-H.C. is also supported by the Henry J.M. Barnett Research Scholarship and New Investigator Award from the Heart and Stroke Foundation of Canada and Early Researcher Awards from the Ontario Ministry of Research and Innovation. A.F.R.S. is supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant), the Canadian Institutes for Health Research (MOP-259068), and Canadian Diabetes Association (OG-3-11-3520-HC). Z.Q. and K.K. are supported by Natural Sciences and Engineering Research Council of Canada Graduate Scholarships. We thank Dr. Mary-Ellen Harper for the use of the metabolic cages and Dr. Gregory Morton for helpful discussion.
The authors declare no competing financial interests.
References
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi T, Kusakawa S, Sanbe A, Hiroyama M, Fujiwara Y, Yamauchi J, Tanoue A. Enhanced effect of neuropeptide Y on food intake caused by blockade of the V(1A) vasopressin receptor. Eur J Pharmacol. 2009;622:32–36. doi: 10.1016/j.ejphar.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488:172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Nicol GD, Schild JH, DiMicco JA. Synaptic and membrane properties of neurons in the dorsomedial hypothalamus. Brain Res. 2003;985:150–162. doi: 10.1016/S0006-8993(03)03047-6. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Brown BS, Yu SP. Modulation and genetic identification of the M channel. Prog Biophys Mol Biol. 2000;73:135–166. doi: 10.1016/S0079-6107(00)00004-3. [DOI] [PubMed] [Google Scholar]
- Cederquist GY, Azim E, Shnider SJ, Padmanabhan H, Macklis JD. Lmo4 establishes rostral motor cortex projection neuron subtype diversity. J Neurosci. 2013;33:6321–6332. doi: 10.1523/JNEUROSCI.5140-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HH, Yip JW, Stewart AFR, Frank E. Differential expression of a transcription regulatory factor, the LIM domain only 4 protein Lmo4, in muscle sensory neurons. Development. 2002;129:4879–4889. doi: 10.1242/dev.129.21.4879. [DOI] [PubMed] [Google Scholar]
- Duquette PM, Zhou X, Yap NL, MacLaren EJ, Lu JJ, Wallace VA, Chen HH. Loss of LMO4 in the retina leads to reduction of GABAergic amacrine cells and functional deficits. PLoS One. 2010;5:e13232. doi: 10.1371/journal.pone.0013232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Srisai D, Cone RD. Multinodal regulation of the arcuate/paraventricular nucleus circuit by leptin. Proc Natl Acad Sci U S A. 2011;108:355–360. doi: 10.1073/pnas.1016785108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm K, Sum EY, Fujiwara Y, Lindeman GJ, Visvader JE, Orkin SH. Defective neural tube closure and anteroposterior patterning in mice lacking the LIM protein LMO4 or its interacting partner Deaf-1. Mol Cell Biol. 2004;24:2074–2082. doi: 10.1128/MCB.24.5.2074-2082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Wittmann G, Turi GF, Liposits Z, Fekete C. Hypophysiotropic thyrotropin-releasing hormone and corticotropin-releasing hormone neurons of the rat contain vesicular glutamate transporter-2. Endocrinology. 2005;146:341–347. doi: 10.1210/en.2004-0856. [DOI] [PubMed] [Google Scholar]
- Hur EE, Zaborszky L. Vglut2 afferents to the medial prefrontal and primary somatosensory cortices: a combined retrograde tracing in situ hybridization study [corrected] J Comp Neurol. 2005;483:351–373. doi: 10.1002/cne.20444. [DOI] [PubMed] [Google Scholar]
- Joshi K, Lee S, Lee B, Lee JW, Lee SK. LMO4 controls the balance between excitatory and inhibitory spinal V2 interneurons. Neuron. 2009;61:839–851. doi: 10.1016/j.neuron.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashani AH, Qiu Z, Jurata L, Lee SK, Pfaff S, Goebbels S, Nave KA, Ghosh A. Calcium activation of the LMO4 transcription complex and its role in the patterning of thalamocortical connections. J Neurosci. 2006;26:8398–8408. doi: 10.1523/JNEUROSCI.0618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol. 2008;22:1723–1734. doi: 10.1210/me.2008-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz KA, Hart SG, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, McLane MP. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring) 2010;18:1516–1523. doi: 10.1038/oby.2009.444. [DOI] [PubMed] [Google Scholar]
- Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC, Tasker JG. Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol. 2002;14:929–932. doi: 10.1046/j.1365-2826.2002.00867.x. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Chapdelaine A, Yang H, Orkin SH. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood. 2001;97:324–326. doi: 10.1182/blood.V97.1.324. [DOI] [PubMed] [Google Scholar]
- Pandey NR, Zhou X, Qin Z, Zaman T, Gomez-Smith M, Keyhanian K, Anisman H, Brunel JM, Stewart AF, Chen HH. The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J Neurosci. 2013;33:12647–12655. doi: 10.1523/JNEUROSCI.0746-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Qin Z, Zhou X, Gomez-Smith M, Pandey NR, Lee KF, Lagace DC, Béïque JC, Chen HH. LIM domain only 4 (LMO4) regulates calcium-induced calcium release and synaptic plasticity in the hippocampus. J Neurosci. 2012;32:4271–4283. doi: 10.1523/JNEUROSCI.6271-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG. Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol. 2003;465:593–603. doi: 10.1002/cne.10860. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, Kageyama H, Kunita S, Takahashi S, Goto K, Koyama Y, Shioda S, Yanagisawa M. Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron. 2005;46:297–308. doi: 10.1016/j.neuron.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Schock SC, Xu J, Duquette PM, Qin Z, Lewandowski AJ, Rai PS, Thompson CS, Seifert EL, Harper ME, Chen HH. Rescue of neurons from ischemic injury by PPARγ requires a novel essential cofactor LMO4. J Neurosci. 2008;28:12433–12444. doi: 10.1523/JNEUROSCI.2897-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternson SM. Hypothalamic survival circuits: blueprints for purposive behaviors. Neuron. 2013;77:810–824. doi: 10.1016/j.neuron.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Simmons JR, Stornetta RL, Toney GM, Guyenet PG. Water deprivation activates a glutamatergic projection from the hypothalamic paraventricular nucleus to the rostral ventrolateral medulla. J Comp Neurol. 2006;494:673–685. doi: 10.1002/cne.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz-Potter EH, Willis LR, DiMicco JA. Muscimol acts in dorsomedial but not paraventricular hypothalamic nucleus to suppress cardiovascular effects of stress. J Neurosci. 1996;16:1173–1179. doi: 10.1523/JNEUROSCI.16-03-01173.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA. Neuropeptide Y receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. J Physiol. 2001;531:67–79. doi: 10.1111/j.1469-7793.2001.0067j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Rogawski MA. T-type calcium channels mediate the transition between tonic and phasic firing in thalamic neurons. Proc Natl Acad Sci U S A. 1989;86:7228–7232. doi: 10.1073/pnas.86.18.7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab. 1995;80:573–579. doi: 10.1210/jc.80.2.573. [DOI] [PubMed] [Google Scholar]
- Tse E, Smith AJ, Hunt S, Lavenir I, Forster A, Warren AJ, Grutz G, Foroni L, Carlton MB, Colledge WH, Boehm T, Rabbitts TH. Null mutation of the Lmo4 gene or a combined null mutation of the Lmo1/Lmo3 genes causes perinatal lethality, and Lmo4 controls neural tube development in mice. Mol Cell Biol. 2004;24:2063–2073. doi: 10.1128/MCB.24.5.2063-2073.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW. Calcium channels in excitable cell membranes. Annu Rev Physiol. 1983;45:341–358. doi: 10.1146/annurev.ph.45.030183.002013. [DOI] [PubMed] [Google Scholar]
- van Loo KM, Schaub C, Pernhorst K, Yaari Y, Beck H, Schoch S, Becker AJ. Transcriptional regulation of T-type calcium channel CaV3.2: bi-directionality by early growth response 1 (Egr1) and repressor element 1 (RE-1) protein-silencing transcription factor (REST) J Biol Chem. 2012;287:15489–15501. doi: 10.1074/jbc.M111.310763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wu Z, Xu Y, Zhu Y, Sutton AK, Zhao R, Lowell BB, Olson DP, Tong Q. An obligate role of oxytocin neurons in diet induced energy expenditure. PLoS One. 2012;7:e45167. doi: 10.1371/journal.pone.0045167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Tong Q. Expanding neurotransmitters in the hypothalamic neurocircuitry for energy balance regulation. Protein Cell. 2011;2:800–813. doi: 10.1007/s13238-011-1112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman T, Lee K, Park C, Paydar A, Choi JH, Cheong E, Lee CJ, Shin HS. Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron. 2011;70:95–108. doi: 10.1016/j.neuron.2011.02.042. [DOI] [PubMed] [Google Scholar]
- Zhou X, Gomez-Smith M, Qin Z, Duquette PM, Cardenas-Blanco A, Rai PS, Harper ME, Tsai EC, Anisman H, Chen HH. Ablation of LMO4 in glutamatergic neurons impairs leptin control of fat metabolism. Cell Mol Life Sci. 2012;69:819–828. doi: 10.1007/s00018-011-0794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP. Distribution of vesicular glutamate transporter mRNA in rat hypothalamus. J Comp Neurol. 2002;448:217–229. doi: 10.1002/cne.10257. [DOI] [PubMed] [Google Scholar]