

Abstract
Tau is a microtubule-associated protein well known for its stabilization of microtubules in axons. Recently, it has emerged that tau participates in synaptic function as part of the molecular pathway leading to amyloid-beta (Aβ)-driven synaptotoxicity in the context of Alzheimer's disease. Here, we report the implication of tau in the profound functional synaptic modification associated with synaptic plasticity. By exposing murine cultured cortical neurons to a pharmacological synaptic activation, we induced translocation of endogenous tau from the dendritic to the postsynaptic compartment. We observed similar tau translocation to the postsynaptic fraction in acute hippocampal slices subjected to long-term potentiation. When we performed live confocal microscopy on cortical neurons transfected with human-tau-GFP, we visualized an activity-dependent accumulation of tau in the postsynaptic density. Coprecipitation using phalloidin revealed that tau interacts with the most predominant cytoskeletal component present, filamentous actin. Finally, when we exposed cortical cultures to 100 nm human synthetic Aβ oligomers (Aβo's) for 15 min, we induced mislocalization of tau into the spines under resting conditions and abrogated subsequent activity-dependent synaptic tau translocation. These changes in synaptic tau dynamics may rely on a difference between physiological and pathological phosphorylation of tau. Together, these results suggest that intense synaptic activity drives tau to the postsynaptic density of excitatory synapses and that Aβo-driven tau translocation to the spine deserves further investigation as a key event toward synaptotoxicity in neurodegenerative diseases.
Keywords: amyloid-beta oligomers, excitatory synapses, LTP, phosphorylation, synaptic plasticity, tau
Introduction
Fibrillar deposits of phosphorylated tau are a characteristic feature of several neurodegenerative diseases, including Alzheimer's disease (AD). Increasing evidence suggests that the presence of neurofibrillary tangles does not cause direct neuronal dysfunction, but rather alterations to physiological tau functions may be key to disease processes (for review, see Jaworski et al., 2010; Morris et al., 2011). Tau is a highly soluble protein with a predominant expression in neurons (Trojanowski et al., 1989). A large proportion of tau is in the axon, where it interacts with microtubules (MTs) through its C-terminal microtubule-binding domain to promote MT polymerization and stabilization (for review, see Götz et al., 2013). Accordingly, tau exerts a critical role in the regulation of microtubule-dependent axonal transport. More recently, numerous studies have revealed novel functions for tau, including in neuronal signaling pathways, DNA protection, and synaptic regulation. These studies were based on the identification of tau interaction with novel molecular partners such as DNA or RNA (Loomis et al., 1990; Sultan et al., 2011), neuronal membranes (Pooler et al., 2012), and synaptic located proteins such as Fyn or PSD-95 (Ittner et al., 2010; Mondragón-Rodríguez et al., 2012). Subsequently, the association of tau with synaptic proteins has raised the intriguing possibility that tau exerts physiological roles in the synapse and that altered tau function may be involved in the synaptic dysfunction that precedes synaptic loss common across neurodegenerative tauopathies.
Although postsynaptic localization of tau was initially identified under pathological conditions (Hoover et al., 2010), there are several lines of evidence suggesting the involvement of tau in physiological synaptic functions. In 2010, Ittner et al. showed that the presence of tau in the postsynaptic compartment mediates the targeting of the src family kinase fyn to glutamatergic NMDA receptors. More recently, Chen et al. (2012) suggested that tau participates in spine edification by demonstrating that reduction of tau expression results in synapse loss. Together, these data are consistent with a functional role for tau in the physiological activation of excitatory synapses.
Dendritic spines are small protuberances observed on the dendrites that are the physical support of postsynaptic sites of excitatory synapses. They are enriched in neurotransmitter receptors, specific scaffolding proteins, and display an abundant actin cytoskeleton responsible for spine morphology (for review, see Hotulainen and Hoogenraad, 2010). Under specific activation patterns, spines undergo profound and long-lasting modifications of morphology and synaptic strength that together underlie synaptic plasticity. Synaptic dysfunction is the best correlate of the cognitive decline that characterizes AD, and tau has been implicated in this synaptotoxic effect triggered by amyloid-beta (Aβ) oligomer exposure. With the emergent idea that tau could penetrate into the synaptic compartment, we investigated the mechanisms of tau recruitment to the spines under synaptic activation and Aβo exposure. To monitor tau localization, we used subcellular fractionation of both ex vivo (i.e., acute hippocampal slices) and in vitro (i.e., cultured cortical neurons) subjected to an electrophysiological or chemical long-term potentiation (LTP). In parallel, we monitored GFP-tagged tau coupled with dynamic fluorescence imaging.
Materials and Methods
Primary cultures of cortical neurons.
Mouse cortical neurons were cultured from 14- to 15-d-old OF1 embryos as described previously (Léveillé et al., 2008). Cerebral cortices were dissected, dissociated, and cultured in Dulbelcco's modified Eagle's medium containing 5% fetal bovine serum, 5% horse serum, and 2 mm glutamine (all from Sigma) on 24-well plates (Falcon; Becton Dickinson) for biochemical experiments. Neurons were seeded on 35 mm glass-bottom dishes (MatTek) at a final concentration of two cortical hemispheres per dish for confocal experiments. All plates, dishes, and coverslips were coated with 0.1 mg/ml poly-d-lysine and 0.02 mg/ml laminin (Sigma). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2/95% air. After 3–4 d in vitro (DIV), cytosine arabinoside (AraC, 10 μm; Sigma) was added to inhibit proliferation of non-neuronal cells in cultures used for biochemistry experiments; 98% of the cells were considered as neuronal. The day before the experiments, cells were washed in DMEM. Treatments were performed on neuronal cultures at 14–15 DIV.
Neuronal transfection.
Transfections were performed on cortical neuron cultures after 12 DIV with calcium phosphate precipitation. Growth medium (DMEM and sera) was removed and kept until the last step of transfection. Cells were washed for 1–1.5 h in DMKY buffer containing the following (in mm): 1 kynurenic acid, 0.9 NaOH, 0.5 HEPES, 10 MgCl2, plus phenol red 0.05%, pH 7.4. Then, 3.5 μg of the plasmids pEGFP-Tau 2N4R (full-length human nervous system tau) and LifeAct-RFP, a peptide that specifically interacts with filamentous actin (Riedl et al., 2008), were mixed with CaCl2 (120 mm) in HBS containing the following (in mm): 25 HEPES, 140 NaCl, and 0.750 Na2HPO4, pH 7.06) and left for 20 min to precipitate the DNA. Plasmids were then applied to cells for 30 min. Transfection medium was replaced with conditioned growth medium and cultures were returned to the incubator until use at DIV 14–15.
Mutagenesis.
Point mutations were performed on pEGFP-Tau 2N4R (full-length human nervous system tau) using the QuikChange kit from Agilent Technologies.
Fluorescence recovery after photobleaching experiments.
Fluorescence recovery after photobleaching (FRAP) was performed on cultured neurons 48 h after transfection. Images were acquired with an inverted Nikon Eclipse Ti C2 confocal microscope with a Nikon 60× water objective with a 1.33 numerical aperture. EGFP-tau was bleached at 405 nm and the fluorescence recovery was measured for 80 s (at 1 s/frame). Fluorescent signal analysis was performed with the Nikon software Nis.
Time-lapse imaging of transfected neurons.
Transfected neurons were placed in HBBSS solution containing the following (in mm): 0.110 NaCl, 0.005 KCl, 2 CaCl2, 0.0008 MgSO4, NaH2PO4 1 NaH2PO4, 12 HEPES, 5 d-glucose, 25 NaHCO3, and 0.01 glycine (all from Sigma) 1.5–2 h before experiments. Neurons were imaged with a Zeiss LSM 710 confocal imaging system using a 63× oil-immersion objective and ZEN 2010 software. Z-stacks were 0.61 μm per step (1 pixel = 0.134 μm) with a 1 AU pinhole at 84 μm. Images were collected immediately before and 15 and 30 min after treatment. Evolution of the fluorescence within spine heads in maximum projection was analyzed with MetaMorph software (Molecular Devices). A total of 20–40 spines per neurons were followed throughout treatment for each experiment.
Slice preparation.
Hippocampal slices were prepared from 2- to 4-month-old male OF1 mice. Mice were cervically dislocated and immediately decapitated. Their hippocampi were dissected out and 400-μm-thick transverse slices were cut in ice-cold cutting solution containing the following (in mm): 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, 234 sucrose, and 11 glucose, saturated with 95% O2 and 5% CO2 with a Leica VT1200 blade microtome. After the dissection, slices were kept in oxygenated artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 11 glucose 11 at a temperature of 27 ± 1°C for at least 1 h.
Electrophysiology recordings.
Slices were visualized in a chamber on an upright microscope with transmitted illumination and continuously perfused at 1 ml/min with the same oxygenated ACSF at 27 ± 1°C. Stimulating electrodes (bipolar microelectrodes) were placed in the stratum radiatum to stimulate the Schaffer collaterals pathway. Field EPSPs (fEPSPs) were recorded in the stratum radiatum using a recording glass pipette filled with ACSF and amplified with an Axon Axopatch 200B amplifier (Molecular Devices). Data were digitized through an Axon Digidata 1440A and acquired with pCLAMP 10 software (Molecular Devices). The initial slope of the fEPSPs was measured to avoid population spike contamination. For LTP experiments, test stimuli (0.2 ms pulse width) were delivered once every 15 s and the stimulus intensity was set to give baseline fEPSP slopes that were 50% of maximal evoked slopes.
Slices that showed maximal fEPSP sizes <1 mV were rejected. LTP was induced by applying 4 trains of 100 stimuli at 100 Hz with an intertrain interval of 5 min. CA1 slices were frozen in liquid nitrogen at the end of experiment for subsequent biochemical analyses.
Purification of tau.
His-tagged human Tau (1N4R Tau-412 isoform) plasmid was kindly provided by Dr. N. Sergeant (Lille, France). Recombinant Tau was purified from E. coli BL21 (Invitrogen) on Talon metal affinity resin (Clontech) according to the manufacturer's instructions. The protein was further processed by a size exclusion chromatography in BRB80 buffer containing the following (in mm): 80 Pipes, 1 EGTA, and 1 MgCl2, pH 6.8, concentrated, and ultracentrifuged at 230,000 × g for 10 min. The protein was frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined by a Bradford assay (Sigma) using BSA as a standard.
In vitro cosedimentation assays.
Actin cosedimentation assays were performed with purified human platelet actin (Cytoskeleton) resuspended in buffer G containing the following (in mm): 2 Tris, pH 8.0, 0.2 ATP, 0.5 DTT, and 0.1 CaCl2. Then, 5 μm G-actin was polymerized for 1 h at room temperature in the absence or in the presence of 1 μm Tau in buffer G containing the following (in mm): 50 KCl, 1 MgCl2,1 EGTA, and 10 imidazole, pH 7.5. The 20 μl reaction mixtures were then centrifuged either for 15 min at 100,000 × g or for 10 min at 15,000 × g. At low-speed centrifugation, filamentous actin remains in the supernatant and only filamentous actin (F-actin) bundles are pelleted, whereas the high-speed centrifugation precipitates all F-actin forms. Supernatants and pellets fractions were recovered and subjected to 10% SDS-PAGE gel, followed by Coomassie blue staining.
Subcellular fractionation.
Cultured neurons or hippocampal CA1 slices were homogenized in cold buffer containing 0.32 m sucrose and 10 mm HEPES, pH 7.4. Samples were maintained at 4°C during all steps of the experiment. Homogenates were cleared at 1000 × g for 10 min to remove nuclei and large debris. The resulting supernatants were concentrated at 12,000 × g for 20 min to obtain a crude membrane fraction, which was then resuspended twice (4 mm HEPES, 1 mm EDTA, pH 7.4, 20 min at 12,000 × g). Then, the pellet was incubated (20 mm HEPES, 100 mm NaCl, 0.5% Triton X-100, pH 7.2) for 1 h at 4°C with mild agitation and centrifuged at 12,000 × g for 20 min to pellet the synaptosomal membrane fraction. The supernatant was collected as the non-postsynaptic density membrane fraction (non-PSD) or Triton-soluble fraction. The pellet was then solubilized (20 mm HEPES, 0.15 mm NaCl, 1% Triton X-100, 1% deoxycholic acid, 1% SDS, pH 7.5) for 1 h at 4°C and centrifuged at 10,000 × g for 15 min. The supernatant contained the PSD or Triton-insoluble fraction. The integrity of non-PSD was verified by immunoblotting for synaptophysin, which was enriched in the non-PSD fraction, and the PSD fraction was confirmed by the immunoblotting of PSD-95 enriched in this compartment.
Aβ oligomerization and fibrillogenesis.
Recombinant Aβ1–42 peptide (Bachem) was resuspended in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to 1 mm until complete resuspension as described previously (Stine et al., 2003). Aβo's were prepared by diluting Aβ to 1 mm in DMSO and then to 100 μm in ice-cold HEPES and bicarbonate-buffered saline solution (HBBSS) with immediate vortexing and bath sonication, and then incubated at 4°C for 24 h with mild agitation.
Synaptic activation and actin cytoskeleton disruption.
Cultures were placed in DMEM 24 h before the experiment. Synaptic activation was induced at 37°C with 50 μm bicuculline (Bic), a GABAa receptor antagonist, and 2.5 mm 4-amino-pyridine, a weak potassium channel blocker (all from Tocris Bioscience) for 15 min (Léveillé et al., 2008). Actin stabilization was induced by jasplakinolide (Santa Cruz Biotechnology) application at 1 μm (Lazaro-Dieguez et al., 2008).
Immunoprecipitation.
After treatment, primary neurons were rapidly transferred on ice to remove the media and immediately extracted with fresh buffer (100 mm Na2HPO4, 100 mm NaH2PO4, pH 7.2, 2 mm ATP, 2 mm MgCl2, phosphatase, and protease inhibitor mixture; Sigma). Equal amounts of protein were incubated with biotin-XX-phalloidin (Molecular Probes) 0.45 unit/condition with gentle rocking for 1 h at room temperature. Thereafter, Dynabeads M-280 Streptavidin (Invitrogen) were added to the complex formed by filamentous actin-biotin-XX phalloidin and incubated for 30 min at room temperature using gentle rotation. The actin-coated beads were separated with a magnet for 2–3 min, washed several times in the buffer, and boiled in 1× loading buffer containing 124 mm Tris, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, and 1% bromophenol blue for 5 min. Samples were stored at −20°C until used.
Immunoblotting.
Samples in loading buffer were boiled for 5 min and equal amounts of proteins (5–10 μg) were resolved on a 4–12% or 4–20% gradient Bis-Tris polyacrylamide precast gels (Bio-Rad) in denaturing conditions. Proteins were transferred to a polyvinylidene difluoride membrane (Millipore) for 2 h at 4°C. Membranes were blocked with Tris-buffered saline (10 mm Tris, 150 mm NaCl, pH 7.4) containing 0.01% Tween 20 and 5% nonfat dry milk for 1 h at room temperature. Membranes were then incubated overnight at 4°C with the primary antibodies tau (1:50,000 dilution, A0024; Dako); tau phospho serine 404 and tau phospho tyrosine 205 (1:1000 dilution, PA1–14422, PA1–14426, PA1–14415; Thermo Scientific); actin (1:2000, A2066, Sigma); GluA1, GluN2B, GluN2A, and synaptophysin (1:1000 dilution, A1504, MAB5778, 04–901, MAB368; Millipore); Fyn (1:500 dilution, 15-sc-434; Santa Cruz Biotechnology); and PSD-95 (1:1000 dilution, clone 6G6–1C9 ab2723; Abcam). Membranes were incubated with the HRP-conjugated secondary antibodies rabbit (1:40,000) and mouse (1:5000) (both from Sigma) for 45 min at room temperature. Specific proteins were visualized with an enhanced chemiluminescence ECL Detection System (Bio-Rad). Chemiluminescence detections were performed with the Bio-Rad Chemidoc system and analyzed with the software Imagelab (Bio-Rad).
Statistics.
Results are expressed as the mean ± SEM from independent biological samples. Statistical analyses were performed with GraphPad Prism 6.0 software by two-way ANOVA followed by Bonferroni's test for confocal analysis, one-way ANOVA for phalloidin precipitation analysis, and Student's t test for Western blot analysis.
Results
Axonal and dendritic localization of tau in primary cortical neurons under resting conditions
To characterize the localization of tau in cultured neurons under resting conditions (14 DIV), we transfected full-length tau 2N4R-EGFP and LifeAct-RFP, the latter of which encodes a small, 17 aa peptide that binds specifically to filamentous actin (Riedl et al., 2008). In neurons, filamentous actin is known to be enriched to the synaptic compartment (Star et al., 2002). By using LifeAct-RFP instead of actin-GFP, we maintained actin stoichiometry. We first verified the localization of LifeAct-RFP in the synaptic compartment by cotransfecting a synaptic scaffold protein, PSD-95-YFP. The merged image showed that filamentous actin and PSD-95 are both localized in dendritic spines (Fig. 1A). We then cotransfected LifeAct-RFP with EGFP-tau in cortical neurons and observed that EGFP-tau fluorescence is restricted to axonal and dendritic shafts in the resting condition (Fig. 1B). Only a minimal number of spines displayed traces of EGFP-tau (Fig. 1B).
Figure 1.
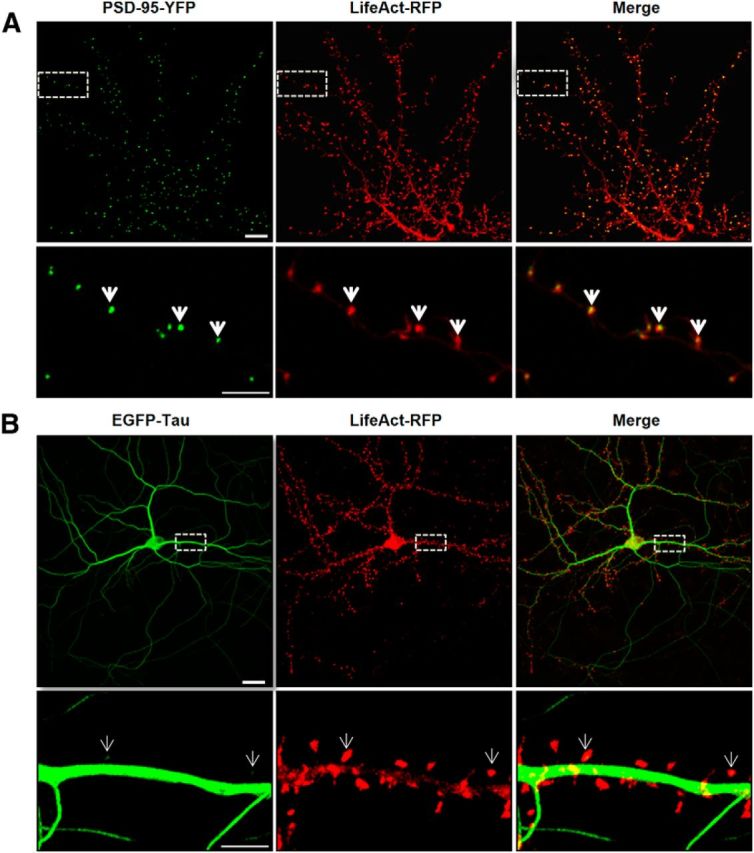
LifeAct colocalizes with PSD-95-YFP in dendritic spines and displays a limited colocalization with EGFP-Tau. A, Confocal imaging of dendritic spines of primary murine cortical neuronal cultures (14 DIV) cotransfected with LifeAct-RFP and PSD-95-YFP 48 h before imaging. Merge images show the colocalization (yellow, white arrows) of PSD-95 and filamentous actin in dendritic spines. B, Dendritic spines of cortical neuron culture cotransfected with LifeAct-RFP and EGFP-Tau. Merged images show only limited colocalization of EGFP-Tau in the indicated dendritic spines (white arrows). Scale bars: top, 10 μm; bottom, 5 μm.
Synaptic activation induces translocation of tau to the synaptic compartment
To determine how and under which circumstances tau enters into the postsynaptic compartment, we monitored EGFP-tau fluorescence under various conditions. First, we induced a long-lasting synaptic activation by exposing cultured cortical neurons to Bic (50 μm), a GABAergic antagonist, in the presence of 4-aminopyridine (4-AP, 2.5 mm), a weak potassium channel blocker that prevents neuronal repolarization (Léveillé et al., 2008), for 30 min (Fig. 2A). In control conditions, EGFP-tau showed a weak but detectable fluorescence in >60% of the spine, but the calculation mode used to quantify the imaging data (ΔF/F0) brings our quantification to zero to highlight changes from base line fluorescence. As early as 15 min after the beginning of the treatment, we measured an increase in EGFP-tau fluorescence in dendritic spines from 0 ± 0.04 to 0.52 ± 0.06, which stabilized after 30 min to 0.64 ± 0.05 (***p < 0.0001, 2-way ANOVA; Bic/4-AP: n = 5, N = 162 spines; control n = 3, N = 40 spines; Fig. 2B) and colocalized with LifeAct-RFP fluorescence. In contrast, the evolution of EGFP-tau fluorescence during a 30 min vehicle application showed no detectable change (Fig. 2B). Recognizing that plasmid-driven expression of EGFP-tau may modify tau cellular distribution, we studied the localization of endogenous tau in neurons subjected to synaptic activation. For this study, we use a primary cortical cultures system at a density that permits biochemical analysis but also renders immunohistochemistry impractical because it is very difficult to distinguish individual immunostained spines. Western blot analysis of tau from total neuronal lysate showed no change after 15 and 30 min Bic/4-AP application (Fig. 2C). Then, we isolated the non-PSD fraction (i.e., Triton-soluble fraction) from the PSD-enriched fraction (i.e., Triton-insoluble fraction) and audited the presence of synaptophysin and PSD-95 in each to verify the fractionation protocol. As expected, synaptophysin appeared only in the non-PSD fraction and PSD-95 only in the PSD-enriched fraction. Next, we performed Western blot analysis of several synaptic proteins to assess the effect of our stimulation protocol (Fig. 2D). No change in tau expression was detected in the non-PSD fraction. In contrast, tau expression in the PSD fraction showed a small amount in resting conditions, which increased 1.5-fold after 15 min of synaptic activation (***p < 0.0001, 2-tailed Student's t test; control 20.62 ± 1.235 vs Bic/4-AP 30.92 ± 1.799, N = 13 independent cultures; Fig. 2E). On the same PSD-positive fractions, we analyzed the variation of synaptic markers that have been identified as potential tau-interacting partners (Lee et al., 1998; Ittner et al., 2010; Mondragón-Rodríguez et al., 2012). After synaptic activation, we detected increases in PSD-95 (*p = 0.0249, 2-tailed Student's t test; control 19.10 ± 2.557 vs Bic/4-AP 26.9 ± 1.894, N = 9; Fig. 2E) and the src family tyrosine kinase Fyn (**p = 0.0023, 2-tailed Student's t test; control 19.42 ± 1.337 vs Bic/4-AP 26.54 ± 1.130, N = 6; Fig. 2E). The AMPA receptor subunit GluA1, which is recruited to the membrane during synaptic plasticity (Malinow and Malenka, 2002), was also increased in the PSD fraction (**p = 0.0026, 2-tailed Student's t test; control 1884 ± 1930 vs Bic/4-AP 2863 ± 1.95, N = 9; Fig. 2E). The actin cytoskeleton is highly reorganized during synaptic activation, as described previously (Star et al., 2002). Here, we saw an increase in actin content restricted to the PSD fraction (*p = 0.0101, 2-tailed Student's t test; control 22.18 ± 1.913 vs Bic/4-AP 29.62 ± 1.517, N = 7 independent cultures; Fig. 2E). Therefore, during a long-lasting synaptic activation, we observed an increase in tau, fyn, actin, GluA1, and PSD-95 content in the PSD-positive fraction, which is consistent with the characteristic features of synaptic plasticity (Ehlers, 2003). These results suggest that tau translocates from the dendritic shaft to the synapse during activation and probably takes part in the activity-driven synaptic reorganization that underlies synaptic plasticity. To generalize these findings, we chose to confirm these results in an ex vivo model of synaptic plasticity: LTP in hippocampal slices (Fig. 3A). We performed synaptosomal fractionation of microdissected CA1 regions taken from potentiated hippocampal slices. We observed a similar LTP-induced increase in tau content within the PSD-enriched fraction from CA1 synaptosomes (29.86 ±4.86 to 70.15 ± 4.86, **p = 0.0011; Fig. 3B). As expected, actin and GluA1 were also increased, strengthening the idea that tau is involved in synaptic reorganization processes necessary for synaptic plasticity (34.31 ± 2.518 to 64.92 ±2.518, ***p = 0.0002; 34.31 ± 8.15 to 65.70±8.15, *p = 0.0346; Fig. 3C).
Figure 2.

Synaptic activation induced by Bic/4-AP triggers translocation of tau into dendritic spines of primary cortical neuron cultures. A, Confocal imaging of cortical neurons (14 DIV) cotransfected with EGFP-Tau and LifeAct-RFP. Left, Spatial repartition of EGF-Tau (green) and LifeAct-RFP (red) in nonactivated neuron. The merged image displays only minor colocalization (yellow). Scale bar, 5 μm. Right, Higher magnification of EGFP-Tau, LifeAct in dendritic spines (designated box in the left) of a neuron after synaptic activation triggered by 15 and 30 min of Bic/4AP exposure. The merged image shows the activity driven colocalization of tau/LifeAct (yellow) in the activated dendritic spine after 15 and 30 min Bic/4-AP exposure. Scale bar, 1 μm. B, Quantification of fluorescence intensity mediated by EGFP-Tau in the head of spines during synaptic activation (Bic/4-AP treatment). The graph represents the evolution of ΔF/F0 generated by EGFP-Tau in neurons subjected to synaptic activation or not. Statistical analysis was performed by two-way ANOVA followed by Bonferroni's posttest (mean ± SEM, ***p < 0.0001, control n = 40 spines on 3 independent cultures; Bic/4-AP n = 162 spines on 5 independent cultures). C, Representative Western blot of endogenous tau expression in a whole lysate extract of cortical neurons (14 DIV) exposed to Bic/4-AP for 15 and 30 min. Quantification of tau expression normalized to actin levels does not display any change. D, Representative Western blots of protein expression from primary cortical cultures after fractionation. Presynaptic protein (synaptophysin), postsynaptic proteins (PSD-95, GluA1, GluN2A, GluN2B, Fyn), cytoskeletal protein (actin), and tau in non-PSD fraction (Triton-soluble fraction) and PSD-fraction (Triton-insoluble fraction) on primary cortical neurons treated 15 min with Bic/4-AP. E, Quantitative analysis of PSD-95, GluA1, GluN2B, GluA1, Fyn, actin, and tau in the PSD fraction under control and Bic/4-AP (15 min) treated conditions is shown in graphs (mean ± SEM, 2-tailed Student's t test,***p < 0.001, **p = 0.0026, *p = 0.0101, N ≥ 7 independent cultures).
Figure 3.

LTP in hippocampal slices triggers translocalization of tau to CA1 dendritic spines. A, Representative curve of fEPSP slope in percentage of baseline illustrating the LTP induction in CA1 region of hippocampus slices. Sample EPSPs are shown before (1) and after (2) LTP induction. B, Representative Western blots of synaptic protein expression from the CA1 region of hippocampal slices subjected to LTP protocol. C, Quantifications of Western blot from GluA1, actin, and tau in control and LTP conditions (mean ± SEM, 2-tailed Student's t test, *p = 0.0346, **p = 0.0011,***p = 0.0002, N = 4 independent slices).
Tau interacts with filamentous actin in the synapse
Although it was initially described as a microtubule-associated protein (MAP), tau has been shown to interact with other molecular partners in the PSD fraction, such as fyn or PSD-95 (Ittner et al., 2010; Mondragón-Rodríguez et al., 2012). Among several synaptically located proteins, actin is of particular interest for its potential capacity to interact with human tau (Fulga et al., 2006; Yu and Rasenick, 2006; He et al., 2009). We investigated whether tau interacts with actin, the most predominant cytoskeleton element of the synapse. Purified G-actin was polymerized with the 1N4R tau isoform and centrifuged at 100,000 × g (Fig. 4A) or 15,000 × g (Fig. 4B). Although tau alone were observed only in the supernatant in both experimental conditions, ruling out a nonspecific coaggregation, we found that tau coprecipitated with the pellet obtained from high- and low-speed centrifugation, illustrating its direct interaction with both F-actin (Fig. 4A) and actin bundles (Fig. 4B). It is worth noting that the filamentous nature of actin in the presence of tau was confirmed by electron cryomicroscopy (data not shown). We also performed transfection with 1N4R-GFP and we observed similar synaptic-activity-dependent translocation as observed with the full-length tau (Fig 4C). Similarly, a phalloidin precipitation of neuronal lysate that selectively collected the neuronal F-actin and its binding partners revealed the presence of tau by Western blotting. This result illustrates that tau interacts with F-actin in neurons (Fig. 5A). After synaptic activation, we observed an increase in the amount of F-actin harvested (***p = 0.0003, 2-tailed Student's t test; control 17.67 ± 2.08; Bic/4-AP 31.76 ± 1.882, N = 7 independent cultures; Fig. 5B) and a concomitant increase in tau (***p = 0.0002, control 16.91 ± 1.843; Bic/4-AP 31.49 ± 2.028, N = 7 independent cultures). These results show that the amount of tau collected is proportional to neuronal F-actin content, suggesting a close link between F-actin and tau. Next, we studied the impact of pharmacological manipulations of actin organization on synaptic EGFP-tau fluorescence localization. For this, we treated our primary cortical neurons with jasplakinolide (1 μm), a compound that promotes actin polymerization (Lazaro-Dieguez et al., 2008), or with a latrunculin A (at 500 nm), a compound that depolymerizes F-actin into soluble globular actin (G-actin; Coué et al., 1987; Fig. 5C). After jasplakinolide application, we observed a large increase in synaptic EGFP-tau fluorescence (***p < 0.0001, 2-way ANOVA; ΔF/F0, 0.64±0.05 after 15 min, 0.59±0.06 after 30 min; control n =40, N = 3; jasplakinolide, n = 106, N = 4; Fig. 4D). Latrunculin treatment produced rapid actin depolymerization and the corresponding disappearance of LifeAct-RFP fluorescence in every spine studied; no synaptic EGFP-tau fluorescence was observed (data not shown).
Figure 4.

Tau interacts with actin in purified system. Cosedimentation of actin filaments with tau. Actin was polymerized at 5 μm for 1 h at room temperature in the absence or in the presence of 1 μm tau. The reaction mixtures were then centrifuged at 100,000 × g to sediment actin filaments (A) or at 15,000 × g to precipitate only actin bundles (B). Supernatants (SN) and pellets (P) were applied to 10% SDS-PAGE gel and proteins were stained with Coomassie blue. C, Confocal imaging of cortical neurons (14 DIV) cotransfected with EGFP-Tau1N4R and LifeAct-RFP. Left, Spatial repartition of EGFP-Tau1N4R (green) and LifeAct-RFP (red) in untreated neurons. Scale bar, 5 μm. Right, Higher magnification of EGFP-Tau1N4R and LifeAct-RFP in a dendritic spine neuron after Bic/4-AP treatment (15 and 30 min, designated box in the left) by 15 and 30 min of Bic/4-AP treatment. The merged image shows colocalization of F-actin and tau. Scale bar, 1 μm. D, Quantification of fluorescence intensity mediated by EGFP-Tau1N4R and Tau2N4R in the head of spines during synaptic activation (Bic/4-AP treatment). The graph represents the evolution of ΔF/F0 generated by EGFP-Tau2N4R and EGFP-Tau1N4R in neurons subjected to synaptic activation or not. Statistical analysis was performed by two-way ANOVA followed by Bonferroni's posttest (mean ± SEM, ***p < 0.0001 vs control, ###p < 0.0001 vs Tau-2N4R; control n = 40 spines on 3 independent cultures; Bic/4-AP Tau2N4R n = 162 spines on 5 independent cultures, Bic/4AP Tau 1N4R n = 54 spines on 2 independent cultures).
Figure 5.

Tau interacts with F-actin in the synaptic compartment. A, Representative Western blot showing F-actin-associated tau in cultured cortical neurons treated for 15 min with Bic/4-AP. Cell lysate was collected after treatment and F-actin precipitated with phalloidin as described in the Materials and Methods. B, Quantifications of actin and tau in control and Bic/4-AP condition (mean ± SEM, 2-tailed Student's t test, ***p < 0.001, N = 7 independent cultures). C, Confocal imaging of cortical neurons (14 DIV) cotransfected with EGFP-Tau and LifeAct-RFP treated with jasplakinolide (1 μm), a specific agent that stabilizes F-actin. Left, Cropped view of dendritic branch of transfected neuron under control condition. Scale bar, 5 μm. Right, Higher magnification of EGFP-Tau and LifeAct-RFP in dendritic spines (cropped view of dotted box in the left) of neuron before and 15 and 30 min after jasplakinolide (1 μm) treatment. The merged image shows colocalization of EGFP-Tau and LifeAct-RFP (white arrows). Scale bar, 1 μm. D, Quantification of the fluorescence intensity variation (ΔF/F0) of EGFP-Tau in the head of spines during actin network stabilization (jasplakinolide treatment). Statistical analysis was performed by two-way ANOVA followed by Bonferroni's posttest (mean ± SEM, ***p < 0.0001, control n = 40, N = 3 independent cultures, jasplakinolide n = 106, N = 4 independent cultures). E, Representative Western blots of protein expression from PSD-enriched fraction primary cortical cultures treated or not with jasplakinolide (1 μm, synaptophysin [SYN], PSD-95, GluA1, Fyn actin, and tau in PSD-fraction (Triton-insoluble fraction)). F, Quantitative analysis of Western blots of PSD-95, GluA1, actin, and tau in PSD fraction (mean ± SEM, 2-tailed Student's t test, **p < 0.01, *p < 0.05, N = 4 independent cultures).
We analyzed actin and tau in the PSD-enriched fraction from primary cortical neurons treated with jasplakinolide (Fig. 5E). We observed that increased neuronal F-actin content promotes concurrent tau enrichment (*p = 0.0150, 2-tailed Student's t test; control 17.49 ± 0.7755 vs jasplakinolide 27.02 ± 2719, N = 4 independent cultures; Fig. 5F). GLUA1, the membrane trafficking of which is known to be actin dependent, was increased (*p = 0.0279, 2-tailed Student's t test; control 16.91 ± 1015 vs jasplakinolide 31.00 ± 4.778, N = 4 independent cultures). The amount of Fyn in the PSD was decreased (*p = 0.0265, 2-tailed Student's t test; control 27.25 ±5.003 vs jasplakinolide 11.71 ± 1.786, N = 4 independent cultures). Together, these results suggest that tau translocation to the synapse depends on the F-actin stabilization that promotes their interaction.
Aβ oligomers disrupt the link between synaptic activation and tau recruitment to the synapse
In the context of AD, recent studies have shown that synaptotoxicity driven by Aβo exposure relies on the presence of tau (Rapoport et al., 2002; Roberson et al., 2007; Ittner et al., 2010). To study this pathological regulation of tau, we exposed EGFP-tau- and LifeAct-RFP-expressing cultures to 100 nm synthetic human Aβo's (1–42; Fig. 6A). We observed a delocalization of tau in the synapse within 15 min after Aβo treatment (***p < 0.001; control n = 40, N = 3, Aβo 100 nm for 15 min 0.52± 0.04, for 30 min 0.71 ± 0.05, n = 79, N = 5 independent cultures; Fig. 6B). We confirmed these results with endogenous tau by Western blot analysis of PSD-enriched fractions (Fig. 6C). Aβo exposure induced a translocation of tau into the PSD fraction (***p = 0.0002, 2-tailed Student's t test; control 20.12 ± 1228 vs Aβo 29.74 ± 1.748, N = 12 independent culture). There was also an increase of PSD-95 (***p = 0.0006, 2-tailed Student's t test; control 19.10 ± 2.557 vs Aβo 33.3 ± 2153, N = 9 independent culture), GluA1 (**p = 0.0078, 2-tailed Student's t test; control 18.84 ± 1.930 vs Aβo 26.22 ± 1.475, N = 9 independent culture) and fyn (**p = 0.0041, 2-tailed Student's t test; control 19.42 ± 1.337 vs Aβo 29.67 ± 2.181, N = 6 independent cultures; Fig. 6D). Then, we evaluated the effect of a synaptic activation after the Aβo treatment (Fig. 7A). Preceded by 15 min Aβo treatment, synaptic activation disrupted LifeAct-RFP fluorescence, suggesting an alteration of F-actin organization and no additional EGFP-tau recruitment at the synapse. We confirmed these results on endogenous proteins by Western blot analysis on PSD-positive fractions (Fig. 7B). After Aβo treatment, synaptic activation did not trigger any increase in synaptic markers and in fact decreased synaptic actin (***p = 0.0009, 2-tailed Student's t test; Aβo 29.64 ± 1.495, Aβo + Bic/4-AP 18.56 ±2.030, N = 7 independent cultures; Fig. 7C), PSD-95 (***p = 0.0007, 2-tailed Student's t test; Aβo 33.37 ± 2.153, Aβo + Bic/4-AP 19.25 ±2.550, N = 7 independent cultures) and tau levels (**p = 0.0014, 2-tailed Student's t test; Aβo 29.74± 1.748, Aβo + Bic/4-AP 20.68 ±1.751, N = 12 independent cultures). If the spines' protein content did not display any significant difference, these treatments are associated with a profound modification of the spine structure, as revealed by the morphological analysis performed on cortical culture transfected with a plasma membrane marker tagged with a GFP (Fig. 7D). Finally, we performed a phalloidin precipitation assay after a 15 min Aβo treatment on our neuron culture (Fig. 8A) and observed that tau/F-actin content was increased (**,*p < 0.05 relative to control, #p < 0.05 relative to Aβo, 1-way ANOVA; control 15.45 ± 1.529, Aβo 32.90 ± 3.181, Aβo+ Bic/4-AP 20.18 ± 2671 for actin; control 16.34 ± 2.618, Aβo 31.77 ± 1.952, Aβo+ Bic/4-AP 17.70 ± 4.080 for tau, N = 5 independent cultures Fig. 8B). A subsequent synaptic activation did alter tau interaction with F-actin.
Figure 6.

Aβo treatment in primary cortical neuron cultures induces synaptic mislocalization of EGFP-Tau. A, Confocal imaging of cortical neurons (14 DIV) cotransfected with EGFP-Tau (full-length human tau) and LifeAct-RFP. Left, Spatial repartition of EGFP-Tau (green) and LifeAct-RFP (red) in nontreated neuron. Scale bar, 5 μm. Right, Higher magnification of EGFP-Tau and LifeAct-RFP in dendritic spine neuron after Aβ treatment (15 and 30 min, designated box in the left) by 15 and 30 min of Aβo 100 nm treatment. The merged image shows colocalization of F-actin and tau (yellow, area indicated with white arrows). Scale bar, 1 μm. B, Quantification of the fluorescence intensity of EGFP-Tau in the head of spines during the Aβo treatment. The graph represents the evolution of ΔF/F0 generated by EGFP-Tau induced by Aβo 100 nm treatment. Statistical analysis was performed by two-way ANOVA followed by Bonferroni's posttest (mean ± SEM, ***p < 0.001, control n = 40, N = 3 independent cultures; Aβo 100 nm n = 79, N = 5 independent cultures). C, Representative Western blots of protein expression from fractionated primary cortical cultures treated or not with Aβo (100 nm) for 30 min. Presynaptic protein (synaptophysin), postsynaptic proteins (PSD-95, GluA1, GluN2A, GluN2B, Fyn), cytoskeletal protein (actin), and tau in PSD-fraction (Triton-insoluble fraction) were analyzed. D, Quantitative analysis of PSD-95, GluA1, GluN2B, GluA1, Fyn, actin, and tau in PSD fractions (mean ± SEM, 2-tailed Student's t test, ***p < 0.001, **p < 0.01, n ≥ 7 independent cultures). E, Percentage of positives and negatives spines in EGFP-Tau-transfected neurons before and during synaptic activation or Aβo treatment (Bic/4-AP N = 3, n = 130 spines; Aβo N = 3, n = 142; control N = 6, n = 272; ***p < 0.0001, **p > 0.01).
Figure 7.

Aβo treatment on primary cortical neuron cultures induces synaptic mislocalization of endogenous tau that is disrupted by synaptic activation. A, Confocal imaging of cortical neurons (14 DIV) cotransfected with EGFP-Tau and LifeAct-RFP. Left, Spatial repartition of EGFP-Tau (in green) and LifeAct-RFP (in red) in untreated neurons. Scale bar, 5 μm. Right, Higher magnification of EGFP-Tau and LifeAct-RFP in a dendritic spine (designated box in the left) neuron showing the effect of 15 min of Aβ on tau localization (green) and actin cytoskeleton (red) followed by 15 min of Bic/4-AP. Scale bar, 1 μm. B, Representative Western blots of protein expression from fractionated primary cortical neurons treated with Aβo (100 nm) and then for 15 min with Bic/4-AP. Presynaptic protein (synaptophysin), postsynaptic proteins (PSD-95, GluA1,GluN2A, GluN2B,Fyn), cytoskeleton protein (actin), and tau in PSD-fraction (Triton-insoluble fraction) were analyzed. C, Quantitative analysis of PSD-95, GluA1, GluN2B, GluA1, Fyn, actin, and tau in PSD fraction (mean ± SEM, 2-tailed Student's t test, N ≥ 7). D, Confocal imaging of cortical neurons (14 DIV) transfected with GFP membrane. Top, Untreated neurons. Scale bar, 10 μm. Middle and Right, higher magnification of GFP membrane in dendrites (scale bar, 5 μm) and dendritic spine (designated box in the middle) from a neuron showing the effect of 15 min of Aβ treatment on spines followed by 15 min of Bic/4-AP. Scale bar, 1 μm.
Figure 8.

Aβo treatment on primary cortical neuron cultures increases the interaction of F-actin with tau, which is reduced after synaptic activation. A, Representative Western blot showing the effect of 30 min Aβo (100 nm) and the effect of 15 min pretreatment with Aβo (100 nm) then 15 min of Bic/4-AP treatment on tau associated with F-actin in cultured cortical neurons. F-actin was collected after treatment and precipitated with phalloidin. B, Western blot analysis of actin and tau in control, Aβo (100 nm), and Aβo (100 nm) + Bic/4-AP 15 min condition (**,*p < 0.05 vs control, 1-way ANOVA; #p < 0.05 vs Aβo, N = 5 independent cultures).
Pool of synaptic tau evoked by Aβo is more dynamic and has a distinct phosphorylation state from that induced by synaptic activation
Because image acquisition for live imaging of EGFP-tau was performed at a relatively slow rate to accumulate fluorescence intensity, we did not have any direct measurement of tau turnover in the spines. To further understand the mechanisms by which Aβo exposure and the synaptic activation induced translocation of tau to the synapse, we investigated tau dynamics using a FRAP technique. On neurons transfected with EGFP-tau and LifeAct-RFP, we induced tau translocation to spine by synaptic activation, and then photobleached the GFP signal of individual spine heads (Fig. 9A) and measured tau fluorescence, which resulted in a recovery half-life of 4.829 s and a plateau of 50.83% (Fig. 9B). The recovery curves indicated 3 pools of tau: a mobile, unbleached fraction comprising 9.72 ± 1.03%, a dynamic fraction at 47.08 ± 3.06%, and a stable, unrecoverable fraction at 42.75 ± 2.78%. This stable fraction suggests that a large portion of tau is anchored in the spine. When we investigated Aβo-driven tau translocation to the synapse, we did not see any change in half-life recovery (4.729 s) from those measured with synaptic activation. However, the plateau value was drastically modified (71.20%), illustrating that, whereas Aβo induced tau translocation and subsequently its interaction with actin filament, the resulting synaptic tau is less stable. Comparison between synaptic activation and Aβo showed a significant difference between their dynamic fractions (***p < 0.0001, 2-tailed Student-t test; Bic/4AP dynamic fraction 47.09± 3.063, n = 21 spines on 4 independent cultures; Aβo 68.49 ± 2.64, n = 24 on 4 independent cultures; Fig. 9C).
Figure 9.

The dynamics of tau in spines changes is distinct between translocation inducing synaptic activation from Aβ exposure and could be phosphorylation dependent. A, Time lapse of a spine pretreated with Bic/4-AP containing EGFP-tau and showing FRAP. B, Representative FRAP curves (%FRAP normalized to the prebleached intensity of the spine of EGFP-Tau-transfected neurons) of Tau WT (EGFP-Tau 2N4R) after 45 min of Bic/4AP (green, n = 21) or 45 min after Aβo application (red, n = 24) and TauT205A after 45 min of Bic/4AP (blue, n = 21). C, Percentage of EGFP-tau fluorescence or EGFP-tauT205A that was stable (white), dynamic (gray), or unbleached (black). Statistical analysis was performed by 2-tailed t test, ***p < 0.0001. D, Representative Western blot of Tau phospho-Threonine 205 (p-Thr205), phospho-Serine 404 (p-Ser 404), and tau total from PSD-enriched fractions for control, Bic/4-AP (30 min), Aβ (30 min), and Aβ (15 min)+Bic/4-AP (15 min) conditions. E, Quantifications of p-Thr 205 and p-Ser 404 under control, Bic/4-AP, Aβ, or Aβ + Bic/4-AP conditions (mean ± SEM, **,*relative to control; # # #relative to Bic/4-AP, +relative to Aβ, p > 0.05, 1-way ANOVA with Bonferroni's posttest, N = 5 independent cultures). F, Confocal imaging of cortical neurons (14 DIV) cotransfected with EGFP-Tau S404A and LifeAct-RFP. Left, Spatial repartition of EGFP-TauS404A (green) and LifeAct-RFP (red) in untreated neurons. Scale bar, 5 μm. Right, Higher magnification of EGFP-Tau S404A and LifeAct-RFP in dendritic spine neuron after Aβ treatment (15 and 30 min, designated box in the left) by 15 and 30 min of Aβo 100 nm treatment. The merged image shows no colocalization of F-actin and tau. Scale bar, 1 μm. G, Quantification of the fluorescence intensity of EGFP-TauS404A in the head of spines during the Aβo treatment. The graph represents the evolution of ΔF/F0 generated by EGFP-Tau under control conditions (black), EGFP-Tau (red), and EGFP-TauS404A (pink) induced by Aβo 100 nm treatment. Statistical analysis was performed by 2-way ANOVA followed by Bonferroni's posttest (mean ± SEM, ***p < 0.001, control n = 40, N = 3 independent cultures; Tau-WT Aβo 100 nm n = 79, N = 5; Tau S404A n = 98, N = 3 independent cultures).
Tau is a phosphorylated protein containing 80 potential phosphorylation sites (Sergeant et al., 2005). Alterations of tau localization in disease states are associated with abnormal phosphorylation of tau (Hoover et al., 2010; Tai et al., 2012; Merino-Serrais et al., 2013); therefore, we investigated the phosphorylation state of tau after synaptic activation or Aβo exposure (Fig. 9D). We chose to investigate the phosphorylation states of two representatives residues that have both been described as sites of physiological and pathological phosphorylation: one located in the proline rich domain, Thr-205, and one located in the C terminus domain, Ser-404 (Sergeant et al., 2005; Merino-Serrais et al., 2013). Thr-205 phosphorylated-tau was only increased under synaptic activation in the PSD fraction (control 24.57 ± 0.9754 vs Bic/4-AP 38.90 ± 1.936; Fig. 9E), whereas it was decreased after Aβo treatment (control 24.57 ± 0.9754 vs Aβ 13.64 ± 2.416). Synaptic activation after Aβo exposure did not produce any significant Thr-205 phosphorylation of tau (Aβ + Bic/4-AP 22.89±2.796 vs Bic/4-AP 38.90 ± 1.93 vs Aβ 13.64±2.50). Conversely, only Aβo exposure promoted significant tau phosphorylation on Ser 404 (**p < 0.05, 1-way ANOVA; control 15.67 ± 2.418 vs Aβo 32.65 ± 3.76 vs Bic/4-AP 26.75 ± 1.17 vs Aβo + Bic/4-AP 24.97 ± 4.48, N = 4). These results revealed that, although synaptic activation or Aβo promote tau translocation to PSD fractions, the synaptic tau displays a different phosphorylation profile that may be responsible for the conditional tau properties observed. Finally, to investigate whether the difference in tau retention in the spine was related to differential phosphorylation, we overexpressed nonphosphorylatable tau mutant EGFP-Tau T205A or EGFP-Tau S404A in neurons exposed to synaptic activation or to Aβo exposure. We observed that synaptic activation promoted EGFP-Tau T205A translocation to the spine but FRAP experiments revealed a shorter tau turnover time in the spine (Fig. 9B), whereas Aβo-driven translocation to the spine was no longer observable in EGFP-Tau S404A-transfected neurons (Fig. 9F,G). These experiments highlight the pivotal role of these phosphorylations in tau translocation features to the spine.
Discussion
Although tau has been extensively studied for its role as an axonal MAP, the discovery of its synaptic localization under pathological conditions has raised extensive questions on the consequence of this mislocation (Ittner and Götz, 2011). Here, we report that tau displays a widespread distribution in the dendrites and responds to synaptic input by trafficking from the dendritic shaft to the postsynaptic compartments. The trafficking of tau occurs under pharmacological and electrophysiological synaptic plasticity stimulation paradigms in primary culture and acute hippocampal slices, respectively. This activity-dependent tau translocation is concomitant with major recompositions of the postsynaptic protein content that results in an increase in PSD-95, glutamatergic AMPA receptors, GluA1, actin, and tau's proposed partner, Fyn kinase. Together, our results are consistent with tau having a postsynaptic role in the organization that sustains synaptic plasticity. Further, we demonstrate that, once translocated to the synapse, tau interacts with the most predominant cytoskeleton element of the postsynaptic structure: F-actin. This interaction is consistent with a possible implication in the complex cascade leading to synaptic potentiation. Further supporting this hypothesis, we observed that pharmacological stabilization of F-actin with jasplakinolide also induces synaptic tau trafficking, which suggests a link between F-actin and tau. Interestingly, we found that by exposing cortical neurons to synthetic Aβo, tau translocates to spines without induced synaptic activation. This mislocation of tau into the spine heads is a characteristic feature observed in AD (Tai et al., 2012) and frontotemporal dementia (FTD) and parkinsonism linked to chromosome 17 (FTDP-17) and tau P301L mutation (Hoover et al., 2010). Synaptic activation subsequent to Aβo did not further increase tau content into the dendritic spines, which suggests that Aβo exposure disrupts the activity-dependent enrichment of tau concomitant with the preclusion of synaptic plasticity.
The identification of activity-driven translocation of tau to the synaptic compartment is consistent with Ittner et al. (2010), who described tau function as carrying the Src kinase Fyn to the synaptic compartment after synaptic activation. The present findings also add to the growing body of evidence consistent with a wide distribution of neuronal tau in the dendrites (Tashiro et al., 1997) and the axon (Mandell and Banker, 1996). Tau is a well known MAP and microtubules have been shown to penetrate into spines (Jaworski et al., 2009; Merriam et al., 2013), which strongly suggests that microtubules are involved in the observed translocation. Different mechanisms may account for the distribution of tau in dendritic spine, for example, cotransport with microtubule fragments or by motor transport or diffusion. The simplest, and therefore most likely, scenario is one in which tau enters spines with microtubules. After being liberated from MTs, tau may bind to actin filaments, thereby anchoring tau within the postsynaptic compartment. This hypothesis is supported by previous work from Ittner et al. (2010), which showed that removing the MT-associated domain from tau prevents the synaptic localization of tau and its binding partner Fyn. When tau is truncated to contain only the N-terminal and proline-rich domains, tau is unable to localize to dendrites. Because this truncated form of tau is still able to bind Fyn, the exclusion of tau from spines also precludes fyn localization to the postsynaptic compartment. As an active transporter of synaptic proteins (e.g., AMPARs; Kneussel and Wagner, 2013), myosin may also carry tau to the spines along a cytoskeletal track. Currently, only one study suggests an interaction between tau and myosin based on their colocalization within neurofibrillary tangles found in AD and FTD patients' brains (Feuillette et al., 2010). Moreover, myosin VI was shown to modulate tau-dependent neurodegeneration in the Thy-Tau22 Drosophila model (Blard et al., 2007). However, these studies provide no direct evidence for a physiological interaction between tau and myosin. Last, tau is unlikely to enter spines through simple diffusion because very little tau is present in spines before synaptic activation or exposure to Aβ. Therefore, we favor a combined mechanism that involves tau diffusion along the microtubule lattice, as demonstrated by Hinrichs et al. (2012). When microtubules are briefly entering the dendritic spines during activation, tau diffusion guided by the microtubule lattice may promote spine distribution independently of directed motor-dependent transport. The importance of tau translocation to spines warrants further investigation of the many potential mechanisms, which may be different depending on the evoking stimuli.
Once located in the spines, tau may contribute to spine morphology. Such a role is supported by previous observations that the silencing of tau expression using small hairpin RNA produced spine loss in cultured neurons (Chen et al., 2012).
The stimuli that led to tau translocation into spine heads also induced the long-lasting modifications of the postsynaptic element that characterize LTP. The tau increase in the PSD-enriched fraction is concomitant with enrichment in PSD95 and GLUA1, both of which are necessary characteristics of synaptic potentiation (Ehlers, 2003; Steiner et al., 2008; Kessels and Malinow, 2009). Following the same behavior of well known plasticity-related synaptic proteins such as GluA1, PSD-95, or actin suggests that tau may contribute to the functional modifications that sustain the enhanced synaptic strength associated with LTP. The role of tau in synaptic plasticity could be due to its ability to transport the Src kinase Fyn to synaptic targets (e.g., GluN2B subunits of the NMDA receptor). In accordance with this hypothesis, tau KO mice or those expressing tau without MT-binding domains display no Fyn in the PSD fraction (Ittner et al., 2010). The importance of Fyn in synaptic plasticity processes is underscored by the decreased LTP observed in Fyn KO mice (Grant et al., 1992; Kojima et al., 1997).
Tau interacts with F-actin in both purified protein assay and spines. This interaction was identified and characterized for the first time in similar purified protein assay involving tau MT-binding domains and the portion of the proline-rich domain that interacts directly with F-actin (He et al., 2009). Similarly, this interaction has been identified in PC12 cells (Yu and Rasenick, 2006) and in a Drosophila model of AD (Fulga et al., 2006). The consequences of the interaction between tau and F-actin are a stabilization of actin filaments and their organization into bundles. Here, we show that interaction between F-actin and tau is increased during synaptic activation. As revealed by LifeAct fluorescence, filamentous actin displays a higher concentration in the spine heads. The potential interaction of tau and filamentous actin in the shaft and then the translocation to the synapse is possible, although we favor a molecular mechanism that involves interaction of tau/actin in the spine because of the presence of this dense spine actin cytoskeleton. The role of actin in the structural reorganization necessary for synaptic plasticity has been studied extensively (for review, see Hotulainen and Hoogenraad, 2010). Indeed, modulation of actin dynamics toward F-actin complex structures can be considered as one of the key elements responsible for the activity-dependent modification of spine morphology during synaptic plasticity (Star et al., 2002). Therefore, tau may be important for synaptic plasticity because of its ability to influence F-actin structure or stability. F-actin as causal to tau translocation is central to our hypothesis. The fact that jasplakinolide treatment induces a translocation of tau to spine heads strongly supports a causal relation between these events (i.e., direct stabilization of F-actin produces tau translocation).
Finally, when we reproduced pathological conditions observed in AD by exposing cortical neurons to Aβo, we observed a translocation of tau to the synapse despite the lack of synaptic activation. This result is consistent with abnormal tau accumulation in dendritic spines (Hoover et al., 2010). Recently, similar synaptic accumulation of tau has been described in AD patients, further strengthening the hypothesis that abnormal synaptic localization of tau may be involved in the synaptic dysfunction observed in AD (Roberson et al., 2011; Tai et al., 2012). Under Aβo exposure, tau still interacts with F-actin once in the postsynaptic compartment. Our FRAP data highlighted a more dynamic pool of synaptic tau under Aβo compared with that evoked by synaptic activation, which suggests that tau interactions with its potential partners in the synapse, such as F-actin, are disturbed. This likely disrupts tau-related modification of the postsynaptic content observed during synaptic plasticity. Therefore, synaptic activation in the presence of Aβo did not lead to increased levels PSD95 or GluA1 in the PSD fraction and precluded the establishment of long-lasting modification of synaptic strength, which is consistent with the exhaustive reports of Aβo impairing LTP (Shankar et al., 2007; Selkoe, 2008; Li et al., 2011).
The difference in tau behavior depending on the stimulus driving it to the synapse is puzzling and may rely on the variation in the phosphorylation profile (Allyson et al., 2010; Mondragón-Rodríguez et al., 2012). It is now well accepted that tau function and localization to the membrane, nucleus, synapse, or axons is modulated by phosphorylation (Mandell and Banker, 1996; Sultan et al., 2011; Pooler et al., 2012; Hoover et al., 2010). We showed that Aβo and synaptic activation display differences in the phosphorylation of two tau phospho-epitopes. Under synaptic activation, PSD-located tau is phosphorylated on threonine 205, whereas it is largely dephosphorylated under Aβo exposure. In contrast, serine 404 is not differentially phosphorylated between these two experimental conditions. The nonphosphorylation incompetent mutated tau exhibited drastic different features, with wild-type tau strengthening the importance of these phosphorylations in tau behavior.
In summary, we provide evidence that, under physiological stimulations, tau may be involved in the molecular cascade leading to synaptic plasticity. Aβo renders the synapse unable to establish plasticity-related modifications of the postsynaptic content. The disruption of the activity-driven relocalization of tau to the synapse by Aβo could be one of the synaptotoxic steps in the progression of AD. The mechanism responsible for the altered localization of tau may represent a novel therapeutic approach for tauopathies, including AD.
Footnotes
This work was supported by the Fondation Neurodis, INSERM, and the Centre National de la Recherche Scientifique joint ATIP-Avenir program and the Agence Nationale de la Recherche (ANR MALAAD program). A.E. is supported by a grant from the Région Rhône-Alpes (program CIBLE 2011). We thank N. Sergeant (Lille, France) for providing us with His-tagged human tau plasmid.
The authors declare no competing financial interests.
References
- Allyson J, Dontigny E, Auberson Y, Cyr M, Massicotte G. Blockade of NR2A-containing NMDA receptors induces tau phosphorylation in rat hippocampal slices. Neural Plast. 2010;2010:340168. doi: 10.1155/2010/340168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blard O, Feuillette S, Bou J, Chaumette B, Frébourg T, Campion D, Lecourtois M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16:555–566. doi: 10.1093/hmg/ddm011. [DOI] [PubMed] [Google Scholar]
- Chen Q, Zhou Z, Zhang L, Wang Y, Zhang YW, Zhong M, Xu SC, Chen CH, Li L, Yu ZP. Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem Int. 2012;60:233–242. doi: 10.1016/j.neuint.2011.12.013. [DOI] [PubMed] [Google Scholar]
- Coué M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Feuillette S, Deramecourt V, Laquerriere A, Duyckaerts C, Delisle MB, Maurage CA, Blum D, Buée L, Frébourg T, Campion D, Lecourtois M. Filamin-A and Myosin VI colocalize with fibrillary Tau protein in Alzheimer's disease and FTDP-17 brains. Brain Res. 2010;1345:182–189. doi: 10.1016/j.brainres.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2006;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Götz J, Xia D, Leinenga G, Chew YL, Nicholas H. What renders TAU toxic. Front Neurol. 2013;4:72. doi: 10.3389/fneur.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- He HJ, Wang XS, Pan R, Wang DL, Liu MN, He RQ. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009;10:81–92. doi: 10.1186/1471-2121-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs MH, Jalal A, Brenner B, Mandelkow E, Kumar S, Scholz T. Tau protein diffuses along the microtubule lattice. J Biol Chem. 2012;287:38559–38568. doi: 10.1074/jbc.M112.369785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Götz J. Amyloid-β and tau–a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di Stefano P, Demmers J, Krugers H, Defilippi P, Akhmanova A, Hoogenraad CC. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Jaworski T, Kügler S, Leuven F. Modeling of tau-mediated synaptic and neuronal degeneration in Alzheimer's disease. Int J Alzheimers Dis. 2010;2010:573138. doi: 10.4061/2010/573138. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Wagner W. Myosin motors at neuronal synapses: drivers of membrane transport and actin dynamics. Nat Rev Neurosci. 2013;14:233–247. doi: 10.1038/nrn3445. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci U S A. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázaro-Diéguez F, Aguado C, Mato E, Sánchez-Ruíz Y, Esteban I, Alberch J, Knecht E, Egea G. Dynamics of an F-actin aggresome generated by the actin-stabilizing toxin jasplakinolide. J Cell Sci. 2008;121:1415–1425. doi: 10.1242/jcs.017665. [DOI] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008;22:4258–4271. doi: 10.1096/fj.08-107268. [DOI] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis PA, Howard TH, Castleberry RP, Binder LI. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci U S A. 1990;87:8422–8426. doi: 10.1073/pnas.87.21.8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rábano A, Avila J, DeFelipe J. The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer's disease. Brain. 2013;136:1913–1928. doi: 10.1093/brain/awt088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW. Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci. 2013;33:16471–16482. doi: 10.1523/JNEUROSCI.0661-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287:32040–32053. doi: 10.1074/jbc.M112.401240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging. 2012;33:431.e27–431.e38. doi: 10.1016/j.neurobiolaging.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Delacourte A, Buée L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–197. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, Colin M, Bonnefoy E, Buée L, Galas MC. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286:4566–4575. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport. 1997;8:2797–2801. doi: 10.1097/00001756-199708180-00029. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–215. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- Yu JZ, Rasenick MM. Tau associates with actin in differentiating PC12 cells. FASEB J. 2006;20:1452–1461. doi: 10.1096/fj.05-5206com. [DOI] [PubMed] [Google Scholar]