

Abstract
Parkinson's disease (PD) is a movement disorder characterized by a progressive loss of nigrostriatal dopaminergic neurons. Restoration of dopamine transmission by l-DOPA relieves symptoms of PD but causes dyskinesia. Trace Amine-Associated Receptor 1 (TAAR1) modulates dopaminergic transmission, but its role in experimental Parkinsonism and l-DOPA responses has been neglected. Here, we report that TAAR1 knock-out (KO) mice show a reduced loss of dopaminergic markers in response to intrastriatal 6-OHDA administration compared with wild-type (WT) littermates. In contrast, the TAAR1 agonist RO5166017 aggravated degeneration induced by intrastriatal 6-OHDA in WT mice. Subchronic l-DOPA treatment of TAAR1 KO mice unilaterally lesioned with 6-OHDA in the medial forebrain bundle resulted in more pronounced rotational behavior and dyskinesia than in their WT counterparts. The enhanced behavioral sensitization to l-DOPA in TAAR1 KO mice was paralleled by increased phosphorylation of striatal GluA1 subunits of AMPA receptors. Conversely, RO5166017 counteracted both l-DOPA-induced rotation and dyskinesia as well as AMPA receptor phosphorylation. Underpinning a role for TAAR1 receptors in modulating glutamate neurotransmission, intrastriatal application of RO5166017 prevented the increase of evoked corticostriatal glutamate release provoked by dopamine deficiency after 6-OHDA-lesions or conditional KO of Nurr1. Finally, inhibition of corticostriatal glutamate release by TAAR1 showed mechanistic similarities to that effected by activation of dopamine D2 receptors. These data unveil a role for TAAR1 in modulating the degeneration of dopaminergic neurons, the behavioral response to l-DOPA, and presynaptic and postsynaptic glutamate neurotransmission in the striatum, supporting their relevance to the pathophysiology and, potentially, management of PD.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is characterized by a progressive loss of nigrostriatal dopaminergic neurons. Restoration of dopamine transmission by l-DOPA relieves symptoms of PD but causes severe side effects. Trace Amine-Associated Receptor 1 (TAAR1) modulates dopaminergic transmission, but its role in PD and l-DOPA responses has been neglected. Here, we report that TAAR1 potentiates the degeneration of dopaminergic neurons and attenuates the behavioral response to l-DOPA and presynaptic and postsynaptic glutamate neurotransmission in the striatum, supporting the relevance of TAAR1 to the pathophysiology and, potentially, management of PD.
Keywords: dyskinesia, l-DOPA, Parkinsonism, TAAR1, trace amine
Introduction
Parkinson's disease (PD) is characterized by a progressive degeneration of dopaminergic neurons projecting from substantia nigra pars compacta to striatum, eventually resulting in bradykinesia, rigidity, resting tremor, and postural imbalance (Lees et al., 2009). There are currently no approved treatments for slowing down the neurodegenerative process in PD. However, at early stages, the motor symptoms can be treated successfully with dopamine replacement therapies, including the dopamine precursor l-DOPA. Unfortunately, its therapeutic effect is gradually shortened and becomes more variable while disabling side effects, including dyskinesia, emerge (Iravani and Jenner, 2011). Strategies for prolonging the beneficial actions of l-DOPA and for precluding the appearance of side effects would be of considerable interest.
Endogenous trace amines, which are structurally related to monoamines and present in low concentrations throughout the mammalian brain, interact with monoamine transmission (Burchett and Hicks, 2006; Grandy, 2007). Altered levels of trace amines have been associated with disorders related to monoaminergic dysfunctions, including schizophrenia, depression, and PD (Branchek and Blackburn, 2003; Berry, 2007; Sotnikova et al., 2010). In 2001, a family of 15 G-protein-coupled trace amine-associated receptors (TAARs) were cloned (Borowsky et al., 2001; Bunzow et al., 2001). It was demonstrated that TAAR1 is not only activated by trace amines, but also by several amphetamine-related psychostimulants (Bunzow et al., 2001). All TAARs except TAAR1 are expressed in the olfactory epithelium (Liberles and Buck, 2006). TAAR1 is widely expressed in monoaminergic nuclei, including the dorsal raphe nucleus, locus ceruleus, ventral tegmental area, and substantia nigra (Borowsky et al., 2001; Miller et al., 2005; Lindemann et al., 2008; Di Cara et al., 2011). Although TAAR1 appears to be more highly expressed in the ventral tegmental area than in the substantia nigra (Lindemann et al., 2008), qPCR, in situ hybridization, and TAAR1 promotor-driven lacZ staining have identified TAAR1 in substantia nigra (Borowsky et al., 2001; Miller et al., 2005; Di Cara et al., 2011). TAAR1 is also moderately expressed in dopaminoceptive regions, including dorsal striatum, nucleus accumbens, amygdala, and prefrontal cortex (Bunzow et al., 2001, Xie et al., 2007; Di Cara et al., 2011). Studies have demonstrated that TAAR1 negatively modulates functional DAT levels and reduces agonist potency at D2-like receptors (Miller et al., 2005; Xie et al., 2007; Bradaia et al., 2009; Espinoza et al., 2011; Revel et al., 2011). Furthermore, it was recently reported that TAAR1 regulates NMDA receptor subunit composition and the NMDA/AMPA receptor ratio in layer V cortical neurons (Espinoza et al., 2015). Because many cortical neurons in layer V project to the striatum, these data raise the possibility that TAAR1 may modulate corticostriatal glutamate transmission. Such a modulation could have implications for PD and l-DOPA responsivity because dysfunctional interactions between glutamate and dopamine in striatum have been implicated in the pathophysiology and neurodegeneration of PD (Calabresi et al., 2007; Lau and Tymianski, 2010; Gerfen and Surmeier, 2011), as well as in the emergence of l-DOPA-induced dyskinesias (LIDs) (Iravani and Jenner, 2011; Sgambato-Faure and Cenci, 2012). Antagonism at glutamate NMDA and/or AMPA receptors is associated with anti-dyskinetic properties (Chase and Oh, 2000) and amantadine, an NMDA receptor antagonist (Greenamyre and O'Brien, 1991), is the only treatment against LIDs recommended by the Movement Disorder Society (International Parkinson and Movement Disorder Society, 2012).
In light of the above observations, we hypothesized that altered TAAR1 activity would affect neurodegeneration in experimental Parkinsonism and/or the response to l-DOPA. Accordingly, the degeneration of dopaminergic neurons was studied after TAAR1 inhibition [by TAAR1 knock-out (KO) mice] and TAAR1 stimulation (by RO5166017) and compared with wild-type (WT) mice using a well established, unilateral intrastriatal 6-OHDA paradigm. Furthermore, using genetic and pharmacological tools, along with biochemical and behavioral analyses, the influence of TAAR1 upon the emergence of l-DOPA supersensitivity and LIDs was also assessed. Finally, because a dysfunctional interplay between dopamine and glutamate is implicated in PD and in the response to l-DOPA, we studied the effects of TAAR1 on presynaptic glutamate release and turnover in the striatum and SNc and upon postsynaptic glutamate transmission as reflected in phosphorylation of AMPA receptors at the GluA1 subunit.
Materials and Methods
Animals.
Mice were housed as groups of littermates in Makrolon III cages under controlled temperature and humidity, a 12 h light/dark cycle, and with access to food and water ad libitum. All animal experiments were approved by the local animal ethics committee (Stockholms Norra Djurförsöksetiska Nämnd; approval numbers: N351/08, N434/10, N40/13, N368/10, N24/12, N269/13, N352/08, N367/10, N23/12, and N41/13). Adult male C57BL/6J mice (Charles River Laboratories), constitutive male TAAR1 KO mice, and WT littermates (Di Cara et al., 2011) and conditional male Nurr1 KO (cNurr1DATCreER KO) mice and WT littermates (Kadkhodaei et al., 2013) were used. All studied mice were backcrossed on a C57BL/6J genetic background. cNurr1DATCreER mice were injected with tamoxifen (2 mg, i.p.) (9:1 sunflower seed oil/ethanol; Sigma-Aldrich) daily for 5 consecutive days starting at postnatal day 35 (P35) to induce Nurr1 ablation.
Intrastriatal 6-OHDA and median forebrain bundle (MFB) 6-OHDA lesioning of nigrostriatal neurons.
Adult male TAAR1 KO and WT mice (2–3 months of age) were pretreated with 25 mg/kg desipramine intraperitoneally (Sigma-Aldrich), a noradrenergic reuptake inhibitor, and 5 mg/kg pargyline intraperitoneally (Sigma-Aldrich), a monoamine oxidase B inhibitor, to protect noradrenergic neurons. After 30 min, they were anesthetized (80 mg/kg ketamine and 5 mg/kg xylazine, i.p.), mounted in a stereotaxic frame, and unilaterally injected either with 10 μg of 6-OHDA (10 μg/μl in saline containing 0.02% ascorbic acid; Sigma-Aldrich) into the striatum of the right hemisphere to produce partial lesions or with 3 μg of 6-OHDA (3 μg/μl in saline containing 0.02% ascorbic acid) into the MFB to produce complete lesions. The coordinates for injections were as follows: AP, +0.5 mm; ML, −2 mm; and DV, −2.8 mm, and: AP, −1.1; ML, −1.1; DV, −4.8; relative to bregma and the dural surface (Franklin and Paxinos, 1997), respectively. In one group of mice with striatal 6-OHDA lesions, the TAAR1 agonist (S)-4-[(ethyl-phenyl-amino)-methyl]-4,5-dihydro-oxazol-2-ylamine (RO5166017; 1 mg/kg, i.p.) or vehicle was injected 5 min before the intrastriatal 6-OHDA injection. The animals were then treated daily with 0.1 mg/kg RO5166017 or vehicle intraperitoneally for 4 weeks and their motor functions were monitored in the beam traversal test once per week. Two weeks after 6-OHDA lesioning, mice were administered 1 mg/kg apomorphine intraperitoneally (Sigma-Aldrich) and their numbers of rotations were counted for 30 min. WT mice with intrastriatal 6-OHDA lesions rotated significantly less than TAAR1 KO mice (59.4 ± 7.3 vs 83.3 ± 8.3; p < 0.05). Likewise, WT mice with MFB 6-OHDA lesions rotated significantly less than TAAR1 KO mice (70.2 ± 8.5 vs 102.3 ± 10.2; p < 0.05). Mice with intrastriatal 6-OHDA lesions were killed 4 weeks after surgery and the brains were dissected out for autoradiography, immunoblotting, and in situ hybridization experiments. Mice with MFB 6-OHDA lesions and >50 rotations during 30 min went on to receive l-DOPA treatment.
Subchronic l-DOPA treatment and behavioral measurements of rotational behavior and dyskinesias.
Subchronic l-DOPA treatment was initiated 1 month after MFB 6-OHDA administration. Mice were treated daily with saline or 10/7.5 mg/kg l-DOPA/benserazide intraperitoneally (Sigma-Aldrich). At days 1, 7, and 12 of the subchronic treatment regimen, animals were placed in individual cages immediately after l-DOPA/benserazide injection and their behavior was recorded for 30 min using a ceiling-mounted video camera. Contralateral rotations, which increase due to supersensitization after chronic l-DOPA administration (Ungerstedt and Arbuthnott, 1970; Von Voigtlander and Moore, 1973), were counted offline. Immediately after the recording of rotational behaviors, the animals were filmed from the side for 5 min and the frequency of abnormal involuntary movements (AIMs), representing LIDs, was scored (Zhang et al., 2008). This procedure was adapted from a validated rodent scale in which AIMs are classified into four subtypes: forelimb, orofacial, axial, and locomotive dyskinesias (Lundblad et al., 2004). The severity of each AIM subtype was assigned a score based on intensity (0: absent; 1: occasional, i.e., present <50% of the time; 2: frequent, i.e., present >50% of the time; 3: continuous, but interrupted by strong sensory stimuli; and 4: continuous, not interrupted by strong sensory stimuli). The four subtypes were pooled into a total AIM score. Increased manifestations of normal behaviors, such as grooming and rearing, were disregarded. A group of C57BL/6J mice were pretreated with RO5166017 (0.5 mg/kg, i.p.; kind gift from M.J.M.) 15 min before l-DOPA/benzerazide injections at days 1, 7, and 12. Animals were killed 15 min after the last injection and striata were rapidly dissected out and snap frozen for immunoblotting experiments of AMPA receptor phosphorylation.
Immunoblotting.
Frozen striatal tissue samples were sonicated in 1% SDS and boiled for 10 min. Small aliquots of the homogenate were retained for protein determination using the bicinchoninic acid protein assay method (Thermo Fisher Scientific). Equal amounts of protein (20 μg) were loaded onto 12% acrylamide gels and the proteins were separated by SDS-PAGE and transferred to Immobilon-P PVDF membranes (Sigma-Aldrich). The membranes were immunoblotted using p-Ser845-GluA1 (Millipore) and antibodies that are not phosphorylation state specific were used to estimate total levels of GluA1 (Millipore), tyrosine hydroxylase (TH) (Merck Millipore), and β-actin (Sigma-Aldrich). Antibody binding was detected by incubation with goat anti-mouse or anti-rabbit horseradish peroxidase-linked IgG (1:6000–8000 dilution) and the ECL immunoblotting detection system (GE Healthcare).
Autoradiographic detection of DAT.
Frozen brains were cryostat cut in 12 μm sections and mounted on SuperFrost microscope slides (Gerhard Menzel) for DAT binding by autoradiography. Sections were preincubated in 50 mm Tris-HCl/120 mm NaCl, pH 7.5, for 20 min and then incubated in binding buffer with 50 pm [125I] RTI-55 (PerkinElmer Life Sciences) with 1 μm fluoxetine (Tocris Bioscience) for 60 min. Next, 100 μm nomifensine (Sigma-Aldrich) was added to the assay to determine nonspecific binding. The slides were washed in ice-cold binding buffer (2 × 10 s) and in deionized water, dried, and exposed to Kodak Biomax MR film (Sigma-Aldrich). Autoradiograms were digitized using a Dia-Scanner (Epson Perfection 4870 PHOTO) and optical density values were measured using ImageJ version 1.29 software.
In situ hybridization.
A DARPP-32 antisense probe (500 bp fragment of DARPP-32 cDNA subcloned into pBluescript KS; Svenningsson et al., 2002) was linearized and transcribed with T3 RNA polymerase together with 6 μl of [35S]UTP (25 mCi/ml; PerkinElmer) and 25 μm each of ATP, CTP, and GTP, along with 10.5 μm unlabeled UTP. The reaction mixture was incubated at 37°C for 2 h. After DNase I digestion for 15 min, the labeled RNA was purified using microspin G25 columns (GE Healthcare). In situ hybridization was performed on cryostat-cut sections for 16 h at 55°C in humid chambers. After coverslips were removed in 4× SSC and 10 mm DTT, the slides were washed at room temperature for 1 h in the same solution and then in 50% formamide, 10 mm Tris-HCl, pH 8, 75 mm NaCl, and 2.5 mm EDTA. Sections were treated with RNase A (20 μg/ml; Sigma-Aldrich) in 400 mm NaCl, 10 mm Tris-HCl, pH 7.5, and 50 mm EDTA for 1 h at 37°C and then rinsed for 15 min at 60°C in 2× SSC, followed by 0.1× SSC. After dehydration, the sections were air-dried and exposed to Biomax-MR films (Kodak). Densitometric measurements of mRNA signals in the dorsal striatum were obtained from autoradiograms using ImageJ software after subtraction of background values.
In vivo amperometry measurements of glutamate levels.
In vivo recordings were performed as described previously (Hascup et al., 2008; Eriksson et al., 2013). Each microelectrode assembly (MEA) was of the S2 model which consisted of four platinum recording sites (15 × 333 μm, Fig. 1A), and was fabricated and assembled according to previous publications (Burmeister et al., 2000). Before each experiment two platinum sites were coated with recombinant glutamate oxidase (Yamasa Corporation) mixed with 1% bovine serum albumin (BSA) and 0.125% glutaraldehyde. Glutamate oxidase causes enzymatic breakdown of glutamate into α-ketoglutarate and peroxide. The second pair of sites was used for self-referencing and was coated only with the BSA/glutaraldehyde solution to form an inactive protein matrix. All four sites were then coated with a protective layer of meta-phenylenediamine dihydrochloride (mPD; Thermo Fisher Scientific) to block interferents. Each MEA was calibrated in vitro before each experiment to determine the selectivity and sensitivity for glutamate against ascorbic acid. During calibration, the MEA tip was placed in a stirred solution of 0.05 m PBS, pH 7.4, at 37°C, after which final buffer concentrations of 20, 40, and 60 μm glutamate were added. Next, 250 μm ascorbic acid and 2 μm dopamine were added to assess the size-exclusion properties of the mPD coating layer and 8.8 μm H2O2 was applied to verify that all four channels responded similarly to the reporter molecule. Finally, 100 μm ropinirole (Sigma-Aldrich), 2.5 μm N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251; Tocris Bioscience), 500 nm RO5166017 (synthesized at Servier Laboratories), and 10 nm N-(3-ethoxy-phenyl)-4-pyrrolidin-1-yl-3-trifluoromethyl-benzamide (EPPTB; synthesized at Servier Laboratories) were added as test substances to ensure that none of these compounds were electrochemically active. A pulled single-barrel glass micropipette (inner tip diameter: 10–15 μm) was aligned with its opening between the four platinum electrode sites of each MEA (50–100 μm from the MEA) and used for local deliveries of 70 mm KCl with or without the pharmacological compound of interest. Stock solutions of ropinirole, AM251, RO5166017, or EPPTB were mixed with isotonic KCl to make final concentrations of 100 μm, 2.5 μm, 500 and 10 nm in 70 mm KCl, respectively. Doses were derived from pilot studies (data not shown) and previous experiments (Revel et al., 2011). The volume of delivery was controlled using a Picospritzer III (Parker Hannifin) monitored via a stereomicroscope fitted with a reticule. A miniature Ag/AgCl reference electrode was prepared by stripping a Teflon-coated silver wire on both ends and soldering one end to a gold-plated socket (Pronexus Analytical). The other end was coated with AgCl by applying a current of +9 V for 15 min while suspended into a 1 m HCl plating bath saturated with NaCl. During in vivo amperometry recordings, mice were anesthetized with isoflurane (3% for induction, 0.7–1% for maintenance) and mounted in a stereotaxic frame (David Kopf Instruments) fitted with a Cunningham mouse adapter (Stoelting). Small cranial windows were drilled over the corresponding coordinates (striatum: AP: +1.3; ML: ±1.5; DV: −2.5–3.0; SNc: AP: −3.0; ML: ±1.2; DV: −4.5 vs bregma) and MEA assemblies were inserted into each region of interest, with the order of insertion randomized between experiments. The FAST-16 mkII system (Quanteon) recorded amperometric data at 2 Hz and created ejection synchronization marks for all four channels. During amperometric recordings, KCl ejections were made for 1 s with 1 min between each ejection and an average of 4–5 amplitudes was used for analysis in each region. A MATLAB graphic user interface developed by Quanteon was used to calculate concentrations of glutamate (Fig. 1B). Treatment-induced effects on tonic glutamate levels were determined as 10 data points preceding each ejection selected for analysis. Responses recorded at the reference sites were subtracted from those generated at the enzyme-coated recording sites, with the resulting signal representing glutamate measurements. Negative baseline values were excluded from the analysis.
Figure 1.

Configuration of MEA, definition of glutamate measurements, and verification of MEA placement. A, Schematic drawing of a MEA tip. The platinum sites used as recording sites (yellow) were coated with glutamate oxidase (GluOx), which cause enzymatic breakdown of glutamate into α-ketoglutarate and peroxide (H2O2). The second pair of sites, labeled “reference sites,” were coated with BSA and glutaraldehyde, forming an inactive protein matrix and used for self-referencing. All four sites were then coated with a protective layer of mPD to block interferents. B, Schematic drawing of a KCl-induced glutamate release event. The glutamate amplitude was defined as the maximum increase in glutamate concentration compared with the baseline value recorded at the event of KCl ejection (arrow) minus the amplitude recorded at the corresponding reference channel. Tonic glutamate concentrations were recorded 10 data points before each KCl ejection minus the concentration recorded at the corresponding reference channel. T80 was defined as the time elapsed from the maximum amplitude until 80% of the glutamate peak had decayed. C, Histological verification of MEA placement in striatum and SNc by local application of methylene blue solution followed by Nuclear Fast Red counterstaining.
Histological verification of MEA placement.
Mice were injected locally with 0.25 μl of methylene blue solution, after which the brain was rapidly removed and frozen. Next, 50 μm coronal brain slices were sectioned in a cryostat and counterstained with Nuclear Fast Red (Eriksson et al., 2013) to determine MEA position during recording (Fig. 1C).
Statistics.
Statistica software (StatSoft) was used for all statistical analyses. Student's unpaired t tests were used when comparing only two groups. Repeated-measures ANOVA was used to investigate the treatment effects of l-DOPA on behavior over time. Two- and three-way ANOVAs were used where appropriate for analyzing immunoblotting and autoradiography data. In amperometry experiments, one-way ANOVAs were used for comparisons between pharmacological treatment and genotype or state of lesion in the striatum. Significant effects of ANOVA tests were followed by post hoc analyses by Fisher's least significant difference (LSD) test as indicated. p < 0.05 was set as the threshold for statistical significance. Data are presented as the mean ± SEM.
Results
Effects of intrastriatal 6-OHDA administration in WT and TAAR1 KO mice
Intrastriatal 6-OHDA, a model to study degeneration of dopaminergic neurons (Sauer and Oertel, 1994; Przedborski et al., 1995), was applied unilaterally to WT and TAAR1 KO mice. Western blotting of striatal tissue from the mice sustaining intrastriatal 6-OHDA injections revealed that TAAR1 KO mice had increased levels of TH both in the intact and lesioned hemispheres compared with the intact and lesioned hemispheres of WT mice, respectively (genotype: F(1,18) = 15.92, p < 0.001; lesion: F(1,18) = 104.07, p < 0.001; interaction: F(1,18) = 0.0013, p = 0.97; Fisher's LSD post hoc test: WT intact vs lesioned, p < 0.001; WT intact vs KO intact, p < 0.05; WT lesioned vs KO lesioned, p < 0.05; KO intact vs KO lesioned, p < 0.001; Fig. 2A). When the lesioned hemispheres of each WT and TAAR1 KO animal were normalized against the respective control hemispheres of each animal, there was a significant difference between the ratios of the genotypes (Student's unpaired t test: p < 0.01; Fig. 2B), suggesting less severe dopaminergic neurodegeneration in TAAR1 KO mice.
Figure 2.

Levels of TH and dopamine transporter in WT and TAAR1 KO mice after intrastriatal or MFB 6-OHDA administration. A, TH levels in striatal tissue from WT and TAAR1 KO mice were reduced by striatal 6-OHDA injections. TH levels were higher in both the intact and the lesioned hemispheres of TAAR1 KO mice compared with WT mice. B, TH ratio between the lesioned and the intact hemispheres after intrastriatal 6-OHDA injections showed a higher TH ratio in TAAR1 KO mice than WT mice. C, Intrastriatal 6-OHDA injections caused a significant reduction of DAT binding, detected by [125I]RTI-55, at the level of striatum in both WT and TAAR1 KO mice. DAT levels were higher in the intact and the lesioned hemispheres in TAAR1 KO mice compared with WT mice. D, DAT ratio between the lesioned and the intact hemispheres after intrastriatal 6-OHDA injections showed a higher DAT ratio in TAAR1 KO mice than WT mice. E, Striatal tissue from WT and TAAR1 KO mice with MFB 6-OHDA injections showed a near-complete reduction of TH in the lesioned side of both genotypes. F, TH ratio between the lesioned and the intact hemisphere after MFB 6-OHDA injections did not differ between WT and TAAR1 KO mice. DAT, Dopamine transporter. #p < 0.05, ##p < 0.01 by Student's unpaired t test; *p < 0.05, ***p < 0.001 by Fisher's LSD post hoc test.
To further study whether dopaminergic markers differ between WT and TAAR1 KO mice at baseline and after intrastriatal 6-OHDA, DAT levels were determined by autoradiography. DAT binding revealed that TAAR1 KO mice displayed higher DAT levels compared with WT mice in both the intact and lesioned hemispheres (genotype: F(1,18) = 24.04, p < 0.001; lesion: F(1,18) = 223.04, p < 0.001, interaction: F(1,18) = 0.78, p = 0.38; Fisher's LSD post hoc test: WT intact vs KO intact, p < 0.001; WT lesioned vs KO lesioned, p < 0.05; WT intact vs lesioned, p < 0.001; KO intact vs lesioned, p < 0.001; Fig. 2C). When the lesioned hemispheres of each WT and TAAR1 KO animal were normalized against their respective control hemispheres, there was significantly less DAT reduction in TAAR1 KO mice (Student's unpaired t test: p < 0.05; Fig. 2D).
There was no difference in DARPP32 mRNA levels between WT and TAAR1 KO mice and intrastriatal 6-OHDA treatment did not affect these levels, indicating that dopaminoceptive medium-sized spiny neurons (MSNs) were not affected by the intrastriatal 6-OHDA injections (genotype: F(1,18) = 0.12, p = 0.72; treatment: F(1,18) = 0.05; p = 0.83; interaction: F(1,18) = 0.0004, p = 0.98).
Effects of MFB 6-OHDA administration in WT and TAAR1 KO mice
Both WT and TAAR1 KO mice showed a near-complete reduction of TH in the lesioned hemispheres, with no difference between the genotypes (genotype: F(1,63) = 0.026, p = 0.87; lesion: F(1,63) = 237.72, p < 0.001; interaction: F(1,16) = 0.54, p = 0.46; Fisher's LSD post hoc test: WT intact vs WT lesioned, p < 0.001; KO intact vs KO lesioned, p < 0.001; Fig. 2E). When injected in the MFB, 6-OHDA decreased TH to a similar extent in WT and TAAR1 KO mice (Fig. 2F).
Effects of intrastriatal 6-OHDA administration in WT mice chronically treated with RO5166017 or vehicle
To further investigate the effects of TAAR1 on intrastriatal 6-OHDA, the TAAR1 agonist RO5166017 (1 mg/kg, i.p.) or vehicle was injected 5 min before the intrastriatal 6-OHDA injection in one group of control mice. The animals were then treated daily with 0.1 mg/kg RO5166017 or vehicle intraperitoneally until they were killed after 4 weeks. During this time their motor functions were monitored in the beam traversal test, revealing a higher number of errors per step in animals treated with RO5166017 compared with animals treated with vehicle after 1 week (vehicle: 5433 ± 0,7004; RO: 8718 ± 0,8452, Student's unpaired t test: p < 0.05), 2 weeks (vehicle: 9647 ± 1,608; RO: 13,62 ± 0,7628, p < 0.05) and 3 weeks (vehicle: 8293 ± 0,8765; RO: 13,80 ± 1,402, p < 0.01). These data suggest a more severe motor phenotype in animals treated with the TAAR1 agonist in response to intrastriatal 6-OHDA lesioning compared with vehicle-treated animals. In agreement, DAT binding revealed that RO5166017-treated mice exhibited lower DAT levels in the lesioned hemisphere compared with vehicle-treated mice (treatment: F(1,19) = 5.76, p < 0.05; lesion: F(1,19) = 173.2, p < 0.001, interaction: F(1,19) = 2.21, p = 0.15; Fisher's LSD post hoc test: vehicle intact vs vehicle lesioned, p < 0.001; vehicle lesioned vs RO lesioned, p < 0.05; RO intact vs RO lesioned, p < 0.001; Fig. 3A). Accordingly, when the lesioned hemispheres of each vehicle- and RO5166017-treated animal were normalized against their respective control hemispheres, there was significantly more reduction in the DAT ratio in RO5166017-treated mice (Student's unpaired t test: p < 0.01; Fig. 3B).
Figure 3.

Effect of TAAR1 agonism on intrastriatal 6-OHDA administration. A, Intrastriatal 6-OHDA injections caused a significant reduction of striatal DAT binding, detected by [125I]RTI-55, in mice chronically administered with RO5166017 compared with vehicle-treated mice. B, DAT ratio between the lesioned and the intact hemispheres after intrastriatal 6-OHDA injections was in mice treated with RO5166017 compared with vehicle-treated mice. VEH, vehicle; RO, RO5166017. ##p < 0.01 by Student's unpaired t test; *p < 0.05, ***p < 0.001 by Fisher's LSD post hoc test.
Behavioral responses to subchronic l-DOPA treatment in WT and TAAR1 KO mice with MFB 6-OHDA lesions
To determine l-DOPA responsivity in WT and TAAR1 KO mice, experiments were pursued in mice with MFB 6-OHDA injections and subchronic l-DOPA treatment. This is a well established procedure for quantitative studies of l-DOPA motor actions (Ungerstedt and Arbuthnott, 1970; Simola et al., 2007) and LIDs (Lundblad et al., 2004; Francardo and Cenci, 2014). Mice with MFB 6-OHDA injections were treated with 10/7.5 mg/kg l-DOPA/benserazide for 12 d to investigate rotational behavior and AIMs. Rotational behaviors were measured on days 1, 7, and 12 of the subchronic treatment regimen. In WT and TAAR1 KO mice, a repeated-measures ANOVA revealed a significant effect of treatment duration on rotational behavior (genotype: F(1,20) = 4.20, p = 0.054; time: F(2,40) = 14.74, p < 0.001; interaction: F(2,40) = 1.51, p = 0.23). Fisher's LSD post hoc test indicated that the WT mice sensitized after 12 d of subchronic l-DOPA treatment (day 1 vs day 12, p < 0.05), whereas the TAAR1 KO mice sensitized already at day 7 (day 1 vs day 7, p < 0.01), an effect that progressed in severity until day 12 (day 1 vs day 12, p < 0.001; Fig. 4A). TAAR1 KO mice performed significantly more contralateral rotations in response to l-DOPA compared with the WT group on day 12 (WT vs KO, p < 0.05).
Figure 4.
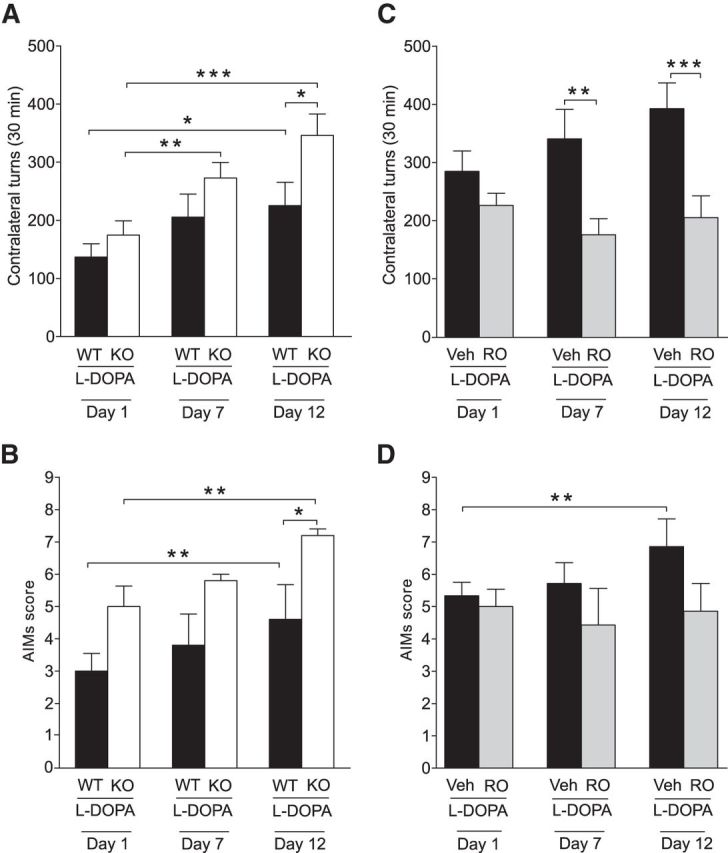
Effects of TAAR1 KO and TAAR1 activation on l-DOPA-induced rotations and AIMs. A, Both 6-OHDA-treated WT (n = 11) and TAAR1 KO (n = 11) animals rotated significantly more after 12 d of subchronic l-DOPA treatment compared with day 1. Supersensitization to l-DOPA appeared already after 7 d of treatment in TAAR1 KO animals. l-DOPA-induced rotations were more frequent in TAAR1 KO animals compared with WT animals after 12 d of subchronic l-DOPA treatment. B, Both 6-OHDA-treated WT and TAAR1 KO animals performed significantly more AIMs after 12 d of subchronic l-DOPA treatment compared with day 1. TAAR1 KO animals performed more AIMs compared with WT animals after 12 d of subchronic l-DOPA treatment. C, In 6-OHDA-treated mice treated daily with l-DOPA (n = 7), pretreatment with RO5166017 on days 1, 7, and 12 reduced l-DOPA induced rotations in TAAR1 KO mice compared with WT on days 7 and 12 (n = 7). D, Mice treated with l-DOPA performed significantly more AIMs after 12 d of subchronic treatment compared with day 1. No similar effect was found in mice treated with l-DOPA and RO5166017. RO, RO5166017. *p < 0.05, **p < 0.01, ***p < 0.001 by Fisher's LSD post hoc test.
To examine LIDs, we also measured AIMs immediately after the scoring of rotational behavior. A repeated-measures ANOVA revealed a significant effect of treatment duration (F(2,16) = 13.82, p < 0.001) and genotype (F(1,16) = 6.17, p < 0.05), but no interaction (F(2,16) = 0.46, p = 0.64). Fisher's LSD post hoc test revealed a higher level of AIMs both in 6-OHDA-treated WT and TAAR1 KO mice after 12 d of subchronic l-DOPA treatment (day 1 vs day 12, p < 0.01) and a higher number of AIMs in TAAR1 KO mice compared with WT at day 12 (p < 0.05; Fig. 4B). These findings indicate that TAAR1 KO mice exhibited more AIMs induced by repeated exposure to l-DOPA compared with WT mice.
Separate experiments were performed with l-DOPA-treated MFB 6-OHDA-lesioned C57BL/6J mice using the selective TAAR1 agonist RO5166017 (0.5 mg/kg, i.p.). RO5166017 was administered 15 min before administration of l-DOPA at days 1, 7, and 12. A repeated-measures ANOVA revealed a significant effect of treatment (F(1,10) = 19.94, p < 0.01), but not of treatment duration (F(2,20) = 0.22, p = 0.80) or interaction (F(2,20) = 1.76, p = 0.20). Pretreatment with the TAAR1 agonist significantly reduced l-DOPA-induced rotations after 7 and 12 d of l-DOPA treatment (Fisher's LSD post hoc test: vehicle day 7 vs RO day 7, p < 0.01; vehicle day 12 vs RO day 12, p < 0.001; Fig. 4C). The number of AIMs were significantly higher in the l-DOPA-treated group after 12 d of treatment compared with day 1 (treatment duration: F(2,24) = 3.00, p = 0.069; treatment: F(1,12) = 0.82, p = 0.38; interaction: F(2,24) = 3.80, p < 0.05; Fisher's LSD post hoc test: l-DOPA day 1 vs l-DOPA day 12, p < 0.01), but this effect was not found in the group pretreated with RO5166017 (Fig. 4D). These findings indicate that RO5166017 counteracted the induction of AIMs induced by l-DOPA.
Effect of TAAR1 on phosphorylation of GluA1 subunits of AMPA receptors
Increased glutamate neurotransmission underlies LIDs (Iravani and Jenner, 2011; Sgambato-Faure and Cenci, 2012). To strictly measure postsynaptic events of glutamate neurotransmission, AMPA receptors rather than NMDA receptors were examined. Enhanced phosphorylation at Ser845 of the GluA1 subunit of AMPA receptors has been correlated with l-DOPA-induced AIMs (Chase, 2004). Here, we found a significant interaction on p-Ser845-GluA1 levels between subchronic l-DOPA treatment and dopamine denervation as revealed by a three-way ANOVA (F(1,61) = 5.58, p < 0.05), whereas there were no individual effects of treatment (F(1,61) = 1.84, p = 0.18), genotype (F(1,61) = 0.76, p = 0.39) or lesion (F(1,61) = 3.17, p = 0.08). Fisher's LSD post hoc test showed a significantly higher level of p-Ser845-GluA1 in the lesioned hemisphere of l-DOPA-treated TAAR1 KO mice compared with the intact hemisphere (p < 0.01) and compared with the lesioned hemisphere of saline-treated TAAR1 KO mice (p < 0.05; Fig. 5A).
Figure 5.

The effect of TAAR1 KO and TAAR1 agonism on l-DOPA-induced phosphorylation of Ser845-GluA1. A, There was a higher level of p-Ser845-GluA1 in the lesioned hemisphere of l-DOPA-treated TAAR1 KO mice compared with the intact hemisphere and compared with the lesioned hemisphere of saline-treated TAAR1 KO mice. B, There was a significantly higher level of p-Ser845-GluA1 in the lesioned hemisphere of C57BL/6J mice treated with l-DOPA compared with saline. This effect was normalized by pretreatment with the TAAR1 agonist RO5166017. RO, RO5166017. *p < 0.05, **p < 0.01 by LSD post hoc test.
In l-DOPA-treated WT mice pretreated with the TAAR1 agonist RO5166017, there was a significant effect of treatment (F(2,34) = 3.71, p < 0.05), but not of lesion (F(1,34) = 1.08, p = 0.31) or interaction (F(2,34) = 2.22, p = 0.13; Fig. 5B). Post hoc analysis revealed a significantly higher level of p-Ser845-GluA1 in the 6-OHDA lesioned hemisphere after subchronic l-DOPA treatment compared with the intact hemisphere (p < 0.05), the 6-OHDA-treated hemisphere of the saline-treated group (p < 0.01), and the 6-OHDA lesioned hemisphere of animals cotreated with l-DOPA and RO5166017 (p < 0.05). These effects indicate that a TAAR1 agonist could attenuate the effect of l-DOPA on GluA1 phosphorylation, mirroring the reduction of l-DOPA-induced AIMs.
Influence of TAAR1 on evoked striatal glutamate release in two models of experimental Parkinsonism
There was a prominent effect of TAAR1 on the phosphorylation of postsynaptic glutamate AMPA receptors and because TAAR1 modulates glutamatergic functions in pyramidal neurons of cortical layer V projecting to striatum (Espinoza et al., 2015), we studied the effects of TAAR1 on extracellular glutamate levels. For this purpose, a biosensor-based methodology was used (Hascup et al., 2008). Mice injected with 6-OHDA into the MFB had a 97 ± 1.3% reduction of DAT binding in the 6-OHDA lesioned hemisphere compared with the intact hemisphere (Fig. 6A). KCl-evoked glutamate amplitudes were enhanced in striatum of the lesioned hemisphere compared with the intact side and inhibited by local administration of RO5166017 as revealed by a one-way ANOVA (F(1,16) = 7.60, p < 0.01; Fig. 6B). These findings indicate that TAAR1 stimulation normalized the elevation of glutamate seen in striatum of MFB 6-OHDA lesioned mice.
Figure 6.
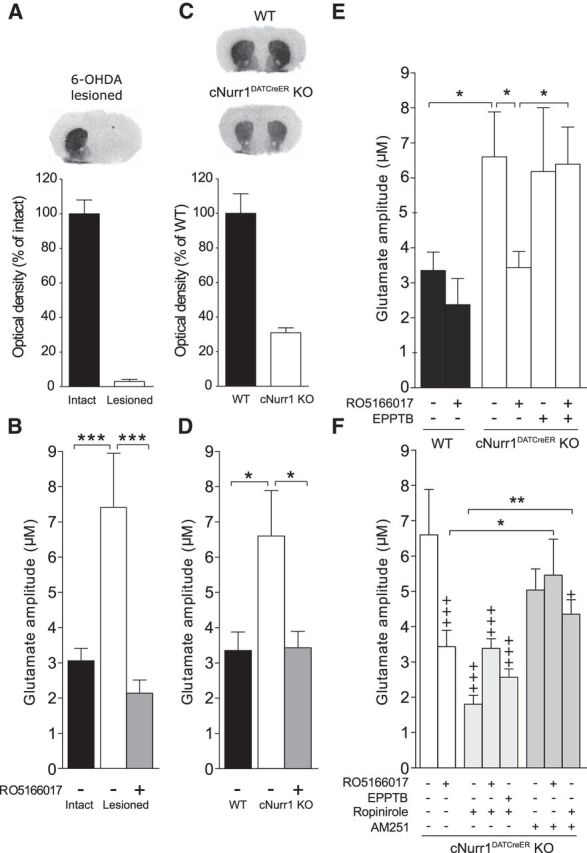
DAT levels and striatal glutamate release under dopamine deficiency and the modulatory effects of TAAR1, ropinirole, and AM251. A, Autoradiographic detection of DAT by [125I]RTI-55 in brain slices revealed a 97 ± 1.3% reduction in the lesioned compared with the intact hemisphere of MFB 6-OHDA-treated C57BL/6J mice (n = 10). B, Real-time in vivo amperometry revealed significantly enhanced glutamate release in the lesioned hemisphere compared with the intact hemisphere. This hyperglutamatergic state was reversed after local application of the TAAR1 agonist RO5155017. C, There was a 67 ± 3.8% reduction in DAT levels in cNurr1DATCreER KO (n = 6) compared with WT mice (n = 6). D, KCl-evoked glutamate concentrations were significantly enhanced in the striatum of KO mice compared with WT mice. This effect was normalized after local application of RO5166017. E, Local administration of EPPTB did not significantly affect KCl-evoked striatal glutamate release in cNurr1DATCreER KO animals, but blocked the effect of RO5166017 when these compounds were coadministered. F, Local administration of ropinirole attenuated KCl-evoked glutamate amplitudes in striatum. Coadministration of ropinirole did not significantly change the effects of RO5166017 or EPPTB in cNurr1DATCreER KO animals. AM251 had no effects by itself, but attenuated the inhibitory effect of RO5166017 on KCl-evoked glutamate release. Likewise, AM251 attenuated the inhibitory effect of ropinirole. The WT vehicle, cNurr1DATCreER KO vehicle, and RO5166017 data have been included in several graphs to facilitate comparisons. Data are presented as the mean ± SEM from 4–5 glutamate peaks from 5–10 animals/treatment. DAT, Dopamine transporter;. +p < 0.05, +++p < 0.001 vs WT vehicle; *p < 0.05, **p < 0.01, ***p < 0.01 vs indicated group by Fisher's LSD post hoc test.
Amperometry experiments were continued in cNurr1DatCreER KO mice in which DAT binding was reduced by 67 ± 3.8% compared with corresponding WT mice (Fig. 6C). cNurr1DatCreER KO mice have the advantage that both hemispheres can be used to study the effect of dopamine depletion and pharmacological interventions on corticostriatal glutamate release. Similar to 6-OHDA-treated animals, KCl-evoked glutamate amplitudes were enhanced in striatum of cNurr1DATCreER KO compared with WT mice (F(5,38) = 3.06, p < 0.05; Fisher's LSD post hoc test: p < 0.05; Fig. 6D). Local administration of RO5166017 normalized the enhanced glutamate release in cNurr1DATCreER KO mice (p < 0.05), an effect that was counteracted by coadministration of the selective TAAR1 antagonist EPPTB (p < 0.05; Fig. 6E). EPPTB alone did not affect corticostriatal glutamate release at this dose. These findings indicate that TAAR1 stimulation normalized the elevation of striatal glutamate, not only in MFB 6-OHDA lesioned mice, but also in cNurr1DATCreER KO mice.
Mechanisms underlying TAAR1-mediated changes in glutamate release
There is evidence for functional interactions between TAAR1 and D2 receptors (Espinoza et al., 2011). Mechanistic studies have also provided indirect evidence that glutamate release is inhibited both by presynaptic D2 heteroreceptors at corticostriatal terminals (Wang and Pickel, 2002; Bamford et al., 2004; Wang et al., 2012) and postsynaptic D2 receptors on striatal MSNs (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006). Electrophysiological recordings indicate that postsynaptic D2 receptors expressed on MSNs reduce corticostriatal glutamate by increasing endocannabinoid (eCB) levels, which retrogradely act on inhibitory CB1 receptors at glutamatergic terminals (Giuffrida et al., 1999; Kreitzer and Malenka, 2005; Yin and Lovinger, 2006).
To investigate the mechanisms by which TAAR1 counteracts the enhanced corticostriatal glutamate release, we examined interactions between TAAR1 and D2 receptors with the endocannabinoid system. Like the TAAR1 agonist RO5166017, the D2-like receptor agonist ropinirole significantly attenuated corticostriatal glutamate release in cNurr1DATCreER KO mice when administered alone (F(7,51) = 5.59, p < 0.001; Fisher's LSD post hoc test: p < 0.001). There was no significant interaction between RO5166017 or EPPTB with ropinirole (Fig. 6F). Conversely, the CB1 receptor antagonist AM251 did not affect corticostriatal glutamate release by itself, but attenuated the effect of RO5166017 (p < 0.05), suggesting that TAAR1 activation counteracts corticostriatal glutamate release through a mechanism that involves CB1 receptors (Fig. 6F). Consistent with previous work (Giuffrida et al., 1999; Kreitzer and Malenka, 2005; Yin and Lovinger, 2006), AM251 also attenuated the effect of ropinirole (p < 0.001), suggesting that retrograde eCB release underlies ropinirole's effect on striatal glutamatergic neurotransmission (Fig. 6F). Notably, AM251 did not mediate a complete reversal of ropinirole's effect, suggesting that a minor part of ropinirole's effect is independent of CB1 receptors. This part may be mediated by presynaptic D2 receptors at glutamatergic terminals or it may represent the blockade of glutamate released from thalamostriatal projections that lack CB1 receptors (Uchigashima et al., 2007).
There were no significant effects of RO5166017, ropinirole, or AM251 alone or in combination on evoked corticostriatal glutamate release in WT animals (F(8,26) = 1.77, p = 0.71).
Tonic striatal glutamate concentrations in two models of experimental Parkinsonism
Tonic extracellular glutamate concentrations in the lesioned side of striatum were not different from the intact side in mice with MFB 6-OHDA injections (Fig. 7A). Tonic extracellular glutamate concentrations in striatum of cNurr1DATCreER KO mice were not affected compared with WT mice or mice after pharmacological TAAR1 targeting (Fig. 7B). Tonic extracellular glutamate concentrations were elevated in striatum of cNurr1DATCreER KO mice after AM251, an effect that was reversed by the TAAR1 agonist RO5166017 (F(7,40) = 2.53, p < 0.05; Fisher's LSD post hoc test: KO vehicle vs KO AM251, p < 0.05; KO AM251 vs KO AM251+RO5166017, p < 0.01; Fig. 7C).
Figure 7.
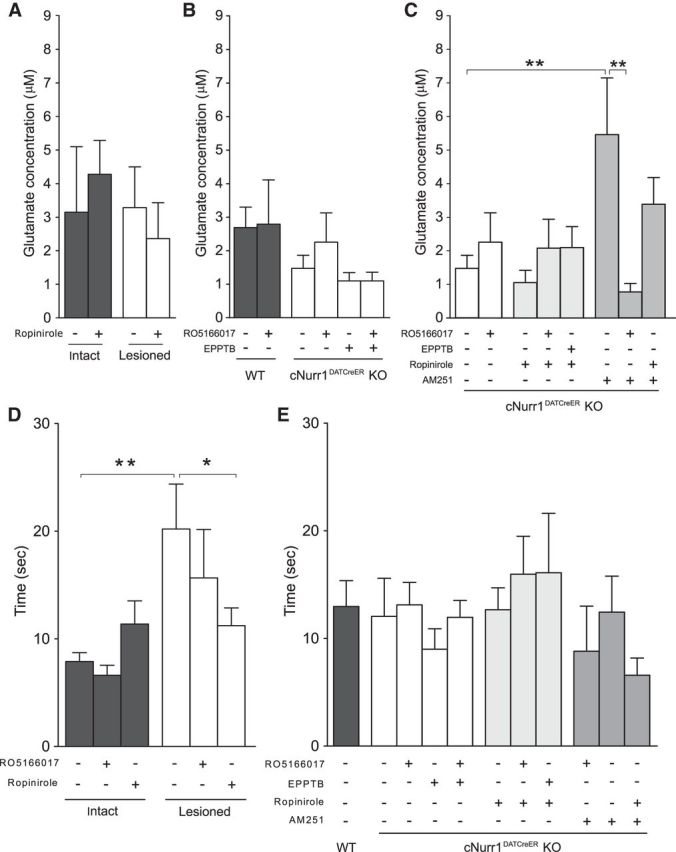
Tonic striatal glutamate release and the reuptake rate (T80) of evoked striatal glutamate in 6-OHDA-treated and cNurr1DATCreER KO mice. A, Tonic extracellular glutamate concentrations in striatum of MFB 6-OHDA-treated mice were not affected by lesion or ropinirole administration. B, Tonic extracellular glutamate concentrations were not significantly affected in cNurr1DATCreER KO mice or by TAAR1 ligands. C, Local administration of AM251 significantly enhanced tonic glutamate concentrations in cNurr1DATCreER KO mice. D, T80 was prolonged in striatum of 6-OHDA-treated mice after ejection of KCl. Local application of ropinirole inhibited this effect. E, No T80 alterations were seen in striatum of cNurr1DATCreER KO mice compared with WT mice or mice after TAAR1 ligands, ropinirole, or AM251. Coadministration with RO5166017 attenuated the AM251-induced hyperglutamatergic state. *p < 0.05, **p < 0.01 by Fisher's LSD post hoc test.
Glutamate reuptake rate (T80) in two models of experimental Parkinsonism
The glutamate reuptake rate (T80) was measured as the time elapsed from the peak maximum amplitude of glutamate release until 80% of the peak had decayed (Fig. 1B). In the striatum of 6-OHDA-treated mice, the T80 was prolonged by 156% in the lesioned compared with the intact hemisphere after administration of KCl alone (lesion: F(1,32) = 9.38, p < 0.001; treatment: F(2,32) = 0.70, p = 0.50; interaction: F(2,32) = 2.63, p = 0.088; Fisher's LSD post hoc test: intact vehicle vs lesioned vehicle, p < 0.01), an effect that was inhibited when ropinirole was coadministered (p < 0.05, lesioned vehicle vs lesioned ropinirole; Fig. 7D). This inhibitory effect could be directly related to a reduced glutamate release observed after administration of ropinirole in the lesioned hemisphere (p < 0.01, lesioned vehicle vs lesioned ropinirole) and thus may not be a true indicator of an altered glutamate clearance rate. Conversely, the TAAR1 agonist RO5166017 did not significantly affect the T80 in 6-OHDA-treated mice. In contrast to 6-OHDA-treated mice, cNurr1DATCreER mice exhibited no significant effects on the T80 between WT and KO, indicating that the altered glutamate release observed in this animal model was associated with a compensatory elevation in the glutamate reuptake rate (Fig. 7E). Local application of ropinirole, AM251, EPPTB, and RO5166017 did not affect the T80 in cNurr1DATCreER KO mice.
Regulation of evoked and tonic glutamate levels by dopamine in SNc in two models of experimental Parkinsonism
Our intrastriatal 6-OHDA administration experiments (Figs. 2A–D) indicated a neuroprotective effect of TAAR1 toward dopaminergic degeneration. The degeneration of midbrain dopaminergic neurons in PD leads to disinhibition of the subthalamic nucleus, which is glutamatergic and projects to the substantia nigra, including SNc, where it is in a position to exert excitotoxic cell damage (Chergui et al., 1994; Rodriguez et al., 1998). This process has been proposed to act in a vicious cycle to further aggravate cell death in PD (Lau and Tymianski, 2010) and led us to perform a series of experiments on glutamate release and turnover in SNc. However, in contrast to the effects seen in striatum, evoked and tonic glutamate concentrations in SNc of 6-OHDA-treated mice and cNurr1DATCreER mice were not significantly affected by dopamine deficiency (evoked: intact vs lesioned, p = 0.78; WT vs KO, p = 0.66; tonic: intact vs lesioned, p = 0.79; WT vs KO, p = 0.13). Likewise, no effects on glutamate reuptake rates were found in SNc (intact vs lesioned, p = 0.83; WT vs KO, p = 0.99).
Discussion
TAAR1 potentiates DAT loss in response to intrastriatal 6-OHDA
Intrastriatal 6-OHDA causes a gradual loss of dopaminergic neurons (Sauer and Oertel, 1994; Przedborski et al., 1995) and is commonly used as a model to study the degeneration of dopaminergic neurons. Our immunoblotting and autoradiography experiments revealed more prominent reductions of TH and DAT after intrastriatal 6-OHDA administration in WT mice than in TAAR1 KO mice. However, after MFB 6-OHDA injections, a near-complete loss of TH levels was found in both genotypes, indicating that the partial survival of dopaminergic neurons is required for this effect of TAAR1. Consistent with our finding of enhanced DAT levels in TAAR1 KO mice, previous in vitro studies in brain synaptosomes and transfected cells have reported that activation of TAAR1 internalizes DAT from the cell membrane, inhibits dopamine reuptake, and enhances dopamine efflux via DAT reversal (Xie and Miller, 2007, 2009; Xie et al., 2008), However, because 6-OHDA elicits its neurotoxic effects by entering the neuron via DAT, the finding of a more pronounced reduction of TH in TAAR1 KO compared with WT mice was unexpected. These studies were extended to experiments examining the effects of a TAAR1 agonist, RO5166017, on reduction of DAT by intrastriatal 6-OHDA administration in WT mice. Chronic treatment with RO5166017 induced more extensive DAT loss, paralleled by a more severe motor phenotype, compared with vehicle-treated animals. Together, these data suggest that TAAR1 could potentiate DAT loss.
TAAR1 inhibits l-DOPA-induced rotational sensitization and the emergence of LIDs
To avoid baseline differences in degree of dopaminergic lesions, l-DOPA treatment experiments were conducted in mice with MFB 6-OHDA lesions. Effects of subchronic l-DOPA treatment was measured with regard to both the motor actions of l-DOPA (Ungerstedt and Arbuthnott, 1970; Simola et al., 2007) and LIDs (Lundblad et al., 2004; Francardo and Cenci, 2014). TAAR1 KO mice developed more contralateral turns and AIMs compared with WT mice after 12 d of subchronic l-DOPA treatment. This increased sensitivity to l-DOPA in TAAR1 KO mice is reminiscent of data found with dopaminergic psychostimulants. Indeed, amphetamine, metamphetamine, and MDMA have repeatedly been shown to have enhanced stimulating activities in TAAR1 KO mice (Bunzow et al., 2001; Di Cara et al., 2011; Achat-Mendes et al., 2012). Conversely, pharmacological activation of TAAR1 via systemic injections of the selective TAAR1 agonist RO5166017 before l-DOPA treatment reduced the number of contralateral rotations in MFB 6-OHDA-injected mice. The opposing effects of TAAR1 KO versus TAAR1 agonism on behavioral responses to subchronic l-DOPA treatment suggest a direct role of TAAR1 rather than compensatory developmental abnormalities in TAAR1 KO mice.
It may appear puzzling that TAAR1 activation potentiates DAT loss but reduces LIDs. However, anatomical sites for these actions are partially separated, with corticostriatal and striatonigral neurons being critical for LIDs (Iravani and Jenner, 2011). Other receptors have also opposing effects on dopamine degeneration versus proneness of developing LIDs. For example, dopamine D3 receptors protects dopamine neurons (Scheller et al., 2007; Li et al., 2010), but stimulate LIDs (Bordet et al., 1997).
TAAR1 counteracts l-DOPA-induced phosphorylation of AMPA receptors
LIDs are associated with enhanced phosphorylation of the AMPA receptor subunit GluA1 (Chase, 2004), which promotes postsynaptic glutamatergic signaling. Subchronic l-DOPA treatment increased phosphorylation of Ser845-GluA1 in the lesioned hemispheres of C57BL/6J mice. This effect was further enhanced in TAAR1 KO mice, but attenuated after acute administration of the TAAR1 agonist RO5166017. These biochemical effects are consistent with the behavioral responses to l-DOPA, suggesting an inhibitory role of TAAR1 activation on postsynaptic glutamate neurotransmission.
Evoked glutamate release is enhanced in striatum, but not in SNc, under dopamine deficiency
To further investigate the role of TAAR1 on glutamate neurotransmission under dopamine-deficient conditions, we studied effects on glutamate release and turnover in striatum and SNc. We used in vivo amperometry coupled with enzyme-coated MEAs, a methodology developed to measure rapid glutamate release and turnover (Burmeister et al., 2000, 2002; Hascup et al., 2008). We also used the cNurr1DATCreER KO mouse, which is characterized by a progressive loss of midbrain dopaminergic system integrity and reduced dopamine levels (Kadkhodaei et al., 2013), more closely resembling PD than 6-OHDA-induced models. Nurr1 is a transcription factor essential for the maintenance of dopaminergic neurons and expression of several genes required for the synthesis and function of dopamine, including TH and DAT (Zetterström et al., 1997; Perlmann and Wallén-Mackenzie, 2004; Jankovic et al., 2005). The cNurr1DATCreER KO mouse model has bilateral dopamine deficiency, a characteristic that reduces the number of experimental animals needed for in vivo amperometry and facilitates comparisons between drugs. Therefore, after an initial set of experiments to verify that both 6-OHDA-treated and cNurr1DATCreER KO animals exhibited elevated striatal glutamate concentrations, experiments were continued exclusively in the cNurr1DATCreER model.
Evoked, but not tonic, extracellular glutamate concentrations were enhanced in striatum in both MFB 6-OHDA-treated and cNurr1DATCreER KO mice compared with corresponding controls. These data are in partial agreement with previous electrophysiological data demonstrating that degeneration of nigrostriatal dopaminergic neurons leads to excess spontaneous corticostriatal glutamatergic activity in striatum (Calabresi et al., 1993; Gubellini et al., 2002; Gerfen and Surmeier, 2011). The glutamate reuptake was slower in striatum of the lesioned hemisphere, but not in striatum of cNurr1DATCreER KO mice. This difference between the two models may be related to the degree of neurodegeneration, which is more severe in the 6-OHDA model, and/or to a neurotoxin-induced downregulation of glutamate transporters GLT1 and GLAST, as shown previously in 6-OHDA-treated mice (Chung et al., 2008).
Degeneration of nigrostriatal dopaminergic neurons in PD leads to enhanced activation of the indirect basal ganglia pathway and disinhibition of the subthalamic nucleus, which sends glutamatergic projections to substantia nigra, including the SNc (Chergui et al., 1994; Hamani et al., 2004), and may thus cause excitotoxic cell damage (Rodriguez et al., 1998). It was therefore somewhat unexpected that we found no alterations in glutamate release and turnover in the SNc in MFB 6-OHDA-treated mice or in cNurr1DATCreER KO mice. This possibly reflects compensatory mechanisms, for example, at the level of glutamate reuptake or other mechanisms regulating glutamate release, a question meriting further study.
TAAR1 as a modulator of evoked corticostriatal glutamate release
Previous studies have reported that TAAR1 stimulation reduces evoked dopamine release (Leo et al., 2014). Here, we found that local administration of the selective TAAR1 agonist RO5166017 inhibited evoked corticostriatal glutamate release in the dopamine-denervated hemisphere of 6-OHDA-treated mice, as well as in cNurr1DATCreER KO mice. These effects of RO5166017 were blocked by the TAAR1 antagonist EPPTB, suggesting that TAAR1 negatively regulates evoked corticostriatal glutamate release after degeneration of dopaminergic pathways. Like RO5166017, the D2-like agonist ropinirole reduced evoked, but not tonic, glutamate concentrations in striatum, an effect resistant to EPPTB and thus exerted independently of TAAR1.
Coadministration of RO5166017 and ropinirole did not further decrease glutamate release, indicating an overlapping or convergent mechanism of action. Consistent with this supposition, the CB1 antagonist AM251 counteracted the effects of both ropinirole and RO5166017 on corticostriatal glutamate release. The precise mechanism(s) underlying the crosstalk between TAAR1 and CB1 receptors in reducing corticostriatal glutamate transmission remains to be elucidated. However, D2-like agonist-induced suppression of glutamate release involves an increase in endocannabinoid activity at presynaptic CB1 receptors on glutamatergic terminals (Yin and Lovinger, 2006; Kreitzer and Malenka, 2008). Whole-cell voltage-clamp experiments in rat brain slices reported that CB1 receptor activation not only inhibits evoked striatal glutamate release, but also the frequency of spontaneous release events (Gerdeman and Lovinger, 2001). Accordingly, we found that blockade of CB1 receptors by AM251 consistently increased tonic glutamate concentrations in cNurr1DATCreER KO mice and, interestingly, this effect was inhibited by RO5166017.
Possible molecular and cellular actions of TAAR1
Despite detailed studies on interactions with DAT and D2 receptors, TAAR1 remains poorly characterized with regard to its cellular distribution and its impact on signal transduction, particularly in neurons in situ. Although expression of TAAR1 mRNA has been found in several brain regions (Borowsky et al., 2001; Di Cara et al., 2011), it is not known precisely which cell types express TAAR1 mRNA. It seems that TAAR1 is expressed, not only in nerve cells, but also in glia because a recent study reported that TAAR1 activation on human astrocytes can decrease glutamate clearance in vitro via downregulation of EAAT-2 expression (Cisneros and Ghorpade, 2014). There is also a need for more precise information on the subcellular localization of TAAR1 in intact cells, especially because several experiments in transfected HEK293 cells have indicated that TAAR1 is mainly localized intracellularly (Bunzow et al., 2001; Miller et al., 2005; Xie et al., 2007, 2008). Future studies are needed to determine whether a cytosolic localization of TAAR1 causes specific effects on intracellular signaling with relevance to neurotransmitter release.
Concluding comments
Together, using a complementary genetic, pharmacological, behavioral, and neurochemical approach, along with two models of experimental Parkinsonism, the present data demonstrate that TAAR1 modulates both the long-term behavioral response to l-DOPA and, likely in an interrelated manner, presynaptic and postsynaptic glutamatergic neurotransmission in the striatum. Therefore, they unveil a novel dimension of the neurobiology of TAAR1 in regard to its potential implication in the treatment of PD and, in particular, the emergence of disabling dyskinesia upon prolonged exposure to l-DOPA.
Footnotes
This work was supported by grants from the Swedish Brain Fund, Swedish Research Council, Karolinska Institutet, and Swedish Strategic Foundation.
The authors declare no competing financial interests.
References
- Achat-Mendes C, Lynch LJ, Sullivan KA, Vallender EJ, Miller GM. Augmentation of methamphetamine-induced behaviors in transgenic mice lacking the trace amine-associated receptor 1. Pharmacol Biochem Behav. 2012;101:201–207. doi: 10.1016/j.pbb.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/S0896-6273(04)00265-X. [DOI] [PubMed] [Google Scholar]
- Berry MD. The potential of trace amines and their receptors for treating neurological and psychiatric diseases. Rev Recent Clin Trials. 2007;2:3–19. doi: 10.2174/157488707779318107. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci U S A. 1997;94:3363–3367. doi: 10.1073/pnas.94.7.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowsky B, Adham N, Jones KA, Raddatz R, Artymyshyn R, Ogozalek KL, Durkin MM, Lakhlani PP, Bonini JA, Pathirana S, Boyle N, Pu X, Kouranova E, Lichtblau H, Ochoa FY, Branchek TA, Gerald C. Trace amines: identification of a family of mammalian G-protein-coupled receptors. Proc Natl Acad Sci U S A. 2001;98:8966–8971. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradaia A, Trube G, Stalder H, Norcross RD, Ozmen L, Wettstein JG, Pinard A, Buchy D, Gassmann M, Hoener MC, Bettler B. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc Natl Acad Sci U S A. 2009;106:20081–20086. doi: 10.1073/pnas.0906522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchek TA, Blackburn TP. Trace amine receptors as targets for novel therapeutics: legend, myth and fact. Curr Opin Pharmacol. 2003;3:90–97. doi: 10.1016/S1471-4892(02)00028-0. [DOI] [PubMed] [Google Scholar]
- Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, Quigley DI, Darland T, Suchland KL, Pasumamula S, Kennedy JL, Olson SB, Magenis RE, Amara SG, Grandy DK. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- Burchett SA, Hicks TP. The mysterious trace amines: protean neuromodulators of synaptic transmission in mammalian brain. Prog Neurobiol. 2006;79:223–246. doi: 10.1016/j.pneurobio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Moxon K, Gerhardt GA. Ceramic-based multisite microelectrodes for electrochemical recordings. Anal Chem. 2000;72:187–192. doi: 10.1021/ac9907991. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119:163–171. doi: 10.1016/S0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, Sancesario G, Bernardi G. Electrophysiology of dopamine-denervated striatal neurons: implications for Parkinson's disease. Brain. 1993;116:433–452. doi: 10.1093/brain/116.2.433. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Chase TN. Striatal plasticity and extrapyramidal motor dysfunction. Parkinsonism Relat Disord. 2004;10:305–313. doi: 10.1016/j.parkreldis.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Chase TN, Oh JD. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends Neurosci. 2000;23:S86–S91. doi: 10.1016/S1471-1931(00)00018-5. [DOI] [PubMed] [Google Scholar]
- Chergui K, Akaoka H, Charléty PJ, Saunier CF, Buda M, Chouvet G. Subthalamic nucleus modulates burst firing of nigral dopamine neurones via NMDA receptors. Neuroreport. 1994;5:1185–1188. doi: 10.1097/00001756-199406020-00006. [DOI] [PubMed] [Google Scholar]
- Chung EK, Chen LW, Chan YS, Yung KK. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J Comp Neurol. 2008;511:421–437. doi: 10.1002/cne.21852. [DOI] [PubMed] [Google Scholar]
- Cisneros IE, Ghorpade A. Methamphetamine and HIV-1-induced neurotoxicity: role of trace amine associated receptor 1 cAMP signaling in astrocytes. Neuropharmacology. 2014;85:499–507. doi: 10.1016/j.neuropharm.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cara B, Maggio R, Aloisi G, Rivet JM, Lundius EG, Yoshitake T, Svenningsson P, Brocco M, Gobert A, De Groote L, Cistarelli L, Veiga S, De Montrion C, Rodriguez M, Galizzi JP, Lockhart BP, Cogé F, Boutin JA, Vayer P, Verdouw PM, Groenink L, Millan MJ. Genetic deletion of trace amine 1 receptors reveals their role in auto-inhibiting the actions of ecstasy (MDMA) J Neurosci. 2011;31:16928–16940. doi: 10.1523/JNEUROSCI.2502-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson TM, Alvarsson A, Stan TL, Zhang X, Hascup KN, Hascup ER, Kehr J, Gerhardt GA, Warner-Schmidt J, Arango-Lievano M, Kaplitt MG, Ogren SO, Greengard P, Svenningsson P. Bidirectional regulation of emotional memory by 5-HT1B receptors involves hippocampal p11. Mol Psychiatry. 2013;18:1096–1105. doi: 10.1038/mp.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza S, Lignani G, Caffino L, Maggi S, Sukhanov I, Leo D, Mus L, Emanuele M, Ronzitti G, Harmeier A, Medrihan L, Sotnikova TD, Chieregatti E, Hoener MC, Benfenati F, Tucci V, Fumagalli F, Gainetdinov RR. TAAR1 modulates cortical glutamate NMDA receptor function. Neuropsychopharmacology. 2015;40:2217–2227. doi: 10.1038/npp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza S, Salahpour A, Masri B, Sotnikova TD, Messa M, Barak LS, Caron MG, Gainetdinov RR. Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol Pharmacol. 2011;80:416–425. doi: 10.1124/mol.111.073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francardo V, Cenci MA. Investigating the molecular mechanisms of L-DOPA-induced dyskinesia in the mouse. Parkinsonism Relat Disord. 2014;20:S20–S22. doi: 10.1016/S1353-8020(13)70008-7. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Grandy DK. Trace amine-associated receptor 1-Family archetype or iconoclast? Pharmacol Ther. 2007;116:355–390. doi: 10.1016/j.pharmthera.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, O'Brien CF. N-methyl-D-aspartate antagonists in the treatment of Parkinson's disease. Arch Neurol. 1991;48:977–981. doi: 10.1001/archneur.1991.00530210109030. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernardi G, Finazzi-Agrò A, Maccarrone M. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamani C, Saint-Cyr JA, Fraser J, Kaplitt M, Lozano AM. The subthalamic nucleus in the context of movement disorders. Brain. 2004;127:4–20. doi: 10.1093/brain/awh029. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Pomerleau F, Huettl P, Gerhardt GA. Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. J Pharmacol Exp Ther. 2008;324:725–731. doi: 10.1124/jpet.107.131698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Parkinson and Movement Disorder Society. Update on treatments for motor symptom of PD. 2012. [Accessed March 1, 2015]. Available from: http://www.movementdisorders.org/MDS-Files1/PDFs/EBM-Papers/update-on-treatments-for-motor-symptoms-of-PD.pdf.
- Iravani MM, Jenner P. Mechanisms underlying the onset and expression of levodopa-induced dyskinesia and their pharmacological manipulation. J Neural Transm. 2011;118:1661–1690. doi: 10.1007/s00702-011-0698-2. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Prog Neurobiol. 2005;77:128–138. doi: 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Kadkhodaei B, Alvarsson A, Schintu N, Ramsköld D, Volakakis N, Joodmardi E, Yoshitake T, Kehr J, Decressac M, Björklund A, Sandberg R, Svenningsson P, Perlmann T. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc Natl Acad Sci U S A. 2013;110:2360–2365. doi: 10.1073/pnas.1221077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Leo D, Mus L, Espinoza S, Hoener MC, Sotnikova TD, Gainetdinov RR. Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: role of D2 dopamine autoreceptors. Neuropharmacology. 2014;81:283–291. doi: 10.1016/j.neuropharm.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Li C, Biswas S, Li X, Dutta AK, Le W. Novel D3 dopamine receptor-preferring agonist D-264: Evidence of neuroprotective property in Parkinson's disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J Neurosci Res. 2010;88:2513–2523. doi: 10.1002/jnr.22405. [DOI] [PubMed] [Google Scholar]
- Liberles SD, Buck LB. A second class of chemosensory receptors in the olfactory epithelium. Nature. 2006;442:645–650. doi: 10.1038/nature05066. [DOI] [PubMed] [Google Scholar]
- Lindemann L, Meyer CA, Jeanneau K, Bradaia A, Ozmen L, Bluethmann H, Bettler B, Wettstein JG, Borroni E, Moreau JL, Hoener MC. Trace amine-associated receptor 1 modulates dopaminergic activity. J Pharmacol Exp Ther. 2008;324:948–956. doi: 10.1124/jpet.107.132647. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2004;16:110–123. doi: 10.1016/j.nbd.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Miller GM, Verrico CD, Jassen A, Konar M, Yang H, Panas H, Bahn M, Johnson R, Madras BK. Primate trace amine receptor 1 modulation by the dopamine transporter. J Pharmacol Exp Ther. 2005;313:983–994. doi: 10.1124/jpet.105.084459. [DOI] [PubMed] [Google Scholar]
- Perlmann T, Wallén-Mackenzie A. Nurr1, an orphan nuclear receptor with essential functions in developing dopamine cells. Cell Tissue Res. 2004;318:45–52. doi: 10.1007/s00441-004-0974-7. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Levivier M, Jiang H, Ferreira M, Jackson-Lewis V, Donaldson D, Togasaki DM. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience. 1995;67:631–647. doi: 10.1016/0306-4522(95)00066-R. [DOI] [PubMed] [Google Scholar]
- Revel FG, Moreau JL, Gainetdinov RR, Bradaia A, Sotnikova TD, Mory R, Durkin S, Zbinden KG, Norcross R, Meyer CA, Metzler V, Chaboz S, Ozmen L, Trube G, Pouzet B, Bettler B, Caron MG, Wettstein JG, Hoener MC. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc Natl Acad Sci U S A. 2011;108:8485–8490. doi: 10.1073/pnas.1103029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol. 1998;44:S175–S188. doi: 10.1002/ana.410440726. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-X. [DOI] [PubMed] [Google Scholar]
- Scheller D, Chan P, Li Q, Wu T, Zhang R, Guan L, Ravenscroft P, Guigoni C, Crossman AR, Hill M, Bezard E. Rotigotine treatment partially protects from MPTP toxicity in a progressive macaque model of Parkinson's disease. Exp Neurol. 2007;203:415–422. doi: 10.1016/j.expneurol.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson's disease. Prog Neurobiol. 2012;96:69–86. doi: 10.1016/j.pneurobio.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Simola N, Morelli M, Carta AR. The 6-hydroxydopamine model of Parkinson's disease. Neurotox Res. 2007;11:151–167. doi: 10.1007/BF03033565. [DOI] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Espinoza S, Masri B, Zhang X, Salahpour A, Barak LS, Caron MG, Gainetdinov RR. The dopamine metabolite 3-methoxytyramine is a neuromodulator. PLoS One. 2010;5:e13452. doi: 10.1371/journal.pone.0013452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Witkin JM, Fienberg AA, Nomikos GG, Greengard P. Involvement of striatal and extrastriatal DARPP-32 in biochemical and behavioral effects of fluoxetine (Prozac) Proc Natl Acad Sci U S A. 2002;99:3182–3187. doi: 10.1073/pnas.052712799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24:485–493. doi: 10.1016/0006-8993(70)90187-3. [DOI] [PubMed] [Google Scholar]
- Von Voigtlander PF, Moore KE. Turning behavior of mice with unilateral 6-hydroxydopamine lesions in the striatum: effects of apomorphine, L-DOPA, amanthadine, amphetamine and other psychomotor stimulants. Neuropharmacology. 1973;12:451–462. doi: 10.1016/0028-3908(73)90061-0. [DOI] [PubMed] [Google Scholar]
- Wang H, Pickel VM. Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J Comp Neurol. 2002;442:392–404. doi: 10.1002/cne.10086. [DOI] [PubMed] [Google Scholar]
- Wang W, Dever D, Lowe J, Storey GP, Bhansali A, Eck EK, Nitulescu I, Weimer J, Bamford NS. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol. 2012;590:3743–3769. doi: 10.1113/jphysiol.2012.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Miller GM. Trace amine-associated receptor 1 is a modulator of the dopamine transporter. J Pharmacol Exp Ther. 2007;321:128–136. doi: 10.1124/jpet.106.117382. [DOI] [PubMed] [Google Scholar]
- Xie Z, Miller GM. A receptor mechanism for methamphetamine action in dopamine transporter regulation in brain. J Pharmacol Exp Ther. 2009;330:316–325. doi: 10.1124/jpet.109.153775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Westmoreland SV, Bahn ME, Chen G-L, Yang H, Vallender EJ, Yao WD, Madras BK, Miller GM. Rhesus monkey trace amine-associated receptor 1 signaling: enhancement by monoamine transporters and attenuation by the D2 autoreceptor in vitro. J Pharmacol Exp Ther. 2007;321:116–127. doi: 10.1124/jpet.106.116863. [DOI] [PubMed] [Google Scholar]
- Xie Z, Westmoreland SV, Miller GM. Modulation of monoamine transporters by common biogenic amines via trace amine-associated receptor 1 and monoamine autoreceptors in human embryonic kidney 293 cells and brain synaptosomes. J Pharmacol Exp Ther. 2008;325:629–640. doi: 10.1124/jpet.107.135079. [DOI] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhang X, Andren PE, Greengard P, Svenningsson P. Evidence for a role of the 5-HT1B receptor and its adaptor protein, p11, in L-DOPA treatment of an animal model of Parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2163–2168. doi: 10.1073/pnas.0711839105. [DOI] [PMC free article] [PubMed] [Google Scholar]