

Abstract
Combining transcranial magnetic stimulation (TMS) and electroencephalography (EEG) constitutes a powerful tool to directly assess human cortical excitability and connectivity. TMS of the primary motor cortex elicits a sequence of TMS-evoked EEG potentials (TEPs). It is thought that inhibitory neurotransmission through GABA-A receptors (GABAAR) modulates early TEPs (<50 ms after TMS), whereas GABA-B receptors (GABABR) play a role for later TEPs (at ∼100 ms after TMS). However, the physiological underpinnings of TEPs have not been clearly elucidated yet. Here, we studied the role of GABAA/B-ergic neurotransmission for TEPs in healthy subjects using a pharmaco-TMS-EEG approach. In Experiment 1, we tested the effects of a single oral dose of alprazolam (a classical benzodiazepine acting as allosteric-positive modulator at α1, α2, α3, and α5 subunit-containing GABAARs) and zolpidem (a positive modulator mainly at the α1 GABAAR) in a double-blind, placebo-controlled, crossover study. In Experiment 2, we tested the influence of baclofen (a GABABR agonist) and diazepam (a classical benzodiazepine) versus placebo on TEPs. Alprazolam and diazepam increased the amplitude of the negative potential at 45 ms after stimulation (N45) and decreased the negative component at 100 ms (N100), whereas zolpidem increased the N45 only. In contrast, baclofen specifically increased the N100 amplitude. These results provide strong evidence that the N45 represents activity of α1-subunit-containing GABAARs, whereas the N100 represents activity of GABABRs. Findings open a novel window of opportunity to study alteration of GABAA-/GABAB-related inhibition in disorders, such as epilepsy or schizophrenia.
Keywords: electroencephalography, GABA, human cortex, inhibition, pharmaco-TMS-EEG, transcranial magnetic stimulation
Introduction
Transcranial magnetic stimulation (TMS) is a noninvasive brain stimulation method that allows to study human cortical function in vivo (Hallett, 2007). However, the potential of TMS as a stand-alone technique is limited. This potential can be enhanced by combining TMS with simultaneous registration of electroencephalography (EEG), thus providing direct information about cortical excitability and connectivity with reasonable spatial and excellent time resolution (Rogasch and Fitzgerald, 2013). A single TMS pulse delivered over the primary motor cortex (M1) results in a sequence of positive and negative EEG deflections. These TMS-evoked cortical potentials (TEPs) last for up to 300 ms in both the vicinity of the stimulation as well as in remote interconnected brain areas (Ilmoniemi et al., 1997; Bonato et al., 2006; Lioumis et al., 2009). TMS-EEG recordings have provided important insights into cortical processing both in health (Massimini et al., 2005; Ferrarelli et al., 2010) and disease (Rosanova et al., 2012; Ragazzoni et al., 2013). However, the neurophysiological underpinnings of TEPs have not been clearly elucidated yet.
Animal studies show that electrical stimulation of a cortical area provokes an initial, brief excitation followed by two phases of inhibition (Connors et al., 1988). The first inhibition (fast IPSP [fIPSP]) occurs at short latencies <50 ms and is linked to activation of GABA-A receptors (GABAARs). The second inhibition (slow IPSP [sIPSP]) has a delayed onset, is long-lasting (50–200 ms), and is generated by an increase in potassium conductance via activation of metabotropic GABA-B receptors (GABABRs) (Connors et al., 1988; Deisz, 1999). Based on these findings, it was hypothesized that activation of GABAARs contributes to the TEP at ∼45 ms after TMS over M1 (N45), whereas activation of GABABRs is involved in the negative deflection at ∼100 ms after TMS (N100) (Nikulin et al., 2003; Ferreri et al., 2011; Bruckmann et al., 2012; Rogasch and Fitzgerald, 2013; Farzan et al., 2013).
Here, we tested these hypotheses in healthy human subjects by conducting two separate double-blind, placebo-controlled pharmaco-TMS-EEG studies. In Experiment 1, we investigated the acute effects of alprazolam, a classical benzodiazepine and allosteric-positive modulator of α1, α2, α3, and α5 subunits of the GABAARs, and zolpidem, which mainly binds at α1-subunit-containing GABAARs, on TEP amplitudes. We aimed to study the role of different α-subunit-containing GABAARs as they underlie different physiological functions (Möhler et al., 2002), and differentially underlie the pathophysiology of epilepsy (Cossette et al., 2002), schizophrenia (Uhlhaas and Singer, 2012), or developmental cortical plasticity (Fagiolini et al., 2004). In Experiment 2, we studied the acute effects of baclofen, a specific GABABR agonist, and diazepam, a classical benzodiazepine, on TEP amplitudes. Alprazolam and diazepam increased the N45 and decreased the N100 amplitude, whereas zolpidem increased the N45 amplitude only. In contrast, baclofen specifically increased the N100 amplitude without affecting earlier TEPs. These data suggest that the N45 represents activity of α1-subunit-containing GABAARs, whereas the N100 represents activity of GABABRs. These TMS-EEG findings can help to further characterize function of the GABAergic inhibitory system in health and disease.
Materials and Methods
Subjects
Twenty-two male subjects 21–32 years of age (mean age, 25.0 ± 2.5 years) and 19 male subjects 22–32 years of age (26.4 ± 3.5 years) participated after giving written informed consent in Experiments 1 and 2, respectively. Female participants were excluded in both studies because of menstrual cycle-related effects on cortical excitability, which can be a potential confound in TMS studies (Smith et al., 1999). All subjects were right-handed according to the Edinburgh Handedness Inventory (laterality score ≥75%) (Oldfield, 1971). Subjects underwent a physical examination before each experiment and were screened for contraindications to TMS (Rossi et al., 2009). Exclusion criteria included the presence of a history of neurological or psychiatric disease, use of CNS active drugs, abuse of any drugs (including nicotine and alcohol), or contraindications to the study medications (alprazolam, zolpidem, diazepam and baclofen).
Experiments were approved by the Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte), and by the local Ethics Committees of the Medical Faculty of Goethe-University Frankfurt (Experiment 1) and the Medical Faculty of Eberhard-Karls-University Tübingen (Experiment 2).
Experimental design
Experiment 1.
The first experiment was designed to investigate the role of GABAA-ergic neurotransmission on TMS-evoked EEG recordings. To address this question, we used a pseudo-randomized, placebo-controlled, double-blinded crossover study to assess the acute effects of a single oral dose of alprazolam versus zolpidem on TEPs. Alprazolam is a classical benzodiazepine and allosteric-positive modulator of α1, α2, α3, and α5 subunit-containing GABAARs, whereas zolpidem is a short-acting nonbenzodiazepine hypnotic that preferentially binds to α1 subunit-containing GABAARs. Subjects participated in three experimental sessions at least 1 week apart. After baseline measurements, subjects received a single oral dose of alprazolam (1 mg, Alprazolam-ratiopharm, ratiopharm), zolpidem (10 mg, Zolpidem-ratiopharm, ratiopharm), or placebo (P-Tabletten Lichtenstein) (Fig. 1). The order of drug allocation was pseudo-randomized and balanced across subjects. Dosages of alprazolam and zolpidem were chosen because they are effective standard daily doses in the treatment of patients with anxiety or sleep disorders, respectively, and according to previously shown sedative effects by these drugs as indexed by a significant slowing of the saccadic peak velocity (SPV) (Blom et al., 1990; de Haas et al., 2010).
Figure 1.

Timeline of experiments. Peak velocity of visually guided saccades (Saccades) and EEG responses evoked by single-pulse TMS over the dominant (left) primary motor cortex (TMS-EEG) were measured before and 90 min after intake of a single oral dose of alprazolam (1 mg), zolpidem (10 mg), or placebo (Experiment 1), or diazepam (20 mg), baclofen (50 mg), or placebo (Experiment 2).
Postdrug measurements were performed 90 min after drug intake according to drug pharmacokinetics with plasma levels peaking at ∼1.5 h after oral intake (Greenblatt and Wright, 1993; Salvà and Costa, 1995) and in line with previous reports, where TMS measures of motor cortical excitability were significantly altered at this time point (Greenblatt and Wright, 1993; Salvà and Costa, 1995; Di Lazzaro et al., 2006; Di Lazzaro et al., 2007).
Experiment 2.
The second experiment aimed at assessing the role of GABAB-ergic neurotransmission for TEPs. To this end, subjects took a single oral dose of baclofen (50 mg Lioresal, Novartis Pharma), diazepam (20 mg Diazepam-ratiopharm, ratiopharm), or placebo (P-Tabletten Lichtenstein). Baclofen is a specific GABABR agonist, whereas diazepam is a classical benzodiazepine binding at α1, α2, α3, and α5 subunit-containing GABAARs. Diazepam was included into Experiment 2 to study the specificity of baclofen-mediated modulation of TEPs (see below). Given its similar pharmacological profile with respect to alprazolam (Experiment 1), it also allowed validation of GABAAR-mediated modulation of TEPs.
As for Experiment 1, subjects participated in three pseudo-randomized experimental sessions, at least 1 week apart, where the effects of the drugs on TEPs were evaluated 90 min after baseline measurements (Fig. 1). Drug doses and time point of postdrug measurements were chosen according to previous experiments that reported a significant modulation of TMS parameters for cortical inhibition 1.5 h after drug intake (Müller-Dahlhaus et al., 2008) and in line with the pharmacokinetics of the two study medications (Shader et al., 1984; McDonnell et al., 2006).
SPV measurements
Visually guided SPV is a biomarker of sedation mediated through α1 subunit-containing GABAARs (de Visser et al., 2003; de Haas et al., 2009, 2010). SPV was measured at baseline and after drug intake (Fig. 1). Subjects sat in front of a screen (eyes to screen distance, 67.5 cm) and were instructed to make visually guided saccades in response to a white dot (subtending an angle of view of 1°) on a black screen while the head was maintained in straight position. The dot jumped at random intertrial intervals of 2–3 s (to prevent anticipation of the next event) horizontally from one lateral edge to the opposite edge of the screen, subtending an angle of view of 40°. Both before and after drug intake, 50 trials were presented. Saccade recordings were obtained by electronystagmography (ENG) using surface electrodes placed at the outer canthus of each eye. The ENG raw signals were amplified and bandpass filtered (0.5–70 Hz; Digitimer D360), digitized at an A/D rate of 10 kHz per channel (CED Micro 1401; Cambridge Electronic Design), and stored in a laboratory computer for online visual display and later offline analysis using customized data collection and conditional averaging software. ENG data were then exported into MATLAB (version 2008b; MathWorks), and in-house-written software was used for manual identification of saccade onset and offset (Velázquez-Pérez et al., 2004). SPV (in °/s) was obtained through third-order polynomial fits of the ENG raw signal. Conditional SPV averages were calculated for leftward and rightward saccades, and a grand mean was calculated for each individual, session, and measurement (predrug/postdrug intake).
TMS
Focal TMS of the hand area of left primary motor cortex (M1) was performed with a figure-of-eight coil (external diameter of each wing, 90 mm) connected to a Magstim 2002 magnetic stimulator (Magstim) with a monophasic current waveform. The optimal coil position over the hand area of left M1 for eliciting MEPs in the right abductor pollicis brevis muscle (APB) was determined as the site where TMS at a slightly suprathreshold intensity consistently produced the largest MEPs. MEP recordings were obtained by surface EMG, using Ag-AgCl cup electrodes in a belly–tendon montage. The EMG raw signal was amplified and bandpass filtered (20 Hz to 2 kHz; D360 amplifier, Digitimer) and digitized at an A/D rate of 10 kHz per channel (CED Micro 1401; Cambridge Electronic Design). The coil was held tangential to the scalp with the handle pointing backwards and 45 degrees away from the midline. This orientation induces a lateral–posterior to medial–anterior current in the brain, activating the corticospinal system preferentially trans-synaptically via horizontal corticocortical connections (Di Lazzaro et al., 2008). Resting motor threshold (RMT) was determined using the relative frequency method (Groppa et al., 2012) and was defined as the minimum intensity that was sufficient to elicit an MEP of >50 μV peak-to-peak amplitude in at least five often subsequent trials. The position of the APB hotspot was marked with a felt pen on the EEG cap to ensure constant coil placement throughout the experiment.
High-density EEG recordings during TMS
TMS-evoked EEG potentials were recorded using a TMS-compatible EEG system (BrainAmp DC, BrainProducts), which prevents EEG amplifier saturation and allows continuous data recording during TMS. The EEG signal was digitized at a sampling frequency of 5 kHz and continuously recorded by 62 electrodes mounted on an elastic cap according to the standard layout (BrainCap-Fast'n Easy 64Ch, Brain Products). Horizontal eye movements were recorded by placing an electrode outside the outer canthus of the eye while an electrode placed below the right eye recorded vertical eye movements and blinks. The impedance of all electrodes was kept <5 kΩ throughout the experiment.
During TMS-EEG recordings, subjects were seated on a comfortable reclining chair and asked to stay awake with eyes open. A masking noise was applied by earphones to avoid contamination of the EEG signal by auditory potentials evoked by the click associated with current discharge through the TMS coil (Massimini et al., 2005). At baseline and at 90 min after drug intake, 150 TMS pulses (Experiment 1) or 125 TMS pulses (Experiment 2) each were applied over the left M1 APB hotspot at an intensity of 100% RMT. Thus, no or only liminal MEPs were elicited during TMS-EEG recordings. Therefore, the TEPs were not contaminated by somatosensory afferent signals from muscle twitches. The interstimulus interval between TMS pulses was, on average, 5 s (random intertrial interval variation of 25% to reduce anticipation of the next trial).
Data processing and TEP analysis
EEG data preprocessing and TEP analysis were performed using the Fieldtrip open source MATLAB toolbox (www.ru.nl/fcdonders/fieldtrip/) (Oostenveld et al., 2011). The EEG signal was first rereferenced to the linked mastoids (channels TP9 and TP10) and down-sampled to 1 kHz. EEG trials were segmented from continuously recorded EEG time series from −500 ms before to 500 ms after TMS pulses. The TMS artifact was removed by applying a linear interpolation for 10 ms before and after the TMS pulse (Thut et al., 2011). Thereafter, each trial was linearly detrended and bandpass filtered between 2 and 80 Hz. A 50 Hz notch filter was applied to reduce line noise contamination. EEG trials were visually scrutinized, and trials containing artifacts resulting from, for example, eye movements or muscle activation, were eliminated. The data from 2 subjects in Experiment 1 and 4 subjects in Experiment 2 had to be excluded from final analysis because of excessive artifact contamination of the EEG traces.
Artifact-free EEG trials for Experiment 1 (averaged number of trials across subjects in before and after alprazolam: 126 ± 5 and 119 ± 4; zolpidem: 125 ± 3 and 109 ± 3; placebo: 123 ± 4 and 128 ± 3) and Experiment 2 (averaged number of trials across subjects in before and after diazepam: 107 ± 3 and 99 ± 3; baclofen: 109 ± 3 and 108 ± 3; placebo: 104 ± 3 and 107 ± 4) were baseline corrected by subtracting the mean amplitude during an interval between −500 ms and −100 ms before the TMS onset. TEPs were then calculated by averaging the EEG signal over all retained trials for each channel. To smooth the TEP, we used data filtered between 1 and 45 Hz. We considered five TEP components (P, positive deflection; N, negative deflection), which were the most reproducible: P25 (time window of interest [TOI]: 20–30 ms), N45 (35–60 ms), P70 (60–80 ms), N100 (85–140 ms), and P180 (150–230 ms) [Experiment 1]; P25 (20–37 ms), N45 (40–60 ms), P70 (65–85 ms), N100 (88–150 ms), and P180 (150–230 ms) [Experiment 2], in accordance with the literature (Lioumis et al., 2009) (see Fig. 3). TOIs were chosen on the basis of grand averaged TEPs, which were slightly different between Experiments 1 and 2 because of intersubject variability of peak latencies of TEP components (Lioumis et al., 2009) and were kept identical for the analysis of each predrug and postdrug condition. The amplitude of each TEP component (measured as peak to baseline) was evaluated before and after drug administration in the specified TOIs.
Figure 3.

TEPs before drug intake. Grand-average TEPs (over all electrodes and artifact-free trials) induced by left M1 TMS before intake of alprazolam (red), zolpidem (black), or placebo (blue) (Experiment 1, A), and diazepam (red), baclofen (black), or placebo (blue) (Experiment 2, B). The time of TMS equals 0 ms. The shaded gray bar represents the part of the EEG trace (±10 ms) that was linearly interpolated to remove the TMS-induced artifact. TEP components are labeled according to their polarity and approximate latency. Bottom line indicates topographical distributions of surface voltages for the most pronounced TEP components (P25, N45, P70, N100, P180; grand-average across predrug measurements in the three drug conditions) for Experiment 1 (A) and Experiment 2 (B). Maps are scaled and color-coded individually according to their respective maximum (red) and minimum (blue).
It has been recently proposed that spontaneous and drug-induced changes of α oscillations can affect somatosensory-evoked potentials (Nikulin et al., 2007; Supp et al., 2011). To rule out an effect of drug-induced changes of neural oscillations on TEPs, a Morlet-wavelet convolution of a width of 5 cycles per wavelet was used to analyze the power spectra of the EEG signal at baseline (i.e., −500 ms to −100 ms before TMS) in the θ (4–8 Hz), α (8–12 Hz), β (13–30 Hz), and γ (30–50 Hz) frequency bands in Experiment 1 and 2 separately. We then extracted the average power values for each spectral band as well as the TEP amplitudes for each subject before and after drug administration in the channels that showed a drug-induced change in TEP amplitude (see Results). Spearman correlation analyses were run between the TEP amplitude change (amplitude postdrug − amplitude predrug) and the baseline power modulation (power postdrug − power predrug) in the different drug conditions.
Statistics
To evaluate drug effects on SPV, a repeated-measures ANOVA was performed independently for Experiments 1 and 2, with drug (3 levels: alprazolam, zolpidem, placebo [Experiment 1]; baclofen, diazepam, placebo [Experiment 2]) and time (2 levels: predrug, postdrug) as within-subject factors.
To assess whether RMT measurements were similar and reproducible in the three predrug conditions, one-way repeated-measures ANOVAs with factor drug condition (3 levels: Experiment 1: alprazolam, zolpidem, placebo; Experiment 2: diazepam, baclofen, placebo) were used.
To analyze drug-induced TEP amplitude modulations, multiple dependent sample t tests (predrug vs postdrug intake) were applied on the level of individual electrodes within each drug condition and each TOI separately. To correct for multiple comparisons (i.e., electrodes, time points within TOIs), we conducted a cluster based permutation analysis (Maris and Oostenveld, 2007) as implemented in FieldTrip (http://fieldtrip.fcdonders.nl/). That is, a paired t test comparing the predrug versus postdrug condition was conducted for each electrode at each time bin within the five different TOIs. t values exceeding an a priori threshold of p < 0.01 were clustered based on adjacent time bins and neighboring electrodes. Cluster-level statistics were calculated by taking the sum of the t values within every cluster. The statistical comparisons were done with respect to the maximum values of summed t values. By means of a permutation test (i.e., randomizing data across predrug and postdrug conditions and rerunning the statistical test 1500 times), we obtained a reference distribution of the maximum of summed cluster t values to evaluate the statistic of the actual data. Clusters in the original dataset were considered to be significant at an α level of 5% if <5% of the permutations used to construct the reference distribution yielded a maximum cluster-level statistic larger than the cluster-level value observed in the original data.
The reported p values are further Bonferroni-corrected for TOIs (n = 5) and predrug and postdrug conditions (n = 2), which is reflected by a corrected α value = 0.05/10.
All data are presented as means ± SEM. if not indicated otherwise.
Results
Experimental procedures and study drugs were generally well tolerated, except for zolpidem, which caused nausea in one subject. Other common adverse events were mild to moderate sedation and dizziness, which did not affect the capability of the subjects to fully comply with the requirements of this study.
Experiment 1
Drug effects on SPV
Repeated-measures ANOVA of drug effects on SPV showed a significant main effect of TIME (F(1,19) = 65.52, p < 0.001) and DRUG (F(2,38) = 17.22, p < 0.001). In addition, there was a significant interaction between TIME and DRUG (F(2,38) = 16.18, p < 0.001), which was explained by a significant reduction of SPV 90 min after intake of alprazolam (p < 0.001) and zolpidem (p < 0.001), whereas placebo had no significant effect on SPV (p > 0.05) (Fig. 2A). Post hoc analysis further showed that reduction of SPV was greater with alprazolam than with zolpidem (p = 0.018). These results are in line with prior reports demonstrating that visually guided SPV is a biomarker of sedation mediated through α1-subunit-containing GABAARs (de Visser et al., 2003; de Haas et al., 2009, 2010). This notion is further supported by the degree of modulation of SPV (after − predrug) by alprazolam and zolpidem, which showed a significant positive correlation (Spearman r2 = 0.20, p = 0.048; data not shown).
Figure 2.

Visually guided saccade peak velocity (SPV). SPV was measured before (Pre, black bars) and 90 min after (Post, white bars) intake of a single oral dose of 1 mg alprazolam, 10 mg zolpidem, or placebo (Experiment 1, A), or 20 mg diazepam, 50 mg baclofen, or placebo (Experiment 2, B). A, Data are means from 20 subjects. B, Data are means from 15 subjects. Error bars indicate ± SEM. *Significant differences predrug versus postdrug (p < 0.05).
Drug effects on TEPs
RMT was not significantly different between the predrug conditions (alprazolam: 47.8 ± 1.8% maximum stimulator output [MSO]; zolpidem: 47.4 ± 1.6% MSO; placebo: 48.7 ± 1.9% MSO; F(2,38) = 1.47, p = 0.24).
The grand-average TMS-evoked EEG response after single-pulse TMS of M1 at baseline (Fig. 3A) showed the typical TEP components (P25, N45, P70, N100, and P180) previously described (Lioumis et al., 2009). Topographical surface voltage maps (Fig. 3A) suggested that early TEPs are located in central (P25) and contralateral frontal regions (N45). The following component (P70) was mainly expressed in the stimulated hemisphere, whereas the later TEPs showed a bilateral distribution over central (N100) and centrofrontal sites (P180) (Bonato et al., 2006; Ferreri et al., 2011; Veniero et al., 2013).
To determine drug effects on specific TEP components, we performed a cluster-based permutation analysis of TEP amplitudes in five nonoverlapping TOIs (P25, 20–30 ms; N45, 35–60 ms; P70, 60–80 ms; N100, 85–140 ms; P180, 150–230 ms) before and after drug intake. Of note, between the predrug conditions, there were no significant differences (all TOIs p > 0.005, corrected for multiple comparisons), which may have contributed to drug-induced modulation of TEPs. The benzodiazepine alprazolam increased the N45 potential (pre: −1.93 ± 0.48 μV; post: −3.07 ± 0.68 μV; p < 0.001) and reduced the N100 (pre: −3.91 ± 0.54 μV; post: −2.27 ± 0.36 μV; p < 0.001) (Fig. 4A). In contrast, zolpidem increased the N45 only (pre: −2.68 ± 0.39 μV; post: −3.87 ± 0.44 μV; p = 0.001) (Fig. 4A). There were no significant differences before versus after placebo in any of the five TOIs (p > 0.005, corrected for multiple comparisons) (Fig. 4A).
Figure 4.
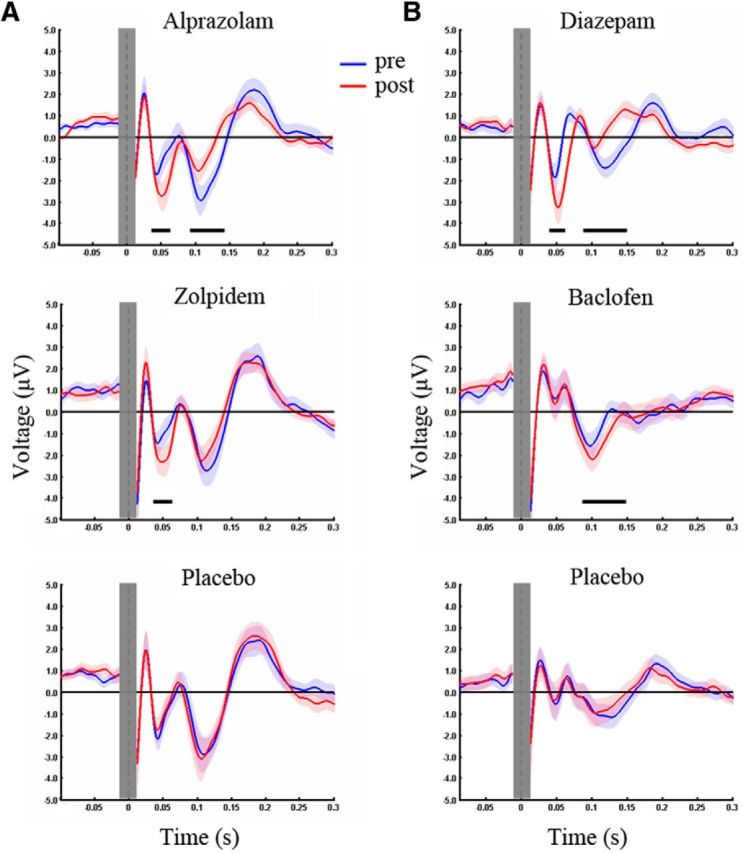
Drug-induced modulation of TEPs. TEPs were recorded before (Pre, blue) and after (Post, red) intake of a single oral dose of alprazolam, zolpidem, or placebo (Experiment 1, A), and diazepam, baclofen, or placebo (Experiment 2, B). Whereas alprazolam increased the N45 and reduced the N100 amplitude, zolpidem increased the N45 only. Diazepam increased the N45 and decreased the N100 similarly to alprazolam, whereas baclofen increased the N100. Black bars underneath represent significant drug-induced changes in TEPs. The baseline was corrected between −500 ms and −100 ms. To illustrate drug-induced changes of TEP components, representative channels were chosen for each drug condition. Shades represent ± SEM. For further information see also Figure 3.
Topographical plots of EEG surface voltages of the drug-induced changes of the N45 (alprazolam, Fig. 5A; zolpidem, Fig. 5C) and N100 (alprazolam, Fig. 5B) showed central and right hemispheric changes (i.e., changes contralateral to the stimulation site over left M1 hand area). Spearman correlation analysis revealed a significant positive correlation between the N45 amplitude modulation (postdrug − predrug intake) in the alprazolam and zolpidem condition (r2 = 0.29, p = 0.014; Fig. 6; average of common significant channels F2, FC2, FC4, FC6, FT8, C4, C6, T8, CP4, CP6; compare Fig. 5A,C), suggesting activation of α1-containing GABAARs as the common mechanism of action.
Figure 5.

Topographical plots for TEP components N45 and N100 before and after intake of alprazolam and zolpidem (Experiment 1). Topographical distributions of the TEP amplitudes (color coded in μV) before (PRE, left column) and after (POST, middle column) alprazolam (A, N45; B, N100) and zolpidem (C, N45). Large crosses indicate site of TMS of the hand area of left-hemispheric M1. Right column, t-statistic maps of the TEP amplitude postdrug versus predrug differences. Crosses indicate significant channels, predominantly in the nonstimulated right hemisphere. Red represents increase in negativity; blue represents decrease in negativity. For further details, see Material and Methods.
Figure 6.

Correlation between modulation of the N45 amplitude by alprazolam and zolpidem (Experiment 1). A significant correlation was found between modulation of the N45 amplitude by alprazolam (x-axis) and modulation of the N45 amplitude by zolpidem (y-axis) (Spearman correlation coefficient, r2 = 0.29, p = 0.014).
These results were confirmed by a cluster-corrected analysis between the postdrug conditions. Compared with placebo, the N45 potential under alprazolam and zolpidem was larger (alprazolam vs placebo: p = 0.004, significant channels: FT8, Cz, C2, C6, T8, CP2, CP4, CP6; zolpidem vs placebo: p < 0.001, significant channels: FC2, FC4, FC6, FT8, C2, C4, C6, CPz, CP2, CP4, CP6, Pz, P2, P6, POz, PO3, PO4, O2). In addition, the N100 potential under alprazolam was smaller compared with placebo (p = 0.001, significant channels: F8, FC2, FC4, FC6, FT8, C2, C4, C6, T8, CP4, CP6). Finally, the N100 potential under zolpidem showed a nonsignificant trend toward a decrease compared with placebo (p > 0.005, corrected for multiple comparisons; see Material and Methods).
In addition, we studied drug-induced changes in N45 and N100 potentials at the level of individual subjects. Although most of the subjects showed increases in the N45 potential after intake of alprazolam and zolpidem (see Fig. 8A) and a reduction in the N100 potential after intake of alprazolam (see Fig. 8B), a few subjects had opposite effects compared with the group on average.
Figure 8.

Drug-induced changes of TEPs (single subject data). Scatter plots of individual amplitude modulations (postdrug − predrug) of the N45 (A, C) and N100 (B, D) TEP components from Experiments 1 and 2, respectively. The amplitudes were extracted for common significant channels in the alprazolam and zolpidem condition (N45: A, compare Fig. 5A,C), significant channels in the alprazolam condition (N100: B, compare Fig. 5B), and significant channels in the diazepam condition (N45: C, compare Fig. 7A). D, Data were taken from significant channels in the diazepam and baclofen condition, respectively (as diazepam changed the N100 contralateral to the stimulation site, whereas baclofen changed the N100 ipsilateral to the stimulation site; compare Fig. 7B,C). For placebo, changes in the N100 were extracted from significant channels in diazepam and baclofen conditions. Error bars indicate group mean ± SEM.
To investigate whether drug-induced fluctuations in oscillatory neural activity underlie the observed modulations in TEPs, we analyzed EEG power spectra at baseline (i.e., −500 to −100 ms before TMS) predrug/postdrug intake and correlated them with drug-induced TEP changes. Results showed no significant correlations between N45 or N100 amplitude modulation and power spectra modulation in any of the frequency bands analyzed (i.e., θ, α, β, and γ; for definition see Material and Methods) by alprazolam, or between N45 amplitude modulation and θ, α, β, or γ power spectra modulation by zolpidem (all p > 0.05), suggesting that TEP changes are not based on drug-induced changes in oscillatory neural activity.
Correlation between drug-induced effects on SPV and TEPs
SPV is a measure of sedation known to be mediated by α1-containing GABAARs (de Visser et al., 2003). Likewise, the above data suggested that modulation of the N45 TEP component by alprazolam and zolpidem is mediated by α1-containing GABAARs. However, we did not find a correlation between alprazolam- and zolpidem-induced changes (predrug − postdrug) of SPV and N45 amplitude (both p > 0.05).
Experiment 2
Drug effects on SPV
Repeated-measures ANOVA of drug effects on SPV showed a significant main effect of TIME (F(1,14) = 15.15, p = 0.002) but not of DRUG (F(2,28) = 1.84, p = 0.110), and a significant interaction between TIME and DRUG (F(2,28) = 10.17, p < 0.001), which was explained by a significant reduction of SPV 90 min after intake of diazepam (p = 0.002) (Fig. 2B). There was no effect of baclofen or placebo on SPV (both p > 0.05). These data confirmed and extended the results from Experiment 1 that reduction of SPV as a biomarker of sedation is mediated by α1-subunit-containing GABAARs (de Visser et al., 2003; de Haas et al., 2009, 2010), whereas reduction of SPV is not mediated by GABABRs.
Drug effects on TEPs
RMT was not significantly different between the predrug conditions (diazepam: 44.6 ± 1.3% MSO; baclofen: 44.1 ± 1.13% MSO; placebo: 44.8 ± 1.18% MSO; F(2,28) = 1.12, p = 0.34).
The grand-average TMS-evoked EEG response after single-pulse TMS of the left M1 confirmed the results from Experiment 1, with the most prominent TEP components being P25, N45, P70, N100, and P180 (Fig. 3B). Likewise, topographical surface voltage maps reproduced that early TEPs are located in the stimulated sensorimotor cortex (P25) and contralateral regions (N45), whereas late TEPs are located in frontocentral sites (Fig. 3B).
As in Experiment 1, a cluster-based permutation analysis in the five nonoverlapping TOIs (P25, 20–37 ms; N45, 40–60 ms; P70, 65–85 ms; N100, 88–150 ms; P180, 150–230 ms) showed no significant difference in TEP amplitudes between the predrug conditions (all TOIs, p > 0.005, corrected for multiple comparisons). Diazepam increased the N45 (pre: −2.19 ± 0.52 μV; post: −3.71 ± 0.54 μV; p < 0.001) and reduced the N100 (pre: −3.01 ± 0.41 μV; post: −1.07 ± 0.16 μV; p < 0.001) (Fig. 4B). In contrast, the GABABR agonist baclofen significantly increased the N100 (pre: −2.21 ± 0.34 μV; post: −2.97 ± 0.30 μV; p = 0.003) but had no effect on the N45 component (p > 0.005, corrected for multiple comparison) (Fig. 4B). In addition, there were no significant differences before versus after placebo in any of the five TOIs (p > 0.005, corrected for multiple comparisons) (Fig. 4B).
Topographical plots of surface voltages of diazepam-induced changes of TEPs showed a right hemispheric modulation of the N45 and N100 potentials (i.e., contralateral to the stimulation site) (Fig. 7A,B), very similar as observed in the alprazolam condition of Experiment 1 (compare Fig. 5A,B). In contrast, baclofen-induced changes in TEPs were predominantly located over the left (i.e., stimulated hemisphere) (Fig. 7C).
Figure 7.

Topographical plots for TEP components N45 and N100 after intake of baclofen and diazepam (Experiment 2). Topographical distributions of the TEP amplitudes (color coded in μV) before (PRE, left column) and after (POST, middle column) diazepam (A, N45; B, N100) and baclofen (C, N100). Large crosses indicate site of TMS of the hand area of left-hemispheric M1. Right column, t-statistic maps of the TEP amplitude postdrug versus predrug differences. Crosses indicate significant channels, predominantly in the nonstimulated right hemisphere for diazepam, and in the stimulated left hemisphere for baclofen. Red represents increase in negativity; blue represents decrease in negativity. For further details, see Material and Methods.
These drug-induced effects on TEPs could be confirmed when comparing the postdrug TEP amplitudes across drug conditions. Compared with placebo, the N45 potential under diazepam was larger (p = 0.001, significant channels: FC2, FT8, Cz, C2, C4, C6, T8, CP2, CP4, CP6, TP8, P2, P4, P6, P8, PO4, PO8, O2) and the N100 potential under diazepam was smaller (p = 0.002, significant channels: FC2, FC4, FC6, FT8, C2, C4, C6, T8, CP4, CP6, TP8). The N100 potential under baclofen showed a nonsignificant trend toward an increase compared with placebo (p = 0.04 in channels: C3, C1, CP5, CP3, P5, P3, and PO3).
In addition, investigation of drug-induced changes in N45 and N100 potentials (postdrug/predrug) at the level of individual subjects showed that most of the subjects showed an increase in the N45 potential (Fig. 8C) and a decrease in the N100 potential (Fig. 8D) after intake of diazepam, whereas most of the subjects showed an increase in the N100 potential after intake of baclofen (Fig. 8D). As in Experiment 1, few subjects showed opposite drug effects compared with the group on average.
Importantly, these drug-induced modulations of TEP components cannot be explained by drug-induced changes in oscillatory neural activity, as we did not find any significant correlation between diazepam- or baclofen-induced changes in θ, α, β, or γ power spectra at baseline (i.e., −500 to −100 ms before TMS) predrug/postdrug intake and diazepam- or baclofen-induced N45 and N100 amplitude changes, respectively (all p > 0.05). These findings thus confirm and extend the results from Experiment 1 that enhancement of neurotransmission through GABAARs by diazepam increases early (N45) and decreases late (N100) TEP components predominantly in the nonstimulated hemisphere, enhancement of neurotransmission through the GABABR by baclofen selectively increases the N100 in the stimulated hemisphere.
Correlation between drug-induced effects on SPV and TEPs
Similar as for alprazolam in Experiment 1, we did not find a significant correlation between diazepam-induced changes of SPV and N45 amplitude (p > 0.05).
Discussion
Using pharmacological modulation of neurotransmission through the GABAAR and GABABR in combination with TMS-EEG, we show here, for the first time, that activity of α1-subunit-containing GABAARs contributes to early TEPs (the N45 potential), whereas activity of GABABRs contributes to a later TEP (the N100 potential). These findings are discussed in detail below.
The role of GABAARs in TEPs
A contribution of GABAAergic neurotransmission to TEPs has been investigated before in two studies using paired-pulse TMS at a short interstimulus interval of 3 ms to measure short-interval intracortical inhibition, a marker of GABAAR activity (Ziemann et al., 1996). MEP amplitudes were in line with short-interval intracortical inhibition in both studies, whereas the effects of paired-pulse TMS on TEPs were inconsistent. In one study, paired-pulse TMS had no significant effect on the N45 and N100 amplitudes compared with single-pulse TMS (Paus et al., 2001), whereas the other study demonstrated a focal increase in N45 and N100 amplitudes in the stimulated hemisphere by paired-pulse TMS (Ferreri et al., 2011).
In this study, we applied subtype-specific modulators at the GABAAR to explore the contribution of GABAAergic neurotransmission on specific TEP components. In Experiment 1, the positive modulators at the GABAAR alprazolam and zolpidem significantly increased the N45 amplitude. Moreover, analysis of common channels of significant drug effects showed that the increase in the N45 potential in both drug conditions was positively correlated, suggesting a common underlying mechanism. In Experiment 2, we replicated the effect of GABAARs on the N45 potential by the classical benzodiazepine diazepam. As alprazolam, zolpidem, and diazepam show a common receptor profile targeting the α1-subunit of the GABAAR, our data strongly suggest that activation of α1-subunit-containing GABAARs contributes to the generation of the N45 potential.
Topographical plots of surface voltages of drug-induced changes in the N45 potential revealed that these changes were predominantly located in the hemisphere contralateral to the site of stimulation. In contrast, previous reports showed a dose-dependent increase in early TEPs at the site of stimulation during midazolam-induced loss of consciousness, whereas later TEPs were suppressed by midazolam along with a breakdown in cortical effective connectivity (Ferrarelli et al., 2010). This discrepancy may be the result of the different sites of stimulation (right premotor cortex in the previous study, left M1 in our study) or the level of consciousness (loss of consciousness in the previous study, slight sedation in our study). However, comparison between the two studies is also limited by the fact that topographical distributions of surface voltages do not allow to make conclusive inferences about the location of changes in the underlying neuronal circuits. To address this issue, source modeling analysis on TMS-evoked EEG responses is needed. Whatever the underlying mechanism, the topography of drug-induced TEP changes matched the topography of the maximum negativity of the N45 before drug intake (compare right vs left columns in Figs. 5A,C and 7A).
Notably, we did not find a correlation between drug-induced changes in our behavioral measure (SPV) and modulation of the N45 potential by alprazolam and zolpidem (Experiment 1) or diazepam (Experiment 2). Because the drug-induced changes in both measures are mediated by activation of α1-subunit-containing GABAARs, this lack of a correlation suggests a different role for GABAAergic neurotransmission in visuomotor processes in specific frontoparietal networks (i.e., frontal, supplementary, and parietal eye fields) (Luna et al., 1998) and signal propagation of the N45 from left M1 stimulation in a widespread distributed cortical network.
In contrast to the similar effect of alprazolam, zolpidem, and diazepam on the N45 amplitude, alprazolam and diazepam, but not zolpidem, reduced the N100 amplitude. Topographical plots of surface voltages showed that the N100 potential, after drug intake, is lateralized to the site of stimulation but distributed across both hemispheres before drug intake (compare Figs. 5B and 7B,C). Drug-induced changes of the N100 potential showed a spatial dissociation: whereas the alprazolam- and diazepam-induced N100 amplitude decreases were only significant in channels in the hemisphere contralateral to the stimulation (compare Figs. 5B and 7B), baclofen-induced N100 amplitude increases were located in close proximity to the stimulation site (compare Fig. 7C).
The mechanisms of these drug effects are not clear. Reduction of the N100 potential in the hemisphere contralateral to the stimulation by alprazolam and diazepam, but not zolpidem, may be explained by a sedation-related suppression of signal propagation from the stimulated to the nonstimulated hemisphere (compare Figs. 5B and 7B). An extensive breakdown of long-range corticocortical effective connectivity as measured by late TEPs has been observed during midazolam-induced loss of consciousness (Ferrarelli et al., 2010) and during NREM sleep (Massimini et al., 2005).
Alternatively, propagation of the N100 from the stimulated to the nonstimulated hemisphere may involve cortico-subcortical-cortical loops (e.g., via the thalamus). The thalamus is a major subcortical hub and essential for sensory processing and motor output (Huguenard and McCormick, 2007). Importantly, thalamocortical cells have an excitatory input but are encapsulated by two inhibitory shell-like nuclei: the thalamic reticular and perigeniculate nuclei (RT/PGN cells). This sophisticated and well-balanced excitatory/inhibitory drive of thalamocortical cells is critical for orchestrating thalamocortical oscillations. It is known from animal experiments that RT and PGN cells provide intrathalamic desynchronizing activity mediated by α3-GABAARs, whereas α1-GABAARs located at the level of thalamocortical cells are pro-oscillatory (Huguenard and McCormick, 2007). Given the higher binding affinity of alprazolam and diazepam than zolpidem to α3-subunit-containing GABAARs, alprazolam/diazepam may favor antioscillatory activity within the thalamocortical loop, thus suppressing thalamocortical communication. This disruption of thalamocortical connectivity by alprazolam/diazepam may alternatively explain reductions in N100 signal propagation from the stimulated to the nonstimulated hemisphere under these conditions. However, further analyses, including source modeling, are needed to clarify precisely the propagation of TEPs and its change by drugs.
Finally, cellular experiments demonstrated that activation of GABAARs inhibits sIPSP generated in neocortical and hippocampal pyramidal neurons by GABABRs (Lopantsev and Schwartzkroin, 1999; Thomson and Destexhe, 1999), probably through an increase in intracellular chloride concentration that inhibits the potassium conductance underlying the GABABR-mediated sIPSP (Lenz et al., 1997). This may be another mechanism to account for the observed decrease of the N100 amplitude by alprazolam and diazepam. This explanation would fit with the observation that diazepam, and lorazepam, another benzodiazepine, resulted in shortening of the cortical silent period duration (Inghilleri et al., 1996; Kimiskidis et al., 2006), a TMS-EMG measure of GABABergic neurotransmission (Siebner et al., 1998).
The role of GABABR-mediated neurotransmission for TEPs
The N100 is the most pronounced and reproducible TEP component and has been related to sIPSP (Nikulin et al., 2003; Bender et al., 2005). Specifically, the N100 amplitude is increased under augmented inhibition, such as during preparation to resist a forthcoming TMS evoked movement (Bonnard et al., 2009), although it is decreased during preparation to assist a TMS-evoked movement (Nikulin et al., 2003), motor response preparation (Bender et al., 2005), or motor performance (Kicić et al., 2008). A recent study showed that the N100 amplitude is related to the duration of the cortical silent period (Farzan et al., 2013). In Experiment 2 of our study, baclofen, but not any of the GABAAR modulators, increased the N100 amplitude, thus strongly corroborating a role of GABABRs in the generation of the N100 potential.
Importantly, none of these drug-induced modulations of TEP components can be explained by drug-induced changes in oscillatory neural activity (Supp et al., 2011), as we did not find any significant correlation between drug-induced changes in θ-, [alpha-], β-, or γ-power at baseline (i.e., −500 to −100 ms before TMS) and drug-induced N45 and N100 amplitude changes.
In clinical practice, it would be highly interesting to use TMS-EEG recordings to study, or even monitor longitudinally, the state of neural circuits in individual patients. Previous work in healthy subjects showed that TMS-evoked cortical responses essentially reflect deterministic properties of the stimulated network and thus, indeed, may be used to detect longitudinal changes in the state of cortical circuits (Casarotto et al., 2010). However, our pharmaco-TMS-EEG experiments showed notable interindividual variability of drug effects on TEPs, with some subjects showing opposite drug effects with respect to the group average. Thus, interpretation of TMS-EEG recordings at the single-subject level is difficult and may be limited. Clearly, further studies are needed to explore the full potential of TMS-EEG recordings for analysis of perturbation-induced (i.e., drug-, training-, noninvasive brain stimulation-, or disease-related) effects on cerebral networks in individual subjects/patients.
In conclusion, here we used a novel pharmaco-TMS-EEG approach to study the effect of CNS-active drugs directly on cortical activity. Our data demonstrate a differential effect of GABAAergic versus GABABergic neurotransmission on specific components of TMS-evoked EEG activity. Findings suggest that TMS-EEG may be used to study GABAAergic and GABABergic inhibition in neurological and psychiatric disorders, in which abnormal cortical inhibition has been implicated in the pathophysiology, such as epilepsy or schizophrenia (Jones-Davis and Macdonald, 2003; Lewis et al., 2005). In addition, pharmaco-TMS-EEG offers the opportunity to study the actions of CNS-active drugs on human cortex in greater detail than it is hitherto possible with TMS or EEG alone.
Footnotes
This work was supported by the Centre of Integrative Neuroscience Tübingen to U.Z. We thank Dr. Mario Rosanova for his technical support, and Dr. Markus Siegel and Professor John R. Huguenard for helpful discussions on the manuscript.
The authors declare no competing financial interests.
References
- Bender S, Basseler K, Sebastian I, Resch F, Kammer T, Oelkers-Ax R, Weisbrod M. Electroencephalographic response to transcranial magnetic stimulation in children: evidence for giant inhibitory potentials. Ann Neurol. 2005;58:58–67. doi: 10.1002/ana.20521. [DOI] [PubMed] [Google Scholar]
- Blom MW, Bartel PR, de Sommers K, Van der Meyden CH, Becker PJ. The effects of alprazolam, quazepam and diazepam on saccadic eye movements, parameters of psychomotor function and the EEG. Fundam Clin Pharmacol. 1990;4:653–661. doi: 10.1111/j.1472-8206.1990.tb00045.x. [DOI] [PubMed] [Google Scholar]
- Bonato C, Miniussi C, Rossini PM. Transcranial magnetic stimulation and cortical evoked potentials: a TMS/EEG co-registration study. Clin Neurophysiol. 2006;117:1699–1707. doi: 10.1016/j.clinph.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Bonnard M, Spieser L, Meziane HB, de Graaf JB, Pailhous J. Prior intention can locally tune inhibitory processes in the primary motor cortex: direct evidence from combined TMS-EEG. Eur J Neurosci. 2009;30:913–923. doi: 10.1111/j.1460-9568.2009.06864.x. [DOI] [PubMed] [Google Scholar]
- Bruckmann S, Hauk D, Roessner V, Resch F, Freitag CM, Kammer T, Ziemann U, Rothenberger A, Weisbrod M, Bender S. Cortical inhibition in attention deficit hyperactivity disorder: new insights from the electroencephalographic response to transcranial magnetic stimulation. Brain. 2012;135:2215–2230. doi: 10.1093/brain/aws071. [DOI] [PubMed] [Google Scholar]
- Casarotto S, Romero Lauro LJ, Bellina V, Casali AG, Rosanova M, Pigorini A, Defendi S, Mariotti M, Massimini M. EEG responses to TMS are sensitive to changes in the perturbation parameters and repeatable over time. PloS One. 2010;5:e10281. doi: 10.1371/journal.pone.0010281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW, Malenka RC, Silva LR. Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol. 1988;406:443–468. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- de Haas SL, Franson KL, Schmitt JA, Cohen AF, Fau JB, Dubruc C, van Gerven JM. The pharmacokinetic and pharmacodynamic effects of SL65.1498, a GABA-A alpha2,3 selective agonist, in comparison with lorazepam in healthy volunteers. J Psychopharmacol. 2009;23:625–632. doi: 10.1177/0269881108092595. [DOI] [PubMed] [Google Scholar]
- de Haas SL, Schoemaker RC, van Gerven JM, Hoever P, Cohen AF, Dingemanse J. Pharmacokinetics, pharmacodynamics and the pharmacokinetic/pharmacodynamic relationship of zolpidem in healthy subjects. J Psychopharmacol. 2010;24:1619–1629. doi: 10.1177/0269881109106898. [DOI] [PubMed] [Google Scholar]
- Deisz RA. GABA(B) receptor-mediated effects in human and rat neocortical neurones in vitro. Neuropharmacology. 1999;38:1755–1766. doi: 10.1016/S0028-3908(99)00136-7. [DOI] [PubMed] [Google Scholar]
- de Visser SJ, van der Post JP, de Waal PP, Cornet F, Cohen AF, van Gerven JM. Biomarkers for the effects of benzodiazepines in healthy volunteers. Br J Clin Pharmacol. 2003;55:39–50. doi: 10.1046/j.1365-2125.2002.t01-10-01714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lazzaro V, Pilato F, Dileone M, Ranieri F, Ricci V, Profice P, Bria P, Tonali PA, Ziemann U. GABAA receptor subtype specific enhancement of inhibition in human motor cortex. J Physiol. 2006;575:721–726. doi: 10.1113/jphysiol.2006.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lazzaro V, Pilato F, Dileone M, Profice P, Ranieri F, Ricci V, Bria P, Tonali PA, Ziemann U. Segregating two inhibitory circuits in human motor cortex at the level of GABAA receptor subtypes: a TMS study. Clin Neurophysiol. 2007;118:2207–2214. doi: 10.1016/j.clinph.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Ziemann U, Lemon RN. State of the art: physiology of transcranial motor cortex stimulation. Brain Stimul. 2008;1:345–362. doi: 10.1016/j.brs.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Fritschy JM, Löw K, Möhler H, Rudolph U, Hensch TK. Specific GABAA circuits for visual cortical plasticity. Science. 2004;303:1681–1683. doi: 10.1126/science.1091032. [DOI] [PubMed] [Google Scholar]
- Farzan F, Barr MS, Hoppenbrouwers SS, Fitzgerald PB, Chen R, Pascual-Leone A, Daskalakis ZJ. The EEG correlates of the TMS-induced EMG silent period in humans. Neuroimage. 2013;83C:120–134. doi: 10.1016/j.neuroimage.2013.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarelli F, Massimini M, Sarasso S, Casali A, Riedner BA, Angelini G, Tononi G, Pearce RA. Breakdown in cortical effective connectivity during midazolam-induced loss of consciousness. Proc Natl Acad Sci U S A. 2010;107:2681–2686. doi: 10.1073/pnas.0913008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreri F, Pasqualetti P, Määttä S, Ponzo D, Ferrarelli F, Tononi G, Mervaala E, Miniussi C, Rossini PM. Human brain connectivity during single and paired pulse transcranial magnetic stimulation. Neuroimage. 2011;54:90–102. doi: 10.1016/j.neuroimage.2010.07.056. [DOI] [PubMed] [Google Scholar]
- Florian J, Müller-Dahlhaus M, Liu Y, Ziemann U. Inhibitory circuits and the nature of their interactions in the human motor cortex a pharmacological TMS study. J Physiol. 2008;586:495–514. doi: 10.1113/jphysiol.2007.142059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt DJ, Wright CE. Clinical pharmacokinetics of alprazolam: therapeutic implications. Clin Pharmacokinet. 1993;24:453–471. doi: 10.2165/00003088-199324060-00003. [DOI] [PubMed] [Google Scholar]
- Groppa S, Oliviero A, Eisen A, Quartarone A, Cohen LG, Mall V, Kaelin-Lang A, Mima T, Rossi S, Thickbroom GW, Rossini PM, Ziemann U, Valls-Solé J, Siebner HR. A practical guide to diagnostic transcranial magnetic stimulation: report of an IFCN committee. Clin Neurophysiol. 2012;123:858–882. doi: 10.1016/j.clinph.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett M. Transcranial magnetic stimulation: a primer. Neuron. 2007;55:187–199. doi: 10.1016/j.neuron.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Ilmoniemi RJ, Virtanen J, Ruohonen J, Karhu J, Aronen HJ, Näätänen R, Katila T. Neuronal responses to magnetic stimulation reveal cortical reactivity and connectivity. Neuroreport. 1997;8:3537–3540. doi: 10.1097/00001756-199711100-00024. [DOI] [PubMed] [Google Scholar]
- Inghilleri M, Berardelli A, Marchetti P, Manfredi M. Effects of diazepam, baclofen and thiopental on the silent period evoked by transcranial magnetic stimulation in humans. Exp Brain Res. 1996;109:467–472. doi: 10.1007/BF00229631. [DOI] [PubMed] [Google Scholar]
- Jones-Davis DM, Macdonald RL. GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Curr Opin Pharmacol. 2003;3:12–18. doi: 10.1016/S1471-4892(02)00015-2. [DOI] [PubMed] [Google Scholar]
- Kicić D, Lioumis P, Ilmoniemi RJ, Nikulin VV. Bilateral changes in excitability of sensorimotor cortices during unilateral movement: combined electroencephalographic and transcranial magnetic stimulation study. Neuroscience. 2008;152:1119–1129. doi: 10.1016/j.neuroscience.2008.01.043. [DOI] [PubMed] [Google Scholar]
- Kimiskidis VK, Papagiannopoulos S, Kazis DA, Sotirakoglou K, Vasiliadis G, Zara F, Kazis A, Mills KR. Lorazepam-induced effects on silent period and corticomotor excitability. Exp Brain Res. 2006;173:603–611. doi: 10.1007/s00221-006-0402-1. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Pitler TA, Alger BE. High intracellular Cl− concentrations depress G-protein-modulated ionic conductances. J Neurosci. 1997;17:6133–6141. doi: 10.1523/JNEUROSCI.17-16-06133.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lioumis P, Kicić D, Savolainen P, Mäkelä JP, Kähkönen S. Reproducibility of TMS-Evoked EEG responses. Hum Brain Mapp. 2009;30:1387–1396. doi: 10.1002/hbm.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopantsev V, Schwartzkroin PA. GABAA-dependent chloride influx modulates GABAB-mediated IPSPs in hippocampal pyramidal cells. J Neurophysiol. 1999;82:1218–1223. doi: 10.1152/jn.1999.82.3.1218. [DOI] [PubMed] [Google Scholar]
- Luna B, Thulborn KR, Strojwas MH, McCurtain BJ, Berman RA, Genovese CR, Sweeney JA. Dorsal cortical regions subserving visually guided saccades in humans: an fMRI study. Cereb Cortex. 1998;8:40–47. doi: 10.1093/cercor/8.1.40. [DOI] [PubMed] [Google Scholar]
- Maris E, Oostenveld R. Nonparametric statistical testing of EEG- and MEG-data. J Neurosci Methods. 2007;164:177–190. doi: 10.1016/j.jneumeth.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Massimini M, Ferrarelli F, Huber R, Esser SK, Singh H, Tononi G. Breakdown of cortical effective connectivity during sleep. Science. 2005;309:2228–2232. doi: 10.1126/science.1117256. [DOI] [PubMed] [Google Scholar]
- McDonnell MN, Orekhov Y, Ziemann U. The role of GABA(B) receptors in intracortical inhibition in the human motor cortex. Exp Brain Res. 2006;173:86–93. doi: 10.1007/s00221-006-0365-2. [DOI] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- Nikulin VV, Kicić D, Kähkönen S, Ilmoniemi RJ. Modulation of electroencephalographic responses to transcranial magnetic stimulation: evidence for changes in cortical excitability related to movement. Eur J Neurosci. 2003;18:1206–1212. doi: 10.1046/j.1460-9568.2003.02858.x. [DOI] [PubMed] [Google Scholar]
- Nikulin VV, Linkenkaer-Hansen K, Nolte G, Lemm S, Müller KR, Ilmoniemi RJ, Curio G. A novel mechanism for evoked responses in the human brain. Eur J Neurosci. 2007;25:3146–3154. doi: 10.1111/j.1460-9568.2007.05553.x. [DOI] [PubMed] [Google Scholar]
- Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia. 1971;9:97–113. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- Oostenveld R, Fries P, Maris E, Schoffelen JM. FieldTrip: open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Comput Intell Neurosci. 2011;2011:156869. doi: 10.1155/2011/156869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus T, Sipila PK, Strafella AP. Synchronization of neuronal activity in the human primary motor cortex by transcranial magnetic stimulation: an EEG study. J Neurophysiol. 2001;86:1983–1990. doi: 10.1152/jn.2001.86.4.1983. [DOI] [PubMed] [Google Scholar]
- Ragazzoni A, Pirulli C, Veniero D, Feurra M, Cincotta M, Giovannelli F, Chiaramonti R, Lino M, Rossi S, Miniussi C. Vegetative versus minimally conscious states: a study using TMS-EEG, sensory and event-related potentials. PloS One. 2013;8:e57069. doi: 10.1371/journal.pone.0057069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogasch NC, Fitzgerald PB. Assessing cortical network properties using TMS-EEG. Hum Brain Mapp. 2013;34:1652–2669. doi: 10.1002/hbm.22016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosanova M, Gosseries O, Casarotto S, Boly M, Casali AG, Bruno MA, Mariotti M, Boveroux P, Tononi G, Laureys S, Massimini M. Recovery of cortical effective connectivity and recovery of consciousness in vegetative patients. Brain. 2012;135:1308–1320. doi: 10.1093/brain/awr340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, Hallett M, Rossini PM, Pascual-Leone A. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol. 2009;120:2008–2039. doi: 10.1016/j.clinph.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvà P, Costa J. Clinical pharmacokinetics and pharmacodynamics of zolpidem: therapeutic implications. Clin Pharmacokinet. 1995;29:142–153. doi: 10.2165/00003088-199529030-00002. [DOI] [PubMed] [Google Scholar]
- Shader RI, Pary RJ, Harmatz JS, Allison S, Locniskar A, Greenblatt DJ. Plasma concentrations and clinical effects after single oral doses of prazepam, clorazepate, and diazepam. J Clin Psychiatry. 1984;45:411–413. [PubMed] [Google Scholar]
- Siebner HR, Dressnandt J, Auer C, Conrad B. Continuous intrathecal baclofen infusions induced a marked increase of the transcranially evoked silent period in a patient with generalized dystonia. Muscle Nerve. 1998;21:1209–1212. doi: 10.1002/(SICI)1097-4598(199809)21:9<1209::AID-MUS15>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Keel JC, Greenberg BD, Adams LF, Schmidt PJ, Rubinow DA, Wassermann EM. Menstrual cycle effects on cortical excitability. Neurology. 1999;53:2069–2072. doi: 10.1212/WNL.53.9.2069. [DOI] [PubMed] [Google Scholar]
- Supp GG, Siegel M, Hipp JF, Engel AK. Cortical hypersynchrony predicts breakdown of sensory processing during loss of consciousness. Curr Biol. 2011;21:1988–1993. doi: 10.1016/j.cub.2011.10.017. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Destexhe A. Dual intracellular recordings and computational models of slow inhibitory postsynaptic potentials in rat neocortical and hippocampal slices. Neuroscience. 1999;92:1193–1215. doi: 10.1016/S0306-4522(99)00021-4. [DOI] [PubMed] [Google Scholar]
- Thut G, Veniero D, Romei V, Miniussi C, Schyns P, Gross J. Rhythmic TMS causes local entrainment of natural oscillatory signatures. Curr Biol. 2011;21:1176–1185. doi: 10.1016/j.cub.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron. 2012;75:963–980. doi: 10.1016/j.neuron.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Velázquez-Pérez L, Seifried C, Santos-Falcón N, Abele M, Ziemann U, Almaguer LE, Martínez-Góngora E, Sánchez-Cruz G, Canales N, Pérez-González R, Velázquez-Manresa M, Viebahn B, von Stuckrad-Barre S, Fetter M, Klockgether T, Auburger G. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol. 2004;56:444–447. doi: 10.1002/ana.20220. [DOI] [PubMed] [Google Scholar]
- Veniero D, Ponzo V, Koch G. Paired associative stimulation enforces the communication between interconnected areas. J Neurosci. 2013;33:13773–13783. doi: 10.1523/JNEUROSCI.1777-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann U, Lönnecker S, Steinhoff BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man. Exp Brain Res. 1996;109:127–135. doi: 10.1007/BF00228633. [DOI] [PubMed] [Google Scholar]