

Abstract
Monocyte-derived macrophages (mo-MΦs) and T cells have been shown to contribute to spinal cord repair. Recently, the remote brain choroid plexus epithelium (CP) was identified as a portal for monocyte recruitment, and its activation for leukocyte trafficking was found to be IFN-γ-dependent. Here, we addressed how the need for effector T cells can be reconciled with the role of inflammation-resolving immune cells in the repair process. Using an acute spinal cord injury model, we show that in mice deficient in IFN-γ-producing T cells, the CP was not activated, and recruitment of inflammation-resolving mo-MΦ to the spinal cord parenchyma was limited. We further demonstrate that mo-MΦ locally regulated recruitment of thymic-derived Foxp3+ regulatory T (Treg) cells to the injured spinal cord parenchyma at the subacute/chronic phase. Importantly, an ablation protocol that resulted in reduced Tregs at this stage interfered with tissue remodeling, in contrast to Treg transient ablation, restricted to the 4 d period before the injury, which favored repair. The enhanced functional recovery observed following such a controlled decrease of Tregs suggests that reduced systemic immunosuppression at the time of the insult can enhance CNS repair. Overall, our data highlight a dynamic immune cell network needed for repair, acting in discrete compartments and stages, and involving effector and regulatory T cells, interconnected by mo-MΦ. Any of these populations may be detrimental to the repair process if their level or activity become dysregulated. Accordingly, therapeutic interventions must be both temporally and spatially controlled.
Keywords: immune resolution, monocyte-derived macrophages, spinal cord injury, T cells, Th1, Treg
Introduction
Healing of the CNS following traumatic injury was considered for decades to be a tissue autonomous process independent of outside intervention. It is now evident that CNS wound healing shares many of the properties and requirements of its peripheral counterparts (Shechter and Schwartz, 2013). As in any other tissue, CNS healing is a multifactorial process, and only a deep understanding and integration of the mechanisms behind CNS repair can lead to the identification of the key limiting factors that hinder recovery.
In response to spinal cord injury (SCI), an intense activation and infiltration of immune cells into the injured parenchyma occurs. Thus, for example, activated microglia and monocyte-derived macrophages (mo-MΦs) play critical and nonredundant roles in the repair process (Elkabes et al., 1996; Bomstein et al., 2003; Glezer et al., 2007; Kigerl et al., 2009; Shechter et al., 2009). Specifically, infiltrating mo-MΦs are essential for the resolution of the resident microglial inflammatory response, via their expression of the anti-inflammatory cytokine, IL-10 (Shechter et al., 2009). In addition, T cells recognizing CNS antigens are pivotal for promoting neuronal survival and functional recovery following acute CNS insults (Moalem et al., 1999; Serpe et al., 1999; Hauben et al., 2000; Yoles et al., 2001; Kipnis et al., 2002a). In particular, passive transfer of CNS-specific T helper 1 (Th1) lymphocytes was shown to be beneficial following injury; however, the beneficial effect of this cell type is dependent on additional populations of CD4+ T cells (Kipnis et al., 2002a).
An autoimmune response specific to CNS antigens was shown to facilitate recruitment of inflammation-resolving mo-MΦs (Shechter et al., 2009), whose entry into the injured CNS was recently demonstrated to take place through a designated selective and educating remote gate, the choroid plexus (CP; Shechter et al., 2013). The activation of the CP epithelium was found to be dependent on IFN-γ signaling (Kunis et al., 2013). Thus, although several types of immune cells were identified to be involved in the recovery process following SCI, their spatial and temporal regulation, and their interrelationships are not yet fully understood.
Here, we demonstrated that peripheral Th1 cells play an important role at the initial stage following injury by activating the remote epithelial blood–cerebrospinal fluid (CSF) barrier to induce expression of molecules essential for leukocyte trafficking. We further showed that mice deficient in Th1 cells exhibited reduced infiltration of resolving mo-MΦs to the lesion site. The infiltrating mo-MΦs were found to orchestrate recruitment of regulatory T cells (Tregs) to the injured parenchyma. Finally, we found that although well controlled reduction of Treg levels at the early stage following the injury had a beneficial effect on the repair process, their ablation at the subacute/chronic phase interfered with tissue remodeling.
Materials and Methods
Animals.
Mouse strains used included C57BL/6J; Tbx21−/− (B6.129S6-Tbx21tm1Glm/J); Cd11cDTR [B6.FVB-Tg Itgax-DTR/GFP 57Lan/J, carrying a transgene encoding human diphtheria toxin receptor (DTR) under control of the murine Cd11c promoter; Jung et al., 2002]; Cx3cr1GFP/+ (B6.129P-Cx3cr1tm1Litt/J, in which one of the Cx3cr1chemokine receptor alleles is replaced with a gene encoding GFP). Cd11cDTR and Cx3cr1GFP/+ transgenic mice were a generous gift from Steffen Jung (Weizmann Institute of Science, Israel); Foxp3GFP mice (Fontenot et al., 2005), a generous gift from Alexander Rudensky (Memorial Sloan Kettering Cancer Center, New York, NY); Foxp3GFP (on the B6 background; Bettelli et al., 2006), a generous gift from Vijay Kuchroo (Harvard Medical School, Boston, MA); Foxp3DTR transgenic mice, carrying a transgene encoding a human DTR under control of the murine Foxp3 promoter (Suffner et al., 2010), were a generous gift from Günter J. Hämmerling (German Cancer Research Center, Heidelberg, Germany). For all experiments, adult males aged 8–10 weeks were used. All animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee.
SCI.
The spinal cords of deeply anesthetized mice were exposed by laminectomy at T12, and a contusive (200 kdynes) centralized injury was performed using the Infinite Horizon spinal cord impactor (Precision Systems), as previously described (Rolls et al., 2008; Shechter et al., 2009). The animals were maintained on twice-daily bladder expression. Animals that were contused in a nonsymmetrical manner were excluded from the experimental analysis.
Assessment of functional recovery from SCI.
Mice were randomly assigned to groups before treatment, while validating similar average starting functional score, which was evaluated 24 h postinjury, in all groups. Recovery was evaluated by hind-limb locomotor performance, assessed according to the open-field Basso Mouse Scale (BMS; Basso et al., 2006), as previously described (Rolls et al., 2008; Shechter et al., 2009), with nonlinear scores ranging from 0 (complete paralysis) to 9 (normal mobility); each score represents a distinct motor functional state. In the Treg-depletion experiments, animals were randomized so that both the control and experimental group were present in the same cage, and both received diphtheria toxin (DTx; the control group consisted of the DTR-negative siblings). In all the BMS experiments, blinded scoring ensured that observers were not aware of the identity of tested animals. Animals that showed a difference of >2 score points between their two hind limbs were excluded from the analysis.
Bone marrow radiation chimeras.
[Cd11cDTR>WT] and [Cx3cr1GFP/+>WT] bone marrow (BM) chimeras were prepared by subjecting gender-matched recipient mice (8- to 10-weeks-old) to lethal whole-body irradiation (950 rad) while shielding the brain, as previously described (Rolls et al., 2008; Shechter et al., 2009). The mice were reconstituted with 5 × 106 BM cells from an appropriate donor, according to a previously described protocol (Rolls et al., 2008; Shechter et al., 2009). The chimeric mice were subjected to spinal cord contusion 8–10 weeks after BM transplantation.
DTx administration.
DTx (Sigma-Aldrich) was injected intraperitoneally. For depletion of mo-MΦs, mice were repeatedly injected at 1 d intervals, starting immediately after the injury, throughout the first week of recovery. For depletion of Tregs after injury, mice received DTx daily from days 6 to 13. Ablation of mo-MΦs and Tregs postinjury was induced by administration of 300 ng of DTx. Depletion of Tregs before injury was performed by administration of 400 ng of DTx, using two different regimens: DTx on days −4, −2, and 0; DTx daily from days −7 to −1.
MC-21 administration.
MC-21 (Mack et al., 2001) was injected intraperitoneally starting immediately after the injury, throughout the first week of recovery (days 0, 1, 2, 4, and 6 postinjury).
Antigens and vaccination.
A MOG-derived altered peptide ligand, MEVGWYRSPFDRVVHLYRNGK (MOG-45D, termed 45D; an analog of MOG35-55), in which aspartic acid is substituted for serine, was prepared by the Peptide Synthesis Unit at the Weizmann Institute. 45D peptide is a weak agonist of the encephalitogenic MOG35-55 peptide and does not induce autoimmune disease (Ford and Evavold, 2004). Ovalbumin (OVA) peptide was purchased from Sigma-Aldrich. Adult mice were vaccinated with 45D or OVA (100 μg) emulsified in an equal volume of complete Freund's adjuvant (Difco) containing Mycobacterium tuberculosis (2.5 mg/ml; Difco), as previously described (Shechter et al., 2009). The emulsion (total volume 0.1 ml) was injected subcutaneously at one site in the flank, 7 d before the spinal cord injury.
Immunohistochemistry.
Due to technical limitations of some of the antibodies that were used, two different tissue preparation protocols (paraffin embedded and microtomed frozen sections) were applied, as previously described (Rolls et al., 2008). Whenever possible, the results were confirmed using both techniques. The following antibodies were used: rabbit anti-GFP (1:100; MBL), goat anti-GFP (1:100; Abcam), rabbit anti-glial fibrillary acidic protein (GFAP; 1:100; DakoCytomation), goat anti-IL-10 (1:20; R&D Systems), hamster anti-TCRβ (1:50; Biolegend), rat anti-CD3 (1:200; Serotec). For microglial/MΦ labeling, FITC-conjugated Bandeiraea simplicifolia isolectin B4 (IB-4; 1:50; Sigma-Aldrich) was added for 1 h to the secondary antibody solution. The slides were exposed to Hoechst stain (1:4000; Invitrogen Probes) for 1 min. GFAP staining was used for demarcation of the lesion site. For microscopic analysis, a Nikon light microscope (Eclipse E800) equipped with a Nikon digital camera (DS-Ri1) or fluorescence microscope (Eclipse 80i) equipped with Nikon digital camera (DXM1200F) were used. Longitudinal sections of the spinal cord were analyzed. Immunoreactivity (density) and lesion size were determined automatically with Image-Pro Plus 4.5 software (Media Cybernetics). To determine lesion size, the damaged site was demarcated based on Luxol Nissl staining, H&E staining, and GFAP reactivity. Evaluation of cell number was performed manually. To avoid overestimation due to counting of partial cells that appeared within the section, special care was taken to count only cells with intact morphology and a nucleus that appeared larger than 4 μm in diameter. The ImagePro quantification was performed on 2 mm2 fields from spinal cord tissue images, centered on the lesion site, and including the lesion site, the margins, and surrounding undamaged parenchyma. Three sections from different depths were assessed for each animal, and between 4 and 14 mice were tested in each group. For immunoreactivity measurements, the values are presented in arbitrary units and indicate total signal intensity in the tissue. The cell number per cubic millimeter was calculated based on the thickness of the sections.
Isolation of spinal cord cells and flow cytometric analysis/fluorescence-activated cell sorting.
Mice subjected to spinal cord injury were killed by an overdose of anesthetic, and their spinal cords were prepared for flow cytometric analysis by perfusion with PBS via the left ventricle. Spinal cord sections were cut from individual mice, including the injury site and adjacent margins, and tissues were homogenized using a software controlled sealed homogenization system (Dispomix; http://www.biocellisolation.com). The following fluorochrome-labeled mAbs were purchased from BioLegend, eBioscience, R&D Systems, or BD Biosciences, and used according to the manufacturers' protocols: PE-conjugated anti-CD11b, NK1.1, GITR, CD103, CD44, CD62L, IL-10 and IL-17 antibodies; PerCP-cy5.5-conjugated anti-Ly6C, CD44, CD103, CXCR3, and TCRβ antibodies; allophycocyanin-conjugated anti-CD115, Ly6G, TCRβ, CCR4, IFN-γ, Foxp3, Nrp1, and CD62L; and Pacific Blue/Brilliant Violet-conjugated anti-TCRβ, CD4, CD11b, and CD45.2 antibodies. Foxp3 staining was performed using the Foxp3 staining buffer set (eBioscience), according to the manufacturer's protocol. For intracellular staining of cytokines, single-cell suspensions were incubated with PMA/ionomycin with Golgi Stop (BD Biosciences) for 5 h at 37°C. Cells were stained for surface markers and then permeabilized using the Golgi Stop kit, according to the manufacturer's instructions, and stained for cytokine expression. Cells were analyzed on a FACS LSRII cytometer using FACSDiva software (both from BD Biosciences). Analysis was performed with FlowJo software (Tree Star). For isolation of CD4+ T cells (CD4+TCRβ+CD45.2+) and Ly6C−monocyte-derived macrophages (Ly6C−CD45.2highCD11bhighLy6G−), cells were labeled and isolated using fluorescence-activated cell sorting. In each experiment, relevant negative and positive control groups were used to identify the populations of interest and to exclude the others.
Statistical analysis.
All parametric analyses of data were preceded by confirmation that the data were normally distributed. Levene's test was used to confirm homoscedasticity. In the case of equal variances, data were analyzed using Student's t test to compare between two groups, or by one-way or two-way ANOVA to compare several groups; repeated measures two-way ANOVA was used in the functional BMS scoring. After the null hypothesis was rejected (p < 0.05), Tukey's Honestly Significant Difference test was used for follow-up pairwise comparison of groups in one-way ANOVA, and the Bonferroni test was used as the post-test for two-way ANOVA. Reported for the two-way ANOVA are the p values for interaction between the two factors (pint), and each of the main effects, such as time (ptime), strain (pstrain, when comparing WT to KO mice) or treatment (ptreat, such as vaccination or DTx). In the case of unequal variances, data were log-transformed to achieve equal variances and normality; otherwise, the Kruskal–Wallis (K–S) test was used to compare several groups, followed by Dunn's test. The specific tests used to analyze each experiment are indicated in the figure legends. The results are presented as mean ± SEM. In the graphs, y-axis error bars represent the SEM.
Quantitative real time PCR.
Spinal cords (8 mm samples flanking the lesion site) were homogenized in Tri reagent (Sigma-Aldrich), and total RNA was extracted using Qiagen RNeasy Mini-kit. For choroid plexus samples, total RNA was extracted using the RNA MicroPrep kit (Zymo Research). For sorted cells, total RNA was extracted using miRNeasy Mini Kit. Random hexamers (AB) were used for first-strand cDNA synthesis. The procedures were performed according to the manufacturer's instructions. The relative amounts of mRNA were calculated using the standard curve method, and were normalized to the housekeeping gene, PpiA (peptidylprolyl isomerase A). Each RNA sample was run in triplicate. The primers used are shown in Table 1.
Table 1.
Primers
| Forward | Reverse | |
|---|---|---|
| Arg1 | 5′-TCCTTAGAGATTATCGGAGCGCCT-3′ | 5′-TTTCCAGCAGACCAGCTTTCCTCA-3′ |
| Ccl1 | 5′-CAGGATGTTGACAGCAAGAG-3′ | 5′-GTTCAGCCTGAATACCACAG-3′ |
| Ccl5 | 5′-GTGCTCCAATCTTGCAGTCGTGTT-3′ | 5′-ACTTCTTCTCTGGGTTGGCACACA-3′ |
| Ccl17 | 5′-GTACCATGAGGTCACTTCAG-3′ | 5′-GACAGTCAGAAACACGATGG-3′ |
| Ccl21 | 5′-CAGGACTGCTGCCTTAAGTA-3′ | 5′-GCACATAGCTCAGGCTTAGA-3′ |
| Ccl22 | 5′-CTCTGATGCAGGTCCCTATG-3′ | 5′-TTTGAGGTCCAGAAGAACTCC-3′ |
| Ccl28 | 5′-CTCAGAAGCCATACTTCCCA-3′ | 5′-ATCCACTGCTTCAAAGTACGA-3′ |
| Cx3cl1 | 5′-ATGTGCGACAAGATGACCTCACGA-3′ | 5′-TTTCTCCTTCGGGTCAGCACAGAA-3′ |
| Cxcl9 | 5′-GAGTTCGAGGAACCCTAGTG-3′ | 5′-AACTGTTTGAGGTCTTTGAGG-3′ |
| Cxcl10 | 5′-AACTGCATCCATATCGATGAC-3′ | 5′-GTGGCAATGATCTCAACAC-3′ |
| Cxcl11 | 5′-CTTCTGTAATTTACCCGAGTAACG-3′ | 5′-TTCTATTGCCTGCATTATGAGG-3′ |
| Gata3 | 5′-AGGCAACCACGTCCCGTCCT-3′ | 5′-TACCTGGCTCCCGTGGTGGG-3′ |
| Icam | 5′-AGATCACATTCACGGTGCTGGCTA-3′ | 5′-AGCTTTGGGATGGTAGCTGGAAGA-3′ |
| Ifnγr2 | 5′-TCCCACACCCATTCACAG-3′ | 5′-AGGTCCAACAGTAACATTCTC-3′ |
| Igf1 | 5′-TGAGCTGGTGGATGCTCTTCAGTT-3′ | 5′-TCATCCACAATGCCTGTCTGAGGT-3′ |
| Il1b | 5′-CCAAAAGATGAAGGGCTGCTT-3′ | 5′-TGCTGCTGCGAGATTTGAAG-3′ |
| Il6 | 5′-TGCAAGAGACTTCCATCCAGTTG-3′ | 5′-TAAGCCTCCGACTTGTGAAGTGGT-3′ |
| Il10 | 5′-TGAATTCCCTGGGTGAGAAGCTGA-3′ | 5′-TGGCCTTGTAGACACCTTGGTCTT-3′ |
| Il12 p35 | 5′-TCACCCTGTTGATGGTCACG-3′ | 5′-AAATGAAGCTCTGCATCCTGC-3′ |
| Il17a | 5′-TCATCTGTGTCTCTGATGCT-3′ | 5′-TTCACATTCTGGAGGAAGTC-3′ |
| Madcam | 5′-AGCACTCCGTGAAGATCCTTGTGT-3′ | 5′-TAGCAGGGCAAAGGAGA GACTGTT-3′ |
| Mcsf | 5′-CCACATGATTGGGAATGGAC-3′ | 5′-GTAGCAAACAGGATCATCCA-3′ |
| Nt5e | 5′-GCTTCAGGGAATGCAACATGGGAA-3′ | 5′-ATGCCACCTCCGTTTACAATGCAC-3′ |
| PpiA | 5′-AGCATACAGGTCCTGGCATCTTGT-3′ | 5′-CAAAGACCACATGCTTGCCATCCA-3′ |
| Tbx21 | 5′-CAACCACCTGTTGTGGTCCAAGTT-3′ | 5′-TCGCCGTCCTTGCTTAGTGATGAT-3′ |
| Tnfα | 5′-GCCTCTTCTCATTCCTGCTT-3′ | 5′-CTCCTCCACTTGGTGGTTTG-3′ |
| Tgfβ2 | 5′-AATTGCTGCCTTCGCCCTCTTTAC-3′ | 5′-TGTACAGGCTGAGGACTTTGGTGT-3′ |
| Vcam | 5′-TGTGAAGGGATTAACGAGGCTGGA-3′ | 5′-CCATGTTTCGGGCACATTTCCACA-3′ |
Results
Th1 cells are needed for controlled recruitment of inflammation-resolving macrophages
T cells, as well as mo-MΦs have been shown to contribute to CNS repair following injury (Moalem et al., 1999; Hauben et al., 2000; Kigerl et al., 2009; Shechter et al., 2009). Recently, our group demonstrated that the brain CP acts as a portal of entry for mo-MΦs (Shechter et al., 2013), and activation of this gate was found to be IFN-γ-dependent (Kunis et al., 2013). These findings led us to consider that effector T cells, in the form of Th1, are needed for the process of recovery, but are active outside the CNS. To test this hypothesis, we took advantage of Tbx21−/− mice, which lack TBX21, a critical transcription factor for the control of IFN-γ production in CD4+ T cells (Szabo et al., 2002).
Quantitative real-time (qRT)-PCR analysis of CP tissue revealed that following injury, key trafficking molecules required for the recruitment of monocytes (Shechter et al., 2013) were not upregulated in the Tbx21−/− mice, as opposed to the WT animals (Fig. 1A). Tbx21−/− mice showed lower expression levels of trafficking molecules in the CP even in the absence of injury Fig. 1A), suggesting the need for Th1 cells during routine immune surveillance (Kunis et al., 2013). As a corollary, genes associated with the Th1 pathway, such as Tbx21 and Ifnγr, were upregulated in the CP of WT mice immediately following the insult, further supporting the involvement of Th1 cells in the activation of the CP for trafficking of mo-MΦs following SCI. Given the role of Th1 cells in activation of the CP for leukocyte trafficking, we compared the parenchymal infiltration of immune cells at different time points following injury, in Tbx21−/− mice relative to WT mice (Fig. 1B). Of note, infiltration of TCRβ+ T cells was decreased in Tbx21−/− mice (Fig. 1C). Importantly, we found significantly lower numbers of CD45highCD11bhigh infiltrating mo-MΦs, specifically on day 7 postinjury (Fig. 1C). In addition, we observed lower numbers of NK and NK T cells, in line with the role of TBX21 in the development and maturation of NK cells (Townsend et al., 2004; Lazarevic et al., 2013); however, these cells represented minute populations at the injured spinal cord (1.33 ± 0.17% and 0.35 ± 0.05%, respectively, on day 14 postinjury, the time point at which their numbers were the highest).
Figure 1.

Th1 cells activate the CP gate and affect infiltration of monocyte-derived macrophages. A, Gene expression determined by qRT-PCR of chemokines and adhesion molecules in the CP of WT and Tbx21−/− noninjured mice, and on day 1 postinjury; n = 4–6 per group, per time point; two-way ANOVA, Tbx21: pint = 0.08, pstrain < 0.0001, ptime = 0.059; Mcsf: pint = 0.83, pstrain < 0.0001, ptime = 0.92; Icam: pint = 0.35, pstrain < 0.0001, ptime = 0.32; Vcam pint = 0.01, pstrain = 0.37, ptime = 0.46; Madcam: pint = 0.15, pstrain < 0.0001, ptime = 0.12; Cx3cl1: pint = 0.08, pstrain < 0.0001, ptime = 0.92; Ifnγr: pint = 0.008, pstrain = 0.01, ptime = 0.19; Nt5e pint = 0.94, pstrain = 0.001, ptime < 0.0001. B, Flow cytometry (FC) gating strategy for quantification of the distinct immune infiltrates: Ly6G+CD45.2+ neutrophils; Ly6G−CD45.2highCD11bhigh infiltrating mo-MΦs; Ly6G−CD45.2lowCD11blow myeloid cells; Ly6G−CD45.2+CD11b−TCRβ+ T cells; Ly6G−CD45.2+CD11b−TCRβ+NK1.1+ NK T cells; Ly6G−CD45.2+CD11b−TCRβ−NK1.1+ NK cells. C, FC quantification of immune cell infiltration at the injured spinal cord parenchyma at different time points following injury, in WT and Tbx21−/− mice; n = 4–5 per group. Numbers of cells per 10,000 live cells are indicated. Two-way ANOVA, neutrophils: pint = 0.34, pstrain = 0.02, ptime < 0.0001; CD11blowCD45.2low: pint = 0.16, pstrain = 0.44, ptime < 0.0001; mo-MΦs: pint = 0.12, pstrain = 0.02, ptime < 0.0001; T cells: pint = 0.75, pstrain = 0.0001, ptime = 0.12; NK cells: pint = 0.001, pstrain < 0.0001, ptime = 0.40; NK T cells: pint = 0.04, pstrain < 0.0001, ptime = 0.007. Asterisks indicate statistically significant differences compared with the WT control; *p < 0.05, **p < 0.01, ***p < 0.001.
As we found a local reduction, relative to WT, of mo-MΦs in Tbx21−/− mice on day 7 postinjury (Fig. 1C), the time of peak infiltration of IL-10+ resolving mo-MΦs to the lesion site via the CP in WT mice (Shechter et al., 2009, 2013), we evaluated how the absence of peripheral Th1 cells affected the recruitment of this specific subset to the injury site. We found that the absence of circulating Th1 cells led to a significant reduction of IL-10-producing cells located at the margins of the lesion site, as determined by costaining for GFAP, at day 7 postinjury (Fig. 2A). IL-10 was mainly expressed by IB4+ activated myeloid cells (Fig. 2B), located outside of the lesion epicenter; as previously shown, the infiltrating mo-MΦs that express IL-10 are restricted to the margins of the lesion and are almost absent from the lesion epicenter (Shechter et al., 2009). Flow cytometry following intracellular staining confirmed that the infiltrating CD45.2highCD11bhigh mo-MΦs were the main source of IL-10 at the lesion site (Fig. 2C), and revealed a significant decrease in IL-10-producing mo-MΦs in the Tbx21−/− mice, when compared with WT controls (Fig. 2D). Notably, this subset of IL-10+ M2-like mo-MΦs was previously shown to produce TGF-β following SCI (Shechter et al., 2013). qRT-PCR analysis of the injured spinal cord lesion site, at distinct time points following SCI, revealed decreased Il10 expression in the Tbx21−/− mice (Fig. 2E). To assess how the absence of Th1 cells would affect the spinal cord microenvironment, as a function of time postinjury, we excised the lesion site and analyzed gene expression by qRT-PCR. We found reduced expression of genes characteristic of an anti-inflammatory milieu (Ccl1, Arg1, and Igf1) at the lesion sites of Tbx21−/− mice, relative to WT mice, whereas genes associated with a proinflammatory response (Il1β, Il12, and Il6) were less strongly affected by the absence of TBX21 (Fig. 2F). Finally, we evaluated functional recovery of the hind limbs of Tbx21−/− and WT spinal cord injured mice, using the BMS (Basso et al., 2006); in this scale, a locomotion score of 0 represents complete paralysis of the hindlimb, whereas a score of 9 reflects normal mobility (Basso et al., 2006). Compared with WT animals, Tbx21−/− mice showed worse recovery (Fig. 2G). To rule out the possibility that the worse outcome was due to changes in the T cell profile of the Tbx21−/− mice, in particular, changes in the encephalitogenic Th17 cells (Park et al., 2005), we evaluated whether the absence of Th1 cells led to an increase of the neurotoxic IL-17-producing cells. To this end, we measured IL-17 expression by the local CD4+ T cells in WT and Tbx21−/− mice at day 14 postinjury, and found a decrease in the numbers of IL-17-expressing CD4+ T cells (Fig. 2H), apparently as a result of the fewer infiltrating T cells in Tbx21−/− mice (Fig. 1C). Together, these data demonstrate that circulating Th1 cells are important players in the spontaneous wound healing response following SCI, by contributing to the recruitment of the M2-like resolving mo-MΦ subset into the injured spinal cord.
Figure 2.
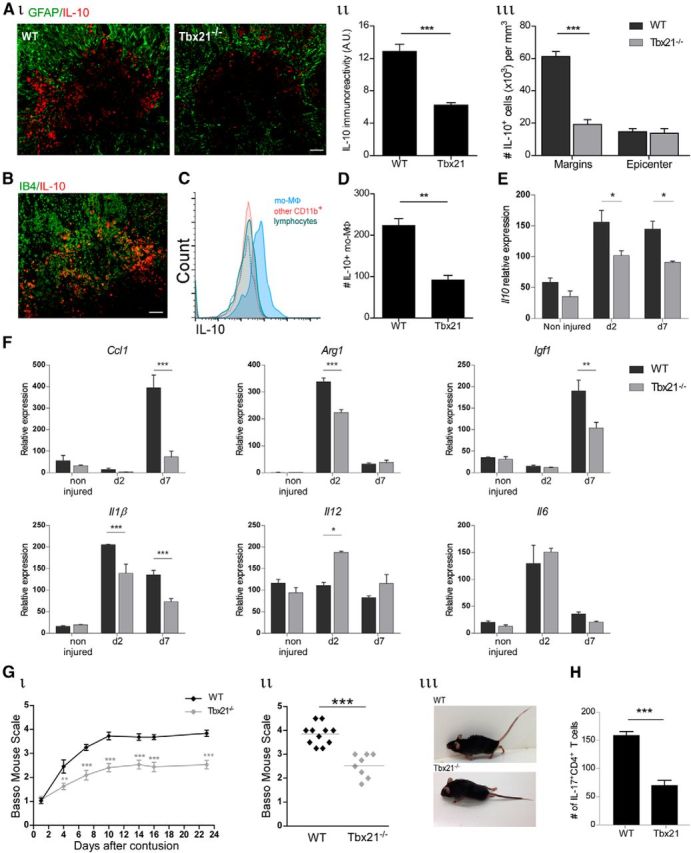
Th1 cells are needed for recovery following SCI and act by facilitating recruitment of resolving monocyte-derived macrophages. Ai, Representative image of IL-10-expressing cells (red) and their location relative to the lesion epicenter, demarcated by GFAP expression (green). Quantitative analysis of (Aii) IL-10 immunoreactivity (Student's t test; p = 0.0003), and (Aiii) number of IL-10-expressing cells (Student's t test; p < 0.001), in isolated tissue sections; n = 5–6 per group. B, Representative image of IL-10-expressing cells (red) and IB4+ activated myeloid cells (green). C, FC histogram of IL-10 expression by distinct immune populations at the lesion site. Isotype control is represented by the dashed line. D, Number of IL-10-expressing CD11bhighCD45.2high mo-MΦs, normalized to 50,000 live gate cells (Student's t test; p = 0.002). Four spinal cord lesion sites were pooled; n = 3 per group. E, qRT-PCR showing expression of Il10, at the lesion site, at different time points following SCI (two-way ANOVA; pint = 0.28, pstrain = 0.0003, ptime < 0.0001). F, Gene expression determined by qRT-PCR of anti-inflammatory and proinflammatory markers in the spinal cord lesion site of WT and Tbx21−/− mice at distinct times postinjury; n = 3–6 per group. Two-way ANOVA, Ccl1: pint = 0.0008, pstrain = 0.001, ptime < 0.0001; Arg1: pint < 0.0001, pstrain < 0.0001, ptime < 0.0001; Igf1: pint = 0.013, pstrain = 0.016, ptime < 0.0001; Il1β: pint = 0.0002, pstrain < 0.0001, ptime < 0.0001; Il12: pint = 0.011, pstrain = 0.02, ptime = 0.01; Il6: pint = 0.14, pstrain = 0.95, ptime < 0.0001. Gi, Motor score analysis according to the BMS (repeated-measures ANOVA; pint = 0.0007, pstrain < 0.0001, ptime < 0.0001) of injured WT and Tbx21−/− mice; n = 8–11 per group. Gii, Individual scorings at day 23 postinjury (Student's t test; p < 0.001). Each dot represents one animal. Giii, Representative picture of injured WT and Tbx21−/− mice at day 23 postinjury. H, Quantification of IL-17+-expressing CD4+ T cells in the injured spinal cord of Tbx21−/− and WT mice, at day 14 postinjury, by FC (Student's t test; p < 0.001), normalized to 10,000 live cells. Two spinal cord lesion sites were pooled; n = 4 per group. Asterisks indicate statistically significant differences compared with the WT control. Scale bar; 50 μm. The functional outcome assessment shown is from a single experiment, representative of two independent experiments performed. Analysis was performed at day 7 postinjury for A–D; *p < 0.05, **p < 0.01, ***p < 0.001.
Heterogeneity of CD4+ T cells at the lesion site
The lesion site contains not only infiltrating mo-MΦs (Shechter et al., 2009) but also CD4+ T cells (Moalem et al., 1999; Hauben et al., 2000). Immunohistochemical staining of spinal cord sections from [Cx3cr1GFP>WT] BM chimeric mice, which enables infiltrating macrophages to be distinguished from the resident microglia (Shechter et al., 2009), revealed the presence of TCRβ+ T cells at the lesion epicenter, as well as at its margins, and in close proximity to CX3CR1+GFP+ infiltrating mo-MΦs (Fig. 3A). This led us to ask which T cells are needed at the lesion site, and to further evaluate their relationship with the macrophages. To this end, we followed the kinetics of both infiltrating mo-MΦs and CD4+ T cells at the injured parenchyma following SCI. Similar to the wound healing response outside the CNS (Wynn, 2004; Shechter et al., 2013), infiltration of CD4+ T cells to the lesion parenchyma was found to occur following the wave of infiltrating mo-MΦs (Fig. 3B). Whereas the mo-MΦs were detected as early as day 1 postinjury (Shechter and Schwartz, 2013), CD4+ T cells arrived with a delay relative to the mo-MΦs. Characterization of the CD4+ T-cell subsets that home to the lesion site revealed heterogeneous parenchymal CD4+ T cell populations, including cytokine producing-effector memory (TEM, Foxp3−CD44highCD62Llow), central memory (TCM, Foxp3−CD44highCD62Lhigh), and Foxp3+ Treg cells. Notably, although the frequency of TCM remained low and unchanged from day 7 to day 14 postinjury, both numbers and frequencies of Tregs and TEM significantly increased during this period (Fig. 3C).
Figure 3.
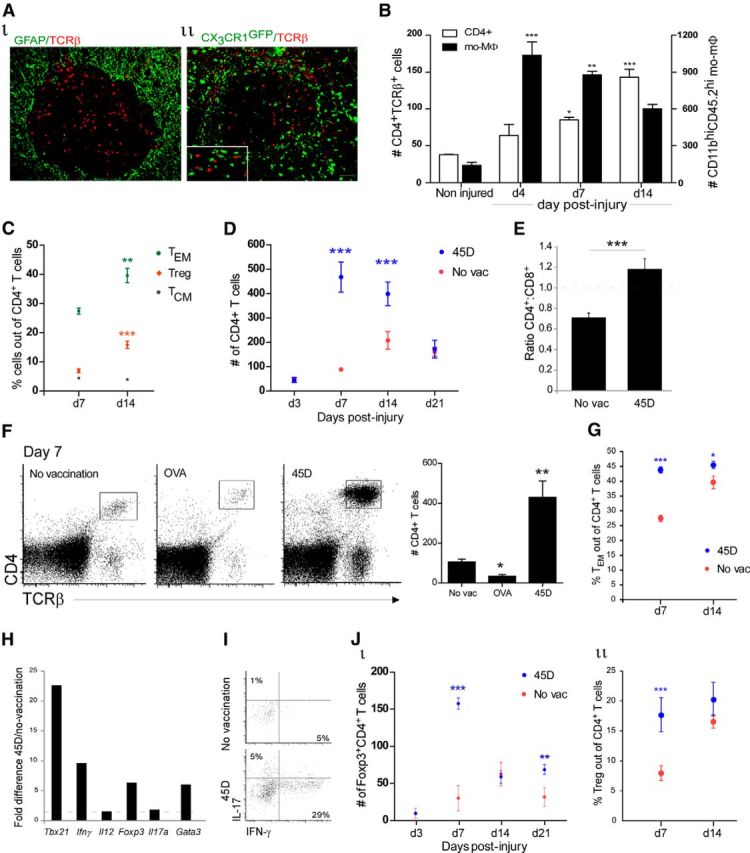
Vaccination with CNS-specific antigen increases CD4+ T cell infiltration into the lesion parenchyma. A, Immunohistochemical staining of T cells (TCRβ, red) on day 14 postinjury, demonstrating (Ai) the spatial organization of T cells at the lesion site (GFAP, green); (Aii) the distribution of T cells relative to the infiltrating mo-MΦs in [Cx3cr1GFP/+>WT] BM chimeric mice (CX3CR1, green; inset highlights the close proximity between the mo-MΦs and T cells). B, Kinetics of accumulation of CD4+ T cells and mo-MΦs at the injury site of WT mice, as measured by FC (K-Smacrophages; p = 0.0002. ANOVACD4+; F = 36.99, p < 0.0001); n = 3–6 per group. C, FC analysis of the frequency of Foxp3−CD44hiCD62Llow (TEM; Student's t test; p = 0.009), Foxp3−CD44hiCD62Lhi (TCM; Student's t test; p = 0.23), and Foxp3+ (Treg; Student's t test; p = 0.0002) cells out of the CD4+ T cells; n = 4–7 per group. D, Kinetics of CD4+ T cell infiltration to the injury site, measured by FC, in 45D-vaccinated (blue) and nonvaccinated (red) WT mice (two-way ANOVA; pint < 0.0001, ptreat < 0.0001, ptime < 0.0001); n = 4–8 per group. E, Ratio of CD4+ to CD8+ T cells (Student's t test; p = 0.0002); n = 4–6 per group. F, FC plot of CD4+ T cells at the injured spinal cord in nonvaccinated, OVA- and 45D-vaccinated mice. Right plot shows quantification of the CD4+ T cells under the different conditions (K-S; p < 0.0001); n = 9–16 per group. G, Foxp3GFP mice were immunized with 45D. Their spinal cord lesion site was excised at various days postinjury, and analyzed by FC for quantification of TEM (two-way ANOVA; pint = 0.005, ptreat < 0.0001, ptime = 0.0006); n = 4–5 per group, per time point. H, qRT-PCR quantification of gene expression of sorted CD4+ T cells derived from pooled spinal cords of 45D-vaccinated and nonvaccinated mice. Pooled sample of 12 mice each. The ratio of 45D:nonvaccinated is presented. I, FC plot demonstrating the percentage of IFN-γ and IL-17-producing CD4+ T cells. Pooled sample from five animals. J, Foxp3GFP mice were immunized with 45D. Their spinal cord lesion site was excised at different days postinjury, and analyzed by FC for quantification of regulatory T cells (Jii, two-way ANOVA: pint < 0.0001, ptreat < 0.0001, ptime < 0.0001; Jii, two-way ANOVA: pint = 0.08, ptreat = 0.0009, ptime = 0.004); n = 5–10 mice per group, per time point. Asterisks indicate statistically significant differences compared with the noninjured group in (B), relative to day 7 in (C), and relative to the nonvaccinated group in the remaining graphs. Numbers of cells per 10,000 live cells are indicated. Scale bar, 50 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
To achieve a better understanding of the relationship between the recruitment of mo-MΦs and T cells at the lesion site, we adopted a protocol that leads to enhanced recruitment of M2-like mo-MΦs and tested its effect on the infiltration of T cells. A CNS-specific autoimmune response was shown to benefit repair (Hauben et al., 2000), at least in part by boosting the spontaneous recruitment of mo-MΦs (Shechter et al., 2009). We therefore adopted this active immunization protocol using a myelin protein-derived peptide, 45D, before injury (Shechter et al., 2009); we observed that the number of infiltrating CD4+ T cells at the lesion site was enhanced compared with nonvaccinated animals (Fig. 3D), in a manner similar to the increase in the numbers of recruited M2 mo-MΦs following this vaccination protocol (Shechter et al., 2009). As a result of the vaccination, a significant shift in the CD4+:CD8+ ratio was observed (Fig. 3E). The increase in CD4+ T cells was dependent on CNS-antigen specificity, as vaccination with the nonrelated peptide, OVA, did not lead to an increase in the numbers of CD4+ T cells (Fig. 3F). Notably, this augmentation was accompanied by an increase in the frequencies of cytokine producing effector memory CD4+ T cells (Fig. 3G), in particular, 45D vaccination promoted an increase in the Th1 response, as found by qRT-PCR of sorted parenchymal CD4+ T cells (Fig. 3H) and flow cytometry of the spinal cord lesion site (Fig. 3I). The infiltration of CD4+ T cells to the lesion site led us to consider that they might be part of the immunological network needed to promote overall repair. Importantly, however, for such self-reactive T cells to support CNS recovery and homeostasis (Moalem et al., 1999; Hauben et al., 2000; Kipnis et al., 2002a), they must be maintained under a tight control (Kipnis et al., 2002a) to avoid autoimmune disease. This led us to hypothesize that recruitment of Treg to the lesion site might be needed at the subacute/chronic phase of SCI repair. We therefore assessed Treg accumulation into the parenchyma following injury, and found increased infiltration of these cells following the insult, and that vaccination with the CNS-derived peptide further augmented their numbers (Fig. 3J).
Regulatory T cells at the lesion site are thymic-derived and express activation markers
To understand the mechanism underlying accumulation of Tregs and their potential role, we followed their levels by flow cytometry analysis at different time points following injury. In the absence of immunization, the Treg population started to increase at the site of injury from day 7 onward, reaching highest numbers at day 14 postinjury, and declined by the third week of recovery (Fig. 4A). At day 14 postinjury, these cells expressed high levels of CD44, CD103, and GITR, and low levels of CD62L, consistent with the phenotype of activated (Huehn et al., 2004; Stephens et al., 2007), effector memory-like Tregs (Koch et al., 2009; Fig. 4B). To address whether the Tregs were recruited from the periphery or were the outcome of a local conversion from effector T cells, we measured the levels of Neuropilin-1 (Nrp1), the expression of which is known to be associated with the thymus-derived Treg subset, but not with the inducible Tregs (Weiss et al., 2012; Yadav et al., 2012). We found that ∼90% of the regulatory cells on day 14 expressed Nrp1 (Fig. 4C), suggesting that Tregs were recruited into the lesion site from the peripheral pool of thymus-derived Foxp3+ Treg cells. In addition, we found that the parenchymal Tregs expressed high levels of the Th1 chemokine receptor, CXCR3 (Fig. 4D). The frequencies of Foxp3+-expressing CXCR3+CD4+ T cells increased over the course of the response to injury (Fig. 4E). Notably, CXCR3+ Tregs have been reported to accumulate at sites of Th1 cell-mediated inflammation and suppress Th1 responses (Koch et al., 2009). In addition, the gene expression profile of the CXCR3-associated chemokines Cxcl9, Cxcl10, and Cxcl11, at the lesion site was changed over time following the insult (Fig. 4F). Overall, these results suggest a potential role of the recruited Treg, for the regulation of the effector Th1 response, during the tissue-remodeling period.
Figure 4.

Tregs express high levels of several activation markers, including the Th1 chemokine receptor, CXCR3. A, Kinetics of accumulation of Foxp3+CD4+TCRβ+ T cells at the injured site, as measured by FC (ANOVA; F = 45.4, p < 0.0001), in Foxp3GFP mice; n = 4–6 per group, for each time point. B, FC analysis of spinal cord lesion site on day 14 postinjury in Foxp3GFP mice. The numbers above the bars refer to the percentage of cells positive for the indicated marker out of Foxp3+CD4+ T cells; n = 4–5 per group, two independent experiments. C, FC plot indicating the expression of Nrp1 by parenchymal Foxp3+CD4+ T cells, on day 14 postinjury, in Foxp3GFP mice. Each sample represents a pool of two animals; n = 4. D, FC analysis of CXCR3+ expression in Foxp3+CD4+ T cells at the injured spinal cord of Foxp3GFP mice; n = 5. E, Frequency of Foxp3+-expressing cells out of the CXCR3+CD4+ T-cell population on days 7 and 14 following SCI (Student's t test; p = 0.006), at the injured parenchyma; n = 4–6 mice per time point. F, Gene expression, determined by qRT-PCR, of CXCR3-associated chemokines at different time points following SCI (ANOVA, Cxcl9: F = 8.14, p = 0.0009; Cxcl11: F = 5.5, p = 0.003; K-S, Cxcl10: p = 0.004); n = 3–4 mice per time point. Asterisks indicate statistically significant differences relative to noninjured mice in A and F and relative to day 7 postinjury in E. *p < 0.05, **p < 0.01, ***p < 0.001.
Monocyte-derived macrophages control Treg numbers at the lesion site
Our observations that parenchymal infiltration of CD4+ T cells following injury is preceded by the recruitment of mo-MΦs, both spontaneously and as a result of CNS-peptide vaccination, led us to hypothesize that the presence of CD4+ T cells at the lesion site might be dependent on the infiltrating monocytic cells. To test this working hypothesis, we assessed whether depletion of mo-MΦs would result in a reduction of T cells at the lesion site. [Cd11cDTR>WT] BM chimeric mice were vaccinated before injury with the 45D peptide, as described above (Fig. 3), and treated with DTx during the first week following the insult. Under this experimental setup, the poor recovery following DTx-mediated depletion of CD11c+ cells in the chimeric mice was shown to be due to specific ablation of the infiltrating mo-MΦs (Shechter et al., 2009). Using this paradigm, we found a significant reduction in the numbers of CD4+ T cells at the injured spinal cord relative to animals that were not subjected to DTx-mediated mo-MΦ ablation (Fig. 5A). Importantly, whereas the numbers of parenchymal CD4+ T cells were dramatically affected by the DTx-mediated depletion of mo-MΦ, the levels of circulating CD4+ T cells remained unchanged (control: 1003 ± 256; DTx: 866 ± 110. Student's t test, p = 0.569, n = 5–6 per group). This observation was further substantiated in nonchimeric mice, using an anti-CCR2 antibody (MC-21) that ablates the Ly6C+(Gr1+)CCR2+ monocytes, the macrophage precursors (Mack et al., 2001). Ablation of CCR2+ monocytes in the circulation (control: 311 ± 15; MC-21: 61 ± 18. Student's t test, ***p = 0.0004, n = 5–6 per group), which results in decreased monocyte recruitment to the lesion site and impaired repair (Shechter et al., 2009; London et al., 2011), led to a local reduction of CD4+ T cells (Fig. 5B), whereas the numbers of circulating CD4+ T cells were unchanged (control: 394 ± 10; MC-21: 397 ± 19. Student's t test, p = 0.8889, n = 5–6 per group).
Figure 5.

Monocyte-derived macrophages are involved in the parenchymal recruitment of Foxp3+ Tregs following SCI. A, [Cd11cDTR>WT] BM chimeras were vaccinated with 45D, and 1 week later were subjected to SCI. FC analysis at day 7 postinjury evaluating the number of CD4+ and CD4− T cells at the lesion site in nonvaccinated and vaccinated, depleted, and nondepleted mice (Ai, ANOVACD4+: F = 35.22, p < 0.0001; Aii, K-SCD4−; p = 0.18); n = 5–6 per group. B–E, Foxp3GFP 45D-vaccinated mice were injured and treated with anti-CCR2 (MC-21) antibody; analysis was performed on day 7 postinjury. The MC-21 nontreated group was used as a control. B, FC plot for CD4+ T cells and quantification (Student's t test; p < 0.0001); the dot plot was pregated on TCRβ+ cells. C, Labeling of spinal cord sections with GFAP (red) and Foxp3 (GFP, green) in MC-21 nontreated (control) and treated mice (MC-21). D, FC quantification of Foxp3+ Tregs (gated on TCRβ+CD4+ cells; ANOVA: F = 9.08, p = 0.0026). E, Histogram of CD44 expression on Foxp3+CD4+TCRβ+ cells (Student's t test; p = 0.003). F, WT mice were subjected to SCI and treated with MC-21 antibody. Mice were killed on day 10 postinjury, and their spinal cord lesion site excised for qRT-PCR analysis; n = 6 per group. G, Kinetics of Ccl22 gene expression at the lesion site throughout recovery, determined by qRT-PCR (K-S; p = 0.002); n = 3–4 per group. H, Histogram showing CCR4 Tregs, pregated on Foxp3+CD4+TCRβ+ cells, in Foxp3GFP mice; n = 4. I, Ccl22 expression in excised spinal cord tissue by qRT-PCR in Tbx21−/− and WT mice (two-way ANOVA; pint < 0.0001, pstrain < 0.0001, ptime < 0.0001); n = 3–5 per group. J, Ccl22 expression in sorted Ly6C−CD45.2highCD11bhigh mo-MΦs on day 7 postinjury. Pooled samples of 12 mice each. K, FC quantification of Foxp3+CD4+ T cells in Tbx21−/− and WT mice, at day 12 postinjury; frequency of Tregs out of CD4+ T lymphocytes (Student's t test; p = 0.002). Each sample represents a pool of three animals; n = 3 per group. L, Quantification of levels of Foxp3+ Tregs at the injured site of vaccinated WT and Tbx21−/− mice (ANOVA: F = 16.8; p = 0.006), at day 12 postinjury. Each sample represents a pool of three animals; n = 2–3 per group. Asterisks indicate statistically significant differences compared with respective controls in A, D, and L; MC-21 nontreated controls in B, E, and F; noninjured animals in G; and WT mice in I and K. Numbers of cells per 10,000 live cells are indicated. Scale bar, 50 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
The results described above raised the question of whether mo-MΦs affect a specific subset of infiltrating CD4+ T cells. To address this issue, we assessed levels of local Tregs under such experimental conditions. We found that systemic monocyte depletion with the anti-CCR2 antibody resulted in a dramatic reduction of the numbers of Foxp3+ Tregs in the injured parenchyma (Fig. 5C,D), but not in the blood (control: 40 ± 3; MC-21: 33 ± 3. Student's t test, p = 0.179, n = 5–6 per group). In addition, we observed that the absence of mo-MΦs resulted in a significant reduction in the percentage of parenchymal CD44high Tregs (Fig. 5E). As CD44 is a marker of T cell activation and it is highly expressed by activated Treg cells that went through extensive division, driven by antigen recognition of tissue self-antigens (Fisson et al., 2003), our results suggest a role for the infiltrating mo-MΦs in regulating the activation state of the Treg cells encountering the injured CNS parenchyma. To rule out the possibility of a direct effect of CCR2 antibodies on circulating effector memory CD4+ T cells and some Tregs that express CCR2 (Brühl et al., 2004), we analyzed their numbers in the circulation following antibody treatment; neither subset of T cells was affected (frequency of CD44high out of CD4+ T cells − control: 10.3% ± 0.9; MC-21: 9.3% ± 1.0; Student's t test, p = 0.529. Frequency of CD44high out of Foxp3+CD4+ T cells − control: 38.3% ± 2.1; MC-21: 35.5% ± 3.7; Student's t test, p = 0.579, n = 5–6 per group), supporting our interpretation that the local reduction of the different CD4+ T cell subsets was an outcome of monocyte depletion, rather than a direct effect on T cells. All together, these results suggested that monocyte-derived macrophages play a local role in orchestrating the levels and activation state of parenchymal Treg cells.
To understand the molecular mechanism by which macrophages could locally orchestrate the accumulation of the Tregs, we determined by qRT-PCR at day 10 postinjury the expression, within the injured spinal cord, of several chemokines known to be involved in recruitment of Tregs. To this end, lesion sites were excised from spinal cord injured, monocyte-depleted and nondepleted mice. Of all the tested molecules, absence of the recruited macrophages most significantly affected the expression of Ccl22 and Ccl17 (Fig. 5F). The kinetics of expression of Ccl22 in WT mice following the injury (Fig. 5G) was similar to the kinetics of accumulation of IL-10-expressing mo-MΦs at the lesion site (Shechter et al., 2013). The expression of CCR4, the CCL22 receptor, by the Tregs at day 14 postinjury (Fig. 5H) further supported the possible involvement of the CCL17/22-CCR4 chemokine axis in the recruitment of Tregs by mo-MΦs. As we found a specific reduction in the resolving subset of mo-MΦs in the absence of Th1 cells (Fig. 2), we took advantage of Tbx21−/− mice to further explore the relationship between recruited mo-MΦs and Tregs. We evaluated local expression of Ccl22 following injury in Tbx21−/− mice, and found a substantial reduction of Ccl22 gene expression on day 7 postinjury (Fig. 5I), the time point at which, in WT mice the frequency of resolving macrophages is at its peak (Shechter et al., 2009, 2013). Importantly, qRT-PCR analysis of Ccl22 expression in sorted Ly6C−CD45.2highCD11bhigh mo-MΦs, which includes the resolving M2-like Ly6C−CX3CR1high mo-MΦ subset, revealed an almost sixfold reduction in the expression of this chemokine in the sorted Ly6C− mo-MΦs of Tbx21−/− mice, compared with WT, on day 7 postinjury (Fig. 5J). Finally, we measured Treg infiltration by flow cytometry in Tbx21−/− mice at day 12 postinjury; Tbx21−/− mice showed decreased frequencies of Tregs at their lesion site relative to their WT counterparts (Fig. 5K), whereas the numbers of circulating Tregs were unchanged (WT: 73 ± 6.7; Tbx21−/−: 76 ± 4.2. Student's t test, p = 0.757, n = 5 per group). Overall, these results support our conclusion that mo-MΦs affect the extent of Treg accumulation in the lesion site, through a mechanism that may depend on the chemokine CCL22. Notably, to further substantiate the importance of the sequential relationship between Th1 cells, infiltrating mo-MΦs and Tregs in response to SCI, we compared Treg accumulation in WT and Tbx21−/− mice following immunization with the myelin-derived MOG-45D peptide. The immunization in Tbx21−/− mice failed to augment Treg accumulation (Fig. 5L).
Treg and the inflammation-resolving macrophages are nonredundant immunoregulatory players at the lesion site
The increased local frequency of Treg cells within the parenchyma following injury, which was temporally and spatially dependent on the infiltrating mo-MΦs, led us to consider that this population of immune regulatory cells might play a complementary role with the resolving mo-MΦs, at the subacute/chronic phase of the recovery process. To assess their function, we used a conditional in vivo ablation strategy that allowed the selective depletion of Foxp3+ Tregs, using Foxp3DTR mice (Suffner et al., 2010). In light of the fact that most of the Tregs at the lesion site seemed to be the outcome of recruitment rather than local conversion, and because their levels peaked at the second week following the injury, we ablated Tregs, using DTx administration, during the second week following SCI (days 6–13 postinjury). We found that such DTx-treatment led to parenchymal immune cell changes including reduction of Tregs (Fig. 6A), and a substantial increase in T lymphocytes (Fig. 6B–D), at day 14 postinjury, mainly of the effector memory subsets (CD44hiCD62Llow) (Fig. 6E). When we examined cytokine expression, we found an increase in the production of IFN-γ by CD4+ T cells in the Treg-depleted group (Fig. 6F). These results suggest that the local Tregs are involved in downregulating the local adaptive immune response, possibly involving a CXCR3-dependent interaction (Fig. 4D,E). Similarly, depletion of Tregs in 45D-vaccinated mice also resulted in the augmentation of IFN-γ-producing CD4+ T cells (Fig. 6G). In the periphery, we observed that these Treg-depleted mice exhibited an almost threefold increase in the expression of IFN-γ by CD4+ T cells in the spleen (Fig. 6H), and displayed splenomegaly, with significant increase in the weight and size of their spleens (Fig. 6I). As a consequence of the dysregulation of the effector T cell response in the absence of Tregs, we found augmented spread of myelin damage (Fig. 6J), indicating impairment of tissue remodeling following the injury. Because the depletion of Tregs occurred after the peak of recruitment of resolving macrophages, it appears that the regulatory T cells display an important and nonredundant role with the resolving mo-MΦs in controlling the immune response following SCI. Accordingly, the resolving macrophages are needed for the resolution of the innate microglial response (Shechter et al., 2009), and the regulatory T cells for resolving the effector adaptive (Teff) inflammatory response, thereby supporting tissue remodeling. Flow cytometry analysis of several populations including the CD11blowCD45.2low microglia, CD11bhighCD45.2high infiltrating mo-MΦs, and Ly6G+ cells (including neutrophils), indicated that depletion of Tregs during the second week of recovery affected mainly the lymphocytic population and not the myeloid cells (Fig. 6K). All together, these results emphasize that the local Teff/Treg balance should be in favor of Treg at the stage of tissue remodeling following SCI.
Figure 6.

Depletion of Tregs increases the local effector T cell response, with negative consequences for tissue remodeling. Foxp3DTR mice were subjected to SCI, and half of them received daily IP injections of DTx from day 6 until day 13 postinjury; analysis was performed on day 14 postinjury. A, FC histogram with frequency of Foxp3+ Tregs, out of CD4+ T cells, at the lesion site (Student's t test; p < 0.0001); n = 5 per group. B, Frequency of TCRβ+ cells (out of the leukocyte spinal cord population) by FC following depletion of Tregs (Student's t test; p = 0.005); n = 5–6 per group. C, FC analysis of levels of T cell subsets at the injured parenchyma (Student's t test, T cells, p = 0.002; CD4+, p = 0.38; CD4−, p = 0.0008); n = 4–5 per group. D, Spinal cord sections were stained for CD3 (brown), and the number of T cells was quantified (Student's t test; p = 0.002); n = 5–7 per group. E, Pie chart representation of the frequencies (of the total TCRβ+ T cell population) of CD44highCD62Llow effector cells in the CD4+ and CD4− compartment, and other T cells (naive CD44− and central memory), with and without depletion; n = 4–5 per group. F, G, FC analysis of IFN-γ production by CD4+ T cells at the lesion site at day 14 postinjury in (F) nonvaccinated mice (each sample represents a pool of four animals) and (G) 45D-vaccinated mice (Student's t test; p = 0.0002; n = 4 per group; each sample represents a pool of two animals). H, FC histogram of IFN-γ expression by CD4+TCRβ+ splenocytes (Student's t test; p = 0.05); n = 5 per group. I, Representative picture of spleens of Treg ablated and nonablated mice, with quantification of spleen weight, length and width. Each dot represents one animal. J, Spinal cord sections were stained by Luxol-Nissl and the lesion size quantified (Student's t test; p = 0.02) in nondepleted mice (left), or depleted (right) mice; n = 5–7 per group. K, FC analysis of levels of different myeloid subsets at the injured parenchyma; n = 4–5 per group. Asterisks indicate statistically significant differences relative to nondepleted mice. Numbers of cells per 10,000 live cells are indicated. Scale bar, 250 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
A peripheral Treg-Teff balance favoring Teff, during the early response following SCI, benefits repair
The observation above that the local Teff/Treg balance was in favor of Tregs at the subacute/chronic stage of tissue remodeling following SCI, raised the question of how this might be reconciled with our present findings that peripheral Th1 cells are essential for the repair immediately after injury (Figs. 1, 2), and with our previous findings that depletion of Tregs promotes recovery following acute CNS axonal injury (Kipnis et al., 2002b). We hypothesized that Tregs play distinct and even opposite roles at different phases of repair, and possibly in distinct compartments. Accordingly, we expected that early reduction of Tregs, unlike late ablation, would contribute to repair by alleviating CNS injury-induced immune depression (Meisel et al., 2005), and thereby allow activation of peripheral effector T cells which can then facilitate monocyte recruitment. To test this hypothesis, we transiently ablated Tregs, by treating Foxp3DTR mice with DTx starting 4 d before the injury (DTx administered on days −4, −2, and 0), and evaluated by flow cytometry how the ablation would affect the circulating immune cell pool at the time of the insult. Although we found a small increase in effector T cells, a robust decrease in the frequencies of blood Foxp3+ Tregs by the time of the injury was noted (Fig. 7A). To understand how the reduction in Tregs, at the time of injury, would affect recovery, we followed the functional recovery of the depleted mice over time; the ablation protocol resulted in improved recovery relative to the spontaneous recovery observed in nondepleted mice (Fig. 7B). Characterization of immune cells at the site of injury during the acute (day 7) and subacute/chronic (day 12) stages revealed that ablation of Tregs promoted increased infiltration of T lymphocytes to the lesion site (Fig. 7C), and specifically of TEM at the acute stage (day 7), which returned to control levels by day 12 (Fig. 7D). Concomitantly, a dramatic local increase of regulatory T cells at the subacute/chronic stage (day 12) was observed (Fig. 7E), the phase at which Tregs were found to contribute to tissue remodeling (Fig. 6). Of note, within the CD4+ T cell population, there was a specific increase in the frequencies of Tregs (control: 14.9% ± 0.97; DTx: 32.73% ± 0.66. Student's t test, ***p < 0.0001); similar high frequencies of parenchymal Tregs at the lesion site, on day 12 postinjury were observed following treatment with the beneficial 45D-vaccination (Fig. 5L). Overall, these results support our hypothesis that effector T cells are needed at the early stage following injury, whereas Tregs are needed in the CNS at the subacute/chronic stage; such immune state could be achieved by transiently weakening peripheral immunosuppression or active vaccination before the injury.
Figure 7.

A well regulated balance between regulatory and effector T cells, in favor of the effector subset, is needed to promote recovery following SCI. Foxp3DTR mice were treated with DTx before the insult (DTx administered IP on days −4, −2, and 0 before the injury), and subjected to SCI. Non-DTR-expressing siblings were used as a control (termed “control”), and received DTx as well (the DTR-positive group, i.e., susceptible to ablation, was termed “DTx”). A, Following depletion and immediately before the injury (day 0), the levels of circulating Foxp3−CD62Llow effector CD4+ T cells and Foxp3+CD4+ T cells were quantified by FC. Total Foxp3+ Tregs were determined using Foxp3+ intranuclear staining. Shown are the frequencies out of the CD4+ T-cell population. B, Hind-limb locomotor performance was assessed according to the BMS. The right plot corresponds to the individual motor scorings at the last day recorded. BMS scoring (repeated-measures ANOVA; pint = 0.0007, ptreat = 0.046, ptime < 0.0001). Right plot (Student's t test; p = 0.003); n = 7–8 per group. C–E, FC analysis of the T cell composition (numbers of T cells, C; frequency of Foxp3−CD44highCD62LlowCD4+ TEM, D, and Foxp3+ Tregs, E, out of the total T cell population) at the lesion site on days 7 (acute) and 12 (subacute/chronic) postinjury, following depletion. Each dot represents a single animal, except on day 12 postinjury plots, where each sample represents a pool of two animals. Asterisks indicate statistically significant differences relative to control. *p < 0.05, **p < 0.01, ***p < 0.001.
Notably, however, because Tregs are keeping effector T cells, primarily autoimmune ones, under control (Rosenblum et al., 2011; Yamaguchi et al., 2013), we also evaluated the fate of the tissue following injury if a more aggressive protocol of depletion before the injury would be adopted. To this end, we depleted Tregs for a longer period of time before the insult (DTx administered daily from days −7 to −1). When we evaluated how this depletion protocol affected recovery from SCI, we found a worse outcome relative to the spontaneous recovery of the nondepleted mice (Fig. 8A). The worse recovery was associated with a dramatic increase in circulating effector and memory T cells at the time of the insult (Fig. 8B,C). Subsequent changes in the T cell infiltration to the lesion site were observed (Fig. 8D), in particular reduced frequency of Tregs at the acute phase (day 7) and increased levels of TEM at the subacute/chronic stage (day 12; Fig. 8E,F). These results suggest that the limited recovery observed upon extensive depletion of Tregs (days −7 to −1) is due to the delayed recruitment of effector cells (Fig. 8E) and limited resolution by Tregs (Fig. 8F), whereas improved recovery is associated with an earlier infiltration of TEM (Fig. 7D) and high levels of Tregs at the resolution phase (Fig. 7E). To rule out the possibility that other cell populations could account for the distinct functional outcome, we measured changes in CD4+ TCM and CD44highCD62Llow CD4− cells. We found that these cell populations underwent similar changes in the two depletion paradigms (Improved recovery, TCM: control 1.2% ± 0.13 vs DTx 1.4% ± 0.07, Student's t test, p = 0.29; CD44highCD62Llow: control 36.1% ± 0.9 vs DTx 49.2% ± 1.2, Student's t test, ***p < 0.0001, n = 4–5 per group, pools of 2 animals; Adverse recovery, TCM: control 0.98% ± 0.2 vs DTx 1.7% ± 0.23, Student's t test, p = 0.051; CD44highCD62Llow: control 32.3% ± 1.8 vs DTx 48.8% ± 1.9, Student's t test, ***p = 0.0004, n = 4–5 per group, pools of 2 animals).
Figure 8.

Overwhelming peripheral inflammation at the time of the insult impairs recovery from spinal cord injury. Foxp3DTR mice were treated with DTx before the insult (DTx administered IP from days −7 to −1 before the injury), and subjected to SCI. Non-DTR-expressing siblings were used as a control, and received DTx as well (the DTR-positive group, i.e., susceptible to ablation was termed DTx). A, Hind-limb locomotor performance was assessed according to the BMS. The right plot corresponds to the individual motor scorings at the last day recorded. BMS scoring (repeated-measures ANOVA; pint < 0.0001, ptreat < 0.0001, ptime < 0.0001). Right plot (Student's t test; p < 0.0001); n = 11 per group. B, C, Following depletion and immediately before the injury (day 0), the levels of circulating (B) Foxp3−CD62Llow effector CD4+ T cells and (C) Foxp3−CD44high CD4+ memory T cells were quantified by FC. Shown are the frequencies out of the CD4+ T-cell population. D–F, FC analysis of the T-cell composition (numbers of T cells, D; frequency of Foxp3−CD44highCD62LlowCD4+ TEM, E; and Foxp3+ Tregs, F; out of the total T cell population) at the lesion site on days 7 (acute) and 12 (subacute/chronic) postinjury, following depletion. Each dot represents a single animal, except on day 12 postinjury plots, where each sample represents a pool of two animals. Asterisks indicate statistically significant differences relative to control. *p < 0.05, **p < 0.01, ***p < 0.001.
Overall, these results demonstrate that a fine balance between Th1 and Tregs is necessary throughout the recovery phase to support proper repair, with the need for skewing the immune cell bias in favor of effector T cells in the blood and in the parenchyma at the initial stages postinjury, and in favor of Treg cells at the subacute/chronic phase, the resolution, and remodeling stages. Such a change in the cellular balance could be achieved by breaking immune suppression at the time of the injury, either by active vaccination or by well controlled depletion of Treg cells.
Discussion
In the present study, using a series of pharmacological and selective ablation techniques together with immunological manipulations, we dissected the complex immune network that is needed to support recovery following CNS injury. We demonstrated that both Th1 effector cells and Foxp3+ Tregs are needed for recovery following SCI, at discrete locations and with distinct kinetics (Fig. 9); with monocyte-derived macrophages playing a functional interconnecting role in this network.
Figure 9.

The immune response following SCI (a model). Recovery from SCI is limited under physiological conditions, but is amenable to boosting. A well controlled effector T-cell (Th1 cells) response at the time of the injury, regulated by the action of Treg cells, is critical for the overall repair. Immediately following the injury (day 1), the circulating Th1 cells activate the blood–CSF barrier, thereby facilitating the active but controlled trafficking of the vital resolving mo-MΦs to the lesion site. These mo-MΦs locally contribute to the resolution of the innate proinflammatory response (mediated by microglia and macrophages) via the secretion of the anti-inflammatory cytokine, IL-10. Furthermore, they recruit regulatory T cells, via the expression of CCL22-CCL17. By downregulating adaptive immunity, the parenchymal Tregs contribute to the repair process, preventing further spread of damage and promoting tissue remodeling.
Previous studies from our group (Kipnis et al., 2002a,b; Shaked et al., 2005; Butovsky et al., 2006) and others (Ishii et al., 2012) demonstrated that Th1 cells and IFN-γ can promote neuronal repair. However, it also became apparent that Th1 cells are not themselves sufficient to mediate this reparative function (Kipnis et al., 2002a), supporting the possibility that a dynamic immune network is required for proper CNS repair.
Here, we found that at the early stage following SCI, the activity of Th1 cells takes place remotely from the injured parenchyma, at the CP epithelium, which was recently shown to form a regulated selective and educative gate for the infiltration of resolving M2-like mo-MΦs (Shechter et al., 2013). The CP epithelium of Tbx21−/− mice was compromised in its expression of trafficking determinants, and thus was poorly activated upon injury, relative to WT mice. In light of the recent findings from our group that effector memory T cells reside in the CP (Baruch et al., 2013), and that injury-induced CP epithelial expression of trafficking components is dependent on IFN-γ receptor signaling (Kunis et al., 2013), these findings emphasize that Th1 cells contribute to recovery immediately following acute CNS injury.
Importantly, deficiency in TBX21 resulted in suboptimal recruitment of inflammation-resolving IL-10+ mo-MΦs. We further found decreased expression at the lesion site of the growth factor, Igf1, previously shown to be produced by macrophages/microglia (Rolls et al., 2008) and to have essential roles in CNS maintenance, repair (Carro et al., 2003), and cell renewal (Aberg et al., 2000; Hsieh et al., 2004; Butovsky et al., 2006). TBX21 has been shown to have essential functions in the development of several innate and adaptive immune cells, including CD8+ T cells and NK cells (Sullivan et al., 2003; Townsend et al., 2004). In light of the pivotal role of IL-10, specifically expressed by mo-MΦs, in recovery from SCI (Shechter et al., 2009), the observed reduced infiltration of resolving mo-MΦs in Tbx21−/− mice highlights the relevance of this relationship to the repair process.
Multiple roles have been attributed to infiltrating mo-MΦs in the recovery process, including regulation of the local innate proinflammatory response (Shechter et al., 2009) and resolution of the glial scar (Shechter et al., 2011). Here, we discovered an additional function for these innate cells in controlling Tregs at the lesion site. Thus, reduction of infiltrating mo-MΦs, following specific depletion of mo-MΦs, or as occurs in Tbx21−/− mice, resulted in a severe reduction in the accumulation of parenchymal Tregs, and also lower levels of Ccl22 expression. A similar mechanism of CCL22-dependent recruitment of Tregs was identified for macrophages in the tumor environment (Curiel et al., 2004; Huang et al., 2006). The parenchymal Tregs identified here appeared to derive from the thymic pool, and were endowed with an effector memory-like phenotype characterized by high expression of CD44, CD103, and GITR, and low levels of CD62L. We suggest that although the monocyte-derived macrophages locally regulate Tregs, the latter are necessary for controlling parenchymal effector T cells at the tissue-remodeling phase and thereby prevent myelin loss. The high percentage of parenchymal CXCR3+ Tregs and the upregulation of CXCR3-associated chemokines at the lesion site are consistent with the role of Tregs in controlling Th1 cells. It is possible that the parenchymal Tregs, through the upregulation of CXCR3, responded to the same chemokine cues as Th1 cells, thereby contributing to the resolution of the local Th1 response. This mechanism of co-opting selective aspects of the T helper differentiation program has been demonstrated in EAE, and in type-1 (Müller et al., 2007; Koch et al., 2009), and Th2 (Zheng et al., 2009) inflammatory responses.
In accordance with previous findings from our group (Kipnis et al., 2002a,b), we propose that a controlled and timely breakdown of tolerance, achieved by Treg ablation, evokes an early transient increase in parenchymal effector memory T cells at the acute stage, followed by augmentation of regulatory T cells at the subacute/chronic stage at the lesion site (when Treg cells are needed to promote tissue remodeling and support repair, as we demonstrated here). Because Foxp3+ Tregs mainly suppress autoimmune T cells (Rosenblum et al., 2011; Yamaguchi et al., 2013), such a response recapitulates the beneficial effect of myelin peptide-derived vaccination (Shechter et al., 2009), which was shown to lead to increased recruitment of resolving macrophages (Shechter et al., 2009). We further found that such a vaccination also elicited the recruitment of Treg cells. Importantly, we underline the dual and nonredundant role of resolving mo-MΦs and Treg cells in controlling innate and adaptive immune responses, respectively, and in promoting restoration of homeostasis following CNS injury. Nevertheless, extensive Treg depletion before the insult led to an uncontrolled immune response, abrogating the potential protective effect of Th1 cells; loss of regulation by the Treg cells resulted in complete loss of recovery and amplified inflammation at the injured CNS.
In summary, we established that sequential participation of immune cells, including Th1 cells, infiltrating resolving mo-MΦs and regulatory T cells, is needed for recovery from CNS injury. This response, spontaneously activated following injury, is apparently not sufficient, but it is amenable to boosting by CNS-specific T cells (Hauben et al., 2001; Shechter et al., 2009), a phenomenon named by us “protective autoimmunity” (Moalem et al., 1999), or as we further show here, by transient reduction of Tregs before the insult. Our results highlight the delicate spatial and temporal dynamics of effector and regulatory T cells, the balance of which is pivotal for the CNS repair process.
Footnotes
This work was supported by a European Research Council Grant Award (M.S.), and by the Seventh Framework Programme (FP7) HEALTH-2011 Grant (M.S.). C.R. was supported by a PhD Fellowship from Foundation for Science and Technology (FCT), Portugal. M.S. holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology. We thank Steffen Jung for his generous gift of Cd11cDTR and Cx3cr1GFP/+ transgenic mice, Alexander Rudensky for his generous gift of Foxp3GFP mice, Vijay Kuchroo for kindly providing the Foxp3GFP mice (on the B6 background), Günter J. Hämmerling for the generous gift of Foxp3DTR mice, Matthias Mack for kindly providing the MC-21 antibody, Dr Shelley Schwarzbaum for editing the manuscript, Dr Hillary Voet for statistical consultation, Margalit Azulai for assistance with animal handling, and Dr Gilad Kunis for assistance with intravenous injections.
The authors declare no competing financial interests.
References
- Aberg MA, Aberg ND, Hedbäcker H, Oscarsson J, Eriksson PS. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, Mirlas-Neisberg N, Cardon M, Vaknin I, Cahalon L, Berkutzki T, Mattson MP, Gomez-Pinilla F, Friedman N, Schwartz M. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci U S A. 2013;110:2264–2269. doi: 10.1073/pnas.1211270110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bomstein Y, Marder JB, Vitner K, Smirnov I, Lisaey G, Butovsky O, Fulga V, Yoles E. Features of skin-coincubated macrophages that promote recovery from spinal cord injury. J Neuroimmunol. 2003;142:10–16. doi: 10.1016/S0165-5728(03)00260-1. [DOI] [PubMed] [Google Scholar]
- Brühl H, Cihak J, Schneider MA, Plachý J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, Schlöndorff D, Mack M. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172:890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Núñez A, Torres-Aleman I. Brain repair and neuroprotection by serum insulin-like growth factor I. Mol Neurobiol. 2003;27:153–162. doi: 10.1385/MN:27:2:153. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisson S, Darrasse-Jèze G, Litvinova E, Septier F, Klatzmann D, Liblau R, Salomon BL. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med. 2003;198:737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Ford ML, Evavold BD. An MHC anchor-substituted analog of myelin oligodendrocyte glycoprotein 35–55 induces IFN-gamma and autoantibodies in the absence of experimental autoimmune encephalomyelitis and optic neuritis. Eur J Immunol. 2004;34:388–397. doi: 10.1002/eji.200324502. [DOI] [PubMed] [Google Scholar]
- Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147:867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, Neeman M, Cohen IR, Schwartz M. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000;20:6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauben E, Agranov E, Gothilf A, Nevo U, Cohen A, Smirnov I, Steinman L, Schwartz M. Posttraumatic therapeutic vaccination with modified myelin self-antigen prevents complete paralysis while avoiding autoimmune disease. J Clin Invest. 2001;108:591–599. doi: 10.1172/JCI200112837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Nakashima K, Gage FH. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol. 2004;164:111–122. doi: 10.1083/jcb.200308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, Niesner U, de la Rosa M, Schmidt CA, Bräuer R, Buer J, Scheffold A, Hamann A. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Jin X, Ueno M, Tanabe S, Kubo T, Serada S, Naka T, Yamashita T. Adoptive transfer of Th1-conditioned lymphocytes promotes axonal remodeling and functional recovery after spinal cord injury. Cell Death Dis. 2012;3:e363. doi: 10.1038/cddis.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/S1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Yoles E, Ben-Nur A, Schwartz M. Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. J Neuroimmunol. 2002a;130:78–85. doi: 10.1016/S0165-5728(02)00219-9. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A. 2002b;99:15620–15625. doi: 10.1073/pnas.232565399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, Schwartz M. IFN-gamma-dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain. 2013;136:3427–3440. doi: 10.1093/brain/awt259. [DOI] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13:777–789. doi: 10.1038/nri3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London A, Itskovich E, Benhar I, Kalchenko V, Mack M, Jung S, Schwartz M. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med. 2011;208:23–39. doi: 10.1084/jem.20101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachý J, Brühl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlöndorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- Müller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, Dreykluft A, Lu B, Gerard C, King NJ, Campbell IL. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. 2007;179:2774–2786. doi: 10.4049/jimmunol.179.5.2774. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Segev Y, Jacob-Hirsch J, Amariglio N, Rechavi G, Schwartz M. Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med. 2008;5:e171. doi: 10.1371/journal.pmed.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. 2011;480:538–542. doi: 10.1038/nature10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J Neurosci. 1999;19:RC7. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x. [DOI] [PubMed] [Google Scholar]
- Shechter R, Schwartz M. CNS sterile injury: just another wound healing? Trends Mol Med. 2013;19:135–143. doi: 10.1016/j.molmed.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Raposo C, London A, Sagi I, Schwartz M. The glial scar-monocyte interplay: a pivotal resolution phase in spinal cord repair. PLoS One. 2011;6:e27969. doi: 10.1371/journal.pone.0027969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GL, Andersson J, Shevach EM. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. J Immunol. 2007;178:6901–6911. doi: 10.4049/jimmunol.178.11.6901. [DOI] [PubMed] [Google Scholar]
- Suffner J, Hochweller K, Kühnle MC, Li X, Kroczek RA, Garbi N, Hämmerling GJ. Dendritic cells support homeostatic expansion of Foxp3+ regulatory T cells in Foxp3.LuciDTR mice. J Immunol. 2010;184:1810–1820. doi: 10.4049/jimmunol.0902420. [DOI] [PubMed] [Google Scholar]
- Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A. 2003;100:15818–15823. doi: 10.1073/pnas.2636938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477–494. doi: 10.1016/S1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, Yang Y, Floess S, Huehn J, Oh S, Li MO, Niec RE, Rudensky AY, Dustin ML, Littman DR, Lafaille JJ. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209:1723–1742. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, Von Schack D, Bluestone JA. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209:1713–1722. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Kishi A, Osaki M, Morikawa H, Prieto-Martin P, Wing K, Saito T, Sakaguchi S. Construction of self-recognizing regulatory T cells from conventional T cells by controlling CTLA-4 and IL-2 expression. Proc Natl Acad Sci U S A. 2013;110:E2116–E2125. doi: 10.1073/pnas.1307185110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A, Kuchroo V, Cohen IR, Weiner H, Schwartz M. Protective autoimmunity is a physiological response to CNS trauma. J Neurosci. 2001;21:3740–3748. doi: 10.1523/JNEUROSCI.21-11-03740.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]