

Abstract
We demonstrate a role of the vitamin D receptor (VDR) in reducing cerebral soluble and insoluble amyloid-β (Aβ) peptides. Short-term treatment of two human amyloid precursor protein-expressing models, Tg2576 and TgCRND8 mice, with 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3], the endogenous active ligand of VDR, resulted in higher brain P-glycoprotein (P-gp) and lower soluble Aβ levels, effects negated with coadministration of elacridar, a P-gp inhibitor. Long-term treatment of TgCRND8 mice with 1,25(OH)2D3 during the period of plaque formation reduced soluble and insoluble plaque-associated Aβ, particularly in the hippocampus in which the VDR is abundant and P-gp induction is greatest after 1,25(OH)2D3 treatment, and this led to improved conditioned fear memory. In mice fed a vitamin D-deficient diet, lower cerebral P-gp expression was observed, but levels were restored on replenishment with VDR ligands. The composite data suggest that the VDR is an important therapeutic target in the prevention and treatment of Alzheimer's disease.
Keywords: Alzheimer's, amyloid beta, blood-brain barrier, P-glycoprotein, vitamin D receptor
Introduction
Aggregation of pathologic forms of amyloid-β (Aβ) peptides is considered to play a key role in Alzheimer's disease (AD). Aβ peptides are formed by cleavage of the amyloid precursor protein (APP) via β- and γ-secretases (Kang et al., 1987; Haass et al., 1992; Hartmann et al., 1997; Weidemann et al., 1999). The comparatively more hydrophobic 42 aa variant, Aβ1–42, displays a greater tendency to form oligomers and insoluble plaques (Jarrett et al., 1993) and is considered to be more pathogenic (Roher et al., 1993; Lesné et al., 2006) compared with the 40 aa variant, Aβ1–40, a major component of cerebrovascular plaques (Miller et al., 1993). Brain Aβ levels are governed by cerebral synthesis and clearance. The amyloid clearance hypothesis asserts that Aβ accumulation is the result of decreased efflux from the brain (Zlokovic et al., 2000; Deane et al., 2009). Aβ clearance at the blood–brain barrier (BBB) is also controlled by the receptor for advanced glycation end products (RAGE) and low-density lipoprotein receptor-related protein 1 (LRP1), balancing influx and efflux, respectively (Deane et al., 2004). In addition, P-glycoprotein (P-gp), an efflux transporter that is encoded by the multidrug resistance protein 1 (MDR1) gene, has been implicated in Aβ excretion (Lam et al., 2001). Aβ accumulation is higher in brains of mdr1a/b knock-out mice (Cirrito et al., 2005), and autopsies of patients with AD showed a strong negative correlation between densities of senile plaque lesions and the expression of P-gp in capillaries (Vogelgesang et al., 2002), whose function is known to decrease with age (Toornvliet et al., 2006).
MDR1 expression at the BBB is regulated by the pregnane X receptor (PXR; Bauer et al., 2004), the constitutive androstane receptor (Miller 2010; Wang et al., 2010), and the vitamin D receptor (VDR; Durk et al., 2012); a response element for VDR has been identified on the human MDR1 gene (Saeki et al., 2008). Mice treated with 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3], the active, physiological ligand of VDR, showed lower brain accumulation of the P-gp substrate digoxin (Chow et al., 2011). In vitro, 1,25(OH)2D3 treatment increased P-gp expression in rat brain endothelial cells (RBE4) and human (hCMEC/D3) cerebral microvessel endothelial cells and reduced intracellular accumulation of rhodamine 6G and Aβ1–42, both P-gp substrates (Durk et al., 2012).
This study investigates the potential of 1,25(OH)2D3 in lowering cerebral accumulation of human Aβ (hAβ) in two transgenic (Tg) mouse models of AD: (1) Tg2576 (at the pre-plaque age of 10 weeks); and (2) TgCRND8 (treated from age 9–17 weeks during the period of plaque formation or at age 20 weeks for 8 d after plaque has already formed). Although both mice overexpress hAPP, the TgCRND8 mouse preferentially forms hAβ1–42 and exhibits faster plaque deposition (Chishti et al., 2001). We identify that the central Vdr-specific mechanism for reduction of Aβ accumulation in Tg2576 and TgCRND8 mice is via Mdr1/P-gp induction and that early and prolonged Vdr activation lowers Aβ accumulation, decreases plaque burden, and improves cognitive function, suggesting that VDR is a potential target in the prevention and treatment of AD.
Materials and Methods
Reagents and chemicals.
1,25(OH)2D3 and all other chemicals were obtained from Sigma-Aldrich. Materials for quantitative real-time PCR (qPCR) were purchased from Applied Biosystems. Elacridar was a kind gift from GlaxoSmithKline. The primary antibodies anti-P-gp (C219), anti-breast cancer resistance protein (Bcrp)/BCRP (BXP-53), anti-Rage/RAGE (DD/A11), anti-Lrp1/LRP1 (5A6), anti-APP (Y188), anti-VDR (9A7), and anti-Gapdh (6C5) were purchased from Abcam. The secondary fluorescent antibodies Alexa Fluor 488 (FITC) goat anti-mouse (A11001) and Alexa Fluor 546 (TRITC) goat anti-rat (A11081) were from Invitrogen. ELISA kits for hAβ1–40 (KHB3481) and hAβ1–42 (KHB3441) were obtained from Invitrogen. The 4G8 antibody to Aβ for plaque staining was purchased from Covance, and resorufin was obtained from Sigma-Aldrich. Vitamin D-sufficient (TD.07370) and vitamin D-deficient (TD.07541) diets were prepared by Harlan Laboratories; the vitamin D-deficient diet was supplemented with calcium and phosphorus (2.5 and 1.5% compared with 0.47 and 0.3%, respectively, in normal diet) to maintain calcium and phosphorus at normal physiological levels.
Animals.
All mice were maintained under a 12 h light/dark cycle, and all protocols were approved by the Faculty of Medicine and Pharmacy Animal Care Committee. Mice from some treatment groups received intraperitoneal doses of 0 or 2.5 μg/kg 1,25(OH)2D3 in filtered, sterile corn oil on alternate days for 8 d [every 2 d, 4 times (q2d × 4)] or every third day for 8 weeks [every 3 d, 19 times (q3d × 19)].
Sampling.
Blood (via cardiac puncture) and brain tissue (after transcardial perfusion with 10 ml of ice-cold saline) were taken from C57BL/6 or Tg mice. Tissues were snap-frozen in liquid nitrogen and stored at −80°C until analysis.
C57BL/6 mice.
Whole brains of untreated 8-week-old C57BL/6 mice (Charles River) were fixed for immunostaining. Two other groups of mice were injected with either vehicle or 1,25(OH)2D3 intraperitoneally on alternate days (2.5 μg/kg, q2d × 4), a previously established dosing regimen that would elicit increased brain P-gp expression (Chow et al., 2011). Group 1 consisted of 8-week-old males whose brains were removed and microdissected to separate the striatum, prefrontal cortex, hippocampus, cerebellum, and olfactory bulb to examine the distribution of Vdr and P-gp and region-specific induction patterns. Group 2 consisted of 8-week-old males receiving 1,25(OH)2D3 to demonstrate temporal changes in brain 1,25(OH)2D3 and mRNA of Mdr1a and Cyp24a1, both Vdr target genes. Two to four mice were killed at each time point to provide plasma and brain tissues throughout the 8 d 1,25(OH)2D3 treatment period (Chow et al., 2013). A third group of mice, housed under incandescent light and receiving either a vitamin D-sufficient or vitamin D-deficient diet was used to assess effects of vitamin D deficiency on P-gp expression. Blood (150 μl) was withdrawn from the saphenous vein 2, 4, and 6 weeks after diet manipulation to measure plasma 1,25(OH)2D3. On week 7, the vitamin D-deficient mice were treated with vehicle, 20 μg/kg dietary vitamin D, or 2.5 μg/kg 1,25(OH)2D3, q2d × 4, intraperitoneally and killed 2 d after the last dose.
Tg mice and Aβ disposition.
Tg2576 mice and littermate controls (Taconic Farms) were treated with 1,25(OH)2D3 (2.5 μg/kg, i.p., q2d × 4) or vehicle at 10 weeks of age. Tg2576 mice possess the Swedish mutation, resulting in hAPP overexpression (Hsiao et al., 1996). At this age, Tg2576 mice are plaque free but exhibit high levels of soluble hAβ in the brain (Hsiao et al., 1996) without displaying signs of cognitive impairment (Kawarabayashi et al., 2001) and were selected to study soluble hAβ efflux from the brain after 1,25(OH)2D3 treatment. TgCRND8 mice (bred and maintained at the University of Toronto), which possess both the Indiana and Swedish mutations, exhibit faster plaque deposition than the Tg2576 model (3 vs 9 months) because of the higher hAβ1–42 versus hAβ1–40 production (Chishti et al., 2001). One group of non-Tg and TgCRND8 mice was treated before and during the period of plaque formation, from a pre-plaque age (9 weeks) until 17 weeks of age when plaques and cognitive deficits are easily detectable (Chishti et al., 2001). To avoid hypercalcemia, a protracted regimen of 2.5 μg/kg 1,25(OH)2D3 or vehicle q3d × 19 intraperitoneally was used for long-term treatment; we verified this treatment did not elicit changes in plasma calcium or phosphorus levels or loss of body weight compared with vehicle-treated controls (data not shown). A second group of TgCRND8 mice was treated acutely at 20 weeks of age, at which time mice would have high levels of soluble and insoluble hAβ and plaques to appraise changes in soluble and insoluble Aβ after treatment with 1,25(OH)2D3 and elacridar (GF120918), a P-gp inhibitor. Here, TgCRND8 mice were treated with vehicle, 2.5 μg/kg 1,25(OH)2D3, q2d × 4 intraperitoneally, or 2.5 μg/kg 1,25(OH)2D3 together with 10 mg/kg elacridar in sterile PEG600/ddH2O, 1:3 (v/v), q12h × 8 intraperitoneally, during the final 4 d of the 1,25(OH)2D3 treatment. This dosing regimen of elacridar, verified to inhibit P-gp successfully in the mouse brain, was estimated to attain >20 nm in plasma, the EC50 for P-gp inhibition (Imbert et al., 2003), according to computer simulations based on a two-compartment model (data not shown) and pharmacokinetic parameters published previously (Hyafil et al., 1993).
Fear conditioning.
Fear conditioning studies were performed with TgCRND8 and non-Tg mice that underwent prolonged treatment with 1,25(OH)2D3 from an early age. At the end of the 8 week 1,25(OH)2D3 treatment, fear conditioning studies were conducted over the course of 3 d according to a published method (Hanna et al., 2012), with modifications. Studies were performed in two Plexiglas chambers (25.4 × 25.4 × 18 cm) with floors made of 32 stainless steel rods, 3.5 mm in diameter and spaced 5 mm apart for the delivery of a 0.6 mA footshock. The containers were placed in sound-attenuated cabinets, and a speaker was used to deliver a loud tone [conditioned stimulus (CS)]. On the first day (training session), each mouse was placed in the apparatus individually and subjected to the 60 s CS into the session, and two consecutive shocks for 2 s each were delivered at 88 (to 90) s and 148 (to 150) s. On the second day, mice were placed in cages for 300 s without the CS or shock. On the third day (probe trial), the appearance of the cage was altered, and the CS was presented for 3 min, 2 min into the trial. During the 2 min before CS and 3 min after CS, movement was monitored by beam breaks of a laser grid, and the number of 1 s intervals in which new beam breaks were absent was measured to determine freezing frequency. This was used to calculate percentage of pre- and post-CS intervals in which the mice exhibited freezing behavior.
Immunostaining.
C57BL/6 mice were transcardially perfused with 25 ml of cold PBS and then 50 ml of 4% paraformaldehyde and were postfixed in 4% paraformaldehyde at 4°C overnight. Brains were embedded in paraffin, and 7 μm sections were prepared. After dewaxing, sections underwent antigen retrieval in 10 mm sodium citrate, pH 6.0, followed by incubation in 2N HCl at 37°C for 30 min. Sections were preblocked with 5% goat serum in PBS containing 0.1% Tween 20 and incubated with primary antibodies to P-gp and Vdr overnight at a dilution of 1:50 v/v in preblock. Sections were rinsed three times with preblock and incubated with the fluorescent secondary antibodies for 2 h at room temperature. After washing, sections were imaged using a Nikon E1000R fluorescence microscope with a 40×/0.75 Nikon PlanFluor lens and Nikon FDX-35 camera. Images were acquired with SimplePCI software at room temperature, and images captured with different filters were superimposed using Adobe Photoshop, with which brightness and contrast were adjusted in a linear manner. The fidelity of the C219 antibody and 9A7 antibody staining was verified in brains of mdr1a/b−/− (Taconic Farms) and vdr−/− (B6.129S4-Vdrtm1Mbd/J) (The Jackson Laboratory) mice, respectively (data not shown).
Plaque staining and quantification.
TgCRND8 brains were prepared for immunostaining as described previously (Chishti et al., 2001). For total plaque staining, every fifth section of 25 paraffin-embedded sections was stained with the 4G8 antibody to Aβ and detected with DAB. Slides were scanned using the Mirax Scan version 1.11 software and Zeiss Mirax Slide Scanner at 20× magnification with a Zeiss 20×/0.8 objective lens and a Marlin F146-C CCD camera, operated at room temperature. The rendered digital images were analyzed using the color deconvolution algorithm in the Aperio Imagescope software, as described previously (Lillard-Wetherell, 2008). Red–green–blue values were determined for both the applied hematoxylin and DAB stains. DAB was chosen as the positive color channel for identifying and quantifying Aβ-stained plaques within different areas of the brain (cortex and hippocampus). Furthermore, recognition and measurement of dense and diffuse plaque-stained areas were achieved by setting the threshold values of color intensity. The strong positive threshold was set to 80, correlating with dense staining. The medium positive threshold was set to 160, correlating with medium/diffuse staining, and the weak positive threshold was set to 0. In this way, the amyloid-positive area, as well as intensity of Aβ staining, was quantified in different brain regions, allowing for the quick, objective comparison between brains from different animals.
For cerebrovascular plaque staining, the method of Han et al., (2011) was used. Briefly 105 sections were cut, and the first four of every 30 sections were retained for analysis. These sections were dewaxed as described previously (Han et al., 2011) and blocked in PBS containing 0.1% Triton X-100, 0.2% skim milk, and 1% BSA at room temperature for 45 min. Sections were then permeabilized in 0.25% Triton X-100 (in PBS) for 30 min at room temperature and then incubated in 1 μm resorufin for 30 min at room temperature in 0.25% Triton X-100 in PBS. After this, sections were washed three times in PBS, then once in 50% EtOH in PBS, followed by three more PBS washes. Sections were coverslipped and imaged using a Nikon E1000R fluorescence microscope with a 40×/0.75 Nikon PlanFluor lens and Nikon FDX-35 camera. Images were acquired with the SimplePCI software. All visible cortical vessels were imaged, and all images used for quantification were obtained with an exposure time of 1 s. Cross-sectional intensity was quantified using the NIH ImageJ software.
Immunoblotting.
Brains were homogenized in 5× (w/v) buffer (in mm: 250 sucrose, 10 HEPES, and 10 Tris) containing protease inhibitor mixture (Sigma-Aldrich) and centrifuged at 3000 × g for 10 min at 4°C. The pellet, a nuclear fraction, was resuspended in buffer containing the following (in mm): 15 Tris-HCl, 60 KCl, 15 NaCl, 5 MgCl2, 0.1 EGTA, 0.5 DTT, 0.1 PMSF, 300 sucrose, and protease inhibitor, which was used for Vdr blotting. A crude membrane fraction was prepared for P-gp protein blotting by further centrifugation of the supernatant at 33,000 × g for 10 min at 4°C, and the pellet was suspended in buffer containing the following (in mm): 50 mannitol, 20 HEPES, and 20 Tris, as well as protease inhibitor mixture. Protein samples were resolved by SDS-PAGE. Protein bands were normalized to the respective Gapdh band of the same sample (Chow et al., 2009).
qPCR.
RNA was isolated from brains using TRIzol (Sigma-Aldrich), according to the protocol of the manufacturer. The method for the determination of relative brain mRNA levels by qPCR has been described previously (Durk et al., 2012). Primers used include murine cyclophillin (forward, 5′-GGAGATGGCACAGGAGGAA-3′; reverse, 5′-GCCCGTAGTGCTTCAGCTT-3′), Cyp24a1 (forward, 5′-CTGCCCCATTGACAAAAGGC-3′; reverse, 5′-CTCACCGTCGGTCATCAGC-3′), and Mdr1a (forward, 5′- CATGACAGATAGCTTTGCAAGTGTAG-3′; reverse, 5′-GGCAAACATGGCTCTTTTATCG-3′).
1,25(OH)2D3 levels by enzyme immunoassay.
Brains were homogenized in 1:2 (v/v) CH2Cl2/MeOH and centrifuged at 3000 × g for 20 min at room temperature. The bottom (organic) phase was recovered with a pipette, and CH2Cl2 was added to the top phase, after which the centrifugation was repeated and the bottom phase was collected again. The pooled organic phase was dried down and resuspended in charcoal-stripped human serum, and 1,25(OH)2D3 levels were determined by an enzyme immunoassay kit from Immunodiagnostics Systems, according to the protocol of the manufacturer. Brain 1,25(OH)2D3 levels were plotted against published plasma levels (Chow et al., 2013). These, together with the temporal data of the mRNA levels of brain Mdr1a and Cyp24a1, were used to characterize the induction of Vdr target genes.
ELISA for hAβ.
hAβ1–40 and hAβ1–42 levels in brains of Tg2576 and non-Tg mice were measured by ELISA according to the protocol of the manufacturer. Briefly, brains were homogenized in eight times (w/v) in buffer containing 50 mm Tris-HCl and 5 m guanidine-HCl and diluted fivefold or 10-fold for total hAβ1–40 and hAβ1–42 measurement, respectively, in PBS containing 5% BSA and 0.03% Tween 20. Soluble hAβ from TgCRND8 and non-Tg brains was extracted according to a published method (Petanceska et al., 2000). Hemibrains were homogenized in 1:10 (w/v) buffer containing 20 mm Tris, 0.25 m sucrose, 1 mm EDTA, and 1 mm EGTA, pH 7.4. Soluble hAβ was extracted by mixing 1:1 homogenate with 0.4% diethylamine/100 mm NaCl in a Dounce homogenizer and centrifuged at 100,000 × g for 1 h at 4°C, followed by neutralization with volume with 0.5 m Tris, pH 6.8. The pellet was used to extract insoluble Aβ after addition of 440 μl of ice-cold 70% formic acid, followed by sonication with a hand-held sonicator for 20 s on ice; then 400 μl was centrifuged for 1 h at 100,000 × g at 4°C. An aliquot of 210 μl of the supernatant was neutralized with 4 ml of 1 m Tris base containing 0.5 m Na2HPO4 and stored at −80°C until analysis by ELISA.
Statistical analysis.
All data are presented as mean ± SEM. A one-way ANOVA was used to evaluate differences among multiple groups, with p values determined by Bonferroni's multiple comparisons test. Student's two-tailed t test was used to evaluate differences only between experiments in which two treatment groups were compared. Differences were considered to be statistically significant at p < 0.05.
Results
Vdr and P-gp in murine brain capillaries and brain regions
The distribution of Vdr and P-gp in brain regions and changes in P-gp after 1,25(OH)2D3 treatment were first studied in C57BL/6 mice. Both Vdr and P-gp were found to be present in cortical capillaries (Fig. 1a). The signals for Vdr and P-gp protein expression were absent in controls when only the secondary antibody was used and in brains of vdr−/− and mdr1a/b−/− mice, respectively (data not shown). In microdissected brain regions, Vdr (Fig. 1b) protein expression was highest in the hippocampus and prefrontal cortex, then the striatum, olfactory bulb, and cerebellum. After 1,25(OH)2D3 treatment (2.5 μg/kg, q2d × 4, i.p.), P-gp induction by 1,25(OH)2D3 was also region specific. The highest induction (2.5-fold) occurred in the hippocampus, in which Vdr expression was high. In the prefrontal cortex, an inductive trend in P-gp was observed, whereas there was no change in other regions of the brain (Fig. 1c).
Figure 1.

Distribution of Vdr and P-gp in 8-week-old C57BL/6 mouse brains and effects of 1,25(OH)2D3 treatment on P-gp levels in brain regions. a, P-gp (green), along with Vdr (red), was expressed in brain capillaries within the cerebral cortex. Scale bars, 100 μm. Vdr levels were highest in the hippocampus and cortex (b), and P-gp levels were elevated mostly in the hippocampal and cortical regions after 1,25(OH)2D3 treatment (c) (for striatum, n = 4; for other regions, n = 6). Data are mean ± SEM. Differences among groups were compared using one-way ANOVA, and p values were determined by Bonferroni's multiple comparisons test. PFC, Prefrontal cortex; STR, striatum; HC, hippocampus; CER, cerebellum; OLB, olfactory bulb.
1,25(OH)2D3 plays a central role in the induction of cerebral Mdr1a/P-gp in C57BL/6 mice
In mice receiving repeated doses of 1,25(OH)2D3, brain 1,25(OH)2D3 levels rose in unison with those in plasma (Fig. 2a). Brain Mdr1a mRNA levels rose temporally and paralleled the induction pattern of Cyp24a1 mRNA, a Vdr target gene encoding an enzyme that catabolizes 1,25(OH)2D3. Patterns of induction of both lagged slightly behind tissue 1,25(OH)2D3 levels (Fig. 2b). In mice given a vitamin D-deficient diet, plasma 1,25(OH)2D3 was reduced to 25% of basal levels by weeks 6–8 of diet manipulation (Fig. 3a), and there was a corresponding significant reduction in cerebral Mdr1a and P-gp expression by week 8 (Fig. 3b). Intervention at week 7 of the vitamin D-deficient diet by administration of 1,25(OH)2D3 (2.5 μg/kg, q2d × 4, i.p.) fully restored Mdr1a mRNA/P-gp protein expression to basal levels at week 8 but not on replenishment with dietary vitamin D (20 μg/kg, q2d × 4, i.p.; Fig. 3b).
Figure 2.

Changes in 1,25(OH)2D3 levels and Vdr target gene expression during the 1,25(OH)2D3 treatment period in control and 1,25(OH)2D3-treated 8-week-old C57BL/6 mice. a, After treatment with repeated doses of 1,25(OH)2D3, brain levels (black symbols) of 1,25(OH)2D3 rose and fell in unison with those in plasma [gray symbols; from a previous publication (Chow et al., 2013)]; open and filled symbols represent levels in brains of untreated and treated mice, respectively. The lines connect the mean levels. b, Basal mRNA levels of Vdr target genes, Cyp24a1 and Mdr1a, were relatively unaltered in vehicle-treated animals but were increased after treatment. Cyp24a1 levels rose and fell sharply, but Mdr1a mRNA levels were sustained during the dosing period (n = 2–4 for each time point).
Figure 3.

Vitamin D (Vit D) deficiency lowers plasma 1,25(OH)2D3 and cerebral P-gp expression. a, C57BL/6 mice (8 weeks old) that were fed a vitamin D-deficient diet exhibited significantly lower (75%) plasma 1,25(OH)2D3 levels after 6 weeks of the diet (n = 4 for samples at 2, 4, and 6 weeks). Replenishment of the vitamin D-deficient mice after week 6 with dietary vitamin D or 1,25(OH)2D3 elevated plasma 1,25(OH)2D3 levels, albeit not to basal levels (n ≥ 6). b, Mdr1a mRNA expression and P-gp protein levels were fully restored on replenishment with 1,25(OH)2D3. For vitamin D-deficient and vitamin D-sufficient mice, n = 6. Data are mean ± SEM; a one-way ANOVA was used to evaluate differences between groups, with p values determined by Bonferroni's multiple comparisons test.
1,25(OH)2D3 induces Mdr1a/P-gp in 10-week-old Tg2576 mice, reducing soluble hAβ levels
Tg2576 mice were treated with 1,25(OH)2D3 to test the hypothesis that Vdr activation of Mdr1a reduces soluble Aβ. After the short-term 1,25(OH)2D3 treatment (2.5 μg/kg, q2d × 4, i.p.), brain Mdr1a mRNA expression was increased (by ∼70%) and P-gp was induced in 1,25(OH)2D3-treated Tg2576 mice (Fig. 4a). The expression of mRNA and protein for Bcrp, Lrp1, and Rage did not differ between non-Tg and Tg2576 mice, and levels remained unchanged with 1,25(OH)2D3 treatment (data not shown). The ratio of total hAβ1–42/hAβ1–40 in vehicle-treated Tg2576 mice was 0.28, indicating that the predominant species is hAβ1–40. After treatment, levels of cerebral hAβ1–40 and hAβ1–42 were nearly halved (39.1 ± 4.25 to 15.4 ± 2.15 and 11.1 ± 0.240 to 5.82 ± 0.891 ng/g wet weight; Fig. 4b), although the relative APP levels, detected by immunoblotting, remained unchanged with treatment (data not shown).
Figure 4.

1,25(OH)2D3 treatment induces P-gp in 10-week-old Tg2576 mice (a) and reduces soluble hAβ1–40 and hAβ1–42 in the brain (b). N/D denotes that levels were below detection limits. For Tg2576 mice, n = 5; for non-Tg mice, n = 6. A one-way ANOVA was used to evaluate differences in Mdr1a/P-gp between groups, with p values determined by Bonferroni's multiple comparisons test. To compare Aβ levels between treated and control Tg2576 mice, p values were determined by Student's two-tailed t test.
Early Vdr induction of brain P-gp leads to decreased plaque-associated, insoluble Aβ in TgCRND8 mice
After verification that short-term Vdr activation could reduce accumulation of soluble Aβ in Tg2576 mice, the TgCRND8 model that is associated with faster plaque formation was used to test the hypothesis that early Vdr activation before the period of plaque formation would reduce the accumulation of both soluble and insoluble plaque-associated Aβ. Because preliminary studies showed no difference in P-gp, Rage, Lrp1, Vdr, or Aβ levels between male and female TgCRND8 mice in response to 1,25(OH)2D3 treatment (data not shown), TgCRND8 mice of both sexes were pooled for study.
For the TgCRND8 mice, we used a longer 1,25(OH)2D3 dosing regimen, spanning from a pre-plaque (9 weeks) to post-plaque (17 weeks) age. For TgCRND8 mice, the ratio of total hAβ1–42/hAβ1–40 was 4.19, demonstrating that hAβ1–42 is the predominant species. TgCRND8 mice received the 1,25(OH)2D3 dose less frequently (2.5 μg/kg for 8 weeks, q3d × 19, i.p.) to avoid hypercalcemia and weight loss (Chow et al., 2013), side effects associated with 1,25(OH)2D3 treatment. At the end of treatment, Mdr1a mRNA expression was higher in both treated non-Tg (20%) and TgCRND8 (25%) mice compared with the corresponding vehicle-treated controls, but there was no change in Rage or Lrp1 mRNA expression (data not shown). P-gp protein levels were increased 2.25-fold in TgCRND8 mice (Fig. 5a). Again, there was no change in Rage or Lrp1 protein with treatment (data not shown). 1,25(OH)2D3 treatment decreased cerebral soluble hAβ1–42 and hAβ1–40 levels by ∼30% each (from 3360 ± 122 to 2240 ± 153 and from 872 ± 33.7 to 618 ± 18.7 ng/g wet weight, respectively; p < 0.05). The plaque-associated, insoluble hAβ1–42 in brain decreased significantly (15%, from 4640 ± 143 to 3870 ± 133 ng/g wet weight; p < 0.05) after 1,25(OH)2D3 treatment, as did the insoluble hAβ1–40 (35%, from 924 ± 66.7 to 514 ± 69.0 ng/g wet weight; p < 0.05; Fig. 5b).
Figure 5.

Relative brain Mdr1a mRNA and P-gp protein levels (a) and brain concentrations of soluble and insoluble hAβ1–40 and hAβ1–42 (b) in non-Tg and TgCRND8 mice (at 4 months) after treatment with 1,25(OH)2D3 or vehicle for 8 weeks. Significant reduction of soluble hAβ1–42 and insoluble hAβ1–40 and hAβ1–42 was observed after 1,25(OH)2D3 treatment. N/D denotes that levels were below detection limits. For TgCRND8 mice, n ≥ 7; for non-Tg mice, n ≥ 4. Data are mean ± SEM; a one-way ANOVA was used to evaluate differences in Mdr1a/P-gp between groups, with p values determined by Bonferroni's multiple comparisons test. To compare Aβ levels between treated and control TgCRND8 mice, p values were determined by Student's two-tailed t test.
Analysis of amyloid plaque distribution by staining with the 4G8 antibody after 1,25(OH)2D3 treatment confirmed these changes. Both dense and diffuse plaques were significantly decreased in the hippocampus and cortex, with reduction being lower in the cortex (Fig. 6a). We attribute the observations to the relatively higher Vdr (Fig. 1b) levels and the greater induction of P-gp in the hippocampus (Fig. 1c). Staining for cerebrovascular plaque revealed the presence of amyloid deposits in cortical vessels, especially the dural vessels (Fig. 6b). We did not observe a significant difference in cerebrovascular plaque staining between the control and treatment groups, but a nonsignificant increase was observed for treated mice compared with controls, suggestive of a trend toward increased deposition of cerebrovascular plaque with prolonged upregulation of P-gp. However, treatment with 1,25(OH)2D3 at an early stage of AD development, namely during the period of plaque formation, was able to reduce both soluble and insoluble Aβ, demonstrating that the optimal treatment should begin before the period of plaque formation.
Figure 6.
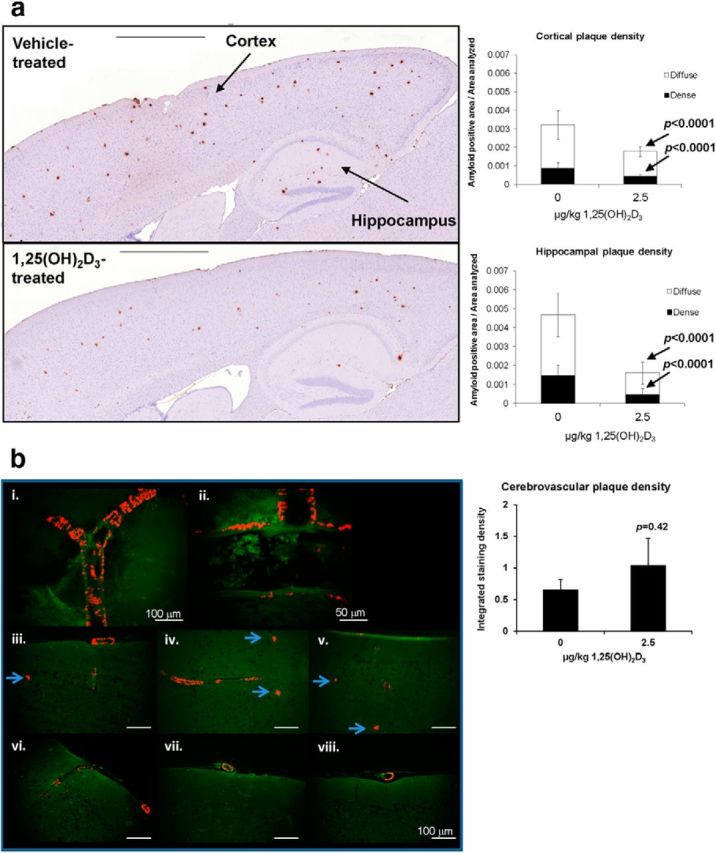
1,25(OH)2D3 treatment decreases dense and diffuse amyloid plaques in TgCRND8 mice. a, Representative images of brains of vehicle-treated and 1,25(OH)2D3-treated TgCRND8 mice after long-term treatment and significant decreases in dense and diffuse amyloid plaques in cortex and hippocampus. For each group, n = 7 mice. Every fifth section of 25 sections from each brain were stained and quantified. Data are mean ± SEM; p values were determined by Student's two-tailed t test. Scale bars, 1 mm. b, Resorufin staining in the cerebral cortex. bi, bii, Patterns of resorufin binding observed in cerebral arteries of TgCRND8 mice at 17 weeks of age. Exterior surface of cerebral artery (bi) and cross-section of cerebral artery demonstrating interspersed laminar stripes of resorufin distribution on abluminal surface (bii). biii–bv, Resorufin staining seen in cortex of vehicle- treated animals; bvi–bviii, resorufin staining observed in cortex of 1,25(OH)2D3-treated animals. Although statistically significant differences in resorufin staining were not observed in cerebral vasculature, a diminution of resorufin-positive neuritic plaques was noted in 1,25(OH)2D3-treated animals (arrowheads). Scale bars (in iii–viii), 100 mm. For each group, n = 5 mice. For 105 sections, the first four of every 30 sections from each brain was stained and quantified. Data are mean ± SEM; p values were determined by Student's two-tailed t test.
Vdr improves conditioned fear memory in TgCRND8 mice after early treatment with 1,25(OH)2D3
We used fear conditioning to assess learning and memory in the 17-week-old TgCRND8 mice after vehicle or 2.5 μg/kg 1,25(OH)2D3 treatment for 8 weeks, q3d × 19 intraperitoneally, because these mice begin to show signs of cognitive deficits at ∼2 months of age (Chishti et al., 2001). During probe trials, no difference was found between any treatment group before the CS, wherein mice exhibited freezing behavior ∼10% of the time. After the CS, non-Tg mice exhibited freezing behavior for ∼40–50% of the time, and there was no difference between vehicle-treated and 1,25(OH)2D3-treated mice. A considerable difference in frequency of freezing behavior was found between vehicle-treated non-Tg and TgCRND8 mice. Expectedly, TgCRND8 mice only exhibited freezing behavior for 15–20% of the time, and 1,25(OH)2D3 treatment restored freezing behavior, albeit not to levels of the non-Tg mouse (only 65%). Treated TgCRND8 mice exhibited freezing behavior 40, 40, and 30% of the time for the first 30, 60, and 180 s after CS, respectively (Fig. 7). These results show that 1,25(OH)2D3 improves conditioned fear memory in TgCRND8 mice that were treated at an earlier age and for a longer period of time (9 weeks old for 8 weeks).
Figure 7.

Fear conditioning studies: frequency of freezing behavior is partially restored in TgCRND8 mice that received 1,25(OH)2D3 treatment. Data are mean ± SEM percentage of time mice exhibited freezing behavior before and after the CS. Vehicle-treated non-Tg mice, n = 10; 1,25(OH)2D3-treated non-Tg mice, n = 8; vehicle-treated TgCRND8 mice, n = 8; and 1,25(OH)2D3-treated TgCRND8 mice, n = 10. A one-way ANOVA was used to evaluate differences between groups, with p values determined by Bonferroni's multiple comparisons test.
Late treatment with 1,25(OH)2D3 induces P-gp in 20-week-old TgCRND8 mice, reducing only soluble hAβ levels
Twenty-week-old TgCRND8 mice were treated with 1,25(OH)2D3 after the onset of plaque formation to test the hypotheses that Vdr activation at a later stage reduces only soluble hAβ and that inhibition of P-gp negates this effect. After 1,25(OH)2D3 treatment (2.5 μg/kg, q2d × 4, i.p.), brain P-gp protein (Fig. 8a) was increased in 1,25(OH)2D3-treated mice compared with vehicle-treated mice, and P-gp induction was observed in mice treated with 1,25(OH)2D3 plus elacridar. There was no change in Rage or Lrp1 mRNA or protein expression after either treatment; the same was observed for Bcrp mRNA (data not shown). The ratio of total (soluble and insoluble) hAβ1–42/hAβ1–40 in vehicle-treated TgCRND8 mice was 4.41, indicating that the predominant species is hAβ1–42. Plasma hAβ levels remained unchanged after all treatments (data not shown). After 1,25(OH)2D3 treatment, brain levels of soluble hAβ1–40 and hAβ1–42 were reduced significantly in TgCRND8 mice, from 1060 ± 25.9 to 675 ± 54.3 and 2920 ± 202 to 1970 ± 130 ng/g wet weight, respectively (Fig. 8b). Levels of insoluble hAβ1–40 and hAβ1–42 in brain remained generally unchanged after treatment with 1,25(OH)2D3 compared with vehicle (Table 1). Treatment with elacridar, together with 1,25(OH)2D3, did not affect the Vdr-mediated induction of P-gp protein but successfully reduced P-gp function. In these TgCRND8 mice, the soluble hAβ1–40 and hAβ1–42 levels were significantly higher than those of controls (from 1060 ± 25.9 to 1240 ± 27.6 and 2920 ± 202 to 4700 ± 214 ng/g wet weight, respectively). Levels of insoluble hAβ remained unchanged, although an increasing trend for hAβ1–40 was observed in the 1,25(OH)2D3-treated mice given elacridar. The combined data confirmed that the Vdr-mediated reduction of brain hAβ occurs with the soluble and not insoluble forms and that the late, short-term treatment with 1,25(OH)2D3 is unable to reverse the insoluble plaque already formed. However, if the 1,25(OH)2D3 treatment is more prolonged, it is foreseeable that less plaque would form with increased brain efflux of the soluble hAβ by P-gp.
Figure 8.

1,25(OH)2D3 treatment increases P-gp in 20-week-old TgCRND8 mice (a) and reduces soluble hAβ1–40 and hAβ1–42 in the brain (b). Data are mean ± SEM. For all groups, n = 4. A one-way ANOVA was used to evaluate differences between groups, with p values determined by Bonferroni's multiple comparisons test.
Table 1.
Comparison of cerebral hAβ levels in vehicle-treated and 1,25(OH)2D3-treated Tg mice
| Vehicle or 1,25(OH)2D3 treatment (μg/kg, i.p.) | Soluble Aβ concentration in brain (ng/g wet weight) |
Insoluble Aβ concentration in brain (ng/g wet weight) |
||||
|---|---|---|---|---|---|---|
| hAβ1–40 | hAβ1–42 | hAβ1–42/hAβ1–40 ratio | hAβ1–40 | hAβ1–42 | hAβ1–42/hAβ1–40 ratio | |
| Tg2576 (5 males per group), began treatment at 10 weeks old, 11 weeks old when killed | ||||||
| 0 (q2d × 4) | 39.1 ± 4.25 | 11.1 ± 0.240 | 0.276 ± 0.0169 | NPb | NP | NP |
| 2.5 (q2d × 4) | 15.4 ± 2.15* | 5.82 ± 0.891* | 0.357 ± 0.0226 | NP | NP | NP |
| TgCRND8 (4 males, 3–4 females per group), began treatment at 9 weeks old, 17 weeks old when killed | ||||||
| 0 (q3d × 19) | 872 ± 33.7 | 3360 ± 122 | 3.65 ± 0.176 | 923 ± 66.7 | 4640 ± 143 | 4.75 ± 0.247 |
| 2.5 (q3d × 19) | 618 ± 18.7* | 2240 ± 153* | 3.39 ± 0.196 | 514 ± 69.0* | 3870 ± 133* | 7.16 ± 0.361* |
| TgCRND8 (4 females per group), began treatment at 20 weeks old, 21 weeks old when killed | ||||||
| 0 (q2d × 4) | 1060 ± 25.9 | 2920 ± 202 | 2.78 ± 0.219 | 3850 ± 931 | 18000 ± 1700 | 3.83 ± 0.166 |
| 2.5 (q2d × 4) | 675 ± 54.3** | 1970 ± 130** | 3.04 ± 0.417 | 4710 ± 282 | 16500 ± 878 | 3.54 ± 0.312 |
| 2.5 (q2d × 4) + 10 mg/kg elacridar (q12h × 8) | 1240 ± 27.6*** | 4700 ± 214*** | 3.78 ± 0.107*** | 6030 ± 238 | 20400 ± 443 | 3.40 ± 0.198 |
hAβ levels were not detected in non-Tg mice. NP, Not present.
*p < 0.05 for 1,25(OH)2D3 treatment compared with vehicle treatment, Student's two-tailed t test.
**p < 0.05 for 1,25(OH)2D3 treatment compared with vehicle treatment, one-way ANOVA with Bonferroni's multiple comparisons test.
***p < 0.05 for elacridar plus 1,25(OH)2D3 treatment compared with vehicle treatment, one-way ANOVA with Bonferroni's multiple comparisons test.
Discussion
Although AD is a multifaceted disease, aggregation of pathologic forms of Aβ peptides is considered key to chronic neural injury and cognitive decline in patients with AD (Kidd 1964), with Aβ plaque formation viewed as a significant clinical hallmark. The amyloid clearance hypothesis suggests that increased Aβ accumulation in brain is attributable to reduced Aβ clearance (Zlokovic et al., 2000) in brains of patients with AD (Mawuenyega et al., 2010) and that increased Aβ clearance across the BBB is beneficial (Castellano et al., 2012), with the periphery serving as a sink (Zhang and Lee, 2011). P-gp was implicated to play a role in brain-to-blood efflux of these cleavage products (Vogelgesang et al., 2004) and functions as an Aβ efflux pump (Tai et al., 2009). Lam et al. (2001) were among the first to demonstrate that P-gp-enriched vesicles transport Aβ in an ATP-dependent manner. The basolateral-to-apical transport of Aβ1–40 and Aβ1–42 in a polarized, MDR1-transfected porcine proximal renal tubule endothelial cell line was inhibited by verapamil, a P-gp inhibitor (Kuhnke et al., 2007). Consistent with these observations, mdr1a/b−/− hAPP Tg mice exhibited decreased clearance of hAβ and increased hAβ levels in various regions of the brain, including the hippocampus, a region that plays a key role in regulating the formation and consolidation of long-term memory (Cirrito et al., 2005), and it is interesting to note that a significant negative correlation exists between the densities of senile plaque lesions and P-gp levels in brain capillaries of patients with AD (Vogelgesang et al., 2002). In contrast, Ohtsuki et al. (2010) inferred that P-gp is not a significant clearance pathway of Aβ, because verapamil failed to alter the brain clearance of [125I]Aβ1–40 injected into rat brain in vivo or in a cell monolayer, findings that contrasted our observations using elacridar and those of Cirrito et al. (2005) using another P-gp inhibitor in vivo. The difference could be explained by the potency of verapamil, a lower-affinity P-gp inhibitor compared with that of elacridar whose EC50 is 20 nm (Imbert et al., 2003). Upregulation of brain Mdr1a/P-gp with pregnenolone-16α-carbonitrile (Hartz et al., 2010) or St. John's Wort (Brenn et al., 2014), ligands of the PXR, also led to increased hAβ1–42 efflux in isolated rat brain capillaries and reduced brain levels of soluble hAβ1–40 and hAβ1–42 in hAPP Tg mice.
Other transporters may also affect Aβ accumulation. It was suggested that BCRP mediates the transport of Aβ in the brain (Xiong et al., 2009). Patients with AD tend to exhibit higher BCRP levels than age-matched nondemented patients, and the same applies to the Tg3x mouse model of AD against its matched littermate control (Xiong et al., 2009). If this were the case, increased BCRP should have promoted Aβ excretion. We showed that murine Bcrp mRNA or protein expression did not differ between non-Tg and Tg mice, patterns that also remained unaltered with 1,25(OH)2D3 or elacridar treatment (data not shown), suggesting that Bcrp is unlikely to alter brain Aβ deposition in our studies. Although Rage and Lrp1 may play crucial roles in Aβ deposition in the brain (Deane et al., 2009), their mRNA and protein expression was unaltered after short-term or long-term 1,25(OH)2D3 treatment of Tg mice (data not shown). Bace1 (β-secretase activity of the β-site APP-cleaving enzyme 1) levels in TgCRND8 mice also remained unchanged with treatment (data not shown). After 1,25(OH)2D3 treatment, only P-gp was found to be induced by Vdr, and this led to a significant reduction in Aβ deposition.
One goal in AD prevention is reduction of the Aβ1–42/Aβ1–40 ratio (Yin et al., 2007), because Aβ1–42 is considered to be the more pathogenic species and the major component of neuritic plaques (Jarrett et al., 1993). With the recognition that Aβ1–40 is a major contributor to cerebrovascular amyloid angiopathy and associated with stroke in patients with AD (Ozawa et al., 2002), it may be beneficial to increase the clearances of both Aβ1–40 and Aβ1–42 rather than just Aβ1–42. At an age when plaques and cognitive impairment are absent in Tg2756 mice (Kawarabayashi et al., 2001), 1,25(OH)2D3-mediated induction of P-gp expression led to reduced soluble hAβ1–40 and hAβ1–42 accumulation almost equally (Fig. 4). In TgCRND8 mice, the more suitable mouse model for the study of plaque formation and AD pathology, both soluble and insoluble hAβ were reduced after early treatment with 1,25(OH)2D3, demonstrating that it is best to begin intervention early on, before the period of plaque formation, because P-gp is believed to directly influence only soluble Aβ levels (Table 1). Furthermore, we are able to demonstrate that vitamin D deficiency reduced cerebral P-gp expression in mice (Fig. 3), thus providing a plausible explanation for the clinical association between vitamin D deficiency and reduced cognitive performance in older adults (Wilkins et al., 2006; Evatt et al., 2008). In addition, a few animal studies demonstrated a link between vitamin D and Aβ. In rats treated with Aβ, vitamin D supplementation nearly restored the suppressed synaptic plasticity to control levels (Taghizadeh et al., 2013), and hAPP Tg mice receiving a vitamin D-deficient diet had higher Aβ plaque load and increased cognitive deficits compared with controls, whereas mice fed a vitamin D-enriched diet had a lower plaque load and increased cognitive function compared with controls (Yu et al., 2011).
It is recognized that activation of P-gp is not the only mechanism whereby Vdr reduces cerebral Aβ. Masoumi et al. (2009) suggested that 1,25(OH)2D3 stimulates macrophages via a genomic pathway for Aβ phagocytosis and clearance in patients with AD, whereas others demonstrate that 1,25(OH)2D3 treatment increased cerebral clearance of [125I]Aβ1–40 by both genomic and nongenomic Vdr actions (Ito et al., 2011). It is further suggested that Aβ may suppress effects of Vdr signaling (Dursun et al., 2011), such as release of nerve growth factor in hippocampal neurons that would otherwise render a neuroprotective effect (Brown et al., 2003). Despite this suggested suppressive mechanism, Vdr-mediated induction of P-gp was found to persist even at high Aβ levels in the present study.
Our data strongly assert a positive link between 1,25(OH)2D3-liganded Vdr, induction of Mdr1a/P-gp, and brain Aβ accumulation. We show that, whereas Vdr increases Mdr1a and P-gp expression, which reduces brain Aβ levels, inhibition of P-gp function with elacridar leads to accumulation of soluble Aβ in the brain. Vdr activation during the period of plaque formation decreases the soluble hAβ load and reduces plaque formation in younger TgCRND8 mice, improving conditioned fear memory, confirming that decreasing cerebral hAβ load in these younger mice delays the progression and intensity of AD-like symptoms, as found by others in Tg2576 mice (Karlnoski et al., 2009). Short-term treatment of older TgCRND8 mice after the period of plaque formation lowered the soluble hAβ but was unable to decrease the plaque burden (Table 1). This study shows unequivocally that manipulation of P-gp by the VDR to increase efflux of soluble Aβ, likely the monomeric form, is a promising mechanism for the prevention and treatment of AD and that maintenance of adequate vitamin D levels in the elderly may be crucial in preventing neurodegeneration.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR) and the Alzheimer Society of Ontario. M.R.D. was supported by a CIHR Strategic Training Grant in Biological Therapeutics and Pfizer Canada Graduate Scholarship in Science and Technology. E.C.Y.C. was supported by the Natural Sciences and Engineering Research Council of Canada Alexander Graham Bell Fellowship. We thank Dr. Bingning Dong (Baylor College of Medicine, Texas Medical Center, Houston, TX) for discussion on immunostaining.
The authors declare no competing financial interests.
References
- Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol. 2004;66:413–419. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Brenn A, Grube M, Jedlitschky G, Fischer A, Strohmeier B, Eiden M, Keller M, Groschup MH, Vogelgesang S. St. John's Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer's disease mouse model: role of P-glycoprotein. Brain Pathol. 2014;24:18–24. doi: 10.1111/bpa.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Bianco JI, McGrath JJ, Eyles DW. 1,25-Dihydroxyvitamin D3 induces nerve growth factor, promotes neurite outgrowth and inhibits mitosis in embryonic rat hippocampal neurons. Neurosci Lett. 2003;343:139–143. doi: 10.1016/S0304-3940(03)00303-3. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A. 2012;109:15502–15507. doi: 10.1073/pnas.1206446109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Chow EC, Maeng HJ, Liu S, Khan AA, Groothuis GM, Pang KS. 1α,25-Dihydroxyvitamin D3 triggered vitamin D receptor and farnesoid X receptor-like effects in rat intestine and liver in vivo. Biopharm Drug Dispos. 2009;30:457–475. doi: 10.1002/bdd.682. [DOI] [PubMed] [Google Scholar]
- Chow EC, Durk MR, Cummins CL, Pang KS. 1α,25-Dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(−/−) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J Pharmacol Exp Ther. 2011;337:846–859. doi: 10.1124/jpet.111.179101. [DOI] [PubMed] [Google Scholar]
- Chow EC, Quach HP, Vieth R, Pang KS. Temporal changes in tissue 1α,25-dihydroxyvitamin D3, vitamin D receptor target genes, and calcium and PTH levels after 1,25(OH)2D3 treatment in mice. Am J Physiol Endrocrinol Metab. 2013;304:E977–E989. doi: 10.1152/ajpendo.00489.2012. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-β peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2009;8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durk MR, Chan GN, Campos CR, Peart JC, Chow EC, Lee E, Cannon RE, Bendayan R, Miller DS, Pang KS. 1α,25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J Neurochem. 2012;123:944–953. doi: 10.1111/jnc.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dursun E, Gezen-Ak D, Yilmazer S. A novel perspective for Alzheimer's disease: vitamin D receptor suppression by amyloid-beta and preventing the amyloid-β induced alterations by vitamin D in cortical neurons. J Alzheimers Dis. 2011;23:207–219. doi: 10.3233/JAD-2010-101377. [DOI] [PubMed] [Google Scholar]
- Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol. 2008;65:1348–1352. doi: 10.1001/archneur.65.10.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- Han BH, Zhou ML, Vellimana AK, Milner E, Kim DH, Greenberg JK, Chu W, Mach RH, Zipfel GJ. Resorufin analogs preferentially bind cerebrovascular amyloid: potential use as imaging ligands for cerebral amyloid angiopathy. Mol Neurodegener. 2011;6:86. doi: 10.1186/1750-1326-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna A, Iremonger K, Das P, Dickson D, Golde T, Janus C. Age-related increase in amyloid plaque burden is associated with impairment in conditioned fear memory in CRND8 mouse model of amyloidosis. Alzheimers Res Ther. 2012;4:21. doi: 10.1186/alzrt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease Aβ40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Miller DS, Bauer B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer's disease. Mol Pharmacol. 2010;77:715–723. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–4602. [PubMed] [Google Scholar]
- Imbert F, Jardin M, Fernandez C, Gantier JC, Dromer F, Baron G, Mentre F, Van Beijsterveldt L, Singlas E, Gimenez F. Effect of efflux inhibition on brain uptake of itraconazole in mice infected with Cryptococcus neoformans. Drug Metab Dispos. 2003;31:319–325. doi: 10.1124/dmd.31.3.319. [DOI] [PubMed] [Google Scholar]
- Ito S, Ohtsuki S, Nezu Y, Koitabashi Y, Murata S, Terasaki T. 1α,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-β peptide(1–40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS. 2011;8:20. doi: 10.1186/2045-8118-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The C-terminus of the beta protein is critical in amyloidogenesis. Ann N Y Acad Sci. 1993;695:144–148. doi: 10.1111/j.1749-6632.1993.tb23043.x. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Karlnoski RA, Rosenthal A, Kobayashi D, Pons J, Alamed J, Mercer M, Li Q, Gordon MN, Gottschall PE, Morgan D. Suppression of amyloid deposition leads to long-term reductions in Alzheimer's pathologies in Tg2576 mice. J Neurosci. 2009;29:4964–4971. doi: 10.1523/JNEUROSCI.4560-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd M. Alzheimer's disease—an electron microscopical study. Brain. 1964;87:307–320. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, Cascorbi I, Walker LC, Kroemer HK, Warzok RW, Vogelgesang S. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer's amyloid-β peptides—implications for the mechanisms of Aβ clearance at the blood-brain barrier. Brain Pathol. 2007;17:347–353. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB. β-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001;76:1121–1128. doi: 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lillard-Wetherell K. Methods for analyzing amyloid plaque burden in mouse models of Alzheimer's disease. Online Aperio Application Note. 2008. www.aperio.com.
- Masoumi A, Goldenson B, Ghirmai S, Avagyan H, Zaghi J, Abel K, Zheng X, Espinosa-Jeffrey A, Mahanian M, Liu PT, Hewison M, Mizwickie M, Cashman J, Fiala M. 1α,25-dihydroxyvitamin D3 interacts with curcuminoids to stimulate amyloid-β clearance by macrophages of Alzheimer's disease patients. J Alzheimers Dis. 2009;17:703–717. doi: 10.3233/JAD-2009-1080. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- Miller DS. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci. 2010;31:246–254. doi: 10.1016/j.tips.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki S, Ito S, Terasaki T. Is P-glycoprotein involved in amyloid-β elimination across the blood-brain barrier in Alzheimer's disease? Clin Pharmacol Ther. 2010;88:443–445. doi: 10.1038/clpt.2010.160. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Tomiyama T, Maat-Schieman ML, Roos RA, Mori H. Enhanced Aβ40 deposition was associated with increased Aβ42–43 in cerebral vasculature with Dutch-type hereditary cerebral hemorrhage with amyloidosis (HCHWA-D) Ann N Y Acad Sci. 2002;977:149–154. doi: 10.1111/j.1749-6632.2002.tb04810.x. [DOI] [PubMed] [Google Scholar]
- Petanceska SS, Nagy V, Frail D, Gandy S. Ovariectomy and 17β-estradiol modulate the levels of Alzheimer's amyloid β peptides in brain. Neurology. 2000;54:2212–2217. doi: 10.1212/WNL.54.12.2212. [DOI] [PubMed] [Google Scholar]
- Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zürcher-Neely HA, Heinrikson RL, Ball MJ, Greenberg BD. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer's disease. J Biol Chem. 1993;268:3072–3083. [PubMed] [Google Scholar]
- Saeki M, Kurose K, Tohkin M, Hasegawa R. Identification of the functional vitamin D response elements in the human MDR1 gene. Biochem Pharmacol. 2008;76:531–542. doi: 10.1016/j.bcp.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Taghizadeh M, Talaei SA, Djazayeri A, Salami M. Vitamin D supplementation restores suppressed synaptic plasticity in Alzheimer's disease. Nutr Neurosci. 2013 doi: 10.1179/1476830513Y.0000000080. doi: 10.1179/1476830513Y.0000000080. Advance online publication. Retrieved April 14, 2014. [DOI] [PubMed] [Google Scholar]
- Tai LM, Loughlin AJ, Male DK, Romero IA. P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-β. J Cereb Blood Flow Metab. 2009;29:1079–1083. doi: 10.1038/jcbfm.2009.42. [DOI] [PubMed] [Google Scholar]
- Toornvliet R, van Berckel BN, Luurtsema G, Lubberink M, Geldof AA, Bosch TM, Oerlemans R, Lammertsma AA, Franssen EJ. Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[11C]verapamil and positron emission tomography. Clin Pharmacol Ther. 2006;79:540–548. doi: 10.1016/j.clpt.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW. Deposition of Alzheimer's β-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002;12:535–541. doi: 10.1097/00008571-200210000-00005. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2004;1:121–125. doi: 10.2174/1567205043332225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sykes DB, Miller DS. Constitutive androstane receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol Pharmacol. 2010;78:376–383. doi: 10.1124/mol.110.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann A, Paliga K, Dürrwang U, Reinhard FB, Schuckert O, Evin G, Masters CL. Proteolytic processing of the Alzheimer's disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem. 1999;274:5823–5829. doi: 10.1074/jbc.274.9.5823. [DOI] [PubMed] [Google Scholar]
- Wilkins CH, Sheline YI, Roe CM, Birge SJ, Morris JC. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14:1032–1040. doi: 10.1097/01.JGP.0000240986.74642.7c. [DOI] [PubMed] [Google Scholar]
- Xiong H, Callaghan D, Jones A, Bai J, Rasquinha I, Smith C, Pei K, Walker D, Lue LF, Stanimirovic D, Zhang W. ABCG2 is upregulated in Alzheimer's brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Aβ(1–40) peptides. J Neurosci. 2009;29:5463–5475. doi: 10.1523/JNEUROSCI.5103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin YI, Bassit B, Zhu L, Yang X, Wang C, Li YM. γ-Secretase substrate concentration modulates the Abeta42/Abeta40 ratio: Implications for Alzheimer disease. J Biol Chem. 2007;282:23639–23644. doi: 10.1074/jbc.M704601200. [DOI] [PubMed] [Google Scholar]
- Yu J, Gattoni-Celli M, Zhu H, Bhat NR, Sambamurti K, Gattoni-Celli S, Kindy MS. Vitamin D3-enriched diet correlates with a decrease of amyloid plaques in the brain of AbetaPP transgenic mice. J Alzheimers Dis. 2011;25:295–307. doi: 10.3233/JAD-2011-101986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lee DH. Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist. 2011;17:163–173. doi: 10.1177/1073858410381532. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B. Clearance of amyloid β-peptide from brain: transport or metabolism? Nat Med. 2000;6:718–719. doi: 10.1038/77397. [DOI] [PubMed] [Google Scholar]