

Abstract
A polyglutamine expansion in huntingtin (HTT) causes the specific death of adult neurons in Huntington's disease (HD). Most studies have thus focused on mutant HTT (mHTT) toxicity in adulthood, and its developmental effects have been largely overlooked. We found that mHTT caused mitotic spindle misorientation in cultured cells by altering the localization of dynein, NuMA, and the p150Glued subunit of dynactin to the spindle pole and cell cortex and of CLIP170 and p150Glued to microtubule plus-ends. mHTT also affected spindle orientation in dividing mouse cortical progenitors, altering the thickness of the developing cortex. The serine/threonine kinase Akt, which regulates HTT function, rescued the spindle misorientation caused by the mHTT, by serine 421 (S421) phosphorylation, in cultured cells and in mice. Thus, cortical development is affected in HD, and this early defect can be rescued by HTT phosphorylation at S421.
Keywords: cortical neurogenesis, Huntington disease, mitosis, spindle orientation
Introduction
Huntington's disease (HD) is a dominant neurodegenerative disorder caused by an abnormal polyglutamine stretch in the huntingtin protein (HTT). Studies of cellular and mouse models of HD have led to the characterization of molecular mechanisms explaining how mutant HTT (mHTT) causes the death of adult neurons (Imarisio et al., 2008). Little is known about the consequences of the presence of mHTT during brain development.
Current knowledge of HTT biology suggests that it acts as a scaffold for diverse cellular functions. These functions include transcription (Zuccato et al., 2003), microtubule (MT)-based transport (Gauthier et al., 2004; Caviston et al., 2007), and ciliogenesis (Keryer et al., 2011). In HD, the function of HTT in these processes is impaired, leading to neuronal toxicity (Zuccato et al., 2003; Gauthier et al., 2004). This is consistent with the hypothesis that a loss of the functions of the wild-type HTT and the gain of new toxic functions contribute to HD pathogenesis. Wild-type HTT is also involved in mitosis, in which it regulates the orientation of the spindle (Godin et al., 2010; Elias et al., 2014). Yet, the effect of the HD mutation on HTT function during mitosis has not been reported.
During embryonic corticogenesis, the neural stem cells initially form a single layer of neuroepithelial progenitors, and this cell population then expands massively, through symmetric divisions (Morin and Bellaïche, 2011). As neurogenesis progresses, the neuroepithelial progenitors switch to an asymmetric mode of division, to produce a self-renewing radial glial progenitor and a daughter cell, which moves away from the ventricular zone to differentiate. We demonstrated that HTT inactivation in murine cortical progenitors changes the nature of the cleavage during division, thereby decreasing the pool of cycling progenitors and increasing neuronal differentiation (Godin et al., 2010).
In this study, we addressed the consequences of HTT mutation for spindle orientation in dividing cortical progenitor cells, and for neocortical development.
Materials and Methods
Cell lines and transfection.
STHdh cells were electroporated using cell line nucleofector kit L (Amaxa). Mouse embryonic fibroblasts (MEFs) were prepared from E14.5 embryos. MEFs and HeLa cells were transfected using Lipofectamine 2000 (Invitrogen).
DNA constructs.
Plasmids encoding Akt c.a. (myristylated-ΔPH; HA tagged), EB3-GFP, and the first 480 amino acids fragment of HTT were previously described (Humbert et al., 2002; Stepanova et al., 2003). To obtain pCAGGS-HTT-480 constructs, the SpeI-XhoI fragment of the corresponding HTT-480 construct was inserted in the pCAGGS plasmid. mCherry-α-tubulin and pBOShistone2B-mCherry-IRES-hygromycin were from C. Janke and M. Piel (Institut Curie, Paris).
Antibodies.
Antibodies used were as follows: anti-dynein intermediate chain (1:100; Millipore Bioscience Research Reagents), anti-p150Glued (1:100; BD Transduction Laboratories), anti-NuMA (1:200; Calbiochem), anti-γ-tubulin (clone GTU88; 1:1000, Sigma; 1:100, Abcam; clone AK-15; 1:300; Sigma), anti-Tuj1 (β-III-tubulin; clone TU-20; 1:100; Millipore Bioscience Research Reagents), anti-CLIP170 (1:300) (Coquelle et al., 2002), anti-nestin (clone Rat401; 1:200; BD Biosciences PharMingen), anti-Tbr1 (1:200; Abcam), and anti-Cux1 (1:200; Millipore). AlexaFluor-conjugated secondary antibodies were from Invitrogen.
Fixation procedures.
Anti-p150Glued and anti-NuMA immunostaining: cells were fixed with 10% trichloroacetic acid (7 min), then cold methanol (−20°C, 10 min). Anti-dynein immunostaining: cells were fixed in methanol at −20°C containing 2% PFA. To analyze +TIP localization at MT plus-end: EB3-GFP electroporated cells were fixed with 1 mm EGTA in cold methanol (−20°C, 10 min) and with 4% PFA in PBS (room temperature, 10 min). To measure spindle orientation, cells were fixed with methanol (−20°C, 5 min). For other experiments, cells were fixed in 4% PFA and permeabilized using 0.5% PBT (5 min). For spindle orientation analyses, 14 μm brain sections were permeabilized in cold acetone (−20°C, 2 min). To analyze cortical thickness, sections were treated with 10 mm sodium citrate solution, pH 6.0, and permeabilized in 1× PBS/0.3% Triton X-100. Serial 35 μm brain slices from P21 mice were obtained as described previously (Ben M'barek et al., 2013).
Image acquisition and analysis.
Images were acquired with a Leica DM-RXA microscope and with a Leica TCS-SP5 laser scanning confocal microscope. Line Scan function of ImageJ was used to reveal fluorescence intensity of p150Glued and NuMA along a line crossing the spindle poles and the cell cortex and to calculate CLIP170, p150Glued, and EB3-GFP fluorescence intensity at MT plus-ends. EB3-comets velocity analyses were generated with KymoToolBox (home-made ImageJ plug-in available upon request). Persistence time and catastrophe rate were calculated as described previously (Peris et al., 2009). Spindle orientation was calculated using home-made ImageJ macros (available upon request); distribution and average of angles are shown.
Micropatterns.
[L]-shaped fibronectin micropatterns were made as described previously (Théry et al., 2005). STHdhQ111/Q111 and STHdhQ7/Q7 cells were electroporated with histone2B-mCherry; positive cells were selected with Hygromycin B (50 μg/ml) and sorted by flow cytometry. The positioning of the metaphasic plate was measured using MetaMorph software (Molecular Devices).
Mice.
All mice were in the CD-1 background. P21 mice were of either sex. In utero electroporations were performed as described previously (Godin et al., 2010). Experiments were performed in strict accordance with the recommendations of the European Community (86/609/EEC) and the French National Committee (87/848) for care and use of laboratory animals (permission 91-448 to S.H.).
Statistical analyses.
Statview 4.5 software (SAS Institute) and GraphPad Prism 6.0 software were used for statistical analysis. The criterion for statistical significance was set at p < 0.05.
Results
mHTT causes misorientation of the mitotic spindle
The previously described role of HTT in mitosis led us to investigate whether mHTT impaired spindle orientation. We examined the position of the spindle pole axis in STHdhQ7/Q7 and STHdhQ111/Q111 cells derived from HdhQ7/Q7 (wild-type) and mutant HdhQ111/Q111 mice (Trettel et al., 2000). In HdhQ111/Q111 mice, exon 1 was replaced with a human exon 1 with a 111 glutamine expansion under the control of a mouse HTT promoter (Wheeler et al., 2002). We measured the angle formed between the pole–pole axis and the substrate plane (Fig. 1A). In most wild-type cells, the spindle axis was oriented parallel to the substratum plane (8.0 ± 0.7°, n = 74) (Fig. 1B). mHTT-expressing cells displayed defective spindle positioning, with a higher mean angle (18.2 ± 1.25°, n = 89). We also analyzed spindle orientation in HeLa cells exogenously expressing a wild-type (17Q) or mutant (68Q) 480-amino acid fragment of HTT (Fig. 1C). Cells transfected with an empty pcDNA or with an HTT-480-17Q vector behaved similarly (control: 11.5 ± 0.9°, n = 107; HTT-480-17Q: 10.9 ± 1.1°, n = 74). In HD conditions, the mean angle was markedly higher (HTT-480-68Q: 16.2 ± 1.16°, n = 89).
Figure 1.

mHTT causes spindle misorientation. A, Measurement of the spindle angle α. STHdhQ7/Q7 (Q7) and STHdhQ111/Q111 (Q111) cells stained as indicated. B, C, Spindle angles of Q7, Q111 (B), and HeLa (C) cells. D, E, Spindle angles of Q7 (D) and Q111 (E) cells grown on micropatterns. Time-lapse pictures were used to measure the positioning of the metaphasic plate (H2B-mCherry). Scale bars, 10 μm. Error bars indicate SEM. **p < 0.01 (unpaired, one-tailed t test). ***p < 0.001 (unpaired, one-tailed t test).
We confirmed these findings with printed [L]-shaped fibronectin micropatterns (Fig. 1D,E). Micropattern approach makes it possible to mimic the geometric and mechanical constraints imposed by the microenvironment (Théry et al., 2005). Most of the mitotic spindles of wild-type cells stably expressing histone2B-mCherry grown on [L]-shaped patterns had metaphasic plates oriented at an angle of 45° (Fig. 1D; n = 156). By contrast, spindle angles were more dispersed in mHTT-expressing cells (Fig. 1E; n = 135). In STHdhQ111/Q111 cells, ∼60% (±1.0; n = 77) of the spindles deviated from the mean angle by >15°, whereas this was the case for only 35% (±0.25; n = 44) of STHdhQ7/Q7 cells (p < 0.05; unpaired, one-tailed t test). Thus, mHTT alters spindle orientation during mitosis.
The localization of dynein, the p150Glued subunit of dynactin, NuMA, and MT plus-end-binding proteins during mitosis is altered in HD
The dynein/dynactin complex is required for the assembly and positioning of the mitotic spindle, and its presence at the cell cortex is essential, to exert pulling forces on astral MTs (Morin and Bellaïche, 2011). NuMA is involved in spindle pole formation; it also links astral MTs to the cell cortex. In wild-type cells, p150Glued and NuMA were detected at the spindle poles and in the cellular cortex facing the spindle poles (Fig. 2A–C). In STHdhQ111/Q111 cells, these proteins were abundant at the spindle poles and their cortical distribution was altered. The dynein localization to the spindle, spindle poles, and astral MTs in STHdhQ7/Q7 cells was also disrupted in STHdhQ111/Q111 cells. In particular, only a small proportion of these cells displayed dynein staining of astral MTs (Fig. 2D).
Figure 2.

mHTT alters the mitotic distribution of p150Glued, NuMA, and Dynein. A, STHdhQ7/Q7 (Q7) and STHdhQ111/Q111 (Q111) cells were stained as indicated. Scale bars, 10 μm. B, Representative line scan analysis showing the distribution of p150Glued and NuMA. C, Percentage of cells with cortical labeling of p150Glued and NuMA. D, Percentage of Q7 and Q111 cells with dynein on astral microtubules. C, D, At least n = 25 cells in three independent experiments. Error bars indicate SEM. **p < 0.01 (unpaired, one-tailed t test). ***p < 0.001 (unpaired, one-tailed t test).
The localization of dynein to MT plus-ends is ensured by its interaction with MT plus-end-binding proteins (+TIPs), including CLIP170 (cytoplasmic linker protein 170), p150Glued, and EB (end-binding) proteins. In STHdhQ7/Q7 cells transfected with EB3-GFP, endogenous CLIP170 and p150Glued colocalized with EB3 in comet-like structures at MT plus-ends (Fig. 3A–D). By contrast, mutant cells were essentially devoid of CLIP170 (Fig. 3A,B) and p150Glued (Fig. 3C,D) comets. In STHdhQ7/Q7 cells, most of the MT plus-ends positive for EB3 were positive for endogenous CLIP170 (83.1 ± 2.6%, n = 17) and for p150Glued (78 ± 2.4%, n = 16) (at least 30 MT plus-ends per cell; p < 0.001; unpaired, one-tailed t test). In STHdhQ111/Q111 cells, the EB3/+TIPs ratio was much lower for CLIP170 (39.4 ± 1.9%, n = 24) and for p150Glued (52 ± 1.7%, n = 20) (at least 30 MT plus-ends per cell; p < 0.001; unpaired, one-tailed t test).
Figure 3.
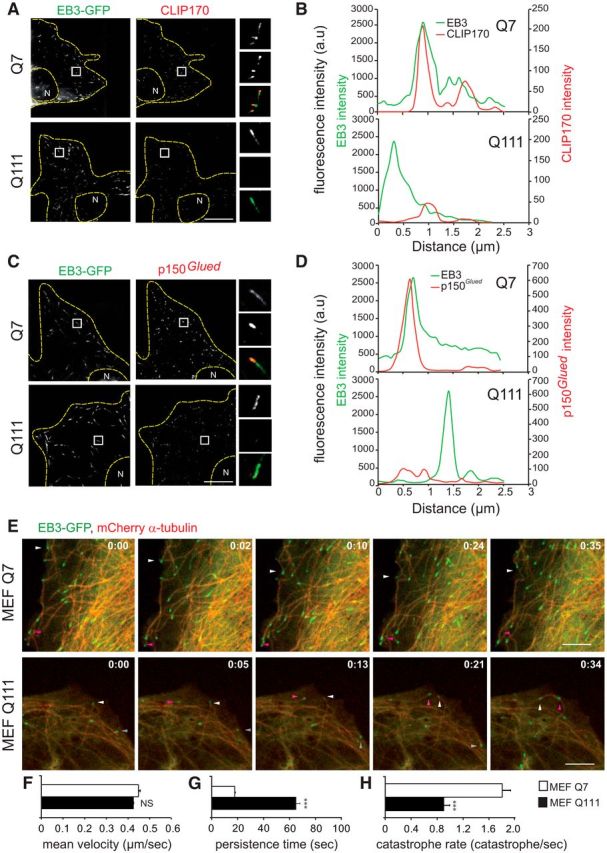
+TIPs localization at microtubules plus-ends and microtubule dynamics are modified in HD. A, C, Immunostainings of EB3-GFP transfected STHdhQ7/Q7 (Q7) and STHdhQ111/Q111 (Q111) cells with anti-CLIP170 (A) and anti-p150Glued (C) antibodies. N, nucleus. B, D, Representative line scan analysis of fluorescence intensity of CLIP170 (B) or p150Glued (D), and EB3, starting at the end of the MT to 3 μm inwards. E, Time-lapse recordings illustrating the MT dynamic instability in HdhQ7/Q7 (MEF Q7) and HdhQ111/Q111 (MEF Q111) MEFs expressing mCherry-α-tubulin and EB3-GFP. Arrowheads indicate localization of the MT tip close to the leading edge. Time is expressed as min:sec. F–H, MT dynamics in Q7 and Q111 MEFs (at least 5 MTs examined in n = 10 cells in two independent experiments). F, Mean velocity of EB3-comets. G, Persistence time. H, MT catastrophe rate. Scale bars, 10 μm. Error bars indicate SEM. NS, Not significant. **p < 0.01 (unpaired, one-tailed t test). ***p < 0.001 (unpaired, one-tailed t test).
We conclude that mHTT modifies the mitotic distribution of dynein, dynactin, and NuMA at the spindle poles and cellular cortex, and that of +TIPs at MT plus-ends.
The dynamic instability of MTs is impaired in mouse embryonic fibroblasts from HD mutant mice
EB proteins, p150Glued and CLIP170, regulate the dynamic instability of MT, characterized by cycles of MT growth and shrinkage (Gouveia and Akhmanova, 2010). We used MEFs; these are of an ideal size for studying MT dynamics (Fig. 3E–H). EB3 comet velocity was similar in wild-type and mutant fibroblasts (Fig. 3F). We analyzed the time that MTs spent exploring the area near the leading edge by live imaging (Fig. 3E,G). In wild-type MEFs, most of the MTs depolymerized on contact with the plasma membrane. By contrast, MTs continued to grow at a tangent to the leading edge after coming into contact with the membrane in mutant MEFs. Consistent with this, the catastrophe rate was lower in mutant MEFs than in wild-type cells (Fig. 3H).
Thus, mHTT decreases the dynamic instability of MT and leads to the abnormal persistence of MTs after membrane contact. Together with the abnormal localization of dynein/dynactin/NuMA and +TIPs proteins during mitosis, this may participate to altered spindle orientation.
mHTT modifies the division of cortical progenitors and embryonic cortical development
We investigated whether mHTT altered cell division in vivo, by analyzing the spindle orientation of dividing cortical progenitors in HdhQ7/Q7 and HdhQ111/Q111 mice on embryonic days 10.5 and 14.5 (E10.5 and E14.5) (Fig. 4A–C). At E10.5 and E14.5, most of the wild-type cells divided with their spindles parallel (horizontal/planar division) to the ventricular surface (E10.5: 18.9 ± 2.7°, n = 49; E14.5: 15.4 ± 1.4°, n = 104) (Fig. 4B,C). By contrast, in mHTT-expressing cells, a smaller percentage of progenitors underwent planar division and a greater proportion of cells divided with an angle of >30° (oblique and parallel divisions; E10.5: 30.5 ± 3.1°, n = 46; E14.5: 42.2 ± 2.1°, n = 121). Similar results were obtained during late anaphase when the final spindle orientation had occurred (wild-type, E10.5: 16.2 ± 2.7°, n = 26; E14.5: 11.4 ± 1.1°, n = 105; mutant, E10.5: 34.8 ± 3°, n = 23; E14.5: 31.1 ± 2°, n = 108) (Peyre et al., 2011).
Figure 4.
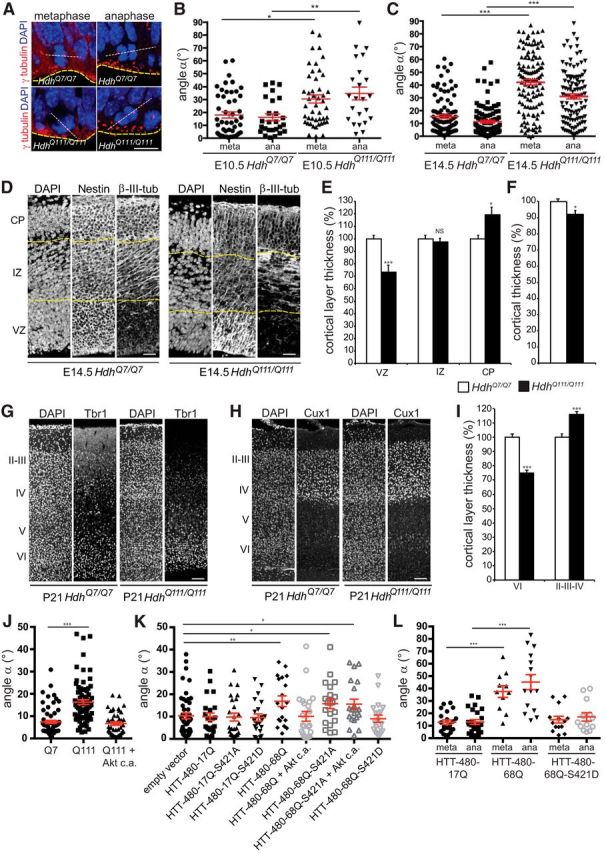
Spindle orientation of dividing progenitors and cerebral cortex thickness are altered in HdhQ111/Q111 embryos. A, Illustration of dividing progenitors. White dashed line indicates spindle; yellow line indicates ventricular surface. γ-tubulin marks centrosomes. Scale bar, 15 μm. B, C, Spindle angles during divisions in E10.5 (B) and E14.5 (C) embryos at metaphase (meta) and anaphase (ana). D, Nestin and β-III-tubulin immunostainings of E14.5 coronal sections. Scale bars, 50 μm. E, F, VZ, IZ, CP (E) and overall cortical (F) thicknesses (n = 3 embryos per genotype). G, H, Tbr1 (G) and Cux1 (H) immunostainings of P21 coronal sections. Scale bars, 100 μm. I, Layers VI and II-IV thicknesses (n = 3 mice per genotype). J, K, Spindle angles of STHdhQ7/Q7 (Q7), STHdhQ111/Q111 (Q111), and STHdhQ111/Q111 cells transfected with Akt c.a. (J) and of HeLa cells transfected with the indicated constructs (K). L, Spindle angles in E16.5 electroporated progenitors. Error bars indicate SEM. NS, Not significant. *p < 0.05 (unpaired, one-tailed t test). **p < 0.01 (unpaired, one-tailed t test). ***p < 0.001 (unpaired, one-tailed t test).
We analyzed whether the changes in spindle orientation observed at E10.5 and E14.5 due to the expression of mHTT had developmental consequences, by analyzing cortical sections at E14.5 and E16.5. We immunolabeled wild-type and mutant brains for Nestin and β-III-tubulin, markers of progenitor cells and neurons, respectively (Fig. 4D). We measured the ventricular zone (VZ), intermediate zone (IZ), and cortical plate (CP) thicknesses. The VZ of mutant E14.5 embryos was found to be ∼30% smaller than that of the wild-type (Fig. 4E). The weaker progenitor labeling was not associated with an increase in cell death rates (data not shown). In contrast, whereas the density of cells in wild-type and mutant embryos was similar (141.5 ± 6.2 vs 144.4 ± 6.04 cells per region of interest, arbitrary unit, nonsignificant), the CP thickness was increased by 20% in mutant condition (Fig. 4E). Consistent with these observations, the cortex was thinner in HdhQ111/Q111 embryos than in HdhQ7/Q7 embryos (Fig. 4F). Similar observations were made at E16.5 (data not shown).
We addressed whether the mHTT-induced defects in corticogenesis were observed at later stages. We immunostained postnatal day 21 (P21) cortical sections for Tbr1, Foxp1, and Cux1, markers of layers VI, IV/V, and II/III/IV, respectively. In HD mice, the thickness of layer VI was reduced, whereas it was increased for layers II/III/IV (Fig. 4G–I). The thickness of layers IV/V was similar in control and mutant mice (data not shown).
Thus, mHTT leads to changes in the cleavage plane of progenitors and in neurogenesis in the developing cortex with impact in young adult brains.
Akt rescues the spindle orientation defect caused by mHTT
Phosphorylation of S421 by Akt rescues mHTT-induced defective function in vesicular transport (Gauthier et al., 2004; Zala et al., 2008). We investigated whether Akt could restore mitotic function in HD. We transfected STHdhQ111/Q111 cells with a construct encoding a constitutively active Akt (Akt c.a.) and analyzed spindle orientation (Fig. 4J). The mean spindle angle was 16.3 ± 1.0° in STHdhQ111/Q111 cells (n = 75) and was similar in STHdhQ7/Q7 and STHdhQ111/Q111/Akt c.a. cells (STHdhQ7/Q7: 7.4 ± 0.7°, n = 107; STHdhQ111/Q111/Akt c.a.: 6.7 ± 0.7°, n = 89). Similarly, the coexpression of Akt with HTT-480-68Q in HeLa cells rescued the defect observed in the presence of HTT-480-68Q alone (10.1 ± 2.2°, n = 23 vs 16.9 ± 2.4°, n = 19) (Fig. 4K).
We addressed whether the effect of Akt involved HTT phosphorylation at S421 by expressing mHTT in which S421 was replaced with an alanine (unphosphorylatable, S421A) or an aspartic acid (phosphomimetic, S421D) (Fig. 4K). In HTT-480-68Q-S421A cells, the spindle was not correctly oriented parallel to the substratum plane (15.8 ± 2.2°, n = 20). By contrast, HTT-480-68Q-S421D behaved like the wild-type, with most cells having a spindle aligned with the substratum plane (9.0 ± 1.6°, n = 20 vs 9.9 ± 1.7°, n = 23). Whereas the mHTT-induced defect was rescued by the expression of Akt c.a. (16.9 ± 2.4°, n = 19 versus 10.1 ± 2.2°, n = 23), Akt c.a. had no effect in cells expressing mHTT that could not be phosphorylated (15.6 ± 2.2°, n = 20) (Fig. 4K). Thus, Akt exerts its effects, at least partly, through mHTT S421 phosphorylation.
We performed in utero electroporation-based transfection of E14.5 cortical progenitors with vectors expressing GFP and HTT-480-17Q, HTT-480-68Q, or HTT-480-68Q-S421D (Fig. 4L). The expression of mHTT resulted in a greater dispersion of spindle angles than observed in the control situation (metaphase: 37.6 ± 4.7°, n = 11 vs 12.8 ± 1.8°, n = 21; anaphase: 45.2 ± 5°, n = 15 versus 13.04 ± 1.9°, n = 25) (Fig. 4L). Mimicking constitutive phosphorylation at S421 resulted in a distribution of angles similar to that in control conditions (metaphase: 14.9 ± 2.7°, n = 14; anaphase: 17.1 ± 3.3°, n = 13). Thus, phosphorylation of S421 of mHTT by Akt rescues spindle misorientation in dividing cortical progenitors in vivo.
Discussion
We propose that mHTT interferes with mitotic spindle orientation in several ways. HTT is required for the recruitment of dynein/dynactin and NuMA to the spindle poles (Godin et al., 2010; Elias et al., 2014); this recruitment is altered by mHTT. Another event regulating spindle positioning is the +TIP-mediated interaction of astral MTs with the cell cortex (Gouveia and Akhmanova, 2010). In HD, CLIP170 and p150Glued are mislocalized from MT plus-ends and MT persistence time after membrane contact and catastrophe rates are affected, providing evidence of the critical nature of HTT for the recruitment of +TIPs. Finally, we found that mHTT disrupts the cortical distribution of NuMA and p150Glued, which form part of the complex linking astral MT plus-ends to the cell cortex.
The effect of mHTT on development has been overlooked. Wild-type HTT is involved in development as illustrated by the embryonic lethality at E7.5 of the knock-out (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995). HTT is essential for the early patterning of the embryo during formation of the anterior region of the primitive streak (Woda et al., 2005). In addition, the inactivation of HTT in Wnt1 cell lineages leads to congenital hydrocephalus in mice (Dietrich et al., 2009). HTT is involved in cortical development. The depletion of HTT from cycling E14.5 mouse cortical progenitors changes their cleavage plane, decreases their maintenance in the VZ, and promotes differentiation (Godin et al., 2010). An earlier knockdown of HTT expression, in the E12.5 neuroepithelium, decreases neuronal survival, proliferation, and migration (Tong et al., 2011). Our findings now indicate that mHTT affects cortical development by changing the nature of the mode of division in cortical progenitors.
Our data support the notion that development may be abnormal in HD carriers. Consistent with this idea, HD mutation carriers have a smaller intracranial adult brain volume before the onset of the disease than controls, probably reflecting abnormal development (Nopoulos et al., 2010). Evaluations of basic anthropometric measurements have shown that children at risk of HD have disproportionately small heads, suggestive of abnormal brain growth (Lee et al., 2012). mHTT post-translational modifications, including S421 phosphorylation, may be involved in compensatory mechanisms accounting for the mild effect of mHTT on development and in early adulthood. These compensatory mechanisms might be overwhelmed in aging organisms.
In conclusion, knock-in mouse models are genetically close to HD patients and express temporally and spatially appropriate levels of mHTT. However, the slow onset of symptoms in these models renders them difficult to use for preclinical trials. Our data provide the proof-of-concept that studies of early phenotypes in mice expressing full-length mHTT constitute an alternative to test molecules of therapeutic interest.
Footnotes
This work was supported by Agence Nationale pour la Recherche Grant ANR-09-BLAN-0080 to S.H., Fondation pour la Recherche Médicale to S.H., Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, and Institut Curie. S.H. is an Institut National de la Santé et de la Recherche Médicale investigator. M.M.-C. was supported by an Institut Curie Ph.D. fellowship. M.B. was supported by an NERF Ile de France fellowship. E.A. was supported by a CIFRE fellowship (2012-0401). We thank F. Saudou, members of the S.H./Saudou laboratories, C. Janke, C. Benstaali, N. Carpi, H. Yu, and staff of the Institut Curie animal, imaging, histology and flow cytometry facilities for help with experiments, reagents, and/or discussion.
The authors declare no competing financial interests.
References
- Ben M'barek K, Pla P, Orvoen S, Benstaali C, Godin JD, Gardier AM, Saudou F, David DJ, Humbert S. Huntingtin mediates anxiety/depression-related behaviors and hippocampal neurogenesis. J Neurosci. 2013;33:8608–8620. doi: 10.1523/JNEUROSCI.5110-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A. 2007;104:10045–10050. doi: 10.1073/pnas.0610628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquelle FM, Caspi M, Cordelières FP, Dompierre JP, Dujardin DL, Koifman C, Martin P, Hoogenraad CC, Akhmanova A, Galjart N, De Mey JR, Reiner O. LIS1, CLIP-170's key to the dynein/dynactin pathway. Mol Cell Biol. 2002;22:3089–3102. doi: 10.1128/MCB.22.9.3089-3102.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich P, Shanmugasundaram R, Shuyu E, Dragatsis I. Congenital hydrocephalus associated with abnormal subcommissural organ in mice lacking huntingtin in Wnt1 cell lineages. Hum Mol Genet. 2009;18:142–150. doi: 10.1093/hmg/ddn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Elias S, Thion MS, Yu H, Sousa CM, Lasgi C, Morin X, Humbert S. Huntingtin regulates mammary stem cell division and differentiation. Stem Cell Reports. 2014;2:491–506. doi: 10.1016/j.stemcr.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, Bellaïche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Gouveia SM, Akhmanova A. Cell and molecular biology of microtubule plus end tracking proteins: end binding proteins and their partners. Int Rev Cell Mol Biol. 2010;285:1–74. doi: 10.1016/B978-0-12-381047-2.00001-3. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/S1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, Rubinsztein DC. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- Keryer G, Pineda JR, Liot G, Kim J, Dietrich P, Benstaali C, Smith K, Cordelières FP, Spassky N, Ferrante RJ, Dragatsis I, Saudou F. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest. 2011;121:4372–4382. doi: 10.1172/JCI57552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Mathews K, Schlaggar B, Perlmutter J, Paulsen JS, Epping E, Burmeister L, Nopoulos P. Measures of growth in children at risk for Huntington disease. Neurology. 2012;79:668–674. doi: 10.1212/WNL.0b013e3182648b65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Bellaïche Y. Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell. 2011;21:102–119. doi: 10.1016/j.devcel.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nopoulos PC, Aylward EH, Ross CA, Johnson HJ, Magnotta VA, Juhl AR, Pierson RK, Mills J, Langbehn DR, Paulsen JS. Cerebral cortex structure in prodromal Huntington disease. Neurobiol Dis. 2010;40:544–554. doi: 10.1016/j.nbd.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris L, Wagenbach M, Lafanechère L, Brocard J, Moore AT, Kozielski F, Job D, Wordeman L, Andrieux A. Motor-dependent microtubule disassembly driven by tubulin tyrosination. J Cell Biol. 2009;185:1159–1166. doi: 10.1083/jcb.200902142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyre E, Jaouen F, Saadaoui M, Haren L, Merdes A, Durbec P, Morin X. A lateral belt of cortical LGN and NuMA guides mitotic spindle movements and planar division in neuroepithelial cells. J Cell Biol. 2011;193:141–154. doi: 10.1083/jcb.201101039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry M, Racine V, Pépin A, Piel M, Chen Y, Sibarita JB, Bornens M. The extracellular matrix guides the orientation of the cell division axis. Nat Cell Biol. 2005;7:947–953. doi: 10.1038/ncb1307. [DOI] [PubMed] [Google Scholar]
- Tong Y, Ha TJ, Liu L, Nishimoto A, Reiner A, Goldowitz D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J Neurosci. 2011;31:14794–14799. doi: 10.1523/JNEUROSCI.2774-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Gutekunst CA, Vrbanac V, Lebel LA, Schilling G, Hersch S, Friedlander RM, Gusella JF, Vonsattel JP, Borchelt DR, MacDonald ME. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet. 2002;11:633–640. doi: 10.1093/hmg/11.6.633. [DOI] [PubMed] [Google Scholar]
- Woda JM, Calzonetti T, Hilditch-Maguire P, Duyao MP, Conlon RA, MacDonald ME. Inactivation of the Huntington's disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev Biol. 2005;5:17. doi: 10.1186/1471-213X-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Colin E, Rangone H, Liot G, Humbert S, Saudou F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 2008;17:3837–3846. doi: 10.1093/hmg/ddn281. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]