

Abstract
Extensive evidence implicates GluN2B-containing NMDA receptors (GluN2B-NMDARs) in excitotoxic-insult-induced neurodegeneration and amyloid β (Aβ)-induced synaptic dysfunction. Therefore, inhibiting GluN2B-NMDARs would appear to be a potential therapeutic strategy to provide neuroprotection and improve cognitive function in Alzheimer's disease (AD). However, there are no reports of long-term in vivo treatment of AD mouse models with GluN2B antagonists. We used piperidine18 (Pip18), a potent and selective GluN2B-NMDAR antagonist with favorable pharmacokinetic properties, for long-term dosing in AD mouse models. Reduced freezing behavior in Tg2576 mice during fear conditioning was partially reversed after subchronic (17 d) Pip18 treatment. However, analysis of freezing behavior in different contexts indicated that this increased freezing likely involves elevated anxiety or excessive memory generalization in both nontransgenic (NTG) and Tg2576 mice. In PS2APP mice chronically fed with medicated food containing Pip18 for 4 months, spatial learning and memory deficits were not rescued, plaque-associated spine loss was not affected, and synaptic function was not altered. At the same time, altered open field activity consistent with increased anxiety and degraded performance in an active avoidance task were observed in NTG after chronic treatment. These results indicate that long-term treatment with a GluN2B-NMDAR antagonist does not provide a disease-modifying benefit and could cause cognitive liabilities rather than symptomatic benefit in AD mouse models. Therefore, these results challenge the expectation of the therapeutic potential for GluN2B-NMDAR antagonists in AD.
Keywords: Alzheimer's disease, cognition, GluN2B, memory, NMDAR, spine
Introduction
The NMDA subtype of glutamate receptors play essential physiological roles in neural development, synaptic plasticity, and circuit function, but can also mediate damage and death of neurons when excessively activated (Paoletti et al., 2013). A large body of evidence from in vitro experiments implicates GluN2B-containing NMDA receptors (GluN2B-NMDARs) in particular as mediators of neuronal damage during excitotoxicity and pathological activation of GluN2B-NMDARs has been demonstrated in preclinical animal models of various neurodegenerative conditions including Huntington's disease and stroke (Reyes et al., 1998; Zeron et al., 2002; Hammond et al., 2006; Liu et al., 2007; Heng et al., 2009; Milnerwood et al., 2010; Tu et al., 2010; Lai et al., 2011). In the context of Alzheimer's disease (AD), studies using selective antagonists have specifically implicated GluN2B-NMDARs in mediating the deleterious effects of amyloid β (Aβ), including synapse loss and impairment of synaptic plasticity (Hu et al., 2009; Li et al., 2011; Rammes et al., 2011; Rönicke et al., 2011; Costa et al., 2012). Together, the evidence that GluN2B-NMDARs mediate neurodegeneration and have a major contribution to AD-relevant pathology suggests that GluN2B-NMDARs are a potential target for neuroprotection in AD. Given that potent and selective GluN2B-NMDARs are available and have entered clinical trials for various other indications, including Parkinson's disease, major depressive disorder, and traumatic brain injury (Yurkewicz et al., 2005; Nutt et al., 2008; Preskorn et al., 2008; Addy et al., 2009; Ibrahim et al., 2012), clinical testing of this hypothesis is now feasible. However, the evidence supporting GluN2B-NMDAR antagonism for treatment of AD comes mainly from in vitro experiments and the effects of chronic GluN2B inhibition in vivo in mouse models of AD are unknown.
Although extrasynaptic GluN2B-NMDARs on excitatory neurons are especially implicated in mediating neurodegeneration (Hardingham et al., 2002; Hardingham and Bading, 2010), GluN2B-NMDARs are also present at synapses on inhibitory GABAergic interneurons and contribute to interneuron excitation and the balance between excitation and inhibition (Hanson et al., 2013). Consistent with a role in normal physiological function, acute treatment with GluN2B-NMDAR antagonists can alter circuit function and prolonged treatment may lead to long-lasting alterations in neural circuitry (Hanson et al., 2013). Therefore, testing chronic treatment with GluN2B-NMDAR antagonists in AD models is essential, not only for evaluating potential neuroprotective benefits such as preventing synapse loss, but also for evaluating potential circuit impacts that could translate into either symptomatic benefits or liabilities.
In this study, we evaluated both subchronic treatment (17 d) and chronic treatment (4 months) in different AD mouse models and assessed both neuroprotection and cognitive function. Overall, chronic treatment failed to protect against spine loss or rescue memory deficits in the AD model mice, whereas both subchronic and chronic treatment disrupted normal cognitive functions. These results suggest GluN2B-NMDAR antagonists could have liabilities related to disruption of normal cognitive function, but do not predict a disease-modifying benefit in AD.
Materials and Methods
Animals and drugs.
Initial in vitro characterization of piperidine18 (Pip18) effects on NMDARs in brain slices was done using 2-week-old and 2-month-old male C57BL/6 mice. Five-month old female Tg2576 mice overexpressing human amyloid precursor protein (APP) with the “Swedish” mutation K670N/M671L under control of the prion protein (PrP) promoter were used for fear-conditioning experiments. Chronic treatment experiments were done using male PS2APP mice that coexpress human APP with the Swedish mutation as well as human Presenilin 2 with the N141I mutation, driven by Thy1 and PrP promoters, respectively (Ozmen et al., 2009). The PS2APP allele was bred to homozygosity for all mice used in this study. PS2APP;GFP-M mice used for behavior and two-photon imaging were generated by crossing Thy1-GFP M line mice (Feng et al., 2000) with PS2APP mice. Pip18 was synthesized at Medicilon and quality control was performed at Genentech. For subchronic dosing studies, the vehicle for Pip18 was 30% hydroxypropyl-β-cyclodextrinin, 25 mm citrate, pH 5. For chronic dosing studies, Pip18 was milled into rodent diet (#5001; Purina) at 150 mg/kg food (Research Diets) and untreated chow was used as the control diet. In all experiments involving drug treatment, experimenters were blind to both genotype and treatment groups during data acquisition and analysis. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at Genentech and are in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
NMDAR calcium influx assay.
Twenty-four hours before assay, HEK cells stably expressing doxycycline-inducible GluN1 along with either GluN2A or GluN2B were seeded into clear-bottom 384-well poly-d-lysine-coated plates (25,000 cells per well) in MEM (w/o l-glutamine) and induced with 7.5 μg/ml doxycycline. To prevent NMDAR-mediated cell toxicity during the overnight culture, 500 μm (+)ketamine was added to the induction medium. For measurement of changes in cytosolic calcium, the seeding medium was removed and the cells were incubated with different concentrations of Pip18 at 37°C for 60 min with 1× Becton Dickinson Calcium Assay Kit reagent in HBSS w/o magnesium, including 1.8 mm calcium, 0.65 mg/ml probenecid, and 10 μm (+)ketamine, pH 7.15), and then allowed to equilibrate to room temperature for 30 min. Well contents were removed and fresh compound in the same solution was reapplied before data acquisition was started. Activation of NMDAR-mediated calcium influx was achieved by adding 500 nm l-glutamate (approximate EC50) and 30 μm glycine with test compound in HBSS. After a 10 s baseline read, maximum relative fluorescence units was measured over a 5 min period on a fluorescence plate reader (FDSS 7000; Hamamatsu). Responses are scaled relative to positive control wells treated with 500 nm l-glutamate and 30 μm glycine (1.0) and negative control wells with 30 μm glycine without glutamate (0). IC50 values were calculated by fitting with a four-parameter Hill equation.
Pharmacokinetic analysis of Pip18.
Liquid chromatographic-tandem mass spectrometry was used for the pharmacokinetic (PK) analysis of Pip18 in brain homogenates.
Tg2576 fear conditioning.
Contextual fear conditioning (CFC) was performed at Charles River Discovery Research Services on 5-month-old female Tg2576 or nontransgenic (NTG) littermates mice during the final 2 d of subchronic treatment (consisting of 17 d of twice daily injection of Pip18 at 5 or 15 mg/kg, i.p.) using a modification of previously published protocols (Comery et al., 2005) and a FreezeFrame system (Coulbourn). Training consisted of placing a mouse in a 30 × 24 × 21 cm chamber with the bright house light on and allowing exploration for 2 min. Afterward, an auditory cue [1700 Hz, 80 dB; the conditioned stimulus (CS)] was presented for 15 s. A foot shock [1.5 mA; the unconditioned stimulus (US)] was administered for the final 2 s of the auditory cue. This procedure was repeated and the mouse was removed from the chamber 30 s later. Freezing behavior was recorded during (2 s) and after (5 s) the shocks by a computerized camera tracking system. The next day, the mouse was returned to the same chamber in which the training occurred and freezing behavior was recorded (memory for context). At the end of the 5 min context test, the mouse was returned to its home cage. One hour later, freezing behavior was recorded in a novel environment (altered context) and in response to the cue (memory for cue). The novel environment used for the altered context used the same modular test chambers where training occurred but modified by different lighting conditions, colors, and textures on the walls and different floor material. The mouse was placed in the novel environment and freezing was scored for 3 min. The auditory cue (1700 Hz, 80 dB, CS) was then presented for 3 min and freezing was scored again. Freezing scores for each subject were expressed as a percentage for each portion of the test (memory for context, altered context, and cue). Reductions in freezing behavior in Tg2576 mice compared with NTG littermates were assessed using a Student's t test. Effects of Pip18 treatment on freezing behavior were assessed using ANOVA followed by the Holm–Sidak method and preplanned comparisons between certain groups.
Aβ levels analysis.
Fresh-frozen right hippocampal and ventral cortical samples were used for biochemical analysis. Aβ 1–40 and 1–42 levels in the soluble fraction of brain tissue samples were analyzed with ELISA using Aβ 1–40 and 1–42 ELISA kits (EZbrain40 and EZbrain42 ELISA, respectively; Millipore) according to the manufacturer's instructions. Soluble Aβ was measured using the supernatant that was collected after initial tissue homogenization and centrifugation. The mean value of NTG vehicle samples was subtracted from all other measurements to normalize the assay background.
Morris water maze.
The Morris water maze consisted of a pool (122 cm diameter) filled with water (18 ± 2°C) made opaque with nontoxic white tempera paint and placed in a room surrounded by distinct extramaze cues. Mice were first given 4 pretraining trials in which they had to swim down a channel (15 × 122 cm) and mount a platform (15 cm) hidden 1.5 cm below the water surface at the end of the channel. Mice were then trained to locate a hidden platform (15 cm) submerged 1.5 cm below the water for 5 consecutive days. Mice received 1 or 2 training sessions per day, with each session consisting of 3 trials (10 min between trials) and separated by 3–4 h for a total of 10 hidden sessions. The platform location remained the same throughout the hidden-platform training, but the drop location was changed semirandomly between trials. Mice had 60 s to locate the platform during training. Mice that did not find the platform were guided to it and placed on it for 10 s. A spatial probe trial was performed 16–18 h after the third day of training (beginning of session 7) and 24 h after the completion of hidden-platform training. The platform was removed and mice were allowed to swim in the pool for 60 s. For cued visible-platform training, the platform (15 cm diameter) was submerged (1.5 cm) but visibly cued with a striped mast (15 cm in height). Over 2 consecutive days, mice received 1 or 2 training sessions per day separated by 3–4 h for a total of 3 cued sessions. Each session consisted of two training trials with an intertrial interval of 10 min. For each session, the platform was moved to a new location and the drop location was changed semirandomly between trials. Escape latencies, swim paths, swim speeds, percentage of time spent in each quadrant, and platform crossings (in a zone 200% the size of the platform) were recorded using a ceiling-mounted camera and tracked using CleverSys TopScan software. Effects of PS2APP genotype and Pip18 treatment on spatial learning and memory were assessed by two-way repeated-measures ANOVA.
Barnes maze.
The Barnes maze consisted of an elevated (90 cm) white circular table (91 cm in diameter) with 20 holes (5 cm diameter) equally spaced around the perimeter (Stoelting). A target escape tunnel (6 × 25 cm) was placed below one of the holes. The maze was placed in the center of a room with prominent extramaze visual cues. During training, mice were released from a cylinder (8 × 10 cm diameter) in the center of the maze as bright lights were turned on. Once the mouse found and entered the escape tunnel, the bright lights were dimmed and the mouse was allowed to stay in the tunnel for 10–20 s. The mice first received familiarization training in which a channel was made such that only the escape hole was available for the animal to enter. Mice received 4 consecutive trials (1 min max) to become familiar with entering the escape tunnel. If the mouse did not reach the goal within 1 min, the experimenter would guide the mouse gently to the escape box and leave the mouse inside for 10–20 s. Spatial training consisted of allowing the mouse to freely explore the entire maze for up to 3 min to locate the target hole (in a new location from familiarization training). During this time, the animal's movements were recorded using a ceiling mounted camera and tracked using CleverSys TopScan software. Immediately after the mouse entered the escape box, the lights were dimmed and the mouse was allowed to stay in the tunnel for 10–20 s. If the mouse did not reach the goal within 1 min, the experimenter would guide the mouse gently to the escape box. Training occurred over 2 consecutive days, with 4 trials per day and an intertrial interval of 15 min. Latency to enter target hole, distance traveled, velocity (in meters per second), and errors (total number of nose pokes into incorrect holes) were analyzed. A spatial probe trial was conducted 24 h after the final training session, in which the escape tunnel was removed and the animal was allowed to explore for 3 min. Number of nose pokes (errors) and percentage time spent exploring in each hole, latency, and path length to reach the former target hole were measured. A measure of percentage accuracy was calculated as (time exploring target hole/time exploring all locations) × 100. Effects of PS2APP genotype and Pip18 treatment on spatial learning and memory were assessed by two-way repeated-measures ANOVA.
Active avoidance.
Mice were trained in standard avoidance boxes for mice (44 × 17 × 25 cm), which consisted of two compartments separated by an automated guillotine door (MedAssociates). The floor of the chamber contained metal rods that delivered an electric shock (0.3 mA, US) by a scrambled shocker. Each compartment contained a speaker and a stimulus light that delivered a 72–73 dB tone and light CS. Each experimental chamber was located in a sound-attenuating cubicle. Shuttling (movement from one compartment to the other) was monitored by two infrared arrays. Mice received 3 consecutive days of avoidance training following a modified protocol (M. Weber, unpublished data, and Foldi et al., 2011). Each training session began with a 5 min acclimation period during which the animal was placed into one side of the shuttle box with the door closed and no stimuli were presented. Avoidance training trials started immediately after this acclimation period. During each avoidance trial, the door between the two sides of the shuttle box opened and a light and tone CS was presented for a total of 7 s on the side of the chamber that contained the animal. The CS was co-terminated with a 2 s foot shock (US), after which the door closed and a new trial began after a varying intertrial interval that averaged 40 s. If the animal shuttled during the CS presentation, the tone and light were terminated immediately, the US was not delivered, and the door closed; this was scored as an avoidance response. If the animal shuttled during the US foot shock presentation, the shock was immediately terminated and the door closed; this was recorded as an escape response. An escape failure was recorded if the animal did not shuttle to the opposite side within the 7 s. Each animal received 100 trials each day. Stimulus delivery and avoidance responses were controlled and collected automatically using MED-PC IV software (MedAssociates). The percentage of avoidance responses was calculated for each block of 20 trials. Effects of PS2APP genotype and Pip18 treatment on activity were assessed by one- or two-way ANOVA.
Open field.
Spontaneous locomotor activity was measured with an automated Photobeam Activity System Open Field system (San Diego Instruments). Mice were placed individually in a clear plastic chamber (41 × 41 × 30 cm) and their horizontal and vertical movements were monitored for 15 min/session with two 16 × 16 photobeam arrays. Total distance traveled (in centimeters), number of rearings, percentage activity in the center, and periphery in the open field were analyzed. Effects of PS2APP genotype and Pip18 treatment on activity were assessed by one- or two-way ANOVA.
Two-photon microscopy imaging and spine/plaque density analysis.
The somatosensory cortex of PS2APP;GFP-M mice was imaged ex vivo. Twenty-four hours before imaging, animals were injected with 10 mg/kg Methoxy-X04 intraperitoneally to visualize individual plaques (Klunk et al., 2002). Animals were killed and transcardially perfused with 10 ml of PBS followed by 10 ml of 4% PFA. Brains were harvested, incubated overnight in 4% PFA, and then washed and embedded in agarose for imaging. Apical dendrites and their spines in somatosensory cortex were imaged via a two-photon laser-scanning microscope (Ultima In Vivo Multiphoton Microscopy System; Prairie Technologies) using a Ti:sapphire laser (MaiTai DeepSee Spectra Physics; Newport) tuned to 840 nm and a 60× numerical aperture 1.0 objective lens (Olympus) with pixel resolution of 0.1 μm/pixel across a 1024 × 1024 pixel field of view (FOV) using 0.5 μm steps. At least five cells were collected per condition per animal. For plaque density measurements, larger image stacks were collected using a 20× objective across a 1024 × 1024 pixel FOV with 2 μm steps. Dendritic spine density and size measurements were generated using custom, semiautomated image analysis routines in MATLAB (MathWorks). Spine density was estimated as the total number of visible dendritic spines divided by the corresponding length of dendrite. Relative spine volumes were estimated for each detected spine based on the number of corresponding GFP+ pixels in x, y, z dimensions above a local threshold applied as part of an automated image segmentation algorithm. For comparison of spine density relative to plaques in PS2APP animals, an FOV containing a dendrite and nearby plaque was considered “near plaque” and an FOV containing only a dendrite with no visible plaque was considered “away from plaque.” To meet the “away from plaque” criteria, we confirmed that no plaque was present in the FOV and at least 100 μm outside of the containing FOV. Plaque density was assessed over the same region as spine density measurements. Larger volumes (∼200 μm depth) were collected and plaque density was quantified by a threshold-based MATLAB routine designed to automatically identify methoxy-X04-labeled plaques.
Electrophysiology.
Horizontal slices (400 μm) from mouse brain containing hippocampus were prepared with a vibrating sectioning system (Leica) and recorded in oxygenated artificial CSF (ACSF) containing the following (in mm): 127 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.25 Na2HPO4, 25 NaHCO3, and 25 glucose. Slices were prepared in ice-cold oxygenated ACSF with the MgSO4 concentration elevated to 7 mm, NaCl replaced with 110 mm choline, and with 11.6 mm Na-ascorbate and 3.1 mm Na-pyruvate added. Field recordings of EPSPs were measured from the stratum radiatum of CA1 in response to stimulation of Schaffer collateral inputs using field recordings. NMDAR EPSPs were measured with the Mg2+ concentration in ACSF reduced to 0.5 mm and in the presence of PTX and NBQX. Paired pulse ratios (PPRs) were measured by dividing the slope of the second EPSP by the slope of the first EPSP evoked using paired stimulations and varying interstimulus intervals. Input–output relationships were measured by stimulating at logarithmically spaced stimulus intensities Significance of the maximum responses was assessed using a two-way ANOVA with genotype and treatment as the factors and the significance of PPRs was assessed using a three-way ANOVA including interstimulus interval as a factor. Post hoc assessment of multiple comparisons was performed using the Holm–Sidak method.
Results
Pip18 is a potent and selective GluN2B antagonist with good brain availability
To chronically inhibit GluN2B-NMDARs, a selective and potent antagonist with favorable PK properties is needed. Therefore, we characterized Pip18, a small-molecule GluN2B-NMDAR antagonist reported to fulfill these requirements (M. E. Layton, ACS ProSpectives conference 2009; Fig. 1). Pip18 is reported to be an antagonist of GluN2B-NMDARs with a Ki of 1.6 nm (cortex) and a cell-based IC50 of 4.6 nm (with >65,000-fold selectivity vs GluN2A; M. E. Layton, ACS ProSpectives conference 2009). To confirm the potency and selectivity of Pip18, we examined the effects of Pip18 on GluN2B or GluN2A containing NMDARs stably expressed in HEK cells using a Ca2+ influx assay (Fig. 1A). We measured an IC50 of 9.5 ± 1.3 nm for GluN2B-NMDARs and no detectable antagonism of GluN2A-NMDARs. For comparison, this assay yielded an IC50 of 53.7 ± 5.7 nm for the GluN2B-NMDAR antagonist Ro25-6981, which has a reported Ki of 9 nm (Fischer et al., 1997), supporting the observation that compounds usually show less potency in cell-based assays than in in vitro receptor occupancy experiments. Overall, these experiments provide confirmation that Pip18 is a potent and selective GluN2B-NMDAR antagonist. To test the efficacy of Pip18 on native NMDARs, we examined synaptic NMDAR EPSPs in hippocampal brain slices. Similar to our previous results with Ro25-6981 (Hanson et al., 2013), 1 μm Pip18 inhibited NMDAR EPSPs in 2-week-old mice, but not in 2-month-old mice (Fig. 1B), which is consistent with a developmental shift of GluN2B-NMDARs from synaptic to extrasynaptic locations. This result is notable because these synapses in mature animals contain a significant population of triheteromeric NMDARs (GluN1/2A/2B composition; Gray et al., 2011; Rauner and Köhr, 2011; Soares and Lee, 2013; Tovar et al., 2013). Therefore, this result demonstrates a lack of impact of Pip18 on triheteromeric NMDARs. This is consistent with evidence from in vitro studies examining isolated triheteromeric receptors that have found greatly reduced potency and efficacy of GluN2B antagonists on GluN1/2A/2B receptors compared with diheteromeric GluN1/2B receptors (Hatton and Paoletti, 2005; Hansen et al., 2014). Therefore, the “GluN2B-NMDARs” that are inhibited in our study (and other studies with GluN2B antagonists) are mainly GluN1/2B diheteromeric receptors with likely little contribution from GluN1/2A/2B triheteromeric receptors.
Figure 1.
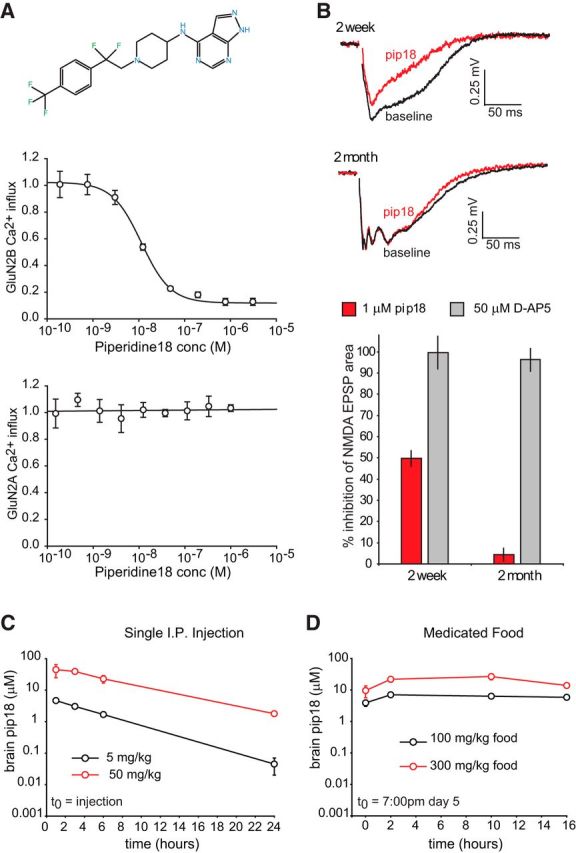
Pip18 is a potent and selective GluN2B antagonist with good brain availability. A, Potency and selectivity of Pip18 in cell-based assays. Top, Structure of Pip18. Center, Example experiment showing the dose–response curve for Pip18 using HEK cells stably expressing GluN1 and GluN2B NMDAR subunits. Bottom, Example experiment showing the lack of antagonism using HEK cells stably expressing GluN1 and GluN2A NMDAR subunits. Data are shown as mean ± SD. B, Top, Examples of isolated NMDAR fEPSPs recorded from CA1 stratum radiatum in response to Schaffer collateral stimulation for 2-week-old and 2-month-old mice before and after 1 μm Pip18 application. Bottom, Quantification of NMDAR EPSP inhibition by Pip18 in 2-week-old and 2-month-old mice (n = 4/group). As a control, inhibition by the nonselective NMDAR antagonist D-AP5 (50 μm) is shown. Data are shown as mean ± SEM. C, PK analysis of Pip18 brain levels after injections of 5 or 50 mg/kg Pip18 (n = 3 mice/time point/dose). Data are shown as mean ± SD. D, PK analysis of Pip18 brain levels during day 5 of treatment with Pip18 via medicated food using 100 or 300 mg/kg food (n = 3 mice/time point/dose). Data are shown as mean ± SD.
We performed PK analysis of the brain levels of Pip18 after a single intraperitoneal injection or ongoing feeding with medicated food and found that both routes of administration provided prolonged brain exposure of Pip18 at high levels (Fig. 1C,D). Even the low dose of 5 mg/kg intraperitoneally was sufficient to achieve brain concentrations >1.7 ± 0.2 μm for 6 h, whereas a 50 mg/kg intraperitoneal dose achieved brain concentrations >1.8 ± 0.1 μm for 24 h. Medicated food at the lowest concentration of 100 mg/kg food achieved sustained brain concentrations of at least 1.8 ± 0.9 μm measured throughout the final day of 5 d of treatment.
Subchronic Pip18 injections increase freezing in Tg2576 mice during CFC testing
To assess therapeutic potential, we tested both the shorter- and longer-term effects of GluN2B-NMDAR inhibition in AD model mice using Pip18. We first examined the more immediate effects of Pip18 using subchronic treatment in Tg2576 AD model mice, which exhibit a robust deficit in CFC (Jacobsen et al., 2006) that is amenable to testing in a short period of time. Mice were treated for 2 weeks with Pip18 (5 or 15 mg/kg, i.p., twice daily to maximize trough brain exposure) and then tested for CFC while continuing treatment. In this experimental paradigm, the vehicle-treated Tg2576 mice exhibited less freezing than NTG littermates when exposed to the training context. They also exhibited less freezing when returned to the same chambers after modifications had been made to introduce select alterations to the context, as well as to subsequent presentation of the cue while in the altered context (Fig. 2A). Tg2576 mice treated with either dose of Pip18 showed more freezing than vehicle-treated Tg2576 in all three test conditions, which would appear to be consistent with increased fear memory (Fig. 2A). However, analysis of the freezing during training showed that the Tg2576 mice froze significantly less than the NTG mice after the second foot shock, suggesting an altered response to the training stimulus (Fig. 2B). This raises the possibility that the impaired freezing behavior in Tg2576 mice during testing may not reflect memory impairment, but rather altered behavioral response to an aversive stimulus. Notably, however, there was no impact of drug treatment on the freezing during training, indicating that the drug-induced increase in freezing during testing was not secondary to altered responsiveness during training. To rule out any direct effects of GluN2B antagonism on Aβ production or clearance, we also examined Aβ40 and Aβ42 levels in the drug-treated Tg2576 mice and found no changes (Fig. 2C).
Figure 2.
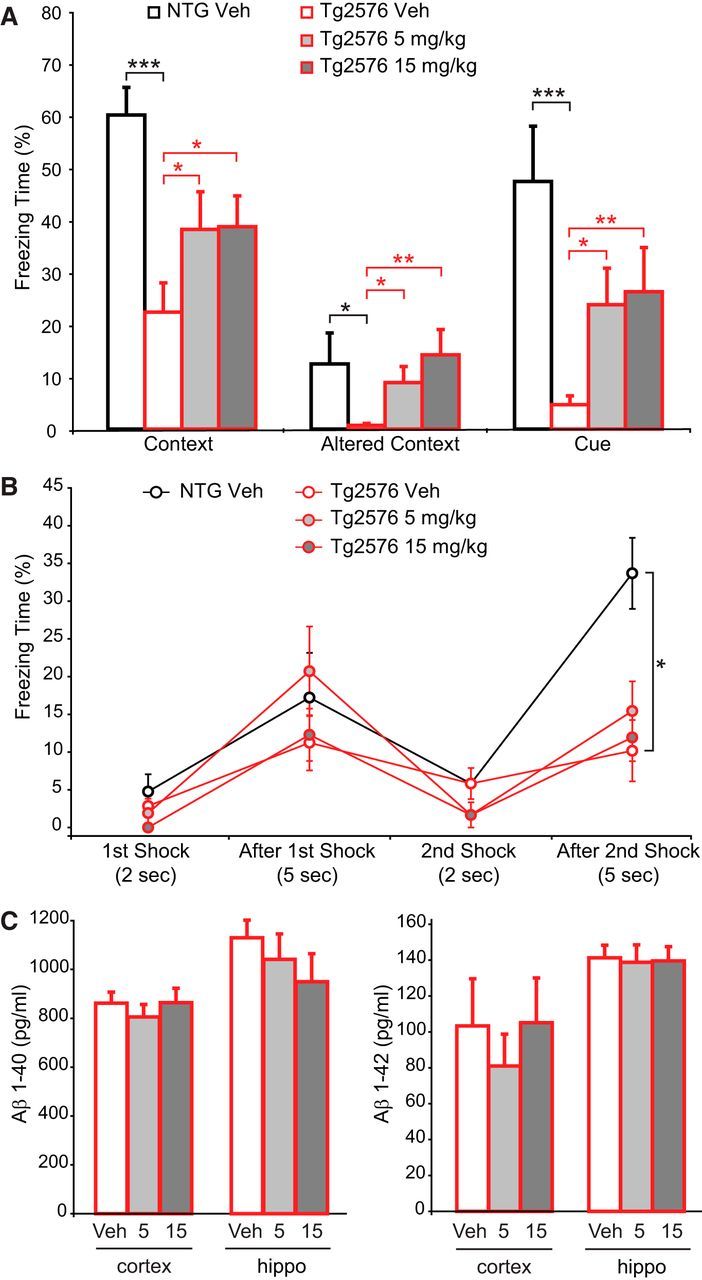
Subchronic Pip18 partially reverses decreased freezing in Tg2576 mice during CFC. A, Freezing in response to the context, altered context, and cue. Planned comparisons showed significantly reduced freezing in vehicle-treated Tg2576 mice compared with NTG mice in response to the context, altered context, and cue (one-tailed t tests, black stars). ANOVA with the NTG group excluded revealed a significant effect of Pip18 treatment within Tg2576 mice for the altered context (F(2,42) = 4.219, p = 0.021) and follow-up tests showed that the 15 mg/kg treatment was significantly different from vehicle (p < 0.05). ANOVA with the NTG group excluded also revealed a significant effect of Pip18 treatment within Tg2576 mice for the cue (F(2,42) = 3.399, p = 0.043). Planned comparisons also showed increased freezing in Pip18-treated versus vehicle-treated Tg2576 mice (one-tailed t test, red stars). n = 14 NTG, n = 15 Tg2576/treatment group. *p < 0.05; **p < 0.01; ***p < 0.001. B, Freezing during training is shown. Comparison between vehicle-treated NTG and Tg2576 mice by ANOVA across time points revealed a significant interaction between genotype and time point (F(3,112) = 4.458, p = 0.005) and follow-up tests showed a significant overall difference between NTG and Tg2576 mice and a significant effect of genotype within the last time point. *p < 0.05. ANOVA of the Tg2576 treatment groups across time points with the NTG group excluded revealed a significant effect of time point (F(3,168) = 13.342, p < 0.001), but not treatment and no interaction between treatment and time point. C, Aβ levels measured by ELISA in the cortex and hippocampus of Tg2576 mice. There was no significant effect of treatment on Aβ40 or Aβ42 in either the cortex or hippocampus (n = 15 per group). All data are shown as mean ± SEM.
Subchronic Pip18 injections increase fear responses in Tg2576 and NTG mice in a manner consistent with excessive memory generalization
To evaluate additional dose levels and to better understand the nature of the Pip18 effects during CFC, we performed a second study using 1 or 10 mg/kg Pip18 twice daily treatment in both NTG and Tg2576 mice. In this experiment, we again saw decreased freezing relative to NTG controls in the vehicle-treated Tg2576 mice during all three test phases (Fig. 3A). We also saw increased freezing relative to vehicle-treated animals in both NTG and Tg2576 mice treated with either dose of Pip18 when tested in the altered context or in response to the cue (Fig. 3A). The increased freezing in the altered context suggests both genotypes had an enhanced general fear response after Pip18 treatment. This could be explained by increased anxiety and/or an overgeneralization of the learned fear response to a somewhat similar context. To quantify the latter possibility, we calculated a discrimination index (based on the relative freezing in the altered vs training context for each animal) as a measure of the degree of memory specificity vs generalization (Xu and Südhof, 2013). This analysis showed that Pip18 treatment decreased the discrimination index in both NTG and Tg2576 mice (Fig. 3B). Therefore, one interpretation of the increased freezing in Pip18-treated mice during CFC testing is that they exhibit an inappropriate anxiety response in situations that are similar to remembered harmful situations (i.e., excessive memory generalization). Therefore, rather than simply reflecting a selective improvement of impaired memory in the Tg2576 mice with Pip18, the overall profile of freezing responses could reflect more complicated impacts on fear-related behavior in both NTG and Tg2576 mice.
Figure 3.
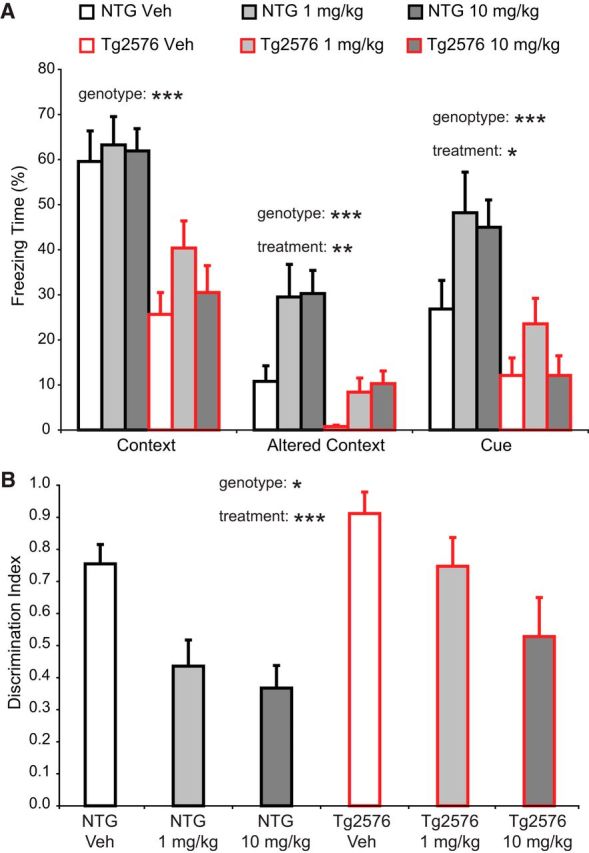
Subchronic treatment increases fear memory generalization in both NTG and Tg2576 mice. A, Freezing during fear memory testing. ANOVA for freezing in the context revealed a significant effect of genotype (F(1,83) = 37.69, p < 0.001), but not treatment (n = 14–15 mice per group). ANOVA for freezing in the altered context revealed a significant effect of treatment (F(2,83) = 6.86, p = 0.002) and genotype (F(1,83) = 23.51, p < 0.001), but no interaction. Follow-up tests showed that both 1 and 10 mg/kg were significantly different from vehicle (p < 0.01). ANOVA for freezing to the cue revealed a significant effect of treatment (F(2,83) = 3.52, p = 0.034) and genotype (F(1,83) = 22.85, p < 0.001), but no interaction. Follow-up tests showed that the 1 mg/kg group was significantly different from the vehicle group (p < 0.05). B, The discrimination index (DI) was calculated from the time spent freezing as follows: DI = (Context − Altered)/(Context + Altered). ANOVA revealed significant effects of treatment (F(2,83) = 8.864, p < 0.001) and genotype (F(1,83) = 5.95, p = 0.017), but no interaction. Follow-up tests showed that both 1 and 10 mg/kg were significantly different from vehicle (p < 0.05 and p < 0.001, respectively). The decreased DI values correspond to increased generalization. *p < 0.05; **p < 0.01; ***p < 0.001.
Chronic Pip18 treatment does not rescue spatial memory deficits in PS2APP mice
To examine the impacts of chronic Pip18 treatment and potential disease-modifying benefits, we focused on PS2APP AD model mice that also carry a transgene expressing GFP under the Thy1 promoter. Our pilot studies in these mice found that, as in other mouse models (Tsai et al., 2004), there was a decrease in the density of dendritic spines, especially near plaques, in PS2APP mice. In agreement with previous studies of PS2APP mice (Richards et al., 2003), we also have observed deficits in spatial memory in these mice. To examine both cognitive and dendritic spine phenotypes after chronic GluN2B inhibition, PS2APP mice and NTG littermates were fed Pip18 (150 mg/kg drug concentration milled into mouse chow pellets) for 16 weeks. Medication started at 7 months of age and mice were subjected to a battery of behavioral tests during the final 4 weeks of treatment before tissue harvest for spine analysis (Fig. 4A). Terminal PK analysis after this chronic treatment showed Pip18 brain levels of 1.4 ± 0.6 μm. This indicates that, despite potential degradation of compound over the course of treatment, the total brain levels remained dramatically higher than the reported Ki of 1.6 nm (M. E. Layton, ACS ProSpectives conference 2009) by the end of the study. Even when protein binding is taken into account (estimated free drug in plasma is 7% of total), the concentration of free Pip18 in brain is predicted to be >60× the Ki. Therefore, this dosing regimen achieved effective target engagement during the treatment period.
Figure 4.
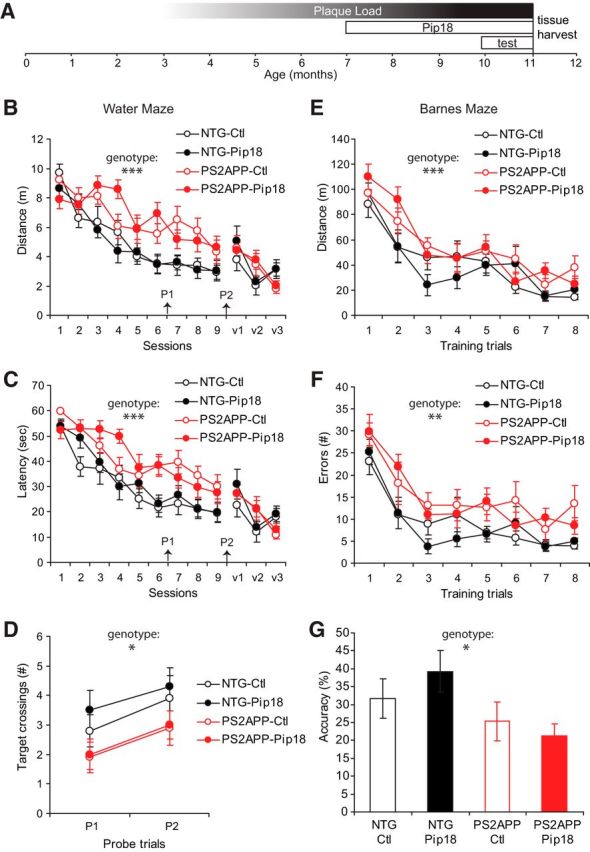
Chronic treatment does not rescue spatial learning and memory deficits in PS2APP mice. A, Course of Pip18 treatment relative to the accumulation of plaque load (approximated as average plaque size × average plaque volume) in PS2APP mice. The timing of the behavioral testing before tissue harvest for subsequent spine analysis is also illustrated. B, Distance traveled to locate a hidden or visible platform in the water maze. Assessment of spatial learning during hidden training sessions (1–9) with a 2-way repeated-measures ANOVA revealed a significant effect of training session (F(8,28) = 22.9; p < 0.0001) and genotype (F(1,35) = 26.1; p < 0.0001), but no treatment effect or interaction (n = 9–10/group). There were no significant genotype or treatment differences during visible training sessions (v1–v3). C, Latency to locate a hidden or visible platform in the water maze. Two-way repeated-measures ANOVA revealed a significant main effect of training session (F(8,28) = 37.5; p < 0.0001) and genotype (F(1,35) = 21.1; p < 0.0001), but no treatment effect or interaction (n = 9–10/group). There were no significant genotype or treatment differences during visible training sessions (v1–v3). D, Spatial memory assessment in the water maze. Probe trials were given before training session 7 and 24 h after the final training session. Two-way ANOVA of the number of crossings over the target zone (200% larger than the platform) revealed a significant main effect of genotype (F(1,35) = 4.3; p < 0.05), but no treatment effect or interaction (n = 9–10/group). All data are shown as mean ± SEM. E, Distance traveled to locate a hidden tunnel in the Barnes maze. Assessment of spatial learning by training trial with a two-way repeated-measures ANOVA revealed a significant main effect of training trial (F(7,31) = 24.1; p < 0.001) and genotype (F(1,37) = 9.4; p < 0.01), but no treatment effect or interaction (n = 10–11/group). F, Errors made during Barnes maze training. Assessment of spatial learning by training trial with a two-way repeated-measures ANOVA revealed a significant main effect of training trial (F(7,31) = 19.7; p < 0.001) and genotype (F(1,37) = 15.7; p < 0.001), but no treatment effect or interaction (n = 10–11/group). G, Spatial memory was tested 24 h after the final Barnes maze training trial by measuring the percentage of time searching over the former target/all hole locations. Two-way ANOVA revealed a significant main effect of genotype (p < 0.05), but no treatment effect or interaction (n = 10–11/group). All data are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To determine whether chronic Pip18 treatment could rescue cognitive deficits in PS2APP mice, we first tested spatial learning and memory using the Morris water maze. PS2APP mice were significantly impaired in the acquisition of the hidden component of this task compared with NTG mice in that they traveled a greater distance (Fig. 4B) and took more time (Fig. 4C) to locate a hidden platform (sessions 1–9) during repeated training sessions. Pip18 treatment did not alter the performance in this task, demonstrating a failure to rescue the learning deficit in PS2APP mice. There were no significant genotype differences in swim speeds (data not shown) or acquisition of the visible platform component of the task (sessions v1–v3; Fig. 4B,C), suggesting that the cognitive deficits in the PS2APP mice are not due to sensorimotor impairments. Spatial memory was assessed by giving a probe trial before session 7 and 24 h after the final training session (Fig. 4D). During these probe trials, PS2APP mice crossed over the target platform zone significantly fewer times than the NTG mice, suggesting impairment in spatial memory, but this effect was not altered by Pip18 treatment, indicating a failure to improve memory in PS2APP mice (Fig. 4D).
As a second test of whether Pip18 affects spatial learning and memory, treated mice were also tested in the Barnes maze, a spatial memory task that does not require swimming. Analysis of spatial learning in the Barnes maze found that, whereas all animals ultimately performed better in the task with training, PS2APP mice showed a significant impairment in acquisition of the task as evidenced by traveling a significantly longer distance (Fig. 4E) and making more errors (nose pokes into incorrect holes; Fig. 4F) over the course of training compared with NTG mice. Chronic Pip18 treatment failed to alter this deficit in learning (Fig. 4E,F). Spatial memory was assessed 24 h after the final training trial and PS2APP mice were found to be significantly less accurate in searching for the location of the previously hidden escape tunnel compared with NTG mice (Fig. 4G). As with the learning deficit, Pip18 treatment did not alter this memory deficit.
Chronic Pip18 treatment induces anxiety-like behavior and impairs active avoidance learning in NTG mice
Although spatial memory was unaffected by chronic treatment, other assays showed evidence of bioactivity of chronic Pip18 treatment. Body weights were significantly lower in Pip18-treated mice (significant main effect of genotype p < 0.001, and treatment p < 0.001 by ANOVA; NTG-control-fed 35.0 ± 0.78 g; NTG-Pip18, 30.6 ± 0.78 g; PS2APP-control 29.0 ± 0.78 g; PS2APP-Pip18, 26.5 ± 0.74 g; n = 10–11/group). In the open field test, there was a genotype-dependent effect of treatment that reduced rearings (Fig. 5A) and activity in the center of the chamber (Fig. 5B) in treated NTG mice. Total activity, however, was not significantly different between any of the treatment groups (Fig. 5C), suggesting that chronic Pip18 treatment did not alter activity in general but instead selectively affected anxiety-related behaviors in the NTG mice. In the active avoidance test, animals learn to either avoid or escape from a foot shock by shuttling to the opposite side of a two-chamber apparatus. PS2APP mice showed a significant impairment in avoidance leaning (Fig. 5D), but were able to display escape responses (Fig. 5E), suggesting that the avoidance deficits are not due to lack of motivation or physical inability to perform the task. Chronic Pip18 treatment, however, resulted in a significant impairment of performance in the NTG mice on day 3. This represents both another failure to rescue behavioral deficits in PS2APP mice and another example of disruption of normal function by chronic Pip18 treatment. Overall, the reduced body weights in both genotypes, along with the increased anxiety-like behavior and worsening of active avoidance performance in NTG mice support the bioactivity and CNS effects of Pip18 during this treatment regimen.
Figure 5.

Chronic treatment alters anxiety-like behavior and impairs active avoidance performance in NTG mice. A, Percent activity in the center of the open field. ANOVA revealed a significant difference between the control and Pip18-treated NTG mice groups (F(3,37) = 5.7; p < 0.01). *p < 0.05 by Tukey's post hoc test. B, Number of rearings in the open field. ANOVA revealed a significant difference between the control and Pip18-treated NTG mice groups (F(3,37) = 10.8; p < 0.0001). ***p < 0.001 by Tukey's post hoc test. C, Total distance traveled in the open field. There were no significant differences between the groups. D, Percent avoidance responses during active avoidance training. Two-way repeated-measures ANOVA revealed a significant effect of genotype (F(1,31) = 9.5; p < 0.01), but no treatment effect or interaction. However, analysis of performance on day 3 of training (blocks 11–15) found a significant effect of treatment within the NTG mice (F(1,15) = 5.1; *p < 0.05; n = 8–9/group). All data are shown as mean ± SEM. E, Percentage of escape responses during active avoidance training. Two-way repeated-measures ANOVA revealed a significant main effect of genotype (F(1,31) = 8.6; p < 0.01), but no treatment effect or interaction. However, analysis of performance on day 3 of training (blocks 11–15) found a significant main effect of treatment in the NTG mice (F(1,15) = 5.2; *p < 0.05; n = 8–9/group). All data are shown as mean ± SEM.
Chronic Pip18 treatment does not rescue spine loss or alter synaptic strength in PS2APP mice
That chronic Pip18 treatment did not rescue spatial memory deficits suggests that synaptic loss, which correlates with cognitive impairment in AD patients (Terry et al., 1991), may not have been rescued in the PS2APP mice. However, it is also possible that functional alterations independent of synapse phenotypes could contribute to the memory deficits in PS2APP mice even if synapse deficits were rescued by Pip18. At the same time, the impact of Pip18 on anxiety-like and active avoidance behavior suggests that there could be an effect of drug treatment on synaptic function. Therefore, to test for any treatment effects, we analyzed dendritic spine density and size, which are morphological correlates of excitatory synapses onto pyramidal neurons, and synaptic function in PS2APP and NTG mice after chronic treatment (Fig. 6).
Figure 6.

Chronic Pip18 treatment does not rescue spine loss in PS2APP mice. A, Example images of GFP-labeled spines that were imaged at the end of the study in NTG mice or PS2APP mice either away from plaques or near plaques (methoxy-X04-labeled in blue). Scale bar is 20 μm and applies to all panels. B, Pip18 treatment did not alter the density of plaques or the average volume of plaques (n = 7–8 mice per group). C, Although there was a significant effect of genotype on spine density, with reduced density in PS2APP mice, there was no effect of treatment (n = 7–8 mice per group). Spine density was significantly reduced in PS2APP mice both near plaques and away from plaques in both the vehicle and Pip18 treatment groups compared with vehicle-treated NTG mice. *p < 0.05; **p < 0.01; ***p < 0.001. D, There was no effect of genotype or treatment on spine volume. All data are shown as mean ± SEM.
Chronic Pip18 treatment had no effect on the density or size of amyloid plaques in PS2APP mice (Fig. 6B). Although PS2APP mice showed a decrease in spine density that was especially dramatic near amyloid plaques, chronic Pip18 treatment failed to rescue this deficit (Fig. 6C). There were also no significant effects of treatment on spine volume (Fig. 6D). Spine density and volume were also unaffected in NTG mice treated with Pip18. We evaluated whether Pip18 treatment altered synaptic function in either genotype using in vitro electrophysiology on brain slices from mice of the same age and treated with same medicated food protocol as those used for spine analysis. These experiments confirmed the previous demonstration of no change to basal synaptic strength as measured by the synaptic input–output relationship in PS2APP mice (Richards et al., 2003; Fig. 7A,B). At the same time, we observed a significant reduction in paired-pulse facilitation at short interstimulus intervals in PS2APP mice (25 and 50 ms; Fig. 7C). This suggests altered presynaptic function could contribute to maintaining total synaptic strength in the face of spine loss in PS2APP mice. In PS2APP mice chronically treated with Pip18, we did not observe any changes to basal synaptic strength or the reduced paired-pulse responses at short intervals (Fig. 7B,C). Therefore, chronic Pip18 treatment failed to alter spine density or synaptic function in either PS2APP mice or NTG controls.
Figure 7.

Chronic treatment does not alter synaptic function in PS2APP mice. A, Input–output relationships for EPSPs measured in response to logarithmically increasing stimulus intensities (n = 26–32 slices from six to seven animals per group). The corresponding stimulus intensities were as follows: 0.004, 0.006, 0.008, 0.012, 0.017, 0.024, 0.034, 0.040, 0.057, 0.082, 0.117, 0.168, 0.240, 0.344, 0.493, 0.706, and 1.000 mA. B, The data from the maximal stimulus intensities in A replotted for clarity. There we no effects of genotype or treatment on the maximal EPSP response. C, EPSPs were measured in CA1 in response to Schaffer collateral stimulation. The PPR is shown as a function of interstimulus interval (n = 26–32 slices from six to seven animals per group). Assessment by 3-way ANOVA found a significant interaction between interval and genotype (DF = 6, F(6,749) = 4.76, p < 0.001), but no effect of treatment. All data are shown as mean ± SEM. ***p < 0.001.
Starting Chronic Pip18 treatment before accumulation of plaque pathology does not prevent spine loss in PS2APP mice
One potential reason for the failure to show efficacy against spine loss could be that the intervention was performed too late to reverse pathology that had already occurred because treatment began at 7 months of age, after significant plaque burden had already accumulated. To test this, we performed an additional study in which PS2APP mice were fed the same level of Pip18 for 2 months starting at 3 months of age, a time point just before the onset of plaque accumulation (Fig. 8A). However, this treatment regimen also failed to alter plaque density, prevent spine loss, or alter spine size (Fig. 8B–D). Therefore, GluN2B antagonism is insufficient to modify the course of spine loss regardless of whether treatment is begun before or after the onset of plaque appearance and concomitant spine loss.
Figure 8.
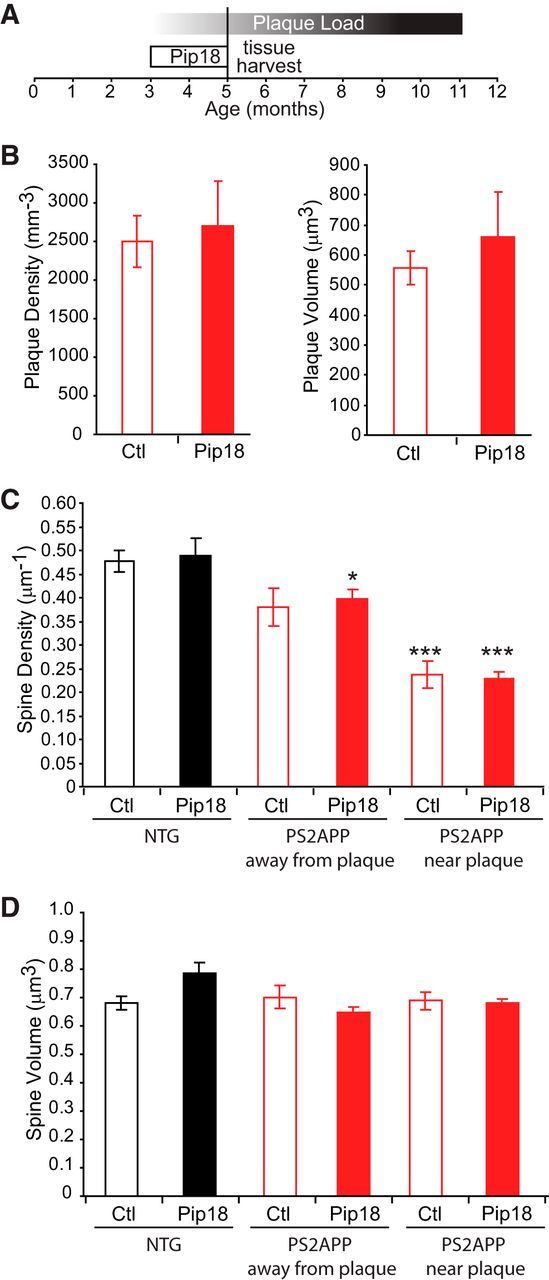
Early treatment with Pip18 fails to prevent spine loss in PS2APP mice. A, Course of Pip18 treatment relative to the accumulation of plaque load (approximated as average plaque size × average plaque volume) in PS2APP mice and the timing of tissue harvest for spine analysis. B, Pip18 treatment did not alter the density of plaques or the average volume of plaques (n = 4–5 mice per group). C, Although there was a significant effect of genotype on spine density with reduced density in PS2APP mice, there was no effect of treatment (n = 4–5 mice per group). Spine density was reduced in PS2APP mice both near plaques and away from plaques. *p < 0.05; **p < 0.01; ***p < 0.001 relative to vehicle-treated NTG mice. D, There was no effect of genotype or treatment on spine volume. All data are shown as mean ± SEM.
Discussion
Based on compelling theoretical rationale derived largely from in vitro experiments, we tested the potential of chronic in vivo GluN2B antagonism to modify the course of disease in mouse models of AD. Amyloid-plaque-associated spine loss and memory deficits were not rescued by chronic GluN2B antagonist treatment. These results therefore do not provide support for potential disease modification (prevention of spine/synapse loss) or symptomatic benefit (amelioration of cognitive deficits) by GluN2B antagonists for Aβ-driven dysfunction. At the same time, we did observe behavioral impacts of GluN2B antagonist treatment with effects that could be related to increasing anxiety and disrupting learning, including increased fear memory generalization, altered open field activity, and impaired active avoidance conditioning.
Neuronal targets of Pip18
Our PK analysis for both subchronic and chronic treatment indicates that brain concentrations well above the level needed to inhibit GluN2B-NMDARs were achieved. Although we did not observe gross alterations to synaptic function in CA1 pyramidal neurons after chronic treatment, the altered behavioral measures could result from impacts on network function mediated via GluN2B-NMDARs on CA1 interneurons, neurons in other parts of the hippocampus, and/or in other brain structures. For example, the potential excessive memory generalization observed during subchronic treatment is predicted to occur after disinhibition of hippocampal circuits (Bennett et al., 1994; Hanson and Madison, 2010), which can be caused by GluN2B-NMDAR antagonist effects on CA1 interneurons (Hanson et al., 2013). Furthermore dentate gyrus GluN2B-NMDARs have been shown to mediate pattern separation because their deletion causes excessive memory generalization (Kheirbek et al., 2012). At the same time, GluN2B-NMDARs in the amygdala are essential for extinction of fear memory (Sotres-Bayon et al., 2007) and effects on these receptors could be responsible for the inappropriate fear behavior that we observed. In addition, prefrontal cortex activity, which is important for both suppressing amygdala function during active avoidance behavior (Moscarello and LeDoux, 2013) and for polysynaptic activation of the hippocampus during pattern separation (Xu and Südhof, 2013), depends on GluN2B receptor activation (Wang et al., 2013). Therefore, broad neurocircuit effects of GluN2B antagonism could contribute to the disrupted behaviors observed during GluN2B antagonist treatment. Although off-target effects could potentially contribute to the observed behavioral effects, reports of altered cognitive behavior in animals models with systemic injection of other GluN2B antagonists (Dalton et al., 2011; Hanson et al., 2013) and cognitive and psychiatric adverse events in patients taking GluN2B antagonists (Nutt et al., 2008; Preskorn et al., 2008) corroborate the interpretation that the disrupted behaviors that we observed are likely due to on-target effects of GluN2B antagonism. Ultimately, future studies using multiple distinct GluN2B antagonists and more diverse behavioral paradigms could help to characterize the role of GluN2B-NMDARs definitively in specific behaviors such as anxiety or context discrimination.
GluN2B antagonists do not appear promising for treating AD
Although accumulation of amyloid plaques and loss of neurons are hallmark features of AD, synaptic loss is the postmortem phenotype that best correlates with cognitive impairment (Terry et al., 1991; DeKosky et al., 1996; Pozueta et al., 2013). Although neuron loss does not occur in most mouse models of AD, the critical phenotype of synaptic loss is reproduced in many AD mouse models and is often assessed by measuring the loss of dendritic spines (Pozueta et al., 2013). Spine loss in mouse models, including the PS2APP mice used in this study, is often most prominent along dendrites in close proximity to plaques, but also occurs in regions devoid of plaques, suggesting that spine loss is driven by soluble Aβ. Our results show that chronic GluN2B antagonism is not effective at rescuing this spine loss when administered after its onset or at preventing spine loss when administered before appearance of plaques. Because spine loss is generally thought to occur as an early event in the cascade of AD disease progression, the ineffectiveness of GluN2B antagonists for blocking this phenotype casts doubt on the potential for disease modification in AD. Although mechanisms independent of spine loss could contribute to the memory impairments in PS2APP mice, the unaltered spine loss is consistent with the ineffectiveness of GluN2B antagonism in improving the spatial memory deficits in these mice. In any case, the failure to improve memory deficits in this model does not support a symptomatic benefit of GluN2B antagonism in AD.
The lack of benefit of GluN2B antagonisms in our study contradicts the prevalent idea that GluN2B-NMDARs (likely at extrasynaptic sites) mediate the deleterious effects of Aβ on synapse number and synaptic function. An idea that is more consistent with our data is a subsynaptic localization hypothesis that posits that extrasynaptic location is critical to the deleterious effects of NMDAR activation regardless of the GluN2A versus GluN2B subunit composition (Zhou and Sheng, 2013). Because GluN2B-NMDARs only make up a portion of the extrasynaptic pool (Harris and Pettit, 2007), GluN2B antagonism alone could therefore be insufficient to eliminate pathological activation of extrasynaptic NMDARs. These could include GluN1/2A diheteromeric receptors and/or GluN1/2A/2B triheteromeric receptors, neither of which are significantly blocked by Pip18. Another explanation for our results is that activation of synaptic NMDARs is actually necessary for the harmful effects of NMDAR activation. For example, a recent study using brain slices has suggested that protection from NMDAR-mediated neurotoxicity (induced by exogenous NMDA application) requires blockade of synaptic NMDARs, whereas blockade of extrasynaptic/GluN2B-NMDARs alone is insufficient for protection (Papouin et al., 2012). It should also be noted that most studies demonstrating a role of GluN2B-NMDARs in synapse loss and dysfunction caused by Aβ typically use acute application of relatively high concentrations of synthetic Aβ (usually in vitro; Hu et al., 2009; Li et al., 2011; Rammes et al., 2011; Rönicke et al., 2011; Costa et al., 2012), whereas in our in vivo studies of AD model mice, the increase in Aβ level is gradual and results in progressive effects on the system. One potential explanation for these disparate results is that distinct mechanisms are engaged by these different paradigms of Aβ insult, with the in vivo paradigm more likely being relevant to AD pathology.
Our study, which for the first time examined the potential for disease modification by chronic GluN2B antagonism in an AD mouse model, addresses two general concerns regarding the therapeutic potential of GluN2B antagonists. First, GluN2B antagonism could have undesirable effects on cognition and mood. Similar conclusions were drawn from our previous work examining in vivo GluN2B antagonism in NTG and Ts65Dn Down syndrome model mice (Hanson et al., 2013). Other studies using genetic and pharmacological disruption of GluN2B-NMDARs also point to the importance of these receptors in normal physiological function and cognitive behavior (von Engelhardt et al., 2008; Mathur et al., 2009; Dalton et al., 2011). Clinical trials using GluN2B antagonists for Parkinson's disease and depression reporting cognitive and psychiatric adverse events further support this conclusion (Nutt et al., 2008; Preskorn et al., 2008). Second, we do not find support for modification of the synapse loss and memory disruptions seen in a mouse model of AD. Although it is possible that synapse loss and memory impairment in sporadic AD in humans are not mediated by the same mechanisms as the plaque-associated synapse loss and memory dysfunction in transgenic mouse models of AD, our results argue against the potential utility of GluN2B antagonists for neuroprotection in Aβ-driven disease.
Footnotes
We thank Jukka Puoliväli for Tg2576 behavioral studies, Xingrong Liu and Gauri Deshmukh for PK analysis, Jacob Schwartz for drug synthesis, Amy Sambrone for formulations, Hilda Solanoy and Heather Kennedy for necropsy support and tissue processing, Martin Weber for developing the active avoidance protocol, and Eric Bourhis and Kimberly Stark for project management.
The authors declare no competing financial interests.
References
- Addy C, Assaid C, Hreniuk D, Stroh M, Xu Y, Herring WJ, Ellenbogen A, Jinnah HA, Kirby L, Leibowitz MT, Stewart RM, Tarsy D, Tetrud J, Stoch SA, Gottesdiener K, Wagner J. Single-dose administration of MK-0657, an NR2B-selective NMDA antagonist, does not result in clinically meaningful improvement in motor function in patients with moderate Parkinson's disease. J Clin Pharmacol. 2009;49:856–864. doi: 10.1177/0091270009336735. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Gibson WG, Robinson J. Dynamics of the CA3 pyramidal neuron autoassociative memory network in the hippocampus. Philos Trans R Soc Lond B Biol Sci. 1994;343:167–187. doi: 10.1098/rstb.1994.0019. [DOI] [PubMed] [Google Scholar]
- Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Jacobsen JS, Marquis KL. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, Oliveira CR, Rego AC, Pereira CM. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell. 2012;11:823–833. doi: 10.1111/j.1474-9726.2012.00848.x. [DOI] [PubMed] [Google Scholar]
- Dalton GL, Ma LM, Phillips AG, Floresco SB. Blockade of NMDA GluN2B receptors selectively impairs behavioral flexibility but not initial discrimination learning. Psychopharmacology (Berl) 2011;216:525–535. doi: 10.1007/s00213-011-2246-z. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25-6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit–characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Foldi CJ, Eyles DW, McGrath JJ, Burne TH. The effects of breeding protocol in C57BL/6J mice on adult offspring behaviour. PLoS One. 2011;6:e18152. doi: 10.1371/journal.pone.0018152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA. Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron. 2011;71:1085–1101. doi: 10.1016/j.neuron.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond MS, Sims C, Parameshwaran K, Suppiramaniam V, Schachner M, Dityatev A. Neural cell adhesion molecule-associated polysialic acid inhibits NR2B-containing N-methyl-D-aspartate receptors and prevents glutamate-induced cell death. J Biol Chem. 2006;281:34859–34869. doi: 10.1074/jbc.M602568200. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Madison DV. Imbalanced pattern completion vs. separation in cognitive disease: network simulations of synaptic pathologies predict a personalized therapeutics strategy. BMC Neurosci. 2010;11:96. doi: 10.1186/1471-2202-11-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Weber M, Meilandt WJ, Wu T, Luu T, Deng L, Shamloo M, Sheng M, Scearce-Levie K, Zhou Q. GluN2B antagonism affects interneurons and leads to immediate and persistent changes in synaptic plasticity, oscillations, and behavior. Neuropsychopharmacology. 2013;38:1221–1233. doi: 10.1038/npp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–519. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Heng MY, Detloff PJ, Wang PL, Tsien JZ, Albin RL. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci. 2009;29:3200–3205. doi: 10.1523/JNEUROSCI.5599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu NW, Klyubin I, Anwy R, Rowan MJ. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci U S A. 2009;106:20504–20509. doi: 10.1073/pnas.0908083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim L, Diaz Granados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, Potter WZ, Zarate CA., Jr A Randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32:551–557. doi: 10.1097/JCP.0b013e31825d70d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirbek MA, Tannenholz L, Hen R. NR2B-dependent plasticity of adult-born granule cells is necessary for context discrimination. J Neurosci. 2012;32:8696–8702. doi: 10.1523/JNEUROSCI.1692-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Lai TW, Shyu WC, Wang YT. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med. 2011;17:266–275. doi: 10.1016/j.molmed.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur P, Graybeal C, Feyder M, Davis MI, Holmes A. Fear memory impairing effects of systemic treatment with the NMDA NR2B subunit antagonist, Ro 25–6981, in mice: attenuation with ageing. Pharmacol Biochem Behav. 2009;91:453–460. doi: 10.1016/j.pbb.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Moscarello JM, LeDoux JE. Active avoidance learning requires prefrontal suppression of amygdala-mediated defensive reactions. J Neurosci. 2013;33:3815–3823. doi: 10.1523/JNEUROSCI.2596-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt JG, Gunzler SA, Kirchhoff T, Hogarth P, Weaver JL, Krams M, Jamerson B, Menniti FS, Landen JW. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov Disord. 2008;23:1860–1866. doi: 10.1002/mds.22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozmen L, Albientz A, Czech C, Jacobsen H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener Dis. 2009;6:29–36. doi: 10.1159/000170884. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer's disease and its models. Neuroscience. 2013;251:51–65. doi: 10.1016/j.neuroscience.2012.05.050. [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Rammes G, Hasenjäger A, Sroka-Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology. 2011;60:982–990. doi: 10.1016/j.neuropharm.2011.01.051. [DOI] [PubMed] [Google Scholar]
- Rauner C, Köhr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J Biol Chem. 2011;286:7558–7566. doi: 10.1074/jbc.M110.182600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Reyes A, Opitz T, Kapin MA, Stanton PK. Eliprodil, a non-competitive, NR2B-selective NMDA antagonist, protects pyramidal neurons in hippocampal slices from hypoxic/ischemic damage. Brain Res. 1998;782:212–218. doi: 10.1016/S0006-8993(97)01280-8. [DOI] [PubMed] [Google Scholar]
- Richards JG, Higgins GA, Ouagazzal AM, Ozmen L, Kew JN, Bohrmann B, Malherbe P, Brockhaus M, Loetscher H, Czech C, Huber G, Bluethmann H, Jacobsen H, Kemp JA. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci. 2003;23:8989–9003. doi: 10.1523/JNEUROSCI.23-26-08989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rönicke R, Mikhaylova M, Rönicke S, Meinhardt J, Schröder UH, Fändrich M, Reiser G, Kreutz MR, Reymann KG. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging. 2011;32:2219–2228. doi: 10.1016/j.neurobiolaging.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Soares C, Lee KF. A prominent role for triheteromeric GluN1/GluN2A/GluN2B NMDARs at central synapses. J Neurosci. 2013;33:14975–14977. doi: 10.1523/JNEUROSCI.3109-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, Westbrook GL. Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci. 2013;33:9150–9160. doi: 10.1523/JNEUROSCI.0829-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JN, Seeburg PH, Bannerman DM, Monyer H. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, Morrison JH, Wang XJ, Arnsten AF. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013;77:736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Südhof TC. A neural circuit for memory specificity and generalization. Science. 2013;339:1290–1295. doi: 10.1126/science.1229534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkewicz L, Weaver J, Bullock MR, Marshall LF. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J Neurotrauma. 2005;22:1428–1443. doi: 10.1089/neu.2005.22.1428. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/S0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Sheng M. NMDA receptors in nervous system diseases. Neuropharmacology. 2013;74:69–75. doi: 10.1016/j.neuropharm.2013.03.030. [DOI] [PubMed] [Google Scholar]