

Abstract
Paranodal axoglial junctions are critical for maintaining the segregation of axonal domains along myelinated axons; however, the proteins required to organize and maintain this structure are not fully understood. Netrin-1 and its receptor Deleted in Colorectal Cancer (DCC) are proteins enriched at paranodes that are expressed by neurons and oligodendrocytes. To identify the specific function of DCC expressed by oligodendrocytes in vivo, we selectively eliminated DCC from mature myelinating oligodendrocytes using an inducible cre regulated by the proteolipid protein promoter. We demonstrate that DCC deletion results in progressive disruption of the organization of axonal domains, myelin ultrastructure, and myelin protein composition. Conditional DCC knock-out mice develop balance and coordination deficits and exhibit decreased conduction velocity. We conclude that DCC expression by oligodendrocytes is required for the maintenance and stability of myelin in vivo, which is essential for proper signal conduction in the CNS.
Keywords: caspr, DCC, myelin, netrin, oligodendrocyte, paranode
Introduction
Axonal myelination is a key mechanism that increases the speed of nerve transmission in vertebrates; however, the complexity of myelin, both structurally and metabolically, renders it particularly vulnerable to damage and deficits that give rise to various forms of dysmyelinating and demyelinating diseases. The rapid saltatory conduction of action potentials along a myelinated axon depends on the precise organization of specialized subcellular domains: the node of Ranvier, the paranode, the juxtaparanode, and the internode.
The paranode, located on both sides of the node, is the region where each layer of the myelin sheath terminates in a cytoplasm-filled membrane loop that tightly adheres to the axon. Several proteins are known to be enriched at paranodes and mediate paranodal axoglial interactions, including Caspr, contactin, neurofascin-155 (nfasc155), αII- and βII-spectrin, protein 4.1B, and ankyrinB (for review, see Buttermore et al., 2013). The axonal proteins Caspr and contactin and the glial protein nfasc155 are key players at the axoglial septate-like junctions, and disruption of the expression of any of these proteins results in disorganized paranodes, behavioral deficits, and reduced axon conduction velocity (Bhat et al., 2001; Boyle et al., 2001; Sherman et al., 2005; Pillai et al., 2009).
Components of noncompact myelin membranes also contribute to paranode and myelin integrity, including CNP (Lappe-Siefke et al., 2003), myelin-associated glycoprotein (MAG; Fruttiger et al., 1995), and galactolipids (Coetzee et al., 1996; Dupree et al., 1998). In addition, several intracellular signaling molecules including cdc42 and rac1 have been demonstrated to influence myelin architecture (Thurnherr et al., 2006). Thus, myelin formation and stability depend on multiple factors acting in concert to maintain this highly specialized cellular structure.
Netrin-1 and the netrin receptor Deleted in Colorectal Cancer (DCC) are enriched in noncompact myelin membranes (Manitt et al., 2001), particularly at paranodes (Jarjour et al., 2008). Conventional DCC or netrin-1 null knock-out mice die within a few hours after birth (Serafini et al., 1996; Fazeli et al., 1997), limiting their utility for studying myelination in vivo. Previous studies performed in vitro using organotypic cerebellar slice cultures provided evidence that DCC and netrin-1 are not required for the initial formation of compact myelin or paranodal junctions, but rather contribute to maintain axonal domain segregation and appropriate paranode ultrastructure (Jarjour et al., 2008). In the adult mammalian CNS, both neurons and oligodendrocytes express netrin-1 and DCC (Manitt et al., 2001, 2004); however, the cellular source of these proteins required for paranode maintenance is not known.
To identify the functional significance of DCC expressed by oligodendrocytes in vivo, we generated mice in which DCC expression was selectively deleted from mature oligodendrocytes. This resulted in progressive disruption of paranodes and compact myelin, reduced conduction velocity, and deficits in coordination and balance. We conclude that DCC expression by oligodendrocytes is essential for the maintenance of myelin in vivo, and that myelin abnormalities caused by the absence of DCC result in deficits in axonal conduction velocity and behavior.
Materials and Methods
Animals.
DCCflox/flox mice were developed to enable cre-mediated excision of exon 23, encoding the DCC transmembrane domain, as described previously (Krimpenfort et al., 2012). PLPcreERT mice (Doerflinger et al., 2003) were obtained from Dr. Samuel David (McGill University, Montreal, Canada). Conventional dcc knock-out mice (Fazeli et al., 1997) were obtained from Dr. Robert Weinberg (Whitehead Institute for Biomedical Research, Cambridge, MA), and extensively backcrossed into a CD-1 genetic background. CMVcre mice (Schwenk et al., 1995) were provided by Dr. David Colman (McGill University, Montreal, Canada). ROSA26-lacZ reporter mice (Soriano, 1999) were provided by Dr. Jean-François Cloutier (McGill University, Montreal, Canada). All procedures were performed in accordance with the Canadian Council on Animal Care guidelines for the use of animals in research.
Tamoxifen induction.
Tamoxifen (Sigma; T5648) was dissolved in a 10:1 mixture of sunflower oil/ethanol at a concentration of 10 mg/ml. An intraperitoneal injection of tamoxifen, 1 mg in 0.1 ml, was delivered twice a day for 5 consecutive days in the test group, as well as the control littermates. Injections were performed between 4.5 and 6 weeks of age. All animals used were males.
Antibodies.
The following antibodies were used in this study: mouse monoclonal anti-Caspr (UCDavis/NIH NeuroMab Facility; catalog #75-001, RRID:AB_2083496), rabbit polyclonal anti-Kv1.2 (Alomone Labs; catalog #APC-010, RRID:AB_2040146), rabbit polyclonal anti-NFM (Millipore; catalog #AB1987, RRID:AB_91201), goat polyclonal anti-DCC (Santa Cruz Biotechnology; catalog #SC6535, RRID:AB_2245770), mouse monoclonal anti-DCC (BD Biosciences; catalog #554223, RRID:AB_395314), rabbit polyclonal anti-calnexin (BioVision; catalog #3811-100, RRID:AB_2069013), rabbit polyclonal anti-MAG and mouse monoclonal anti-PLP (a gift from Dr. David Colman, McGill University), rabbit polyclonal anti-nfasc (Tait et al., 2000; a gift from Dr. Peter Brophy, University of Edinburgh), rat polyclonal anti-MBP (Millipore; catalog #MAB386, RRID:AB_94975), rabbit polyclonal anti-GAPDH (Santa Cruz Biotechnology; catalog #Sc-25778, RRID:AB_641106), mouse monoclonal anti-CNP (Abcam; catalog #ab24566, RRID:AB_2260805), rabbit polyclonal anti-olig2 (Abcam; catalog #ab81093, RRID:AB_1640746), and mouse monoclonal anti-APC (CC1; Abcam; catalog #ab16794, RRID:AB_443473). Secondary antibodies used were Alexa 546-conjugated goat anti-mouse (Invitrogen; catalog #A11003, RRID:AB_141370) and goat anti-rabbit (Invitrogen; catalog #A11010, RRID:AB_143156), Alexa 488-conjugated goat anti-rabbit (Invitrogen; catalog #A11008, RRID:AB_143165), HRP-conjugated donkey anti-goat (Cedarlane; catalog #705-035-147), donkey anti-rat (Cedarlane; catalog #712-035-153), donkey anti-mouse (Cedarlane; catalog #715-035-150), and donkey anti-rabbit (Cedarlane; catalog #711-035-152).
Immunohistochemistry.
For the analysis of adult PLPcreERTDCCflox/flox mice, animals were deeply anesthetized and perfused with phosphate buffered saline, pH 7.4, (PBS) followed by 4% PFA. Brain and spinal cord were dissected, postfixed in 4% PFA for 1 h, and equilibrated in 30% sucrose at 4°C. After embedding in optimal cutting temperature compound (Sakura Finetek), 16 μm sections were cut on a cryostat. For the study of CMVcreDCCflox/flox mice, embryos were fixed in Carnoy's solution (60% ethanol, 30% chloroform, and 10% acetic acid), dehydrated, and embedded in paraffin. Eight micrometer sections were cut and processed for staining. After blocking in 3% BSA, 0.3% Triton X-100 in PBS, primary antibodies were added (diluted in blocking solution), incubated overnight at 4°C, and rinsed with PBS. Sections were then incubated with secondary antibodies, along with phalloidin and/or Hoechst when needed, before washing and mounting with Fluoromount-G (Southern Biotech). Images were captured with a MagnaFire CCD camera (Optronics) and an Axiovert 100 microscope (Carl Zeiss). For paranode analysis, images were captured using a Zeiss LSM 510 confocal microscope and quantified using LSM510 software by an experimenter blind to experimental conditions. The length of the Caspr-immunoreactive domain was normalized to the width of Caspr at the paranode. The average width of Caspr-immunoreactive domains did not differ between groups.
X-gal staining.
Ten days after tamoxifen administration, PLPcreERT+ROSA26 mice were anesthetized and perfused with PBS, followed by 4% PFA containing 2 mm MgSO4, and 5 mm EGTA. Brain and spinal cord sections, 16 μm thick, were then rapidly processed for X-gal staining as described previously (Mombaerts et al., 1996).
Electron microscopy.
Mice were deeply anesthetized and perfused with 0.1 m phosphate buffer, pH 7.4, followed by 2.5% glutaraldehyde with 2% PFA, and then by 2% PFA. Tissue was dissected and postfixed for 1 week at 4°C, rinsed with buffer, and postfixed in 2% osmium tetroxide for 40 min. Fixed tissue was then dehydrated through a graded ethanol series and embedded in Epon. Ultrathin sections (70–100 nm) were prepared, placed onto 200 mesh copper grids, and stained with 4% uranyl acetate for 5 min, followed by Reynold's lead citrate for 3 min. Images were captured using a transmission electron microscope at 120 kV equipped with a Tecnai 12 (FEI) Gatan BioScan CCD camera.
Measurement of g-ratios.
The g-ratios (axon diameter/fiber diameter) were calculated with a g-ratio calculator plug-in for ImageJ (available on-line at http://gratio.efil.de/) applied to electron microscopy images of coronal sections of optic nerves. Sixty myelinated axons per animal were randomly selected for measurement (six animals per group, for a total of 360 axons per genotype). Mice were 6 months or 9 months post induction. No effect of age was detected.
Outfolding quantification.
The number of axons with myelin outfoldings was counted from 10 electron micrograph images taken at random from cross sections of optic nerve. Percentages were calculated based on the total number of myelinated axons in these images. In total, between 5000 and 6000 axons were examined per animal with three animals per group. The length of the myelin outfoldings were measured using ImageJ software (RRID:nif-0000–30467; http://imagej.nih.gov/ij/).
Behavioral testing.
Open-field tests were performed in a square open field (50 × 50 cm) surrounded by 30 cm high walls. After a 20 min habituation period, recordings of individual mouse movements were performed for 2 h using VideoTrack software (ViewPoint Life Sciences). The hanging wire grip test was performed, as described previously (Sango et al., 1996), to test overall muscle strength. Briefly, mice were put on a cage lid held upside down and the time that each mouse was able to grip to the wires without falling was recorded. A cutoff time was set at 60 s. The balance beam test was performed as previously described (Carter et al., 1999) to assess general balance and coordination. Briefly, mice were trained for 4 consecutive days to cross a narrow beam (0.8 cm diameter, 50 cm long) and reach an enclosed safety platform. On the fifth day, the time taken by each mouse to cross the beam was recorded; with a cutoff time set at 60 s. Accelerating rotarod (Rotamex; 3 cm rod diameter, gray PVC with a knurled finish) testing was performed after 3 consecutive days of training. During each training session, the mice received five trials on an accelerating rotarod (1–24 rpm) for a maximum of 150 s. On the test day, mice were tested on an accelerating rod (1 rpm/5 s) that reached a maximum speed of 24 rpm. Mice were allowed between three and five trials with a rest of at least 20 min between each trial. The latency to fall was recorded, with a cutoff time of 300 s.
Brain lysates and Western blotting.
Embryonic day 14 (E14) brains were lysed in RIPA buffer (10 mm phosphate buffer, pH 7.2, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing protease inhibitors. Adult brain tissue was lysed in 3% Triton X-100 buffer (20 mm Tris, pH 8, 150 mm NaCl, 10 mm EGTA, 10 mm EDTA, and 3% Triton X-100) containing protease inhibitors, to maximize myelin membrane solubilization. Proteins were separated by SDS-PAGE, transferred onto nitrocellulose membrane (GE Healthcare), and assayed by Western blotting. Densitometric analysis was performed using Adobe Photoshop. Values were normalized to loading control values for GAPDH. To minimize variability, each PLPcreERT+DCCflox/flox mouse was compared with a control littermate. Five mice of each genotype were used in the analysis.
Cell number count.
Sections of optic nerve and spinal cord from animals 9 months post induction were immunolabeled for CC1 and Olig2, both markers of oligodendrocytes. Pictures of randomly selected fields were taken (three fields of view per animal; three animals per genotype) and CC1/Olig2 double-positive cells counted.
Conduction velocity analysis.
For terminal surgical experiments mice were anesthetized with a single intraperitoneal injection of a solution of 10% urethane at 2 g/kg. Mice were then transferred to a stereotaxic frame with body temperature maintained at 37°C using a feedback-controlled heating blanket. To assess the somatosensory response, EMG electrodes on the right hamstring were used for stimulation, with a current of 0.5–2 mA delivered in three pulses of 0.1 ms duration and a 1 ms delay between pulses. Stimulation intensity was set at two times muscle threshold. A hole was made in the skull over the left somatosensory cortex that responds to the hindlimb region and a ±1 MΩ tungsten microelectrode inserted through the dura to record field potential amplitudes following the hamstring stimulation. A reference electrode was positioned under the skin on the posterior portion of the neck. Spike 2 neuronal software (CED) was used to acquire and analyze the data. EEG field potential and EMG responses were measured for latency, at the start of the first stimulating pulse to the first deflection of the responses from baseline in either direction.
Results
Validating an inducible, oligodendrocyte-specific DCC knock-out
To identify the consequences of loss of DCC function on myelin maintenance in vivo, we used a cre/loxP gene-targeting strategy to eliminate DCC from mature oligodendrocytes. Since signaling downstream of DCC regulates oligodendrocyte precursor cell (OPC) migration (Jarjour et al., 2003; Tsai et al., 2003), OPC process retraction (Rajasekharan et al., 2010), and process branching by oligodendrocytes (Rajasekharan et al., 2009), we avoided disrupting initial oligodendrocyte development by crossing DCCflox/flox mice to the PLPcreERT mouse line. This allowed temporal regulation of the excision of the floxed DCC sequence by injection of tamoxifen to induce cre translocation into the nucleus (Doerflinger et al., 2003). To study loss of function specifically in oligodendrocytes, the age at which recombination is induced is critical when using the PLPcreERT mouse line. Before P16, the proteolipid protein (PLP) promoter is active in oligodendrocytes, Schwann cells, and some neurons; however, after 3 weeks of age, the PLP promoter induces cre expression exclusively in oligodendrocytes (Michalski et al., 2011). In our study, recombination was induced between 4.5 and 6 weeks of age, when myelination is nearly complete in the rodent CNS (Foran and Peterson, 1992; Hamano et al., 1998; Richardson et al., 2011).
To confirm that the injection protocol (1 mg tamoxifen twice a day for 5 consecutive days) induced efficient recombination, PLPcreERT mice were crossed with ROSA26-lacZ mice, in which the β-gal reporter is expressed as a result of cre-mediated recombination (Araki et al., 1995). In PLPcreERT+ROSA26-lacZ mice, high expression of the reporter protein was detected in cells of all CNS white matter tracts examined within 10 d after the end of the tamoxifen injection regimen, but not following injection with vehicle (Fig. 1A). Consistent with Michalski et al. (2011), lacZ expression was not detected by Schwann cells in peripheral nerve. In all subsequent experiments, control littermates (PLPcreERT−DCCflox/flox and PLPcreERT+DCCw/w) were submitted to the same tamoxifen regimen as the PLPcreERT+DCCflox/flox group to account for any indirect effects of tamoxifen. Only males were used to avoid possible variability due to gender.
Figure 1.

Validation of mouse lines. Cre-mediated recombination of the floxed DCC allele ablates expression of a functional DCC protein. A, Validation of the tamoxifen-induction protocol (1 mg, i.p.; twice a day for 5 consecutive days) was performed by crossing PLPcreERT mice to ROSA26-lacZ mice. Successful cre recombination was observed based on robust β-gal expression in white matter tracts. Scale bar, 200 μm. B, DCCflox/flox mice were bred to CMVcre mice to induce ubiquitous recombination. Phenotypes were compared with conventional DCC null mice. No DCC protein was detected in brain lysates of E14 CMVcre+DCCflox/flox and DCC−/− embryos. C, Neurofilament M staining showing that CMVcre+DCCflox/flox embryos have a thinner spinal cord ventral commissure compared with littermate controls at E14. A similar phenotype was observed in E14 DCC−/− spinal cord. Scale bar, 100 μm. D, DCC protein expression in cerebellar lysates. Two weeks after induction, no differences in DCC protein levels were detected. Two months after induction, a reduction in DCC protein levels was observed, which remained decreased 8 months after induction.
To confirm that cre-mediated recombination of DCC loxP sites disrupts DCC function, we initially crossed DCCflox/flox mice with CMVcre mice in which cre expression is regulated by a human CMV promoter. The CMV promoter results in ubiquitous cre expression before the implantation stage of embryonic development (Schwenk et al., 1995). Thus, loss of DCC expression is anticipated in all cells and CMVcre+DCCflox/flox embryos are expected to phenocopy conventional DCC−/− embryos. At E14, DCC protein was not detected in CMVcre+DCCflox/flox brain lysates (Fig. 1B). To confirm the cre-mediated loss of DCC function, we examined the formation of the spinal cord ventral commissure in the CMVcre+DCCflox/flox embryos, which is reduced in conventional DCC knock-out mice (Fazeli et al., 1997). At E14, the spinal ventral commissure of CMVcre+DCCflox/flox embryos was substantially thinner than in CMVcre−DCCflox/flox littermates, phenocopying the deficit in conventional E14 DCC knock-out mice (Fig. 1C). These results demonstrate that cre-mediated deletion of DCC results in loss of DCC protein function.
DCC expression and turnover in PLPcreERT+DCCflox/flox mice
To assess the loss of DCC expression by oligodendrocytes in PLPcreERT+DCCflox/flox mice, Western blots were used to assay protein lysates of cerebellum, a brain region rich in white matter. At 2 weeks after induction DCC protein levels did not differ between PLPcreERT+DCCflox/flox mice and their control littermates. In contrast, 2 months after induction, DCC protein levels had decreased in PLPcreERT+DCCflox/flox mice (Fig. 1D), indicating that DCC made by oligodendrocytes has a relatively slow turnover, similar to other proteins localized to paranodal junctions (Hedstrom et al., 2008). DCC protein levels remained low 8 months after induction (Fig. 1D). As expected, not all DCC protein was eliminated, since DCC is also expressed by neurons in the adult CNS (Manitt et al., 2001, 2004; Shatzmiller et al., 2008). In light of the apparent long half-life of DCC in myelin and because we were interested in studying the effect of the absence of DCC on myelin maintenance, in subsequent experiments mice were examined at least 6 months after tamoxifen induction.
Disruption of paranodal junctions in PLPcreERT+DCCflox/flox mice
The absence of DCC expression in vitro causes a defect in the maintenance of paranodal ultrastructure (Jarjour et al., 2008), which is significantly more severe than the disorganization commonly observed with aging (Mierzwa et al., 2010; Shepherd et al., 2012). We examined paranodal ultrastructure by electron microscopy in sagittal sections of optic nerve from PLPcreERT+DCCflox/flox mice 6 months after tamoxifen induction. The degree of disorganization was assessed by scoring each paranode according to the presence or absence of the following four faults: lack of transverse bands, lack of interloop densities, membrane loops detached from the axon, or everted membrane loops (Fig. 2A).
Figure 2.

Paranodal architecture becomes disrupted in PLPcreERT+DCCflox/flox mice. A, Paranodes were scored according to four faults: absence of transverse bands (1), absence of interloop densities (2), detached loops (3), and everted loops (4). B, Example of paranodes in control and PLPcreERT+DCCflox/flox optic nerve. The majority of control paranodes exhibit normal loops contacting the axon and visible transverse bands (black arrows), whereas the majority of knock-out paranodes were abnormal. White arrowheads display glial loops that do not contact the axon. Scale bar, 0.1 μm. C, At 6 months after induction, PLPcreERT+DCCflox/flox paranodes display more faults per paranode than control paranodes (***p = 0.0002; two-tailed Student's t test). D, The majority of control paranodes are normal while the majority of PLPcreERT+DCCflox/flox paranodes are abnormal, bearing one or more faults. E, Prevalence of each type of fault in PLPcreERT+DCCflox/flox paranodes compared with controls. For all graphs n = 44–47 paranodes; three different animals were assessed per genotype.
Paranodes in PLPcreERT+DCCflox/flox mice had twice the number of faults compared with control mice of the same age (Fig. 2B,C). While only 33% of control paranodes analyzed exhibited any fault, 70% of PLPcreERT+DCCflox/flox paranodes were scored as abnormal, with one or more faults (Fig. 2D). Detachment of glial loops from the axon was the most common fault detected with 52% of paranodes in PLPcreERT+DCCflox/flox mice exhibiting at least one glial loop that did not contact the axon, compared with15% of paranodes in controls (Fig. 2B,E). In contrast, the absence of DCC expression by oligodendrocytes did not lead to a dramatic increase in the proportion of paranodes that displayed abnormal interloop densities or spacing between the loops (Fig. 2E; 19% for controls versus 25% for PLPcreERT+DCCflox/flox). Thus, loss of DCC function primarily disrupts interactions between the axon and oligodendrocyte rather than between oligodendrocyte membrane loops. We conclude that the CNS of PLPcreERT+DCCflox/flox mice exhibits paranodal ultrastructural defects within 6 months of loss of DCC gene expression.
Impaired maintenance of paranodal domain organization in PLPcreERT+DCCflox/flox mice
Caspr is an axonal transmembrane protein, highly enriched at paranodes, which interacts with contactin in cis and with glial nfasc155 in trans (Bonnon et al., 2007). The absence of DCC in vitro resulted in diffusion of Caspr along the axon (Jarjour et al., 2008). This is thought to result from degeneration of the integrity of paranodal axoglial contacts, which allows unbound Caspr to diffuse along the axonal membrane away from the paranode.
To investigate axonal domain organization in PLPcreERT+DCCflox/flox mice, we measured the length of the Caspr-immunoreactive domain at paranodes in corpus callosum, cerebellum, spinal cord, and optic nerve at both 6 and 9 months after tamoxifen induction (Fig. 3). We were surprised to find that despite the ultrastructural abnormalities detected at paranodes, Caspr domain length was not measurably affected 6 months post induction in any of the four regions examined. In contrast, 9 months following induction, we detected a significant increase in the length of Caspr immunoreactivity in all regions analyzed (Fig. 3C). This diffusion along the axon frequently resulted in overlap between juxtaparanodal Kv1.2 channels and Caspr (Fig. 3B; white arrowhead). These findings indicate that the absence of DCC expression by oligodendrocytes results in the disorganization of axonal domains. Notably, this occurs after defects could be detected in paranodal ultrastructure, revealing a progressive increase in the severity of paranodal disorganization.
Figure 3.
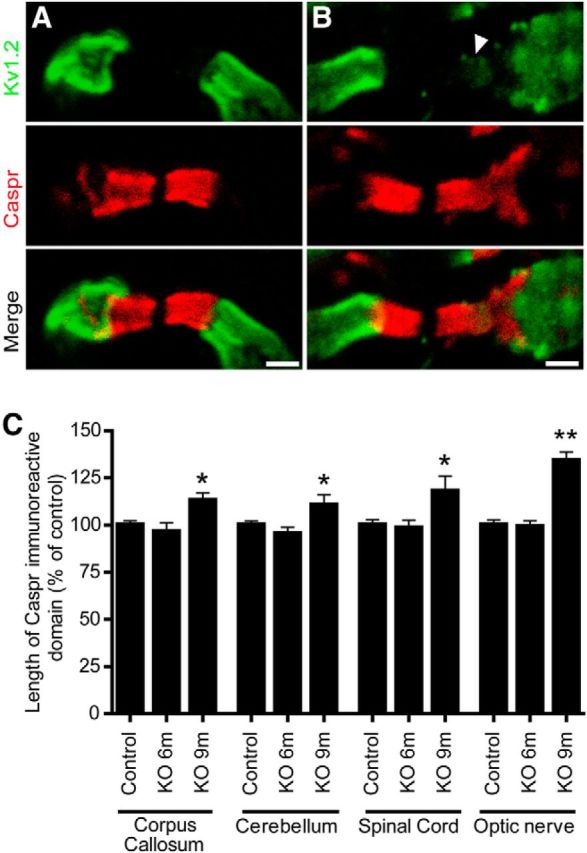
Progressive disorganization of paranodal domains in PLPcreERT+DCCflox/flox mice. Paranodal domain organization was assessed by measuring the length of the paranodal marker Caspr. Examples of spinal cord paranodes seen in control (A) and PLPcreERT+DCCflox/flox (B) mice 9 months after tamoxifen induction are depicted. Paranodal domains were visualized with Caspr antibodies and juxtaparanodal domains were visualized with potassium channel (Kv1.2) antibodies. Some paranodes displayed Caspr leaking out of the paranodes and Kv1.2 leaking into the paranodes (B; white arrowhead). C, Quantification of the length of the Caspr-immunoreactive domain. Caspr-immunoreactive domains are similar to controls 6 months after induction of PLPcreERT+DCCflox/flox mice (KO 6 m), but lengthened 9 months after tamoxifen induction (KO 9 m) in the corpus callosum (n = 52–138), cerebellum (n = 84–205), spinal cord (n = 84–194), and optic nerve (n = 150; *p < 0.05, **p < 0.01; ANOVA with Tukey's post hoc test; three animals per genotype were analyzed at each time point). Scale bar, 2 μm. KO, knock-out.
Abnormalities in compact myelin architecture and protein content in PLPcreERT+DCCflox/flox mice
We next investigated whether the paranodal defects observed in PLPcreERT+DCCflox/flox mice were accompanied by defects in compact myelin. Using electron microscopy, we examined myelin architecture in optic nerve of PLPcreERT+DCCflox/flox mice and their control littermates 6 months after tamoxifen induction. In cross sections of optic nerves, we detected a >3-fold increase in the number of aberrant myelin outfoldings (Fig. 4A; control = 0.60% ± 0.15%, PLPcreERT+DCCflox/flox = 1.93% ± 0.26%, p = 0.0115, n = 3), indicating that in addition to defects in paranodal ultrastructure, compact myelin exhibits abnormalities in PLPcreERT+DCCflox/flox mice.
Figure 4.
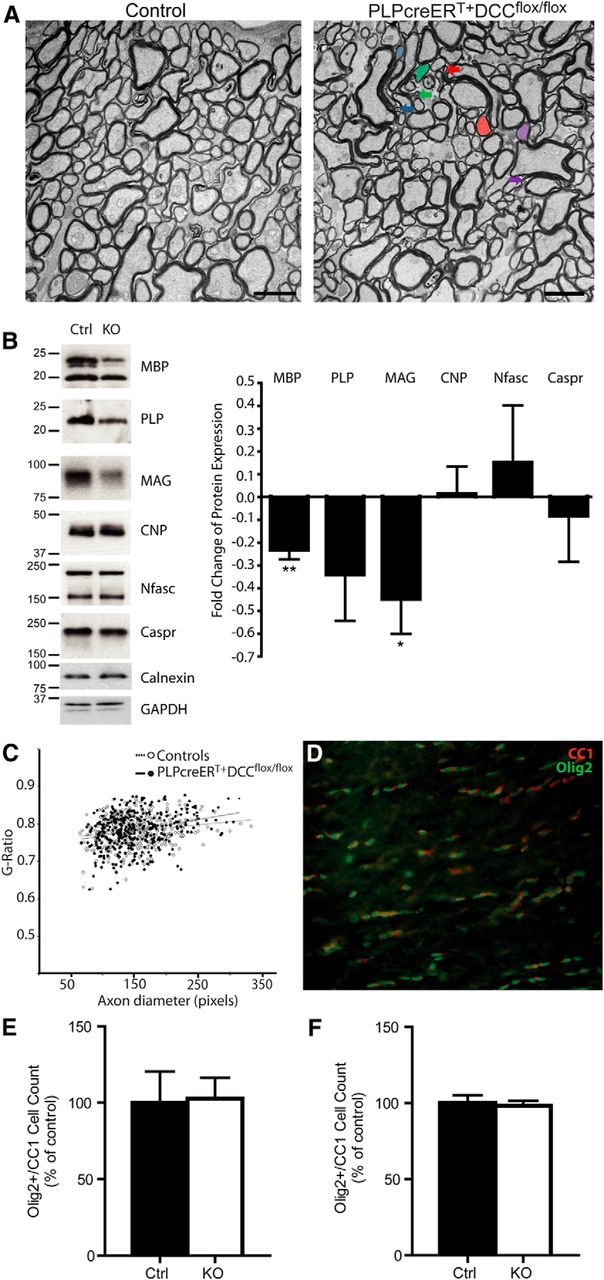
Abnormalities in compact myelin architecture and protein content. A, Cross sections of optic nerve were examined 6 months after tamoxifen induction in control and PLPcreERT+DCCflox/flox mice. Three times more myelin outfoldings were observed in PLPcreERT+DCCflox/flox myelin. The colored arrows point to the end of myelin outfoldings corresponding to the same colored axon. Scale bar, 2 μm. B, Representative Western blots and quantification reveal decreased levels of MBP and MAG protein in the cerebellum of PLPcreERT+DCCflox/flox mice compared with their control littermates 7–9 months after induction. Levels of CNP, neurofascin (Nfasc), and Caspr were unaffected (n = 5; **p < 0.01, *p < 0.05; t test). C, Compared with controls, PLPcreERT+DCCflox/flox mice did not exhibit changes in optic nerve myelin g-ratios 6–9 months after tamoxifen induction (n = 6, no effect of age was detected). D, No difference in the number of Olig2/CC1-positive oligodendrocytes was observed in either the spinal cord (E) or the optic nerve (F) of PLPcreERT+DCCflox/flox mice compared with controls 9 months after induction (n = 3). KO, knock-out.
Given the defects in paranode and compact myelin ultrastructure, we speculated that the absence of DCC might lead to alterations in myelin protein content. We therefore examined myelin proteins in the cerebellum of PLPcreERT+DCCflox/flox mice and control littermates 7–9 months post induction. MBP and MAG levels were significantly decreased in PLPcreERT+DCCflox/flox mice, while paranodal and nodal proteins, Caspr and nfasc155/186, were not affected. Although the mean values of PLP protein detected were lower in knock-outs, this change was not significant (Fig. 4B).
To determine whether the altered levels of myelin proteins were due to a change in the thickness of compact myelin, we measured the g-ratios of myelin in the optic nerve, excluding axons surrounded by myelin with abnormal outfoldings. We did not detect a significant change in the thickness of myelin sheaths in the absence of DCC expression by oligodendrocytes (Fig. 4C; control = 0.7815 ± 0.009, PLPcreERT+DCCflox/flox = 0.7839 ± 0.012. n = 6 animals/group, 60 axons measured per animal, p = 0.87, Student's t test). To determine whether a possible loss of oligodendrocytes in PLPcreERT+DCCflox/flox mice might account for the alterations in myelin protein content, cell counts were performed in the optic nerve and spinal cord; however, the number of olig2/CC1 double-positive cells did not change in either region 9 months after induction (Fig. 4D–F). We did not detect an increase in the percentage of unmyelinated axons in electron micrograph cross sections of optic nerve in PLPcreERT+DCCflox/flox mice (control = 2.77% ± 0.54%, PLPcreERT+DCCflox/flox = 4.08% ± 0.89%; n = 3; p = 0.28, Student's t test).
Reduced axon conduction velocity in the CNS of PLPcreERT+DCCflox/flox mice
To determine whether the myelin defects detected in PLPcreERT+DCCflox/flox mice resulted in a measurable impairment of the capacity of CNS axons to conduct action potentials, we assessed axonal conduction velocity along sensory pathways in mice 16 months following induction. An EMG electrode was placed to stimulate the right hamstring muscle of the hind leg and a recording electrode placed in the hindlimb response region of the left primary somatosensory cortex. Figure 5A illustrates a representative somatosensory-evoked EEG and the delay measured between stimulation of the right hamstring muscle and the somatosensory response in the left somatosensory cortex. PLPcreERT+DCCflox/flox mice exhibited a significant increase in latency compared with control littermates (Fig. 5B), indicating that the structural and molecular defects observed in the absence of oligodendroglial DCC have functional consequences.
Figure 5.

Reduced conduction velocity in PLPcreERT+DCCflox/flox mice. Axonal conduction velocity along sensory pathways was assessed in mice 16 months following induction. A, A representative somatosensory-evoked EEG following stimulation of the right hamstring muscle and recording the response in the left somatosensory cortex. B, Quantification of the delay in somatosensory conduction showed that PLPcreERT+DCCflox/flox mice exhibit a significant increase in latency compared with their control littermates (n = 7–9; **p = 0.007; t test). KO, knock-out.
PLPcreERT+DCCflox/flox mice develop deficits in coordination and balance
Mutations in genes encoding proteins enriched in compact myelin or at paranodal junctions often result in some degree of motor deficit (Bhat et al., 2001; Boyle et al., 2001; Mierzwa et al., 2010). PLPcreERT+DCCflox/flox mice did not exhibit an overt behavioral phenotype. In an open-field test performed 6 months post induction, PLPcreERT+DCCflox/flox mice exhibited normal spontaneous locomotor activity. In a 2 h test period, they traveled a mean distance that was not significantly different from control littermates, with no difference in duration of activity or average velocity detected (Fig. 6A). Muscle strength assessed with the grip test was also normal in PLPcreERT+DCCflox/flox mice compared with controls (Fig. 6B). However, defects in balance and coordination were observed 6 months post induction. PLPcreERT+DCCflox/flox mice took more time to cross a narrow balance beam and reach an enclosed safety platform than control littermates (Fig. 6C). In contrast, when tested 1 month post induction, PLPcreERT+DCCflox/flox mice performed as well as controls in this task (Fig. 6C). This indicates that the mice develop normally and that this behavioral deficit results from the loss of DCC expression by oligodendrocytes. We also challenged PLPcreERT+DCCflox/flox mice using the rotarod, another behavioral test that assays motor coordination and balance. Again, PLPcreERT+DCCflox/flox mice demonstrated decreased motor performance when tested on an accelerating rotarod 6 months after induction, exhibiting reduced latency to fall compared with control littermates (Fig. 6D). We conclude that the absence of DCC expression by oligodendrocytes results in the development of deficits in coordination and balance.
Figure 6.

PLPcreERT+DCCflox/flox mice develop balance and coordination deficits. Different aspects of locomotion were tested 6 months after tamoxifen induction. A, Gross motility was normal in PLPcreERT+DCCflox/flox mice as tested in the open-field test, assessed by total distance traveled and time spent walking in a 2 h testing session (n = 7–8). B, Motor strength was also comparable, as tested with the hanging wire grip test (n = 19–20). C, PLPcreERT+DCCflox/flox mice exhibited impaired balance and motor coordination 6 months after induction, as tested in the balance beam test (n = 22). Motor coordination and balance were normal 1 month after induction, consistent with normal initial development (n = 14–17). D, PLPcreERT+DCCflox/flox mice exhibited decreased motor performance 6 months after induction when tested on an accelerating rotarod (n = 9—10; *p < 0.05; one-tail t test). KO, knock-out.
Discussion
DCC is required for myelin maintenance
DCC and its ligand netrin-1 are expressed by neurons and oligodendrocytes (Manitt et al., 2001, 2004), and enriched at paranodes in the mature mammalian CNS (Jarjour et al., 2008). Our earlier studies using organotypic slices provided evidence that neither netrin-1 nor DCC are essential for the initial formation of CNS paranodal junctions, but that both are required to maintain paranodal structure and the organization of the axonal domains associated with nodes of Ranvier (Jarjour et al., 2008). Here, using PLPcreERT mice to selectively induce cre expression in mature oligodendrocytes in vivo, we addressed the significance of DCC expressed specifically by myelin-forming oligodendrocytes, while sparing effects on OPC migration, oligodendrocyte maturation, and initial myelination. Our findings demonstrate that oligodendroglial DCC contributes to the maintenance of myelin. Selective loss of DCC expression from mature oligodendrocytes resulted in a progressive disruption of paranodal structure and organization, as well as disorganization of compact myelin. Importantly, deletion of DCC expression from oligodendrocytes was functionally relevant, resulting in reduced conduction velocity and deficits in coordination and balance.
Although substantial defects were detected, it is likely that ongoing myelination and oligodendrocyte turnover reduced the severity of the loss-of-function phenotype. The PLPcreERT+DCCflox/flox mice were induced with tamoxifen at 4.5–6 weeks of age; however, oligodendrocytes differentiating from OPCs in these mice later in life would express DCC. It has been reported that between 6.5 and 30% of oligodendrocytes in the adult mouse are generated after 9 weeks of age, depending on the brain region analyzed (Rivers et al., 2008; Zhu et al., 2011; Young et al., 2013), and myelin turnover in the adult CNS has been described (Lasiene et al., 2009). Despite this turnover, our findings reveal that DCC is essential for the maintenance of paranodes and compact myelin.
DCC as an adhesion molecule at the paranode
The Caspr–contactin–nfasc155 complex is a key player in the organization of paranodal axoglial junctions (Buttermore et al., 2013). Caspr binds in cis to contactin on the axonal membrane, while nfasc155 is expressed by oligodendrocytes on paranodal membrane loops (Rios et al., 2000; Charles et al., 2002). Loss of any of these three proteins prevents the formation of axoglial paranodal junctions, as observed by the absence of transverse bands and failure of paranodal myelin membrane loops to maintain contact with the axon (Bhat et al., 2001; Boyle et al., 2001; Pillai et al., 2009). Mislocalization of these proteins, particularly Caspr, is commonly observed in mice exhibiting paranodal abnormalities (Marcus et al., 2002, 2006; Schaeren-Wiemers et al., 2004; Buttermore et al., 2011). We report that deletion of DCC from myelinating oligodendrocytes results in loss of adhesion between the paranodal loops and the axon, which is followed by a lengthening of the Caspr-immunoreactive domain. Based on these findings, we hypothesize that the absence of DCC results in disruption of the adhesive complex between oligodendrocyte membrane loops and the axonal plasma membrane, freeing a portion of Caspr molecules to diffuse laterally along the axon. Notably, we did not detect defects in the interaction between oligodendrocyte membrane loops, indicating that DCC is not essential for adhesion between the glial loops at paranodes. Furthermore, our findings indicate that the nfasc155–Caspr–contactin complex is not sufficient to maintain paranodal organization. We propose that DCC and netrin-1 may function in parallel as an adhesive complex that links the oligodendroglial loops to the axon. It is also possible that DCC may influence the localization of nfasc155 or signal to facilitate nfasc155 function to promote paranodal organization. Since relatively little is known of DCC or nfasc155 signaling in oligodendrocytes, additional studies are required to elucidate the underlying mechanisms involved.
Our findings reveal that the loss of DCC expression by oligodendrocytes results in readily detectable changes in the ultrastructural organization of paranodal loops. Notably, the ultrastructural defects were first detected at 6 months post-induction, a time point at which the paranodal distribution of Caspr appeared normal. Later, at 9 months post-induction, lengthening of the distribution of Caspr was detected. The ultrastructural changes that precede the mislocalization of Caspr indicate that loss of DCC function by mature oligodendrocytes results in defects that become progressively worse with age. These findings also highlight the importance of maintaining tight adhesion between the paranodal myelin loops and the axon to maintain axonal domain organization, reinforcing the model in which the paranode functions as a molecular barrier that limits the lateral diffusion of proteins localized to the node, paranode, and juxtaparanode (Pedraza et al., 2001; Poliak and Peles, 2003).
Multiple studies have identified short-range roles for DCC and netrin-1 regulating cell—cell interactions and adhesion (for review, see Baker et al., 2006). Although netrin-1 is a secreted protein, most netrin-1 in the CNS is not freely soluble, but membrane associated (Manitt et al., 2001), consistent with the restricted distribution of netrin-1 at paranodes (Jarjour et al., 2008). Disrupting DCC function blocks cell adhesion to substrate bound netrin-1 (Shekarabi et al., 2005; Moore et al., 2008), and recent studies indicate that mechanical attachment of netrin-1 to a substrate is necessary to generate the force required for chemoattraction (Moore et al., 2009, 2012). These findings are consistent with netrin-1 and DCC contributing directly to adhesion between the axonal plasma membrane and oligodendrocyte paranodal loops.
Beyond the paranode: evidence for a broader role of DCC in myelin
In addition to disorganized paranodal loops, we detected compact myelin outfoldings and reduced amounts of MAG and MBP protein in aged PLPcreERT+DCCflox/flox mice. The underlying cause of these internodal myelin deficits is not clear. While DCC is concentrated at the paranode, it may also be present and influence the organization of other compartments of noncompact CNS myelin, such as the adaxonal or abaxonal membrane. For example, in PNS myelin, DCC was detected outside Schwann cell paranodes at Schmidt–Lantermann incisures (Webber et al., 2011). Surprisingly, the decreased levels of MBP and MAG protein detected in aged PLPcreERT+DCCflox/flox mice did not correlate with changes in g-ratio or in the percentage of myelinated axons. While this may seem unexpected, a previous study reported no effect on the percentage of myelinated axons or myelin thickness until >50% of MBP protein was lost (Shine et al., 1992). Still, the decreased levels of protein detected may have consequences for myelin stability and contribute to the structural defects observed at paranodes and compact myelin in PLPcreERT+DCCflox/flox mice.
It is possible that the myelin outfoldings and changes in myelin protein levels detected result from a primary defect in paranodal organization, which subsequently spreads to the internode to disrupt the stability of compact myelin. Alternatively, loss of DCC signaling may directly affect myelin stability via regulation of myelin protein translation or turnover, which could lead to the paranodal defects identified. DCC regulates src family kinase signaling and activation of the RhoGTPases cdc42, rac1, and rhoA to direct cytoskeletal organization (Lai Wing Sun et al., 2011). Loss of cdc42 or rac1 in oligodendrocytes results in the disorganization of compact and noncompact myelin membranes, including the formation of aberrant myelin outfoldings (Thurnherr et al., 2006), while loss of the src family kinase fyn results in hypomyelination (Sperber et al., 2001). Although the outfolded myelin found in the PLPcreERT+DCCflox/flox mice appears similar to what was detected in the cdc42 and rac1 conditional knock-outs, previous work has shown that the DCC-dependent morphological response of oligodendrocytes to netrin-1 during maturation in vitro is independent of cdc42 or rac1 activation (Rajasekharan et al., 2009). However, changes in Rho GTPase signaling mediated by DCC have been described during oligodendrocyte maturation (Rajasekharan et al., 2010), and it remains possible that DCC may regulate cdc42 and rac1 in mature myelinating oligodendrocytes in vivo.
Signaling downstream of DCC may also be linked to the detected decreased levels of MBP and MAG protein observed. Fyn, which is recruited to and activated downstream of DCC in oligodendrocytes (Rajasekharan et al., 2009), regulates the local translation of MBP (Lu et al., 2005; White et al., 2008). Interestingly, netrin-1 and DCC directly regulate local translation in other cell types (Campbell and Holt, 2001; Leung et al., 2006; Tcherkezian et al., 2010; Goldman et al., 2013) and deregulation of such signal transduction mechanisms in mature myelinating oligodendrocytes may contribute to the phenotypes reported here.
Implication in aging and disease
Deficits in the maintenance of paranodal junctions and the stability of compact myelin are thought to underlie the onset of a number of pathological conditions. For example, myelin instability has been proposed to contribute to cognitive deficits associated with aging (Bartzokis, 2004). Paranodal defects have also been proposed to constitute a predisposition to neuroinflammation leading to the development of demyelinating diseases such as multiple sclerosis (MS; Mastronardi and Moscarello, 2005). The disruption of paranodal junctions has been identified as an early sign of demyelination around MS lesions (Wolswijk and Balesar, 2003; Howell et al., 2006). Notably, a recent proteomic analysis of CSF samples collected from children during their initial presentation of CNS inflammation, who were subsequently diagnosed as having MS, identified elevated levels of proteins enriched at the node of Ranvier and paranodal junction, including elevated levels of DCC, but did not detect compact myelin proteins (Dhaunchak et al., 2012). These studies suggest that the paranode is a particularly vulnerable point along myelinated axons susceptible to degeneration and immune attack, and that damage at the paranode may precede degeneration of compact myelin and demyelination (Desmazières et al., 2012). Identifying and understanding the molecular mechanisms that promote the stability and maintenance of myelin is thus tremendously important to developing therapeutic strategies to treat and ultimately prevent demyelinating disease.
Footnotes
This work was supported by grants from the Multiple Sclerosis Society of Canada and Canadian Institutes of Health Research. S.-J.B. was supported by a Multiple Sclerosis Society of Canada studentship, J.M.B. by a Vanier Canada Graduate scholarship, and T.E.K. by a Scholarship from the Killam Trust and by a Chercheur Nationaux Award from the Fonds de la Recherche en Santé du Québec. We thank Sathy Rajasekharan for comments on this manuscript and Thomas Stroh and Jeannie Mui for guidance related to electron microscopy.
The authors declare no competing financial interests.
References
- Araki K, Araki M, Miyazaki J, Vassalli P. Site-specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A. 1995;92:160–164. doi: 10.1073/pnas.92.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KA, Moore SW, Jarjour AA, Kennedy TE. When a diffusible axon guidance cue stops diffusing: roles for netrins in adhesion and morphogenesis. Curr Opin Neurobiol. 2006;16:529–534. doi: 10.1016/j.conb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer's disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. 2004; author reply 49–62. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/S0896-6273(01)00294-X. [DOI] [PubMed] [Google Scholar]
- Bonnon C, Bel C, Goutebroze L, Maigret B, Girault JA, Faivre-Sarrailh C. PGY repeats and N-glycans govern the trafficking of paranodin and its selective association with contactin and neurofascin-155. Mol Biol Cell. 2007;18:229–241. doi: 10.1091/mbc.E06-06-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/S0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Buttermore ED, Dupree JL, Cheng J, An X, Tessarollo L, Bhat MA. The cytoskeletal adaptor protein band 4.1B is required for the maintenance of paranodal axoglial septate junctions in myelinated axons. J Neurosci. 2011;31:8013–8024. doi: 10.1523/JNEUROSCI.1015-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttermore ED, Thaxton CL, Bhat MA. Organization and maintenance of molecular domains in myelinated axons. J Neurosci Res. 2013;91:603–622. doi: 10.1002/jnr.23197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/S0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, Lubetzki C. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol. 2002;12:217–220. doi: 10.1016/S0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/S0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Desmazières A, Sol-Foulon N, Lubetzki C. Changes at the nodal and perinodal axonal domains: a basis for multiple sclerosis pathology? Mult Scler. 2012;18:133–137. doi: 10.1177/1352458511434370. [DOI] [PubMed] [Google Scholar]
- Dhaunchak AS, Becker C, Schulman H, De Faria O, Jr, Rajasekharan S, Banwell B, Colman DR, Bar-Or A, Canadian Pediatric Demyelinating Disease Group Implication of perturbed axoglial apparatus in early pediatric multiple sclerosis. Ann Neurol. 2012;71:601–613. doi: 10.1002/ana.22693. [DOI] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B. Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci. 1998;18:1642–1649. doi: 10.1523/JNEUROSCI.18-05-01642.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M, Weinberg RA. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J Neurosci. 1992;12:4890–4897. doi: 10.1523/JNEUROSCI.12-12-04890.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M, Montag D, Schachner M, Martini R. Crucial role for the myelin-associated glycoprotein in the maintenance of axon-myelin integrity. Eur J Neurosci. 1995;7:511–515. doi: 10.1111/j.1460-9568.1995.tb00347.x. [DOI] [PubMed] [Google Scholar]
- Goldman JS, Ashour MA, Magdesian MH, Tritsch NX, Harris SN, Christofi N, Chemali R, Stern YE, Thompson-Steckel G, Gris P, Glasgow SD, Grutter P, Bouchard JF, Ruthazer ES, Stellwagen D, Kennedy TE. Netrin-1 promotes excitatory synaptogenesis between cortical neurons by initiating synapse assembly. J Neurosci. 2013;33:17278–17289. doi: 10.1523/JNEUROSCI.1085-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano K, Takeya T, Iwasaki N, Nakayama J, Ohto T, Okada Y. A quantitative study of the progress of myelination in the rat central nervous system, using the immunohistochemical method for proteolipid protein. Brain Res Dev Brain Res. 1998;108:287–293. doi: 10.1016/S0165-3806(98)00063-7. [DOI] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell OW, Palser A, Polito A, Melrose S, Zonta B, Scheiermann C, Vora AJ, Brophy PJ, Reynolds R. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain. 2006;129:3173–3185. doi: 10.1093/brain/awl290. [DOI] [PubMed] [Google Scholar]
- Jarjour AA, Manitt C, Moore SW, Thompson KM, Yuh SJ, Kennedy TE. Netrin-1 is a chemorepellent for oligodendrocyte precursor cells in the embryonic spinal cord. J Neurosci. 2003;23:3735–3744. doi: 10.1523/JNEUROSCI.23-09-03735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarjour AA, Bull SJ, Almasieh M, Rajasekharan S, Baker KA, Mui J, Antel JP, Di Polo A, Kennedy TE. Maintenance of axo-oligodendroglial paranodal junctions requires DCC and netrin-1. J Neurosci. 2008;28:11003–11014. doi: 10.1523/JNEUROSCI.3285-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimpenfort P, Song JY, Proost N, Zevenhoven J, Jonkers J, Berns A. Deleted in colorectal carcinoma suppresses metastasis in p53-deficient mammary tumours. Nature. 2012;482:538–541. doi: 10.1038/nature10790. [DOI] [PubMed] [Google Scholar]
- Lai Wing Sun K, Correia JP, Kennedy TE. Netrins: versatile extracellular cues with diverse functions. Development. 2011;138:2153–2169. doi: 10.1242/dev.044529. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lasiene J, Matsui A, Sawa Y, Wong F, Horner PJ. Age-related myelin dynamics revealed by increased oligodendrogenesis and short internodes. Aging Cell. 2009;8:201–213. doi: 10.1111/j.1474-9726.2009.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KM, van Horck FP, Lin AC, Allison R, Standart N, Holt CE. Asymmetrical beta-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat Neurosci. 2006;9:1247–1256. doi: 10.1038/nn1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Ku L, Chen Y, Feng Y. Developmental abnormalities of myelin basic protein expression in fyn knock-out brain reveal a role of Fyn in posttranscriptional regulation. J Biol Chem. 2005;280:389–395. doi: 10.1074/jbc.M405973200. [DOI] [PubMed] [Google Scholar]
- Manitt C, Colicos MA, Thompson KM, Rousselle E, Peterson AC, Kennedy TE. Widespread expression of netrin-1 by neurons and oligodendrocytes in the adult mammalian spinal cord. J Neurosci. 2001;21:3911–3922. doi: 10.1523/JNEUROSCI.21-11-03911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manitt C, Thompson KM, Kennedy TE. Developmental shift in expression of netrin receptors in the rat spinal cord: predominance of UNC-5 homologues in adulthood. J Neurosci Res. 2004;77:690–700. doi: 10.1002/jnr.20199. [DOI] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J Cell Biol. 2002;156:567–577. doi: 10.1083/jcb.200111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372–381. doi: 10.1002/glia.20292. [DOI] [PubMed] [Google Scholar]
- Mastronardi FG, Moscarello MA. Molecules affecting myelin stability: a novel hypothesis regarding the pathogenesis of multiple sclerosis. J Neurosci Res. 2005;80:301–308. doi: 10.1002/jnr.20420. [DOI] [PubMed] [Google Scholar]
- Michalski JP, Anderson C, Beauvais A, De Repentigny Y, Kothary R. The proteolipid protein promoter drives expression outside of the oligodendrocyte lineage during embryonic and early postnatal development. PLoS One. 2011;6:e19772. doi: 10.1371/journal.pone.0019772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa AJ, Arevalo JC, Schiff R, Chao MV, Rosenbluth J. Role of transverse bands in maintaining paranodal structure and axolemmal domain organization in myelinated nerve fibers: effect on longevity in dysmyelinated mutant mice. J Comp Neurol. 2010;518:2841–2853. doi: 10.1002/cne.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Wang F, Dulac C, Chao SK, Nemes A, Mendelsohn M, Edmondson J, Axel R. Visualizing an olfactory sensory map. Cell. 1996;87:675–686. doi: 10.1016/S0092-8674(00)81387-2. [DOI] [PubMed] [Google Scholar]
- Moore SW, Correia JP, Lai Wing Sun K, Pool M, Fournier AE, Kennedy TE. Rho inhibition recruits DCC to the neuronal plasma membrane and enhances axon chemoattraction to netrin 1. Development. 2008;135:2855–2864. doi: 10.1242/dev.024133. [DOI] [PubMed] [Google Scholar]
- Moore SW, Biais N, Sheetz MP. Traction on immobilized netrin-1 is sufficient to reorient axons. Science. 2009;325:166. doi: 10.1126/science.1173851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Zhang X, Lynch CD, Sheetz MP. Netrin-1 attracts axons through FAK-dependent mechanotransduction. J Neurosci. 2012;32:11574–11585. doi: 10.1523/JNEUROSCI.0999-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza L, Huang JK, Colman DR. Organizing principles of the axoglial apparatus. Neuron. 2001;30:335–344. doi: 10.1016/S0896-6273(01)00306-3. [DOI] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–1793. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Rajasekharan S, Baker KA, Horn KE, Jarjour AA, Antel JP, Kennedy TE. Netrin 1 and Dcc regulate oligodendrocyte process branching and membrane extension via Fyn and RhoA. Development. 2009;136:415–426. doi: 10.1242/dev.018234. [DOI] [PubMed] [Google Scholar]
- Rajasekharan S, Bin JM, Antel JP, Kennedy TE. A central role for RhoA during oligodendroglial maturation in the switch from netrin-1-mediated chemorepulsion to process elaboration. J Neurochem. 2010;113:1589–1597. doi: 10.1111/j.1471-4159.2010.06717.x. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, Peles E, Salzer JL. Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci. 2000;20:8354–8364. doi: 10.1523/JNEUROSCI.20-22-08354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sango K, McDonald MP, Crawley JN, Mack ML, Tifft CJ, Skop E, Starr CM, Hoffmann A, Sandhoff K, Suzuki K, Proia RL. Mice lacking both subunits of lysosomal beta-hexosaminidase display gangliosidosis and mucopolysaccharidosis. Nat Genet. 1996;14:348–352. doi: 10.1038/ng1196-348. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Bonnet A, Erb M, Erne B, Bartsch U, Kern F, Mantei N, Sherman D, Suter U. The raft-associated protein MAL is required for maintenance of proper axon–glia interactions in the central nervous system. J Cell Biol. 2004;166:731–742. doi: 10.1083/jcb.200406092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/S0092-8674(00)81795-X. [DOI] [PubMed] [Google Scholar]
- Shatzmiller RA, Goldman JS, Simard-Emond L, Rymar V, Manitt C, Sadikot AF, Kennedy TE. Graded expression of netrin-1 by specific neuronal subtypes in the adult mammalian striatum. Neuroscience. 2008;157:621–636. doi: 10.1016/j.neuroscience.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Shekarabi M, Moore SW, Tritsch NX, Morris SJ, Bouchard JF, Kennedy TE. Deleted in colorectal cancer binding netrin-1 mediates cell substrate adhesion and recruits Cdc42, Rac1, Pak1, and N-WASP into an intracellular signaling complex that promotes growth cone expansion. J Neurosci. 2005;25:3132–3141. doi: 10.1523/JNEUROSCI.1920-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd MN, Pomicter AD, Velazco CS, Henderson SC, Dupree JL. Paranodal reorganization results in the depletion of transverse bands in the aged central nervous system. Neurobiol Aging. 2012;33 doi: 10.1016/j.neurobiolaging.2010.08.001. 203.e213–e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Shine HD, Readhead C, Popko B, Hood L, Sidman RL. Morphometric analysis of normal, mutant, and transgenic CNS: correlation of myelin basic protein expression to myelinogenesis. J Neurochem. 1992;58:342–349. doi: 10.1111/j.1471-4159.1992.tb09316.x. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, Stein PL, Scherer SS, McMorris FA. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol. 2000;150:657–666. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkezian J, Brittis PA, Thomas F, Roux PP, Flanagan JG. Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell. 2010;141:632–644. doi: 10.1016/j.cell.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurnherr T, Benninger Y, Wu X, Chrostek A, Krause SM, Nave KA, Franklin RJ, Brakebusch C, Suter U, Relvas JB. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J Neurosci. 2006;26:10110–10119. doi: 10.1523/JNEUROSCI.2158-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Tessier-Lavigne M, Miller RH. Netrin 1 mediates spinal cord oligodendrocyte precursor dispersal. Development. 2003;130:2095–2105. doi: 10.1242/dev.00424. [DOI] [PubMed] [Google Scholar]
- Webber CA, Christie KJ, Cheng C, Martinez JA, Singh B, Singh V, Thomas D, Zochodne DW. Schwann cells direct peripheral nerve regeneration through the Netrin-1 receptors, DCC and Unc5H2. Glia. 2011;59:1503–1517. doi: 10.1002/glia.21194. [DOI] [PubMed] [Google Scholar]
- White R, Gonsior C, Krämer-Albers EM, Stöhr N, Hüttelmaier S, Trotter J. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J Cell Biol. 2008;181:579–586. doi: 10.1083/jcb.200706164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolswijk G, Balesar R. Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain. 2003;126:1638–1649. doi: 10.1093/brain/awg151. [DOI] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–753. doi: 10.1242/dev.047951. [DOI] [PMC free article] [PubMed] [Google Scholar]