

Abstract
Activity-based therapies are routinely integrated in spinal cord injury (SCI) rehabilitation programs because they result in a reduction of hyperreflexia and spasticity. However, the mechanisms by which exercise regulates activity in spinal pathways to reduce spasticity and improve functional recovery are poorly understood. Persisting alterations in the action of GABA on postsynaptic targets is a signature of CNS injuries, including SCI. The action of GABA depends on the intracellular chloride concentration, which is determined largely by the expression of two cation-chloride cotransporters (CCCs), KCC2 and NKCC1, which serve as chloride exporters and importers, respectively. We hypothesized that the reduction in hyperreflexia with exercise after SCI relies on a return to chloride homeostasis. Sprague Dawley rats received a spinal cord transection at T12 and were assigned to SCI-7d, SCI-14d, SCI-14d+exercise, SCI-28d, SCI-28d+exercise, or SCI-56d groups. During a terminal experiment, H-reflexes were recorded from interosseus muscles after stimulation of the tibial nerve and the low-frequency-dependent depression (FDD) was assessed. We provide evidence that exercise returns spinal excitability and levels of KCC2 and NKCC1 toward normal levels in the lumbar spinal cord. Acutely altering chloride extrusion using the KCC2 blocker DIOA masked the effect of exercise on FDD, whereas blocking NKCC1 with bumetanide returned FDD toward intact levels after SCI. Our results indicate that exercise contributes to reflex recovery and restoration of endogenous inhibition through a return to chloride homeostasis after SCI. This lends support for CCCs as part of a pathway that could be manipulated to improve functional recovery when combined with rehabilitation programs.
Keywords: complete transection, exercise, H-reflex, KCC2, NKCC1, spinal cord injury
Introduction
There has been significant progress in the acute care of individuals with spinal cord injury (SCI) that increases quality of life and life expectancy. Activity-based therapies have undeniably proven to play a role in the recovery of locomotion, respiration, and cardiovascular and bladder function after SCI. However, the extent of recovery remains limited with substantial and permanent disabilities.
Persisting alteration in the action of GABA on postsynaptic targets is a signature of CNS injuries and disorders, including the development of spasticity after SCI. Spasticity is a debilitating condition that affects ∼75% of individuals with SCI. Several of the incapacitating symptoms of spasticity result from hyperreflexia, which ultimately interferes with residual motor function and hampers motor recovery. Exercise is routinely integrated into rehabilitation programs to induce repetitive activation of the neuromuscular system, to prevent further functional impairment, and to facilitate motor recovery (Edgerton et al., 2008; Harkema et al., 2012; Hubli and Dietz, 2013). However, the mechanisms by which exercise regulates activity in spinal pathways to reduce spasticity and improve motor function are poorly understood. Recent reports give insight into one of the mechanisms involved in the development of spasticity after SCI: downregulation of the chloride cotransporter KCC2 in the spinal cord resulting in a positive shift in the equilibrium potential of chloride (Boulenguez et al., 2010).
The inhibitory action of GABA in the normal adult spinal cord depends on the intracellular chloride concentration ([Cl−]i), which is largely determined by the expression of KCC2 and NKCC1. Both KCC2 and NKCC1 belong to the cation chloride cotransporter (CCC) family as part of the K+-Cl− and Na+-K+-Cl− cotransporter families, respectively (Delpire and Mount, 2002; Payne et al., 2003). Changing the density or activity of these transporters affects [Cl−]i directly and ultimately determines whether GABA has an inhibitory or excitatory effect. This has been shown during CNS development, when regulation of the expression of the cation chloride importer NKCC1 and exporter KCC2 determines the depolarizing action of GABA (Plotkin et al., 1997). Evidence suggests that changes in chloride homeostasis via a modulated expression of chloride cotransporters are critical in setting the polarity in GABAergic signaling in a number of systems (Rivera et al., 1999; Payne et al., 2003; Blaesse et al., 2006) and disorders, including seizure, epilepsy, Alzheimer's disease, autism spectrum disorders, neuropathic pain, and, importantly, the development of spasticity after SCI (Kahle et al., 2008; Ben-Ari et al., 2012).
We hypothesized that the beneficial effect of exercise on the modulation of spinal reflexes depends on the restoration of endogenous inhibition to a mature state. We show that acute pharmacological blockade of KCC2 or NKCC1 indicate a role for chloride homeostasis in reflex recovery after SCI. We further illustrate that the exercise-dependent recovery of reflexes after SCI occurs through an increased level of the chloride cotransporter KCC2 combined with a decreased level of NKCC1 in the lumbar spinal cord. These results demonstrate that a return to chloride homeostasis is integral to reflex recovery after SCI.
Materials and Methods
Protocols.
All procedures were performed in accordance with protocols approved by Drexel University College of Medicine Institutional Animal Care and Use Committee and followed National Institutes of Health guidelines for the care and use of laboratory animals.
Surgical procedures and postoperative care.
Adult female Sprague Dawley rats (n = 87, RRID: RGD_737891) underwent a complete spinal transection at the low thoracic level (T12) as described previously (Côté et al., 2011). Briefly, rats were anesthetized with isoflurane (1–2%) in O2 and a laminectomy was performed at the T10–T11 vertebral level under aseptic conditions. The dura was carefully slit open, a 2 mm cavity created by aspiration, and saline-soaked gel foam was inserted in the cavity to achieve hemostasis. The completeness of the spinal cord lesion was confirmed by the retraction of the rostral and caudal portions of the cord and by examining the ventral floor of the spinal canal. Back muscles were sutured and the skin incision closed with wound clips. Upon completion of the surgery, animals received buprenorphine (0.05 mg/kg, i.m.) for 3 d as an analgesic and saline (5 ml, s.c.) and ampicillin (100 mg/kg, s.c.) for 7 d to prevent dehydration and infection. Bladders were expressed manually twice a day until voiding reflex returned.
Rats were randomly assigned to one of three groups: intact (n = 12) or SCI with or without exercise. Untrained SCI groups were further subdivided in subgroups with the terminal experiment taking place at different time points: 1 week (n = 20), 2 weeks (n = 10), 4 weeks (n = 18), or 8 weeks (n = 6). Trained animals were killed after ∼2 weeks (n = 6) or 4 weeks (n = 18).
Exercise regimen.
Beginning on days 4–5, exercised groups received 60 min of daily exercise 5 d/week until they reached 14 or 28 d after SCI. Animals undergoing bicycling exercise were secured in a support harness with the hindlimbs hanging with the feet secured to pedals with surgical tape. The hindlimbs went through a complete range of motion during pedal rotation (45 rpm) generated by a custom-built motor-driven apparatus (Houlé et al., 1999; Côté et al., 2011).
H-reflex recordings and analysis.
Rats were anesthetized with ketamine (60 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). The tibial nerve was dissected free and mounted on a bipolar hook electrode (FHC). A pool of mineral oil was created with skin flaps to prevent dessication of the nerve. Bipolar wire electrodes (Cooner Wire) were inserted in the interosseus muscles of the hindpaw for EMG recordings and the ground electrode inserted in the skin of the leg. H-reflexes were evoked via an isolated pulse stimulator delivering single bipolar pulses (100 μs) to the tibial nerve. H-waves and M-waves in the interosseus muscle of the hindpaw were recorded in response to a range of increasing stimulus intensities to determine the threshold and the maximal response amplitude for both M-waves and H-waves (Mmax and Hmax, respectively). The stimulation intensity that elicited Hmax response was then used for 3 series of 20 consecutive stimulation pulses delivered at different frequencies (0.3, 5, or 10 Hz). Before termination, the 0.3 Hz series was repeated to verify that the M-wave was still within 95% of the initial trial, otherwise the trial was discarded.
EMG recordings were amplified (1000×; A-M Systems) and band-pass filtered (10–5 kHz). Signal was digitized (10 kHz) and fed to custom software. Response latency (onset of response) and peak-to-peak amplitude were measured for the H and M responses evoked by single pulses. The recruitment curve was plotted by expressing the amplitude of the response as a function of stimulus intensity. Mmax, the response of all motor units with supramaximal stimulation of axons of the tibial nerve, and Hmax, the maximal H-reflex amplitude, were determined. The Hmax/Mmax ratio was calculated to assess the relative proportion of motoneurons recruited through the monosynaptic reflex loop compared with the activation of the entire motor pool. This measure is used to give an estimate of motoneuronal excitability. The Hmax/M ratio was also calculated to estimate the relative activation of the motor pool required to reach maximal reflex amplitude. For the analysis of low-frequency-dependent depression (FDD) of the H-reflex, the first five responses to a train of stimulation were discarded to allow reflex stabilization. The 12 remaining responses were then averaged. The averaged H-reflex amplitude was normalized to Mmax for each animal and frequency and the change in H-reflex response at 5 and 10 Hz was calculated as a percentage of the response measured at 0.3 Hz.
Drug preparation.
In a subgroup of untrained SCI animals (n = 4, SCI-28d; n = 2, SCI-56d), reflex recordings were performed before and after the application of the NKCC1 antagonist bumetanide (Sigma-Aldrich). The NKCC1 antagonist bumetanide was prepared in saline containing 0.25% NaOH and injected intraperitonally (30 mg/kg; Cramer et al., 2008). In another subgroup of SCI-28d+exercise animals (n = 9), reflex recordings were performed before and after the application of the KCC2 antagonist [(dihydroindenyl)oxy]alkanoic acid (DIOA; Sigma-Aldrich) onto the exposed lumbar enlargement (30 mm). DIOA was prepared in saline containing 0.1% DMSO. After drug delivery, H-reflex was measured every 15 min for up to 100 min after injection. The specific time points reported in Figures 2 and 7 reflect the maximal effect of the drug on FDD (i.e., 60 min for bumetanide and 90 min for DIOA) and are consistent with previous reports (Cramer et al., 2008; Boulenguez et al., 2010).
Figure 2.

Blocking NKCC1 mimics recovery after SCI. A, Representative averages of H-reflex recordings after a train of stimulation at 0.3 Hz (black) and 10 Hz (gray) in the same animal before (top) and 1 h after (bottom) bumetanide. The amplitude of the H-reflex is smaller at 10 Hz compared with 0.3 Hz with or without drugs, but the depth of modulation is much larger after bumetanide. B, Bumetanide improves FDD in chronic SCI animals. *p < 0.05; **p = 0.001.
Figure 7.

Blocking KCC2 suppresses exercise-dependent reflex recovery after SCI. A, Representative averages of H-reflex recordings after a train of stimulation at 0.3 Hz (black) and 10 Hz (gray) in the same SCI-28d+exercise animal before (top) and after (bottom) DIOA. Before drugs, the H-reflex is almost totally depressed at 10 Hz compared with 0.3 Hz, whereas DIOA dramatically decreases the depth of modulation. B, Overall, DIOA “impaired” FDD in SCI-7d animals and masked FDD recovery in SCI-28d+exercise animals.
Western blots.
Rats (n = 6/group) were killed with an overdose of Euthasol (390 mg/kg sodium pentobarbital and 50 mg/kg phenytoin, i.p.) 7 or 28 d after SCI. Fresh spinal cord was rapidly removed, frozen in dry ice, and stored at −80°C. Blocks of spinal tissue (L4–L6) were immersed in ice-cold homogenization buffer (modified RIPA; 50 mm Tris buffer, pH 6.8, 1% Triton X-100, 0.1% SDS, 1 mm DTT, 0.5% deoxycholate, 150 mm NaCl) containing a mixture of protease and phosphatase inhibitors (Roche), 2 mm phenyl-methyl sulfonyl-fluoride, and 1 mm sodium fluoride. Homogenates were sonicated and centrifuged at 14,000 × g for 40 min at 4°C. Collected supernatants were aliquoted and protein concentration determined using a BCA protein assay kit (Pierce). Samples were then heated in Laemmli sample buffer for 30 min at 37°C for KCC2 and NKCC1 or boiled for 5 min for BDNF and TrkB, which are known to be upregulated by exercise in the lumbar spinal cord. An equal amount of total protein was loaded in every well and separated on 10–16% SDS-PAGE gels (BDNF and TrkB) or 4–20% Mini Protean TGX gels (Bio-Rad) (KCC2 and NKCC1). Proteins were transferred onto PVDF membranes in transfer buffer and nonspecific sites blocked with 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20. Membranes were probed with primary rabbit polyclonal antibodies against KCC2 (1:1000, RRID: AB_310611, catalog #07-432; Millipore), BDNF (1:1000, RRID: AB_630940, catalog #sc-546; Santa Cruz Biotechnology), TrkB (1:1000, RRID: AB_2155274, catalog #sc-8316; Santa Cruz Biotechnology), or mouse T4 monoclonal antibody against NKCC1 (1:1000, RRID: AB_528406, catalog #t4; Developmental Studies Hybridoma Bank), followed by incubation with the horseradish peroxidase-conjugated donkey anti-rabbit/mouse secondary antibody (RRID: AB_10015282, catalog #711-035-152; Jackson ImmunoResearch). Each sample was run two or three times to ensure replication of results. To confirm equal loading of protein, blots were stripped and reprobed with mouse monoclonal anti-actin antibody (1:10,000, RRID: AB_476744, catalog #A544; Sigma-Aldrich). Immunoreactivity was detected using an enhanced chemiluminescence kit (HyGlo; Denville Scientific). The optical densities of protein bands revealed in the Western blots corresponding to KCC2 (∼140 kDa), NKCC1 (∼145 kDa), BDNF (∼18–27 kDa) and TrkB (∼95–150 kDa) were further quantified using ImageJ Analysis software (RRID: nif-0000–30467). Values for each sample were averaged, normalized to actin, and combined for each group. Final data (mean ± SEM) are presented as a ratio to the intact group, which was assigned a value of 1.0.
Immunofluorescent staining.
Rats (n = 6/group) were killed using an overdose of Euthasol (390 mg/kg sodium pentobarbital and 50 mg/kg phenytoin, i.p.) 28 d after SCI and perfused transcardially with a 4% paraformaldehyde solution. The lumbar spinal cord (L1–L6) was harvested and stored in fixative overnight. After incubation with 30% sucrose/PBS for cryoprotection, transverse sections (30 μm) were prepared on a cryostat for free-floating immunostaining. They were first permeated and preincubated with PBS and BSA (1%), goat or donkey normal serum (5%), and 0.1% Triton X-100 and lysine. Sections were then incubated with these primary antibodies diluted in BSA (1%), normal serum (5%), and 0.1% Triton X-100: rabbit anti-KCC2 (1:800, RRID: AB_310611, catalog #07-432; Millipore), goat anti-ChAT (1:100, RRID: AB_2079751, catalog #AB144P; Millipore) and mouse T4 monoclonal antibody against NKCC1 (1:600, RRID: AB_528406, catalog #t4; Developmental Studies Hybridoma Bank). Species-specific secondary antibodies (1:400, donkey or goat anti-mouse/rabbit conjugated to FITC, rhodamine, or Texas Red; Jackson ImmunoResearch) were applied for 2 h at room temperature and the slides were coverslipped with Vectashield (Vector Laboratories).
Images were acquired using a Leica DM550B fluorescent microscope equipped with a Retiga-SRV digital color camera (QImaging) controlled by Slidebook imaging software version 5 (Olympus). The fluorescence intensity measurements for the gray matter in the ventral horn of the L4–L5 segments (Figs. 5B, 6G) were performed using ImageJ software. The mean pixel intensities were normalized to those for intact animals. The fluorescence intensity for the plasma membrane of motoneurons (identified by ChAT+ labeling, typical large size, and location within the ventral horn) was measured by averaging the integrated area of the density curve obtained by drawing three lines across each motoneurons (six data points) using ImageJ software (Fig. 5C). A minimum of three lumbar motoneurons were averaged per animal.
Figure 5.

Exercise restores KCC2 expression level in the lumbar spinal cord after SCI. A, Digital images showing KCC2 (green) and ChAT (red) immunoreactivity in the lumbar spinal cord of intact, SCI-28d, and SCI-28d+exercise rats. B, Labeling intensity was measured in the ventral horn area delineated by the dotted circle and normalized to intact. KCC2 expression is significantly decreased in the ventral gray matter of the lumbar spinal cord after SCI, but returns toward intact values with exercise. C, Labeling intensity was measured by averaging the integrated density of three lines drawn across the motoneuron cell body to assess KCC2 immunoreactivity along the motoneuronal membrane. Overall, KCC2 was decreased in the motoneuronal membrane after SCI and recovered with exercise. Scale bars: left: 100 μm; middle and right: 25 μm.
Figure 6.
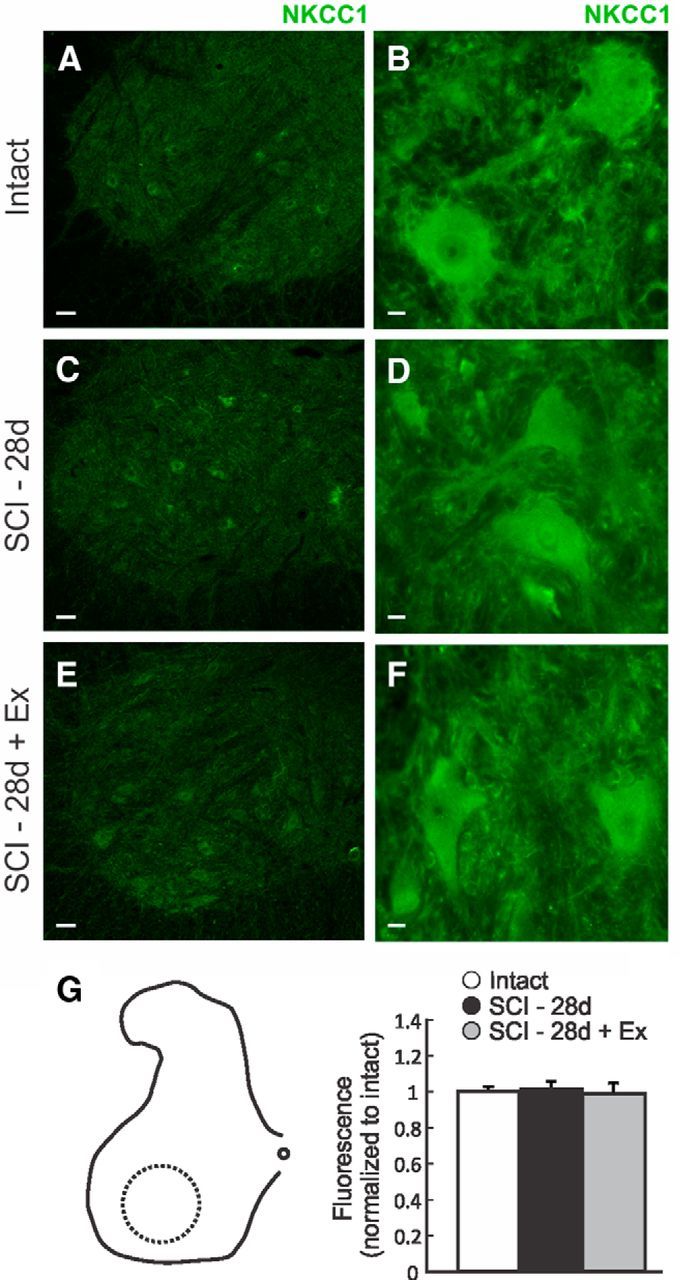
Exercise does not modify NKCC1 protein expression level in the lumbar spinal cord after SCI. A–F, Immunolocalization of NKCC1 protein in the ventral lumbar spinal cord in the lumbar spinal cord of intact, SCI-28d, and SCI-28d+exercise rats. G, Labeling intensity was measured in the ventral horn area delineated by the dotted circle and normalized to intact. NKCC1 expression level was not changed after SCI nor with exercise. Scale bars: A, C, E, 50 μm; B, D, F, 10 μm.
Statistical analysis.
Significant differences across groups were determined using a one-way ANOVA followed by Holm-Sidak post hoc test unless stated otherwise. If the sample variables did not fit a normal distribution or were not equally variant, a one-way ANOVA on ranks followed by Dunn's post hoc test was performed. A two-way ANOVA (or two-way repeated-measures ANOVA for the effect of bumetanide or DIOA) followed by Holm-Sidak post hoc test was used to assess whether stimulation frequency and treatment group had a significant effect on the amplitude of the H-reflex and to evaluate whether the interaction of these factors affected the variable. A paired t test was used to address the changes in H-reflex and M-waves features before and after bumetanide injection or DIOA application (Table 1). All data are reported as mean ± SEM. Linear regression analysis was used to correlate KCC2 to NKCC1, BDNF to KCC2, and NKCC1 protein levels obtained by Western blotting. This analysis was also performed between KCC2 and NKCC1 protein levels and the depth of H-reflex modulation. Statistical analysis was performed using SigmaPlot software version 11.0 (SYSTAT Software). For all statistical tests, the significance level was set to p < 0.05.
Table 1.
Properties of the M-wave and H-reflex: latency, amplitude, and threshold
| MT (mA) | Mmax (mV) | Hmax (mV) | M latency (msec) | H latency (msec) | H-reflex threshold (× MT) | Hmax (× MT) | |
|---|---|---|---|---|---|---|---|
| Intact (n = 12) | 0.013 ± 0.002 | 6.68 ± 1.05 | 2.26 ± 0.53 | 2.46 ± 0.11 | 9.35 ± 0.51 | 1.13 ± 0.06 | 1.84 ± 0.20 |
| SCI 1 wk (n = 20) | 0.027 ± 0.004 | 7.62 ± 1.11 | 3.52 ± 0.67 | 2.60 ± 0.09 | 9.59 ± 0.30 | 1.04 ± 0.03 | 1.42 ± 0.07 |
| SCI 2 wk (n = 10) | 0.025 ± 0.005 | 5.83 ± 0.97 | 2.97 ± 0.53 | 2.83 ± 0.10 | 9.67 ± 0.27 | 1.08 ± 0.07 | 1.33 ± 0.06 |
| SCI 2 wk + Ex (n = 6) | 0.017 ± 0.005 | 6.89 ± 1.15 | 3.44 ± 0.93 | 2.83 ± 0.12 | 9.49 ± 0.32 | 1.10 ± 0.04 | 1.56 ± 0.21 |
| SCI 4 wk (n = 18) | 0.026 ± 0.010 | 6.93 ± 0.92 | 4.41 ± 0.75 | 2.45 ± 0.07 | 8.70 ± 0.18 | 1.05 ± 0.03 | 1.31 ± 0.10 |
| SCI 4 wk + Ex (n = 18) | 0.023 ± 0.003 | 7.94 ± 0.95 | 3.46 ± 0.67 | 2.40 ± 0.06 | 8.83 ± 0.22 | 1.03 ± 0.03 | 1.38 ± 0.07 |
| SCI 8 wk (n = 6) | 0.024 ± 0.013 | 7.00 ± 1.20 | 3.64 ± 0.62 | 2.54 ± 0.11 | 8.49 ± 0.33 | 0.85 ± 0.10 | 1.26 ± 0.12 |
| SCI Pre-Bu (n = 6) | 0.030 ± 0.013 | 6.83 ± 1.27 | 3.52 ± 0.55 | 2.47 ± 0.10 | 8.45 ± 0.32 | 0.86 ± 0.08 | 1.26 ± 0.09 |
| SCI Post-Bu (n = 6) | 0.083 ± 0.030 | 5.14 ± 0.92 | 2.81 ± 0.22 | 2.51 ± 0.09 | 8.88 ± 0.26 | 0.91 ± 0.06 | 1.12 ± 0.12 |
| SCI + Ex Pre-DIOA (n = 9) | 0.016 ± 0.001 | 8.97 ± 1.25 | 4.17 ± 1.14 | 2.96 ± 0.13 | 10.49 ± 0.38 | 0.96 ± 0.06 | 1.32 ± 0.15 |
| SCI + Ex Post-DIOA (n = 9) | 0.020 ± 0.004 | 7.24 ± 1.58 | 4.42 ± 1.06 | 2.74 ± 0.17 | 10.87 ± 0.43 | 0.97 ± 0.04 | 1.31 ± 0.13 |
Values are mean ± SEM. MT, motor threshold; Bu, bumetanide; Ex, exercise.
Results
FDD of the H-reflex after SCI
The H-reflex is commonly used to assess motoneuronal excitability and synaptic transmission from Ia afferents to motoneurons in individuals with spasticity. The advantage of using this reflex resides in the fact that it is monosynaptic. Plasticity arising from injury and/or treatment can therefore be attributed to only two specific locations, primary afferent from group Ia fibers and α-motoneuron, or to synaptic transmission between them. A stimulus to the tibial nerve evokes two successive responses in the muscle: the M-wave and the H-reflex (Fig. 1A, insert). The M-wave is evoked by the direct activation of motor axons without a synaptic component and the H-reflex results from the activation of Ia afferents that monosynaptically synapse onto motoneurons. The general features of the M-wave and H-reflex such as latency, amplitude, and threshold were not significantly altered compared with intact or with time post-SCI up to 56 d (Table 1). SCI animals transition to a state of hyperreflexia between 7 and 28 d post-SCI (Bennett et al., 1999; Yates et al., 2008) and the reduction in the FDD of the H-reflex is widely accepted as an estimate of spasticity after SCI (Thompson et al., 1992). Figure 1A shows representative recordings of the H-reflex at 0.3, 5, and 10 Hz to illustrate that increasing the frequency of stimulation leads to a dramatic decrease in the H-reflex amplitude in intact and acute SCI animals compared with subchronic and chronic SCI time points.
Figure 1.

Spinal excitability gradually increases after SCI. A, FDD gradually decreases over time, reaching a plateau ∼28 d after SCI. Shown are significant differences at 5 Hz (intact and SCI-7d vs SCI-14d, SCI-28d, and SCI-56d; SCI-14d vs SCI-28d and SCI-56d) and at 10 Hz (intact and SCI-7d vs SCI-28d and SCI-56d; SCI-14d vs SCI-28d and SCI-56d). The enlarged recordings depict representative averages of H-reflex recordings after a train of stimulation at 0.3 Hz (light gray), 5 Hz (dark gray) and 10 Hz (black) when the FDD is impaired (top) or not (bottom). B, Hmax/Mmax ratio gradually increases with time after SCI and also plateaus at ∼28 d.
A two-way ANOVA revealed statistically significant differences across stimulation frequency (p < 0.001) and across experimental groups (p < 0.001) and the interaction between frequency and groups was also significant (p < 0.001). Post hoc comparisons showed that 5 and 10 Hz values were different from 0.3 Hz in all groups. Overall, intact and SCI-7d animals displayed a better modulation of the H-reflex compared with all other groups. Seven days after SCI, the FDD was not significantly different from intact and gradually decreased up to 28 d, when it plateaued (Fig. 1A). No further difference was observed beyond this point. The Hmax/Mmax ratio, which estimates the fraction of motoneurons recruited via the H-reflex relative to the activation of the entire motor pool, also increased over time after SCI (Fig. 1B) with values of ∼0.27 in intact animals up to ∼0.66 in SCI-28d (p < 0.05) and SCI-56d (p < 0.05).
Blocking NKCC1 mimics the effect of exercise on reflex recovery after SCI
If a positive shift in chloride equilibrium—that is, a reduction in the expression or activity of KCC2 and/or increase in the chloride importer NKCC1—led to an attenuation of the FDD after SCI, then a pharmacological blockade of NKCC1 would return FDD values toward the intact condition. We therefore investigated the effect of acute bumetanide application on a subgroup of chronic SCI rats that showed impaired FDD. Data from SCI-28d and 56d rats were merged because they showed no difference in Hmax/Mmax and FDD before or after drugs (data not shown). Bumetanide did not significantly modulate the latency, amplitude, or threshold of the M-wave and H-reflex at 0.3 Hz (Table 1). However, an effect was clearly present with increasing stimulus frequencies (Fig. 2A,B). Figure 2A illustrates the H-reflex recorded from the same animal at 0.3 and 10 Hz before and 1 h after bumetanide injection. At 10 Hz, the depression of the H-reflex is larger after drug than before. A two-way repeated-measures ANOVA revealed statistically significant differences across stimulation frequency (p < 0.001) and drug status (p < 0.001) and the interaction between frequency and drug status was also significant (p < 0.001). Overall, the FDD increased after drug with an H-reflex amplitude (normalized to M-wave) from 61 ± 12% to 22 ± 5% at 5 Hz (p = 0.01) and from 47 ± 10% to 19 ± 6 at 10 Hz (p = 0.014).
SCI decreases KCC2 and increases NKCC1 protein levels in the lumbar spinal cord
The level of KCC2 (Fig. 3A) in the lumbar enlargement (L4–L6) is modulated as early as 7 d after SCI, decreasing to 68 ± 4% (p < 0.05) of intact values, whereas NKCC1 expression tended to increase (p = 0.078) but did not reach significance until 28 d, when it reached 161 ± 10% of intact values (p < 0.01; Fig. 3B). The KCC2 to NKCC1 ratio was significantly decreased to 49 ± 6% in SCI-7d and 56 ± 2% in SCI-28d compared with intact (p < 0.01; Fig. 3C). Consistent with their opposing directionality in chloride transport, NKCC1 and KCC2 protein levels are reciprocally regulated after SCI. Linear regression analysis yielded a significant negative relationship between KCC2 and NKCC1 protein levels (p = 0.08, R2 = 0.302) in the lumbar enlargement (Fig. 3D).
Figure 3.

Exercise prevents the modulation of the expression of the cation chloride cotransporters KCC2 and NKCC1. Western blot analysis showing KCC2 (A) and NKCC1 (B) protein levels in the lumbar spinal cord (L4–L6) of intact, SCI-7d, SCI-28d, and SCI-28d+exercise animals. KCC2 decreased significantly after a complete SCI transection, whereas NKCC1 levels increased. Exercise returned KCC2 and NKCC1 expression levels toward intact. KCC2 and NKCC1 proteins were detected and quantified by Western blots using actin as a standard control (*compared with intact; §compared with SCI-28d+exercise). C, KCC2/NKCC1 ratio is decreased after SCI, but not in exercised animals. D, Regression analysis showing a negative relationship between KCC2 and NKCC1 protein levels in the lumbar enlargement (p = 0.008).
Exercise can restore chloride homeostasis and contribute to functional recovery
If the reduction in KCC2 expression in the lumbar enlargement plays a role in the establishment and maintenance of spasticity, as measured by a decrease in FDD after SCI, then the recovery of FDD with exercise could rely on a prevention or limitation of its downregulation. No difference in the features of the H-reflex and M-wave was observed in exercised animals compared with nonexercised (Table 1). A two-way ANOVA revealed statistically significant differences across stimulation frequency (p < 0.001) and across experimental groups (p < 0.001) and the interaction between frequency and groups was also significant (p < 0.001). Post hoc comparisons showed that 5 and 10 Hz values were different from 0.3 Hz in all groups. Overall, exercise significantly improved FDD whether animals were 14 d (p < 0.05) or 28 d postinjury (p < 0.001; Fig. 4A). The depth of modulation of the FDD for the SCI-14d+exercise group was significantly greater than for the SCI-28d+exercise group at both 5 and 10 Hz (p < 0.05). The Hmax/Mmax ratio tended to decrease with exercise in the SCI-14d group, but reached significance only 28 d after SCI (Fig. 4B) from 0.66 ± 0.07 in nonexercised animals down to ∼0.48 ± 0.07 in exercised animals (p < 0.05). The SCI-14d group was only exercised for ∼10 d compared with ∼20 d for the SCI-28d group, but their FDD was not statistically different from intact (as shown in Fig. 1A). This supports the fact that not only does hyperreflexia gradually emerge after SCI, but also that exercise acts more as a limiting than a recovery process in this instance, because exercise started before the establishment of hyperreflexia. Because the SCI-14d animals showed a much wider range of results for all outcomes measured, we chose to continue with the SCI-28d group only.
Figure 4.

Exercise restores spinal excitability 14 and 28 d after SCI. A, Exercise increased the depth of modulation of the FDD of the H-reflex in SCI animals at 14 and 28 d post-SCI. B, The Hmax/Mmax ratio is significantly decreased by 28 d post-SCI.
Further Western blot analysis revealed that KCC2 and NKCC1 protein levels in the lumbar spinal cord of exercised animals did not differ from uninjured animals (94 ± 6% and 88 ± 2% of intact, respectively). Exercise upregulated KCC2 (Fig. 3A) and downregulated NKCC1 (Fig. 3B) compared with the SCI nonexercised group. A linear regression analysis did not yield any significant relationship between KCC2 or NKCC1 levels in the lumbar spinal cord and the depth of modulation of the H-reflex (data not shown).
KCC2 and NKCC1 immunoreactivity is widely distributed throughout the gray and white matter. KCC2 labeling is strong in spinal interneurons and motoneurons and dendrites are strongly labeled, creating a mesh-like structure in the gray matter and a centrifugal appearance in the white matter, especially in the ventral and lateral funiculi (Fig. 5A, left). KCC2 labeling is also strong along the surface membrane of the neurons in the ventral horn defining the contour of the cell somata (Fig. 5A, arrows). After injury, KCC2 decreases in the ventral gray matter area (Fig. 5B), primarily in the association with the motoneuron outer membrane (Fig. 5C), but returns to intact levels with exercise (Fig. 5A–D). NKCC1 has a paler and more diffuse profile without a pattern as distinctive as KCC2, being more evenly distributed in cell bodies, dendrites, axons, and glia (Fig. 6A–F). NKCC1 immunoreactivity was evident in the cell body and processes of neurons in the intermediate gray and ventral horn, but was lacking any clear association with the somatic membrane. Quantitative analysis did not show density change in the ventral gray matter after SCI or after exercise (Fig. 6G).
Blocking KCC2 activity with DIOA “inhibits” reflex recovery
If a lasting, activity-dependent upregulation of KCC2 levels in the lumbar spinal cord led to a recovery of the FDD in exercised animals, then a pharmacological blockade of KCC2 in trained animals would likely return FDD to values close to the nonexercised condition. To test this possibility, we acutely applied DIOA, a KCC2 antagonist, to the lumbar enlargement of animals with a “normal” FDD [i.e., SCI-7d animals (n = 5) or SCI-28d+exercise (n = 4)]. DIOA did not significantly modulate the latency, amplitude, or threshold of the M-wave and H-reflex at 0.3 Hz (Table 1), but increasing stimulus frequencies disclosed a clear effect. Figure 5A illustrates a H-reflex recorded from an exercised animal at 0.3 and 10 Hz before and after DIOA was applied to the lumbar enlargement. At 10 Hz, the depression of the H-reflex is almost complete, whereas the depression is greatly reduced after DIOA. A two-way repeated-measures ANOVA revealed statistically significant differences across stimulation frequency (p < 0.001) and drug status (p < 0.001) and the interaction between frequency and drug status was also significant (p < 0.001). FDD drastically decreased after DIOA and the H-reflex amplitude (normalized to M-wave) went from 23 ± 6% to 66 ± 9% at 5 Hz (p < 0.001) and from 15 ± 4% to 72 ± 11 at 10 Hz (p < 0.001) in the SCI-28d+exercise group (Fig. 7A). A similar decrease was observed in the SCI-7d group. Overall, DIOA reduced FDD in the SCI-7d group and masked recovery in SCI-28d+exercise group (Fig. 7B).
Chloride homeostasis, exercise, and BDNF
Western blot results show that the BDNF (Fig. 8A) protein level in the lumbar spinal cord was decreased by SCI up to 28 d and returned toward intact values in exercised animals, whereas TrkB was transiently decreased 7 d after SCI (Fig. 8B). Exercise tended to increase TrkB expression, but values for this group were highly variable. A linear regression analysis was performed to determine whether there was a relationship between chloride cotransporters and BDNF/TrkB levels and yielded a significant relationship between BDNF and KCC2 (p = 0.028, R2 = 0.229; Fig. 8C), but not NKCC1 (p = 0.264, R2 = 0.0564; Fig. 8D), suggesting a role for BDNF in modulating KCC2 but not NKCC1 expression.
Figure 8.

Relationship between KCC2 and BDNF expression. Expression of BDNF (A) and TrkB (B) in the lumbar spinal cord (L4–L6) of intact, SCI-7d, SCI-28d, and SCI-28d+exercise animals. The expression of BDNF and TrkB decreased significantly after a complete spinal cord transection. Exercise returned BDNF expression levels toward intact. BDNF and TrkB proteins were detected and quantified by Western blots using actin as a standard control (*p < 0.05 vs intact; § p < 0.05 vs SCI-28d+exercise). There is a significant relationship between BDNF and KCC2 (C, p = 0.028) but not NKCC1 expression (D, p = 0.264).
Discussion
Our results show that exercise contributes to restoring chloride homeostasis after SCI. Chloride homeostasis plays a pivotal role in the inhibitory action of GABA and glycine. Fast inhibitory transmission is mediated by GABA and glycine via ligand gated Cl− channels. Before CNS maturation, high [Cl−]i results in Cl− efflux at resting membrane potential, which depolarizes the neurons and can even evoke action potentials (Fig. 9, left; Zhang et al., 1991; Ehrlich et al., 1999; Kakazu et al., 1999). After maturation, low [Cl−]i causes a chloride influx during channel opening that results in hyperpolarization (Fig. 9, right; Eccles, 1966; Kaila, 1994). SCI increases [Cl−]i, which causes a depolarizing shift in ECl. This likely contributes to functional impairment and hypereflexia (Fig. 9, left; Boulenguez et al., 2010) because the inhibitory control of both firing rate and excitatory inputs are greatly compromised (Prescott et al., 2006; Jean-Xavier et al., 2007; Doyon et al., 2011). Our results support that exercise restored the KCC2/NKCC1 ratio (Fig. 9, right).
Figure 9.

Schematic diagram of an immature CNS neuron (left, high [Cl−]i) and an adult CNS neuron (right, low [Cl−]i). Opening GABAAR leads to Cl− exit and depolarization in immature neurons, whereas it causes Cl− entry and hyperpolarization in the adult neuron. The switch from excitatory effect of GABA to inhibition induced by modulation in KCC2 and NKCC1 expression during development is reversed after SCI. We suggest that this change in chloride cotransporter expression that is involved in the development of hyperreflexia after SCI is reverted by exercise.
The impact of SCI on reflex transmission is reduced by exercise, which modifies inhibitory transmission, returning several markers of inhibition [gephyrin, GAD67, GABAA receptors (GABAARs), etc.] toward normal values (Tillakaratne et al., 2000, 2002, Edgerton et al., 2001, Khristy et al., 2009). Our results support a role for exercise in reflex recovery through restoration of inhibitory transmission in the spinal cord. Both presynaptic and postsynaptic mechanisms contribute to functional impairment and spasticity after SCI and are likely to be restored by exercise. Current antispastic treatments include GABAA and GABABR agonists such as baclofen and benzodiazepines (Lapeyre et al., 2010; Simon and Yelnik, 2010). Most GABABRs in the spinal cord are presynaptic and located on incoming primary sensory afferent terminals (Price et al., 1984; Yang et al., 2001). GABABR density is decreased after SCI and recovers with exercise (Bose et al., 2012), which is likely to enhance presynaptic inhibition in a fashion similar to the effect of baclofen (Abbruzzese, 2002; Li et al., 2004). The activity-dependent GABABR upregulation (Bose et al., 2012) could also contribute to increasing BDNF levels to promote GABAergic synapse maturation, as was reported for the hippocampus (Fiorentino et al., 2009).
GABAARs are mostly located postsynaptically and exercise decreases their density after SCI in soleus motoneurons (Khristy et al., 2009). Normal GABAAR function is critically dependent on [Cl−]i, which is largely determined by the relative expression of KCC2 and NKCC1. By restoring the preinjury KCC2/NKCC1 ratio, exercise returns appropriate inhibitory control of FDD, likely through a decrease in [Cl−]i to the appropriate level.
Exercise increases KCC2 levels in the spinal cord
KCC2 plays a pivotal role in the development of inhibition during maturation (Blaesse et al., 2009; Ben-Ari et al., 2012): its upregulation is critical to the switch in GABAA/glycine signaling that results in the hyperpolarization prevalent in mature neurons (Rivera et al., 1999; Hübner et al., 2001). Blocking KCC2 activity with DIOA increases polysynaptic responses in neonatal rat spinal cord in vitro and reduces FDD in vivo (Boulenguez et al., 2010). Therefore, KCC2 activity is critical for proper expression of motor function. We showed that exercise returned KCC2 levels toward pre-SCI values. This would likely contribute to keeping low [Cl−]i and restoring chloride equilibrium and endogenous inhibition. Accordingly, exercise improves spinal reflexes after SCI and returns FDD modulation (Côté et al., 2003, 2011; Côté and Gossard, 2004). Blocking KCC2 in exercised animals greatly reduced FDD close to nonexercised values. This strongly suggests that exercise decreased hyperreflexia, at least partly through a shift in ECl toward more negative values triggered by the restoration of KCC2 expression on motoneuronal membranes.
KCC2 is present as monomer and oligomer in the mature brainstem and spinal cord (Blaesse et al., 2006; Boulenguez et al., 2010). The shift from GABAA-mediated depolarization to hyperpolarization can be determined by oligomerization of and increased gene expression (Blaesse et al., 2006). The relative amount of KCC2 included in the membrane as an oligomeric structure decreases after SCI (Boulenguez et al., 2010; Bos et al., 2013). Recent evidence also suggests that the intrinsic ion transport rate, cell surface stability, and membrane trafficking of KCC2 are rapidly and reversibly modulated by (de)phosphorylation of serine, threonine, and tyrosine residues in the C terminus (Blaesse et al., 2009; Kahle et al., 2013). In the present study, neither KCC2 nor NKCC1 levels in the lumbar enlargement was correlated with the depth of FDD. This may be explained by the fact that protein samples were electrophoretically separated in the presence of β-mercaptoethanol, which results in the disassembly of the oligomers (Blaesse et al., 2006). It remains to be determined whether isolating KCC2 oligomer or measuring phosphorylation would have revealed a relationship between KCC2 levels and the depth of modulation of FDD.
Mechanisms of activity-dependent downregulation of KCC2 protein/mRNA and depolarizing shift in EGABAA include TrkB activation by BDNF (Rivera et al., 2002, 2004; Coull et al., 2005; Miletic and Miletic, 2008). In the intact spinal cord, BDNF delivery decreases plasmalemmal KCC2 and impairs FDD (Boulenguez et al., 2010). Posttraumatic events resemble development in the interplay between BDNF and KCC2: BDNF effect on KCC2 becomes upregulatory, as first illustrated in axotomized cortical neurons (Shulga et al., 2008). Similarly, exogenous administration of BDNF increased KCC2 levels and restored FDD after SCI (Boulenguez et al., 2010). The activity-dependent processes triggered by exercise include an increase in BDNF expression that is associated with an increased effect of descending drive on the tibial motoneuronal pool, normalization of motoneuronal properties, diminished allodynia, restoration of normal sensation, and enhanced motor recovery (Hutchinson et al., 2004; Beaumont et al., 2008; Rojas Vega et al., 2008; Ying et al., 2008). BDNF expression in the spinal lumbar enlargement is also correlated with the depth of modulation of FDD after SCI (Côté et al., 2011). Our results also show a positive relationship between BDNF and KCC2 protein levels in the lumbar spinal cord, suggesting that BDNF contributes to KCC2 upregulation after SCI in an exercise-dependent manner. This is likely to occur specifically, but not exclusively, in motoneurons because KCC2 is neuron specific (Payne et al., 1996) and rat motoneurons synthetize BDNF in response to exercise to contribute to synaptic plasticity (Joseph et al., 2012).
BDNF is a multifaceted neurotrophin. After SCI, an increase in BDNF signaling is associated with many changes that are detrimental, such as maladaptive plasticity (e.g., pain, autonomic dysreflexia), but also beneficial (e.g., promotion of cell survival, regeneration of injured fibers, myelination, exercise-induced functional improvements; for review, see Weishaupt et al., 2012). BDNF-mediated TrkB activation has been shown to regulate KCC2 expression differentially in adult and early postnatal neurons (Rivera et al., 2002, 2004; Ludwig et al., 2011), resulting in a decrease in the adults and an increase in early postnatal neurons. Although the early decrease in KCC2 expression after SCI can be prevented by sequestering BDNF at the time of SCI, BDNF upregulates KCC2 and restores FDD at later time points post-SCI (Boulenguez et al., 2010). Together, these results suggest that, after chronic SCI, BDNF regulates KCC2 as in immature neurons. Interestingly, the downregulation of KCC2 in the adult requires the activation of both Shc and PLC-γ signal pathways by BDNF: if PLC-γ binding is disrupted, then BDNF upregulates the expression of KCC2 (Rivera et al., 2002, 2004). Whether these molecular pathways regulate the activity-dependent expression of KCC2 expression remains to be determined.
Although KCC2 function primarily influences the efficacy of GABAergic signaling, its abundant presence in dendritic spines has attracted growing interest (Gulyás et al., 2001). KCC2 recently emerged as having an ion-transport-independent role in spine genesis (Fiumelli et al., 2013). Therefore, KCC2 is expressed in dendritic spines that receive excitatory synapses, which indicates a potential role in synaptic plasticity. Suppressing KCC2 in mature neurons influences synaptic efficacy at glutamatergic synapses independently of GABAergic function (Gauvain et al., 2011), whereas an increase in KCC2 levels promotes dendritic spine development (Li et al., 2007; Horn et al., 2010). Interestingly, motoneurons display marked atrophy with loss of dendritic membrane and branching after SCI, neither of which is observed in exercised animals (Gazula et al., 2004). It is likely that the activity-dependent increase in KCC2 expression plays a role, not only in in restoring endogenous inhibition after SCI, but also in stabilizing or remodeling the dendritic tree of motoneurons.
Exercise decreases NKCC1 levels in the lumbar spinal cord
Our Western blot results show that exercise prevents the increase in NKCC1 levels in the lumbar enlargement that were observed after SCI in parallel with restored FDD. After SCI, FDD was decreased, confirming that hyperreflexia onset occurs between 7 and 14 d to reach a plateau at ∼28d (Yates et al., 2008). An increased level of NKCC1 associated with a decrease in KCC2 has the potential to shift ECl toward more positive values, resulting in a less hyperpolarized or even depolarized response to GABA and increased neuronal activity (Nabekura et al., 2002; Coull et al., 2003; Payne et al., 2003; Toyoda et al., 2003). Contrary to a previous report, NKCC1 levels in the lumbar spinal cord were increased in our animals after SCI. Boulenguez et al. (2010) saw no change in NKCC1 levels after SCI, but rather found a slight tendency that did not reach significance. Perhaps differences arise from “dilution” of their results based on merging 7–45 d post-SCI animals because NKCC1 expression transiently increases after contusive SCI (Cramer et al., 2008; Lee et al., 2014) or from their including rostral lumbar levels (L2–L3), which we did not.
Blocking NKCC1 with bumetanide “mimicked” values obtained in exercised SCI animals. Although bumetanide may also inhibit KCC2 and other anion transport processes at high concentrations, it is unlikely it had this effect because we used a relatively low dose specific to NKCC1 (Russell, 2000). Exercise likely decreased hyperreflexia at least partly through a negative shift in ECl, as suggested by the increased KCC2/NKCC1 ratio and negative relationship between KCC2 and NKCC1 levels. Interestingly coincident presynaptic and postsynaptic activity downregulates NKCC1 and leads to a similar hyperpolarized ECl (Balena and Woodin, 2008). Whether increased KCC2 with exercise is sufficient to restore reflex modulation after SCI or if a decrease in the NKCC1 level synergistically contributes to restore [Cl-]i remains to be determined. Immunostaining for NKCC1 failed to show any change in the ventral gray area. Further studies using more sensitive quantitative techniques are necessary to verify whether the exercise-dependent decrease in NKCC1 levels measured by immunoblotting reflect changes occurring on motoneurons specifically or in the intermediate gray and dorsal horn. Modulation in KCC2/NKCC1 and BDNF expression play an important role in neuropathic pain after SCI (Coull et al., 2005; Cramer et al., 2008). As we are moving toward clinically relevant models of incomplete SCI, pain-related outcomes will become critical to validate that motor output is not improved at the cost of increasing central sensitization and neuropathic pain.
Using rehabilitation to reestablish chloride homeostasis
There is a pressing need for an intervention that, unlike antispasticity medication, can alleviate hyperreflexia without significantly limiting functional recovery (Dietz and Young, 2003; Angeli et al., 2012). The intent of most clinically approved approaches is to attenuate reflex activity and reduce muscle tone to relieve muscle spasms, which often leads to paresis (Abbruzzese, 2002; Dietz and Young, 2003). Our results support the idea that therapeutic avenues that manipulate GABAergic systems through the modulation of neuronal Cl− gradients via KCC2 and NKCC1 can be successful.
Footnotes
This study was supported by the Craig H. Neilsen Foundation (M.-P.C.) and a Drexel University College of Medicine Student Summer Research Fellowship (S.G.). We thank J. Bouyer, S. Choyke, and H. Schulze for technical assistance.
The authors declare no competing financial interests.
References
- Abbruzzese G. The medical management of spasticity. Eur J Neurol. 2002;9:30–34. doi: 10.1046/j.1468-1331.2002.0090s1030.x. discussion 53–61. [DOI] [PubMed] [Google Scholar]
- Angeli C, Ochsner J, Harkema S. Effects of chronic baclofen use on active movement in an individual with a spinal cord injury. Spinal Cord. 2012;50:925–927. doi: 10.1038/sc.2012.93. [DOI] [PubMed] [Google Scholar]
- Balena T, Woodin MA. Coincident pre- and postsynaptic activity downregulates NKCC1 to hyperpolarize E(Cl) during development. Eur J Neurosci. 2008;27:2402–2412. doi: 10.1111/j.1460-9568.2008.06194.x. [DOI] [PubMed] [Google Scholar]
- Beaumont E, Kaloustian S, Rousseau G, Cormery B. Training improves the electrophysiological properties of lumbar neurons and locomotion after thoracic spinal cord injury in rats. Neurosci Res. 2008;62:147–154. doi: 10.1016/j.neures.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist. 2012;18:467–486. doi: 10.1177/1073858412438697. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Gorassini M, Fouad K, Sanelli L, Han Y, Cheng J. Spasticity in rats with sacral spinal cord injury. J Neurotrauma. 1999;16:69–84. doi: 10.1089/neu.1999.16.69. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Guillemin I, Schindler J, Schweizer M, Delpire E, Khiroug L, Friauf E, Nothwang HG. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J Neurosci. 2006;26:10407–10419. doi: 10.1523/JNEUROSCI.3257-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bos R, Sadlaoud K, Boulenguez P, Buttigieg D, Liabeuf S, Brocard C, Haase G, Bras H, Vinay L. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci U S A. 2013;110:348–353. doi: 10.1073/pnas.1213680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose PK, Hou J, Parmer R, Reier PJ, Thompson FJ. Altered patterns of reflex excitability, balance, and locomotion following spinal cord injury and locomotor training. Front Physiol. 2012;3:258. doi: 10.3389/fphys.2012.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Côté MP, Gossard JP. Step training-dependent plasticity in spinal cutaneous pathways. J Neurosci. 2004;24:11317–11327. doi: 10.1523/JNEUROSCI.1486-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté MP, Menard A, Gossard JP. Spinal cats on the treadmill: changes in load pathways. J Neurosci. 2003;23:2789–2796. doi: 10.1523/JNEUROSCI.23-07-02789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté MP, Azzam GA, Lemay MA, Zhukareva V, Houlé JD. Activity-dependent increase in neurotrophic factors is associated with an enhanced modulation of spinal reflexes after spinal cord injury. J Neurotrauma. 2011;28:299–309. doi: 10.1089/neu.2010.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cramer SW, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D, Resnick D. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Mount DB. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu Rev Physiol. 2002;64:803–843. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]
- Dietz V, Young R. The syndrome of spastic paresis. In: Brandt T, Caplan L, Dichgans J, Diener C, Kennard C, editors. Neurological disorders–course and treatment. Amsterdam: Academic; 2003. pp. 1247–1257. [Google Scholar]
- Doyon N, Prescott SA, Castonguay A, Godin AG, Kröger H, De Koninck Y. Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Comput Biol. 2011;7:e1002149. doi: 10.1371/journal.pcbi.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC. The ionic mechanisms of excitatory and inhibitory synaptic action. Ann N Y Acad Sci. 1966;137:473–494. doi: 10.1111/j.1749-6632.1966.tb50176.x. [DOI] [PubMed] [Google Scholar]
- Edgerton VR, Leon RD, Harkema SJ, Hodgson JA, London N, Reinkensmeyer DJ, Roy RR, Talmadge RJ, Tillakaratne NJ, Timoszyk W, Tobin A. Retraining the injured spinal cord. J Physiol. 2001;533:15–22. doi: 10.1111/j.1469-7793.2001.0015b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton VR, Courtine G, Gerasimenko YP, Lavrov I, Ichiyama RM, Fong AJ, Cai LL, Otoshi CK, Tillakaratne NJ, Burdick JW, Roy RR. Training locomotor networks. Brain Res Rev. 2008;57:241–254. doi: 10.1016/j.brainresrev.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Lohrke S, Friauf E. Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurones is due to age-dependent Cl- regulation. J Physiol. 1999;520:121–137. doi: 10.1111/j.1469-7793.1999.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino H, Kuczewski N, Diabira D, Ferrand N, Pangalos MN, Porcher C, Gaiarsa JL. GABAB receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses. J Neurosci. 2009;29:11650–11661. doi: 10.1523/JNEUROSCI.3587-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Briner A, Puskarjov M, Blaesse P, Belem BJ, Dayer AG, Kaila K, Martin JL, Vutskits L. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex. 2013;23:378–388. doi: 10.1093/cercor/bhs027. [DOI] [PubMed] [Google Scholar]
- Gauvain G, Chamma I, Chevy Q, Cabezas C, Irinopoulou T, Bodrug N, Carnaud M, Lévi S, Poncer JC. The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci U S A. 2011;108:15474–15479. doi: 10.1073/pnas.1107893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazula VR, Roberts M, Luzzio C, Jawad AF, Kalb RG. Effects of limb exercise after spinal cord injury on motor neuron dendrite structure. J Comp Neurol. 2004;476:130–145. doi: 10.1002/cne.20204. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Sík A, Payne JA, Kaila K, Freund TF. The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci. 2001;13:2205–2217. doi: 10.1046/j.0953-816x.2001.01600.x. [DOI] [PubMed] [Google Scholar]
- Harkema SJ, Hillyer J, Schmidt-Read M, Ardolino E, Sisto SA, Behrman AL. Locomotor training: as a treatment of spinal cord injury and in the progression of neurologic rehabilitation. Arch Phys Med Rehabil. 2012;93:1588–1597. doi: 10.1016/j.apmr.2012.04.032. [DOI] [PubMed] [Google Scholar]
- Horn Z, Ringstedt T, Blaesse P, Kaila K, Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci. 2010;31:2142–2155. doi: 10.1111/j.1460-9568.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- Houle JD, Morris K, Skinner RD, Garcia-Rill E, Peterson CA. Effects of fetal spinal cord tissue transplants and cycling exercise on the soleus muscle in spinalized rats. Muscle Nerve. 1999;22:846–856. doi: 10.1002/(SICI)1097-4598(199907)22:7<846::AID-MUS6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Hubli M, Dietz V. The physiological basis of neurorehabilitation–locomotor training after spinal cord injury. J Neuroeng Rehabil. 2013;10:5. doi: 10.1186/1743-0003-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/S0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Hutchinson KJ, Gómez-Pinilla F, Crowe MJ, Ying Z, Basso DM. Three exercise paradigms differentially improve sensory recovery after spinal cord contusion in rats. Brain. 2004;127:1403–1414. doi: 10.1093/brain/awh160. [DOI] [PubMed] [Google Scholar]
- Jean-Xavier C, Mentis GZ, O'Donovan MJ, Cattaert D, Vinay L. Dual personality of GABA/glycine-mediated depolarizations in immature spinal cord. Proc Natl Acad Sci U S A. 2007;104:11477–11482. doi: 10.1073/pnas.0704832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph MS, Tillakaratne NJ, de Leon RD. Treadmill training stimulates brain-derived neurotrophic factor mRNA expression in motor neurons of the lumbar spinal cord in spinally transected rats. Neuroscience. 2012;224:135–144. doi: 10.1016/j.neuroscience.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khristy W, Ali NJ, Bravo AB, de Leon R, Roy RR, Zhong H, London NJ, Edgerton VR, Tillakaratne NJ. Changes in GABAA receptor subunit gamma2 in extensor and flexor motoneurons and astrocytes after spinal cord transection and motor training. Brain Res. 2009;1273:9–17. doi: 10.1016/j.brainres.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapeyre E, Kuks JB, Meijler WJ. Spasticity: Revisiting the role and the individual value of several pharmacological treatments. NeuroRehabilitation. 2010;27:193–200. doi: 10.3233/NRE-2010-0596. [DOI] [PubMed] [Google Scholar]
- Lee HK, Ahmed MM, King KC, Miranpuri GS, Kahle KT, Resnick DK, Sun D. Persistent phosphorylation of NKCC1 and WNK1 in the epicenter of the spinal cord following contusion injury. Spine J. 2014;14:777–781. doi: 10.1016/j.spinee.2013.06.100. [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Li Y, Li X, Harvey PJ, Bennett DJ. Effects of Baclofen on spinal reflexes and persistent inward currents in motoneurons of chronic spinal rats with spasticity. J Neurophysiol. 2004;92:2694–2703. doi: 10.1152/jn.00164.2004. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Uvarov P, Soni S, Thomas-Crusells J, Airaksinen MS, Rivera C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J Neurosci. 2011;31:644–649. doi: 10.1523/JNEUROSCI.2006-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miletic G, Miletic V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain. 2008;137:532–539. doi: 10.1016/j.pain.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain–a neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 1997;33:781–795. doi: 10.1002/(SICI)1097-4695(19971120)33:6<781::AID-NEU6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Prescott SA, Sejnowski TJ, De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol Pain. 2006;2:32. doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price GW, Wilkin GP, Turnbull MJ, Bowery NG. Are baclofen sensitive GABAB receptors present on primary afferent terminals of the spinal cord? Nature. 1984;307:71–74. doi: 10.1038/307071a0. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas Vega S, Abel T, Lindschulten R, Hollmann W, Bloch W, Strüder HK. Impact of exercise on neuroplasticity-related proteins in spinal cord injured humans. Neuroscience. 2008;153:1064–1070. doi: 10.1016/j.neuroscience.2008.03.037. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Shulga A, Thomas-Crusells J, Sigl T, Blaesse A, Mestres P, Meyer M, Yan Q, Kaila K, Saarma M, Rivera C, Giehl KM. Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J Neurosci. 2008;28:6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon O, Yelnik AP. Managing spasticity with drugs. Eur J Phys Rehabil Med. 2010;46:401–410. [PubMed] [Google Scholar]
- Thompson FJ, Reier PJ, Lucas CC, Parmer R. Altered patterns of reflex excitability subsequent to contusion injury of the rat spinal cord. J Neurophysiol. 1992;68:1473–1486. doi: 10.1152/jn.1992.68.5.1473. [DOI] [PubMed] [Google Scholar]
- Tillakaratne NJ, Mouria M, Ziv NB, Roy RR, Edgerton VR, Tobin AJ. Increased expression of glutamate decarboxylase (GAD67) in feline lumbar spinal cord after complete thoracic transection. J Neurosci Res. 2000;60:219–230. doi: 10.1002/(SICI)1097-4547(20000415)60:2<219::AID-JNR11>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Tillakaratne NJ, de Leon RD, Hoang TX, Roy RR, Edgerton VR, Tobin AJ. Use-dependent modulation of inhibitory capacity in the feline lumbar spinal cord. J Neurosci. 2002;22:3130–3143. doi: 10.1523/JNEUROSCI.22-08-03130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Ohno K, Yamada J, Ikeda M, Okabe A, Sato K, Hashimoto K, Fukuda A. Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J Neurophysiol. 2003;89:1353–1362. doi: 10.1152/jn.00721.2002. [DOI] [PubMed] [Google Scholar]
- Weishaupt N, Blesch A, Fouad K. BDNF: the career of a multifaceted neurotrophin in spinal cord injury. Exp Neurol. 2012;238:254–264. doi: 10.1016/j.expneurol.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Yang K, Wang D, Li YQ. Distribution and depression of the GABAB receptor in the spinal dorsal horn of adult rat. Brain Res Bull. 2001;55:479–485. doi: 10.1016/S0361-9230(01)00546-9. [DOI] [PubMed] [Google Scholar]
- Yates C, Charlesworth A, Allen SR, Reese NB, Skinner RD, Garcia-Rill E. The onset of hyperreflexia in the rat following complete spinal cord transection. Spinal Cord. 2008;46:798–803. doi: 10.1038/sc.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Roy RR, Zhong H, Zdunowski S, Edgerton VR, Gomez-Pinilla F. BDNF-exercise interactions in the recovery of symmetrical stepping after a cervical hemisection in rats. Neuroscience. 2008;155:1070–1078. doi: 10.1016/j.neuroscience.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Spigelman I, Carlen PL. Development of GABA-mediated, chloride-dependent inhibition in CA1 pyramidal neurones of immature rat hippocampal slices. J Physiol. 1991;444:25–49. doi: 10.1113/jphysiol.1991.sp018864. [DOI] [PMC free article] [PubMed] [Google Scholar]