

Abstract
Emerging evidence has shown that miRNA-mediated gene expression modulation contributes to chronic pain, but its functional regulatory mechanism remains unknown. Here, we found that complete Freund's adjuvant (CFA)-induced chronic inflammation pain significantly reduced miRNA-219 (miR-219) expression in mice spinal neurons. Furthermore, the expression of spinal CaMKIIγ, an experimentally validated target of miR-219, was increased in CFA mice. Overexpression of spinal miR-219 prevented and reversed thermal hyperalgesia and mechanical allodynia and spinal neuronal sensitization induced by CFA. Concurrently, increased expression of spinal CaMKIIγ was reversed by miR-219 overexpression. Downregulation of spinal miR-219 in naive mice induced pain-responsive behaviors and increased p-NMDAR1 expression, which could be inhibited by knockdown of CaMKIIγ. Bisulfite sequencing showed that CFA induced the hypermethylation of CpG islands in the miR-219 promoter. Treatment with demethylation agent 5′-aza-2′-deoxycytidine markedly attenuated pain behavior and spinal neuronal sensitization, which was accompanied with the increase of spinal miR-219 and decrease of CaMKIIγ expression. Together, we conclude that methylation-mediated epigenetic modification of spinal miR-219 expression regulates chronic inflammatory pain by targeting CaMKIIγ.
Keywords: CaMKIIγ, epigenetics, microRNA, pain, spinal cord
Introduction
miRNAs are post-transcriptional regulators of gene expression that play an important role in a variety of neurophysiological and neuropathological processes, such as neuronal development, plasticity, and CNS diseases. Emerging evidence has shown that miRNA-mediated gene expression modulation in nociceptive pathways is involved in development and maintenance of chronic pain. Recently, several screening studies have shown dysregulated expression of miRNAs in pain pathways from primary afferent nociceptors, DRG, spinal cord, and brain areas associated with pain perception in different inflammatory, neuropathic, and cancer pain models. Manipulation of miRNA expression prevents and reverses persistent inflammatory, neuropathic, and cancer pain behavior (Favereaux et al., 2011; Imai et al., 2011; Bali et al., 2013). In addition, the functional consequences of miRNA regulation of downstream targets and intrinsic neuronal excitability have also been demonstrated. For example, GABAergic and opioidergic synaptic signals related to endogenous pain control are downregulated by miRNAs, such as miR-134 or miR-181a (Ni et al., 2013; Sengupta et al., 2013). miR-103 was shown to regulate neuropathic chronic pain by targeting voltage-gated calcium channels Cav2.1 and Cav2.2 (Favereaux et al., 2011). Despite the fact that miRNAs contributed to pain-related neuronal or synaptic modification and manipulating miRNAs had shown to be a potential pain therapeutic tool, the novel miRNAs for involvement of pain and its functional regulatory mechanisms still need to be determined.
Increasing studies have demonstrated that miR-219, expressed preferentially in the CNS, was involved in neurological and psychiatric disorders, including Alzheimer's disease (Lukiw, 2007), schizophrenia (Kocerha et al., 2009), and depression (Saus et al., 2010), suggesting a potential functional role of miR-219 in dysfunctional CNS diseases. In the present study, we investigated the role and its mechanism of spinal miR-219 in chronic pain with the following findings: (1) Our miRNA microarray profiling study identified spinal miR-219 to be downregulated in chronic pain status. (2) miR-219 negatively regulates the function of NMDA receptors, and to be a negative regulator of calcium/calmodulin-dependent protein kinase II γ (CaMKIIγ) (Kocerha et al., 2009). (3) Previous studies have shown that NMDA receptor and CaMKII played an important role in central sensitization related to chronic pain. Here, we show that, in a complete Freund's adjuvant (CFA)-induced chronic inflammatory pain model, alterations of spinal miR-219 expression regulate central sensitization and chronic pain behavior by targeting CaMKIIγ. Methylation-mediated epigenetic modification in the miR-219 promoter is an important regulatory mechanism of miR-219 expression.
Materials and Methods
Animals.
Adult male Kunming mice (20–25 g) were used in the present study. Chronic inflammatory pain was induced by subcutaneous administration of CFA (40 μl, Sigma-Aldrich) into the plantar surface of the hindpaw. Chronic constrictive injury model, formalin-induced acute pain model, intrathecal injection, paw withdrawal latency to thermal stimulus (a measure of hyperalgesia), and paw withdrawal thresholds to mechanical stimulus (a measure of allodynia) were performed as described previously (Ruan et al., 2010). All animal procedures were approved by the animal care committee of Xuzhou Medical College.
RNA extraction and microarray analysis.
Total RNA, including miRNA, was harvested using TRIzol (Invitrogen) and RNeasy mini kit (QIAGEN); 1 μg RNA from each sample was used for expression quantification with miRCURY LNA Array-version 18.0 (Exiqon).
MicroRNA-specific qRT-PCR.
A total of 1 μg RNA was reverse-transcribed to cDNA using specific primers (U6 RNA as an internal control) and TaqMan microRNA reverse transcription kit (Applied Biosystems). Expression analysis was performed using the TaqMan microRNA assay kit (Applied Biosystems).
Lentivirus system.
Lenti-miR-219, miR-219-sponge, and control lentivirus with empty vector were purchased from GenePharma. Design of miRNA sponges was performed according to the protocols by Ebert et al. (2007). Lentivirus titers are >108 TU. Lentivirus transfection was performed according to the protocol supplied by GenePharma. Briefly, 20 μl lentivirus and 1.5 μl polybrene (1.4 mg/ml) were added in a 24-well plate containing 1 × 105 HEK293T cells and 400 μl DMEM without FBS; after 24 h, transfection medium was replaced with 500 μl fresh complete medium containing 10% FBS; after 48 h culture, cells were collected. Each time, 1 μl lentivirus was intrathecally injected in vivo.
Construction of miR-219 target.
3′-UTR sequence of miR-219 targeting CaMKIIγ was synthesized: wtF, 5′-P-TCGAACTGAAGTATAGACTTTTCTGCTGgacaatcTGCATGGGCATCACCCCCTCACC-3′, wtR, 5′-P-GGCCGGTGAGGGGGTGATGCCCATGCAgattgtcCAGCAGAAAAGTCTATACTTCAGT-3′; mutF, 5′-P-TCGAACTGAAGTATAGACTTTTCTGCTGgtcaaacTGCATGGGCATCACCCCCTCACC-3′, mutR, 5′-P-GGCCGGTGAGGGGGTGATGCCCATGCAgtttgacCAGCAGAAAAGTCTATACTTCAGT-3′. wtF/wtR and mutF/mutR were, respectively, inserted into a dual-reporter psiCHECK2 plasmid digested by Xhol and NotI, and named as wild-type pCHK-wt-CaMKIIγ and mutant-type pCHK-mut- CaMKIIγ.
Construction of promoter reporter vector.
The defined region of miR-219 promoter was amplified from mouse genomic DNA (forward: 5′-CCGCTCGAGCCGTGGAAGTCAGTGGTATGAGG-3′; reverse: 5′-CCCAAGCTTAGAATTGCGTTTGGACAATCAGGA-3′) and cloned into pGL6 plasmid (Beyotime) via XhoI and HindIII digestion. Empty pGL6 vector was used as control plasmid.
Luciferase reporter assay.
HEK293T cells were cultured in DMEM with 10% FBS. HEK293T cells were seeded at 1 × 105 cells per 24 well. Identification of miR-219 targets was performed by transfecting pCHK-wt-CaMKIIγ or pCHK-mut-CaMKIIγ plasmids (50 ng) and miR-219 mimics (80 ng) into HEK293T cells. Identification of miR-219 promoter was performed by transfecting pGL6-219 (50 ng) and pRL-TK plasmid (2 ng) as an internal control (Promega). Cell lysates were prepared and subjected to luciferase assays using the Dual-luciferase reporter kit (Promega) at 24 h after transfection.
miRNA mimics or siRNA delivery.
miRNA mimics or siRNA was synthesized by GenePharma. miRNA or scrambled (Scr) mimics (20 μm, 5 μl) were intrathecally injected. CaMKIIγ siRNA (sense, 5′-GUAGAGUGCUUACGCAAAUTT-3′; antisense, 5′-AUUUGCGUAAGCACUCUACTT-3′) or a control siRNA (sense, 5′-UUCUCCGAACGUGUCACGUdTdT-3′; antisense, 5′-ACGUGACACGUUCGGAGAAdTdT-3′) was designed and validated in vitro and in vivo. CaMKIIγ siRNA or control siRNA (5 μm, 5 μl) was used for intrathecal injection.
FISH, immunofluorescence, and immunohistochemistry.
Spinal cords were rapidly dissected from perfused mice with 4% PFA, fixed with 4% PFA, cryoprotected in 30% sucrose, and then sectioned into 35 μm slices. miR-219 miRCURYTM LNA probes and a scrambled probe (5′-digoxin-TACGAGTCGAAACACCTATGCGCCT-digoxin-3′) were synthesized (Exiqon). FISH was performed using the FISH kit (Guangzhou Exon), and then FISH sections were incubated with NeuN antibody (MAB377, Millipore) or CaMKIIγ antibody (1:250, Abcam), finally incubated with fluorescent-conjugated secondary antibody (Alexa-594, Cell Signaling Technology). Fos immunohistochemistry was performed as described previously (Ruan et al., 2010).
Western blot analysis.
The spinal cord in the lumbar enlargement was rapidly removed, and ipsilateral dorsal spinal cord was separated and collected for Western blot analysis. Protein extractions were performed as described previously (Ruan et al., 2010). Protein (20–50 μg per sample) was separated by 10% SDS-PAGE gel and transferred onto nitrocellulose membrane. Membranes were probed with antibody against CaMKIIγ (1:500, Abcam), p-NR1 (S896) (1:500, Millipore), or control GAPDH (1:5000, Abcam). The immune complexes were detected by use of a NBT/BCIP assay kit (Sigma).
Analysis of CpG islands and bisulfite sequencing.
CpG islands, CpG dinucleotide-rich regions, were predicted using Methprimer (Li and Dahiya, 2002) for the upstream region of precursor miR-219 (pre-miR-219) (Chr 17B1). Mouse genomic DNA was extracted using QIAamp DNA Mini Kit (QIAGEN), then subjected to bisulfite conversion using EZ DNA methylation-Gold kit (Zymo). Bisulfite-modified DNAs were amplified using primers BSCF (5′-GAGATAGGATATTTTTTGGTAGT-3′; −310/−288) and BSCR (5′-CTCAAAAATTACATTTAAACAATC-3′; 21/45).
Statistical analysis.
Data are presented as mean ± SEM and analyzed using GraphPad Prism version 5.00 (GraphPad). The data for behavioral tests were analyzed with two-way ANOVA with two repeated factors followed by Tukey multiple-comparison tests. Unpaired comparisons were assessed by two-tailed Student's t test for protein, image, luciferase unit, or qRT-PCR analysis.
Results
Chronic pain reduces spinal miR-219 expression
To identify spinal miRNAs involved in chronic pain, we first compared spinal miRNA expression profiling between CFA-injected and control mice. Microarray analysis identified 12 miRNAs that were upregulated and 20 miRNAs that were downregulated by >40% in CFA animals. Three upregulated miR-219 and other two downregulated miRNAs in microarray analysis were randomly selected and were further confirmed by qRT-PCR (Fig. 1A,B). As spinal NMDA receptors and CaMKII play a critical role in chronic pain modulation (Latremoliere and Woolf, 2009) and miR-219 negatively regulates the function of NMDA receptors and the expression of CaMKIIγ (Kocerha et al., 2009), we further investigated the role of miR-219 in the chronic pain process. We collected the spinal tissue and examined the changes of spinal miR-219 expression at 3 d after CFA injection, 7 d after chronic constrictive injury surgery, and 2 h after formalin injection. The results showed that miR-219 expression was downregulated in both chronic inflammatory and chronic neuropathic pain, but not in acute formalin pain model (Fig. 1C). We then studied the time course of miR-219 expression in chronic inflammatory pain. No significant alteration of miR-219 expression was found in the acute phase of this pain model (2 h after CFA injection). However, miR-219 expression was significantly decreased from 1 to 10 d after CFA injection and then gradually returned to basal levels at 21 d after CFA injection (Fig. 1D). Combining miR-219 FISH and cell-type-specific immunofluorescence staining, we found that miR-219 was colocalized with NeuN (a neuronal maker) in spinal cord and that miR-219 expression was decreased in the CFA group versus the control group (Fig. 1E). These findings implicate the downregulation of spinal miR-219 to be involved in the development of chronic pain.
Figure 1.

Profiling of miRNAs and miR-219 expression in mouse spinal cord. A, The expression profiling of highly enriched miRNAs (upregulated and downregulated by >40%) was generated from microRNA microarray. Samples were collected from ipsilateral spinal cord at 3 d after CFA or vehicle injection mice (n = 3). B, Six differentially expressed miRNAs were subjected to qRT-PCR verification. C, Quantitative analysis of spinal miR-219 expression at 3 d after CFA injection, 7 d after chronic constrictive injury (CCI) surgery, and 2 h after formalin injection. *p < 0.05, versus control group. **p < 0.01, versus control group. n = 5. D, Time course of spinal miR-219 expression in CFA-induced chronic inflammation pain. *p < 0.05, versus control group. **p < 0.01, versus control group. n = 5. E, Combined miR-219 FISH (green) and NeuN (a neuronal marker, red) immunofluorescence staining in spinal cord at 3 d after CFA or saline injection. Scale bar, 25 μm.
Upregulation of miR-219 reverses CFA-induced pain behavior and spinal neuronal sensitization
Intrathecal injections of 2 tools, miRNA mimics (miR-219-mimics) and lentivirus (Lenti-miR-219), were used to upregulate spinal miR-219. The transfection efficiency of Lenti-miR-219 was validated in the HEK293T cell line and mouse spinal cord by qRT-PCR (Fig. 2A). Upregulation of miR-129 via injection of miR-219-mimics or Lenti-miR-219, but not scrambled miRNA or Lenti-vector, for 3 consecutive days significantly reversed CFA-induced thermal hyperalgesia and mechanical allodynia (Fig. 2B,C). Spinal Fos expression was used as a marker of spinal neuronal sensitization related to pain modulation. To further explore the effect of miR-219 upregulation on CFA-induced spinal neuronal sensitization, we investigated the change in Fos expression at 7 d after CFA injection (2 d after 3 continuous days of mimic or lentivirus injection). Intrathecal injection of miR-219-mimics or Lenti-miR-219, but not scrambled miRNA or Lenti-vector, inhibited the CFA-induced increase in Fos expression (Fig. 2D), suggesting that miR-219 upregulation reversed inflammatory pain by inhibiting spinal neuronal sensitization. To further determine the contribution of miR-219 in initiation of chronic inflammation pain, we pretreated animals with Lenti-miR-219 for 3 d before CFA injection and then assessed the preventive effect on CFA-induced pain behavior. We found that this pretreatment inhibited CFA-induced pain behavior and spinal Fos expression (Fig. 2E,F). Finally, we found that knockdown of spinal miR-219 by miR-219-sponge, a “miRNA-loss-of-function” strategy, produced thermal hyperalgesia and mechanical allodynia and increased Fos protein expression in naive mice (Fig. 2G,H). These findings suggested that spinal miR-219 contributes to the modulation of chronic pain processing.
Figure 2.
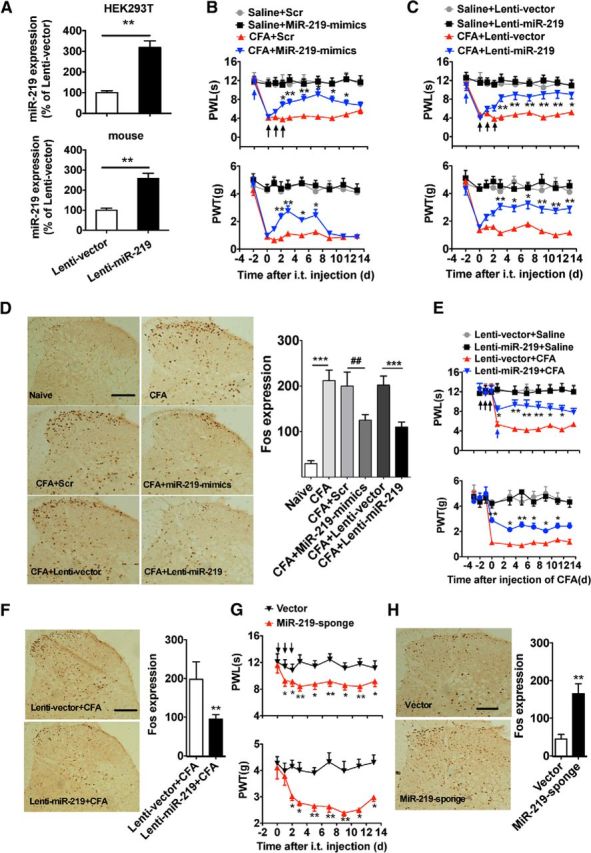
Manipulating spinal miR-219 regulates pain behavior and spinal neuronal sensitization. A, The validation of lenti-miR-219 transfection efficiency in cell lines and in vivo. **p < 0.01, versus Lenti-vector group. n = 6. The expression of miR-219 was measured at 72 h after Lenti-miR-219 transfection in HEK293T cell, or at 2 d after 3 consecutive days of intrathecal injection of Lenti-miR-219 in naive mice. B–D, Intrathecal injection of miR-219-mimics (B) or Lenti-miR-219 (C) for 3 consecutive days reversed CFA-induced thermal hyperalgesia (paw withdrawal latency) and mechanical allodynia (paw withdrawal threshold) and reversed the increase of spinal Fos expression (D). B, C, *p < 0.05, versus CFA + Scr or CFA + Lenti-vector group. **p < 0.01, versus CFA + Scr or CFA + Lenti-vector group. n = 8. D, ***p < 0.001, versus naive or CFA + Lenti-vector group. ##p < 0.01, versus CFA + Scr group. n = 6. E, F, Pretreatment with Lenti-miR-219 for 3 consecutive days prevented CFA-induced thermal hyperalgesia and mechanical allodynia (E) and the increase of spinal Fos expression (F). *p < 0.05, versus Lenti-vector + CFA group. **p < 0.01, versus Lenti-vector + CFA group. n = 6–10. Blue arrow indicates CFA or saline injection; black arrow indicates miR-219-mimics/Scr or lenti-miR-219/Lenti-vector injection. G, H, Intrathecal injection of miR-219-sponge for 3 consecutive days produced thermal hyperalgesia and mechanical allodynia (G) and increased the spinal Fos expression in naive mice (H). *p < 0.05, versus vector group. **p < 0.01, versus vector group. n = 6–8. Black arrow indicates miR-219-sponge or vector injection.
miR-219 targeting CaMKIIγ regulates pain
To identify a potential pain-relevant target gene of miR-219, three independent prediction programs, including TargetScan, miRanda, and PicTar, were performed in TarMir (http://www.tarmir.rgcb.res.in/). A total of 56 genes were predicted to be miR-219 targets (data not shown). Some of these target genes (e.g., Kcna4 and Epha4) were reported to have contributed to pain processes (Cruz-Orengo et al., 2006; Langeslag et al., 2014). As CaMKIIγ has conserved miR-219 binding sites within its 3′ UTR (bp, 1227–1233) and its involvement in modulation of neuronal plasticity through the NMDAR pathway in CNS (Kocerha et al., 2009), we chose CaMKIIγ to evaluate its potential role as a target of miR-219 in pain modulation. To experimentally validate the in silico predictions, we first generated reporter vectors containing 3′ UTR with miR-219-recognized sequences from CaMKIIγ (Fig. 3A). Cotransfection of miR-219 mimics with the reporter pCHK-wt-CaMKIIγ resulted in significantly reduced luciferase activity compared with pCHK-mut-CaMKIIγ. As a control for miR-219, mimics of miR-34a (an enriched miRNA in spinal cord) did not decrease the luciferase activities of wild-type or mutation vector (Fig. 3A). Furthermore, we determined the regulatory role of miR-219 in CaMKIIγ expression at the in vivo level. We found that spinal CaMKIIγ protein expression was increased in CFA mice, which was reversed by intrathecal injection of miR-219 mimics or lentivirus-mediated miR-219 overexpression, but not by scrambled miRNA or lentivirus vector control (Fig. 3B,C). In addition, spinal CaMKIIγ expression was increased by knockdown of miR-219 with intrathecal injection of miR-219-sponge in naive mice (Fig. 3D). FISH-immunofluorescent costaining revealed that miR-219 and CaMKIIγ were coexpressed in spinal cells and that the decrease of miR-219 expression was accompanied by the increase of CaMKIIγ-positive expression in the CFA group compared with the control group (Fig. 3E). These in vitro and in vivo findings suggested that CaMKIIγ is a direct target of miR-219.
Figure 3.

miR-219 targeting CaMKIIγ regulates chronic inflammation pain. A, Validation of miR-219 targeting CaMKIIγ. A mutation was generated in the CaMKIIγ-3′-UTR sequence in the complementary site for the seed region of miR-219 as indicated (pCHK-mut-CaMKIIγ). **p < 0.01, versus pCHK-mut-CaMKIIγ or empty vector group. n = 6. B, C, The increased spinal CaMKIIγ expression in CFA mice was reversed by intrathecal injection of miR-219 mimics or Lenti-miR-219. **p < 0.01, versus adjacent groups. n = 5. CaMKIIγ was measured at 4 d after miR-219 mimic injection (6 d after CFA injection) or at 5 d after Lenti-miR-219 injection (7 d after CFA injection), respectively. D, Spinal CaMKIIγ expression was increased by intrathecal injection of miR-219-sponge in naive mice. **p < 0.01, versus adjacent groups. n = 5. CaMKIIγ was measured at 5 d after injection of miR-219-sponge. E, FISH (miR-219, green) immunofluorescent (CaMKIIγ, red) costaining in spinal cord at 3 d after CFA injection. Scale bar, 25 μm. F, G, Knockdown of CaMKIIγ significantly inhibited or reversed thermal hyperalgesia and mechanical allodynia induced by miR-219-sponge in naive mice. *p < 0.05, versus CaMKIIγ siRNA + miR-219-sponge or miR-219-sponge + CaMKIIγ siRNA group. n = 8. Blue arrow indicates CaMKIIγ siRNA or control siRNA injection; black arrow indicates miR-219-sponge injection. H, The increased expression of spinal p-NR1 in CFA-injected mice (left) or in miR-219-sponge-injected naive mice (right) was reversed by intrathecal injection of CaMKIIγ siRNA. **p < 0.01, versus adjacent groups. *p < 0.05, versus adjacent groups. n = 5. p-NR1 was measured at 4 d after CaMKIIγ or control siRNA injection (6 d after CFA injection), or at 5 d after CaMKIIγ or control siRNA injection (7 d after miR-219-sponge injection). I, MK-801 markedly inhibited thermal hyperalgesia and mechanical allodynia induced by miR-219-sponge in naive mice. *p < 0.05, versus miR-219-sponge + saline group. n = 8. MK-801 (10 nmol) was intrathecally injected at 30 min after pain behavior test. Blue arrow indicates MK-801 or saline injection; black arrow indicates miR-219-sponge injection.
Our data have shown that knockdown of spinal miR-219 induced pain behavior. To explore the role of CaMKIIγ in the mediation of pain modulation by miR-219 at behavioral level, we pretreated or post-treated the animals with an siRNA to knock down CaMKIIγ before or after intrathecal injection of miR-219-sponge, then measured the behavioral response. We found that knockdown of CaMKIIγ significantly inhibited or reversed thermal hyperalgesia and mechanical allodynia induced by downregulation of spinal miR-219 in naive mice, suggesting that the increase of CaMKIIγ expression mediates miR-219 downregulation-induced pain behavior (Fig. 3F,G).
To further determine an association between CaMKIIγ and NMDAR activity under chronic inflammatory pain, we first examined whether knockdown of CaMKIIγ with siRNA could decrease the expression of phosphorylation NMDAR1 (p-NR1). We found that spinal p-NR1 expression was increased not only in CFA mice, but also in naive mice with miR-219-sponge injection, which was reversed by spinal CaMKIIγ knockdown through intrathecal injection of CaMKIIγ siRNA (Fig. 3H). Finally, behavioral testing showed that thermal hyperalgesia and mechanical allodynia induced by downregulation of spinal miR-219 in naive mice were reversed by intrathecal injection of the NMDA receptor antagonist MK-801 (Fig. 3I). Together, these results suggested that spinal miR-219 expression regulates pain behaviors by targeting CaMKIIγ, and NMDAR may participate in this processes.
DNA methylation regulates miR-219 expression and CFA-induced pain behavior
As epigenetic modification has recently been implicated in DNA methylation-mediated silencing of miRNAs (Lujambio et al., 2007), we further investigated whether DNA methylation of miR-219 promoter was involved in the process of chronic pain. Because CpG islands are often colocalized in the promoter region and cytosine methylation of CpG dinucleotides frequently leads to transcriptional silencing, CpG islands have been recognized as a crucial epigenetic regulation region in DNA methylation modification. Our analysis of the 2000 bp upstream sequence of pre-miR-219 revealed the presence of a well-defined CpG island from −266 to −57 (relative to first nucleotide of pre-miR-219) (data not shown). Next, we cloned a 1.359 kb segment with the predicted CpG island into the pGL6 firefly luciferase reporter (pGL6-219) and examined its ability to drive luciferase expression in HEK293T cells. We found that this promoter region produced high luciferase activities compared with no promoter in pGL6 control (Fig. 4A), indicating that this region of the promoter contains the regulatory elements and controls miR-219 expression. However, the pGL6-219 in vitro methylated by methyltransferase M.SssI before being introduced into cells abolished the enhanced activities of luciferase (Fig. 4B). No significant alterations were found in pGL6 control. These results suggested a direct role of DNA methylation of this promoter containing the CpG island in transcriptional regulation of miR-219 gene expression.
Figure 4.

DNA methylation of miR-219 promoter regulates spinal miR-219 expression and chronic pain behavior. A, The activity of the cloned miR-219 promoter was identified by luciferase reporter assay. **p < 0.01, versus adjacent groups. n = 6. B, The activity of methylated or demethylated promoter of miR-219 encompassing CpG islands were detected by firefly luciferase reporter assays in HEK293T cells. **p < 0.01, versus adjacent groups. n = 6. C, CFA increased CpG island methylation of miR-219 promoter, which could be reversed by treatment with demethylation agent Aza. White and black dots represent demethylated and methylated CpG dinucleotide, respectively. Each line indicates an individual sequence. D–G, Aza treatment increased the expression of spinal miR-219 (D) and reversed CFA-induced pain behavior (E) and central sensitization (F), which is accompanied with the decrease in CaMKIIγ expression (G). *p < 0.05, versus adjacent groups. **p < 0.01, versus adjacent groups. n = 5–10. Scale bar, 25 μm. Methylation, Fos, CaMKIIγ, or miR-219 was measured at 4 d after 3 continuous days of intrathecal injection of Aza (875 μmol/d) (at 6 d after CFA injection). Blue arrow indicates CFA or saline injection; black arrow indicates Aza or vehicle injection.
To evaluate whether chronic inflammatory pain caused the hypermethylation of miR-219 promoter, we performed the bisulfite sequencing to analyze the methylation status of the CpG island. The results showed that the fragment surrounding the CpG island was densely methylated in CFA mice compared with the control group, which could be reversed by treatment with the demethylation agent 5′-aza-2′-deoxycytidine (Aza) (Fig. 4C). Furthermore, treatment with Aza also increased spinal miR-219 expression in CFA mice (Fig. 4D), which was accompanied by alleviation of pain behavior and inhibition of spinal Fos and CaMKIIγ expression (Fig. 4E–G). Collectively, these results suggest the functional relevance of DNA demethylation of miR-219 and chronic pain behavior.
Discussion
Central sensitization, characterized by increase in the excitability of neurons and enhancement of response to nociceptive or/and non-nociceptive stimulus, plays a critical role in the pathogenesis of chronic pain. The induction and maintenance of central sensitization are dependent on maladaptive alterations in the expression, distribution, and activity of ion channels, receptors, and intracellular signal transduction pathways. Aberrant pain-related gene expression is one of most prominent contributors to these maladaptive processes. Therefore, the unraveling of the genetic basis and its regulatory mechanisms involved in central sensitization will improve our understanding of chronic pain and provide potential targets for developing novel therapeutic strategies. miRNAs, as a regulatory mechanism of gene expression, attract widespread attention on their potential role in complicated biological processes and human diseases. For example, emerging evidence indicates that miRNA is a key regulator not only in neurodegenerative disorders, such as Alzheimer's, Parkinson's, amyotrophic lateral sclerosis, and polyglutamine disorders (Bicchi et al., 2013), but also in neuroinflammatory diseases, such as rheumatoid arthritis, psoriasis, and multiple sclersosis (Ksiazek-Winiarek et al., 2013).
Recent reports have shown a strong connection between miRNA modulation and chronic pain processes. Conditional deletion of Dicer, a central enzyme in miRNA processing, in DRG sensory neurons leads to deficits in inflammatory pain (Zhao et al., 2010). miRNA expression profiling study in DRG tissue identified 63 and 57 miRNAs whose expression is significantly altered after neuropathic pain (von Schack et al., 2011) and metastatic bone-cancer pain model (Bali et al., 2013). Furthermore, upregulation of spinal miR-124 or miR-103 is reported to prevent and treat persistent inflammatory and neuropathic pain (Favereaux et al., 2011; Willemen et al., 2012). Inhibiting the tumor-induced upregulation of miR-146a and miR-183 in sensory neurons markedly attenuates tumor-mediated hyperalgesia (Li et al., 2013). In contrast, augmenting the expression of miR-370 in DRGs leads to exaggerated tumor-mediated hyperalgesia (Bali et al., 2013). These findings demonstrate miRNAs as key regulators of physiologic and pathologic processes underlying chronic pain and suggest that specific miRNAs may act as new molecular targets for pain prevention and relief. In this study, our results reveal that chronic, but not acute, inflammation pain had a significant correlation with miR-219 expression and that the different correlation may be associated with inconsistent mechanisms underlying chronic pain and those underlying acute pain. In addition, the relationships between maladaptive cellular and behavioral changes and miR-219 expression in chronic pain are consistent with previous findings that miR-219 is involved in NMDAR-mediated synaptic plasticity and psychiatric disorders.
Furthermore, in agreement with a previous report (Kocerha et al., 2009), we demonstrated that CaMKIIγ, a target gene for miR-219, mediates central sensitization in chronic pain. To our knowledge, few studies investigate the functional significant of CaMKIIγ in physiological and pathological processes in the CNS. Recent studies have implicated CaMKIIγ in various physiological and pathological conditions, including bipolar spindle formation, survival and proliferation, apoptosis, and differentiation of cancer (Si and Collins, 2008). In this study, we found that CFA-induced chronic inflammation pain was accompanied with an increase in CaMKIIγ expression. Based on the core regulatory role of CaMKII family of kinases in NMDAR signaling and central sensitization related to chronic pain, we propose that CaMKIIγ mediated the role of spinal miR-219 in pain modulation through negatively regulating the function of NMDA receptors.
Despite a large body of molecular and functional evidence supporting the alteration of miRNA expression in several physiological and pathological processes, little is known about how the miRNA gene expression itself is regulated in these processes. DNA methylation is an important epigenetic modification in the mammalian genome. The growing evidence has linked DNA methylation with some human diseases, including cancer, neurological, and psychiatric disorders, such as Alzheimer disease and depression (Lujambio et al., 2007; Jowaed et al., 2010). Recent studies found methyl-CpG-binding protein 2, a transcriptional repressor complex that induces remodeling and gene silencing, has an important role in gene expression in spinal cord and DRG neurons after CFA-induced inflammation pain (Géranton et al., 2007). Induction of chronic pain is accompanied by decreased global methylation in the prefrontal cortex and amygdala (Tajerian et al., 2013). Therefore, DNA methylation might be involved in mediating the pathologies associated with chronic pain. In vitro and in vivo studies showed that DNA CpG methylation specifically silences miRNA expression in human cancer cells (Lujambio et al., 2007). Considering that ∼50% of miRNA promoters have CpG islands, miRNA methylation might be an important mechanism by which miRNA transcription is regulated. In this study, we found chronic inflammatory pain induced the hypermethylation of CpG islands in the miR-219 promoter. Treatment with demethylation agent markedly increased the spinal miR-219 expression, which is accompanied with alleviation of pain behavior, inhibition of central sensitization, and decrease of spinal CaMKIIγ expression in CFA mice. This is the first report on methylation-mediated epigenetic modification of miR-219 expression and its functional significance in chronic pain in vivo.
In conclusion, this study expands our knowledge about the functional role of miR-219 in CNS and elucidates a novel mechanism for regulation of individual miR-219 expression in the development of chronic pain.
Footnotes
This work was supported by National Natural Science Foundation of China Grants 81070888 and 81230025 to J.-L.C. and Grant 81271231 to Z.P., Key Project of the Natural Science Foundation of Jiangsu Education Department 11KJA320001 to. J.-L.C., China Postdoctoral Science Foundation 2012M511322 to Z.P., Natural Science Foundation of Jiangsu Province BK2011196 to Z.P., and a Project Funded by the Qing Lan Project of Jiangsu, Jiangsu Provincial Six Talent Summit Action Plans, and the Priority Academic Program Development of Jiangsu Higher Education Institutions. We thank Dr. Ming-Hu Han and Barbara Juarez (Department of Pharmacology and Systems Therapeutics, Icahn School of Medicine at Mount Sinai) for their helpful discussion on the manuscript.
The authors declare no competing financial interests.
References
- Bali KK, Selvaraj D, Satagopam VP, Lu J, Schneider R, Kuner R. Genome-wide identification and functional analyses of microRNA signatures associated with cancer pain. EMBO Mol Med. 2013;5:1740–1758. doi: 10.1002/emmm.201302797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicchi I, Morena F, Montesano S, Polidoro M, Martino S. MicroRNAs and molecular mechanisms of neurodegeneration. Genes (Basel) 2013;4:244–263. doi: 10.3390/genes4020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Orengo L, Figueroa JD, Velázquez I, Torrado A, Ortíz C, Hernández C, Puig A, Segarra AC, Whittemore SR, Miranda JD. Blocking EphA4 upregulation after spinal cord injury results in enhanced chronic pain. Exp Neurol. 2006;202:421–433. doi: 10.1016/j.expneurol.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favereaux A, Thoumine O, Bouali-Benazzouz R, Roques V, Papon MA, Salam SA, Drutel G, Léger C, Calas A, Nagy F, Landry M. Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103: role in pain. EMBO J. 2011;30:3830–3841. doi: 10.1038/emboj.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Saeki M, Yanase M, Horiuchi H, Abe M, Narita M, Kuzumaki N, Suzuki T, Narita M. Change in microRNAs associated with neuronal adaptive responses in the nucleus accumbens under neuropathic pain. J Neurosci. 2011;31:15294–15299. doi: 10.1523/JNEUROSCI.0921-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains. J Neurosci. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocerha J, Faghihi MA, Lopez-Toledano MA, Huang J, Ramsey AJ, Caron MG, Sales N, Willoughby D, Elmen J, Hansen HF, Orum H, Kauppinen S, Kenny PJ, Wahlestedt C. MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc Natl Acad Sci U S A. 2009;106:3507–3512. doi: 10.1073/pnas.0805854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek-Winiarek DJ, Kacperska MJ, Glabinski A. MicroRNAs as novel regulators of neuroinflammation. Mediators Inflamm. 2013;2013:172351. doi: 10.1155/2013/172351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeslag M, Malsch P, Welling A, Kress M. Reduced excitability of gp130-deficient nociceptors is associated with increased voltage-gated potassium currents and Kcna4 channel upregulation. Pflugers Arch. 2014 doi: 10.1007/s00424-014-1443-0. doi: 10.1007/s00424-014-1443-0. Advance online publication. Retrieved Jan. 25, 2014. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Li X, Kroin JS, Kc R, Gibson G, Chen D, Corbett GT, Pahan K, Fayyaz S, Kim JS, van Wijnen AJ, Suh J, Kim SG, Im HJ. Altered spinal microRNA-146a and the microRNA-183 cluster contribute to osteoarthritic pain in knee joints. J Bone Miner Res. 2013;28:2512–2522. doi: 10.1002/jbmr.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setién F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Git A, Spiteri I, Das PP, Caldas C, Miska E, Esteller M. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- Ni J, Gao Y, Gong S, Guo S, Hisamitsu T, Jiang X. Regulation of mu-opioid type 1 receptors by microRNA134 in dorsal root ganglion neurons following peripheral inflammation. Eur J Pain. 2013;17:313–323. doi: 10.1002/j.1532-2149.2012.00197.x. [DOI] [PubMed] [Google Scholar]
- Ruan JP, Zhang HX, Lu XF, Liu YP, Cao JL. EphrinBs/EphBs signaling is involved in modulation of spinal nociceptive processing through a mitogen-activated protein kinases-dependent mechanism. Anesthesiology. 2010;112:1234–1249. doi: 10.1097/ALN.0b013e3181d3e0df. [DOI] [PubMed] [Google Scholar]
- Saus E, Soria V, Escaramís G, Vivarelli F, Crespo JM, Kagerbauer B, Menchón JM, Urretavizcaya M, Gratacòs M, Estivill X. Genetic variants and abnormal processing of pre-miR-182, a circadian clock modulator, in major depression patients with late insomnia. Hum Mol Genet. 2010;19:4017–4025. doi: 10.1093/hmg/ddq316. [DOI] [PubMed] [Google Scholar]
- Sengupta JN, Pochiraju S, Kannampalli P, Bruckert M, Addya S, Yadav P, Miranda A, Shaker R, Banerjee B. MicroRNA-mediated GABA Aalpha-1 receptor subunit down-regulation in adult spinal cord following neonatal cystitis-induced chronic visceral pain in rats. Pain. 2013;154:59–70. doi: 10.1016/j.pain.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si J, Collins SJ. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008;68:3733–3742. doi: 10.1158/0008-5472.CAN-07-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, Szyf M, Stone LS. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One. 2013;8:e55259. doi: 10.1371/journal.pone.0055259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schack D, Agostino MJ, Murray BS, Li Y, Reddy PS, Chen J, Choe SE, Strassle BW, Li C, Bates B, Zhang L, Hu H, Kotnis S, Bingham B, Liu W, Whiteside GT, Samad TA, Kennedy JD, Ajit SK. Dynamic changes in the microRNA expression profile reveal multiple regulatory mechanisms in the spinal nerve ligation model of neuropathic pain. PLoS One. 2011;6:e17670. doi: 10.1371/journal.pone.0017670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemen HL, Huo XJ, Mao-Ying QL, Zijlstra J, Heijnen CJ, Kavelaars A. MicroRNA-124 as a novel treatment for persistent hyperalgesia. J Neuroinflammation. 2012;9:143. doi: 10.1186/1742-2094-9-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lee MC, Momin A, Cendan CM, Shepherd ST, Baker MD, Asante C, Bee L, Bethry A, Perkins JR, Nassar MA, Abrahamsen B, Dickenson A, Cobb BS, Merkenschlager M, Wood JN. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J Neurosci. 2010;30:10860–10871. doi: 10.1523/JNEUROSCI.1980-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]