

Abstract
Protein phosphatase-1 (PP1), anchored by regulatory or targeting proteins at excitatory glutamatergic synapses, controls the phosphorylation of postsynaptic substrates and regulates the neurotransmission and plasticity. Here, we found that spinophilin, an actin-binding protein that targets PP1 at postsynaptic density, served as a scaffold for extracellular signal-regulated kinase (ERK) signaling components. Through the C-terminal PDZ domain, spinophilin directly interacted with ERK and its upstream mitogen-activated protein kinase kinase (MEK). PP1, recruited by spinophilin, gained access to and dephosphorylated these kinases, exerting a tonic inhibition of ERK signaling. The removal of PP1 inhibition by disturbing spinophilin/PP1 interaction allowed a restricted activation of MEK/ERK at synapses, which in turn augmented the synaptic transmission specifically mediated by GluN2B subunit-containing N-methyl-d-aspartate subtype of glutamate receptors. We provided evidence that in pain-related spinal cord dorsal horn, the scaffolding function of spinophilin played an important role in the negative control of ERK-dependent and GluN2B-dependent pain sensitization. Expression of wild-type spinophilin produced an effective analgesic action against chronic inflammatory pain induced by complete Freund's adjuvant in rats.
SIGNIFICANCE STATEMENT Extracellular signal-regulated kinase (ERK) relays the signals from multiple transmembrane receptors to a wide range of downstream effectors critical for the regulation of neuronal excitability and plasticity. The strength and duration of ERK signaling is spatiotemporally controlled by protein phosphatases. Sustained activation of ERK has been implicated in a variety of pathological processes. The current study revealed that spinophilin, a well characterized protein phosphatase 1 (PP1) synaptic targeting protein, was able to scaffold mitogen-activated protein kinase kinase (MEK) and ERK for dephosphorylation and inactivation by PP1. The loss of PP1 inhibition, as a result of spinophilin/PP1 dissociation, led to aberrant activation of MEK/ERK signaling, which had important implications for the exaggeration of NMDA receptor-dependent nociceptive synaptic transmission in spinal cord dorsal horn.
Keywords: extracellular signal-regulated kinase, NMDA receptor, pain, protein phosphatase-1, spinophilin
Introduction
Extracellular signal-regulated kinase (ERK) is a point of convergence of many intracellular signaling cascades (Ji et al., 2009). The typical pathway for ERK activation involves Raf-1 kinase-mediated phosphorylation of mitogen-activated protein kinase kinase (MEK) at Ser218/Ser222, which subsequently catalyzes ERK phosphorylation at Thr183/Tyr185 in the activation loop. These components of ERK-signaling cascades are organized and compartmentalized by scaffolding proteins to regulate diverse biological responses (Kolch, 2005). In CNS, the proper targeting and activation of ERK signaling in dendritic and postsynaptic sites regulate the synaptic trafficking of ionotropic glutamate receptors and mediate several forms of long-term potentiation of glutamatergic neurotransmission (Thomas and Huganir, 2004). The termination of ERK signaling is attributed to dephosphorylation events catalyzed by protein phosphatases. At the level of ERK, several dual-specificity phosphatases, serine/threonine phosphatases, and tyrosine phosphatases can remove the phosphates from either one or both of threonine and tyrosine residues within the ERK activation loop, resulting in the complete enzymatic inactivation (Zhou et al., 2002; Paul et al., 2003; Patterson et al., 2009). The upstream MEK and Raf-1 kinases contain several regulatory phosphorylation sites on serine and threonine residues (Matallanas et al., 2011; Roskoski, 2012a), dephosphorylation of which produces different changes of their enzymatic activities. Some scaffolding proteins for ERK signaling components can also recruit protein phosphatases to achieve a spatiotemporal control over the strength and duration of ERK signaling (Morrison and Davis, 2003; Ory et al., 2003; Kolch, 2005; Dard and Peter, 2006).
The protein phosphatase-1 (PP1) is a ubiquitous serine/threonine phosphatase that regulates a number of cellular processes through the interaction of its catalytic subunit with regulatory subunits (Cohen, 2002). The majority of these regulatory subunits are scaffolding or targeting proteins, which direct PP1 to different subcellular compartments and govern PP1 substrate specificity and functional diversity (Cohen, 2002). Spinophilin (SPN) is one of the well characterized PP1 synaptic targeting proteins, with an N-terminal actin-binding domain, a PP1-binding motif, a PSD-95/discs large/zona occludens-1 (PDZ) domain, and a C-terminal coiled-coil region (Sarrouilhe et al., 2006). Through its actin-binding domain, SPN anchors a pool of PP1 at postsynaptic density, where it modulates the glutamatergic neurotransmission and plasticity (Allen et al., 1997; Satoh et al., 1998; Yan et al., 1999; Feng et al., 2000). Here we identified MEK and ERK as the novel binding partners for SPN, and revealed a tonic inhibition of ERK signaling by PP1 in spinal cord dorsal horn of rats.
Materials and Methods
Animals and drugs.
All experimental procedures were in accordance with the guidelines of the Animal Care and Use Committee of Lanzhou University. Male adult Sprague Dawley rats (180–220 g) were purchased from the Experimental Animal Center of Lanzhou University and housed three to four per cage with ad libitum access to food and water. For intrathecal drug delivery, the rats were held firmly by a pelvic girdle and a 30 gauge needle attached to a 25 μl microsyringe was inserted between vertebrae L5 and L6. A sudden advancement of the needle accompanied by a slight flick of the tail was used as the indicator for proper insertion into the subarachnoid space. The chemical drugs and virus were intrathecally injected slowly in 10 μl volume. The replication-defective recombinant adenovirus type 5 (1010 pfu/ml) expressing green fluorescent protein (GFP)-tagged wild-type (WT) SPN [SPN(WT)] or SPN(F451A) mutant (phenylalanine to alanine mutation at residue 451) was commercially obtained from Yingrun Biotechnologies. d-APV, CNQX, and tetrodotoxin (Sigma-Aldrich) were dissolved in artificial CSF (ACSF; in mm: 119.0 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1.0 NaH2PO4, 26.0 NaHCO3, 11.0 d-glucose, pH 7.4). GluN2B-selective antagonist ifenprodil, myosin light chain kinase (MLCK) inhibitor ML-7, MEK inhibitor PD98059, and U-0126 (Sigma-Aldrich) were dissolved in dimethyl sulfoxide, which was diluted with ACSF or internal solution just before use. The final concentration of dimethyl sulfoxide was <0.1%. All the experiments were conducted blindly to the experimenters without knowledge of the manipulations that the animals received. The inflammatory pain was induced by subcutaneous injection of complete Freund's adjuvant (CFA; 50 μl; Sigma-Aldrich) into the plantar surfaces of hindpaws.
Subcellular fractionation.
The rats were anesthetized with sodium pentobarbital (60–90 mg/kg, i.p.) and the lumbar enlargements of spinal cords were quickly removed into ice-cold ACSF (bubbled with 95% O2 and 5% CO2). The dorsal quadrants of spinal cords were dissected out and homogenized in the lysis buffer [in mm: 10.0 Tris-HCl, pH 7.6, 320.0 sucrose, 5.0 EDTA, and proteases/phosphatases inhibitors (10.0 mm NaF; 1.0 mm orthovanadate; 1.0 mm phenylmethylsulfonyl fluoride; 1.0 mg/ml each of aprotinin, chymostatin, leupeptin, antipain, and pepstatin)]. The homogenates were centrifuged at 1000 × g for 10 min at 4°C to remove the nuclei and large debris (P1). The supernatant (S1) was collected and centrifuged at 10,000 × g for 15 min to obtain P2 pellet that contained crude synaptosomal fraction. P2 was incubated for 30 min with the lysis buffer containing 0.5% Triton X-100, and then centrifuged at 32,000 × g for 20 min to harvest synaptosomal membrane fraction (P3; Smith et al., 2006; Jaworski et al., 2009; Sanz-Clemente et al., 2010; Li et al., 2015). To assay the protein expression and phosphorylation (Li et al., 2015), the spinal dorsal horn was homogenized in radio-immunoprecipitation assay (RIPA) buffer (50.0 mm Tris-HCl, pH 8.0, 150.0 mm NaCl, 1.0 mm EDTA, 1.0% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, and proteases/phosphatases inhibitors). After centrifugation at 14,000 × g for 10 min, the supernatant was collected and protein concentration was measured by using bicinchoninic acid assay kit (Pierce).
Coimmunoprecipitation and Western blot.
For coimmunoprecipitation (Li et al., 2015), the P2 fraction was extracted in 50.0 mm Tris-HCl, pH 9.0, 10.0 mm EDTA, 1.0% sodium deoxycholate, and proteases/phosphatases inhibitors at 37°C for 30 min. Equal volume of the dilution buffer (50.0 mm Tris-HCl, pH 7.4, 150.0 mm NaCl, 0.1% SDS, 1.0% Triton X-100, and proteases/phosphatases inhibitors) was added into the extract above. After centrifugation at 14,000 × g, the supernatant was collected and incubated at 4°C with primary antibodies overnight. The protein A/G-agarose beads were incubated with the immune complexes for 4 h. After three washes, the immunoprecipitates were resuspended in SDS sample buffer and boiled for 5 min before Western blot analysis.
The protein samples were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes. After blocking with 5% nonfat milk, the membranes were incubated with primary antibody overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories). The blots were visualized by enhanced chemiluminescence (Beyotime Institute of Biotechnology).
Antibodies.
The primary antibodies used in the present study included the mouse anti-GluN1 antibody from BD PharMingen; rabbit anti-GluN2B, rabbit anti-GluN2A, rabbit anti-SPN, rabbit anti-PP1γ1, and mouse anti-Raf-1-pSer338 antibody from Millipore; mouse anti-β-actin antibody from Sigma-Aldrich; mouse anti-His, mouse anti-GST, rabbit anti-Raf-1, and rabbit anti-Raf-1-pSer259 antibody from Anbo Biotechnology; mouse anti-SPN antibody from BD Transduction Laboratories; rabbit anti-MEK1 antibody from Santa Cruz Biotechology; rabbit anti-ERK1/2, mouse anti-ERK1/2-pThr183/Tyr185, rabbit anti-MEK1/2-pSer218/Ser222, and rabbit anti-MLC-pThr18/Ser19 antibody from Cell Signaling Technology.
Immunohistochemistry.
Rats were perfused through the ascending aorta with PBS (0.01 m) followed by 4% paraformaldehyde in 0.1 m phosphate buffer. The L4–L5 spinal cord segments were dissected and fixed in the same fixative for 4 h. The tissues were cryoprotected in 30% sucrose overnight. Transverse sections (16 μm) were cut and incubated with rabbit anti-GFP antibody (Clontech) and mouse anti-NeuN antibody (Millipore) or mouse anti-ERK1/2-pThr183/Tyr185 antibody and rabbit anti-NeuN antibody (Millipore) at 4°C for 72 h. After several washes, the sections were incubated with Alexa Fluor 488-conjugated and Cy3-conjugated secondary antibodies for 2 h before image capture (Li et al., 2015).
Expression constructs.
The rat full-length SPN cDNA in pEGFP-N1 vector was purchased from Genechem and used as a template to generate SPN(F451A) mutant by PCR. The cDNA encoding SPN(WT), SPN(F451A), SPN(1–648), SPN(1–494), SPN(1–446), SPN(1–154), or SPN(494–648) was PCR subcloned and ligated into pGEX-4T-1 or pcDNA3.1/myc-HisA vector. The human full-length MEK1 cDNA, rat ERK2 cDNA, and human PP1γ catalytic subunit cDNA in pcDNA3.1/myc-HisA or pGEX-6p-1 vector were obtained from Genewiz. The pBabe-puro vector encoding MEK1(S218D/S222D) was a gift from William Hahn (Addgene plasmid #15268). The pBabe-puro-Raf-1 was a gift from Matthew Meyerson (Addgene plasmid #51124), which was used to generate Raf-1(Y340D/Y341D) mutant by Genewiz. All the constructs were confirmed by DNA sequencing.
Transfection.
HEK293T cells were grown in DMEM containing 10% fetal bovine serum in an incubator with humidified air (5% CO2) at 37°C. Cells were transiently transfected with plasmid DNA (10 μg) using a standard calcium phosphate method. Cells were harvested 48 h after transfection.
GST pull-down assay.
The GST fusion proteins were expressed in Escherichia coli BL21 cells and affinity purified by glutathione agarose beads (Sigma-Aldrich) according to the manufacturer's instructions. The His-tagged proteins were expressed in HEK293T cells, purified by nickel-nitrilotriacetic acid (Ni-NTA) column (Roche), and eluted with 0.25 m imidazole in 300 mm NaCl and 50 mm NaH2PO4, pH 8.0. The eluted proteins were concentrated and desalted through Amicon Ultra Centrifugal Filters (Millipore). The protein purity was assessed by Western blot and Coomassie blue staining. For pull-down assays, the purified His proteins (0.5 μm) or lysates from spinal dorsal horn (500 μg) were incubated with GST proteins bound to glutathione agarose beads in RIPA buffer and rotated for 4 h at 4°C. The beads were washed six times with RIPA buffer before immunoblotting analysis.
Dephosphorylation assays.
The HEK293T cells were cotransfected with pBabe-puro-Raf-1(Y340D/Y341D) and pcDNA3.1/myc-HisA-MEK1. The phosphorylated His-tagged MEK1 was purified from cell lysates by using Ni-NTA column. The His-tagged ERK2 was obtained from HEK293T cells coexpressing pBabe-puro-MEK1(S218D/S222D) and pcDNA3.1/myc-HisA-ERK2. These phosphorylated His proteins (0.5 μm) were used as the substrates for dephosphorylation by His-PP1 (0.05 μm; Zhou et al., 2002; Wu et al., 2009). The phosphatase buffer contained 50 mm Tris-HCl, pH 7.5, 0.1 mm EDTA, 2 mm MnCl2, and 10 mm DTT with or without GST-SPN(WT) and GST-SPN(F451A) bound to glutathione agarose beads. After incubation for 30 min at 30°C, the reaction was stopped by adding SDS sample buffer, followed by Western blot analysis of protein phosphorylation. For pull-down assays, the phosphorylated His proteins were incubated with GST-SPN(WT) and His-PP1. The dephosphorylation reaction was conducted at 30°C for 0, 10, and 30 min. At each time point, the beads were collected and washed six times with RIPA buffer before immunoblotting analysis.
Behavioral test.
The pain thresholds were measured blindly. The rats were habituated in a cage with wire mesh floor for ≥30 min before pain sensitivity was measured. A set of von Frey filaments (Stoelting) was applied perpendicularly to the plantar surfaces of hindpaws and 50% paw withdrawal thresholds (PWTs) were calculated by using the up-down method as described previously (Li et al., 2015). To measure the paw withdrawal latency (PWL), the animals were placed on a clear glass plate and a beam of light was focused on the midplantar surfaces of hindpaws to deliver heat stimuli, with the cutoff of 20 s. The time between the onset of heat application and paw withdrawal was recorded automatically as PWL values.
Preparation of spinal cord slices.
The rats (4–5 weeks) were deeply anesthetized with sodium pentobarbital. The lumbar spinal cords were quickly removed into ice-cold sucrose solution (in mm: 50.0 sucrose, 95.0 NaCl, 1.8 KCl, 0.5 CaCl2, 7.0 MgSO4, 1.2 KH2PO4, 26.0 NaHCO3, 15.0 d-glucose, bubbled with 95% O2 and 5% CO2, pH 7.4; Ikeda et al., 2006; Fan et al., 2014). A transverse slice (600 μm thickness) with an intact L4 or L5 dorsal root was cut on a vibratome stage, transferred to the recording chamber, and perfused with oxygenated ACSF at 30–32°C for ≥1 h before electrophysiological recordings or drug treatment.
Electrophysiological recordings.
Whole-cell patch-clamp recordings were performed with an Axon700B amplifier (Molecular Devices) at day 3 after intrathecal viral injection or intraplantar CFA injection. The GFP-expressing lamina II neurons were visually identified by using an Olympus BX51WIF microscope fitted with a 40× water-immersion objective under fluorescence and transmitted light illumination. The glass pipettes (4–8 MΩ) were filled with the internal solution containing (in mm) the following: 115 Cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na2ATP, 0.4 Na-GTP, 0.6 EGTA, and 10 sodium phosphocreatine, pH 7.25 (295–300 mOsm). The attached dorsal roots were stimulated (0.1 Hz, 0.1 ms duration, 2–5 mA) through a suction electrode (Ikeda et al., 2006; Fan et al., 2014). To calculate the AMPAR/NMDAR ratios, the evoked EPSCs were recorded at the holding potential of −70 and +40 mV with the perfusate containing GABAA receptor antagonist picrotoxin (50.0 μm) and glycine receptor antagonist strychnine (2.0 μm). The averaged AMPAR peak currents (15–30 consecutive events) recorded at −70 mV were divided by averaged NMDAR currents that were measured 49–51 ms after the stimulus at +40 mV (Jurado et al., 2013). To pharmacologically isolate the NMDAR component of synaptic responses, AMPAR antagonist CNQX (10.0 μm) was also added into the perfusate (Zhou et al., 2010; Fan et al., 2014). The monosynaptic EPSCs were identified on the basis of the constant latency and the absence of conduction failure in response to high-frequency electrical stimulation (20 Hz; Zhou et al., 2010; Fan et al., 2014). To plot the input–output curves, the synaptic responses were elicited at six different stimulation intensities (0.1, 0.2, 0.5, 1.0, 2.0, and 5.0 mA; Luo et al., 2012). For miniature EPSCs (mEPSCs), the neurons were held at −70 mV, and the recordings were performed in the external solution containing tetrodotoxin (0.5 μm), picrotoxin (50.0 μm), and strychnine (2.0 μm). The paired-pulse ratios were measured by delivering two stimuli at different interstimulus intervals (ISIs; Jurado et al., 2013). The series and input resistances were monitored on-line throughout each experiment (Hu et al., 2007). The recordings were abandoned if any resistance changed >15%. The current signals were filtered at 2 kHz and sampled at 10 kHz.
Statistics.
All data were represented as mean ± SEM. The mEPSC signals were analyzed by minianalysis software, while the peak amplitudes of evoked EPSCs were analyzed by Clampfit 8.0 software. For Western blot, the scanned digital images were quantified by National Institutes of Health ImageJ software. The relative immunoreactive intensity of one protein was calculated by the ratio of its signal to β-actin signal or its nonphosphorylation signal. For display purposes, these ratios ware normalized to control values. Differences were compared by using Student's t test or one-way ANOVA followed by post hoc Tukey's honestly significant difference test. The criterion for statistical significance was p < 0.05.
Results
SPN directly interacted with MEK and ERK through its PSD-95/discs large/zona occludens-1 domain
SPN is one of the PP1 synaptic targeting proteins (Sarrouilhe et al., 2006). The specific antibody against SPN coimmunoprecipitated PP1 from synaptosomal fraction of spinal cord dorsal horn (Fig. 1a). In addition to PP1, anti-SPN antibody also pulled down MEK1 and ERK1/2 (Fig. 1a), the key signaling components involved in synaptic plasticity (Thomas and Huganir, 2004; Ji et al., 2009). The Raf-1 kinase that acted upstream of MEK/ERK was not detectable in SPN precipitates (Fig. 1a). Reciprocal experiments verified that anti-MEK1 (Fig. 1b) or anti-ERK1/2 (Fig. 1c) antibody stably precipitated SPN and PP1. Anti-Raf-1 antibody failed to precipitate SPN, although PP1 was detected in Raf-1 precipitates (Fig. 1d).
Figure 1.

SPN interacted with MEK and ERK. a–d, Coimmunoprecipitation (Co-IP) was performed from synaptosomal fraction of spinal dorsal horn of rats with specific antibody against SPN (a), MEK1 (b), ERK1/2 (c), or Raf-1 (d). The precipitates were immunoblotted with antibodies indicated on the left of panels. Molecular weight markers were shown in kilodalton on the right of panels.
To test the possible direct interaction of SPN with MEK and ERK, we constructed a family of GST fusion proteins of SPN (Fig. 2a), and performed GST pull-down assays. GST fusion of WT SPN [SPN(WT)] precipitated MEK1 (Fig. 2b) and ERK1/2 (Fig. 2c) from lysates of spinal dorsal horn of rats. GST-SPN(1–648), which lacked the C-terminal coiled-coil domain, also pulled down MEK1 and ERK1/2 (Fig. 2b,c). Further truncation of PSD-95/discs large/zona occludens-1 (PDZ) domain [GST-SPN(1–494)], however, abrogated SPN interaction with MEK1 and ERK1/2 (Fig. 2b,c), suggesting that the PDZ domain was likely responsible for MEK/ERK binding. To confirm these results, we purified the recombinant His-tagged MEK1 or ERK2 as the prey proteins for in vitro pull-down assays. GST-SPN(WT), but not GST, interacted with His-MEK1 (Fig. 2d). GST-SPN(494–648), which contained only the PDZ domain, was sufficient to precipitate His-MEK1 (Fig. 2d). GST-SPN(1–494), which was devoid of PDZ domain, did not precipitate His-MEK1 (Fig. 2d). Reciprocally, GST-tagged MEK1 pulled down the purified His-SPN(WT) (Fig. 2e). GST-SPN(WT), GST-SPN(1–648), and GST-SPN(494–648) also pulled down purified His-ERK2, while PDZ domain-defective GST proteins failed to interact with His-ERK2 (Fig. 2f).
Figure 2.

The PDZ domain of SPN was responsible for MEK/ERK binding. a, GST fusion proteins containing different domains of SPN were constructed. ABD, Actin-binding domain; RBD, receptor-binding domain. b, c, MEK1 (b) and ERK1/2 (c) pulled down by GST proteins from lysates of spinal dorsal horn of rats were analyzed by immunoblotting (top). The total amounts of MEK1/ERK1/2 (middle) and GST proteins (bottom) used for pull-down assays were also shown. d–f, The purified recombinant His-MEK1 (d), His-SPN (e), or His-ERK2 (f) pulled down by GST proteins in vitro was analyzed by anti-His antibody (top). The total amounts of His (middle) and GST proteins (bottom) used for pull-down assays were also shown.
The scaffolding function of SPN enabled the direct dephosphorylation of MEK/ERK by PP1
The full activation of MEK1 and ERK1/2 requires Ser218/Ser222 and Thr183/Tyr185 phosphorylation, respectively. Since SPN recruited MEK/ERK to the close vicinity of PP1, we hypothesized that SPN-bound PP1 might regulate MEK/ERK activities by dephosphorylating their serine/threonine residues in the activation loops. To test this, we expressed the constitutively active Raf-1(Y340D/Y341D) mutant in HEK293T cells, which simultaneously increased the phosphorylation of MEK and ERK (Fig. 3a). When SPN(WT) was cotransfected into HEK293T cells, the phosphorylation levels of MEK and ERK were significantly suppressed (Fig. 3a). SPN might take effect by targeting MEK alone or both MEK and ERK. To investigate whether SPN acted at the level of ERK, the constitutively active MEK1(S218D/S222D) mutant was transfected in HEK293T cells to enhance ERK phosphorylation (Fig. 3b). We found that coexpression of SPN(WT) also decreased the phosphorylation of ERK induced by MEK1(S218D/S222D; Fig. 3b), suggesting that SPN was likely to act at the levels of both MEK and ERK.
Figure 3.

SPN facilitated PP1 dephosphorylation of MEK and ERK. a, b, Effects of SPN(WT) on MEK/ERK phosphorylation induced by constitutively active Raf-1(Y340D/Y341D) [Raf-1(DD)] (a) or MEK1(S218D/S222D) [MEK1(DD)] (b) in HEK293T cells. *p < 0.05 relative to control cells. #p < 0.05 relative to Raf-1(DD)-expressing or MEK1(DD)-expressing cells. n = 6 experiments. c, d, The purified, phosphorylated His-MEK1 (His-pMEK1; c) or His-ERK2 (His-pERK2; d) was incubated with His-PP1 and GST-SPN(WT) or GST-SPN(F451A) in vitro at 30°C for 30 min before immunoblotting analysis of MEK1 and ERK2 phosphorylation (top). The total amount of His-pMEK1 (c) or His-pERK2 (d) used for analysis was also shown (bottom). *p < 0.05 relative to the phosphorylation levels without His-PP1. n = 6 experiments. e, f, The dephosphorylated MEK and ERK reduced their bindings to SPN. The dephosphorylation reaction was conducted by incubating His-pMEK1 or His-pERK2 with GSP-SPN(WT) and His-PP1 at 30°C for 0, 10, and 30 min. At each time point, MEK1 (e, top) or ERK2 (f, top) was pulled down by GST-SPN(WT). The graph showed the percentage changes of GSP-SPN(WT)-precipitated MEK1 (e) and ERK2 (f). *p < 0.05 relative to the values at 0 min. n = 6 experiments.
To examine whether SPN facilitated the direct dephosphorylation by PP1 of MEK1, we performed in vitro dephosphorylation assays. The purified, phosphorylated His-MEK1 displayed no significant dephosphorylation when incubated with recombinant PP1 alone (Fig. 3c). The presence of GST-SPN(F451A), a SPN mutant devoid of PP1 binding (Hsieh-Wilson et al., 1999; Yan et al., 1999), also failed to influence His-MEK1 phosphorylation levels (Fig. 3c). When GST-SPN(WT) was added into the reaction buffer, however, a significant dephosphorylation of His-MEK1 was observed (Fig. 3c). Similarly, the purified, phosphorylated His-ERK2 exhibited no detectable dephosphorylation in the presence of PP1 and GST-SPN(F451A (Fig. 3d). A significant reduction of His-ERK2 phosphorylation was observed only when coincubated with GST-SPN(WT) and PP1 (Fig. 3d). With the dephosphorylation by PP1, MEK1 and ERK2 reduced their bindings to SPN. As shown in Figure 3e,f, GSP-SPN(WT) pulled down less MEK1 (Fig. 3e) or ERK2 (Fig. 3f) at 10 and 30 min after in vitro incubation with PP1 and phosphorylated His-MEK1 or His-ERK2 at 30°C. These data suggested that SPN enabled PP1 to dephosphorylate and release MEK/ERK.
Tonic inhibition of synaptic MEK/ERK activities by SPN
To interfere with SPN/PP1 interaction, the recombinant adenovirus encoding GFP-tagged SPN(F451A) or SPN(WT) was intrathecally injected (Mannes et al., 1998; Li et al., 2015). The adenovirus successfully infected dorsal horn neurons, as evidenced by GFP signals that coincided with neuronal marker NeuN (Fig. 4a; infection rate: 70.0 ± 10.5%, n = 6 slices from 6 rats). Compared with GFP control, SPN(F451A) significantly enhanced the basal phosphorylation of MEK and ERK in intact rats, while SPN(WT) had no effect (Fig. 4b). Raf-1 phosphorylation at Ser338 and Ser259, two key phosphorylation sites that regulate Raf-1 catalytic activity, was insensitive to SPN(F451A) or SPN(WT) (Fig. 4c). Consistent with previous reports that the active ERK following noxious stimulation or peripheral inflammation is expressed in dorsal horn neurons (Ji et al., 1999, 2002; Karim et al., 2001; Xu et al., 2008), the phosphorylated ERK caused by SPN(F451A) was also colocalized with NeuN (Fig. 4d). Biochemical analysis demonstrated that MEK/ERK activation in SPN(F451A)-expressing rats was attributed to spontaneous NMDAR activity and restricted to synaptosomal fraction. Treatment of spinal cord slices with tetrodotoxin (1 μm), which blocked spontaneous neuronal activity, or d-APV (100 μm), which blocked NMDAR, abolished the increase of MEK and ERK phosphorylation induced by SPN(F451A) in synaptosomal fraction, while AMPAR antagonist CNQX (10 μm) had no effect (Fig. 4e). No significant changes of cytosolic MEK/ERK phosphorylation were observed after SPN(F451A) expression (Fig. 4e), suggesting that SPN-targeted PP1 was active in the sustained restriction of synaptic MEK/ERK signaling from activation.
Figure 4.
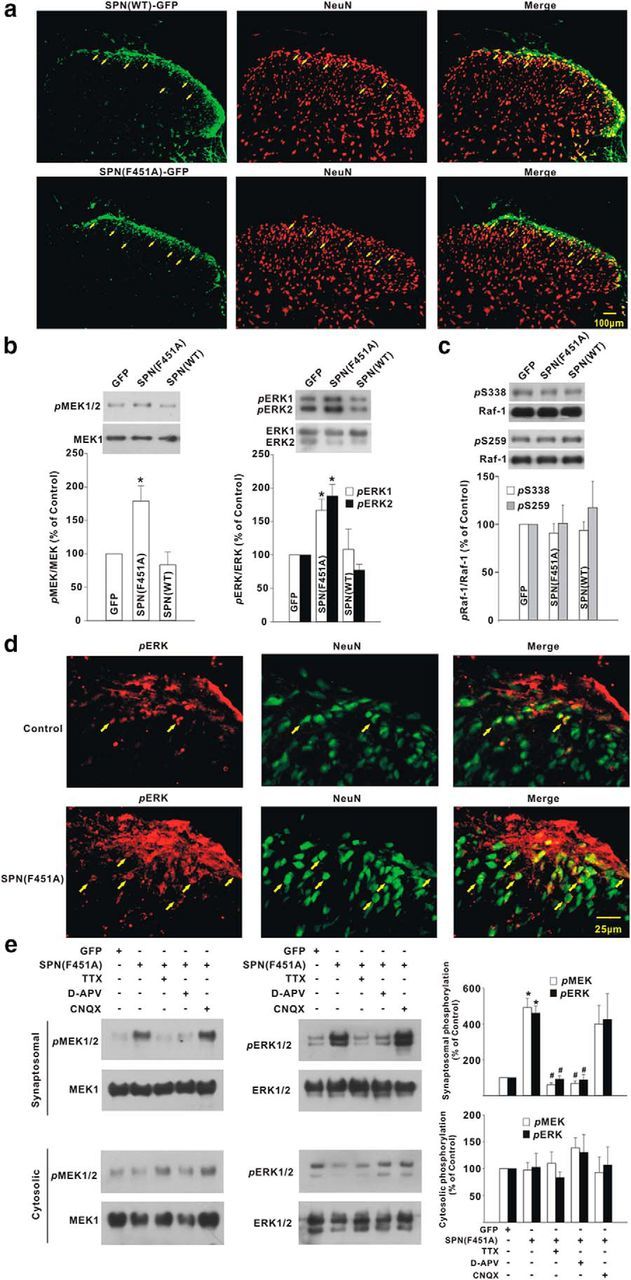
SPN-bound PP1 negatively controlled MEK and ERK phosphorylation at synaptosomal fraction of spinal dorsal horn of rats. a, GFP signals (green) in spinal dorsal horn coincided with neuronal marker NeuN (red) after intrathecal injection of recombinant adenovirus encoding GFP-tagged SPN(WT) or SPN(F451A). The arrows indicated the neurons positive for GFP. b, c, The phosphorylation levels of MEK/ERK (b) and Raf-1 (c) were probed at day 3 after viral expression of GFP, SPN(WT), and SPN(F451A). *p < 0.05 relative to GFP control. n = 6 experiments in each group. d, Immunostaining illustrated the colocalization of phosphorylated ERK (pERK; red) with NeuN (green) at day 3 after intrathecal administration of adenovirus encoding SPN(F451A). The arrows indicated the neurons positive for phosphorylated ERK. e, The spinal cord slices were prepared at day 3 after GFP or SPN(F451A) expression and incubated with tetrodotoxin (TTX; 1 μm), d-APV (100 μm), or CNQX (10 μm) for 2 h before immunoblotting analysis of phosphorylated MEK or ERK at synaptosomal and cytosolic fraction. *p < 0.05 relative to GFP, #p < 0.05 relative to SPN(F451A). n = 4 experiments in each group.
Specific regulation of GluN2B receptor-mediated synaptic transmission by SPN through MEK/ERK signaling
The effects of SPN on synaptic transmission was investigated by recording primary afferent-evoked EPSCs in spinal cord slices at day 3 after viral expression of GFP, GFP-tagged SPN(WT), or SPN(F451A). SPN(WT) or GFP alone had no effect on the ratios of the amplitudes of AMPAR-mediated EPSCs to NMDAR-mediated EPSCs (AMPAR/NMDAR ratios; Fig. 5a). In neurons expressing SPN(F451A), however, the AMPAR/NMDAR ratios were substantially decreased relative to those in GFP-expressing neurons (Fig. 5a). There was no significant difference in the amplitude and frequency of AMPAR-mediated mEPSCs among intact neurons and GFP-expressing, SPN(F451A)-expressing, and SPN(WT)-expressing neurons (Fig. 5b), implicating a selective potentiation of NMDAR synaptic responses after disturbing SPN-mediated PP1 targeting. The input–output curves confirmed this by showing a substantial increase of NMDAR-EPSC amplitudes in neurons expressing SPN(F451A), but not SPN(WT), when compared with GFP control (Fig. 5c). The paired-pulse ratios of EPSCs, which correlate with the neurotransmitter release probability, were comparable at each ISI among GFP-expressing, SPN(F451A)-expressing, and SPN(WT)-expressing neurons (Fig. 5d), suggesting that the presynaptic function was unchanged.
Figure 5.

SPN-bound PP1 regulated NMDAR-mediated EPSCs in spinal cord slices from rats. a, The ratios of AMPAR-EPSC amplitudes to NMDAR-EPSC amplitudes (AMPAR/NMDAR ratios) were recorded in control neurons and GFP-, SPN(WT)-expressing or SPN(F451A)-expressing neurons. n = 12 neurons in each group. *p < 0.05 relative to GFP neurons. b, Comparison of the amplitudes and frequencies of AMPAR-mediated mEPSCs. n = 6 neurons in each group. c, NMDAR EPSCs were elicited at six different stimulation intensities (left) and the input (stimulation intensity)–output (synaptic response) curves were plotted (right). n = 10 neurons in each group. *p < 0.05 relative to GFP. d, NMDAR EPSCs were evoked by a pair of electrical stimuli at different ISIs in GFP-expressing, SPN(WT)-expressing, or SPN(F451A)-expressing neurons (top), and the paired-pulse ratios (PPR) were plotted against ISI (bottom). n = 6 neurons in each group.
Most NMDARs are composed of GluN1 and GluN2B or GluN2A subunits in the spinal cord. Immunoblotting analysis demonstrated that, compared with GFP control, SPN(F451A) significantly increased the immunoreactive intensities of NMDAR GluN2B and GluN1 subunits, but not GluN2A subunits, at the synaptosomal membrane fraction (Fig. 6a). The total protein level of each NMDAR subunit exhibited no difference among GFP-expressing, SPN(F451A)-expressing, and SPN(WT)-expressing rats (Fig. 6b). To test whether the GluN2B receptor was the specific target for SPN/PP1 regulation, we recorded NMDAR EPSCs and extracellularly perfused ifenprodil, a GluN2B-selective antagonist. In SPN(F451A)-expressing neurons, a dramatic decrease in the amplitudes of NMDAR EPSCs was observed after ifenprodil (3.0 μm) application (Fig. 6c). The synaptic responses were insensitive to ifenprodil in GFP-expressing or SPN(WT)-expressing neurons (Fig. 6c).
Figure 6.

SPN specifically regulated the synaptic accumulation of GluN2B receptors. a, b, The protein contents of NMDA receptor GluN1, GluN2B, and GluN2A subunits at synaptosomal membrane fraction (P3; a) or homogenates (b) were examined at day 3 after intrathecal injection of recombinant adenovirus encoding GFP, SPN(F451A), and SPN(WT) in rats. Equal protein loadings were indicated by β-actin signals. The graph summarized the percentage changes of NMDAR subunits. n = 6 experiments for each subunit. *p < 0.05 relative to GFP control. c, Effects of GluN2B-selective antagonist ifenprodil on NMDAR-EPSC amplitudes. The horizontal bar indicated the period of extracellular ifenprodil perfusion. n = 6 neurons in each group.
Our data showed that MEK/ERK signaling was likely responsible for GluN2B synaptic accumulation. Bath application of MEK inhibitor U-0126 (0.5 μm) suppressed NMDAR-EPSC amplitudes in neurons expressing SPN(F451A) (Fig. 7a). No reduction of NMDAR EPSCs was produced by U-0126 in SPN(WT)-expressing neurons (Fig. 7a). Previous studies have indicated that ERK can phosphorylate and activate MLCK (Klemke et al., 1997), an important postsynaptic component that phosphorylates myosin light chains (MLCs) to boost the synaptic currents specifically mediated by NMDAR (Lei et al., 2001). We found that SPN(F451A)-expressing rats displayed a significant increase of MLC phosphorylation when compared with GFP rats (Fig. 7b). Spinal treatment with U-0126 (5 μg) repressed MLC phosphorylation (Fig. 7b), implicating an ERK-dependent MLCK activation. A similar suppression of MLC phosphorylation was also observed with MLCK inhibitor ML-7 (50 μg; Fig. 7b). Postsynaptic loading of ML-7 (10.0 μm) through the glass pipettes mimicked MEK inhibition by decreasing NMDAR-EPSC amplitudes in SPN(F451A)-expressing neurons, but not in those expressing GFP or SPN(WT) (Fig. 7c), suggesting that SPN-targeted PP1 might regulate NMDAR synaptic currents through the ERK/MLCK pathway.
Figure 7.

MEK/ERK signaling mediated the synaptic modification by SPN-associated PP1. a, Effects of MEK inhibitor U-0126 on NMDAR-EPSC amplitudes in neurons expressing SPN(WT) or SPN(F451A). The horizontal bar indicated the period of extracellular U-0126 perfusion. n = 6 neurons in each group. b, MLC phosphorylation (pMLC) significantly increased at day 3 after SPN(F451A) expression, which, however, could be repressed by spinal treatment with U-0126 (5 μg) or MLCK inhibitor ML-7 (50 μg) for 30 min. *p < 0.05 relative to GFP rats, #p < 0.05 relative to SPN(F451A) rats. n = 6 experiments. c, Postsynaptic loading of ML-7 through the recording pipettes inhibited NMDAR EPSCs in neurons expressing SPN(F451A). n = 6 neurons in each group.
Functional significance of SPN/PP1 complex in pain-related spinal cord dorsal horn
Intraplantar injection of CFA evokes long-lasting pain sensitization, which requires ERK activation in spinal dorsal horn (Xu et al., 2008; Alter et al., 2010). CFA did not affect the total protein levels of SPN and PP1 at spinal homogenates (Fig. 8a). Nevertheless, the PP1 contents coimmunoprecipitated by anti-SPN antibody from synaptosomal fraction of inflamed rats were much less than those from saline-injected control littermates (Fig. 8b). The MEK and ERK contents in SPN precipitates were comparable between saline-injected and CFA-injected rats (Fig. 8b). SPN/PP1 dissociation might exert a permissive role for peripheral inflammation to initiate MEK/ERK signaling, because overexpression of SPN(WT) significantly suppressed MEK and ERK phosphorylation induced by CFA (Fig. 8c). The hyperactivity of MEK/ERK as a result of the deficiency of PP1 inhibition contributed to the potentiation of NMDAR-mediated synaptic transmission during inflammatory pain. Viral expression of SPN(WT) (Fig. 8d) or direct application of U-0126 (Fig. 8e) repressed the magnitudes of NMDAR EPSCs in CFA rats.
Figure 8.

Intraplantar injection [intradermal (i.d.)] of CFA reduced the inhibitory effects of SPN on MEK/ERK signaling in spinal dorsal horn of rats. a, CFA had no effects on total protein levels of SPN and PP1. n = 6 experiments. b, Coimmunoprecipitation (Co-IP) was performed with anti-SPN antibody from synaptosomal fraction of spinal dorsal horn. The precipitates were immunoblotted with antibodies indicated on the left of panels. n = 6 experiments, *p < 0.05 relative to saline control. c, Effects of SPN(WT) on CFA-induced MEK/ERK phosphorylation. Intrathecal viral injection (i.t.) was performed at 2 h post-CFA and immunoblotting was conducted at day 3 post-CFA. n = 6 experiments. d, NMDAR EPSCs were elicited at six different stimulation intensities (left) and the input–output curves were plotted (right). n = 10 neurons in each group. *p < 0.05 relative to saline-injected GFP-expressing control rats. #p < 0.05 relative to CFA-injected GFP-expressing rats. e, Effects of U-0126 on NMDAR EPSCs recorded in slices from saline-injected or CFA-injected rats. The horizontal bar indicated the period of U-0126 perfusion. n = 6 neurons in each group.
Behavioral tests showed that in intact rats, neither SPN(WT) nor GFP affected the PWTs in response to innocuous mechanical stimuli (Fig. 9a). The PWLs to noxious thermal stimuli were also unchanged after SPN(WT) and GFP expression (Fig. 9a). Interference with SPN/PP1 association by SPN(F451A), however, caused a persistent pain hypersensitivity, which lasted for ≥7 d (Fig. 9a). The pronociceptive action of SPN(F451A) was attributed to the activation of MEK/ERK signaling, because spinal treatment with MEK inhibitor U-0126 (5 μg) or PD98059 (7 μg) attenuated the reduction of pain thresholds (Fig. 9b). Intrathecal application of ifenprodil (5 μg) and nonspecific NMDAR antagonist d-APV (5 μg) also elevated the pain thresholds in SPN(F451A)-expressing rat (Fig. 9c). SPN/PP1 dissociation played an important role in chronic inflammatory pain. Reinstating PP1 inhibition by SPN(WT) expression generated an effective analgesic action against the mechanical allodynia and thermal hyperalgesia induced by CFA (Fig. 9d).
Figure 9.
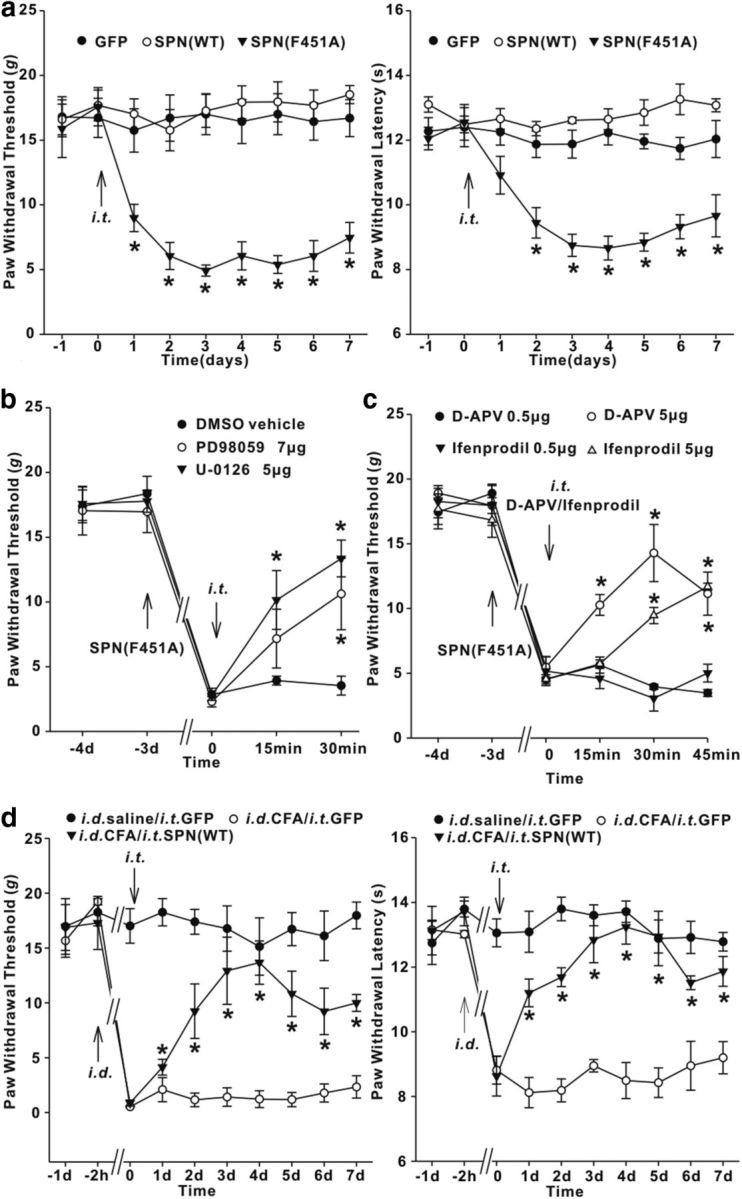
SPN-targeted PP1 was involved in spinal nociceptive processing. a, Time-dependent changes of PWTs (left) and PWLs (right) after intrathecal injection (i.t.) of adenovirus encoding GFP, SPN(WT), or SPN(F451A) in intact rats. n = 6 rats in each group. *p < 0.05 versus baseline values. b, c, Effects of MEK inhibitor U-0126/PD98059 (b) or NMDAR antagonist ifenprodil/d-APV (c) on the pain thresholds of SPN(F451A)-expressing rats. The upward and downward arrows indicated the time points when adenovirus encoding SPN(F451A) or chemical drugs were intrathecally injected, respectively. n = 6 rats in each group. *p < 0.05 relative to predrug values. d, Intrathecal injection of adenovirus encoding SPN(WT) alleviated tactile allodynia (left) and thermal hyperalgesia (right) induced by intradermal injection (i.d.) of CFA. n = 6 rats in each group. *p < 0.05 relative to the values previral injection.
Discussion
A growing number of signaling molecules has been identified as the binding partners for SPN (Sarrouilhe et al., 2006; Baucum et al., 2010). These molecules include transmembrane receptors, actin cytoskeleton-associated proteins, protein kinases, and phosphatases (Sarrouilhe et al., 2006; Baucum et al., 2010). The complex formation with distinct signaling components confers to SPN diverse biological properties (Sarrouilhe et al., 2006). The major finding in the present study was the identification of MEK and ERK as the novel partner proteins of SPN. The PDZ domain, located immediately adjacent to the PP1-binding motif (Hsieh-Wilson et al., 1999), mediated the direct interaction of SPN with MEK1. The same PDZ domain was also responsible for SPN binding to ERK2. Given that SPN can form the homomeric complex through its coiled-coil region (MacMillan et al., 1999; Oliver et al., 2002), the dimerization of SPN might provide >1 binding site for MEK and ERK, organize them into a macromolecular complex, and constitute a platform for the cross talk between distinct signaling pathways. Consistent with this notion, anti-SPN antibody simultaneously precipitated MEK and ERK from spinal dorsal horn of rats, and SPN(WT) expression caused a synchronous dephosphorylation of MEK and ERK in HEK293T cells.
Many studies have investigated the role of PP1 in the regulation of ERK signaling (Westermarck et al., 2001; Zhou et al., 2002; Mitsuhashi et al., 2003; Hédou et al., 2008), which, however, obtain quite different results. Compared with protein phosphatase 2A, which is able to dephosphorylate either MEK or ERK (Zhou et al., 2002), PP1 seems to be less effective in the direct dephosphorylation of MEK and ERK (Zhou et al., 2002). Consistent with these reports, our data showed that PP1 alone produced minimal effects on the phosphorylation of MEK1 and ERK2 in vitro. However, the catalytic efficacy of PP1 could be substantially enhanced once SPN(WT) was supplemented in the reaction buffer. This phenomenon was not observed with SPN(F451A), suggesting that the scaffolding function of SPN was prerequisite for PP1 to access MEK/ERK and conduct the efficient dephosphorylation. In spinal cord dorsal horn in vivo, endogenous SPN might serve as a scavenger for the active MEK and ERK caused by spontaneous neuronal activity. By recruiting the phosphorylated MEK/ERK to its bound PP1 for dephosphorylation, SPN restricted ERK signaling from aberrant activation at physiological conditions. Specific disturbance of SPN/PP1 interaction yielded a significant increase of MEK/ERK phosphorylation in intact rats.
There have been some studies showing that PP1 is a positive regulator of Raf-1, because PP1 inhibition by chemical reagents suppresses Raf-1 activity (Mitsuhashi et al., 2003). We found that, although PP1 physically interacted with Raf-1, specific disruption of SPN/PP1 complex generated no effect on Raf-1 phosphorylation. The fact that Raf-1 did not interact with SPN suggested that the scaffolding function of SPN might determine the substrate specificity of PP1.
Electrophysiological recordings have illustrated that nonspecific inhibition of PP1 activity or its interaction with regulatory/scaffolding proteins generates no influence on the basal synaptic transmission (Morishita et al., 2001). However, the synaptic strength is definitely altered when PP1 interaction with some of these scaffolding proteins is selectively disrupted (Yan et al., 1999; Hu et al., 2007), suggesting that PP1, targeted by different scaffolding proteins, might play distinct or even opposite roles in synaptic responses. In acutely dissociated striatal or neostriatal neurons, disturbing SPN/PP1 interaction blocks the rundown of AMPAR-mediated inward currents generated by application of exogenous kainate (Yan et al., 1999; Feng et al., 2000). However, comparison of mEPSCs mediated by synaptic AMPARs displays no difference between SPN knock-out mice and their WT counterparts (Allen et al., 2006). Our data also showed that the amplitudes and frequencies of mEPSCs in neurons expressing SPN(F451A) were comparable to those in GFP neurons. Distinct from AMPAR responses, the synaptic currents mediated by GluN2B receptors were substantially enhanced by SPN(F451A), implicating a specific regulation of GluN2B receptors by SPN-targeted PP1 in spinal sensory neurons (Feng et al., 2000).
The N-terminal region of SPN interacts with F-actin, which directs SPN along with its associated PP1 into postsynaptic density (Allen et al., 1997; Satoh et al., 1998). This synaptic targeting process is critical for PP1 to accomplish a variety of synaptic modifications (Yan et al., 1999; Feng et al., 2000; Morishita et al., 2001). As is the case for PP1 synaptic targeting, SPN interacted with MEK and ERK at synaptosomal fraction and engaged them in the regulation of postsynaptic substrates. The removal of PP1 from SPN led to a restricted activation of MEK/ERK signaling at synaptosomal fraction. ERK phosphorylation of MLCK has been shown to promote myosin ATPase activity by phosphorylating MLC, the only known substrate for MLCK (Klemke et al., 1997). The enhanced MLCK activity plays an important role in spinal synaptic plasticity (Luo et al., 2012). Consistent with previous reports that MLCK specifically regulates the synaptic responses mediated by NMDAR (Lei et al., 2001; Amparan et al., 2005), postsynaptic perfusion of MLCK inhibitor ML-7 significantly depressed GluN2B synaptic currents in SPN(F451A)-expressing neurons.
ERK activation by peripheral tissue or nerve injuries is pivotal for spinal sensitization of nociceptive behaviors (Ji et al., 2009). ERK1 and ERK2 have been shown to share high structural homology (Roskoski, 2012b). However, ERK2 appears to play a more dominant role than ERK1 in synaptic plasticity and nociceptive sensitization (Satoh et al., 2007; Xu et al., 2008; Alter et al., 2010). ERK1 deletion generates no effect on spinal sensitization (Alter et al., 2010), no matter whether peripheral lesions regulate ERK1 phosphorylation or not (Ji et al., 2002; Xu et al., 2008; Alter et al., 2010; Weyerbacher et al., 2010). Distinct from ERK1, manipulation of ERK2 activity greatly attenuates chronic pathological pain (Xu et al., 2008; Weyerbacher et al., 2010). The reversal of MEK1 hyperactivity yields a similar antinociceptive action as ERK2 inhibition (Karim et al., 2006). The activation of ERK signaling might exacerbate the inflammatory pain through multiple pathways. In addition to boosting NMDAR-mediated nociceptive conveyance, the transcriptional upregulation of prodynorphin and neurokinin-1 expression by ERK signaling has been shown to maintain the persistent inflammatory pain (Ji et al., 2002). The reduction of transient outward (A-type) potassium currents by ERK contributes to dorsal horn neuron hyperexcitability (Hu et al., 2006). The active ERK is also involved in the long-lasting modification of nociceptive synaptic strength (Xin et al., 2006). Our data demonstrated that PP1 hypofunction, as a result of SPN/PP1 dissociation, might represent one of the important mechanisms underlying ERK activation. PP1 affinity for the conserved PP1-docking sequence (R/K-R/K-V/I-X-F) on scaffolding proteins can be dynamically regulated (Hsieh-Wilson et al., 1999; McAvoy et al., 1999; Oliver et al., 2002). For example, neurabin, another PP1-binding and actin-binding protein that is structurally related to SPN, reduces its binding to PP1 in response to the cAMP signaling pathway (McAvoy et al., 1999; Oliver et al., 2002). Intraplantar CFA injection disrupted PP1 interaction with SPN, implicating the loss of PP1 inhibition during inflammatory pain. Overexpression of SPN(WT) to enhance SPN-mediated PP1 targeting repressed MEK/ERK phosphorylation in CFA-injected rats and alleviated the inflammatory pain. Together, these data delineated an intimate interaction of ERK signaling components with SPN, and revealed a critical role of proper PP1 scaffolding for the tight and precise regulation of MEK/ERK activity.
Footnotes
This work was supported by the National Natural Science Foundation of China (30870837, 31271186).
The authors declare no competing financial interests.
References
- Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci U S A. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen PB, Zachariou V, Svenningsson P, Lepore AC, Centonze D, Costa C, Rossi S, Bender G, Chen G, Feng J, Snyder GL, Bernardi G, Nestler EJ, Yan Z, Calabresi P, Greengard P. Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience. 2006;140:897–911. doi: 10.1016/j.neuroscience.2006.02.067. [DOI] [PubMed] [Google Scholar]
- Alter BJ, Zhao C, Karim F, Landreth GE, Gereau RW., 4th Genetic targeting of ERK1 suggests a predominant role for ERK2 in murine pain models. J Neurosci. 2010;30:11537–11547. doi: 10.1523/JNEUROSCI.6103-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amparan D, Avram D, Thomas CG, Lindahl MG, Yang J, Bajaj G, Ishmael JE. Direct interaction of myosin regulatory light chain with the NMDA receptor. J Neurochem. 2005;92:349–361. doi: 10.1111/j.1471-4159.2004.02869.x. [DOI] [PubMed] [Google Scholar]
- Baucum AJ, 2nd, Jalan-Sakrikar N, Jiao Y, Gustin RM, Carmody LC, Tabb DL, Ham AJ, Colbran RJ. Identification and validation of novel spinophilin-associated proteins in rodent striatum using an enhanced ex vivo shotgun proteomics approach. Mol Cell Proteomics. 2010;9:1243–1259. doi: 10.1074/mcp.M900387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PT. Protein phosphatase 1—targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- Dard N, Peter M. Scaffold proteins in MAP kinase signaling: more than simple passive activating platforms. Bioessays. 2006;28:146–156. doi: 10.1002/bies.20351. [DOI] [PubMed] [Google Scholar]
- Fan QQ, Li L, Wang WT, Yang X, Suo ZW, Hu XD. Activation of alpha2 adrenoceptors inhibited NMDA receptor-mediated nociceptive transmission in spinal dorsal horn of mice with inflammatory pain. Neuropharmacology. 2014;77:185–192. doi: 10.1016/j.neuropharm.2013.09.024. [DOI] [PubMed] [Google Scholar]
- Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci U S A. 2000;97:9287–9292. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hédou GF, Koshibu K, Farinelli M, Kilic E, Gee CE, Kilic U, Baumgärtel K, Hermann DM, Mansuy IM. Protein phosphatase 1-dependent bidirectional synaptic plasticity controls ischemic recovery in the adult brain. J Neurosci. 2008;28:154–162. doi: 10.1523/JNEUROSCI.4109-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Wilson LC, Allen PB, Watanabe T, Nairn AC, Greengard P. Characterization of the neuronal targeting protein spinophilin and its interactions with protein phosphatase-1. Biochemistry. 1999;38:4365–4373. doi: 10.1021/bi982900m. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW., 4th The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Hu XD, Huang Q, Yang X, Xia H. Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus. J Neurosci. 2007;27:4674–4686. doi: 10.1523/JNEUROSCI.5365-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di Stefano P, Demmers J, Krugers H, Defilippi P, Akhmanova A, Hoogenraad CC. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Südhof TC, Malenka RC. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron. 2013;77:542–558. doi: 10.1016/j.neuron.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Hu HJ, Adwanikar H, Kaplan D, Gereau RW., 4th Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen activated protein kinase kinase (MEK) Mol Pain. 2006;2:2. doi: 10.1186/1744-8069-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Lei S, Czerwinska E, Czerwinski W, Walsh MP, MacDonald JF. Regulation of NMDA receptor activity by F-actin and myosin light chain kinase. J Neurosci. 2001;21:8464–8472. doi: 10.1523/JNEUROSCI.21-21-08464.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi L, Xu YM, Yang X, Suo ZW, Hu XD. GABAergic inhibition regulated pain sensitization through STEP61 signaling in spinal dorsal horn of mice. Anesthesiology. 2015;122:686–697. doi: 10.1097/ALN.0000000000000532. [DOI] [PubMed] [Google Scholar]
- Luo C, Gangadharan V, Bali KK, Xie RG, Agarwal N, Kurejova M, Tappe-Theodor A, Tegeder I, Feil S, Lewin G, Polgar E, Todd AJ, Schlossmann J, Hofmann F, Liu DL, Hu SJ, Feil R, Kuner T, Kuner R. Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol. 2012;10:e1001283. doi: 10.1371/journal.pbio.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan LB, Bass MA, Cheng N, Howard EF, Tamura M, Strack S, Wadzinski BE, Colbran RJ. Brain actin-associated protein phosphatase 1 holoenzymes containing spinophilin, neurabin, and selected catalytic subunit isoforms. J Biol Chem. 1999;274:35845–35854. doi: 10.1074/jbc.274.50.35845. [DOI] [PubMed] [Google Scholar]
- Mannes AJ, Caudle RM, O'Connell BC, Iadarola MJ. Adenoviral gene transfer to spinal-cord neurons: intrathecal vs. intraparenchymal administration. Brain Res. 1998;793:1–6. doi: 10.1016/S0006-8993(97)01422-4. [DOI] [PubMed] [Google Scholar]
- Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, Kolch W. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAvoy T, Allen PB, Obaishi H, Nakanishi H, Takai Y, Greengard P, Nairn AC, Hemmings HC., Jr Regulation of neurabin I interaction with protein phosphatase 1 by phosphorylation. Biochemistry. 1999;38:12943–12949. doi: 10.1021/bi991227d. [DOI] [PubMed] [Google Scholar]
- Mitsuhashi S, Shima H, Tanuma N, Matsuura N, Takekawa M, Urano T, Kataoka T, Ubukata M, Kikuchi K. Usage of tautomycetin, a novel inhibitor of protein phosphatase 1 (PP1), reveals that PP1 is a positive regulator of Raf-1 in vivo. J Biol Chem. 2003;278:82–88. doi: 10.1074/jbc.M208888200. [DOI] [PubMed] [Google Scholar]
- Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC. Regulation of synaptic strength by protein phosphatase 1. Neuron. 2001;32:1133–1148. doi: 10.1016/S0896-6273(01)00554-2. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Oliver CJ, Terry-Lorenzo RT, Elliott E, Bloomer WA, Li S, Brautigan DL, Colbran RJ, Shenolikar S. Targeting protein phosphatase 1 (PP1) to the actin cytoskeleton: the neurabin I/PP1 complex regulates cell morphology. Mol Cell Biol. 2002;22:4690–4701. doi: 10.1128/MCB.22.13.4690-4701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13:1356–1364. doi: 10.1016/S0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/BJ20082234. [DOI] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem Biophys Res Commun. 2012a;417:5–10. doi: 10.1016/j.bbrc.2011.11.145. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012b;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Matta JA, Isaac JT, Roche KW. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron. 2010;67:984–996. doi: 10.1016/j.neuron.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrouilhe D, di Tommaso A, Métayé T, Ladeveze V. Spinophilin: from partners to functions. Biochimie. 2006;88:1099–1113. doi: 10.1016/j.biochi.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Satoh A, Nakanishi H, Obaishi H, Wada M, Takahashi K, Satoh K, Hirao K, Nishioka H, Hata Y, Mizoguchi A, Takai Y. Neurabin-II/spinophilin. An actin filament-binding protein with one pdz domain localized at cadherin-based cell-cell adhesion sites. J Biol Chem. 1998;273:3470–3475. doi: 10.1074/jbc.273.6.3470. [DOI] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Westermarck J, Li SP, Kallunki T, Han J, Kähäri VM. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol Cell Biol. 2001;21:2373–2383. doi: 10.1128/MCB.21.7.2373-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyerbacher AR, Xu Q, Tamasdan C, Shin SJ, Inturrisi CE. N-Methyl-d-aspartate receptor (NMDAR) independent maintenance of inflammatory pain. Pain. 2010;148:237–246. doi: 10.1016/j.pain.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Guo JY, Tang W, Yang CS, Freel CD, Chen C, Nairn AC, Kornbluth S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat Cell Biol. 2009;11:644–651. doi: 10.1038/ncb1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin WJ, Gong QJ, Xu JT, Yang HW, Zang Y, Zhang T, Li YY, Liu XG. Role of phosphorylation of ERK in induction and maintenance of LTP of the C-fiber evoked field potentials in spinal dorsal horn. J Neurosci Res. 2006;84:934–943. doi: 10.1002/jnr.21013. [DOI] [PubMed] [Google Scholar]
- Xu Q, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the neuronal extracellular signal-regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund's adjuvant-induced pain hypersensitivity. J Neurosci. 2008;28:14087–14096. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- Zhou B, Wang ZX, Zhao Y, Brautigan DL, Zhang ZY. The specificity of extracellular signal-regulated kinase 2 dephosphorylation by protein phosphatases. J Biol Chem. 2002;277:31818–31825. doi: 10.1074/jbc.M203969200. [DOI] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci. 2010;30:4460–4466. doi: 10.1523/JNEUROSCI.5857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]