

Abstract
Systemic inflammatory reactions have been postulated to exacerbate neurodegenerative diseases via microglial activation. We now demonstrate in vivo that repeated systemic challenge of mice over four consecutive days with bacterial LPS maintained an elevated microglial inflammatory phenotype and induced loss of dopaminergic neurons in the substantia nigra. The same total cumulative LPS dose given within a single application did not induce neurodegeneration. Whole-genome transcriptome analysis of the brain demonstrated that repeated systemic LPS application induced an activation pattern involving the classical complement system and its associated phagosome pathway. Loss of dopaminergic neurons induced by repeated systemic LPS application was rescued in complement C3-deficient mice, confirming the involvement of the complement system in neurodegeneration. Our data demonstrate that a phagosomal inflammatory response of microglia is leading to complement-mediated loss of dopaminergic neurons.
Keywords: complement, microglia, neurodegeneration, neuroinflammation, phagosome, transcriptome
Introduction
Bacterial infections leading to systemic inflammation are commonly observed in elderly patients with neurodegenerative diseases and are often associated with exacerbation of clinical symptoms (Perry et al., 2007; Wyss-Coray and Rogers, 2012). Systemic inflammation is known to activate microglia within the CNS, and it was postulated that the activated microglial cells contribute to the progression of neurodegenerative diseases (Perry et al., 2007; Glass et al., 2010; Hirsch et al., 2012).
Systemic intraperitoneal application of bacterial lipopolysaccharides (LPS) in single or repeated challenges in experimental animal models can induce or even exacerbate neurodegeneration (Dutta et al., 2008; Cunningham, 2013). In an animal model of prion disease, it was shown that systemic application of a single dose of 100 ng LPS per gram body weight (gbw) communicated to the brain and induced behavioral changes, microglial activation, and local production of inflammatory cytokines that contributed to neurodegeneration (Cunningham et al., 2009). Furthermore, mice receiving a single systemic application of LPS (5 μg/gbw, i.p.) presented loss of dopaminergic tyrosine hydroxylase (TH)-positive neurons in the substantia nigra (SN) 7 months after injection (Qin et al., 2007). In parkin-deficient mice challenged repeatedly twice a week with 750 EU/gbw LPS intraperitoneally, loss of TH-positive neurons in the SN was already observed after 3 months, whereas loss of TH-positive neurons in control treated parkin-deficient mice was detected only after 6 months (Frank-Cannon et al., 2008). Thus, all these studies showed that the dopaminergic neurons in the SN are susceptible to inflammatory damage after systemic LPS exposure, but the exact molecular mechanism of neurodegeneration is unclear.
Here, we compared in mice the effects of two systemic LPS treatment paradigms: a single and a repeated application. We observed that the repeated LPS treatment induced a distinct prolonged inflammatory microglial phenotype with an enrichment of the complement–phagosome pathway that triggered degeneration of dopaminergic neurons in the SN. Loss of dopaminergic neurons induced by repeated systemic LPS challenges was prevented in complement C3-deficient mice.
Materials and Methods
Experimental animals and treatments.
All animal experiments have been approved by the authors' institutional review boards and by the local government and have been conducted according to the principles expressed in the Helsinki Declaration. C57BL/6J mice and B6;129S4-C3tm1Crr/J mice, backcrossed for at least 8 generations into C57BL/6 (referred as C3 KO in the text) were obtained from The Jackson Laboratory via Charles River and housed in specific pathogen free environment with water and food ad libitum. For the experiments, 3-month-old male mice were injected intraperitoneally with 100 μl LPS (concentrations accordingly to the text; Enzo LifeSciences) or with 100 μl PBS (Invitrogen) vehicle as control. For the single treatment, mice were injected once with 4 μg LPS/gbw or vehicle and killed on the second, fifth, or 19th day after the start of the treatment, according to the text. For the repeated treatment, the protocol from Cardona et al. (2006) was adapted. It consisted of four daily injections with 1 μg/gbw LPS or vehicle, and the analysis was performed on the fifth or 19th day since the start of the treatment, according to the text.
ELISA analysis.
Plasma was isolated from aortic blood by centrifugation in heparin-containing tubes (Ratiopharm) to sediment the blood cells. To prepare brain supernatant, mice were intracardially perfused with 1× PBS, brains were isolated and homogenized in radioimmunoprecipitation assay buffer (RIPA buffer, Sigma) containing proteases and phosphatases inhibitor cocktails (Sigma). After incubation on ice for 15 min, the homogenates were centrifuged and brain supernatants were collected. Tumor necrosis factor α (TNFα) and interleukin 1 β (IL1β) protein expression levels in the plasma or brain were quantified by using corresponding mouse Quantikine ELISA Kits (R&D Systems) according to the manufacturer's instructions.
Real-time RT-PCR analysis.
RNA was isolated from brains of PBS transfused mice using the RNeasy Mini Kit (QIAGEN) according to the manufacturer′s protocol. Reverse transcription of 50 μg total RNA was performed with SuperScript III reverse transcriptase (Invitrogen) and hexamer random primers (Roche Molecular Biochemicals). Semiquantitative RT-PCR was performed using 1 μl cDNA diluted 1:10, SYBR GreenER qPCRSuperMix Universal (Invitrogen) and specific oligonucleotides into a final reaction volume of 25 μl. For amplifications, a Mastercycler epigradient S (Eppendorf) was used, and the results were evaluated with the manufacturer's software. Amplification specificity was confirmed by melting curve analysis, and the quantification was performed using the ΔΔCt method.
Immunohistochemistry.
Animals were perfused intracardially with 1× PBS followed by 4% PFA (Sigma); brains were collected and kept in 30% sucrose (Sigma) supplemented with 0.1% sodium azide (Sigma) until processed into frozen sections. Fixed brains were sectioned in coronal plane (20 μm extent in the rostal-caudal plane) and directly mounted onto superfrost slides. Ventral midbrain containing slides were blocked with 5% normal goat serum (Invitrogen) in PBS and incubated overnight at 4°C with the primary antibody. After washing with PBS, the slices were incubated for 2 h with the corresponding secondary antibodies (Cy3-conjugated antibody, 1:500, Dianova or Alexa-488-conjugated antibody, 1:500, Invitrogen). Mowiol (Sigma) was used as mounting medium, and the slices were kept at −20°C until pictures were taken. Stainings were visualized using AxioObserver Z1 inverted microscope with Apotome (Carl Zeiss), and images were collected with AxioCam Rm (AxioVision software). All images were analyzed using the ImageJ software. For neuronal quantification, slides were double-stained with antibodies directed against the dopaminergic neuronal marker TH (1:1000; Sigma) and with neuronal neuclei marker NeuN (1:500; Millipore). SN pars compacta (SNpc) was demarcated based on TH-positive immunostaining as previously described (Baquet et al., 2009). Optical sections of four SNpc matched levels per animal were used for cell counting (McCormack et al., 2002). The TH-positive neurons were only counted if the whole nucleus was visible within the section. SNpc neurons were distinguished from the ones of the ventral tegmental area based on size and orientation (Baquet et al., 2009). For microglia evaluation, TH immunostaining (1:1000; Millipore) was used for positioning on the slice, together with an antibody directed against the microglial marker ionized calcium binding adaptor molecule1 (Iba1; 1:1000; Wako). Iba1-positive cells were counted in the Substania nigra pars reticulata (SNpr) in the above mentioned section levels. For microglial activation, lysosomal CD68 was quantified in microglial cells from SNpr, which were double-labeled with Iba1 (1:1000, Wako) and CD68 (1:500, Serotec). Confocal z-stack images were acquired, and the area occupied by CD68-positive staining was analyzed using ImageJ software (National Institutes of Health). At least 6 images per animal were analyzed.
Microarray transcriptome analysis.
Mice were challenged with LPS or PBS according to the single or repeated treatment protocol. For the single-treated group, the analysis was conducted on experimental day 2 (24 h after the injection) and day 5; for the repeatedly challenged group, the analysis was performed on day 5 (24 h after the last injection). Brains were collected after PBS perfusion of mice and immediately homogenized in cooled down QIAzol reagent (QIAGEN). Total RNA was extracted according to QIAzol manufacturer's recommendations with −20°C overnight incubation for small RNA precipitation. Total RNA integrity and purity were assessed using the Agilent 2100 Bioanalyzer and RNA 6000 Nano LabChip kits (Agilent Technologies). Only good-quality RNA (no contamination or degradation, RIN > 9) was used and further processed. Total RNA samples were reverse-transcribed to double-stranded cDNA using specific primers, which reduce the priming of rRNA. cRNA was generated by in vitro transcription and reverse transcribed into a sense single-stranded cDNA. The cDNA was fragmented, labeled, and hybridized onto Affymetrix GeneChip Mouse Gene 1.0 ST Arrays according to the Ambion Whole Transcript Expression kit for Affymetrix GeneChip Whole Transcript Expression Array Protocol (P/N 4425209 Rev.B 05/2009) and GeneChip WT Terminal Labeling and Hybridization User Manual for use with the Ambion Whole Transcript Expression kit (P/N 702808 Rev.6). Microarrays were then washed, stained, and scanned according to the manufacturer's instructions. The 36 CEL files generated by the scanner were then imported into Partek Genomics Suite (GS) 6.4 for preprocessing and quality control. Preprocessing aims at estimating transcript cluster expression values from probe signal intensities. Thus, Partek options were set up for GC-content adjustment, robust multiarray background correction, quantile normalization, log2 transformation, and mean summarization. Quality control was assessed through different methods available in Partek GS and did not reveal any outlier. Transcript cluster expression values were then imported into R statistical environment (R Core Team, 2014R: A Language and Environment for Statistical Computing. R Found Stat Comput) for further analysis. Transcript clusters that do not target any gene were removed so that each transcript cluster is associated with a gene. Then, Limma (Smyth, 2004) (R/Bioconductor) was used to detect differentially expressed (DE) genes between LPS and PBS within each group (single/repeated, paradigm, D2/D5). Analysis was done according to “time course experiment,” and the p values were adjusted for multiple testing errors using the Benjamini and Hochberg' false discovery rate (FDR) (Benjamini and Hochberg, 1995). Genes with FDR < 0.05 and absolute log2FC ≥ 0.5 were considerate as DE. For Ingenuity Pathway Analysis (IPA), lists of significantly DE genes between (single or repeated) LPS treatment versus (single or repeated) PBS treatment were uploaded in the IPA tool (Ingenuity Systems, www.ingenuity.com). The significance of the association between each list and function or canonical pathway was measured by Fisher's exact test. As a result, a p value was obtained, determining the probability that the association between the genes in our dataset and function or canonical pathway can be explained by chance alone. Microarray expression data are available at ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-1770.
Statistical analysis.
Data are presented as mean ± SEM of at least three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni post hoc test comparing all columns using GraphPad computer software. Results are considered as significant if p < 0.05, p < 0.01, or p < 0.001.
Results
Loss of dopaminergic neurons in the SN after repeated systemic application of LPS
Dopaminergic neurons in the SN have been shown to be susceptible to systemically applied LPS, but the extent of dopaminergic neurodegeneration was strongly dependent on the dose and kinetics of application (Cunningham, 2013). Therefore, we first compared the effect of systemically applied LPS given to C57BL/6J mice either within a single intraperitoneal injection or repeated intraperitoneal injections of the same cumulative dose over four consecutive days (Fig. 1A). On experimental day 19 after the first application, immunohistochemistry of the SNpc was performed and histological sections were analyzed using antibodies directed against TH and NeuN (Fig. 1B). Although a single dose of LPS (1× 4 μg/gbw) did not induce a significant change of TH-positive or NeuN-positive cell number, repeated systemic application of LPS (4× 1 μg/gbw) triggered a reduction in the number of TH-positive and NeuN-positive neurons (Fig. 1C). The quantification of SNpc serial sections after repeated LPS (4× 1 μg/gbw) application showed a relative reduction of the number of TH-positive cell bodies from an average of 134.6 ± 6.24 to 80.28 ± 6.20 cells per section on experimental day 19 (p < 0.001), and the number of NeuN-positive cell bodies was decreased from 277.9 ± 14.30 to 178.4 ± 12.25 cells per section (p < 0.001; Fig. 1C).
Figure 1.

Loss of dopaminergic neurons in the SN after repeated systemic challenges with LPS. A, Treatment and analysis paradigms. For the single treatment (left), mice were injected intraperitoneally once with 4 μg LPS/gbw and were analyzed on day 19. For the repeated treatment (right), mice were injected intraperitoneally on four consecutive days with 1 μg LPS/gbw and were analyzed on day 19. B, Immunofluorescence staining of the SN with antibodies directed against the dopaminergic cell specific marker protein TH (green) and neuronal nuclei marker protein NeuN (red) on experimental day 19 after repeated treatment with PBS control or LPS. Reduced staining of TH was detected in the SNpc after repeated systemic LPS challenge. Higher-magnification inset as indicated. Scale bar, 50 μm. Representative images for n ≥ 3 mice shown. C, Quantification of TH-positive (left) and NeuN-positive cells (right) in the SNpc of mice on experimental day 19 after systemic single or repeated challenge with PBS control or LPS. Only systemic repeated LPS application induced significant loss of TH-positive and NeuN-positive neurons. n ≥ 3 mice. n.s., Not significant. *p < 0.05. **p < 0.01. ***p < 0.001.
Data show that a single LPS application had no obvious neurotoxic effect within 19 d, whereas four daily applications of the same cumulative dose induced loss of TH- and NeuN-positive neurons in SN.
Microglial activation after systemic LPS application
Detrimental changes in brain parenchyma are quickly sensed by microglial cells (Ransohoff and Perry, 2009). To assay the microglial response in the brain to peripheral application of LPS, we performed semiquantitative real-time PCR (sqRT-PCR) for gene transcripts of Aif1 (Allograft inflammatory factor 1, gene encoding microglial protein marker Iba1). We analyzed brain tissue on day 5 and day 19 after systemic single or repeated application of LPS or PBS vehicle control (Fig. 2A). On experimental day 5, the levels of Aif1 transcripts were slightly increased after single LPS application (from an mRNA fold change (FC) of 1.03 ± 0.15 in PBS controls to a FC of 1.66 ± 0.28) and ∼3-fold increased after repeated LPS challenges (from 1.06 ± 0.23 FC in PBS to 3.08 ± 0.18 FC, p < 0.001; Fig. 2B). However, on experimental day 19, the Aif1 mRNA levels were comparable in the brains of both groups (Fig. 2B).
Figure 2.
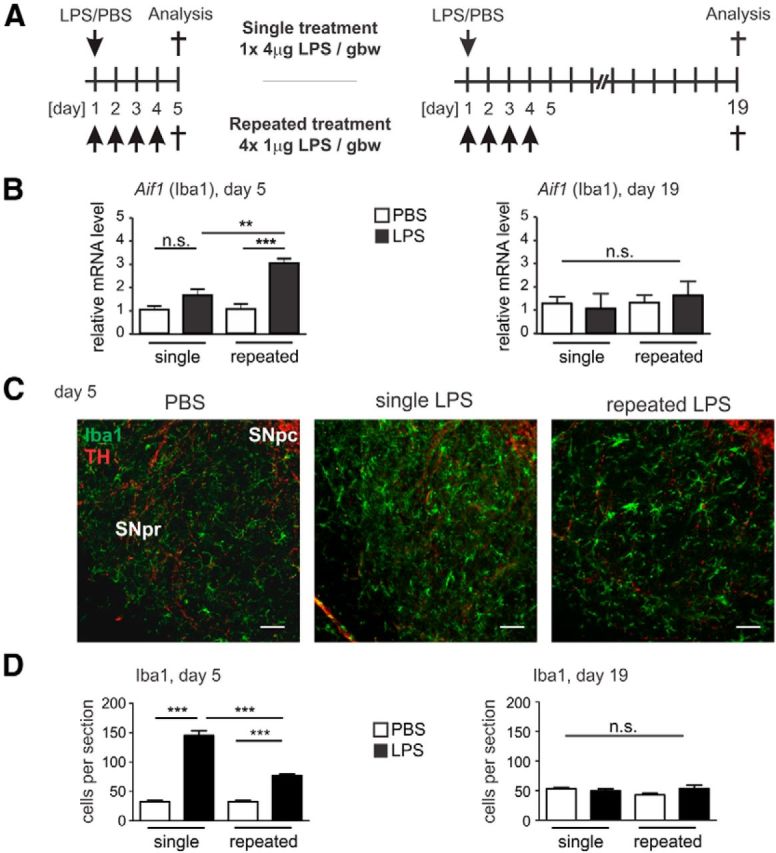
Prolonged microglial activation after repeated systemic challenge with LPS. A, Mice treatment and analysis paradigms. For the single treatment, mice were intraperitoneally injected once with 4 μg LPS/gbw and were analyzed on experimental day 5 or 19 (top). For the repeated treatment, mice were injected intraperitoneally on four consecutive days with 1 μg LPS/gbw and were analyzed on experimental day 5 or 19 (bottom). B, sqRT-PCR determination of Allograft inflammatory factor 1 (Aif1, the microglial marker ionized calcium binding adaptor molecule 1 [Iba1] encoding gene) from the whole brain of mice on experimental day 5 or 19 after single or repeated treatment with PBS control or LPS. Aif1 gene transcripts were increased on day 5 only after repeated application of LPS, and the transcript levels returned to the levels of PBS treatments control on day 19. n ≥ 3 mice. n.s., Not significant. **p < 0.01. ***p < 0.001. C, Immunofluorescence staining of the microglial marker Iba1 (green) in SNpr on experimental day 5 after single or repeated treatment with PBS control or LPS. TH immunostaining (red) was used to match the level of analysis. Microglial Iba1 immunoreactivity was highly increased after single and repeated LPS application. Scale bar, 50 μm. Representative images of n ≥ 3 mice shown. D, Quantification of Iba1 immunostaining in the SNpr sections of mice on experimental day 5 and 19 after single or repeated PBS control or LPS treatment. Microglial Iba1 immunoreactivity was increased on day 5 after both single and repeated systemic LPS challenge and returned to PBS control levels on day 19. n ≥ 3 mice. ***p < 0.001.
Next, we studied the microglial Iba1 immunoreactivity in the SNpr, a brain region that is characterized by a relative high density of microglial cells (Lawson et al., 1990; Kim et al., 2000). Therefore, mice systemically challenged with LPS given in a single or in four repeated injections were analyzed on experimental day 5 (Fig. 2C) or day 19 by double immunostaining with the microglial marker Iba1 on levels matched by TH-positive immunoreactivity. Immunohistochemical analysis on day 5 showed an increase in the Iba1 staining after a single injection of LPS (from 32.53 ± 2.11 cells per section in PBS controls to 145.3 ± 8.35 cells per section, p < 0.001) and to a lesser extent after repeated LPS challenges (from 32.53 ± 2.11 cells per section in PBS controls to 77.07 ± 2.24, p < 0.001; Fig. 2D). However, immunoreactivity of Iba1 on day 19 returned to initial levels (Fig. 2D).
In addition, the level of the lysosomal marker CD68 was quantified in SNpr sections of mice treated with LPS or PBS (Fig. 3). On day 5, an unchanged level of Cd68 mRNA after the single treatment was observed, whereas after the repeated treatment the level was increased from 0.68 ± 0.06 FC (single LPS treatment group) to 3.82 ± 0.54 FC (repeated LPS treatment group, p < 0.001; Fig. 3A). Similarly, on day 19, the single treatment showed no change in the mRNA level, whereas the repeated treatment was increased from 0.83 ± 0.20 FC (single LPS treatment group) to 4.11 ± 0.62 FC (repeated LPS treatment group, p < 0.001; Fig. 3A). Accordingly, immunohistochemistry of the SNpr of mice challenged with repeated LPS treatment showed an increase of CD68 protein staining compared with the single LPS-treated group or PBS-treated group (Fig. 3B). Quantification of the area occupied by CD68 staining confirmed an increase of CD68 immunoreactivity after repeated LPS challenge on day 5, from 147.1 ± 32.2 pixels (single LPS treatment) to 366.7 ± 54.25 pixels (repeated LPS treatment, p < 0.01 Fig. 3C). However, on experimental day 19, the CD68 activation marker was back to control levels for the repeated LPS-treated group (Fig. 3C), and the single LPS-treated animals showed a decrease of the CD68 staining on day 19 from 190.0 ± 29.07 pixels (single PBS-treated group) to 70.65 ± 10.63 pixels (single LPS-treated group, p < 0.05; Fig. 3C).
Figure 3.

Enhanced microglial activation after repeated treatment with LPS. A, Microglial activation marker CD68 mRNA levels in the whole brain of mice on experimental day 5 or 19 after single or repeated treatment with PBS control or LPS. Cd68 gene transcripts were increased on both experimental days after repeated treatment with LPS. n ≥ 3 mice, n.s., Not significant. ***p < 0.001. B, Immunofluorescence staining of CD68 (red) and Iba1 (green) in SNpr on experimental day 5 or 19 after repeated treatment with PBS control or LPS. CD68 immunoreactivity was increased after repeated LPS application on day 5. Scale bar, 10 μm. Representative images of n ≥ 3 mice are shown. C, Quantification of CD68 immunoreactivity area in the SNpr sections of mice on experimental day 5 and 19 after single or repeated PBS control or LPS treatment. Microglial CD68 immunoreactivity was increased on day 5 after repeated systemic LPS challenge. n ≥ 3 mice. n.s., Not significant. *p < 0.05. **p < 0.01.
Thus, we observed increased gene transcription and immunoreactivity of the microglial markers Iba1 or CD68 5 d after LPS application that returned back to almost normal levels after 19 d.
Increased proinflammatory molecule production in the brain after repeated systemic application of LPS
Systemic bacterial infections have been postulated to contribute to the progression of neurodegenerative diseases in elderly patients by triggering an inflammatory response of microglia (Perry et al., 2007). To study the local inflammatory response in the brain after peripheral challenge, we compared cytokine levels after single or repeated intraperitoneal application of LPS (Fig. 4). As determined on day 5 by ELISA, plasma levels of TNFα and IL1β proteins after single or repeated application of LPS showed an increase compared with PBS treated controls but no differences between the treatment paradigms (Fig. 4A). In contrast, higher TNFα and IL1β protein levels were observed in the brain tissue on day 5 after repeated systemic challenge with LPS compared with the single LPS challenge (Fig. 4B). In detail, on experimental day 5, TNFα in the brain was increased ∼12-fold, from 3.5 ± 1.4 pg/ml after single LPS application to 44.4 ± 12.1 pg/ml after repeated LPS application (p < 0.001; Fig. 4B). IL1β was increased ∼4-fold on day 5 from 21.4 ± 4.8 pg/ml (single LPS) to 84.2 ± 12.4 pg/ml (repeated LPS; p < 0.05; Fig. 4B).
Figure 4.

Increased production of proinflammatory cytokines in the brain after repeated systemic challenge with LPS. A, ELISA measurement of TNFα and IL1β proteins in the plasma of mice on experimental day 5 of single or repeated treatment with PBS control or LPS. Plasma levels of the cytokines showed similar response to LPS after single or repeated treatment. n.d., Not detected. B, ELISA measurement of TNFα and IL1β proteins in brains of mice on experimental day 5 of single or repeated treatment with PBS control or LPS. LPS challenge induced more pronounced elevation in the levels of the TNFα and IL1β after repeated treatment. n.d., Not detected. *p < 0.05. ***p < 0.001. C, sqRT-PCR for TNFa (gene transcript for TNFα protein), IL1b (gene transcripts for IL1β), and Nos2 (gene transcript for inducible nitric oxide synthase) in the whole brain of mice on experimental day 5 after single or repeated treatment with PBS control or LPS. The repeated LPS treatment induced increased transcription levels of TNFa, IL1b, and Nos2. n.s., Not significant. *p < 0.05. **p < 0.01. ***p < 0.001. D, sqRT-PCR for TNFa, IL1b, and Nos2 in the whole brain of mice on experimental day 19 after single or repeated intraperitoneal treatment with PBS control or LPS. The analyzed transcript levels were unchanged after single or repeated LPS challenge compared with the control. n.s., Not significant.
Next, we analyzed the TNFα and IL1β gene transcription in the brain along with Nos2 (encoding for the inducible nitric oxide synthase) to evaluate any CNS production of the proinflammatory molecules. Indeed, mice challenged once with LPS showed slight, but not significant, activation of TNFα and IL1β gene transcriptions in the brain tissue on day 5 but no modulation of the Nos2 mRNA (Fig. 4C). After repeated challenge with LPS, increased TNFα, IL1β, and Nos2 gene transcriptions were observed compared with a single LPS challenge (Fig. 4C). In detail, TNFα gene transcription was almost threefold increased, from 3.26 ± 1.17 FC (single LPS challenge) to 8.88 ± 2.19 FC after repeated LPS application on day 5 (p < 0.05), IL1β gene transcription was ∼4-fold increased from 7.27 ± 1.46 FC (single LPS) to 32.47 ± 12.5 FC, and Nos2 was also ∼4-fold induced from 0.8 ± 0.08 FC (single LPS) to 3.2 ± 0.4 FC (repeated LPS; p < 0.001; Fig. 4C). Further, we quantified TNFα, IL1β, and Nos2 mRNA on day 19 after a single or repeated LPS challenge. No significant increase in the levels of these transcripts was observed on day 19 after both single and repeated LPS challenge compared with the PBS control (Fig. 4D). Thus, after repeated challenges with LPS, the inflammatory markers TNFα, IL1β, and Nos2 mRNA levels are increased on day 5 in the brain, along with the levels of TNFα and IL1β proteins.
Repeated systemic LPS application triggers an inflammatory transcriptome in the brain
To better understand the molecular mechanisms leading to neurodegeneration after repeated systemic LPS challenge, whole-brain transcriptome analysis was performed in mice that received either single or repeated intraperitoneal treatments with LPS or PBS vehicle. Because our data showed that the proinflammatory reactions resolve by day 19, total RNA was isolated from brain hemispheres either on the experimental day 2 or day 5 (single LPS challenge) or day 5 (repeated LPS challenge; Fig. 5A). Microarray analysis was performed to identify common and specific transcriptomic changes after the single and repeated LPS treatment paradigms. As seen in heatmaps, the single treatment showed an increased number of DE genes on day 2 (24 h after the injection), which subsided within 5 d (Fig. 5B). When the repeated treatment effect was analyzed on the experimental day 5 (24 h after the last injection), it revealed a lower number of genes upregulated or downregulated compared with the results obtained on the experimental day 2 after the high-dose single LPS challenge but higher than those observed after the single treatment on day 5 (Fig. 5B).
Figure 5.
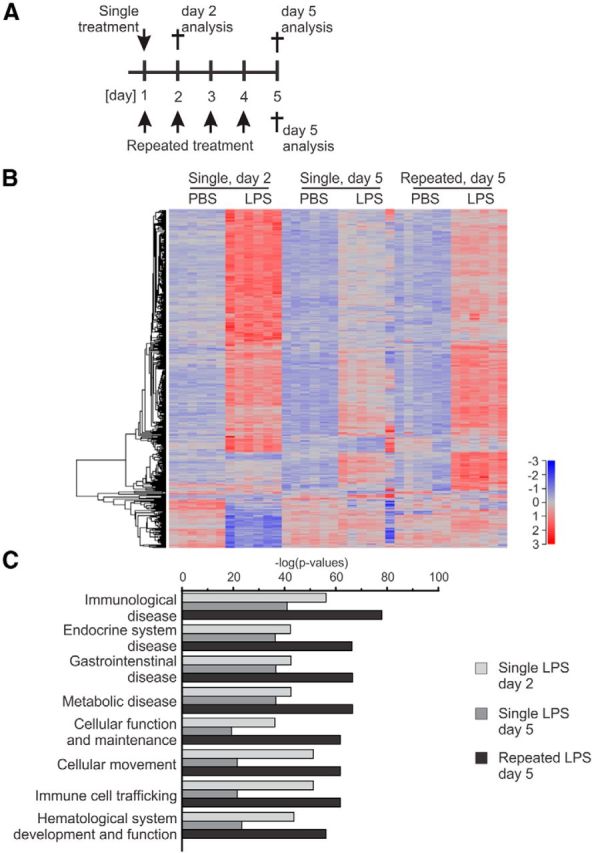
Enriched inflammatory response pathways in the brain after systemic repeated treatment with LPS. A, Treatment and analysis paradigms. For the single treatment (top), mice were injected intraperitoneally once with 4 μg LPS/gbw and were analyzed on experimental day 2 or 5. For the repeated treatment (bottom), mice were injected intraperitoneally on four consecutive days with 1 μg LPS/gbw and were analyzed on day 5. B, Heatmap of the transcriptome in mice after intraperitoneal treatments with LPS versus PBS on experimental day 2 and 5 (single treatment) or on day 5 only (repeated treatment); n > 3 mice. LPS treatment generates an increase of DE genes after both the single or repeated treatment. C, Enriched pathways of the DE genes of mice after intraperitoneal treatments with LPS versus PBS on experimental day 2 and 5 (single treatment) or only on day 5 (repeated treatment). Ingenuity Pathway Analysis was performed on significantly DE genes (FDR < 0.01) in each condition, and the top 12 enriched functions were presented. Score is obtained from the −log(p value) transformation; n > 3 mice.
The analysis of the most significant biological functions underlined by the DE genes after LPS challenges revealed a high correlation with immunological functions within the treatment groups (Fig. 5C).
Repeated systemic challenge with LPS activates the complement–phagosome pathway in the brain
To identify a mechanistic link between the transcriptome analysis and the faster neurodegeneration observed after the repeated LPS challenges, we further performed a more detailed analysis of DE genes on day 5 (single vs repeated LPS). In this regard, we identified specific genes that were selectively and significantly (FDR < 0.05; FC ≥ 2) upregulated in the brain tissue after repeated or single LPS treatment compared with the PBS treated controls (Table 1). In total, 21 genes were found to be commonly induced by both treatment paradigms. These were mostly immune related genes (e.g., complement components: C1qa, C1qc, C3, C3ar1, C4b; chemokine Ccl5; major histocompatibility complex (MHC) molecule H2-k1; Table 1). Only 2 genes were selectively upregulated after single LPS treatment, both indirectly linked to immune function (glutathione peroxidase 3 [Gpx3] and neutrophilic granule protein [Ngp]). Interestingly, 60 genes (mainly immune-related genes, e.g., complement components, Fc receptors, MHC molecules) were selectively upregulated after repeated LPS treatment, therefore being potential causal regulators for the inflammatory neurodegeneration (Fig. 6A; Table 1). Thus, whole-genome transcriptome analysis showed a unique inflammatory molecular signature of the brain after repeated systemic challenge with LPS.
Table 1.
Differentially expressed and common genes after repeated treatment (4× LPS vs 4× PBS) or single treatment (1× LPS vs 1× PBS)a
| 4× LPS versus 4× PBS (60 genes) | Common (21 genes) | 1× LPS versus 1× PBS (2 genes) | ||
|---|---|---|---|---|
| AI607873 | Eltd1 | Lcp1 | C1qa | Gpx3 |
| AW112010 | Eng | Lcp2 | C1qc | Ngp |
| Acer2 | Fcer1g | Lgals3bp | C3 | |
| Aoah | Fcgr2b | Lilrb4 | C3ar1 | |
| Apobec3 | Fcgr3 | Ly6a | C4b | |
| C1qb | Fcgr4 | Ly86 | Ccl5 | |
| Cav1 | Fn1 | Mpeg1 | Cd52 | |
| Ccl3 | Gbp2 | Ms4a6b | Ctss | |
| Cd33 | H2-D1 | Ms4a6d | Emr1 | |
| Cd53 | H2-Q6 | Ms4a7 | Fyb | |
| Cd68 | H2-Q7 | Pglyrp1 | Gm11428 | |
| Cd72 | Hba-a1 | Pilra | H2-K1 | |
| Cd93 | Hba-a2 | Prg4 | Ifitm3 | |
| Clec4a1 | Hbb-b1 | Ptprc | Igsf6 | |
| Clec4a3 | Ifi204 | Stab1 | Lcn2 | |
| Cp | Ifi30 | Tgtp1 | Lyz2 | |
| Ctsc | Ifit1 | Trim30 | Ms4a6c | |
| Ctsh | Ifit3 | Tyrobp | Pld4 | |
| Cxcl13 | Igfbp7 | Saa3 | ||
| Cyba | Il2rg | Serpina3n | ||
| Cybb | Itgam | Vwf | ||
alog2FC ≥ 1(FC ≥ 2); FDR < 0.05.
Figure 6.

The phagosome pathway is activated in the brain after systemic repeated LPS application. A, Venn diagram of the DE genes (FDR < 0.05, FC ≥ 2) in mice after single or repeated intraperitoneal treatments with LPS versus PBS on experimental day 5. Apart from a common group of 21 DE genes, 2 genes are specific to the single LPS treatment, whereas after the repeated challenges, 60 specific DE genes can be observed; n > 6 mice. B, KEGG pathway analysis of the DE microarray elements (FC > 2, FDR < 0.05) in the whole brain after single or repeated LPS challenge on day 5. By comparing the pathways formed by the 81 genes upregulated after the repeated LPS treatment with the pathways of the 23 genes upregulated by the single LPS treatment, the phagosome process shows the most selectively represented pathway in the repeated LPS challenge (12 genes from the repeated treatments and 3 genes from the single treatment). Microorganism response-related pathways were also enriched in the brain after repeated LPS challenge. Additionally, the complement and coagulation cascade were found to be enriched in the brain after repeated treatments (7 genes) and single treatment (6 genes); n > 3 mice. C, Gene transcript levels of elements involved in phagocytosis or phagocytosis-related oxidative stress analyzed via sqRT-PCR in mouse brain on day 5 of the single or repeated treatment paradigm. The genes of adaptor molecules implicated in phagocytosis Tyrobp (encoding DAP12) and Fcer1 g (common γ chain) were analyzed, with Fcer1 g being significantly upregulated by the repeated LPS challenges. NADPH oxidase components Cyba (transcript for p22phox protein) and Cybb (gp91) mRNA levels were upregulated by the single and even higher by the repeated LPS treatment. n > 3 mice. n.s., Not significant. *p < 0.05. **p < 0.01. ***p < 0.001.
To investigate further the intimate mechanism of LPS-triggered dopaminergic degeneration, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) to evaluate the interconnectivity of the elements of the repeated or single LPS treatment paradigm. The pathways underlined by KEGG in the 81 (60 specific and 21 common) genes upregulated after repeated LPS treatment or the 23 (2 specific and 21 common) genes specific to the single LPS treatment were represented by the phagosome and the complement/coagulation cascades (Fig. 6B). The phagosome pathway contained the maximum number of DE genes enriched by the repeated treatment followed by pathways specific to infections with microorganisms (Fig. 6B). KEGG analysis showed that the complement cascade was the strongest common inflammatory enriched pathway from both single and repeated LPS challenge (Fig. 6B). Importantly, several complement components are also part of the KEGG phagosome pathway. As presented in Table 1, elements in the KEGG phagosome pathway common to both single and repeated LPS challenge were thus represented by the complement cascade member C3 or the lysosomal protease cathepsin S (Ctss). Elements unique to the repeated LPS challenges were composed of MHC class I members (H2-d1, H2-q6, H2-q7), complement cascade members (Integrin αMβ2- Itgam, part of the complement receptor 3 (CR3), phagocytosis-related molecules (Fcer2b, Fcgr3, Fcgr4), and p22phox (Cyba) and gp91 (Cybb) members of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex (Table 1).
Next, we performed a validating sqRT-PCR for selected molecules of the phagosome-related pathway enriched by LPS treatment (Fig. 6C), like the TYRO protein tyrosine binding protein gene (Tyrobp, encoding the DAP12 protein), the common γ-chain gene (Fcer1 g), and the NADPH components Cyba and Cybb. Of note, Tyrobp and Fcer1 g, which here were induced in the brain after repeated LPS challenge as found in the microarray, were recently described to be crucially involved in complement-phagosomal signaling during neurodegeneration (Zhang et al., 2013). The Cyba gene transcription was significantly increased after both single LPS treatment (from 1.25 ± 0.41 FC to 2.91 ± 0.27 FC, p < 0.01) and repeated LPS treatment (from 1.00 ± 0.10 FC to 3.64 ± 0.24 FC, p < 0.001), whereas Cybb was showing a significant increase between the repeated versus single LPS treatments, from 4.33 ± 0.94 FC (single LPS) to 9.8 ± 0.83 FC (repeated LPS; p < 0.001). However, Tyrobp mRNA levels were not modulated, whereas after the repeated LPS challenge Fcer1 g was increased to 3.1 ± 0.19 FC from 1.91 ± 0.28 FC (single LPS; p < 0.05; Fig. 6C).
Therefore, data indicate that a complement-phagocytosis process involving Fcer1 g and Cyba/Cybb is linked to the neurodegenerative process.
Loss of dopaminergic neurons is rescued in complement C3-deficient mice
Microglia clear synapses and neurites during development and in neurodegenerative processes via a C1q, C3, CR3, and Dap12/Tyrobp cascade (Stevens et al., 2007; Wakselman et al., 2008; Schafer et al., 2012). Based on these facts and on the microarray results confirmed by sqRT-PCR, we hypothesized that complement is causally involved in neurodegeneration. Detailed analysis of the microarray data for the complement pathway showed that the first components of the classical/alternative complement system (e.g., SerpinG1, C4, C3, HF1) were upregulated in the brain after repeated LPS challenge (Fig. 7A). Validating sqRT-PCR revealed that C1q transcripts were not modulated by LPS challenges (single or repeated), whereas challenges with LPS led to a strong increase in C3 (from 1.08 ± 0.24 to 5.58 ± 1.02 FC, p < 0.01 in the single LPS-treated group and from 1.19 ± 0.40 to 7.75 ± 0.82 FC, p < 0.001 in the repeated treated group). C3-related Itgam gene transcripts were unchanged by the single LPS challenge but highly increased by the repeated challenge (from 0.98 ± 0.2 FC after the single LPS to 4.32 ± 0.14FC after the repeated challenge, p < 0.001). C4b transcript levels were also showing significant differences between the LPS treatment groups (from 2.26 ± 0.45, single LPS, to 9.84 ± 2.60 FC, repeated LPS, p < 0.01; Fig 7B).
Figure 7.
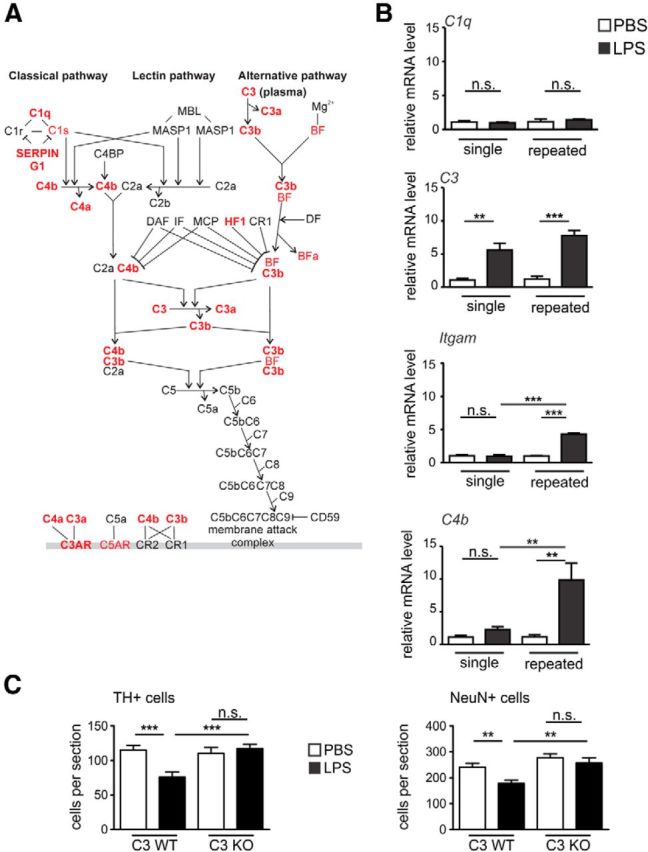
The complement pathway is activated and involved in neurodegeneration after repeated LPS application. A, The complement system canonical pathway of mice after single or repeated intraperitoneal treatments with LPS versus PBS on experimental day 5 shows DE genes. Ingenuity Pathway Analysis was performed on significantly DE genes (FDR < 0.01) in each condition (single and repeated LPS treatment). The canonical complement pathway showed significance in both conditions (p < 1E−06). The overlay represented corresponds to repeated treatment, and significant upregulated genes are shown in red; n > 3 mice. B, Gene transcript levels of complement pathway analyzed via sqRT-PCR in mouse brain on day 5 of the single or repeated treatment paradigm. The classical pathway initiator C1q is unresponsive to the LPS challenge. C3 is upregulated by both single and repeated LPS treatment. Itgam (complement receptor 3 [CR3]) and C4 transcripts are significantly upregulated only by the repeated treatment with LPS. n > 3 mice. n.s., Not significant. **p < 0.01. ***p < 0.001. C, Quantification of TH-positive and NeuN-positive cells in the SNpc of C3-deficient (C3 KO) or wild-type (C3 WT) mice challenged repeatedly with PBS vehicle or LPS. The C3 KO mice showed no loss of TH-positive or NeuN-positive neurons compared with C3 WT animals; n ≥ 3 mice. n.s., Not significant. **p < 0.01. ***p < 0.001.
Within the complement cascade, C3 is in a crucial bottle-neck position (Fig. 7A). Activation of C3 is leading to the formation of the powerful C3 convertase initiating further downstream signaling. Based on the central position of C3 within the complement pathway, we hypothesized that the absence of C3 might block the neurodegenerative process. Thus, we analyzed C3 KO mice challenged with LPS after the repeated treatment paradigm. Quantification of the TH-positive neurons in the SNpc showed that the C3 KO mice were protected against the inflammation-mediated loss (Fig. 7C). In detail, the wild-type (WT) animals showed decreased neuronal numbers (from 115.1 ± 6.86 after PBS to 75.76 ± 7.27 cells per section after LPS treatment, p < 0.001) on experimental day 19 after the repeated LPS treatment, whereas the C3 KO mice were protected against the loss of dopaminergic neurons (110.1 ± 8.63 after PBS vs 117.0 ± 6.22 cells per section after LPS treatment; Fig. 7C). These findings were confirmed by NeuN staining, which showed no reduction in the NeuN-positive cells in the C3 KO animals treated repeatedly with LPS (Fig. 7C). In detail, after repeated injections, NeuN-positive cells were reduced in C3 WT mice from 241.2 ± 14.64 in the PBS group to 178.4 ± 12.25 cells per section in the LPS group (p < 0.01) and kept at similar levels in C3 KO animals (277.9 ± 14.3 in the PBS group vs 257.3 ± 20.11 cells per section in the LPS group).
Thus, complement C3 was an essential intermediate molecule in the loss of dopaminergic neurons triggered by systemic repeated LPS application.
Discussion
In this study, we examined the mechanisms of dopaminergic neuronal loss triggered by repeated systemic challenge with LPS over four consecutive days. Dopaminergic neurons of the SN are more susceptible to inflammatory damage compared with other neuronal subtypes (Block et al., 2007). This susceptibility might involve stronger microglial activation in the midbrain (Perry et al., 2007), higher neuronal sensitivity of dopaminergic neurons to impaired mitochondrial energy metabolism (Lannuzel et al., 2003), and increased sensitivity to oxidative stress resulting from pro-oxidant properties of dopamine and TH (Di Giovanni et al., 2012). To identify the molecular mechanism of systemically applied LPS-triggered degeneration of dopaminergic neurons, we compared a single versus a repeated LPS treatment paradigm in mice. We observed that only the repetitive challenge induced loss of dopaminergic neurons within 19 d as stained with the specific marker TH. These results were validated by NeuN staining that also showed a decrease in the total number of neurons. However, we cannot exclude that other subtypes of neurons also were affected by the repeated LPS challenge. Despite the fact that repeated LPS injections caused a significant loss of dopaminergic neurons in the SN, levels of striatal dopamine were found to be similar between control animals and LPS-treated mice (data not shown). This dissociation between nigral and striatal effects has been reported also in other models of toxicant-induced injury as well as in transgenic models of nigrostriatal pathology (Liberatore et al., 1999; McCormack et al., 2002). Most likely, in both our present and in other previous studies, dopamine levels after challenges were detected as unaffected because of biochemical compensatory mechanisms (e.g., TH upregulation and/or decreased dopamine turnover).
Microglial reaction to systemic LPS challenges showed a strong increase of the microglial specific marker Iba1 or CD68 on experimental day 5 after repeated LPS treatment at both mRNA and protein levels. However, on experimental day 19, both Iba1 mRNA and the protein levels were lowered back to PBS control levels, whereas for CD68, the mRNA level was found to be kept at increased level, in contrast to the protein expression. Similar results were observed in another study that used a comparable repeated LPS treatment protocol and analyzed the time course of Iba1 and microglial activation within a 5 d period (Chen et al., 2012).
To identify causal elements and molecular pathways enriched inside the CNS by systemic repeated LPS application, we performed whole-genome transcriptome analysis of the brain tissue and examined the early changes in gene transcription triggered by the LPS challenges. For this, we collected brain material on experimental day 2 (24 h after the treatment) and day 5 after the single LPS treatment and compared the DE genes with day 5 of the repeated LPS treatment (24 h after the last treatment), when we observed a higher proinflammatory response inside the brain. Transcriptome data pointed toward a sustained higher increase of genes for at least 5 d after the repeated treatment. In contrast, the response obtained after a single LPS challenge peaked on the experimental day 2 and subsided within the following days. Transcriptome analysis revealed that top DE genes were members of the immunological system. More rigorous analysis showed that approximately three times more genes were specifically induced by the repeated LPS challenge (60 genes) compared with the group of common genes (21 genes), and 30 times more genes compared with the single LPS treatment (2 genes). KEGG analysis identified the complement–phagosome linked to the NADPH oxidase as the most representative pathways induced by repeated LPS treatments. Major upregulated components of the complement pathway were complement C3, the integrin subunit Itgam (also known as CD11b or MAC1) of the CR3, and the immunoreceptor tyrosine-based activation motif (ITAM)-signaling common γ-chain subunit (coded by the Fcer1 g gene). The phagosome pathway was mainly represented by the lysosomal activation marker CD68, the NADPH family member p22phox (coded by the Cyba gene), and the NADPH family member gp91phox (coded by the Cybb gene).
Although we identified the complement–phagosome pathway, including the NADPH oxidase, as major elements enriched in the brain after repeated systemic challenge with LPS, it is still unclear whether LPS acts systemically or locally after reaching the CNS. Several studies presumed that the blood–brain barrier remains intact after systemic LPS challenge (for review, see Cunningham, 2013), but LPS might reach the brain parenchyma also directly through the circumventricular organs (Rivest, 2003). Furthermore, leukocytes might infiltrate the brain under LPS application and other severe inflammatory conditions (Ji et al., 2008), thus potentially contributing to the increase of proinflammatory cytokines in the brain parenchymal milieu. It was proposed that the effect of peripheral administration of LPS to the brain is mediated via nervous and/or humoral routes, with an involvement of the proinflammatory cytokines TNFα and IL1β passing the blood–brain barrier (Perry, 2004; Dantzer et al., 2008). In agreement with a previous report (Perry, 2010), our study identified a local production of proinflammatory cytokines inside the brain after repeated intraperitoneal LPS administration.
Our data show that selective components of both classical and alternative complement pathways were enriched in the brain after LPS treatments. Even though C1 transcripts did not show a significant change in our sqRT-PCR validation, C3, CR3 subunit Itgam, and C4 presented increased mRNA levels after the treatment with LPS. However, genes downstream of C3/C4 were not enriched, suggesting no local production of components of the complement membrane attack complex. In our study, C3, as amplifier of the complement response, was instrumental for the loss of dopaminergic neurons after repeated systemic challenge with LPS. Both C3 and its receptor CR3 have been involved in microglial clearance mechanisms before. It was shown that complement-marked neuronal structures are removed by microglial CR3 (Linnartz et al., 2012b; Schafer et al., 2012). In addition, it was suggested that complement C3 contributes directly to “priming of microglia” toward a proinflammatory profile (Ramaglia et al., 2012). The absence of the C3 convertase regulator complement receptor 1-related protein (Crry) leads to increased cleavage of C3 into the active products C3b and iC3b. Deletion of Crry triggered microglia to become “sensitized” to systemic LPS (Ramaglia et al., 2012). This “priming of microglia” was abolished in C3-deficient mice. In our experiments, C3 KO mice were resistant to loss of dopaminergic neurons induced by repeated systemic LPS challenge. Results suggest that the complement C3 is interposed between the activated microglia and the neurodegenerative process. After single or repeated LPS challenge, the gene transcription of complement C3 was upregulated. Of note, transcription was higher after repeated challenge with LPS compared with single LPS treatment. Moreover, Itgam, part of the CR3, has been implicated in dopaminergic neurodegeneration previously (Pei et al., 2007). Loss of dopaminergic neurons was attenuated in CR3-deficient mice challenged with LPS, together with a lower TNFα response and abrogation of superoxide production in Itgam-deficient microglial cultures treated with LPS (Pei et al., 2007). In other myeloid cells, it was shown that CR3 signals via Dap12 to induce phagocytosis and the associated respiratory burst (Ivashkiv, 2009), but the detailed signaling cascade of CR3 in microglia is not fully understood. We failed to see upregulation of Dap12 after systemic and repeated LPS challenges. However, a highly homolog molecule of Dap12, namely, the common γ chain (coded by the Fcer1 g gene) was upregulated. The common γ chain can substitute Dap12 functions in deficient mice, and both molecules present homolog ITAM-signaling motifs. The common γ chain has been shown to be upregulated in the brain during chronic neurodegeneration (Lunnon et al., 2011), whereas common γ chain-deficient mice were partially protected against dopaminergic damage (Cao et al., 2010). Moreover, common γ chain-deficient mice treated with the toxin MPTP showed less dopaminergic neuronal loss compared with control mice (Lira et al., 2011). Developmental microglia-triggered neuronal apoptosis was previously described as being linked with ITAM signaling (Wakselman et al., 2008). Interestingly, activation of ITAM-bearing molecules, such as the common γ chain or Dap12, can lead to NADPH oxidase-mediated release of reactive oxygen species causing progressive degeneration of dopaminergic neurons (Wu et al., 2002; Choi et al., 2012; Qin et al., 2013). Of note, transcripts of Cyba and Cybb encoding for NADPH oxidase members p22phox and gp91 were also found to be upregulated by LPS challenges in our study. Another member of the phagosome pathway specifically found to be enriched by the repeated LPS treatment was the transcript of CD68, a lysosomal protein highly expressed in activated phagocytes (Neher et al., 2013).
Thus, we propose a mechanism by which systemic LPS leads to a complement-mediated response that activates microglia via CR3, releasing ITAM-associated signaling and culminating with the formation of the NADPH oxidase that was previously associated with the production of neurotoxic reactive oxygen species (Qin et al., 2013). Under both normal and pathological conditions, silent phagocytosis is one of the main beneficial functions of microglia (Neumann et al., 2009; Linnartz et al., 2012a; Noda and Suzumura, 2012). However, microglia can also induce delayed neuronal cell death by primary phagocytosis, a process that recently was named “phagoptosis” (Brown and Neher, 2012, 2014; Neher et al., 2013). Our data now bring new insights in this process of primary phagocytosis. By activating a distinct microglial complement–phagosome pathway, the repeated treatment with LPS induced dopaminergic neurodegeneration. Furthermore, we show that complement C3 is causally involved in the inflammatory neurodegeneration.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (KFO177, SFB704, FOR1336) and the Hertie-Foundation. H.N. is member of the DFG-funded excellence cluster ImmunoSensation (EXC 1023). We thank Dr. Colm Cunningham for helpful discussions; and Jessica Schumacher and Rita Hass from the Institute of Reconstructive Neurobiology and Nathalie Nicot from the CRP-Sante Genomics Research Unit for excellent technical support.
The authors declare no competing financial interests.
References
- Baquet ZC, Williams D, Brody J, Smeyne RJ. A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience. 2009;161:1082–1090. doi: 10.1016/j.neuroscience.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg T. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: “phagoptosis.”. Trends Biochem Sci. 2012;37:325–332. doi: 10.1016/j.tibs.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- Cao S, Theodore S, Standaert DG. Fcγ receptors are required for NF-κB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson's disease. Mol Neurodegener. 2010;5:42. doi: 10.1186/1750-1326-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–11715. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DH, Cristóvão AC, Guhathakurta S, Lee J, Joh TH, Beal MF, Kim YS. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson's disease. Antioxid Redox Signal. 2012;16:1033–1045. doi: 10.1089/ars.2011.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61:71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni G, Pessia M, Di Maio R. Redox sensitivity of tyrosine hydroxylase activity and expression in dopaminergic dysfunction. CNS Neurol Disord Drug Targets. 2012;11:419–429. doi: 10.2174/187152712800792938. [DOI] [PubMed] [Google Scholar]
- Dutta G, Zhang P, Liu B. The lipopolysaccharide Parkinson's disease animal model: mechanistic studies and drug discovery. Fundam Clin Pharmacol. 2008;22:453–464. doi: 10.1111/j.1472-8206.2008.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Treviño I, O'Brien DE, Casey B, Goldberg MS, Tansey MG. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson's disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S210–S212. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB. Cross-regulation of signaling by ITAM-associated receptors. Nat Immunol. 2009;10:340–347. doi: 10.1038/ni.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji KA, Eu MY, Kang SH, Gwag BJ, Jou I, Joe EH. Differential neutrophil infiltration contributes to regional differences in brain inflammation in the substantia nigra pars compacta and cortex. Glia. 2008;56:1039–1047. doi: 10.1002/glia.20677. [DOI] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannuzel A, Michel PP, Höglinger GU, Champy P, Jousset A, Medja F, Lombès A, Darios F, Gleye C, Laurens A, Hocquemiller R, Hirsch EC, Ruberg M. The mitochondrial complex I inhibitor annonacin is toxic to mesencephalic dopaminergic neurons by impairment of energy metabolism. Neuroscience. 2003;121:287–296. doi: 10.1016/S0306-4522(03)00441-X. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-W. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Linnartz B, Bodea LG, Neumann H. Microglial carbohydrate-binding receptors for neural repair. Cell Tissue Res. 2012a;349:215–227. doi: 10.1007/s00441-012-1342-7. [DOI] [PubMed] [Google Scholar]
- Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci. 2012b;32:946–952. doi: 10.1523/JNEUROSCI.3830-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira A, Kulczycki J, Slack R, Anisman H, Park DS. Involvement of the Fc gamma receptor in a chronic N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of dopaminergic loss. J Biol Chem. 2011;286:28783–28793. doi: 10.1074/jbc.M111.244830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunnon K, Teeling JL, Tutt AL, Cragg MS, Glennie MJ, Perry VH. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J Immunol. 2011;186:7215–7224. doi: 10.4049/jimmunol.0903833. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- Neher JJ, Emmrich JV, Fricker M, Mander PK, Théry C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A. 2013;110:E4098–E4107. doi: 10.1073/pnas.1308679110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Suzumura A. Sweepers in the CNS: microglial migration and phagocytosis in the Alzheimer disease pathogenesis. Int J Alzheimers Dis. 2012;2012:891087. doi: 10.1155/2012/891087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Z, Pang H, Qian L, Yang S, Wang T, Zhang W, Wu X, Dallas S, Wilson B, Reece JM, Miller DS, Hong JS, Block ML. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia. 2007;55:1362–1373. doi: 10.1002/glia.20545. [DOI] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010;120:277–286. doi: 10.1007/s00401-010-0722-x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;462:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Y, Hong JS, Crews FT. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia. 2013;61:855–868. doi: 10.1002/glia.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I, Baas F, Neal JW, Morgan BP. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A. 2012;109:965–970. doi: 10.1073/pnas.1111924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/S0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Richard M, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease: a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:1–23. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]