

Abstract
Netrin and its receptor, Frazzled, dictate the strength of synaptic connections in the giant fiber system (GFS) of Drosophila melanogaster by regulating gap junction localization in the presynaptic terminal. In Netrin mutant animals, the synaptic coupling between a giant interneuron and the “jump” motor neuron was weakened and dye coupling between these two neurons was severely compromised or absent. In cases in which Netrin mutants displayed apparently normal synaptic anatomy, half of the specimens exhibited physiologically defective synapses and dye coupling between the giant fiber (GF) and the motor neuron was reduced or eliminated, suggesting that gap junctions were disrupted in the Netrin mutants. When we examined the gap junctions with antibodies to Shaking-B (ShakB) Innexin, they were significantly decreased or absent in the presynaptic terminal of the mutant GF. Frazzled loss of function mutants exhibited similar defects in synaptic transmission, dye coupling, and gap junction localization. These data are the first to show that Netrin and Frazzled regulate the placement of gap junctions presynaptically at a synapse.
Keywords: Drosophila, electrophysiology, Frazzled, gap junctions, giant fiber system, Netrin
Introduction
Netrin expression in different glia and neurons creates a hierarchy of Netrin cues necessary for CNS assembly in flies and worms. Netrin was first identified in Caenorhabditis elegans, in which mutants displayed axonal guidance and pioneer cell migration defects (Hedgecock et al., 1990; Wadsworth et al., 1996). Similarly, in the larval CNS of a Drosophila Netrin deficiency line, commissural axons were either missing or thinner compared with wild-type (Mitchell et al., 1996). Specifically, when both NetrinA and NetrinB were missing (homozygous NetAΔBΔ double mutants), commissural neurons displayed midline crossing defects (Brankatschk and Dickson, 2006). A similar phenotype was observed in commissural neurons when Frazzled, Netrin's receptor, was deleted in fra3 mutants (Kolodziej et al., 1996).
In both C. elegans and Drosophila, there is evidence that Netrin is also involved in synapse formation. In Drosophila, there are two Netrin genes, NetA and NetB, and their gene products, NetrinA and NetrinB, are crucial instructive cues for target recognition at the fly neuromuscular junction (NMJ). Axons in Netrin deficiency mutants were able to leave the CNS and orient themselves to the correct group of muscle fibers. However, the growth cones grew past their normal targets (Mitchell et al., 1996). These defects were rescued when one Netrin protein, either A or B, was expressed and secreted from a muscle fiber (Winberg et al., 1998).
Similarly, in C. elegans, glia-secreted Netrin promoted local synaptogenesis. Synaptic vesicle clustering was disrupted when Netrin-Frazzled signaling was interrupted (Colón-Ramos et al., 2007). It was later found that Mig-10, a Rac GTPase, downstream of Netrin's synaptic cue, was responsible for synaptic vesicle clustering (Stavoe and Colón-Ramos, 2012). Finally, in temperature-sensitive Netrin mutants, investigators silenced Netrin signaling in C. elegans adults and demonstrated mislocalization of synaptic components after circuit formation was complete. This showed the importance of communication between Netrin and Frazzled/Unc-40 from development through adulthood (Killeen, 2009).
Here, we demonstrate that, in addition to serving as a local guidance cue, Netrin-Frazzled signaling has a new role in electrical synapse formation in the Drosophila CNS. We show that Netrin-Frazzled signaling plays a crucial role in localization of Innexins, the invertebrate gap junction proteins (Phelan et al., 1996, 2008; Blagburn et al., 1999), in the Drosophila giant fiber (GF) presynaptic terminal.
Materials and Methods
Drosophila lines.
All lines and crosses were raised on standard medium at 22°C. All experimental animals were male (“Y” chromosome indicated with “>”) unless otherwise noted. The following stocks were used: loss-of-function (LOF) mutants NetAΔ, NetBΔ, NetABΔ (hereafter called NetAΔBΔ), and NetAΔBmyc-TM (Brankatschk and Dickson, 2006); fra3 and fra4 (Kolodziej et al., 1996); UAS-NetA and UAS-NetB (Mitchell et al., 1996); UAS-NetBCD8 (; hereafter called UAS-NetBCD8-TM; Timofeev et al., 2012), UAS-fra (Kolodziej et al., 1996); and UAS-fra-ΔC (Hiramoto et al., 2000). For labeling the components of the GFS and for rescue experiments, the UAS-GAL4 system (Brand and Perrimon, 1993) was used to express gene constructs and a green fluorescent protein (GFP) reporter gene. The following P[GAL]4 drivers were used to express constructs in the giant fiber system (GFS): P[GAL4] A307 expressed robustly presynaptically (GF) and postsynaptically (tergotrochanteral motor neuron [TTMn]) throughout pupal development (PD) into adulthood (Allen et al., 1998); P[GAL4] c17 GAL4 expressed in midline glia from 0% to 31% of PD and presynaptically in the GF from 32% of PD through adulthood; 380-slit-GAL4 (Wharton and Crews, 1993; Wheeler et al., 2012) expressed in midline glia from 0 to 75% of PD; and shakB(lethal)-GAL4 expressed postsynaptically throughout PD to adult (TTMn; Jacobs et al., 2000). Rescue experiments using the A307 GAL4 driver were limited due to the need to recombine A307 GAL4 and the frazzled mutation onto a single chromosome. Unfortunately, they are located close to one another on the second chromosome (A307 GAL4 at 50C12 and fra3 or fra4 alleles at 49A10), making it difficult to obtain the appropriate recombination event.
Electrophysiology and dye injection.
Flies were anesthetized with CO2, placed in dental wax ventral side down, and recordings were obtained from known muscles by inserting a glass microelectrode through the cuticle of the animal directly into the tergotrochanteral jump muscle (TTM) or dorsal longitudinal flight muscle (DLM; Tanouye and Wyman, 1980; Allen and Godenschwege, 2010; Augustin et al., 2011). Glass microelectrodes were filled with O'Dowd's Drosophila saline (Gu and O'Dowd, 2006) with a resistance of 40–60 ΜΩ and signals were amplified with a Getting Instruments Model 5A amplifier. GFs were stimulated extracellularly through tungsten wire electrodes placed in the eyes (Fig. 1A) using a Grass Technologies Model S48 stimulator. An Axon Digidata 1440A Data Acquisition System was used to digitize the data and recordings were collected with Clampex software (Molecular Devices). Muscle response latencies and following frequencies at 100 Hz were the two parameters recorded to test the fidelity of the circuit in control and experimental animals (Figs. 1, 2). Latency was reported in milliseconds ± SD. Wild-type TTM response latency is <0.95 ms. The following frequencies were reported in average percentage of successful muscle responses per total stimuli applied when stimulating at 100 Hz. Wild-type TTM following frequency occurs when the impaled muscle responds to ≥90% of stimuli at 100 Hz.
Figure 1.

Physiological recording from the GFS and an analysis of circuit response to repetitive stimulation. A, Schematic of the electrode placement for physiological recording. B, The circuit is composed of a pair of presynaptic giant interneurons, the GF (magenta) and the postsynaptic motor neurons (TTMn, green; DLMn, not shown). The GF projects its axons into the mesothoracic neuromere, where it synapses with the TTMn (inset). C, Wiring diagram of the circuit. The electrical synapses are shown as resistors and the chemical synapses as triangles (“G” for glutamate and “A” for acetylcholine). The giant synapse between the GF and TTMn is composed of both chemical and electrical components. D, The circuit's response at high-frequency stimulation for control, mutant, and rescue experiments. Control specimens (top red trace) responded 1:1 for every stimulation at 100 Hz. Netrin LOF mutants (bottom red trace) did not follow at 100 Hz stimulation. In each animal we tested, we calculated the probability of recording a muscle response on stimulus 1–10 within a single train of stimuli. Netrin heterozygous controls followed nearly 100% for every stimulus within a train. The Netrin LOF mutants exhibited response depression with the probability of response rate falling to ∼45% on stimuli 2–10 of a train. We shifted the following frequencies of the circuit back into near normal ranges in the rescue experiments in which we expressed Netrin under the control of various GAL4 drivers.
Figure 2.
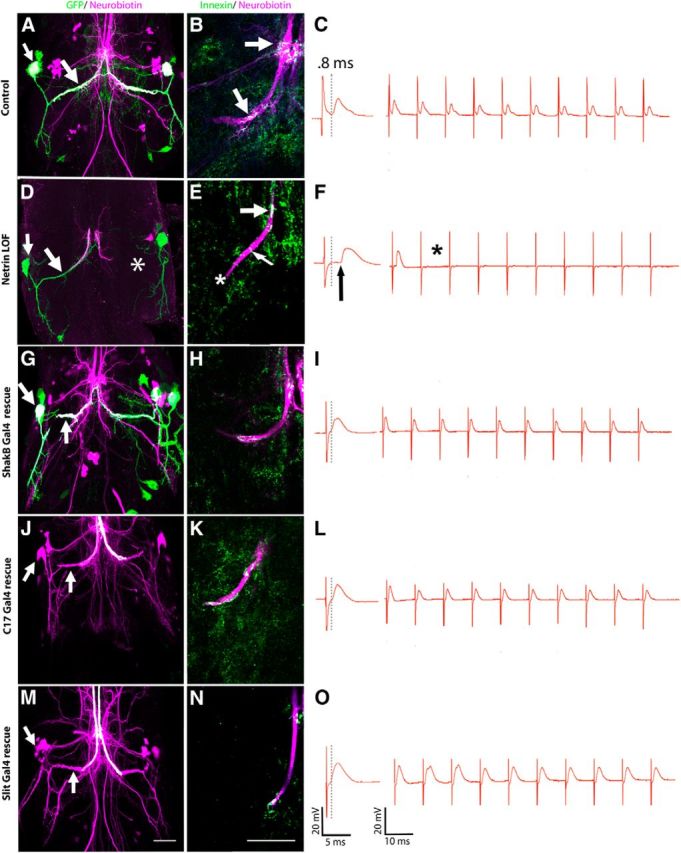
The role of Netrin in GFS synaptogenesis. First column (A, D, G, J, M) is the compressed z-stack of the GFS in the mesothoracic neuromere. Second column (B, E, H, K, N) shows Innexin labeling in the presynaptic terminal from the region indicated in Figure 1B. All panels are single images from a z-stack with 0.5 μm steps. Third column (C,F,I,L,O) is the corresponding physiological response for the left TTMn (green) during stimulation of the GFs (magenta). A, In Netrin LOF control animals, GFs exhibited terminals that dye coupled (white) to the TTMn medial dendrite and cell body (arrows). B, GFs (magenta) exhibited normal levels of Innexin (green), which colocalized (arrows) within the GF terminal (white). C, Control physiology was normal, with a latency of 0.82 ms, and the circuit responded to each stimulus at 100 Hz. D, Netrin LOF males displayed mutant phenotypes: dye coupling was lost (left GF, arrows) and the right TTMn medial dendrite (asterisk) was absent. E, Innexin labeling (green, colocalized white) at the terminal was absent or reduced (asterisk) in the Netrin LOF GF (magenta). In some cases, one to five Innexin puncta could be identified in the axon cytoplasm (thin arrow). Dye coupling was not present at the synapse. Innexin colabeling was present in the axon at the GF-PSI synaptic region (wide arrow). F, Netrin LOF mutants exhibited mutant physiology, including long latencies (arrow) and the inability of the circuit to respond 1:1 at 100 Hz stimulation (asterisk). G, When UAS-NetB was expressed in the TTMn (shakB-GAL4) of Netrin LOF mutants, GFs exhibited a rescue of dye coupling (white) in the medial dendrite and soma of TTMn (arrows). GFS anatomy was also rescued. We simultaneously expressed UAS-GFPCD8 in this experiment to label the postsynaptic cells. H, Innexin labeling in the GF terminal (white) was rescued when UAS-NetB was expressed postsynaptically. I, Physiological function was also restored by UAS-NetB expression postsynaptically. J, When rescuing the Netrin LOF mutant by driving UAS-NetB with c17 GAL4 (in midline glia), we observed a full rescue of anatomy and dye coupling to the TTMn medial dendrite and soma (arrows). TTMns are shown only in magenta (neurobiotin) because genetics would not allow simultaneous GFP expression in the TTMn. K, L, Presynaptic Innexin labeling (K) and physiological function of the circuit (L) was also rescued by midline glial expression. M, 380-slit-GAL4, a midline glial driver that expresses during pupation, rescued anatomy, and dye coupling in the Netrin LOF mutant. Dye-coupled TTMn cell body and medial dendrite are labeled in magenta and indicated with arrows. N, O, Innexin labeling (N) and physiological function (O) were also rescued by 380-slit-GAL4 expression of UAS-NetB. Scale bar, 20 μm for all images.
To measure the function of the circuit's NMJ, tungsten-stimulating electrodes were placed in the thorax of the animal directly behind the head. When a stimulus was applied, the motor neurons were stimulated directly and EPSPs were recorded from the muscle, bypassing the GF axon. Both latency and following frequency of the circuit were measured. Normal thoracic stimulation latency ranges from 0.55 to 0.6 ms. Normally, the muscle responds 1:1 for every stimulus applied at 100 Hz.
To examine the morphology of the GFS, the adult nervous system was removed and mounted in a bubble of O'Dowd's saline on a glass slide with Vectabond (Vector Laboratories; Gu and O'Dowd, 2006). Using a 40× objective and differential interference contrast optics, the GF in the neck connective was located, impaled with a glass electrode, and injected with a mixture of neurobiotin/rhodamine-dextran using depolarizing current. Glass electrodes were backfilled with 3 m potassium acetate. The dextran labeled the GF, whereas the neurobiotin labeled the GF, crossed gap junctions, and also labeled postsynaptic cells dye coupled to the GF. After dye injection, specimens were fixed in 4% paraformaldehyde. To detect the neurobiotin signal, streptavidin (1:1000) with a conjugated fluorophore Cy2, Cy3, or Dylight 649 (Vector Laboratories) was applied and specimens were examined with confocal microscopy (Boerner and Godenschwege, 2010). In some experiments, dye injections were performed with Lucifer yellow using a hyperpolarizing current and electrodes backfilled with 3 m LiCl (Uthaman et al., 2008; Boerner and Godenschwege, 2011).
To examine Shaking-B (ShakB) Innexin in GF terminals, we used a polyclonal antibody kindly provided by Pauline Phelan (University of Kent, Kent, United Kingdom; Blagburn et al., 1999; Phelan et al., 2008). This antibody recognizes the gene products of shakB that are expressed in the GFS; ShakB (n+16) and ShakB (lethal; Phelan et al., 2008). The ShakB Innexin will be referred to herein as simply Innexin. To quantify Innexin signal in GF terminals, Innexin was labeled with anti-ShakB primary antibody (1:100; Phelan et al., 2008), and Dylight 649 secondary anti-rabbit antibody was used for confocal detection. Next, samples were scanned with a Nikon C1si fast spectral confocal system with an AOTF (Nikon) laser unit using EZ-C1 software to acquire a z-stack. Images were acquired at 1024 × 1024 pixel resolution and z-step sizes were 0.05 μm. The two channels were overlaid in a TIFF stack format. Using an ImageJ colocalization plug-in, voxels that represented colocalized Innexin signal and GF signal were identified. The number of colocalized voxels of Innexin and GF signal was measured in each z-section of the TIFF stack. The number of colocalized voxels was divided by the total voxels making up the GF terminal to determine a percentage of the GF terminal occupied by colocalized voxels. The region of the GF axon that we measured Innexin staining consisted of the volume posterior to the GF-peripheral synapsing interneuron (PSI) synapse.
Pupal development.
The pupal CNS was removed at various milestones during development and the expression patterns of Netrin and Frazzled were examined. Labeling in the pupal CNS was performed with rabbit anti-Frazzled (1:200; Kolodziej et al., 1996), guinea pig anti-NetrinA or B (1:500; Albrecht et al., 2011), mouse anti-myc (1:250; Vector Laboratories), and rat or rabbit anti-GFP (1:500; Vector Laboratories) antibodies. Membrane tethered NetrinB (NetAΔB™) (Brankatschk and Dickson, 2006) was tagged with four copies of a myc epitope. Anti-myc-tag labeling was used to detect membrane-tethered NetrinB. The following secondary antibodies with conjugated fluorophores were used to detect labeling under confocal microscopy: anti-rabbit Cy2, anti-rabbit Cy3, anti-guinea pig 488, anti-mouse 488, anti-mouse 561, and anti-rat 488 (Invitrogen).
Imaging and analysis.
To perform TTMn structure analysis, 3D models of the TTMn were constructed in Neuron Studio. Single channel TIFF stacks were used to make a 3D rendering. The starburst algorithm was used to reconstruct the neuron's dendritic tree. Finally, a haulstrum layering over the starburst algorithm created a smooth surface and allowed measurement of the structures possibly missed by the starburst algorithm (Wearne et al., 2005).
Statistics.
When comparing the muscle response latencies of two genotypes, a heteroscedastic Student's two-tailed t test was used because the data were not normalized and experimental groups had unequal n's. A Kruskal–Wallis one-way ANOVA on ranks was used to identify significance between genotypes when comparing Innexin signal and dendritic parameters described in the Results section. To isolate groups that differed from one another, the Dunn's method was used to make multiple comparisons versus the control group. The α value was 0.01 for all statistical tests to ensure confidence in results. SigmaPlot was used for statistical analysis; n signifies the number of GFs (or TTMns) that were examined.
Results
Netrin LOF mutants exhibit a disrupted giant synapse
Electrophysiology of the GF-motor neuron synapse
Netrin homozygous double mutants (NetAΔBΔ/Net AΔBΔ), also referred to as LOF, died as larva. Occasional escapers of the lethal phenotype were observed in males (NetAΔBΔ/>), but not in females (NetAΔBΔ/Net AΔBΔ; Brankatschk and Dickson, 2006; Brierley et al., 2009). In our Netrin LOF mutant stock (NetAΔBΔ/FM6), the ratio of genotypes that eclosed was as follows: 1 (NetAΔBΔ/>): 3 (Fm6/>): 27 (NetAΔBΔ/FM6). Homozygous mutant females never eclosed (NetAΔBΔ/Net AΔBΔ).
To assess the role of Netrin in the GF circuit, we examined Netrin LOF escapers and showed that they exhibited a variety of physiological and anatomical defects. Physiologically, 14.5% of Netrin hemizygous mutant males exhibited no TTM response to a stimulus and 51.5% displayed long latencies and poor following frequencies. The remaining 34% displayed normal function (Figs. 1D, 2, Table 1). We defined normal function as a muscle response latency of <0.95 ms and the ability of the circuit to respond 1:1 for >90% of stimuli applied at 100 Hz. As a group, the hemizygous males exhibited significantly longer latencies than heterozygous controls (two-tailed Student's t test, p = 0.00001). Control (NetAΔBΔ/+, n = 32) latencies averaged 0.85 ms (SD 0.05) and the hemizygous males (NetAΔBΔ/>, n = 208) had a mean latency of 1.26 ms (SD 0.53; Fig. 2C,F). The animals that did not respond to stimulus were not used in the reported statistics. When controls (NetAΔBΔ/+) were stimulated repetitively at 100 Hz, the TTM responded to each stimulus 92% of the time (Figs. 1D, 2C). The following frequency of Netrin LOF mutant TTMs at 100 Hz was 52%. The probability of muscle response in Netrin LOF mutants was between 70% and 80% on the first stimulus in the train. The probability of response to stimulus dropped to 45% over the remaining nine trials of the train (Figs. 1D, 2C,F). We next tested the NMJ to ensure that the synaptic defects were at the GF-TTMn synapse and were not an artifact of NMJ defects. When the TTMn was stimulated directly through thoracic stimulation, the resulting EPSPs recorded from the TTM had a normal response for thoracic stimulation, with a latency of 0.55–0.6 ms and a following frequency of 100% at 100 Hz (data not shown). This demonstrates that the NMJ functioned normally. We concluded that the synaptic defect existed at the GF-TTMn synapse within the CNS (Fig. 1C, black circle) and not at the NMJ of the TTMn.
Table 1.
Comparison of physiological responses across genotypes
| Genotype | GFs (n) | Physiology (% wild-type) | Average latency, ms (SD) |
|---|---|---|---|
| Net AΔBΔ/+ | 32 | 100% | 0.850 (0.5) |
| Net AΔBΔ/> | 208 | 34% | 1.26 (0.53) |
| Rescue (Net AΔBΔ/>; shakB-GAL4 UAS- GFP/+; UAS- NetA/+) | 30 | 87% | 0.84 (0.25) |
| Rescue (Net AΔBΔ/>; shakB-GAL4 UAS- GFP/+; UAS- NetB/+) | 28 | 82% | 0.9239 (0.32) |
| Rescue (NetAΔBΔ/>; c17/+; UAS-NetA/+) | 26 | 92% | 0.73 (0.19) |
| Rescue (NetAΔBΔ/>; c17/+; UAS-NetB/+) | 16 | 100% | 0.76 (0.02) |
| Rescue (NetAΔBΔ/>; +/+; UAS-NetA/380-slit-GAL4) | 16 | 100% | 0.85 (0.06) |
| Rescue (NetAΔBΔ/>; +/+; UAS-NetB/380-slit-GAL4) | 16 | 88% | 0.77 (0.05) |
| fra3/+ | 32 | 100% | 0.85 (0.04) |
| fra3/fra4 | 18 | 22% | 1.24 (0.36) |
We also examined the DLM motor output of the circuit responsible for flight. All DLM recordings showed normal responses. The DLM response latency was 1.26 ms (SD 0.36) and 100 Hz following frequency was 69% (SD 24.98), which were not different from control DLM muscle response latencies (1.34 ms, SD 0.23) or following frequencies (72%, SD 24.85). This supports the theory that the Netrin cue is responsible for GF-TTMn synaptogenesis, but not GF-PSI synaptogenesis.
Anatomy of the presynaptic terminal
When we examined the anatomy of GF in adult Netrin LOF mutants (NetAΔBΔ/>, n = 52), 91% of GF axons reached the mesothoracic neuromere and were in the correct synaptic region. This suggested that Netrin was not responsible for guiding GFs to their synaptic region; however, the phenotypes also suggested local guidance defects (Fig. 3A–C). Ectopic terminal branching was observed in 38% (n = 20/52) of GF terminals. Some GFs (n = 4/52, 8%) reached their synaptic region, ignored the TTMn target, and extended their terminals past the synaptic region. We interpreted the local ectopic branching as a target recognition defect. Of the total GFs examined, 64% exhibited mutant anatomy, mutant physiology, and failed to dye couple to their TTMns.
Figure 3.

A spectrum of GF and TTMn phenotypes was observed in the Netrin LOF (NetAΔBΔ/>). A, B, GFs in the Netrin LOF mutants exhibited ectopic terminal branching (arrows). C, Some GFs ignored their synaptic partners and did not form terminals, but continued to project past the synaptic region. D, E, The TTMns of Netrin LOF mutants exhibited absent medial dendrites at the midline (left TTMn, asterisk). In the same animal, the other TTMn projected its medial dendrite toward the midline (right TTMn, arrow). Scale bar, 20 μm. Schematic illustrates giant fibers (magenta) and TTMns (green) in relation to prothoracic and mesothoracic neuromeres.
Of the 52 GFs in the Netrin LOF mutant background, 28 made anatomically normal synaptic contact with the TTMn. Of these 28 GFs with anatomically normal terminals, 61% exhibited mutant physiology and lacked dye coupling between the GF and TTMn (Fig. 2D, arrow). This demonstrated that, even when the GF and TTMn appeared to make normal morphological contact, synaptic defects were present. The data suggested that Netrin contributed to synaptic function and synaptic morphology. These functions of Netrin are independent of each other. It is important to note that anatomy is not perfectly correlated with function, because anatomically normal GFs displayed both wild-type and mutant function and anatomically mutant GFs could exhibit either wild-type or mutant function.
Anatomy of the postsynaptic dendrite
We also observed anatomical defects in the dendrites of the postsynaptic TTMn. We expressed GFP in the TTMn using the shakB-GAL4 driver in mutants and controls and examined the TTMn dendrites. We observed that the medial dendrite of TTMn was missing in 34% of adult Netrin LOF mutants (NetAΔBΔ/>, n = 50). In Netrin LOF mutants missing TTMn medial dendrites, no physiological response could be recorded (Figs. 2D, 3D,E, asterisk). To quantify the effects of Netrin on the TTMn dendrite, we reconstructed the dendritic trees in adult control (NetAΔBΔ/+) and Netrin LOF mutant backgrounds (NetAΔBΔ/>) with Neuron Studio and measured three parameters for comparison: total dendrite volume, dendrite surface area, and dendrite length. There was no significant difference between control and mutant total dendrite length (one-way ANOVA on ranks, H = 0.015, p = 0.90), total dendrite volume (one-way ANOVA on ranks, H = 1.816, p = 0.18), or total dendrite surface area (one-way ANOVA on ranks, H = 0.96, p = 0.327). These results showed that, despite the absence of the medial branch of the dendritic tree, the TTMn maintained normal dendritic tree size by redistributing the cellular membrane to the lateral dendrite of the TTMn dendritic tree.
Dye coupling is disrupted in Netrin mutants
Because this mixed electrical-chemical synapse is dominated by the electrical component, we examined dye coupling between the GF and the TTMn in Netrin LOF mutants (n = 41) by injecting GFs with a neurobiotin/rhodamine-dextran mixture and determining whether the TTMn was labeled trans-synaptically with neurobiotin. In control animals (NetAΔBΔ/+), neurobiotin crossed from GF to TTMn via gap junctions in 100% of the GFs examined (Fig. 2A, arrows). In Netrin LOF mutants (NetAΔBΔ/>), dye coupling was lost in cases in which the GF was disconnected from the TTMn (13 of 41 synapses; Fig. 2D, asterisk). In specimens in which the GF and TTMn appeared to make synaptic contact (28 of 41 synapses), 54% of the 28 GF-TTMn synapses were not dye coupled (Fig. 2D, arrows). These same synapses were physiologically mutant and therefore dye coupling correlated with synaptic function.
Netrin regulates Innexins
To determine whether Netrin LOF mutants exhibited disrupted gap junctions as suggested by the dye-coupling data, we used anti-ShakB antibody to label Innexin in the GF. The GF forms electrical synapses with two different targets: the PSI and TTMn (Fig. 1C). There was a clear reduction of gap junctions in the presynaptic terminals of Netrin LOF mutants (NetAΔBΔ/>) at the GF-TTMn synapse compared with controls (NetAΔBΔ/+; Fig. 2B,E). We used ImageJ to quantify colocalization of injected rhodamine and anti-Innexin antibody labeling in GF terminals. Quantification is reported as the percentage of GF terminal volume occupied by Innexin (refer to Materials and Methods for quantification of Innexin). Innexin volume was significantly reduced in mutants (1.54%, SD 0.48, n = 15) compared with controls (14.72%, SD 3.99, n = 9) animals (Dunn's method, Q = 4.36; Table 2). Dye coupling was never observed in animals that completely lacked Innexin labeling in the terminal. When animals had a severe reduction in Innexin labeling, dye coupling was variable; some synapses were weakly dye coupled and others were not coupled. Finally, in the specimens with severe reductions, Innexin labeling was often detected in the cytoplasm of the axon, not on the perimeter of the axon or in the axon membrane (Fig. 2B, arrows, E, thin arrow).
Table 2.
Comparison of Innexin levels in the presynaptic terminal of various genotypes
| Genotype | GFs (n) | Innexin quantification, % of terminal occupied by Innexin (SD) |
|---|---|---|
| Net AΔBΔ/+ | 9 | 14.72% (3.99) |
| Net AΔBΔ/> | 16 | 1.54% (0.48) |
| Rescue (Net AΔBΔ/>; shakB-GAL4 UAS- GFP/+; UAS- NetB/+) | 7 | 10.34% (2.58) |
| Rescue (NetAΔBΔ/>; c17/+; UAS-NetA/+) | 17 | 11.58% (1.50) |
| Rescue (NetAΔBΔ/>; c17/+; UAS-NetB/+) | 8 | 9.32% (3.82) |
| Rescue (NetAΔBΔ/>; +/+; UAS-NetB/380-slit-GAL4) | 9 | 8.51% (1.74) |
| fra3/+ | 8 | 10.93% (3.96) |
| fra3/fra4 | 12 | 3.00% (0.57) |
| UAS-fra-ΔC x2 | 9 | 1.73% (0.86) |
The results suggest that there is a threshold level of Innexin that produces a wild-type physiological response. In control animals (NetAΔBΔ/+), 6.5–17% of a GF's terminal volume was occupied by Innexin. When Innexin levels fell below ∼6.5% of terminal volume occupied, muscle response latency was outside of normal ranges (≥0.95 ms). In Netrin LOF mutants that were physiologically mutant (NetAΔBΔ/>), 0.03–4.44% of the terminal volume was occupied by Innexins. In Netrin LOF mutants (NetAΔBΔ/>) that did not function physiologically, Innexin staining was absent or nearly absent. This demonstrated that a threshold between 4.4% and 6.5% of terminal volume occupied by Innexin was required to support normal physiological function. It was also possible to identify postsynaptic Innexin, but it was not possible to reliably measure it because of the small size of the TTMn's dendritic structures. Interestingly, we could not detect Innexin in the lateral dendrite branch of TTMn, which demonstrated that not all dendritic branches of the TTMn made electrical connections using ShakB/Innexin.
In contrast, Innexin signal was always detected in Netrin LOF mutants at the site where the GF makes an electrical synapse with the PSI, a neuron in the DLM flight circuit (Fig. 2E, thick arrow). Innexin staining at the GF-PSI synapse correlated with DLM physiological recordings, which were normal, and Innexin localization, dye coupling, and function were not disrupted for the GF-PSI synapses. The GF-PSI synapse did not use Netrin-Frazzled signaling for electrical synapse assembly. In Netrin LOF mutants, Innexin was apparently produced and trafficked to the GF-PSI synapse (Fig. 2E, thick arrow), but not to the GF-TTMn synapse (Fig. 2E, asterisk). The data suggest that the GF uses multiple signaling mechanisms to build two different electrical synapses only ∼20 μm apart within the same axon terminal. Innexin levels were decreased specifically in the GF terminal and disrupted the GF-TTMn synapse function while leaving the GF-PSI-DLMn pathway unaffected.
Rescue of the Netrin LOF mutant
To confirm the role of Netrin in synaptogenesis of the GFS, we attempted to rescue Netrin LOF mutants by expressing secreted Netrin in the synaptic area under the control of various GAL4 drivers. When we expressed NetrinA (or B) in the postsynaptic TTMn, using the shakB-GAL4 driver, in Netrin LOF animals (Net AΔBΔ/>; shakB-GAL4 UAS-GFP/+; UAS-NetA (or B)/+) the GFS reverted to normal circuit structure and function (Figs. 1D, 2G,I, Table 1). When UAS-NetA was expressed, the latency was restored to 0.84 ms (SD 0.25); for UAS-NetB, the latency was restored to 0.92 ms (SD 0.32). In both UAS-NetA or B rescues, the following frequency was restored to normal levels. Anatomically, the GF and the TTMn defects were corrected in all specimens. Finally, expressing UAS-NetA (or B) postsynaptically in the Netrin LOF mutants rescued dye coupling between the GF and TTMn in 100% of specimens (Fig. 2G, arrows).
Because Netrin is expressed by midline glia in embryos, we also attempted to rescue the Netrin LOF mutants by expressing Netrin in midline glia. The c17 GAL4 driver shows a strong expression in midline glia during the period of synaptogenesis from 0% to 31% PD (see below and Fig. 4). Expression of UAS-NetA under the control of the c17 GAL4 driver (NetAΔBΔ/>; c17/+; UAS-NetA/+, n = 26) rescued anatomy, physiology, and dye coupling in all specimens observed (Figs. 1D, 2J,L, Table 1). The latency was restored to normal and the TTM followed when stimulated at 100 Hz. Similarly, when UAS-NetB was expressed with c17 GAL4 (NetAΔBΔ/>; c17/+; UAS-NetB/+, n = 16), all GFs examined were physiologically normal. The latency was restored and TTMs responded at normally when stimulated at 100 Hz. Anatomically, 92% of the 16 GFs were wild-type and all 16 GFs were dye coupled to TTMn.
Figure 4.

Midline glia secrete Netrin during the GF-TTMn synapse formation. Expression of tethered NetrinB (magenta) under its endogenous promoter was detected throughout early PD in midline glia. UAS-GFPCD8 (Green) was driven by the c17 GAL4 driver and was also localized to glia. A–C, Tethered Netrin and GFP colocalized (white) in glial cells at the midline from 1% up to 31% of PD. A, Inset, There were Netrin-labeled appendages projecting off of midline glia (arrows), which were not included in the population of cells labeled by c17 GAL4 during the early stages of development. D, NetrinB was available at the midline during a critical period of GFS synaptogenesis from 7% to 32% of PD. Expression of NetrinB in midline glia was strong at 32% of PD. However, c17 GAL4 expression in midline glia was absent at 32%, whereas GFP expression in the GF (arrow) began. E, By 78% of PD, expression of NetrinB at the midline was severely reduced. Anatomical development of the GF's presynaptic terminal was complete at this stage (arrows). Each panel is a single image from a collected z-stack (0.5 μm step size). Arrows indicate processes in mesothoracic neuromere in each panel. Scale bar, 20 μm.
To confirm the glial rescue, we expressed UAS-NetA (or B) with a well characterized midline glial driver, 380-slit-GAL4, which is expressed in midline glia of the embryo (Wharton and Crews, 1993; Scholz et al., 1997); we have also demonstrated its expression throughout PD (Fig. 5). When UAS-NetA was expressed with the 380-slit-GAL4 (n = 16) in a Netrin LOF mutant background (NetAΔBΔ/>; +/+; UAS-NetA/380-slit-GAL4), the physiology, anatomy, and dye coupling were rescued in 100% of animals tested. Muscle response latency was normal and muscle responded at nearly 100% when stimulated at 100 Hz (Fig. 1D, Table 1). All TTMn medial dendrites were present in the specimens observed. When we expressed UAS-NetB with 380-slit-GAL4 (n = 16) in the Netrin LOF mutant background (NetAΔBΔ/>; +/+; UAS-NetB/380-slit-GAL4), 88% of the GFs displayed normal physiological function (Figs. 1D, 2O, Table 1). Animals exhibited normal circuit latencies and followed at 84% during 100 Hz stimulation. All GF exhibited wild-type anatomy and were dye coupled to their postsynaptic partners in the correct synaptic region (Fig. 2M, arrows). All TTMns appeared anatomically wild-type, displaying normal medial dendrite extension toward the midline.
Figure 5.

Expression of 380-slit-GAL4/UAS-GFPCD8 in midline glia. The 380-slit-GAL4 has been described previously to drive expression in midline glia in embryo and larva of Drosophila. Here, we showed that the 380-slit-GAL4 driver is expressed in midline glia during PD. A, At 7% of PD, midline glia were clearly defined with UAS-GFPCD8 expression. This expression pattern is representative of the 380-slit-GAL4 driver from 7% to 46% of PD. B, At 47% of PD, the glia were clearly visible with GFP expression. This expression pattern is representative of the 380-slit-GAL4 driver from 47% to 75% of PD. Schematic of early pupal CNS, with midline glia shown in green. Scale bar, 20 μm.
Innexin levels recovered in Netrin LOF rescue experiments
To show directly that Netrin-Frazzled signaling instructs Innexin localization in the GF terminal, we labeled Innexin protein in our rescue experiments. When Netrin LOF mutant animals were rescued by driving expression of UAS-NetB with any of the three drivers (shakB-GAL4, c17 GAL4, and 380-slit-GAL4), Innexins were properly localized at the terminal at normal levels (Fig. 2H,K,N, Table 2). It is important to note that when Innexin localization in the presynaptic terminal was rescued, it correlated with rescued physiology and dye coupling of the circuit. This confirmed Netrin's importance in synapse formation and function. There was no significant difference in Innexin levels between controls (Net AΔBΔ/+; GAL4/+) and rescue animals (Dunn's method, 380-slit-GAL4 rescue Q = 1.27, shakB-GAL4 rescue Q = 0.55, c17 GAL4 rescue Q = 0.31).
Development of the GFS and its requirement for Netrin
Netrin expression by the TTMn
To determine the source of Netrin during GF development, we labeled Netrin and the GFS in two ways. First, we used antibodies against NetrinA (or B) protein (Albrecht et al., 2011). Second, we labeled a myc-tagged membrane tethered version of NetrinB (NetAΔBmyc-TM/>) expressed under the control of the endogenous Netrin promoter (Brankatschk and Dickson, 2006). With Netrin antibodies, we identified NetrinA (or B) expression in the TTMn cell body and medial dendrite (Fig. 6C), between 9% and 27% of PD, a critical period of synaptogenesis (Phelan et al., 1996; Allen et al., 1998). When we labeled Netrin using membrane tethered Myc-tagged NetrinB (NetAΔBmyc-TM/>) in a NetrinA LOF background, we observed NetrinB on the TTMn cell body during the same period of PD (9–27%). However, there was no detectable tethered NetrinB labeling on the medial dendrite.
Figure 6.

Netrin and Frazzled expression in GFS. A–C, Netrin and Frazzled production in the GFS between 9% and 27% of PD. A, TTMn cell body (arrow) was labeled with UAS-GFPCD8 (magenta) and Frazzled was labeled with anti-Frazzled antibody (green). Figure is representative of 27% of PD. Scale bar, 20 μm. B, Frazzled signal (green) was also detected and colocalized (white) in the GF axon (magenta) terminal (arrow) of the same specimen in the mesothoracic neuromere. Scale bar, 50 μm. Panels are single images from a z-stack with 0.5 μm steps. C, anti-Netrin antibody was detected on the TTMn medial dendrite and in the cell body. Figure is representative of 18% of PD. The Lateral dendrite of TTMn did not exhibit anti-Netrin signal because it had not projected from the TTMn at this time in PD. Panel is a compressed z-stack. Schematic of adult CNS with box indicates region examined. Scale bar, 20 μm. Dotted lines indicate location of the midline.
Netrin expression on the pupal midline
A variety of genetic experiments showed that Netrin was present on the embryonic midline and instructive in commissure formation (Bashaw and Goodman, 1999; Hummel et al., 1999; Furrer et al., 2003). More recently, Netrin was shown to be expressed in the midline of Drosophila during embryonic development (Brankatschk and Dickson, 2006; Garbe and Bashaw, 2007) and PD (Brierley et al., 2009). To demonstrate NetrinB labeling of midline glia of pupae, we expressed GFP in midline glia with the c17 GAL4 driver and simultaneously labeled NetrinB using the membrane-tethered Myc-tagged NetrinB mutant (NetAΔBmyc-TM/>; Brankatschk and Dickson, 2006; Fig. 4). The results showed that NetrinB was expressed by midline glia from 0% to 78% of PD. From 0% to 30% of PD, NetrinB signal is colocalized with the GFP reporter gene (Fig. 4A–C). After 31% of PD, the c17 GAL4 discontinued expression of GFP on the midline (Fig. 4D), but NetrinB expression by the midline glia continued through most of PD (Fig. 4E). This demonstrated that the c17 rescue of the Netrin LOF mutant coincided with NetrinB expression in glia during early PD. It also shows that we rescued the Netrin LOF mutant animals by expressing UAS-NetA (or B; c17 GAL4) in midline glia that endogenously express NetrinB.
To examine the relationship between midline glial expression of NetrinB and the medial dendrites of TTMn, we labeled membrane-bound NetrinB (NetAΔBmyc-TM/>; Brankatschk and Dickson, 2006) and simultaneously expressed GFP in the TTMn with shakB-GAL4. At the beginning of PD, NetrinB was expressed in the midline glia (Fig. 7A). By 9% of PD, we could identify the TTMn medial dendrite extending toward the midline glia (Fig. 7B, arrow). By 18%, the filopodia of the TTMn medial dendrite had reached the midline glia and appeared to contact them (Fig. 7C, arrows). After 32% of PD, the anatomical structure of TTMn was essentially complete (Fig. 7D,E). After the major developmental events of the TTMn were complete in the first third of development, the midline glia reduced the production of NetrinB. By 78% of PD, the NetrinB signal was dramatically reduced and totally lost by eclosion (Figs. 4E, 7E). This developmental series demonstrates that the TTMn dendrite projected toward the midline during midline glia expression of NetrinB.
Figure 7.

Development of TTMn's medial dendrite during NetrinB expression in midline glia. During PD, we labeled tethered Myc-tagged NetrinB and expressed UAS-GFPCD8 in TTMn with the shakB-GAL4 driver. A, At 0% of PD, the NetrinB labeling (magenta) was detected on the midline glia (arrows) and longitudinal tracks. B, By 9% of PD, NetrinB signal was observed at the midline and the medial dendrite of TTMn (green) extended (arrow) toward the midline. C, At 18% of PD, the TTMn filopodia (arrows) were at the midline. D, At 32% of PD, the medial dendrite (arrows) was anatomically mature. E, NetrinB production at the midline was no longer observed by 80% of PD. The panels focus on the development of TTMn's medial dendrite. Therefore, the TTMn cell bodies (arrowheads) were absent from the plane of focus in all panels except for E. All panels are single images from a z-stack with 0.5 μm steps. Schematic illustrates TTMns (green) in relation to prothoracic and mesothoracic neuromeres. Scale bar, 20 μm.
Netrin is required for normal development of TTMn dendrites
To determine whether the medial dendrite defects in Netrin LOF mutants occurred early in development, we dissected pupa expressing UAS-GFPCD8 in the TTMn at various stages from 47% to 98% of PD. The earliest stage that we could identify the Netrin LOF mutant escapers was 47% of PD (Fig. 8). In mutant pupal specimens of various ages, the medial dendrites of TTMn were often not present at the midline, revealing an early defect in TTMn medial dendrite extension due to the loss of the Netrin locus. At 47% of PD, 60% of TTMn medial dendrites were present at the midline (n = 10). By 73% of PD, 64% of TTMn medial dendrites were present at the midline (n = 14). At 78% of PD, 43% of TTMn medial dendrites were present at the midline (n = 16). In the adult CNS, 66% of TTMn medial dendrites were present at the midline (n = 50). There was no significant difference in the prevalence of medial dendrites at the midline between the stages of PD and adult observed (two-tailed Fisher's exact test, 73% of PD, p = 1.00; 78% of PD, p = 0.68; adult, p = 0.44).The results suggest that the lack of TTMn medial dendrites at the midline in Netrin LOF mutants occurs early in development and that the prevalence of the defect does not increase as the animal develops. It is possible that these dendrites fail to project toward the midline because the Netrin guidance cue was absent.
Figure 8.
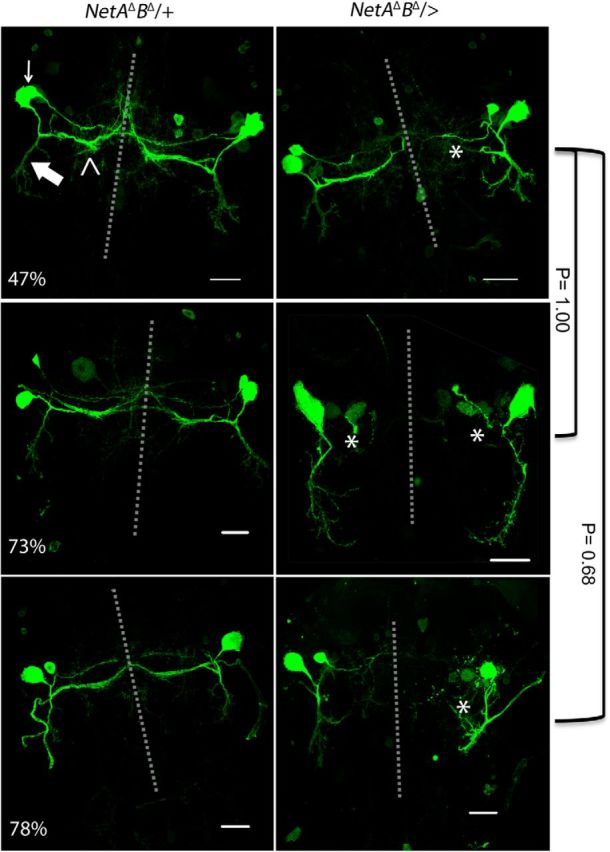
Pupal development of TTMn medial dendrites in control and Netrin LOF mutants. Pupa from 47% to 78% of PD were used to examine TTMn's medial dendrite anatomy. Top left, TTMn soma (arrow), medial dendrite (open arrowhead), and lateral dendrite (large arrow). The left column depicts the control mesothoracic neuromere at three stages of PD. The right column depicts the Netrin LOF mutant mesothoracic neuromere at three stages of PD. Medial dendrite extension toward the midline was absent in 60% of TTMns at 47% of PD (asterisk). At 73% of PD, medial dendrites at the midline were absent in 64% of TTMns (asterisk). Medial dendrites were absent at the midline in 43% of TTMns at 78% of PD (asterisk). There was no significant difference between the presences of medial dendrites at the midline between developmental groups (two-tailed Fisher's exact test). Dotted lines represent the midline. UAS-GFPCD8 was expressed with the ShakB-GAL4 driver in these preparations. Scale bar, 20 μm.
Is localization of Netrin, or secretion of Netrin, the critical factor for synapse assembly in the GFS?
In a previous section, we used the tethered myc-tagged NetAΔBmyc-TM mutant of Brankatschk and Dickson (2006), which is driven by its normal promoter, to describe the localization of NetrinB in the TTMn and midline glia (Fig. 7). We also tested this mutant to determine whether it would allow for proper synapse formation. In both female homozygotes (NetAΔBmyc-TM/NetAΔBmyc-TM) and male hemizygotes (NetAΔBmyc-TM/>), there was no detectable anatomical or physiological phenotype and we observed normal dye coupling in all preparations examined. Apparently, tethering NetrinB to the membrane in this manner while it is expressed under its endogenous promoter supported normal synaptogenesis.
To further assess the role of tethered NetrinB, we attempted to rescue the Netrin LOF mutant using a different tethered NetrinB construct: the UAS-NetBCD8-TM construct of Timofeev et al. (2012). We expressed tethered NetrinB in midline glia (NetAΔBΔ/>; c17/UAS-NetBCD8-TM) to determine whether it could rescue the Netrin LOF mutants, as we observed with the secreted UAS-Netrin constructs (Figs. 1, 2J–L). This experiment was not successful in rescuing the Netrin LOF mutant and new defects were observed. In the example shown, the right TTMn medial dendrite extended toward the midline, made contact with the GF near the PSI synaptic region (Fig. 9C, thick arrow), and exhibited weak dye coupling when the GF was injected with neurobiotin (Fig. 9C, open arrowhead). Anti-Innexin (ShakB) antibodies demonstrated that there were gap junctions in this region of the GF axon (Fig. 9C, thick arrow), as well as in the postsynaptic dendrite at the point of contact. However, gap junction labeling was not detected in the region of the GF terminal or TTMn medial dendrite, where the giant synapse is normally formed (Fig. 9C, bracket). Physiologically, the GF-TTMn synapse functioned poorly and displayed long response latencies of 1.7 ms (SD 0.42, n = 16) and followed 100 Hz stimuli <10% of the time. Finally, there were path-finding errors where the GFs crossed the midline in the brain and synapsed with the contralateral TTMn, as in the specimen shown in Fig. 9, A and B (thin arrow). Other GFs failed to project out of the brain entirely, making no contact with either TTMn (Fig. 9A,B, open arrowhead).
Figure 9.

Expressing tethered NetrinB in glia or the TTMn did not rescue Netrin LOF defects. A–C, When we expressed tethered NetrinB in midline glia with the c17 driver, Netrin LOF mutants were not rescued. B, The GFs displayed midline crossing defects in the brain (arrow and open arrowhead). In this example, both GFs crossed the midline in the brain, but only one GF projected toward the mesothoracic neuromere (arrow). C, The GF that reached the thorax displayed multiple branches (fuchsia) and Innexin (green) was undetectable in the GF axon terminal (bracket). The TTMn cell body (open arrowhead) and dendrites were visible due to dye coupling (red neurobiotin). The TTMn did not synapse with the GF in its normal synaptic area (bracket), but instead at the GF-PSI synaptic region (large arrow). In this region, Innexin staining was observed. Coloring in C is different from A and B because there are three colors used for labeling in C, whereas only two colors are used in A and B. D, E, When we expressed tethered NetrinB in the TTMn with the shakB-GAL4 driver, Netrin LOF mutant defects were not rescued. D, GFs (fuchsia and red) did not make normal terminals or project toward their synaptic targets (arrows). TTMns (green) did not produce dendritic trees (asterisks). E, Some TTMns did project lateral dendrites (arrows), but did not project medial dendrites toward the midline (asterisks). Scale bar, 20 μm.
We also attempted to rescue Netrin mutants by expressing tethered UAS-NetBCD8-TM postsynaptically (NetAΔBΔ/>; shakB-GAL4 UAS-GFP/UAS-NetBCD8-TM), but rather than rescuing the defects, there was an enhancement of the phenotypes. The occurrence of TTMns lacking medial dendrites increased from 34%, seen in Netrin LOF mutants, to 90%. Often, both the medial and lateral dendrites were missing (Fig. 9D,E, asterisks). In the 10% of cases in which a TTMn medial dendrite did project to the midline, none of the specimens displayed dye coupling of the GF with the TTMn. Physiologically, the circuit was disconnected in all specimens, because no muscle response could be recorded when the GF was stimulated. Our interpretation is that the TTMn could not present the tethered NetrinB to the GF because the TTMn's medial dendrite did not project to the midline. We hypothesize that the absence of Netrin expression on the midline could have disrupted TTMn medial dendrite projection.
The very different results for the two tethered Netrin constructs suggest that they are different in some yet to be determined manner. There are at least two possible differences between these tethered protein experiments that may be important to our analysis. The first is pattern of expression. The tethered NetrinB mutant (Brankatschk and Dickson, 2006) is expressed under the control of its endogenous promoter and therefore is expressed in the normal locations, as shown in Figures 4 and 7. The UAS-NetBCD8-TM tethered construct (Timofeev et al., 2012) is expressed under the control of a GAL4 driver and our GAL4 constructs are unlikely to completely match the endogenous expression pattern of the Netrin locus.
The second difference is based on the structure of the two different tethered NetrinB proteins. The normally expressed tethered NetrinB mutant protein has a series of extracellular myc tags between the transmembrane domain and the C domain of the NetrinB protein (Brankatschk and Dickson, 2006). These domains could hold the NetrinB protein further from the cell membrane than the resulting protein of the UAS-NetBCD8-TM-tethered construct, which does not contain these domains (Timofeev et al., 2012). It is possible that these molecular differences contribute to the different phenotypes observed in these experiments. An anonymous reviewer has suggested that these extracellular myc domains may introduce conformational flexibility that allows contact with the receptor on the same cell or other cells or may allow some cleavage of the mutant membrane-bound protein, releasing NetrinB into the extracellular milieu and allowing it to function as a normal secreted ligand. In contrast, the tethered NetrinB construct (UAS-NetBCD8-TM) obtained from Timofeev et al. (2012), does not contain the series of myc tags, and did not rescue the mutants, suggesting that it is not released and cannot function normally. Presumably, the tethered NetrinB construct (UAS-NetBCD8-TM) was restricted to the site where it was expressed and could not interact with Frazzled in the normal manner. In contrast, when we expressed secreted Netrin in either TTMn or midline glia of Netrin LOF mutants, the GFS was rescued to normal function (Fig. 2).
Role of Frazzled in assembly of the GFS
frazzled LOF mutants exhibited synaptic defects at the GF-TTMn synapse. To demonstrate that Netrin signaling was mediated through the Frazzled receptor in the GFS, we examined adult GFs of known frazzled LOF mutants. Two frazzled LOF mutants, fra3 and fra4, have been well characterized (Kolodziej et al., 1996). Both are homozygous lethal and wild-type as heterozygotes (Kolodziej et al., 1996). The trans-heterozygotes (fra3/fra4) are viable and have been used as LOF mutants (Kolodziej et al., 1996). The trans-heterozygote produces some protein product, whereas expression levels are lower than normal and residual protein function is unknown (Yang et al., 2009). The data suggest that fra3 is a hypomorphic allele and fra4 is a null.
Numerous synaptic defects were observed in the GFS of the trans-heterozygotes (fra3/fra4) compared with controls. The GF-TTMn synapse displayed an average latency of 1.27 ms (SD 0.36; n = 18), significantly different from controls (fra3/+), 0.85 ms (SD 0.04; n = 30; two-tailed Student's t test, p = 0.0001). This genotype was also unable to reliably follow 100 Hz stimulation, because it responded to only 59% of stimuli (n = 18; Fig. 10C,F) compared with 92% for controls (fra3/+). It is of great interest that 66% of frazzled trans-heterozygote GFs exhibited bilateral terminals (Fig. 10D). One branch of the bifurcated axon crossed the midline and contacted the ipsilateral target and the other branch contacted the contralateral TTMn. When we evaluated dye coupling, bifurcated terminals dye coupled to both TTMn targets 75% of the time. Because the frazzled trans-heterozygotes are hypomorphs, we hypothesize that there must be enough Frazzled in the system to ensure that all TTMns properly projected their medial dendrites toward the midline and often contacted the presynaptic terminal.
Figure 10.

frazzled LOF mutant anatomy, physiology, and presynaptic Innexin levels. A, Heterozygous frazzled control animals (fra3/+) exhibited normal GF terminal anatomy. B, Innexin labeling (green) in frazzled heterozygous mutant GFs (magenta) colocalized in GF terminals (white) at the GF-PSI synapse and the GF terminal (image is a single slice of a TIFF stack with 0.5 μm steps and dotted lines represent the terminal in other sections of the TIFF stack). C, Control physiological recordings were normal. D, frazzled LOF trans-heterozygous animals (fra3/fra4) often exhibited bifurcated axon terminals (left giant fiber shown). E, Innexin labeling (green) in the GF (magenta) was greatly reduced and colocalized Innexin in the terminal was undetectable (image is a single slice of a TIFF stack as in B). F, This correlated with the mutant physiological phenotype shown by long response latency and inability of the circuit to follow high-frequency stimulation of 100 Hz. Scale bars, 5 μm.
Frazzled expression in the GFS
We labeled Frazzled in specimens expressing GFP in the GFS (GF and TTMn) during the early period of GFS synaptogenesis, 9–27% of PD (Fig. 6A,B). We detected Frazzled in the TTMn cell body, but not in the dendrites (Fig. 6A). Frazzled signal was also detected in the terminal of the GF axon (Fig. 6B). This showed that both presynaptic and postsynaptic cells expressed Frazzled protein during critical periods of development of the GF-TTMn synapse. We performed the same antibody labeling of Frazzled in adult nervous systems and were unable to detect Frazzled signal in the GF or TTMn of adult animals.
Frazzled regulates Innexin
Frazzled mutants were also labeled with the anti-Innexin antibody to assess the levels of Innexin in the GF-TTMn synapse (Fig. 10B,E). The volume of the GF terminal occupied by Innexin was significantly smaller in frazzled LOF mutants (fra3/fra4; n = 12) than controls (fra3/+, n = 8; one-way ANOVA on ranks, H = 14.727, P=<0.0001; Table 2). frazzled LOF mutants exhibited an average of 3.00% (SD 0.57) of the terminal occupied by Innexin signal. In four specimens, the GFs contained almost no detectable Innexin signal, did not dye couple to their TTMn targets, and displayed poor physiological function. In the remaining eight cases, Innexin signal was greatly reduced in the presynaptic terminal and the GFs exhibited weak dye coupling. In GFs that were weakly dye coupled, neurobiotin could be detected in the TTMn medial dendrite very close to the synaptic region, but not in the soma or lateral dendrite of the TTMn. Controls (fra3/+) exhibited an average of 10.93% (SD 3.96) of the terminal occupied by Innexin, which falls within the normal range of Innexin levels required for the proper function of the GF-TTMn synapse.
Because some frazzled mutants dye coupled to the TTMn, we could identify postsynaptic Innexins by examining colocalization of Innexins and the injected dyes. For example, in one frazzled mutant (fra3/fra4) that exhibited weak dye coupling and lowered Innexin levels, Innexin signal was detected outlining the presynaptic terminal of the GF, which was labeled with rhodamine-dextran (Fig. 11A, thick arrow). The Innexin signal did not colocalize with the rhodamine-dextran signal and therefore was outside, not inside, the presynaptic terminal. The same specimen was also injected with neurobiotin, which showed weak dye coupling to the TTMn and normal dye coupling to the PSI (Fig. 11B, small arrow and arrowhead). Because neurobiotin crossed gap junctions and labeled the postsynaptic cell, any Innexin that colocalized in the neurobiotin channel, but not in the rhodamine-labeled signal, was identified as postsynaptic (Fig. 11B, thick arrow). This showed that although the Innexin signal was severely decreased presynaptically in the absence of Frazzled, we still observed Innexin signal in the postsynaptic dendrite. Although we were unable to reliably quantify postsynaptic Innexins, we reliably differentiated between presynaptic and postsynaptic Innexin to ensure high consistency of our presynaptic Innexin measurements.
Figure 11.

Frazzled LOF mutants selectively disrupted presynaptic Innexins. A, In frazzled LOF mutants, presynaptic Innexins were reduced in the terminal and weak dye coupling occurred. Innexin labeling (green) did not colocalize (large closed arrow) with the GF terminal (magenta) rhodamine-dextran signal. B, In the same GF (magenta), the neurobiotin signal crossed gap junctions and labeled parts of the postsynaptic PSI (arrowhead) and TTMn (thin arrow). When the postsynaptic dendrites were labeled with neurobiotin (magenta), Innexin (green) colocalized with neurobiotin (white, thick arrow). The data suggest that presynaptic Innexin was reduced, whereas postsynaptic Innexin was clearly visible. Scale bar, 20 μm. All panels are single images from a z-stack with 0.5 μm steps. Dotted lines represent the GF terminal in the slices of the stack not shown.
Expression of Frazzled lacking its cytoplasmic domain disrupts the giant synapse
A Frazzled construct missing its intracellular domain UAS-fra-ΔC was described as a Frazzled dominant-negative in embryos (Hiramoto et al., 2000) and required Netrin to produce a phenotype (Garbe et al., 2007). Expressing one copy of this dominant-negative construct presynaptically and postsynaptically (in the GF and TTMn; A307/CyO; UAS-fra-ΔC/TM6β, n = 16) had no effect. However, expressing two copies (A307/CyO; UAS- fra-ΔC/UAS-fra-ΔC, n = 16) was deleterious to the circuit. All GFs exhibited mutant physiological responses, with an average TTM response latency of 1.24 ms (SD 0.34), and responded to <50% of stimuli delivered at 100 Hz. GFs also failed to dye couple to the TTMn and Innexin levels were significantly reduced in animals expressing two copies of UAS-fra-ΔC (n = 9) compared with those expressing one copy (n = 8; two-tailed Student's t test, p = 0.001; Fig. 12A,B,E,F, Table 2). These data show that UAS-fra-ΔC disrupts Netrin-Frazzled signaling and that Frazzled function is critical for Innexin localization in the GF presynaptic terminal.
Figure 12.

UAS-fra-ΔC disrupted gap junction localization in the GF terminal and the structure of the TTMn dendrites. A, When one copy of UAS-fra-ΔC was expressed presynaptically and postsynaptically with A307 GAL4, the GFS exhibited wild-type anatomy and the GFs were dye coupled to the TTMn medial dendrites and somata (arrowheads). B, Expressing one copy of UAS-fra-ΔC did not affect gap junction levels in the GF terminal (bracket) and the TTMn soma was dye coupled to the GF (arrowhead). C, The TTMn dendritic anatomy was normal when one copy of UAS-fra-ΔC was expressed postsynaptically. D, Inset of C. The TTMn medial dendrite terminal appeared normal when expressing one copy of UAS-fra-ΔC postsynaptically. E, Expressing two copies of UAS-fra-ΔC presynaptically and postsynaptically produced mutant anatomy. The right GF exhibited a bifurcated terminal, whereas the other was unilateral. This is difficult to see in a collapsed z-stack because the two terminals where both labeled with rhodamine-dextran (magenta). Dye coupling was disrupted (asterisk) and the TTMn's medial dendrite did not properly synapse with the GF terminal. The PSIs were dye coupled to the GF (arrowhead) and the DLM flight pathway was unaffected. F, When the GF terminal was examined, the Innexin labeling was undetectable (asterisk). G, When driving expression of two copies of UAS- fra-ΔC postsynaptically, TTMns exhibited ectopic branching (bracket) and absent lateral dendritic trees (asterisk). H, Inset of G. TTMn medial dendrites displayed ectopic branching at the midline compared with control dendrites (shown in D). They also appeared to have blebbing and filopodia-like structures. TTMn dendrites exhibited immature morphology. Scale bar, 20 μm.
To explore the effect of Netrin-Frazzled signaling on the postsynaptic cell, we expressed UAS-fra-ΔC in the TTMn. When we expressed one copy of UAS-fra-ΔC postsynaptically (shakB-GAL4/CyO; UAS-fra-ΔC/TM6β), the circuit functioned normally and TTMn morphology was wild-type (Fig. 12C,D). When we expressed two copies of the UAS-fra-ΔC postsynaptically (shakB-GAL4/CyO; UAS-fra-ΔC/UAS-fra-ΔC), the GF-TTMn synapse functioned normally and the GF was dye coupled to TTMn, but the TTMn dendritic structure was disrupted. We expressed both UAS-GFPCD8 and UAS-fra-ΔC to more carefully examine the TTMn dendrites (shakB-GAL4 UAS-GFPCD8/CyO; UAS-fra-ΔC/UAS-fra-ΔC) and observed two mutant postsynaptic anatomical phenotypes (n = 14, Fig. 12G). First, the TTMn's medial dendrite appeared to have an immature morphology. Seventy-nine percent of TTMns exhibited numerous ectopic branches and countless filopodia-like structures with blebbing at the tips (Fig. 12G,H). It appeared that normal pruning and refinement of the TTMn medial dendrite morphology did not occur (Fig. 12H). Despite this immature structure, the GF-TTMn synapse functioned normally. The second anatomical phenotype occurred in 57% of cases, where the lateral dendrite stalled near the TTMn soma and did not terminate in its proper synaptic region (Fig. 12G, asterisk). These morphological phenotypes were absent in the sibling controls (shakB-GAL4 UAS-GFPCD8/CyO; UAS-fra-ΔC/TM6β).
These results suggest distinct, cell-autonomous roles for Frazzled in the presynaptic and postsynaptic cells at this synapse. Presynaptic expression of UAS-fra-ΔC disrupted synaptic transmission and presynaptic Innexin in the GF-TTMn synapse. Postsynaptic expression disrupted dendritic morphology, but did not disrupt synaptic transmission or Innexin levels. We hypothesize that postsynaptic expression of UAS-fra-ΔC does not affect synaptic transmission of the GF-TTMn synapse because UAS-fra-ΔC could bind Netrin and present it to the presynaptic cell, permitting normal synapse assembly. However, UAS-fra-ΔC disrupted TTMn lateral dendrite extension and medial dendrite maturation due to the inability to correctly signal intracellularly. This phenotype corresponds with the morphological defect seen with the attempted postsynaptic rescue of Net mutants with UAS-NetBCD8-TM, demonstrating that the postsynaptic cell uses Netrin-Frazzled signaling to regulate dendritic structure. We attempted to show these cell-specific roles with Frazzled RNAi, but expression had no effect either presynaptically or postsynaptically. We suspect that RNAi did not reduce Frazzled to levels low enough to affect the GFS. Further, traditional rescue experiments were not possible due to the genetic limitations described in the Materials and Methods.
We next performed a genetic interaction experiment to determine whether UAS-fra-ΔC enhances the Netrin LOF heterozygote in the GFS. Heterozygous Netrin mutants (NetAΔBΔ/+) were wild-type in every respect. Expression of one copy of UAS-fra-ΔC with the A307 GAL4 driver also yielded wild-type animals. However, when we expressed UAS-fra-ΔC in a heterozygous Netrin LOF background (NetAΔBΔ/+; A307/+; UAS-fra-ΔC/+), the number of anatomically wild-type GFs decreased from 100% in parental lines to 39% in experimental animals. Mutant GFs exhibited increased branching and misguided terminals. Physiologically, response latency increased to 1.44 ms (SD 0.07) and a 51% response frequency at 100 Hz (n = 24). This average latency was significantly longer compared with wild-type responses from heterozygous Netrin LOF mutants (two-tailed Student's t test, p = 0.0001) or expression of one UAS-fra-ΔC copy alone (two-tailed Student's t test, p = 0.0001). Only 25% of GFs tested had a normal physiological response. This interaction result supported the idea that UAS-fra-ΔC functions as a sink for Netrin.
Discussion
Netrin-Frazzled signaling regulates gap junctions in the GFS
Our results show for the first time that Netrin-Frazzled signaling is specifically responsible for localizing gap junctions presynaptically at the GF-TTMn synapse. In the absence of Netrin, the gap junctions are not assembled in the presynaptic terminal and dye coupling is weak or absent in otherwise anatomically normal synapses. Similarly, Frazzled LOF mutants disrupted gap junctions and synaptic transmission. Finally, presynaptic expression of the dominant-negative Frazzled construct that is missing the intracellular domain also disrupts gap junction assembly, dye coupling, and synaptic transmission. We also show that, in Netrin LOF mutants, axonal pathfinding is normal because the GF always projects into the target region and occasionally branches ectopically in the target region. However, dendritic path finding is dependent on Netrin-Frazzled signaling. In Netrin LOF mutants, the TTMn dendrite that normally projects toward the midline is often missing, as observed in other motor neurons (Mauss et al., 2009). Finally, Netrin-Frazzled signaling is implicated in target selection, because GFs that reach the target area often do not build synapses, as seen in other model systems (Winberg et al., 1998).
We hypothesize that the physiological defect seen in Netrin and frazzled mutants arises from a reduction in trans-synaptic coupling between presynaptic and postsynaptic Innexins (Fig. 13). Similar phenotypes, long latency, and lack of dye coupling have been observed in the shakB2 mutant, which lacks gap junctions at the GF-TTMn synapse (Phelan et al., 1996, 2008; Allen et al., 2006). The data suggest that when presynaptic and postsynaptic cells make contact, Netrin-Frazzled signaling is instructive for presynaptic localization of Innexins in the GF terminal to form trans-synaptic gap junctions (Fig. 13).
Figure 13.

Model of Netrin-Frazzled synaptogenic mechanism in the GFS. We identified two roles for Netrin-Frazzled signaling in assembly of the giant fiber system. First, we showed that Netrin (blue squares) acts as a cue to direct the GF to select a target. Netrin-Frazzled signaling was also a local guidance cue for the GF and the medial dendrite of TTMn. The TTMn medial dendrite grows toward the midline glia (orange cells), which we showed was a source of Netrin. Second, we hypothesize that Netrin bound on the postsynaptic Frazzled receptors served as a synaptogenic cue for presynaptic Frazzled located on the GF. We propose that the bound Frazzled receptors directed presynaptic synaptogenesis and Innexin localization in the presynaptic terminal.
The frazzled dominant-negative construct supports the hypothesis that Netrin-Frazzled signaling is instructive in GF-TTMn synaptogenesis and function. Expression of fra-ΔC presynaptically disrupts the circuit by interrupting wild-type Netrin-Frazzled signaling. We demonstrate this through disruption of GF-TTMn synaptogenesis and the absence of gap junctions in the presynaptic terminal. However, the expression of UAS-fra-ΔC postsynaptically did not disrupt function, but did disrupt the morphology of the postsynaptic neuron. Postsynaptic expression of UAS-fra-ΔC disrupted dendritic maturation, resulting in medial dendrite pruning defects and lateral dendrite extension defects. We interpret the fra-ΔC experiments as providing some evidence for Frazzled's cell autonomous role in building this giant synapse. More direct evidence would require rescue experiments. Unfortunately, the relevant genes are located very close to one another, making it difficult to obtain the appropriate recombination event. Future experiments will use recently acquired GAL4 drivers on the third chromosome to clarify this issue. The Frazzled RNAi experiments were uninformative, possibly because RNAi is not a strong enough disruption of frazzled to cause effects in the GFS. In brief, the cell autonomous function of Frazzled warrants further investigation.
When UAS-fra-ΔC was expressed in the embryo in the Netrin LOF background, it revealed that the disruption of commissures was Netrin dependent (Garbe et al., 2007). Our interaction experiment (NetAΔBΔ/+; A307/+; UAS-fra-ΔC/+) revealed a different mechanism by which the dominant-negative fra-ΔC obstructed synaptogenesis. In a heterozygous Netrin LOF background, we expressed the mutant version of Frazzled, further knocking down Netrin-Frazzled signaling to disrupt synaptogenesis. The results suggested that fra-ΔC was acting as a Netrin sink by binding to secreted Netrin, limiting the amount of Netrin that could bind to wild-type Frazzled receptors.
We also observed the chemical synaptic component of the GF-TTMn synapse in the Net LOF mutants using antibodies against the presynaptic density protein bruchpilot (T-bars) with anti-NC82 staining. However, the bruchpilot labeling was not informative. We made no further effort because the cholinergic component has no effect on synaptic circuit function in the adult (Allen and Murphey, 2007 Phelan et al., 1996, 2008; Sun and Wyman, 1996; Blagburn et al., 1999).
In contrast to the GF-TTMn synapse, the GF-PSI synapse is unaffected by the absence of Netrin, Frazzled, or the expression of the dominant-negative Frazzled dominant-negative. This shows that the GF-TTMn synapse specifically is dependent on Netrin-Frazzled signaling for function. This mechanism for gap junction insertion is so specific that neighboring electrical synapses that share the same presynaptic terminal (GF) use different mechanisms for gap junction localization.
Non-cell-autonomous regulation of Netrin-Frazzled signaling
Netrin regulates Innexins in the GF presynaptic terminal from an external source. Netrin is secreted from two known sources, the midline glia and the postsynaptic target TTMn. We suggest a model (Fig. 13) for Netrin localization and function in which Netrin is captured on the surface of one neuron (TTMn) by Frazzled and is then presented to Frazzled receptors on another neuron (GF) to transmit signaling (Hiramoto et al., 2000; Smith et al., 2012; Timofeev et al., 2012). During development, the TTMn extends its medial dendrite toward a source of Netrin, the midline glia. After the TTMn dendrite has grown into the synaptic area by 9% of PD, both the midline glia and TTMn are labeled with Netrin. We hypothesize that this is important in the induction of synaptic maturation of this synapse.
Rescuing Netrin LOF mutants by expressing a secreted form of Netrin specifically in either TTMn or midline glia supports our model that Netrin is presented to the GF to promote synapse formation. The secreted Netrin rescue experiments were effective because Netrin could localize where it would normally as long as it was secreted by a nearby endogenous source. This could explain why we are able to rescue the Netrin LOF mutants in a non-cell-autonomous fashion by expressing secreted Netrin in either midline glia or the TTMn independently. Postsynaptic expression of the Frazzled dominant-negative also supports the presentation model. When we expressed two copies of Frazzled lacking its intracellular domain on the TTMn, Netrin could bind to the mutant Frazzled, be presented to the GF, and support normal synaptic function regardless of disrupted intracellular signaling in the TTMn by the deletion of the intracellular domain (Fig. 13).
In contrast, expressing membrane-tethered UAS-NetBCD8-TM (Timofeev et al., 2012) on either the midline glia or TTMn failed to rescue function of the circuit because localization and secretion of Netrin was disrupted. When we attempted to rescue the Netrin LOF mutants by expressing membrane-tethered NetrinB postsynaptically, the defects were enhanced and the medial dendrite did not extend to the midline in 90% of specimens. However, in the tethered NetB mutant (NetAΔBmyc-TM/>), tethered NetrinB was expressed in both of its endogenous sources, midline glia and TTMn, and the synapse functioned normally (Brankatschk and Dickson, 2006). While being expressed under its endogenous promoter, tethered Netrin supported normal synaptogenesis. It is possible that, through the endogenous expression pattern, cells not identified in this study could contribute to the normal phenotype seen in the mutants in a nonlocal manner. However, we hypothesize that the Brankatschk and Dickson (2006) tethered NetrinB mutant does not behave in a predictable way. We suggest that this protein is not as tightly membrane bound as the UAS-NetBCD8-TM protein product due to the added extracellular myc domains in the tethered mutant (NetAΔBmyc-TM/>). The tethered mutant's additional myc domains may account for differences in phenotypes due to increased protein flexibility or possible cleavage and secretion from the cell of origin. Considering this, non-cell-autonomous expression of a secreted Netrin rescued Netrin LOF defects, whereas expression of the tethered version using the same GAL4 drivers could not rescue the defects. We recognize this as evidence for the importance of Netrin secretion in GFS synaptogenesis.
Footnotes
This work was supported by a Florida Atlantic University Research Priority Grant from the Office of the Vice President for Research. We thank Pauline Phelan for her support and generosity toward this project—without the ShakB antibody she provided, we would not have been able to measure and quantify the regulation of gap junctions described in this report; Brandon Lloyd for contributing anatomical images for developmental experiments; Jana Börner for providing excellent guidance in confocal imaging; the Jan laboratory at UCSF for providing Frazzled antibody; and Benjamin Altenhein at the University of Mainz for providing NetrinA and B antibody.
The authors declare no competing financial interests.
References
- Albrecht S, Altenhein B, Paululat A. The transmembrane receptor Uncoordinated5 (Unc5) is essential for heart lumen formation in Drosophila melanogaster. Dev Biol. 2011;350:89–100. doi: 10.1016/j.ydbio.2010.11.016. [DOI] [PubMed] [Google Scholar]
- Allen M, Godenschwege TA. Electrophysiological recordings from the Drosophila giant fiber system (GFS) Cold Spring Harb Protoc. 2010;2010:pdb.prot5453. doi: 10.1101/pdb.prot5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MJ, Murphey RK. The chemical component of the mixed GF-TTMn synapse in Drosophila melanogaster uses acetylcholine as its neurotransmitter. Eur J Neurosci. 2007;26:439–445. doi: 10.1111/j.1460-9568.2007.05686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MJ, Drummond JA, Moffat KG. Development of the giant fiber neuron of Drosophila melanogaster. J Comp Neurol. 1998;397:519–531. doi: 10.1002/(SICI)1096-9861(19980810)397:4<519::AID-CNE5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Allen MJ, Godenschwege TA, Tanouye MA, Phelan P. Making an escape: Development and function of the Drosophila giant fibre system. Semin Cell Dev Biol. 2006;17:31–41. doi: 10.1016/j.semcdb.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Augustin H, Allen M, Partridge L. Electrophysiological recordings from the giant fiber pathway of D. melanogaster. J Vis Exp. 2011;pii:2412. doi: 10.3791/2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, Goodman CS. Chimeric axon guidance receptors: the cytoplasmic domains of slit and netrin receptors specify attraction versus repulsion. Cell. 1999;97:917–926. doi: 10.1016/S0092-8674(00)80803-X. [DOI] [PubMed] [Google Scholar]
- Blagburn JM, Alexopoulos H, Davies JA, Bacon JP. Null mutation in shaking-B eliminates electrical, but not chemical, synapses in the Drosophila giant fiber system: a structural study. J Comp Neurol. 1999;404:449–458. doi: 10.1002/(SICI)1096-9861(19990222)404:4<449::AID-CNE3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Boerner J, Godenschwege TA. Application for the Drosophila ventral nerve cord standard in neuronal circuit reconstruction and in-depth analysis of mutant morphology. J Neurogenet. 2010;24:158–167. doi: 10.3109/01677063.2010.489624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerner J, Godenschwege T a. Whole mount preparation of the adult Drosophila ventral nerve cord for giant fiber dye injection. J Vis Exp. 2011;pii:3080. doi: 10.3791/3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brankatschk M, Dickson BJ. Netrins guide Drosophila commissural axons at short range. Nat Neurosci. 2006;9:188–194. doi: 10.1038/nn1625. [DOI] [PubMed] [Google Scholar]
- Brierley DJ, Blanc E, Reddy OV, Vijayraghavan K, Williams DW. Dendritic targeting in the leg neuropil of Drosophila: the role of midline signalling molecules in generating a myotopic map. PLoS Biol. 2009;7:e1000199. doi: 10.1371/journal.pbio.1000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colón-Ramos DA, Margeta MA, Shen K. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science. 2007;318:103–106. doi: 10.1126/science.1143762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer MP, Kim S, Wolf B, Chiba A. Robo and Frazzled/DCC mediate dendritic guidance at the CNS midline. Nat Neurosci. 2003;6:223–230. doi: 10.1038/nn1017. [DOI] [PubMed] [Google Scholar]
- Garbe DS, Bashaw GJ. Independent functions of Slit-Robo repulsion and Netrin-Frazzled attraction regulate axon crossing at the midline in Drosophila. J Neurosci. 2007;27:3584–3592. doi: 10.1523/JNEUROSCI.0301-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe DS, O'Donnell M, Bashaw GJ. Cytoplasmic domain requirements for Frazzled-mediated attractive axon turning at the Drosophila midline. Development. 2007;134:4325–4334. doi: 10.1242/dev.012872. [DOI] [PubMed] [Google Scholar]
- Gu H, O'Dowd DK. Cholinergic synaptic transmission in adult Drosophila Kenyon cells in situ. J Neurosci. 2006;26:265–272. doi: 10.1523/JNEUROSCI.4109-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;4:61–85. doi: 10.1016/0896-6273(90)90444-K. [DOI] [PubMed] [Google Scholar]
- Hiramoto M, Hiromi Y, Giniger E, Hotta Y. The Drosophila Netrin receptor Frazzled guides axons by controlling Netrin distribution. Nature. 2000;406:886–889. doi: 10.1038/35022571. [DOI] [PubMed] [Google Scholar]
- Hummel T, Schimmelpfeng K, Klämbt C. Commissure formation in the embryonic CNS of Drosophila. Dev Biol. 1999;209:381–398. doi: 10.1006/dbio.1999.9235. [DOI] [PubMed] [Google Scholar]
- Jacobs K, Todman MG, Allen MJ, Davies JA, Bacon JP. Synaptogenesis in the giant-fibre system of Drosophila: interaction of the giant fibre and its major motorneuronal target. Development. 2000;127:5203–5212. doi: 10.1242/dev.127.23.5203. [DOI] [PubMed] [Google Scholar]
- Killeen MT. The dual role of the ligand UNC-6/Netrin in both axon guidance and synaptogenesis in C. elegans. Cell Adh Migr. 2009;3:268–271. doi: 10.4161/cam.3.3.8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej PA, Timpe LC, Mitchell KJ, Fried SR, Goodman CS, Jan LY, Jan YN. frazzled encodes a Drosophila member of the DCC immunoglobulin subfamily and is required for CNS and motor axon guidance. Cell. 1996;87:197–204. doi: 10.1016/S0092-8674(00)81338-0. [DOI] [PubMed] [Google Scholar]
- Mauss A, Tripodi M, Evers JF, Landgraf M. Midline signalling systems direct the formation of a neural map by dendritic targeting in the Drosophila motor system. PLoS Biol. 2009;7:e1000200. doi: 10.1371/journal.pbio.1000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ, Doyle JL, Serafini T, Kennedy TE, Tessier-Lavigne M, Goodman CS, Dickson BJ. Genetic analysis of Netrin genes in Drosophila: Netrins guide CNS commissural axons and peripheral motor axons. Neuron. 1996;17:203–215. doi: 10.1016/S0896-6273(00)80153-1. [DOI] [PubMed] [Google Scholar]
- Phelan P, Nakagawa M, Wilkin M, Moffat KG, O'Kane CJ, Davies JA, Bacon JP. Mutations in shaking-B prevent electrical synapse formation in the Drosophila giant fiber system. J Neurosci. 1996;16:1101–1113. doi: 10.1523/JNEUROSCI.16-03-01101.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Goulding LA, Tam JL, Allen MJ, Dawber RJ, Davies JA, Bacon JP. Molecular mechanism of rectification at identified electrical synapses in the Drosophila giant fiber system. Curr Biol. 2008;18:1955–1960. doi: 10.1016/j.cub.2008.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz H, Sadlowski E, Klaes A, Klämbt C. Control of midline glia development in the embryonic Drosophila CNS. Mech Dev. 1997;62:79–91. doi: 10.1016/S0925-4773(96)00652-1. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Watson JD, VanHoven MK, Colón-Ramos DA, Miller DM., 3rd Netrin (UNC-6) mediates dendritic self-avoidance. Nat Neurosci. 2012;15:731–737. doi: 10.1038/nn.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AKH, Colón-Ramos DA. Netrin instructs synaptic vesicle clustering through Rac GTPase, MIG-10, and the actin cytoskeleton. J Cell Biol. 2012;197:75–88. doi: 10.1083/jcb.201110127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YA, Wyman RJ. Passover eliminates gap junctional communication between neurons of the giant fiber system in Drosophila. J Neurobiol. 1996;30:340–348. doi: 10.1002/(SICI)1097-4695(199607)30:3<340::AID-NEU3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Tanouye MA, Wyman RJ. Motor outputs of giant nerve fiber in Drosophila. J Neurophysiol. 1980;44:405–421. doi: 10.1152/jn.1980.44.2.405. [DOI] [PubMed] [Google Scholar]
- Timofeev K, Joly W, Hadjieconomou D, Salecker I. Localized netrins act as positional cues to control layer-specific targeting of photoreceptor axons in Drosophila. Neuron. 2012;75:80–93. doi: 10.1016/j.neuron.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uthaman SB, Godenschwege TA, Murphey RK. A mechanism distinct from highwire for the Drosophila ubiquitin conjugase bendless in synaptic growth and maturation. J Neurosci. 2008;28:8615–8623. doi: 10.1523/JNEUROSCI.2990-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth WG, Bhatt H, Hedgecock EM. Neuroglia and pioneer neurons express UNC-6 to provide global and local netrin cues for guiding migrations in C. elegans. Neuron. 1996;16:35–46. doi: 10.1016/S0896-6273(00)80021-5. [DOI] [PubMed] [Google Scholar]
- Wearne SL, Rodriguez A, Ehlenberger DB, Rocher AB, Henderson SC, Hof PR. New techniques for imaging, digitization and analysis of three-dimensional neural morphology on multiple scales. Neuroscience. 2005;136:661–680. doi: 10.1016/j.neuroscience.2005.05.053. [DOI] [PubMed] [Google Scholar]
- Wharton KA, Jr, Crews ST. CNS midline enhancers of the Drosophila slit and Toll genes. Mech Dev. 1993;40:141–154. doi: 10.1016/0925-4773(93)90072-6. [DOI] [PubMed] [Google Scholar]
- Wheeler SR, Pearson JC, Crews ST. Time-lapse imaging reveals stereotypical patterns of Drosophila midline glial migration. Dev Biol. 2012;361:232–244. doi: 10.1016/j.ydbio.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winberg ML, Mitchell KJ, Goodman CS. Genetic analysis of the mechanisms controlling target selection: complementary and combinatorial functions of netrins, semaphorins, and IgCAMs. Cell. 1998;93:581–591. doi: 10.1016/S0092-8674(00)81187-3. [DOI] [PubMed] [Google Scholar]
- Yang L, Garbe DS, Bashaw GJ. A frazzled/DCC-dependent transcriptional switch regulates midline axon guidance. Science. 2009;324:944–947. doi: 10.1126/science.1171320. [DOI] [PMC free article] [PubMed] [Google Scholar]