

Abstract
From the early periods of neurogenesis and migration, up until synaptogenesis, both nitric oxide (NO) and its downstream messenger, cGMP, are thought to influence the development of neurons. The NO/cGMP/cGMP-dependent protein kinase (cGK) pathway regulates the clustering and recruitment of synaptic proteins and vesicles to the synapse, adjusting the exoendocytic cycle to the intensity of activity and accelerating endocytosis following large-scale exocytosis. Here, we show that blockage of the N-methyl-d-aspartate receptor impairs the cycling of synaptic vesicles in a subset of boutons on cerebellar granule cells, an effect that was reversed by increasing cGMP. Furthermore, we demonstrate that presynaptic cGK type II (cGKII) plays a major role in this process. Using the FM1-43 dye to track vesicle recycling, we found that knockdown of cGKII and/or the application of a cGK inhibitor reduced the efficiency of synaptic vesicle recycling to a similar extent. Likewise, in cerebellar granule cells transfected with vGlut1-pHluorin to follow the exoendocytotic cycle, application of a cGK inhibitor slowed vesicle endocytosis when exocytosis was accelerated through strong and sustained stimulation. Additionally, ultrastructural analysis showed that cGKII knockdown or inhibition favored the formation of endosomal-like structures after strong and sustained stimulation. We conclude that cGKII controls the homeostatic balance of vesicle exocytosis and endocytosis in synaptic boutons of rat cerebellar granule cells.
Keywords: cerebellar granule cells, cGMP signaling, endocytosis, retrograde messenger, vesicle recycling
Introduction
Presynaptic nerve terminals at chemical synapses contain many synaptic vesicles (SVs), these being filled with neurotransmitter that is released into the synaptic cleft when the presynaptic neuron is stimulated, thereby eliciting postsynaptic responses (Couteaux and Pecot-Dechavassine, 1970; Sudhof, 2004). After fusion with the membrane and neurotransmitter release, new SVs are formed by endocytosis and once recharged with neurotransmitter they are ready to undergo a new round of exocytosis (Sudhof, 2004; Kavalali, 2006). This vesicular cycle is a finely tuned sequential process that requires the coordination of different proteins and cytoskeletal elements. Moreover, there is evidence that phosphorylation/dephosphorylation fulfils an important role in regulating this cycle (Cousin et al., 2001; Wenk and De Camilli, 2004).
Nerve transmission is not strictly unidirectional and a variety of feedback signals of postsynaptic origin flow toward presynaptic targets. One such retrograde signal is provided by the nitric oxide (NO) synthesized in neurons in a Ca2+-dependent manner by NO synthase (NOS; Mayer et al., 1992; Panda et al., 2001). Indeed, the NO activated cGMP pathway fulfils an important role in coordinating functional and structural alterations at presynaptic and postsynaptic sites in response to neuronal activity (Wang et al., 2005; Ota et al., 2010).
At presynaptic sites the NO/cGMP pathway regulates the clustering and recruitment of synaptic proteins and vesicles (Ratnayaka et al., 2012), and it adjusts the exoendocytic cycle to the intensity of activity (Micheva et al., 2003; Petrov et al., 2008; Tegenge et al., 2009; Eguchi et al., 2012). Although these effects are mostly mediated by activation of a cGMP-dependent protein kinase (cGK; Xue et al., 2004; Wang et al., 2005), the nature of this kinase (type I or type II) remains unclear. Activation of cGK upregulates phosphatidylinositol-4,5-bisphosphate (PIP2), thereby accelerating the endocytosis that follows the large-scale exocytosis caused by a strong and sustained stimulation (Micheva et al., 2003; Eguchi et al., 2012). PIP2 binds to the adaptor protein AP2, which helps localize it to the plasma membrane where it retrieves SV components through clathrin-mediated endocytosis (CME), the dominant mode of SV retrieval during mild stimulation (Granseth et al., 2006). Conversely, during intense stimulation, activity-dependent bulk endocytosis (ADBE) becomes the dominant mode, involving other adaptor proteins (Clayton et al., 2008; Dittman and Ryan, 2009; Cheung and Cousin, 2012). These two forms of endocytosis can coexist in the same synaptic terminal, and competition between them can regulate the rate and extent of SV recycling (Voglmaier et al., 2006; Bartolomé-Martín et al., 2012).
In juvenile cerebellar granule cells, inhibition of soluble guanylyl cyclase (sGC) impairs SV recycling during strong and sustained stimulation (López-Jiménez et al., 2009). In these cells, the key enzymes in cGMP metabolism and signaling are expressed strongly, and their activation is linked to NMDA receptor (NMDAR) stimulation (Jurado et al., 2003). The results we present here show that blockage of the NMDAR slows SV recycling at cerebellar granule cell synaptic boutons during a strong and sustained stimulation, an effect that is prevented by pharmacologically increasing cGMP and that is mimicked by inhibition and/or knockdown of cGK type II (cGKII), a downstream cGMP effector.
Materials and Methods
Cell culture.
Cells were dissociated from the cerebellum of 7-d-old (P7) Wistar albino rat pups of either sex, as described previously (Jurado et al., 2003), and the primary cerebellar cells isolated were diluted in neurobasal A supplemented with B27 (Life Technologies), 20 mm KCl, 0.5 mm glutamine and the stabilized antibiotic antimycotic solution (Sigma-Aldrich). The cells were seeded onto poly-l-lysine-coated coverslips at a density of 1–3 × 105 cells/coverslip, or in 6-well tissue culture plates at 3.5 × 106 cells/well. The cultures were maintained in a humidified incubator at 37°C in an atmosphere of 5% CO2 and after 24 h in culture, 10 μm cytosine-β-d-arabinofuranoside (Sigma-Aldrich) was added to restrict glial cell growth. The cultures were routinely used for experiments between 7 and 9 d in vitro (DIV). All procedures with animals were conducted under the ethical guidelines for animal experiments and the regulations established in the European Council (Directive 2010/63/EU). The experimental protocol was approved by the Commission of Animal Experimentation of the Complutense University. All efforts were made to minimize the number of animals used and their suffering.
Cell transfection.
After 4 DIV, a siRNA designed to selectively silence cGKII (Accell siRNA Ref no. A-092722-15 from GE Healthcare Dharmacon) or an Accell Red Nontargeting siRNA (Ref no. D-001960 from GE Healthcare Dharmacon) was added to the cells, which were incubated in Accell siRNA delivery media (Ref no. B-005000 from GE Healthcare Dharmacon) supplemented with B27, 20 mm KCl, 0.5 mm glutamine, and the stabilized antibiotic antimycotic solution. The transfection efficiency was verified by exciting the cells at 595 nm and visualizing them by fluorescence microscopy. The silencing of cGKII was verified by measuring its mRNA transcripts 24 h and 4 d after transfection by real-time PCR using the TaqMan Gene Expression Assay Rn00435938_m1 for cGKII (Applied Biosystems; Life Technologies) and Rn01775763_g1 to assay GAPDH as an endogenous control (Jurado et al., 2003). The levels of cGKII mRNA were reduced by 81.36 ± 3.39% 24 h after transfection with the specific siRNA and by 81.00 ± 1.65% 4 d after transfection, whereas they were not affected by the scrambled control siRNA. Functional studies were performed on cells 4 d after transfection.
Dissociated neurons were transfected by electroporation with the chicken actin pCAGGs vector (1 μg) containing the VGlut1-pHluorin (VGlut1-pH) construct using an Amaxa Nucleofector II device and the Rat Neuron Amaxa Nucleofector Kit (Lonza), according to the manufacturer's instructions (program O-03). The cells were then grown as described previously until 7–8 DIV (López-Jiménez et al., 2009).
Vesicle recycling (FM1-43).
Recycled SVs were labeled with the cationic styrylpyridinium dye FM 1-43 (Invitrogen), as described previously (López-Jiménez et al., 2009; Bartolomé-Martín et al., 2012). Briefly, the cells were incubated for 10 min in a calcium-free buffer (140 mm NaCl, 5 mm KCl, 5 mm NaHCO3, 1.2 mm NaH2PO4, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES: López-Jiménez et al., 2009), and then incubated for 3 min with FM1-43 dye (10 μm) in high-potassium buffer (95 mm NaCl, 50 mm KCl, 5 mm NaHCO3, 1.2 mm NaH2PO4, 1 mm MgCl2, 1.33 mm CaCl2, 10 mm glucose, 10 mm HEPES, pH 7.4). The coverslips were mounted in a rapid-switching, pH5 laminar-flow perfusion chamber (Warner Instruments) on the stage of a Nikon Eclipse TE2000-S inverted epifluorescence microscope, and they were continuously perfused (1 ml/min) using a VC6 perfusion system (Warner Instruments). First, they were washed for 10 min with a calcium-free low-potassium buffer to remove the surface-bound dye, and then baseline measurements were acquired over 30 s. Subsequently, the cells were stimulated for 3 min with high-potassium medium to produce dye unloading. Alternatively, the cells were stimulated for 10 s with high-potassium medium, followed by perfusion of low-potassium medium for 1 min, and then again with high-potassium medium for 2 min. Temperature was clamped at 37°C with a TC-344B temperature controlling system (Warner Instruments) to minimize the effects of temperature fluctuations. Images were acquired at a rate of 1 Hz using an iXonEM + EMCCD camera (iXonEM+DU885, Andor Technology) and fluorescence emission was recorded through a Nikon CFI Plan Apo VC 60× Oil objective 1.4 (NA). Excitation was provided by a 479 nm monochromator and the light emitted was collected through a FITC filter.
Analysis of FM1-43.
Different fields were selected randomly and individual synaptic boutons were analyzed as described previously (Bartolomé-Martín et al., 2012). Regions-of-interest (ROIs) were identified with Igor Pro software to indicate the synaptic boutons. This analysis was performed according to the automated method described by Bergsman et al. (2006), with a minimum quality criterion for analysis of 0.1. To determine the extent of dye release, the mean background fluorescence of several cell-free regions was subtracted and the values were normalized to the initial fluorescence (F/F0). When the rate of dye release was calculated, we subtracted the remaining fluorescence of each bouton at the end of the stimulation period. Dye release responses were heterogeneous but nevertheless, two main subpopulations of synaptic boutons were distinguished (Bartolomé-Martín et al., 2012).
VGlut1-pH imaging.
We used a direct optical presynaptic readout based on the pH-sensitive GFP pHluorin (Miesenböck et al., 1998) tagged to the luminal domain of the vesicular glutamate transporter to analyze the distribution of vesicles in the recycling and resting pool. The size of the recycling vesicle pool was measured as the ΔF elicited by a large stimulus (50 s) with 50 mm KCl in the presence of bafilomycin A1 (0.5 μm), a V-type ATPase inhibitor that blocks vesicle reacidification (Sankaranarayanan and Ryan, 2001). This saturation indicates that all recycling vesicles underwent at least one round of fusion, and thus the ΔF in this experiment offers a measure of the size of the recycling pool. The total vesicle population was visualized by the subsequent application of NH4Cl saline. Bafilomycin A1 (baf) blocks reacidification of endocytosed SVs, making the VGlut1-pH fluorescence indicative of exocytosis alone. These curves were adjusted to the equation: (F) = Fmax*t/(τ + t), where τ is the time constant (time to reach half Fmax). In the absence of baf, stimulation elicits exocytosis (an increase in fluorescence) followed by endocytosis (fluorescence decay). The decay phase of the VGlut1-pH signal reflects the rate of synaptic vesicle reacidification that follows vesicle retrieval. Reacidification is fast (τ ∼ 4 s), such that the decline in the VGlut1-pH signals is a measure of endocytosis close to real-time (Atluri and Ryan, 2006; Granseth et al., 2006). The curves were fit to single exponential decay curves beginning at a time point offset from the end of the stimulus that is approximately equal to twice the reacidification time constant, where the exponential time course is dominated by endocytosis. F = F0+Ae−t/τ, where τ is the time constant for endocytosis. Images were acquired at a rate of 4 Hz and averaged into one single frame per second to enhance the signal-to-noise ratio, resulting in an actual readout of 1 Hz.
Electron microscopy.
To visualize the effect of the different treatments or experimental conditions on the distribution or recycling of SVs within the presynaptic terminals, cultured cerebellar granule cells subjected to different treatments were maintained under basal conditions or stimulated for 3 min with 50 mm KCl in HEPES-buffered medium (HBM), and then maintained at rest for 10 min in 5 mm KCl in HBM, emulating our protocol for the loading and washing of the FM1-43 dye. After stimulation, the cells were fixed at 4°C for 2 h with 4% paraformaldehyde/2.5% glutaraldehyde in Milloning sodium phosphate buffer (0.1 m, pH 7.3). The cells were then washed twice and kept overnight at 4°C in Milloning buffer. At this point, the cells were postfixed in 1% OsO4 for 1 h at room temperature and dehydrated in ethanol. To detach the cell layer from the plate, cells were treated with propylene oxide and the entire monolayer was collected and embedded using the SPURR embedding kit (TAAB). Alternatively, cells were maintained on coverslips until they were embedded in Spurr (Spurr Embedding Kit, TAAB) and after 24 h, the coverslips were removed with liquid N2. Ultrathin sections (70 nm) were routinely stained with uranyl acetate and lead citrate, and images were obtained on a JEOL 1200 EX II transmission electron microscope. Synapses were identified as containing a cluster of synaptic vesicles in close proximity to a membrane with docked (from a morphological point of view) vesicles. Synapses were also considered to contain mitochondria and to have an apposed postsynaptic cell or dendrite. Measurements were taken with the aid of ImageJ software and two parameters were analyzed: the diameter of the SVs and the rest of the organelles in the bouton, and the number of each organelle within the active zone (AZ).
Statistical analyses.
The data were analyzed with Statgraphic, OriginPro 8.0 or SigmaPlot 10 statistical software. Comparison of two independent groups was performed using the unpaired t test, Mann–Whitney or Kolmogorov–Smirnov test. Comparison of several independent groups was achieved by one-way ANOVA, the latter revealing significant general group effects. In all analyses, a familywise 95% confidence level (p < 0.05) was applied: n indicates the number of individual experiments performed from at least three different cell cultures, and N indicates the number of specimens analyzed (ROIs, boutons, etc.).
Results
NMDAR blockage hinders SV recycling in a subset of boutons under strong and sustained stimulation
To visualize SV recycling, we examined the ability of rat cerebellar granule cells to incorporate the fluorescent styryl dye FM1-43 into SVs when stimulated with 50 mm KCl (Fig. 1A, top), and their capacity to unload it after another round of stimulation with 50 mm KCl (Fig. 1A, middle). Cell stimulation with 50 mm KCl in the presence of 1.33 mm external Ca2+ provokes a depolarization that is associated with the massive influx of Ca2+, resulting in a typical biphasic profile of dye unloading (Klingauf et al., 1998; López-Jiménez et al., 2009; Bartolomé-Martín et al., 2012). The initial phase of this dye loss (≤20 s) can be attributed to dye release from vesicles in a rapidly mobilizable pool (Pyle et al., 2000; Mozhayeva et al., 2002), whereas the slow phase (20–180 s) reflects the replenishment of this rapidly mobilizable pool with the reserve pool (Pyle et al., 2000; Mozhayeva et al., 2002). According to the dye unloading parameters, two populations of boutons could be distinguished on granule cells, although both displayed a biphasic profile (López-Jiménez et al., 2009; Bartolomé-Martín et al., 2012): Group 1 with strong (>40% of the initial fluorescence) and rapid unloading (τ1 = 9.8 ± 1.2 s, τ2 = 96.2 ± 11.4 s; Fig. 1B); and Group 2 with milder (<40% of the initial fluorescence) and slower unloading (τ1 = 16.4 ± 3.0 s, τ2 = 120.4 ± 26.7 s; Fig. 1C).
Figure 1.
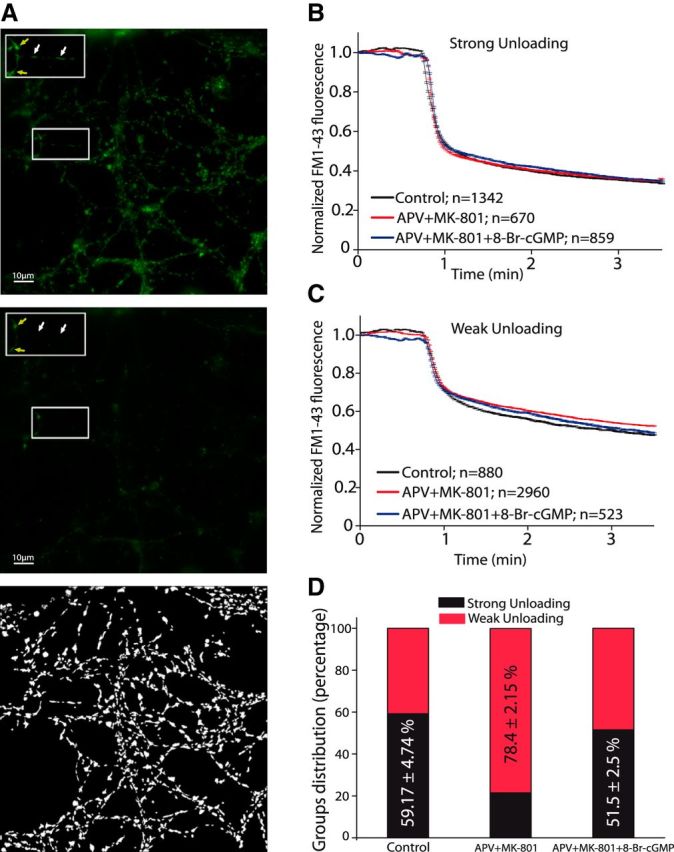
Long-lasting NMDAR blockade hindered FM1-43 unloading in a subset of boutons under strong and sustained stimulation. A, Fluorescence image of cells loaded with FM1-43 (top) and unloaded (middle), and amplified images of the boxed areas showing group1 (white arrows) and Group 2 boutons (yellow arrows). Bottom, Mask of terminals obtained with Igor Pro software that fulfils the quality requirement of 0.1 with the Bergsman routine (Materials and Methods). Scale bars, 10 μm. B, Mean traces of boutons from control cells that undergo strong FM1-43 unloading (Group 1: N = 1342, n = 4 coverslips from 2 different cell cultures), from APV- (50 μm) + MK801- (1 μm) treated cells (N = 670, n = 7 coverslips from 2 cell cultures), and from APV+MK801 + 8-BrcGMP- (0.5 mm) treated cells (N = 859, n = 5 coverslips from 2 cell cultures). C, Mean traces of weak FM1-43 unloading boutons (Group 2) from control cells (N = 880, n = 4 coverslips from 2 different cell cultures), APV+MK801-treated cells (N = 2960, n = 7 coverslips from 2 cell cultures), and APV+MK801 + 8-BrcGMP-treated cells (N = 523, n = 5 coverslips from 2 cell cultures). D, Distribution of boutons into the two groups (strong or weak unloading).
The differences in unloading between synaptic terminals displaying strong and weak dye unloading may indicate that they have distinct recycling capacities, possibly due to the different contributions of the two main endocytotic mechanisms: CME or ADBE (Voglmaier et al., 2006; Bartolomé-Martín et al., 2012). The distribution of boutons into these two groups could be modified by treatments that accelerate or slow SV recycling, such as prolong inhibition of sGC with ODQ or of synaptic activity with tetrodotoxin (López-Jiménez et al., 2009; Bartolomé-Martín et al., 2012). In control cells 7 DIV, 59.17 ± 4.74% of boutons belonged to Group 1, yet when NMDARs were blocked by a 24 h exposure to a mixture of APV (50 μm) and MK-801 (1 μm), this percentage dropped to 21.45 ± 2.19% (p < 0.00001; Fig. 1D). Moreover, this effect was reverted by the presence of 8-Br-cGMP (51.5 ± 2.5%; p < 0.00001 compared with APV+MK-801-treated cells and p = 0.20198 compared with controls), indicating that the intrinsic activity in the culture activated NMDAR and cGMP synthesis, presumably via the NO synthesis (Jurado et al., 2003) that regulates SVs recycling (López-Jiménez et al., 2009).
cGKII knockdown (KD) or inhibition impairs vesicle recycling
To assess whether the cGMP effector cGK is involved in SV recycling, cells were incubated with a specific inhibitor of these kinases. Like NMDAR blockers, prolonged exposure (24 or 96 h) to KT5823 (1 μm) decreased the proportion of Group 1 nerve terminals that displayed strong dye loss under control conditions (60.70 ± 2.72%) to 23.12 ± 2.55% after 24 h (p < 0.00001) and 27.56 ± 4.26% after 96 h (p < 0.00001; Fig. 2A). Conversely, KT5823 increased the proportion of synaptic terminals that only weakly unloaded the dye (Group 2) from 39.30 ± 2.73% in control cells to 76.88 ± 2.54% (p < 0.00001) after 24 h in the presence of KT5823, and 72.44 ± 4.26% (p < 0.00001) after 96 h (Fig. 2A). KT5823 did not alter the extent of dye release by Group 1 boutons (Fig. 2B), whereas the magnitude of dye loss of Group 2 boutons after 24 h in the presence of the inhibitor was reduced by 25% after a 2.5 min KCl perfusion (Fig. 2C). However, the velocities, measured as the slopes of the two components of unloading, were unchanged by these inhibitors.
Figure 2.

Prolonged inhibition of cGKs or cGKII KD increases the proportion of nerve terminals with weak unloading. A, Distribution of boutons into the strong or weak unloading groups in control cells and those exposed to KT5823 (1 μm) for 24 or 96 h. B, Mean traces of strong FM1-43 unloading boutons from control cells (N = 4439, n = 9 coverslips from 4 different cell cultures) or cells exposed to KT5823 (1 μm) for 24 (N = 806, n = 5 coverslips from 3 cell cultures) or 96 h (N = 563, n = 4 coverslips from 2 cell cultures). C, Mean traces of weak FM1-43 unloading boutons from control cells (N = 2874, n = 9 coverslips from 4 cell cultures), or cells exposed to KT5823 (1 μm) for 24 (N = 2768, n = 5 coverslips from 3 cell cultures) or 96 h (N = 1577, n = 4 coverslips from 2 cell cultures). D, Distribution of boutons in function of strong or weak unloading in control cells and in cells transfected with a scrambled siRNA or cGKII siRNA (cGKII KD). E, Mean traces of strong FM1-43 unloading boutons from control cells (N = 4439, n = 9 coverslips from 4 different cell cultures), or cells transfected with a scrambled siRNA (N = 930, n = 3 coverslips from 1 cell culture) and cGKII KD cells (N = 689, n = 6 coverslips from 3 cell cultures). F, Mean traces of weak FM1-43 unloading boutons from control cells (N = 2874, n = 9 coverslips from 4 cell cultures), scrambled siRNA transfected cells (N = 583, n = 3 coverslips from 1 cell culture) and cGKII KD cells (N = 3198, n = 6 coverslips from 3 cell cultures). G, Distribution of strong or weak unloading boutons in control cells and in cGKII KD cells exposed to KT5823 for 24 h. H, Mean traces of strong FM1-43 unloading boutons from control cells (N = 4439, n = 9 coverslips from 4 different cell cultures) and from cGKII KD cells exposed to KT5823 for 24 h (N = 313, n = 4 coverslips from 2 different cell cultures). I, Mean traces of weak FM1-43 unloading boutons from control cells (N = 2874, n = 9 coverslips from 4 cell cultures) and in cGKII KD cells exposed to KT5823 for 24 h (N = 1427, n = 4 coverslips from 2 cell cultures).
Our analysis strongly suggested that cGK activation is necessary for efficient recycling of SVs under strong and sustained stimulation, as occurred in other neurons (Eguchi et al., 2012; Taoufiq et al., 2013). Both cGKI and cGKII are expressed by cerebellar granule cells, both in vivo and in vitro, (Jurado et al., 2004, 2005), and thus we assessed the specific involvement of cGKII in these events by silencing its expression using a specific siRNA. This approach reduced cGKII mRNA levels by 81.36 ± 3.39% within 24 h and by 81.00 ± 1.65% 4 d after transfection. By contrast, the levels of cGKI assessed 4 d after transfection in Western blots were equivalent in control and in siRNA transfected cells (data not shown). Although cGKII expression (and therefore recycling) was not modified by a scrambled siRNA, cGKII KD notably increased the proportion of slow recycling boutons (80.74 ± 3.12% in cGKII KD vs 39.30 ± 2.73% in control or 38.5 ± 2.29% in scrambled siRNA transfected cells, p < 0.00001; Fig. 2D). In addition, whereas the extent of dye released by Group 1 boutons was slightly altered by cGKII silencing (Fig. 2E), the extent of dye release by Group 2 boutons was largely reduced but without affecting the velocity of unloading (Fig. 2F).
The silencing of cGKII effectively occluded the effect of KT5823 and no additional effect was observed when it was added to cGKII KD cells 24 h before studying SV behavior (Fig. 2G–I), indicating that KT5823 did target cGKII when impairing vesicle cycling. The proportion of boutons that displayed strong dye loss was 15.15 ± 1.51% (p < 0.00001 vs control, and p = 0.39556 vs cGKII KD), whereas the percentage of those that displayed weak unloading was 84.85 ± 1.50% (p < 0.00001 vs control, and p = 0.39983 vs cGKII KD). The velocity of the slow component of release by Group 1 boutons was significantly enhanced by this combined treatment (0.32 ± 0.03 to 0.52 ± 0.01 a.u./min, p < 0.00001).
Acute inhibition of cGK also impairs vesicle recycling
We next addressed whether long lasting inhibition of the enzyme was necessary to impair vesicle recycling. Accordingly, we assessed the effects of an acute 30 min exposure to KT5823 (1 μm) before the FM1-43 loading of cells, which was maintained until the unloading step (Fig. 3A). The presence of KT5823 caused a reduction in the proportion of boutons with strong dye loss (Group 1) similar to that produced by the long-term inhibition or knockdown of this protein (Group 1: control 51.80 ± 7.07%; KT5823, 30 min, 20.45 ± 1.30%; p = 0.00614; Fig. 3B), and with a concomitant increase in the boutons with weak unloading (control 48.2 ± 7.07%; KT5823, 30 min, 79.55 ± 1.30%; p = 0.00614). Moreover, the extent of unloading was strongly modified in the two populations of boutons (Fig. 3C,D).
Figure 3.

Acute inhibition of cGKs decreases the proportion of nerve terminals that undergo strong unloading. A, Experimental protocol. B, Distribution of strong or weak unloading boutons from control cells and those exposed to KT5823 (1 μm) for 30 min before the loading step and during the washing period. C, Mean traces of strong FM1-43 unloading boutons from control cells (N = 1158, n = 5 coverslips from 3 different cell cultures) and those exposed to KT5823 (1 μm) for 30 min (N = 263, n = 5 coverslips from 2 cell cultures). D, Mean traces of weak FM1-43 unloading boutons from control cells (N = 902, n = 5 coverslips from 3 different cell cultures) and those exposed to KT5823 (1 μm) for 30 min (N = 1060, n = 5 coverslips from 2 cell cultures).
Vesicular cycle is a finely tuned process of sequential events. Thus, to analyze which part of vesicle cycling (approach and membrane fusion or membrane retrieval) was hindered by cGK inhibition, the cells were exposed to the inhibitor before or after dye loading and unloading was then stimulated in two steps: a 10 s perfusion of a high-potassium buffer followed by 1 min of low-potassium to evoke the release of the rapidly mobilizable pool; and a 2 min perfusion of high-potassium to evoke the release of the total mobilizable vesicles (Fig. 4A). When the average responses of all the responding boutons from cells maintained in the presence or absence of KT5823, both before or after the loading step, were compared, a clear reduction in unloading was observed when cGK was inhibited before FM1-43 loading (∼30%; Fig. 4B–D). However, when the inhibitor was added after the loading step, no significant effect on the unloading caused by the first 10 s stimulus (Fig. 4B,C), and only a small reduction in unloading provoked by the second stimulus (Fig. 4B,D), was observed. This observation suggests that cGK activity is necessary for the first steps of SV retrieval after the massive exocytosis caused by a strong and sustained stimulation, whereas the inhibition of cGK has minimal effects on SVs once they are formed. The reduction in unloading observed when cGK was inhibited before dye loading may be due to enhanced endosome formation (Bartolomé-Martín et al., 2012), making the endocytosed fluorescence less mobilizable.
Figure 4.

cGKII inhibition after the loading step does not impair SV recycling. A, Experimental protocol showing the two unloading stimuli. B, Mean traces of FM1-43 unloading boutons from control cells (N = 3668 from n = 7 coverslips from 3 different cultures), cells exposed to KT5823 (1 μm, 30 min) after FM1-43 loading (N = 1382 from n = 4 coverslips from 3 different cultures) and cells exposed to KT5823 (1 μm, 30 min) before FM1-43 loading (N = 1686 from n = 5 coverslips from 3 different cultures). C, Cumulative probability plots of unloading after the first stimulus (10 s, 50 mm KCl + 1 min, 5 mm KCl) for control cells or cells incubated with KT5823 before (p = 0.000, Kolmogorov–Smirnov) or after (p = 0.155, Kolmogorov–Smirnov) loading. D, Cumulative probability of unloading after the second stimulus (2 min, 50 mm KCl) in control cells or cells incubated with KT5823 before (p = 0.000, Kolmogorov–Smirnov) or after (p = 0.000, Kolmogorov–Smirnov) loading.
cGKII KD or inhibition increases the number of organelles with a diameter >40 nm within the bouton after strong and sustained stimulation
To examine the proportions of the distinct organelles in the boutons, cells were maintained at rest or stimulated for 3 min with 50 mm KCl to mimic the FM1-43 dye loading protocol, and then washed and fixed as described before. The average number of organelles (small synaptic vesicles, sSVs; organelles <40 nm) and endosomes (organelles >40 nm), and also the endosome/sSV ratio was assessed in 22–55 AZs in each state. At rest, AZs contained sSVs (37.86 ± 3.56) and only very few larger organelles (1.36 ± 0.32; Fig. 5A,D). Knockdown or prolonged inhibition of cGKII increased the number of organelles with a diameter <40 nm (58.45 ± 5.79, p = 0.0099 for cGKII KD; 55.03 ± 5.50, p = 0.0172 for KT5823 24 h; and 47.02 ± 4.39, p = 0.32473 for KT5823 30 min), whereas it did not modify the number organelles >40 nm (0.96 ± 0.38, p = 0.7654 for cGKII KD; 0.48 ± 0.15, p = 0.2266 for KT5823 24 h and 1.05 ± 0.21, p = 0.6775 for KT5823 30 min). Moreover, the percentage of AZs with large organelles was very similar in all experimental conditions (Fig. 5B).
Figure 5.

cGKII KD or inhibition increased the presence of endosomal-like structures under strong and sustained stimulation. In electron microscopy experiments, cells were stimulated (50 mm KCl, 3 min) to mimic the FM1-43 loading step or they were maintained at rest. A, Electron micrographs of cerebellar granule cells at rest (left) or after stimulation (right). B, Distribution of AZs (in percentages) with organelles >40 nm (endosomal-like structures) at rest or after stimulation in cells under different experimental conditions. C, The ratio of organelles >40 nm/organelles <40 nm at rest or after stimulation in cells under different experimental conditions: #represents significance of each condition with respect to the control, and *represents the significance between the basal and stimulated cells in each condition. Data are mean ± SEM of control AZs (at rest, n = 22), stimulated control AZs (n = 27, *p = 0.0223, Mann–Whitney), KT 30 min AZs at rest (n = 43), KT 30 min stimulated AZs (n = 55, *p < 0.0001, #p = 0.0162, Mann–Whitney), KT 24 h AZs at rest (n = 26), KT 24 h stimulated AZs (n = 38, *p < 0.0001, #p = 0.0321, Mann–Whitney), cGKII KD AZs at rest (n = 31), and cGKII KD-stimulated AZs (n = 27, *p < 0.0001, #p = 0.0077, Mann–Whitney). D, sSVs/AZ in AZs with an organelles >40 nm/organelles <40 nm ratio above the average value: *represents the significance between the basal and stimulated cells in each condition. At rest: n = 10, control; n = 12, KT 30 min; n = 10, KT 24 h; n = 10, cGKII KD. Stimulated: n = 9, control (p = 0.12, Mann–Whitney); n = 12, KT 30 min (p = 0.09, Mann–Whitney); n = 6, KT 24 h (p = 0.004, Mann–Whitney); and n = 8, cGKII KD (p = 0.0029, Mann–Whitney).
When cells were stimulated there were more AZs that contained large organelles (Fig. 5B) and these organelles were more numerous, reflected in the increased organelles >40 nm/organelles <40 nm ratio (Fig. 5C). In control cells, 23 of 27 AZs contained large organelles, 3.15 ± 0.40 on average, which corresponded to an organelles >40 nm/organelles <40 nm ratio of 0.087 ± 0.014. The number of large organelles and their proportion with respect to sSVs increased further in cGKII KD cells, almost doubling (0.261 ± 0.062, p = 0.0077, M-W), and in cells incubated with KT5823 for 30 min (0.213 ± 0.063, p = 0.0162, M–W) or 24 h (0.230 ± 0.076, p = 0.0321, M–W). These results are consistent with the coexistence of clathrin-dependent bulk endocytosis following strong and sustained stimulation, and with the fact that cGKII KD or inhibition favors the formation of endosome-like structures. In stimulated AZs with an endosome/sSV ratio equal or above the average value in each experimental condition, the number of sSVs was lower than that observed in resting conditions, especially in cGKII KD cells or those exposed to KT5823 for 24 h AZs (Fig. 5D). These data further emphasize that endocytosis mediated by large structures is ineffective in maintaining the pool of small SVs, consistent with the dramatic decrease in FM1-43 unloading under these conditions.
Prolonged cGK inhibition increases the mobilizable and total vesicular pools, and slows endocytosis following strong and sustained stimulation
The formation of endosome-like structures is associated with the exocytosis of more SVs, and hence their increase in cGKII KD cells, or when this enzyme is inhibited, might be associated with the exocytosis of more SVs. To assess this possibility, the size of the mobilizable (recycling) pool was monitored in cells transfected with vGlut1-pHluorin by measuring the increase in fluorescence (Voglmaier et al., 2006) following KCl depolarization when the interior of the acidified vesicle comes in contact with the neutral pH of the external medium in the presence of bafilomycin. As a measure of the total population of fluorescently labeled SVs, KCl depolarization was followed by ammonium chloride application to produce vesicle deacidification without exocytosis. We found that the time necessary to reach half-maximal exocytosis was lower in cells exposed to KT5823 for 24 h (τcontrol = 5.74 ± 0.19 s; τKT5823 = 3.97 ± 0.17 s; p < 0.00001), and although there were no significant differences in the relative size of the mobilizable pool between control and KT5823-treated cells (Fig. 6A), the absolute values of fluorescence indicate that the size of both the mobilizable and total pools was larger in KT5823-treated cells (Fig. 6B).
Figure 6.
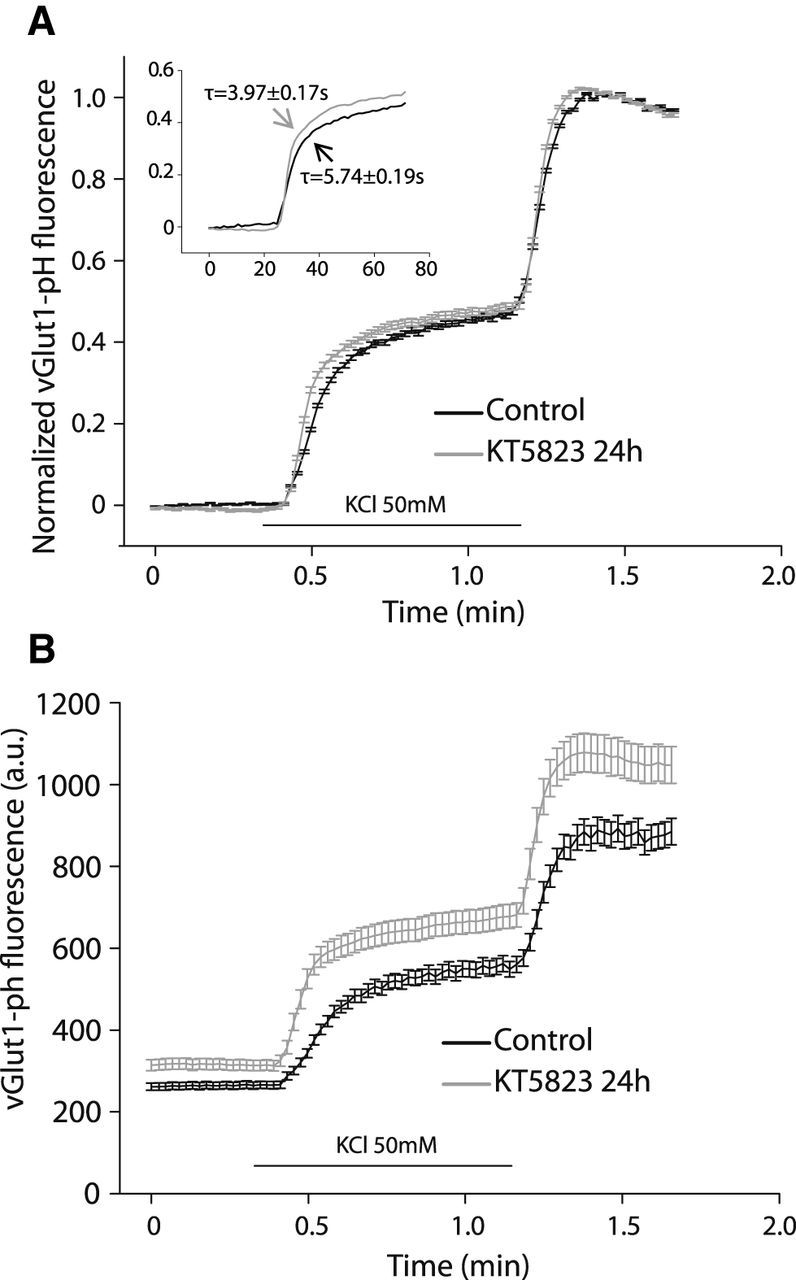
The recycling and resting pool sizes are modified by cGK inhibition. A, Mean traces of 50 mm KCl (50 s) elicited vGlut1-pHluorin fluorescence in the presence of bafilomycin normalized to 50 mm NH4Cl-elicited fluorescence (maximal fluorescence) in control (n = 11 coverslips from 4 different cultures) and KT5823-treated neurons (n = 6 coverslips from 4 different cultures). Inset, Kinetics of the KCl-elicited fluorescence increase in the presence of bafilomycin, indicating the τ value (time to reach half-maximal fluorescence). B, Mean traces of vGlut1-pHluorin fluorescence in the presence of bafilomycin (absolute values, a.u.) in control (n = 11 coverslips from 4 different cultures) and KT5823-treated neurons (n = 6 coverslips from 4 different cultures) showing the membrane fluorescence before stimulation, as well as the increase elicited by 50 mm KCl (50 s) and NH4Cl perfusion.
Two additional experimental approaches were used to analyze how these boutons recycled their membranes. First, we used the protocol described by Ryan et al. (1993), in which vesicle recycling is triggered by a depolarization and FM1-43 is added after distinct periods (Δt; Fig. 7A). Vesicles that undergo endocytosis during the delay period escape labeling, whereas vesicles that are endocytosed after Δt incorporate the dye. Under these experimental conditions, cell pretreated with KT5823 showed greater FM1-43 incorporation than the controls at most of the times analyzed (Fig. 7B,C), suggesting that the endocytosis takes place at longer times after exocytosis ceased when cGK is inhibited.
Figure 7.

cGK inhibition by KT5823 treatment slows down endocytosis. A, Experimental protocol: cells were stimulated with 50 mm KCl for 1 min and the progression of endocytosis was assessed by applying the dye after a variable delay (delay time, Δt). As Δt is prolonged, more endocytosis occurs during this period, reducing the amount of dye subsequently taken up. Dye uptake was quantified by measuring release signals evoked by later KCl stimuli. B, Ensemble averages of measurements of FM l-43 labeled with the fluorescence intensity (a.u.) shown for both control and KT5823-treated cells. C, Ensemble averages of measurements of the FM l-43-labeled showing synaptic vesicle pool size normalized to Δt = 0. The number of individual boutons measured for each condition, where: Δt = 0 s (N = 1105 control, N = 1246 KT 24 h), Δt = 30 s (N = 1816 control, N = 1660 KT 24 h), Δt = 60 s (N = 1200 control, N = 2250 KT 24 h), Δt = 90 s (N = 1216 control, N = 2324 KT 24 h), and Δt = 120 s (N = 893 control, N = 1606 KT 24 h). Data were obtained from n = 3 coverslips obtained from two different cell cultures.
In a second approach, we used cells transfected with vGlut1-pHluorin and stimulated in the absence of bafilomycin. In these conditions the individual responses of the boutons analyzed were quite heterogeneous but they could be grouped into four profiles, although the distribution of the responses into the four profiles was slightly different in the control cells and those exposed to KT5823 for 24 h (Fig. 8A). In more than half of the boutons in control cells or a third of those in KT5823-treated cells, the rate of endocytosis was higher than the rate of exocytosis for a brief period after the first exocytotic step. This disequilibrium was evident through the fluorescent decay, until a steady-state plateau was reached that was maintained for a few seconds after the end of the stimulus (Fig. 8B). The time constants (τ) to reach the steady-state were similar in both the control and experimental cells (Table 1), and this steady-state was the consequence of averaging the behavior of more than one hundred boutons (222 in control and 134 in KT5823-treated cells), given that the transitory increases in fluorescence observed in the individual responses indicate that new exocytotic events took place (results not shown). The fluorescence decayed to basal levels faster in control cells and it decayed below basal levels in the cells exposed to KT5823 (Table 1). The inhibition of cGK diminished the percentage of boutons that exhibited this profile, whereas increasing the proportion of boutons with a slower recovery of basal fluorescence that could be adjusted to a single-phase decay (Profile 2; Fig. 8C), a similar time constant being displayed in both control and experimental conditions (Table 1). There was also a trend (although not significant) toward an increase in the proportion of boutons that recovered the basal fluorescence in the presence of the stimulus and that decayed below basal levels after the end of the stimulus when exposed to KT5823 (Profile 3; Fig. 8D). These boutons recovered slowly when exposed to KT5823 (Table 1). Finally, there was a small proportion of boutons (Profile 4; Fig. 8E) in which the fluorescence increased rapidly and remained high while the stimulus persisted, subsequently decaying but without reaching basal levels within the next 30 s.
Figure 8.

Heterogeneous profiles for vGlut1-pHluorin fluorescence recovery after strong and sustained stimulation (50 s, 50 mm KCl) in control cells and those exposed to KT5823 for 24 h. A, Proportions of the four types of profiles found in control (n = 10 coverslips from 4 cultures) and KT5823-treated cells (n = 8 coverslips from 4 cultures). B, Profile 1: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 222 boutons) and KT5823-treated neurons (N = 134 boutons). C, Profile 2: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 116 boutons) and KT5823-treated neurons (N = 168 boutons). D, Profile 3: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 60 boutons) and KT5823-treated neurons (N = 96 boutons). E, Profile 4: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 21 boutons) and KT5823-treated neurons (N = 14 boutons).
Table 1.
Time constants for the exocytosis and endocytosis associated with the different response profiles after strong and sustained stimulation
| Profiles | Time to reach half-maximal response (τ, s) |
Time recovery constants τ1 and τ2 (s) |
||||
|---|---|---|---|---|---|---|
| Control | KT5823 | p | Control | KT5823 | p | |
| 1 | 6.41 ± 0.10 | 5.99 ± 0.41 | >0.05 | 13.50 ± 1.71 | 10.24 ± 0.69 | 0.113492 |
| 9.32 ± 0.76 | 19.71 ± 4.34* | <0.00001 | ||||
| 2 | 6.06 ± 0.05 | 6.27 ± 0.13 | >0.05 | 35.39 ± 1.41 | 37.62 ± 1.38 | 0.261021 |
| 3 | 6.73 ± 0.20 | 5.92 ± 0.18 | >0.05 | 14.57 ± 0.46 | 19.07 ± 0.72* | <0.00001 |
| 4 | 4.93 ± 0.10 | 4.50 ± 0.19 | >0.05 | 32.91 ± 6.62 | 37.77 ± 6.59 | 0.610233 |
Cells were stimulated with 50 mm KCl for 50 s and the increase in fluorescence was adjusted to (F) = Fmax*t/(τ + t), where τ is the time necessary to reach half-maximal fluorescence. The fluorescence decay was adjusted to F = F0+Ae−t/τ, where τ is the time constant for the basal recovery. Data are mean ± SEM from individual experiments control (control, n = 10; KT-5823-treated cells, n = 8).
* represents the significance between the control and the KT5823-treated cells.
When the stimulus was reduced to 15 s and the mean of all the responding boutons was analyzed, the rate of fluorescence recovery was faster following exposure to KT5823 (τ = 7.99 ± 0.14 vs 12.60 ± 0.4 s in control cells, p = 0.0195; Fig. 9A). KT5823 also altered the distribution of the heterogeneous responses into the different profiles (Fig. 9B). These responses were grouped into five different profiles (Fig. 9C–G; Table 2) and the τ value was significantly reduced by KT5823 in most of these. These findings suggest that long lasting inhibition of cGK only impairs vesicle cycling under sustained stimulation and that it augments the proportion of boutons with a slow rate of membrane retrieval, which is compatible with endosomal-mediated endocytosis under strong and sustained stimulation.
Figure 9.
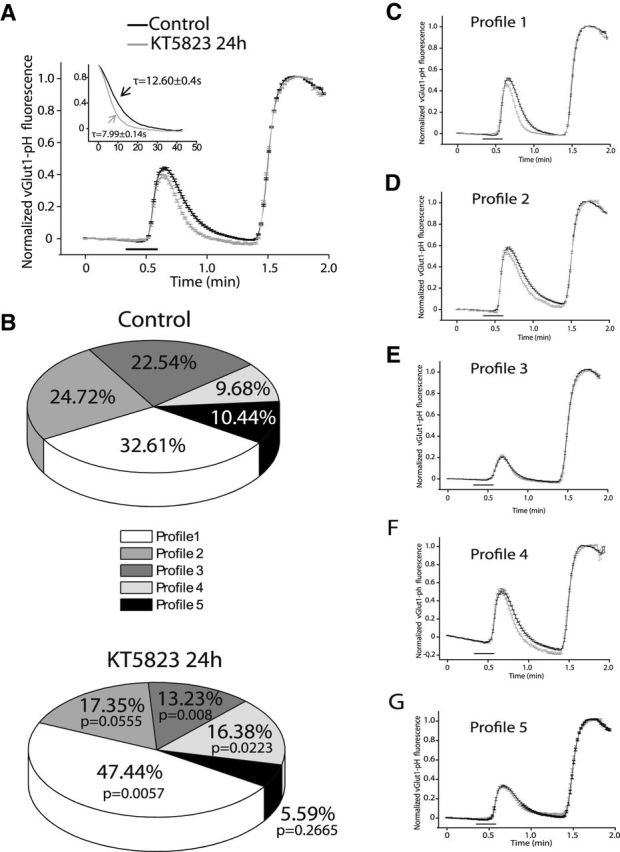
Heterogeneous profiles for vGlut1-pHluorin fluorescence recovery after strong stimulation (15 s, 50 mm KCl) in control cells and those exposed to KT5823 for 24 h. A, Mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence for the total number of boutons analyzed in control (N = 646 boutons, n = 10 coverslips from 4 different cultures) and KT5823-treated neurons (N = 362 boutons, n = 8 coverslips from 4 different cultures). Inset: Normalized fluorescence recovery adjusted to monoexponential decay, τ (time constant for decay in seconds). B, Percentages of the five types of profiles found in control (n = 10 coverslips) and KT-treated cells (n = 8 coverslips). C, Profile 1: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 214 boutons) and KT5823-treated neurons (N = 165 boutons). D, Profile 2: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 180 boutons) and KT5823-treated neurons (N = 62 boutons). E, Profile 3: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 128 boutons) and KT5823-treated neurons (N = 46 boutons). F, Profile 4: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 57 boutons) and KT5823-treated neurons (N = 56 boutons). G, Profile 5: mean traces of KCl-elicited vGlut1-pHluorin fluorescence normalized to NH4Cl-elicited fluorescence in control (N = 67 boutons) and KT5823-treated neurons (N = 33 boutons).
Table 2.
Time constants for the exocytosis and endocytosis associated with the different response profiles after brief stimulation
| Profiles | Time to reach half-maximal response (τ, s) |
Time recovery constant (τ, s) |
||||
|---|---|---|---|---|---|---|
| Control | KT5823 | p | Control | KT5823 | p | |
| 1 | 3.76 ± 0.07 | 4.19 ± 0.08 | >0.05 | 12.30 ± 0.83 | 7.45 ± 0.44* | <0.00001 |
| 2 | 4.75 ± 0.04 | 4.42 ± 0.04 | >0.05 | 19.20 ± 1.10 | 14.69 ± 0.68* | 0.027342 |
| 3 | 6.08 ± 0.14 | 5.77 ± 0.09 | >0.05 | 10.95 ± 1.04 | 9.42 ± 0.76 | 0.306624 |
| 4 | 4.07 ± 0.15 | 2.24 ± 0.73 | >0.05 | 13.29 ± 0.41 | 11.01 ± 0.27* | <0.00001 |
| 5 | 3.63 ± 0.17 | 3.60 ± 0.07 | >0.05 | 15.07 ± 0.89 | 10.58 ± 0.63* | 0.000855 |
The cells were stimulated with 50 mm KCl for 10 s and the increase in fluorescence was adjusted to (F) = Fmax*t/(τ + t), where τ is the time necessary to reach half-maximal fluorescence. The fluorescence decay was adjusted to F = F0+Ae−t/τ, where τ is the time constant for the basal recovery. Data are mean ± SEM from individual experiments control (control, n = 10; KT-5823-treated cells, n = 8).
* represents significance between the control and the KT5823-treated cells.
Discussion
We previously reported certain heterogeneity in the efficiency of vesicle recycling in synaptic boutons on cerebellar granule cells after strong and sustained stimulation (Bartolomé-Martín et al., 2012), and a slowing down of vesicle recycling as a consequence of sGC inhibition (López-Jiménez et al., 2012). Indeed, when cGMP synthesis was inhibited, more boutons exhibited slower kinetics and less dye loss, suggesting that most of the loaded vesicles do not complete the exoendocytosis cycle within the time studied (López-Jiménez et al., 2009). Here, we identified the upstream influence of NMDARs and that of cGKII downstream, both functional components of the cGMP signaling pathway that regulates vesicle recycling following the extensive exocytosis elicited by a strong and sustained stimulus. Blockage of NMDARs impairs SV recycling, which was overcome by the addition of exogenous cGMP, indicating that the intrinsic activity of activated NMDARs and cGMP synthesis regulates SV recycling (López-Jiménez et al., 2009), probably via NO formation as demonstrated previously (Jurado et al., 2003). Similar effects on SVs are produced by a cGK inhibitor or when the expression of cGKII is silenced, suggesting that the activation of this kinase accelerates vesicular cycling under our experimental conditions. Although the effects were similar, the magnitude seems to be stronger when cGKII was knocked down, which could reflect differences in the strength or reversibility of the suppression/inhibition of cGKII activity. KT5823 is an ATP-competitive and reversible inhibitor of cGK, such that the degree of inhibition greatly depends on its concentration and the concentration of ATP. Indeed, the effects of this compound may diminish over time (as its concentration falls) as it was added to the culture media at 1 μm only once and the effects provoked were analyzed after 30 min, 24 h, or 96 h.
Although SV recycling involves the fine tuning of sequential steps, the results obtained here through different experimental approaches seem to indicate that cGKII activity is important for efficient endocytosis during strong and sustained stimulation. On the one hand, FM1-43 unloading was only impaired when cGK was inhibited before the loading step, whereas inhibition of cGK after loading, and when endocytosis had finished, only had a minimal effect on unloading. On the other hand, the endocytotic rate after a strong and sustained stimulus was slower when cGK was inhibited. These results are consistent with observations at synapses in hippocampal cultures (Micheva et al., 2003), and at the calyx of Held synapses in rodent brainstem slices (Eguchi et al., 2012; Taoufiq et al., 2013), where retrograde activation of cGK by the NO released from postsynaptic cells accelerates vesicle endocytosis, strengthening exoendocytic coupling at high-frequency stimulation. In these studies, cGK activation increased the amount PIP2 and favored CME after high-frequency stimulation (Micheva et al., 2003; Eguchi et al., 2012). Interestingly, the slowing of endocytosis due to cGK inhibition in cerebellar granule cells was only observed under strong and sustained stimulation, suggesting that cGK activation favors CME and probably increases PIP2 levels, as occurs in other synapses (Meijering et al., 2004; Eguchi et al., 2012). Increased PIP2 levels may favor endocytosis by recruiting AP2 to the membrane, subsequently promoting clathrin-coated vesicle formation (Slepnev and De Camilli, 2000).
It is accepted that although CME is the dominant mode of SV retrieval during mild stimulation (Granseth et al., 2006), ADBE is the dominant mode during intense stimulation (Clayton et al., 2008; Cheung and Cousin, 2012). These two forms of SV retrieval can coexist in the same synaptic terminal, although their contributions may differ from one bouton to the next (Voglmaier et al., 2006; Bartolomé-Martín et al., 2012), and competition between them can regulate the rate and extent of SV protein recycling (Voglmaier et al., 2006; Foss et al., 2013). The lowering of PIP2, and thus an impairment of CME, might favor recycling through the endosomal-mediated pathway. Alternatively, the formation of endosomes has been linked to large-scale exocytosis (Clayton et al., 2008; Cheung and Cousin, 2012) and to the immaturity of the presynaptic endocytotic machinery (Shetty et al., 2013). These two situations could explain the existence of endosomal-like structures in stimulated cells, given that the cells used in this study (7 or 8 DIV) are not fully mature and 3 min stimulation with 50 mm KCl produces large-scale exocytosis. Indeed, more endosomal-like structures were evident after inhibition or knockdown of cGKII, suggesting a stronger contribution to endosomal recycling that could be at least partially attributed to an increase in exocytosed vesicles because the recycling or the total pool sizes were larger in KT5823-treated cells, even though no differences in the relative size were observed. The increases in the total vesicle pool caused by prolonged exposure to KT5823 or by cGKII KD were also observed at the ultrastructural level. Moreover, enhanced endosomal recycling might be a consequence of impaired CME, possibly due to a reduction in PIP2 levels after inhibition or knockdown of cGKII, as discussed previously.
Although cGK was previously shown to regulate the rate of endocytosis, the nature of this protein was not identified (Micheva et al., 2003; Eguchi et al., 2012; Taoufiq et al., 2013). Two cGK isoforms (cGKI and cGKII) have been identified in mammalian tissue (Butt et al., 1993; Francis and Corbin, 1994; Vaandrager et al., 1996; Vaandrager et al., 1997) and both are expressed in cerebellar granule cells (Jurado et al., 2004, 2005). Myristoylation of Gly-2 after removal of the N-terminal methionine residue anchors cGKII to the plasma membrane (Vaandrager et al., 1996), two events that are required for it to phosphorylate several proteins. Given this subcellular localization of cGKII, it is understandable that many of the processes regulated by it occur in membrane domains. In neurons, cGKII is mainly located in the neuropil (de Vente et al., 2001), suggesting that it may play an important role in synaptic transmission. We have shown here that in cerebellar granule cells, cGKII activity at presynapses is necessary to maintain efficient SV recycling under strong and persistent stimulation, affecting the rate of endocytosis. Indeed, both cGKII knockdown and pharmacological inhibition with KT5823 (which inhibits both cGK isoforms) impair recycling, and no additional effects were observed when the two were combined. On the other hand, it has also been shown previously that cGKII phosphorylates GluA1 at S845 after NMDAR stimulation, augmenting the surface expression of AMPARs at both synaptic and extrasynaptic sites in such cells (Incontro et al., 2013) and in hippocampal neurons (Serulle et al., 2007), indicative of an important role for this kinase at the postsynapse. Thus, as shown previously at hippocampal synapses (Wang et al., 2005) or at rat lateral amygdale synapses following fear conditioning (Ota et al., 2010), the NO-cGMP pathway may serve to coordinate presynaptic and postsynaptic events in cerebellar granular cells in response to a strong and sustained stimulus, with cGKII representing a key protein in these mechanisms.
In conclusion, we have identified novel physiological functions for cGKII, which positively regulates synaptic vesicle endocytosis in cultured cerebellar granule cells. While further work is required to define the molecular mechanisms that may underlie this activity, our data establish cGKII as an important element in maintaining high-fidelity synaptic transmission under strong and sustained stimulation.
Footnotes
This work was supported by Grants from the Ministerio de Economía y Competitividad (M.T.; BFU2012-32105) and the Comunidad de Madrid (J.S.-P. and M.T.; S2011/BMD-2349). A.C.-A. was supported by the Ministerio de Economía y Competitividad and J.R.-F. was supported by the Comunidad de Madrid. The vGlut1-pH was a generous gift from Rob Edwards and Susan Voglamaier (University of California, San Francisco, CA). We thank Dr. M. Sefton for editorial assistance, Agustín Fernández and Marisa García from the electron microscopy facility (Universidad Complutense de Madrid), and Ma Carmen Zamora for technical assistance.
The authors declare no competing financial interests.
References
- Atluri PP, Ryan TA. The kinetics of synaptic vesicle reacidification at hippocampal nerve terminals. J Neurosci. 2006;26:2313–2320. doi: 10.1523/JNEUROSCI.4425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomé-Martín D, Ramírez-Franco J, Castro E, Sánchez-Prieto J, Torres M. Efficient synaptic vesicle recycling after intense exocytosis concomitant with the accumulation of non-releasable endosomes at early developmental stages. J Cell Sci. 2012;125:422–434. doi: 10.1242/jcs.090878. [DOI] [PubMed] [Google Scholar]
- Bergsman JB, Krueger SR, Fitzsimonds RM. Automated criteria-based selection and analysis of fluorescent synaptic puncta. J Neurosci Methods. 2006;152:32–39. doi: 10.1016/j.jneumeth.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Butt E, Geiger J, Jarchau T, Lohmann SM, Walter U. The cGMP-dependent protein kinase: gene, protein, and function. Neurochem Res. 1993;18:27–42. doi: 10.1007/BF00966920. [DOI] [PubMed] [Google Scholar]
- Cheung G, Cousin MA. Adaptor protein complexes 1 and 3 are essential for generation of synaptic vesicles from activity-dependent bulk endosomes. J Neurosci. 2012;32:6014–6023. doi: 10.1523/JNEUROSCI.6305-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, Evans GJ, Cousin MA. Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J Neurosci. 2008;28:6627–6632. doi: 10.1523/JNEUROSCI.1445-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Tan TC, Robinson PJ. Protein phosphorylation is required for endocytosis in nerve terminals: potential role for the dephosphins dynamin I and synaptojanin, but not AP180 or amphiphysin. J Neurochem. 2001;76:105–116. doi: 10.1046/j.1471-4159.2001.00049.x. [DOI] [PubMed] [Google Scholar]
- Couteaux R, Pécot-Dechavassine M. Synaptic vesicles and pouches at the level of “active zones” of the neuromuscular junction (in French) C R Acad Sci Hebd Seances Acad Sci D. 1970;271:2346–2349. [PubMed] [Google Scholar]
- de Vente J, Asan E, Gambaryan S, Markerink-van Ittersum M, Axer H, Gallatz K, Lohmann SM, Palkovits M. Localization of cGMP-dependent protein kinase type II in rat brain. Neuroscience. 2001;108:27–49. doi: 10.1016/S0306-4522(01)00401-8. [DOI] [PubMed] [Google Scholar]
- Dittman J, Ryan TA. Molecular circuitry of endocytosis at nerve terminals. Annu Rev Cell Dev Biol. 2009;25:133–160. doi: 10.1146/annurev.cellbio.042308.113302. [DOI] [PubMed] [Google Scholar]
- Eguchi K, Nakanishi S, Takagi H, Taoufiq Z, Takahashi T. Maturation of a PKG-dependent retrograde mechanism for exoendocytic coupling of synaptic vesicles. Neuron. 2012;74:517–529. doi: 10.1016/j.neuron.2012.03.028. [DOI] [PubMed] [Google Scholar]
- Foss SM, Li H, Santos MS, Edwards RH, Voglmaier SM. Multiple dileucine-like motifs direct VGLUT1 trafficking. J Neurosci. 2013;33:10647–10660. doi: 10.1523/JNEUROSCI.5662-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Corbin JD. Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol. 1994;56:237–272. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Incontro S, Ciruela F, Ziff E, Hofmann F, Sánchez-Prieto J, Torres M. The type II cGMP dependent protein kinase regulates GluA1 levels at the plasma membrane of developing cerebellar granule cells. Biochim Biophys Acta. 2013;1833:1820–1831. doi: 10.1016/j.bbamcr.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Sanchez-Prieto J, Torres M. Differential expression of NO-sensitive guanylyl cyclase subunits during the development of rat cerebellar granule cells: regulation via N-methyl-d-aspartate receptors. J Cell Sci. 2003;116:3165–3175. doi: 10.1242/jcs.00620. [DOI] [PubMed] [Google Scholar]
- Jurado S, Sánchez-Prieto J, Torres M. Elements of the nitric oxide/cGMP pathway expressed in cerebellar granule cells: biochemical and functional characterisation. Neurochem Int. 2004;45:833–843. doi: 10.1016/j.neuint.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Jurado S, Sánchez-Prieto J, Torres M. Expression of cGMP-dependent protein kinases (I and II) and neuronal nitric oxide synthase in the developing rat cerebellum. Brain Res Bull. 2005;65:111–115. doi: 10.1016/j.brainresbull.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Kavalali ET. Synaptic vesicle reuse and its implications. Neuroscientist. 2006;12:57–66. doi: 10.1177/1073858405281852. [DOI] [PubMed] [Google Scholar]
- Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- López-Jiménez ME, Bartolomé-Martin D, Sánchez-Prieto J, Torres M. Suppression of guanylyl cyclase (beta1 subunit) expression impairs neurite outgrowth and synapse maturation in cultured cerebellar granule cells. Cell Death Differ. 2009;16:1266–1278. doi: 10.1038/cdd.2009.57. [DOI] [PubMed] [Google Scholar]
- López-Jiménez ME, González JC, Lizasoain I, Sánchez-Prieto J, Hernández-Guijo JM, Torres M. Functional cGMP-gated channels in cerebellar granule cells. J Cell Physiol. 2012;227:2252–2263. doi: 10.1002/jcp.22964. [DOI] [PubMed] [Google Scholar]
- Mayer B, Klatt P, Böhme E, Schmidt K. Regulation of neuronal nitric oxide and cyclic GMP formation by Ca2+ J Neurochem. 1992;59:2024–2029. doi: 10.1111/j.1471-4159.1992.tb10090.x. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Buchanan J, Holz RW, Smith SJ. Retrograde regulation of synaptic vesicle endocytosis and recycling. Nat Neurosci. 2003;6:925–932. doi: 10.1038/nn1114. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KT, Monsey MS, Wu MS, Schafe GE. Synaptic plasticity and NO-cGMP-PKG signaling regulate pre- and postsynaptic alterations at rat lateral amygdala synapses following fear conditioning. PLoS One. 2010;5:e11236. doi: 10.1371/journal.pone.0011236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda K, Ghosh S, Stuehr DJ. Calmodulin activates intersubunit electron transfer in the neuronal nitric-oxide synthase dimer. J Biol Chem. 2001;276:23349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- Petrov AM, Giniatullin AR, Sitdikova GF, Zefirov AL. The role of cGMP-dependent signaling pathway in synaptic vesicle cycle at the frog motor nerve terminals. J Neurosci. 2008;28:13216–13222. doi: 10.1523/JNEUROSCI.2947-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Rentería ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/S0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Ratnayaka A, Marra V, Bush D, Burden JJ, Branco T, Staras K. Recruitment of resting vesicles into recycling pools supports NMDA receptor-dependent synaptic potentiation in cultured hippocampal neurons. J Physiol. 2012;590:1585–1597. doi: 10.1113/jphysiol.2011.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Sytnyk V, Leshchyns'ka I, Puchkov D, Haucke V, Schachner M. The neural cell adhesion molecule promotes maturation of the presynaptic endocytotic machinery by switching synaptic vesicle recycling from adaptor protein 3 (AP-3)- to AP-2-dependent mechanisms. J Neurosci. 2013;33:16828–16845. doi: 10.1523/JNEUROSCI.2192-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepnev VI, De Camilli P. Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat Rev Neurosci. 2000;1:161–172. doi: 10.1038/35044540. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Taoufiq Z, Eguchi K, Takahashi T. Rho-kinase accelerates synaptic vesicle endocytosis by linking cyclic GMP-dependent protein kinase activity to phosphatidylinositol-4,5-bisphosphate synthesis. J Neurosci. 2013;33:12099–12104. doi: 10.1523/JNEUROSCI.0730-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegenge MA, Stern M, Bicker G. Nitric oxide and cyclic nucleotide signal transduction modulates synaptic vesicle turnover in human model neurons. J Neurochem. 2009;111:1434–1446. doi: 10.1111/j.1471-4159.2009.06421.x. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, Ehlert EM, Jarchau T, Lohmann SM, de Jonge HR. N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J Biol Chem. 1996;271:7025–7029. doi: 10.1074/jbc.271.12.7025. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, Edixhoven M, Bot AG, Kroos MA, Jarchau T, Lohmann S, Genieser HG, de Jonge HR. Endogenous type II cGMP-dependent protein kinase exists as a dimer in membranes and can be functionally distinguished from the type I isoforms. J Biol Chem. 1997;272:11816–11823. doi: 10.1074/jbc.272.18.11816. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, Walter U, Lohmann SM, Hawkins RD, Antonova I. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron. 2005;45:389–403. doi: 10.1016/j.neuron.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Wenk MR, De Camilli P. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc Natl Acad Sci U S A. 2004;101:8262–8269. doi: 10.1073/pnas.0401874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Milburn PJ, Hanna BT, Graham ME, Rostas JA, Robinson PJ. Phosphorylation of septin 3 on Ser-91 by cGMP-dependent protein kinase-I in nerve terminals. Biochem J. 2004;381:753–760. doi: 10.1042/BJ20040455. [DOI] [PMC free article] [PubMed] [Google Scholar]