

Abstract
Activation of presynaptic adenosine A1 receptors (A1Rs) causes substantial synaptic depression during hypoxia/cerebral ischemia, but postsynaptic actions of A1Rs are less clear. We found that A1Rs and GluA2-containing AMPA receptors (AMPARs) form stable protein complexes from hippocampal brain homogenates and cultured hippocampal neurons from Sprague Dawley rats. In contrast, adenosine A2A receptors (A2ARs) did not coprecipitate or colocalize with GluA2-containing AMPARs. Prolonged stimulation of A1Rs with the agonist N6-cyclopentyladenosine (CPA) caused adenosine-induced persistent synaptic depression (APSD) in hippocampal brain slices, and APSD levels were blunted by inhibiting clathrin-mediated endocytosis of GluA2 subunits with the Tat-GluA2–3Y peptide. Using biotinylation and membrane fractionation assays, prolonged CPA incubation showed significant depletion of GluA2/GluA1 surface expression from hippocampal brain slices and cultured neurons. Tat-GluA2–3Y peptide or dynamin inhibitor Dynasore prevented CPA-induced GluA2/GluA1 internalization. Confocal imaging analysis confirmed that functional A1Rs, but not A2ARs, are required for clathrin-mediated AMPAR endocytosis in hippocampal neurons. Pharmacological inhibitors or shRNA knockdown of p38 MAPK and JNK prevented A1R-mediated internalization of GluA2 but not GluA1 subunits. Tat-GluA2–3Y peptide or A1R antagonist 8-cyclopentyl-1,3-dipropylxanthine also prevented hypoxia-mediated GluA2/GluA1 internalization. Finally, in a pial vessel disruption cortical stroke model, a unilateral cortical lesion compared with sham surgery reduced hippocampal GluA2, GluA1, and A1R surface expression and also caused synaptic depression in hippocampal slices that was consistent with AMPAR downregulation and decreased probability of transmitter release. Together, these results indicate a previously unknown mechanism for A1R-induced persistent synaptic depression involving clathrin-mediated GluA2 and GluA1 internalization that leads to hippocampal neurodegeneration after hypoxia/cerebral ischemia.
Keywords: adenosine A1 receptors, AMPA receptor trafficking, clathrin-mediated endocytosis, persistent synaptic depression, pial vessel disruption, stroke
Introduction
Adenosine, a ubiquitous purine nucleoside, plays a putative role as a neuromodulator in both physiological and pathological conditions. Endogenous adenosine is known to be released from neurons and glial cells; and to date, four adenosine receptors have been identified: A1, A2A, A2B, and A3 receptors (Michaelis et al., 1988; Dunwiddie and Masino, 2001; Fredholm et al., 2001). In cerebral ischemia, adenosine levels rise, rapidly inducing synaptic depression through adenosine A1 receptor (A1R) activation (Fowler, 1990; Fowler et al., 2003; Gervitz et al., 2003), which inhibit presynaptic neurotransmitter release (Lupica et al., 1992) by decreasing calcium influx into presynaptic nerve terminals (Dunwiddie and Masino, 2001). Adenosine's postsynaptic actions include inhibition of NMDAR-mediated currents (de Mendonça et al., 1995), inhibition of adenylate cyclase, and stimulation of potassium conductances, all through A1R actions (Siggins and Schubert, 1981; Segal, 1982; Proctor and Dunwiddie, 1983; Haas and Greene, 1984). Microscopy evidence shows A1Rs located on somatodendritic hippocampal structures (Ochiishi et al., 1999), and shown to be highly localized to the active zone and postsynaptic density in hippocampal synapses (Rebola et al., 2003a), suggesting that actions of adenosine are not confined to presynaptic membranes. Some reports also suggest that postsynaptic adenosine receptors (e.g., adenosine A2A receptors [A2ARs] and A3R) regulate glutamatergic receptor function and surface distribution (Dennis et al., 2011; Dias et al., 2012).
AMPARs are glutamate receptors that form functional tetramers of subunits GluA1-GluA4 (Hollmann and Heinemann, 1994; Wenthold et al., 1996) and have been implicated in ischemic brain damage, which reflects increased expression of GluA2-deficient (Ca2+-permeable) AMPARs on postsynaptic membranes, causing increased Ca2+ permeability (Hollmann et al., 1991; Pellegrini-Giampietro et al., 1994; Gorter et al., 1997; Liu et al., 2006; Liu and Zukin, 2007; Kumar and Mayer, 2013). Despite this knowledge, it is still unclear whether postsynaptic A1Rs regulate AMPARs in stroke.
During acute administration of the A1R agonist N6-cyclopentyladenosine (CPA), we observed a profound adenosine-induced persistent synaptic depression (APSD) in hippocampal CA3-CA1 synapses and elevation of phosphorylated p38 MAPK (mitogen-activated protein kinase) and JNK (c-Jun N-terminal kinase) in hippocampal membrane fractions. Notably, Rap1 and Rap2, GTPases dependent on p38 MAPK and JNK, respectively, mediate NMDAR-dependent AMPAR removal during LTD (Zhu et al., 2002, 2005). LTD in mouse primary visual cortex was accompanied by activation of p38 MAPK and clathrin-mediated endocytosis of GluA2 AMPARs (Xiong et al., 2006).
We therefore propose that A1R-mediated p38 MAPK and JNK activation plays a crucial role in regulating AMPAR trafficking during prolonged hypoxia or an in vivo focal cortical small vessel stroke model using Type II pial vessel disruption (Wang and Walz, 2003; Hua and Walz, 2006b; Cayabyab et al., 2013). Our results reveal a previously unknown mechanism for APSD involving clathrin-mediated GluA2 internalization via p38 MAPK and JNK signaling observed after hypoxic/ischemic insult. Our results also modify the original GluA2 hypothesis of excitotoxicity (Pellegrini-Giampietro et al., 1997) in that selective activation of A1Rs can mediate GluA2-containing AMPAR internalization in vulnerable regions, including the hippocampus, representing an important mechanism of ischemic damage with therapeutic potential.
Materials and Methods
Hippocampal slice preparation and treatments.
Hippocampal slices from male Sprague Dawley rats (P21-P28 d) were anesthetized with halothane and rapidly decapitated according to protocols approved by the University Committee of Animal Care and Supply at the University of Saskatchewan. The brains were extracted and immediately placed into ice-cold oxygenated dissection medium. Hippocampal slices (400 μm thick) were cut using a vibrating tissue slicer (Vibram VT1200S, Leica) and maintained for 60–90 min in ACSF before performing electrophysiological recordings or biochemical analysis. Recipes for ACSF and dissection solutions and details of recording conditions were described previously (Brust et al., 2006, 2007). All experiments were conducted at room temperature.
Pial vessel disruption (PVD) as a model of small-vessel stroke.
Class II size pial vessel disruption (PVD) has been shown to induce a focal cortical lesion that, within 3 weeks of surgery lesion, leads to lacunar infarction-like fluid-filled cyst that does not extend to the corpus callosum. This fluid-filled cavity is tightly surrounded by a barrier consisting of processes from reactive astrocytes, the hallmark of lacunar infarctions (Hua and Walz, 2006, 2008). The genesis of such a lacuna (cavitation) has been studied in more detail previously. It has been found that treatment with minocycline or the specific matrix metalloproteinase inhibitor batimastat, an experimental anticancer drug, prevents cavitation and leads to a lesion filled with reactive astrocytes and no barrier (Cayabyab et al., 2013). The procedure is described in detail in previous studies (Wang and Walz, 2003; Hua and Walz, 2006a, b; Cayabyab et al., 2013). Briefly, Sprague Dawley rats <2% isoflurane anesthesia and buprenorphine treatment for pain management received a craniotomy with 5 mm-diameter on the right and rostral side of the bregma adjacent to the coronal and sagittal sutures. After opening of the dura, the Class II pial vessels were disrupted with fine-tipped forceps. The piece of bone was placed back, and the wound was closed with a clip. Sham animals received the same treatment with dura removal but no vessel disruption. This procedure, including the recovery period of the animal, was approved under permit 20020024 by the Animal Research Ethics Board of the University of Saskatchewan.
To investigate the impact of a remote focal ischemic injury on hippocampal signaling, we used this modified pial vessel disruption model. The modification consists of the disruption of the Class II medium vessels only and not the Class I large vessels. We used this in vivo animal stroke model because it has distinct advantages over other models. For example, this PVD model is a small-vessel stroke model that produces permanent damage to Class II size vessels (i.e., a nonreperfusion model), and the cortical lesion volumes can be reliably reproduced and have similarities to a lacunar infarction (Wang and Walz, 2003; Hua and Walz, 2006b). In contrast, most focal or global stroke models involve transient occlusion of large vessels, such as the middle cerebral arteries or carotid arteries (Pellegrini-Giampietro et al., 1992; Gorter et al., 1997; McBean and Kelly, 1998; Prosser-Loose et al., 2010; Tu et al., 2010) and the cerebral ischemic damage often encompasses large volumes of brain regions. Our PVD model results in an ∼1 mm3 cortical lesion volume and is a more subtle small vessel injury in the cerebral cortex (Wang and Walz, 2003). Previous cortical devascularization studies have shown that focal cortical ischemia affects the hippocampus by altering hippocampal synaptic transmission (Ramos et al., 2004) and increasing expression of both c-fos (Herrera and Robertson, 1990) and nerve growth factor (Figueiredo et al., 1995), and adenosine has been implicated in the increased expression of both of these regulatory factors in other brain regions (Rudolphi and Schubert, 1995; Svenningsson et al., 1999). Because stroke in humans (Laghi Pasini et al., 2000) and transient middle cerebral artery occlusion in large-vessel animal stroke models (Matsumoto et al., 1992) have been associated with transient surges in global brain adenosine levels, we hypothesized that brain adenosine elevation occurring in our nonreperfusion PVD stroke model could affect AMPAR trafficking. Interestingly, 2 d after PVD, we found MMP-2 elevation on both the ipsilateral and contralateral side of the PVD cortical lesion (Cayabyab et al., 2013). Therefore, in the current study, we performed morphological, biochemical, and electrophysiological analyses of hippocampal tissue taken from both ipsilateral and contralateral sides of the lesion in sham- and PVD-treated animals. We analyzed the effects of PVD on neurodegeneration using Fluoro-Jade B staining and confocal imaging, on the changes in adenosine tone using field EPSP (fEPSP) recordings, and on alterations in the surface levels of both GluA2 and GluA1 AMPARs and adenosine A1 and A2A receptors using biotinylation and Western blot analyses as described below. Subsequent results are consistent with PVD, inducing elevation of adenosine tone, downregulation of AMPARs and A1Rs, upregulation of A2ARs, and increased Fluoro-Jade B staining in hippocampus.
Biochemical studies.
For biotinylation experiments, hippocampal slices or 7 day cultured hippocampal neurons were incubated with 1.2 μm TTX Thermo-Fisher) to prevent glutamate release induced by treatments with the A1R antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), which could confound the direct effects of CPA on GluA2 and GluA1 AMPAR internalization. In addition to TTX, bicuculline (50 μm, Thermo Fisher), strychnine (1 μm, Thermo Fisher), and d-APV (50 μm, Thermo Fisher) were applied for 20–30 min, to block GABAA, glycine, and NMDA receptors, respectively. After CPA treatments (500 nm, 45 min), slices or neurons were cooled to 4°C (20–30 min) and washed with ice-cold ACSF before biotinylation. Hippocampal brain slices or neurons were incubated with 1 mg/ml NHS-SS-Biotin (Pierce, Thermo Fisher) at 4°C for 45 min. Quenching with glycine buffer containing 192 mm glycine, 25 mm Tris, pH 8.3, stopped the reaction. Slices were then transferred into lysis buffer containing protease inhibitors and 1% NP-40 detergent. Lysis buffer contained 50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm NaF; and the following protease inhibitors: 1 mm PMSF, 10 μg/μl aprotinin, 10 μg/ml pepstatin A, 10 μg/ml leupeptin, 2 mm Na3VO4, 20 mm sodium pyrophosphate, 3 mm benzamidine hydrochloride, and 4 mm glycerol 2-phosphate. After determining the protein concentrations using the Bradford Assay with the DC Protein assay dye (Bio-Rad), equal amounts of protein lysates (200–500 μg) were diluted in lysis buffer and biotinylated proteins were incubated overnight with the streptavidin beads (Thermo-Fisher). The beads were then washed 4–6 times the next day with lysis buffer containing 0.1% NP-40. The proteins were eluted by adding 50 μl of 2× Laemmli sample buffer (Bio-Rad) and boiling the samples at 95°C for 5 min. The proteins were the separated by running the samples through 10% polyacrylamide gels, and the resulting blots were probed with the appropriate primary and secondary antibodies. Enhanced chemiluminescence (ECL) reagent (Santa Cruz Biotechnology) was used to visualize the labeled proteins.
Coimmunoprecipitation was performed to examine interactions between adenosine receptors and AMPARs by incubating 500 μg of extract from hippocampal homogenates with mouse, goat, or rabbit IgG (1 h, 4°C). Then mouse, goat, or rabbit IgG agarose beads (Sigma) were added to the homogenates for an additional 1 h or overnight. After this preclearing stage, the agarose beads were removed by pulse spinning at 6000 rpm for 5 s, and the supernatant was subsequently reacted with the appropriate immunoprecipitating antibody overnight at 4°C. The A1 receptor and other proteins were immunoprecipitated with a polyclonal goat anti-A1 receptor (5 μg, Santa Cruz Biotechnology), a polyclonal rabbit anti-A1 receptor (5 μg, Sigma), a mouse monoclonal GluA2 antibody (2 μg, EMD Millipore), a rabbit anti-A2A receptor antibody (5 μg, Sigma), or a rabbit anti-GluA1 antibody (5 μg, Millipore). After overnight incubation of lysates with a polyclonal rabbit or monoclonal mouse antibody for the select target listed above, the select antigen was captured by incubation of immune complexes for 4 h at 4°C with agarose beads conjugated to secondary antibody (rabbit, mouse, or goat anti-IgG). Agarose beads were then collected by pulse spins and washed four times with wash buffer (lysis buffer listed above containing 0.1% NP-40). Proteins from the agarose beads were detected by Western blotting. The antibody dilutions were as follows: polyclonal rabbit anti-A1 receptor or anti-A2A receptor (1:1000, Sigma), rabbit anti-GluA1 (1:1000, Millipore), rabbit anti-GluA1 (pSer845) (1:1000, Millipore), and mouse anti-GluA2 (1:1000, Millipore). To normalize the protein bands from the membrane fractions, we used a monoclonal mouse anti-GAPDH (1:2000, Millipore) to quantify signals of GluA2 and GluA1 bands. Whole hippocampal lysate blots or blots containing biotinylated proteins were reprobed with anti-β actin antibody (1:1000, Sigma). Labeled protein bands were visualized using ECL (Santa Cruz Biotechnology).
In some experiments, the membrane fractions from hippocampal slices were separated by centrifugation at 13,000 × g for 1 h at 4°C by omitting the detergent (NP-40) from the solubilization buffer. The proteins from the particulate (membrane) fraction were resolved in normal solubilization buffer after removal of the cytosolic fraction. Hippocampal homogenates were diluted with Laemmli sample buffer, boiled for 5 min and resolved in 10% polyacrylamide gel; then they were electrotransferred to PVDF membranes (Millipore). The amount of protein loaded into the gels was consistent across all experiments where 50 μg were loaded for total lysates and 300–500 μg of total lysates were used for immunoprecipitation. After blocking with 5% nonfat milk in TBST for 1 h or overnight, the membranes were incubated with primary antibody in 5% nonfat milk in TBST containing 0.025% sodium azide overnight at 4°C. The PVDF membranes were washed four times with TBST for 15 min and then incubated with a mouse, goat, or rabbit horseradish peroxidase-conjugated secondary antibody against IgG (1:1000; Santa Cruz Biotechnology) in 5% nonfat milk. After three or four 15 min washes with TBST, proteins were visualized using ECL (Santa Cruz Biotechnology). After ECL, the following molecular weights were used to determine protein bands: A1R (37 kDa), A2AR (45 kDa), GluA1 (106 kDa), GluA2 (102 kDa), GAPDH (37 kDa), and β actin (42 kDa).
Hippocampal neuron culture, immunocytochemistry, and confocal imaging.
Rat hippocampal neurons were cultured as described previously (Kaech and Banker, 2006) and used for immunocytochemistry 12–15 d after plating. In brief, low-density hippocampal neurons (5 × 104 cells/35 mm culture dishes) from 17- to 18-day-old embryonic rat brains were grown on polylysine-coated coverslips, which were suspended above a 1-week-old astrocyte feeder layer. For immunocytochemistry, the hippocampal neurons were treated with pharmacologic agents TTX (01.2 μm), bicuculline (50 μm), strychnine (1 μm), and d-APV (50 μm) for 20–30 min, to block neural activity, GABAA currents, glycine receptors, and NMDA receptors, respectively, and then incubated for 1 h with the A1R antagonist DPCPX (500 nm, Sigma), the A2A receptor antagonist SCH 58261 (30 nm), the inhibitor of GluA2 endocytosis Tat-GluA2–3Y peptide (2 μm), or the scrambled Tat-GluA2–3Y peptide (2 μm) before a final 45 min incubation with the A1R agonist CPA (500 nm). The active Tat-GluA2–3Y peptide consists of the following amino acid sequence: YGRKKRRQRRR-869YKEGYNVYG877, where Tat is YGRKKRRQRRR (the cell penetrating amino acid peptide sequence contained within the protein transduction domain of HIV gene called Tat), and 869YKEGYNVYG877 represents a GluA2 C-terminal amino acid sequence that interacts with the endocytic protein AP2, thus preventing GluA2 internalization (Ahmadian et al., 2004). The Tat-GluA2–3Y peptide and its scrambled version (scrambled Tat-GluA2–3Y: YGRKKRRQRRR-VYKYGGYNE) were purchased from GL Biochem. The A2A receptor agonist CGS 21680 (10 nm) was applied to investigate the relationship between A2A receptor and possible internalization of AMPARs. After fixation, neurons were blocked for 1 h at room temperature with PBS containing 5% BSA (Sigma).
To assess the effects of A1R or A2AR stimulation on AMPAR surface expression, hippocampal neurons were washed three times with ice-cold PBS, fixed with 4% PFA, and then blocked for 1 h at room temperature with PBS containing 5% BSA. Surface proteins of neurons were labeled by overnight incubation (at 4°C) with rabbit anti-GluA1 (extracellular epitope, Alomone Labs) or mouse anti-GluA2 (extracellular epitope, Millipore) antibody diluted at 1:250 in blocking buffer followed by three brief washes (10 min each) and then incubated with AlexaFluor-555-conjugated goat anti-rabbit or AlexaFluor-488-conjugated donkey anti-mouse secondary antibodies (Invitrogen) at 1:1000 for 1 h at room temperature. All the neurons were labeled with chicken anti-rat MAP2 antibody (1:2000, Abcam) and AlexaFluor-633-conjugated anti-chicken secondary antibody at 1:1000 and Hoechst (Sigma) after permeabilization with 0.25% Triton X-100 and blocking with 5% BSA. Last, the coverslips were mounted on newly cleaned slides using Prolong Gold Antifade Reagent (Invitrogen) and observed with an LSM700 laser scanning confocal microscope (Carl Zeiss). To demonstrate possible colocalization between GluA2 and GluA1 AMPARs and either A1Rs or A2ARs, hippocampal neurons were permeabilized with 0.25% Triton X-100 before subsequent incubation with primary and secondary antibodies (for further details of primary and secondary antibody combinations used, see Fig. 2 legends). Rabbit polyclonal A1R or A2AR antibodies (1:100, Sigma), goat anti-A1R (1:100, Santa Cruz Biotechnology), and mouse anti-A2AR (1:100, Santa Cruz Biotechnology) were used. All neurons were also colabeled with chicken anti-MAP2 (1:2000).
Figure 2.
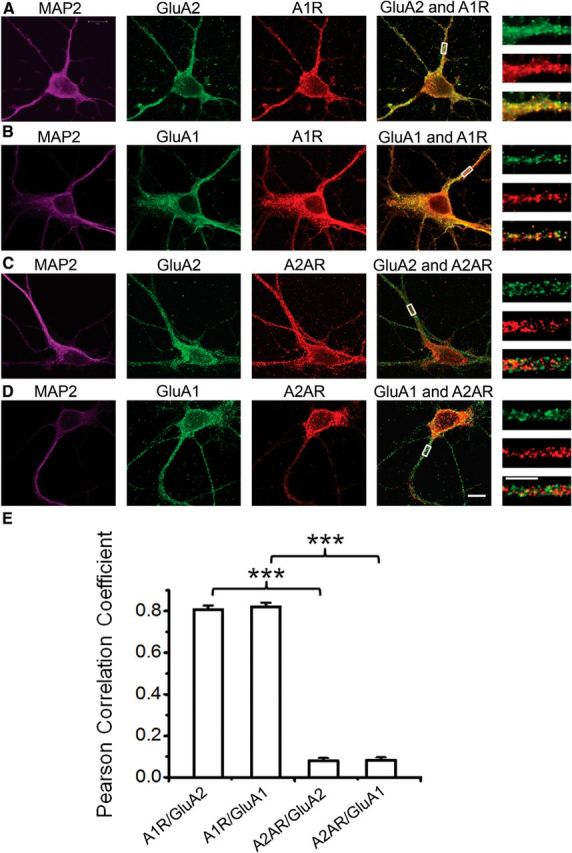
Colocalization of GluA2 and GluA1 with A1R, but not with A2AR, in rat hippocampal neurons. After membrane permeabilization, hippocampal neurons were triple colabeled with the following primary antibodies: (1) chicken anti-MAP2 (Abcam); (2) mouse anti-GluA2 or rabbit anti-GluA1 (both from Millipore); and (3) goat anti-A1R (Santa Cruz Biotechnology), mouse anti-A2AR (Santa Cruz Biotechnology), rabbit anti-A1R (Sigma), or rabbit anti-A2AR (Sigma). Secondary antibodies used were conjugated to AlexaFluor-633 (for MAP2, magenta panels), AlexaFluor-488 (for GluA2 or GluA1, green panels), and AlexaFluor-555 (for A1R or A2AR, red panels). A, B, Immunolabeling of MAP2 (magenta panels), GluA2 (Millipore) (A) or GluA1 (Millipore) (B) (green panels), A1R (Sigma) (A) or A1R (Santa Cruz Biotechnology) (B) (red panels), GluA2 or GluA1 merged with A1R (fourth panels from left), and magnified views of the dendrites indicated by the respective rectangular regions (last panels). A1Rs colocalized with GluA2 (A) or GluA1 (B) around the somas and dendrites, as shown by the highly intense yellow pixels in the merged fourth and fifth panels. C, D, Immunolabeling of MAP2 (magenta panels), GluA2 (Millipore) (C), or GluA1 (Millipore) (D) (green panels), A2AR (Sigma) (C) or A2AR (Santa Cruz Biotechnology) (D) (red panels), GluA2 or GluA1 merged with A2AR (fourth panels from left), and magnified views of the dendrites indicated by the respective rectangular regions (last panels). A2AR did not colocalize with either GluA2 (C) or GluA1 (D) around the somas and dendrites, as shown by the absence of yellow pixels in the merged fourth and fifth panels. Scale bars, large and small, 10 and 5 μm, respectively. E, Colocalization of adenosine receptors with AMPARs was quantified by determining the Pearson correlation coefficients. Dendritic lengths (10 μm) taken 5 μm away from somas from different staining experiments were used for colocalization analysis. The values in bars represent the mean ± SEM; N = 14 neurons each column (from 4 independent hippocampal neuronal cultures, 3 or 4 representative neurons included per culture). ***p < 0.001.
The images were acquired using a Zeiss Plan-Apochromat 63×/1.4 oil objective lens and analyzed with the Zeiss Zen 2009 software (version 5.5 SPI). The 10 μm dendritic lengths located 5 μm away from the cell soma were included in the analysis of GluA2 and GluA1 surface expression, and identical acquisition parameters were used for a given set of labeled neurons without oversaturation or undersaturation of the acquired signals. For analyses of AMPAR and adenosine receptor colocalization in dendrites, the image window of ∼10 μm dendritic length by ∼2 μm dendritic width at 0.06 μm pixel resolution was used for the regions of interest comparisons for the A1R/GluA2, A1R/GluA1, A2AR/GluA2, and A2AR/GluA1 groups. Images were exported as 8 bit TIFF files, and the degree of colocalization between fluorescent probes was quantified by using the Intensity Correlation Analysis plug-in of ImageJ software (downloaded from National Institutes of Health, version 1.44f), which reported the Pearson's correlation coefficients. Negative Pearson's correlation coefficients indicate the two signals do not colocalize, whereas values closer to 1 indicate strong colocalization between the two particles. The average signal intensities or Pearson's correlation coefficients from two to four dendritic processes from a given neuron were determined, and the n values reported in the summary bar charts refer to the number of cells analyzed from at least three different experiments. Different laboratory personnel were involved in preparing the immunocytochemical slides and performing confocal analyses to reduce bias. Data are presented as mean ± SEM. Group results were analyzed by one-way ANOVA with Student-Neuman-Keuls post hoc test comparing more than two treatment groups. p > 0.05 was considered not significant. For transfection experiments, hippocampal neurons were transfected with 1 μg of p38α MAPK shRNA, JNK1 shRNA, or control plasmid A (Santa Cruz Biotechnology) and 2 μl Lipofectamine 2000 (Invitrogen). Two days transfection, hippocampal neurons were treated with 500 nm CPA for 45 min followed by immunocytochemistry as described above.
Fluoro-Jade B staining.
Sprague Dawley rat brains were prepared and sectioned as described previously (Cayabyab et al., 2013). In brief, anesthetized rats were intracardially perfused with 4% PFA in PBS for 30 min. After perfusion, brains were removed and postfixed in 4% PFA in PBS overnight. Brains were then stored in 30% sucrose (w/v) in 0.1 m PBS for additional 3 d. The brains were then frozen in liquid nitrogen in Tissue-Tek OCT mounting medium, and 30 μm coronal sections of hippocampus were cut with cryostat. Sections were subsequently collected on paraffin-coated slides and allowed to air dry. Sections were immersed in 70% ethanol, washed three times with ultrapure water (1 min each), and then soaked in 0.06% KMNO4 (15 min). After three 1 min washes with ultrapure water, sections were subsequently stained with 0.001% Fluoro-Jade B (Millipore Bioscience Research Reagents) for 20 min. Slides were subsequently washed three times in ultrapure water (1 min each) and allowed to dry overnight. Slides were then rinsed in xylene, and coverslips were mounted using Prolong Gold Antifade Reagent (Invitrogen). Digital images were obtained with Zeiss LSM 700 (Carl Zeiss) using a 20× objective for the hippocampal montages and 63×/1.4 oil-immersion objective lens for the magnified regions of the hippocampal pyramidal body layers. Three sham and three PVD rat brains were used for Fluoro-Jade B staining.
Electrophysiological studies.
fEPSPs were evoked by orthodromic stimulation of the Schaffer collateral pathway using a bipolar tungsten-stimulating electrode. Glass micropipettes filled with ACSF (resistance 1–3 mΩ) were used to measure CA1 fEPSPs in stratum radiatum. The fEPSP signals were amplified 1000 times with an AC amplifier, bandpass filtered at 0.1–100 Hz, digitized at 10 kHz using a Digidata 1320A interface board (Molecular Devices), and transferred to a computer for analysis. Data were analyzed using Clampfit 9.0 (Molecular Devices). Baseline synaptic responses were established by evoking fEPSPs every 30 s (0.03 Hz) for at least 20 min. Paired pulses separated by 50 ms were also evoked every 30 s to assess changes in presynaptic function in control, CPA-treated, sham, or PVD hippocampal slices. The fEPSP slope was normalized to the mean of the 20 sweeps (10 min) immediately preceding drug perfusion. The mean normalized fEPSP slope was plotted as a function of time with error bars representing the SEM. Sample traces are the average of 5 sweeps from a recording that was included in the plot of the mean normalized fEPSP slope. All bar graphs show the mean normalized percentage inhibition from baseline ± SEM. Statistical significance was assessed using one-way ANOVA with Student-Neuman-Keuls post hoc test.
Drug inhibitors.
CPA and DPCPX were obtained from Sigma-Aldrich. Baclofen, 3-[4-[2-[[6-amino-9-[(2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxy-oxolan-2-yl]purin-2-yl]amino]ethyl]phenyl]propanoic acid (CGS 21680), okadaic acid, fostriecin, and 5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo(4,3-e)-1,2,4-triazolo(1,5-c)pyrimidine (SCH 58261) were obtained from Tocris Bioscience. SB203580, SB202474, JNK II inhibitor (also called SP600125), and JNK inhibitor II negative control (N1-methyl-1,9 pyrazoloanthrone) were obtained from Calbiochem. All drugs were dissolved in DMSO (Sigma) before being added to ACSF. The final concentration of DMSO was always <0.1%.
Statistical analysis.
Densitometry was performed using Quantity 1 (Bio-Rad) and ImageJ (public domain). A single ANOVA was performed to obtain the overall significance of the treatments followed by a post hoc Student-Newman-Keuls. Student's paired t test was also used when comparing two treatment groups. All statistical tests were performed with GraphPad Instat3 version 3.00 for Windows 97 (GraphPad Software).
Results
GluA2 and GluA1 AMPA receptors physically interact with adenosine A1 receptors, but not with A2A receptors
Physical interactions between different transmembrane GPCRs and ionotropic glutamate receptors are known to exist (Salter, 2003; Lee and Liu, 2004). For example, D2 dopamine receptors exhibit an indirect biochemical interaction with GluA2-containing AMPARs, which causes downregulation of AMPAR surface expression (Zou et al., 2005). Direct interactions between two or more GPCRs are also possible (Angers et al., 2002). For example, it has been shown that A1Rs may form heterodimers with A2ARs (Ciruela et al., 2006). Thus, it is reasonable to propose a possible association of AMPARs with the GPCR A1Rs and A2ARs, which are the most abundant of the four known adenosine receptors in the brain (Dunwiddie and Masino, 2001). We initially sought to characterize this interaction in hippocampal slices and cultured hippocampal neurons.
We performed coimmunoprecipitation experiments to determine whether the adenosine A1R exists in the same signaling protein complex as GluA2 and GluA1 in the rat hippocampus. We found that both GluA2 and GluA1 formed stable complexes with A1Rs (Fig. 1A,B, left), and the reverse immunoprecipitation confirmed the interaction of GluA2 and GluA1 in the A1R immunoprecipitates (Fig. 1A,B, right). However, our coimmunoprecipitation studies did not reveal an association between adenosine A2A receptors with either GluA2 or GluA1 AMPARs (Fig. 1C,D). These results indicated that the inhibitory adenosine A1 receptors, but not the excitatory A2A receptors, are specifically localized in the same protein complex as GluA2-containing AMPARs. These interactions could certainly contribute to the modulation of the AMPAR function and subcellular distribution.
Figure 1.

AMPA receptors physically interact specifically with A1Rs. A, GluA2-containing AMPAR immunoprecipitated complexes from rat hippocampus contained A1Rs, and A1R-immunoprecipitated complex contained GluA2 subunits. B, GluA1 immunoprecipitate from rat hippocampal tissue also contained A1Rs, and the A1R immunoprecipitate likewise contained GluA1 subunits. C, Coimmunoprecipitation of GluA2 did not include A2ARs, and A2AR antibody did not immunopreciptitate GluA2-containing AMPARs. D, GluA1 immunoprecipitate did not contain A2ARs, and A2AR immunoprecipitate did not contain GluA1 subunits. These forward and reverse coimmunoprecipitation studies are from at least three independent experiments, using hippocampal brain lysates from P18–P28 d rats. The molecular weights of the specific bands on the blots were estimated from prestained protein standards and are as follows: A1R (37 kDa), A2AR (45 kDa), GluA1 (106 kDa), and GluA2 (102 kDa). Amount of protein loaded into gels, antibody sources, and dilutions are indicated in the Materials and Methods.
GluA2- and GluA1-containing AMPA receptors colocalize with adenosine A1 receptors, but not with A2A receptors, in cultured hippocampal neurons
To further confirm the potential interaction of A1Rs with GluA2 and GluA1 AMPARs, we used immunocytochemistry and confocal imaging to test whether A1Rs and GluA2-containing AMPARs colocalized in cultured hippocampal neurons. Hippocampal neurons were cultured for 12 d, then fixed with 4% PFA and permeabilized with detergent to label surface and intracellular localizations of proteins. Mouse anti-GluA2 or mouse anti-GluA1 was used with rabbit anti-A1R antibodies, or mouse anti-GluA1 and rabbit anti-A2AR antibodies, followed by incubation with appropriate fluorescent secondary antibodies for double staining of cultured neurons. Visualization of neuronal morphology was facilitated by subsequent immunolabeling with chicken anti-MAP2 antibody and secondary antibody. Immunocytochemical identification of GluA2/GluA1 is shown in green, and A1R/A2AR labeling is in red. Merging the GluA2 or GluA1 AMPAR with the A1R images revealed overlapping regions of colocalization, which is shown as yellow pixels (Fig. 2A,B). In contrast, merging GluA2 and GluA1 with A2ARs produced very few yellow pixels were visible, suggesting little colocalization of A2ARs with GluA2 or GluA1 AMPARs in hippocampus (Fig. 2C,D). Quantification of overlapping A1Rs and GluA2 or A1Rs and GluA1 AMPARs revealed a significant colocalization of A1Rs with GluA2 and GluA1 AMPARs (Fig. 2E). For example, the Pearson correlation coefficients for A1R/GluA2 colocalization were 0.81 ± 0.02 (arbitrary units) (n = 14) compared with 0.08 ± 0.01 for A2AR/GluA2 colocalization (n = 14, p < 0.001). Together, the above biochemical and confocal imaging results indicated that A1Rs, but not A2ARs, specifically formed a physical complex with GluA2 and GluA1 AMPARs and are localized in similar dendritic and somatic compartments of hippocampal neurons.
Stimulation of adenosine A1 receptor triggers GluA2 and GluA1 AMPAR internalization via clathrin-mediated and dynamin-dependent endocytosis
Next, we tested whether this specific physical association of A1Rs with GluA2 and GluA1 AMPARs can functionally modify AMPAR trafficking, which is important for excitation of neurons (Bredt and Nicoll, 2003; Malinow, 2003). To determine whether selective activation of A1Rs could alter the trafficking of GluA2 and GluA1 AMPARs, we performed surface biotinylation of primary cultures of hippocampal neurons followed by Western blotting to track changes in GluA2 and GluA1 surface levels. The results showed that stimulation of A1Rs with the A1R-selective agonist CPA (500 nm, 45 min) caused a significant decrease in GluA2 and GluA1 surface levels (Fig. 3A,B).
Figure 3.

A, B, Prolonged A1R stimulation caused clathrin-mediated AMPAR endocytosis. Surface expression of GluA2 decreased by A1R stimulation in cultured hippocampal neurons. A, Cell-surface biotinylation of hippocampal neurons showed a significant decrease in GluA2 surface expression after CPA treatment (500 nm, 45 min) compared with DMSO (Control). This effect was abolished with Tat-GluA2–3Y (2 μm) peptide, which specifically blocks clathrin-mediated GluA2 endocytosis, but the scrambled Tat-GluA2–3Y peptide did not prevent the inhibitory effects of CPA. Histograms show the densitometric quantification of Western blots for surface-expressed GluA2 AMPARs. B, Biotinylation of cultured hippocampal neurons showed a significant decrease in GluA1 surface expression after prolonged CPA treatment. This effect was abolished by the presence of Tat-GluA2–3Y peptide, but not by scrambled Tat-GluA2–3Y peptide. Note the absence of β actin bands in blots containing the biotinylated GluA2 or GluA1. Bar chart summaries represent biotinylated GluA2 or GluA1 signals normalized to their respective whole hippocampal neuronal lysate signals. Data are mean ± SEM; N = 4 from four independent experiments. *p < 0.05. C, D, Activation of A1R-induced AMPAR endocytosis is dynamin-dependent. C, Immunoblots are of streptavidin precipitates probed with mouse anti-GluA2 antibody. Dynasore (100 μm), an inhibitor of dynamin GTPase, prevented the CPA-induced GluA2 endocytosis in hippocampal brain slices. GluA2 levels remained constant in whole hippocampal slice lysates. D, GluA1 surface proteins detected with rabbit anti-GluA1 were also significantly reduced by prolonged CPA application. Dynasore prevented the CPA-induced decrease in GluA1 surface expression. Beta actin was absent in surface biotinylation blots, confirming little or no contamination of biotinylated AMPARs with cytosolic proteins. Biotinylated signals were normalized to GluA2 or GluA1 signals detected in whole hippocampal brain lysates. Data are mean ± SEM; N = 3 from three independent experiments using a specific GluA2 or GluA1 antibody. *p < 0.05. **p < 0.01.
To determine whether this inhibitory effect requires the clathrin-mediated endocytosis pathway, we preincubated the neurons with Tat-GluA2–3Y peptide, which has been used by other laboratories to block the clathrin-mediated endocytosis of GluA2 (Ahmadian et al., 2004; Brebner et al., 2005; Xiong et al., 2006). Preincubation with Tat-GluA2–3Y peptide prevented the A1R-induced decrease in GluA2 and GluA1 surface expression in cultured hippocampal neurons (Fig. 3A,B, third column). The scrambled Tat-GluA2–3Y peptide did not alter CPA-induced GluA2 and GluA1 surface levels. Similar results were obtained using membrane fractionation of hippocampal brain slices. That is, GluA2 levels in membrane fractions were as follows: Control (DMSO) 100%, CPA alone 78.6 ± 2.7% (p < 0.01 compared with control), CPA + Tat-GluA2–3Y 93.5 ± 3.1% (p > 0.05), and CPA + Scrambled Tat-peptide 72.4 ± 4.9% (p < 0.01, compared with control). All signals were normalized to GAPDH, with N = 5 independent experiments (p < 0.0001, one-way ANOVA). Not surprisingly, we observed a similar pattern of changes in the GluA2 and GluA1 surface distribution in hippocampal neuronal cultures after A1R stimulation (Fig. 3A,B) because both GluA2 and GluA1 are known to form heteromers, and GluA2 and GluA1 heteromeric AMPARs are the most widely expressed subunits in the hippocampus (Wenthold et al., 1996; Sans et al., 2003; Cull-Candy et al., 2006). However, upon closer inspection, it is apparent that the levels of CPA-induced GluA1 internalization were higher (∼50%; Fig. 3B, bottom) compared with those for GluA2 internalization (∼30%; Fig. 3A, bottom). When PVDF membranes of biotinylated proteins and total lysates were reprobed with β-actin antibody, the β-actin was only found in total lysates and that cytosolic proteins were undetectable in blots containing biotinylated surface proteins. Thus, these results suggest that stimulation of A1R induces clathrin-mediated endocytosis of GluA2-containing AMPARs in hippocampal neuronal cultures.
Previous reports also suggested that AMPAR internalization could be mediated by dynamin-dependent endocytosis (Carroll et al., 1999; Luscher et al., 1999; Man et al., 2000; Xiong et al., 2006). Dynamin is critical for the vesicle formation during clathrin-mediated endocytosis (Henley et al., 1999). Because the use of the Tat-GluA2–3Y peptide implicated the clathrin-mediated pathway for A1R-mediated AMPAR endocytosis, we then determined whether inhibition of dynamin function with Dynasore (Macia et al., 2006; Newton et al., 2006) would also blunt the level of CPA-induced AMPAR endocytosis. Preincubation of hippocampal slices with Dynasore (100 μm) for 1 h before CPA stimulation and subsequent biotinylation of surface-expressed proteins revealed that Dynasore did indeed prevent A1R-induced internalization of both GluA2 and GluA1, as summarized in bar charts in Figure 3C, D. Together, the results above demonstrate that prolonged A1R stimulation led to a clathrin-mediated and dynamin-dependent internalization of GluA2 and GluA1 AMPARs in both hippocampal neurons and hippocampal brain slices.
Prolonged A1R stimulation causes A1R-induced persistent synaptic depression (APSD)
Previously, we reported that short-term application of the selective A1R agonist CPA (50 nm, 10 min) caused significant synaptic depression; however, a persistent synaptic depression of ∼25% remained after 1 h of withdrawal of the agonist and was accompanied by a recovery of paired pulse ratio back to baseline levels (Brust et al., 2007). We have subsequently used higher CPA concentration (500 nm) and longer incubation period (30–45 min) because we have found that this experimental condition produced significant effects in GluA2 and GluA1 surface expression. A concentration response experiment has been performed (data not shown) showing that both concentration and longer time periods of drug incubation produced more robust increase in internalization of GluA2 and GluA1, which facilitated our exploration of these mechanisms. This also correlates with previous studies showing that high concentration of CPA for 30 min induced robust activation of both p38 and JNK (Brust et al., 2006, 2007), which we subsequently show in the present study to be crucial for A1R-induced GluA2 internalization. These previous studies also showed that prolonged treatment with high CPA concentration induced robust activation of protein phosphatase 2A (PP2A). Although we used a much higher CPA concentration to induce GluA2 and GluA1 subunit internalization than that required for inhibition of synaptic transmission, we showed in this study that high concentration of CPA was still selective for A1R and did not cause any aberrant activation of the A2AR.
In contrast to our earlier study (Brust et al., 2006, 2007), a 30 min application of 500 nm CPA produced an even greater APSD (∼50–60%; Fig. 4A). Based on our coimmunoprecipitation findings described above, we tested the hypothesis that the biochemical associations of GluA2 and GluA1 with A1Rs could facilitate the expression of persistent synaptic depression, which reflects alterations of AMPAR levels or function. Using the Tat-GluA2–3Y peptide to inhibit GluA2 endocytosis, we found that the APSD levels are lower (∼30% vs 55%–60%) when slices were preincubated with 2 μm Tat-GluA2–3Y peptide compared with no peptide treatment or treatment with the scrambled Tat-GluA2–3Y peptide (Fig. 4A, bottom right). This indicated that the induction of APSDs (i.e., after ∼1 h CPA washout) is mediated in part by clathrin-mediated GluA2-internalization at a postsynaptic locus. Consistent with this idea, the paired-pulse facilitation observed during CPA application returned to baseline levels during APSD despite the presence of significant and persistent synaptic depression during extended CPA washout period (Fig. 4B). Interestingly, the Tat-GluA2–3Y peptide also inhibited the short-term synaptic depression during CPA application (∼50% vs ∼70%; Fig. 4A, bottom left), possibly indicating that acute application of the A1R agonist rapidly activated signaling pathways that contributed to GluA2 and GluA1 endocytosis. It is noteworthy that both p38 MAPK and JNK were shown to be maximally activated within the first 10 min of CPA application (Brust et al., 2007), and both protein kinases have been implicated in glutamate receptor trafficking. The small but significant attenuation of fEPSPs by Tat-GluA2–3Y peptide during a 30 min CPA application suggested that the molecular mechanisms of acute CPA-mediated synaptic depression may involve changes at both presynaptic sites (Brust et al., 2007) as well as postsynaptic sites.
Figure 4.

Prolonged stimulation of A1Rs caused A1R-induced persistent synaptic depression (APSD) in part via clathrin-mediated GluA2 internalization. A, Top, Representative fEPSP traces from hippocampal CA1 region in the absence of Tat-peptides (Control), in scrambled Tat-GluA2–3Y peptide (2 μm), and in Tat-GluA2–3Y peptide (2 μm). The numbers associated with the fEPSP traces correspond to baseline control (1), 30 min after CPA application (2), and 55 min after CPA washout (3), and also apply to middle panel. The time course of CPA-induced synaptic depression is summarized in the middle panel, showing that Tat-GluA2–3Y peptide, but not its scrambled version, partially inhibited the CPA-induced APSD. Bottom, Control (no peptide) and scrambled Tat-GluA2–3Y produced similar levels of synaptic depression during CPA application (left) and after CPA washout (right), whereas the Tat-GluA2–3Y peptide significantly attenuated these responses. *p < 0.05 versus Control (no peptide) or scrambled Tat-GluA2–3Y (Student-Neuman-Keuls post hoc test). ***p < 0.001 versus Control (no peptide) or scrambled Tat-GluA2–3Y (Student-Neuman-Keuls post hoc test). B, Paired-pulse stimulation shows that synaptic depression during CPA was accompanied by significant paired pulse facilitation (bottom panel, *p < 0.05 vs Control), but synaptic depression during APSD showed paired pulse ratios similar to baseline control levels. Numbers inside summary bar charts refer to the number of brain slices from different animals. Data are mean ± SEM. Vertical calibration: 0.5 mV. Horizontal calibration: A, 5 ms; B, 10 ms. These results indicate a functional interaction between A1Rs and AMPARs, leading to clathrin-mediated internalization of GluA2-containing AMPARs and subsequent induction of APSDs.
In addition, we determined that the APSD levels observed after ∼1 h washout of CPA was not likely the result of a persistent binding of CPA to A1Rs in hippocampal slices because a subsequent 30 min application of the A1R antagonist DPCPX (500 nm) did not modify the levels of APSD. The normalized fEPSP slope values in this control (no peptide) group differed significantly (one-way ANOVA p < 0.0001, n = 7 animals) as follows: control (100 ± 0%), 30 min CPA (17 ± 6.1%, p < 0.001 vs control), 1 h CPA washout (48.4 ± 9.4%, p < 0.001 vs control), and 30 min DPCPX (57 ± 5.6%, p < 0.001 vs control, p > 0.05 vs 1 h CPA washout). These results further indicate that functional interactions between A1Rs and GluA2-containing AMPARs in postsynaptic sites facilitate the clathrin-mediated endocytosis of AMPARs and the induction of APSDs.
Confocal imaging analysis revealed A1R stimulation mediates clathrin-mediated internalization of GluA2 and GluA1 AMPARs in hippocampal neurons
To confirm our biochemical findings that A1R stimulation decreases GluA2 and GluA1 surface levels, we used confocal imaging and antibodies that recognized the extracellular epitopes of GluA2 and GluA1 proteins to quantify the surface expression of GluA2 and GluA1 AMPARs from primary cultured hippocampal neurons. As shown in Figure 5, stimulation of A1Rs with CPA produced a similar decrease in surface levels of GluA2 (∼25% decreased, Fig. 5A,B) and GluA1 (∼20% decreased, Fig. 5C,D) expressed on dendritic surfaces located 5 μm away from the cell somas. These CPA-induced reductions in surface AMPARs were blocked by either Tat-GluA2–3Y peptide or the A1R antagonist DPCPX, but not by the scrambled Tat-GluA2–3Y peptide (Fig. 5B,D).
Figure 5.

AMPAR surface levels were decreased by activation of A1Rs with CPA. A, Confocal imaging of surface GluA2 (green) in primary hippocampal neurons. GluA2 receptors were first labeled without membrane permeabilization, and subsequent immunolabeling of MAP2 was performed after permeabilization with 0.25% Triton X-100. These images show that most dendritic processes, when exposed to prolonged A1R agonist CPA (500 nm), demonstrate reduced surface GluA2, whereas preincubation of hippocampal neurons with Tat-GluA2–3Y (2 μm) peptide prevented activation of A1R-induced GluA2 internalization. B, Summary bar chart showing that activation of A1R-induced GluA2 internalization requires clathrin-mediated endocytosis (shown with Tat-GluA2–3Y) and functional A1Rs (shown with DPCPX, 100 nm). The A2A receptor antagonist, SCH 58261 (30 nm), did not prevent activation of A1R-induced GluA2 internalization, whereas the A2AR agonist CGS 21680 (10 nm) did not mimic the effect of CPA. C, D, Similar to GluA2, the surface GluA1 levels were decreased by activation of A1R (with CPA). C, Representative confocal images show that CPA decreased surface levels of GluA1 (red), but not in the presence of Tat-GluA2–3Y peptide or DPCPX. D, Summary bar chart showing that activation of A1Rs induced surface GluA1 internalization, which was prevented by Tat-GluA2–3Y peptide and DPCPX, but not by scrambled Tat-GluA2–3Y peptide and SCH 58261. However, CGS 21680 significantly increased surface levels of GluA1. Average intensity values in bars represent the mean ± SEM, and n values of the number of neurons used are indicated in the bar charts. *p < 0.05 (one-way ANOVA, followed by post hoc Student-Newman-Keuls test). **p < 0.01 (one-way ANOVA, followed by post hoc Student-Newman-Keuls test). ***p < 0001 (one-way ANOVA, followed by post hoc Student-Newman-Keuls test). NS, Not significant (p > 0.05).
These results indicate that functional A1Rs are required for stimulation of A1R-induced GluA2 and GluA1 internalization. Moreover, because the concentration of CPA (500 nm) used in this study may very well be causing significant occupation and subsequent activation of A2A receptors, we also determined whether the CPA effects involved functional A2ARs. However, preincubation of hippocampal neurons with the A2A receptor antagonist SCH 58261 did not prevent CPA-induced GluA2 and GluA1 internalization, and the A2A receptor agonist stimulation with CGS 21680 did not mimic the inhibitory effect of the A1R agonist CPA on GluA2 and GluA1 surface expression (summarized in Fig. 5B,D). The A2A receptor agonist significantly potentiated surface levels of GluA1 but not GluA2 (Fig. 5D). Together, these findings suggest that GluA2 and GluA1 AMPARs selectively and functionally interact with A1Rs, but not with A2ARs, to promote clathrin-mediated endocytosis of GluA2-containing AMPARs.
GluA2-containing AMPARs are regulated by A1R-mediated activation of p38 MAPK, JNK, and PP2A in hippocampal brain slices
In previous studies, we showed that activation of A1Rs by CPA leads to increased activity of p38 MAPK and JNK, and that A1Rs and the p38 MAPK were found in the same protein complex (Brust et al., 2007). We hypothesized that p38 MAPK and JNK activation converged on signaling pathway(s) activated by A1Rs and cause internalization of GluA2-containing AMPARs. To determine whether A1R-p38 MAPK and A1R-JNK signaling pathways are involved in A1R-induced internalization of GluA2, rat hippocampal slices were preincubated with the p38 MAPK inhibitor SB203580 (20 μm) alone or in combination with the JNK inhibitor II (5 μm) for 1 h before CPA applications. After separating the membrane from cytosolic fractions, immunoblotting was performed to quantify the levels of GluA2 in hippocampal membrane fractions. We found that the p38 MAPK inhibitor fully inhibited the CPA-induced attenuation of GluA2 levels in membrane fractions (Fig. 6A). CPA treatment alone caused a ∼48% decrease in membrane GluA2 levels. However, preincubation of hippocampal slices with both p38 MAPK and JNK inhibitors not only prevented the A1R-induced decrease in GluA2 membrane levels, this drug combination also significantly increased the GluA2 membrane levels by threefold (Fig. 6A, bottom panel).
Figure 6.

A1R-mediated internalization of GluA2-containing AMPARs in hippocampal slices is differentially regulated by p38 MAPK, JNK, and PP2A. A, Levels of GluA2 (top) and GAPDH (bottom) in the membrane fraction with the indicated treatments. The summary bar chart shows that activation of A1Rs by CPA (500 nm, 45 min) produced a 48% decrease in GluA2 membrane expression. Preincubation with a p38 MAPK inhibitor (SB203580, 20 μm) before CPA application (500 nm, 45 min) significantly reduced GluA2 internalization. Preincubation with both p38 MAPK and JNK II (5 μm) inhibitors produced GluA2 levels above; N values are as follows: for Control (n = 8 independent blots), CPA (n = 8), SB203580 (n = 5), and SB203580 + JNK inhibitor II (n = 7). B, In surface biotinylation studies, p38 MAPK inhibitor SB203580 (20 μm), but not the inactive analog SB202474 (20 μm) and JNK II inhibitor (5 μm), but not its inactive analog JNK II-negative control (5 μm), individually prevented CPA-mediated GluA2 internalization. Hippocampal slices were preincubated with SB203580, SB202474 (20 μm), JNK II inhibitor, and JNK II-negative inhibitor (5 μm) for 1 h before CPA treatment (500 nm,45 min). C, Surface biotinylation study of hippocampal slices preincubated for 1 h before CPA treatment (500 nm, 45 min) in DMSO (control), or one of the PP2A inhibitors okadaic acid (20 nm) or fostriecin (20 nm). Surface levels of GluA2 after CPA treatment were significantly reduced, as shown before, and this surface reduction was prevented by treatment with PP2A inhibitor treatment. Intensity values in summary bar chart represent the mean ± SEM from n = 4 independent experiments. *p < 0.05. ***p < 0.001.
To confirm these results obtained from membrane fractions and to begin to address potential side effects of drug inhibitors, we also preincubated hippocampal slices with SB203580 (20 μm, p38 MAPK inhibitor), SB202474 (20 μm, negative control of SB203580), JNK II inhibitor (5 μm), or JNK II-negative inhibitor (5 μm) before CPA incubation (500 nm, 45 min). The surface proteins were isolated using biotinylation and quantified by Western blotting. Incubation of the slices with either SB203580 or JNK II inhibitor prevented A1R-induced internalization of GluA2-containing AMPARs, whereas their respective inactive analogs were ineffective in blocking the GluA2 internalization (Fig. 6B).
Phosphorylation of AMPARs is important for trafficking of AMPARs (Shepherd and Huganir, 2007). Activation of phosphorylated p38 MAPK by A1 receptor stimulation induced translocation of PP2A to the cell membrane (Brust et al., 2007). Therefore, modulation of AMPARs in the brain by phosphorylation may play a role in APSD. To determine whether PP2A is involved in A1R-induced internalization of GluA2, rat hippocampal slices were preincubated with the PP2A inhibitor fostriecin (20 nm) or okadaic acid (20 nm) for 1 h before CPA applications (500 nm, 45 min). Both PP2A inhibitors prevented the internalization of GluA2-containing AMPARs induced by CPA (Fig. 6C), suggesting that PP2A is involved in A1R-induced GluA2 AMPARs internalization. Together, these data indicate that p38 MAPK, JNK, and PP2A are involved in clathrin-mediated endocytosis of GluA2 AMPARs.
GluA1-containing AMPARs are regulated by A1R-mediated activation of PP2A, but not p38 MAPK and JNK in hippocampal brain slices
To determine whether p38 MAPK, JNK, and PP2A are involved in A1R-induced GluA1 internalization, rat hippocampal slices were preincubated with the p38 MAPK inhibitor SB203580 (20 μm) alone or in combination with the JNK inhibitor II (5 μm) for 1 h before CPA application (500 nm, 45 min). After separating the membrane from cytosolic fractions, immunoblotting was performed to quantify the levels of GluA1 in hippocampal membrane fractions. Preincubation of p38 MAPK and JNK inhibitors did not inhibit the CPA-induced attenuation of GluA1 levels in membrane fractions (Fig. 7A, summarized in bottom panel).
Figure 7.

A1R-mediated internalization of GluA1-containing AMPARs in hippocampal slices is not regulated by p38 MAPK or JNK but is regulated by PP2A, and robust A1R activation reduced GluA1-Ser845 phosphorylation. A, Levels of GluA1 (top) and GAPDH (bottom) in the membrane fraction of hippocampal slice lysates with preincubation in DMSO (control), SB203580 (20 μm), or SB203580 and JNK II inhibitor (5 μm) together followed by CPA treatment (500 nm, 45 min). Preincubation in either SB203580 or SB203580 and JNK II inhibitor together did not prevent A1R-induced GluA1 internalization. B, Surface biotinylation of hippocampal slices showing that A1R-mediated GluA1 internalization did not depend on the activity of p38 MAPK and JNK. Compared with GluA2, drug treatments with SB203580 (20 μm), but not the inactive analog SB202474 (20 μm), or JNK II inhibitor (5 μm), or the inactive analogs of SB203580 (SB202474, 20 μm), or JNK II inhibitor (JNK II neg. inhibitor, 5 μm), did not prevent A1R-induced internalization of GluA1. C, Preincubation of hippocampal slices in the PP2A inhibitors okadaic acid (20 nm) or fostriecin (20 nm) for 1 h followed by CPA treatment (500 nm, 45 min) prevented A1R-induced internalization of GluA1. D, Whole lysates of hippocampal slices treated with CPA (50 or 500 nm, 45 min) or the GABAB receptor agonist baclofen (10 μm, 45 min) and probed for the C-terminal phosphorylation site GluA1-pSer845. The antibody used was specific for phosphorylated Ser845 (pSer845) of GluA1. CPA treatment of 500 nm caused a robust decrease in pSer845, whereas baclofen and 50 nm CPA did not. All values in summary bar charts are mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
Biotinylation experiments were also performed to confirm the results obtained from membrane fractions. We preincubated hippocampal slices with SB203580 (20 μm), SB202474 (20 μm), JNK II inhibitor (5 μm), or JNK II-negative inhibitor (5 μm) before CPA incubation (500 nm, 45 min). Incubation of the slices with either SB203580 or JNK II inhibitor did not prevent the A1R-induced internalization of GluA1 AMPARs. Their respective inactive analogs were also ineffective in blocking the GluA1 internalization (Fig. 7B). Similar to the results obtained from hippocampal membrane fractions (Fig. 7A), A1R-mediated GluA1 internalization was also unaltered by active or inactive analogs of p38 MAPK and JNK inhibitors (Fig. 7B).
To determine whether PP2A is involved in the A1R-induced GluA1 internalization, hippocampal slices were preincubated with the PP2A inhibitors fostriecin (20 nm) or okadaic acid (20 nm) for 1 h before CPA applications. The PP2A inhibitors prevented the internalization of GluA1 induced by CPA alone (Fig. 7C), suggesting that PP2A is involved in A1R-induced GluA1 internalization. Because CPA induces PP2A translocation to hippocampal membrane fractions and PP2A can dephosphorylate GluA1 at Serine845 (Ser845) (Snyder et al., 2000), we predicted that CPA application decreases the phosphorylation of GluA1 at Serine 845. The resultant blots showed that incubation with a high concentration of CPA (500 nm) significantly decreased phosphorylation of GluA1 at Ser845 (Fig. 7D). In contrast, GluA1 Ser845 phosphorylation levels were unaltered in the presence of a lower CPA concentration (50 nm) or the GABAB receptor agonist baclofen (10 μm) (Mezler et al., 2001). The results suggest that dephosphorylation levels of GluA1 at Ser845 specifically are induced by the activation of A1R but not activation of GABAB receptors. The results also show that the activation of A1Rs by 500 nm CPA, but not 50 nm CPA, induced robust reductions of GluA1-pSer845. Together, these data indicate that PP2A, but not p38 MAPK and JNK, is involved in clathrin-mediated endocytosis of GluA1 AMPARs and that the internalization of GluA1 is correlated with a reduction of phosphorylated GluA1 at Ser845.
Selective inhibition of p38 MAPK and JNK by shRNA transfections prevented A1R-mediated GluA2 internalization in cultured hippocampal neurons
To further address the dependence of GluA2-containing AMPAR internalization on A1R-mediated p38 MAPK and JNK activation, we used confocal imaging of cultured hippocampal neurons and compared the effects of pharmacological inhibitors and genetic knockdown of p38 MAPK and JNK. Cultured hippocampal neurons were pretreated with the p38 MAPK inhibitor SB203580, SB202474 (inactive p38 MAPK inhibitor), JNK II inhibitor or JNK II-negative inhibitor (concentrations same as above) and then stimulated with 500 nm CPA for 45 min. As shown in Figure 8A, surface levels of GluA2 were significantly decreased by CPA, but not in the presence of SB203580 or JNK II inhibitor. Indeed, the JNK II inhibitor not only blunted CPA-induced GluA2 internalization, it also potentiated GluA2 surface levels (Fig. 8A, right). In contrast, the respective negative control compounds SB202474 (for p38 MAPK) or the JNK II-negative inhibitor (for JNK) did not prevent CPA-induced GluA2 internalization.
Figure 8.

A1R-mediated decrease in GluA2 surface levels in primary hippocampal neurons was prevented by pharmacological inhibitors and genetic knockdown of p38 MAPK and JNK. A, Intensity levels of surface-expressed GluA2 were determined by confocal imaging and analyzing 10 μm dendritic lengths located 5 μm away from cell somas. Results showed that A1R-induced GluA2 endocytosis was inhibited by p38 MAPK inhibitor SB203580 (20 μm), but not by inactive analog SB202474, and JNK II inhibitor (5 μm), but not by its inactive analog JNK II-negative control. The compounds SB203580, SB202474, JNK II inhibitor, and JNK II-negative inhibitor were applied to hippocampal neurons for 1 h before CPA treatment (500 nm, 45 min). Surface GluA2 (green) was detected by using an antibody directed against the extracellular epitope of GluA2 in nonpermeabilized conditions, then subsequently permeabilized and stained with chicken anti-MAP2 antibody (red). The p38 MAPK and JNK inhibitors did not significantly affect CPA-mediated GluA1 internalization (see Results). B, Using an shRNA knockdown strategy, the shRNAs p38α MAPK and JNK1 prevented A1R-induced GluA2 internalization. Cultured neurons transfected with the control plasmid A (GFP-fluorescent), p38α MAPK, or JNK1 shRNA were treated with DMSO or CPA (500 nm, 45 min) and subsequently labeled with GluA2 and MAP2 as in A. CPA-induced GluA2 internalization was prevented by transfections of p38α MAPK and JNK1 shRNA plasmids. Average GluA2 intensity values in summary bar charts represent the mean ± SEM from 3 transfections, with the number of neurons indicated inside brackets. *p < 0.05 versus control. **p < 0.01 versus control. ***p < 0.001 versus control. Statistical significance was assessed using one-way ANOVA, followed by post hoc Student-Newman-Keuls test. Scale bars: A, B, 2 μm.
We also tested the effects of p38 MAPK and JNK inhibitors on A1R-mediated GluA1 internalization and found that these pharmacological inhibitors (SB203580 and JNK II inhibitor) did not significantly alter the levels of surface-expressed GluA1, using an antibody directed against an extracellular epitope in nonpermeabilized condition. The GluA1 intensity values (arbitrary units) obtained using a similar analysis performed for GluA2 were as follows: Control GluA1 (691.5 ± 55.4, n = 36), CPA (310.6 ± 24.9, n = 15, p < 0.001 vs control), SB203580 + CPA (433.1 ± 63.4, n = 16, p < 0.05 vs control), SB202474 + CPA (287.6 ± 44.0, n = 22, p < 0.001 vs control), JNK II inhibitor + CPA (503.9 ± 39.5, n = 33, p < 0.01 vs control), and JNK II-negative control + CPA (468.1 ± 29.6, n = 41, p < 001 vs control). These imaging results are in agreement with our biochemical studies (Figs. 6 and 7), indicating that GluA2 and GluA1 internalizations are differentially regulated by A1R-mediated p38 MAPK and JNK activation.
In our previous report (Brust et al., 2007), we found that both SB203580 and JNK II inhibitor (also called SP600125) inhibited A1R-mediated phospho-JNK2/3 elevation, raising the possibility that these drugs may have narrow specificity for p38 MAPK and JNK or that JNK activation is dependent on p38 MAPK activation. We therefore compared the effects of pharmacological inhibitors of p38 MAPK and JNK on CPA-mediated GluA2 internalization with those effects using genetic knockdown of p38 MAPK and JNK. We transfected hippocampal neurons with one of the following plasmids: p38α MAPK shRNA, JNK1 shRNA, or control shRNA Plasmid A. Using Western blotting, we confirmed that, 2 d after transfection, the expression level of p38α MAPK was decreased by 45% and the level of JNK1 was decreased by 46% compared with control shRNA transfections (data not shown). Confocal imaging analysis revealed that, 2 d after transfection of hippocampal neurons with control Plasmid A, p38 MAPK shRNA or JNK shRNA, and subsequent A1R stimulation with CPA (500 nm for 45 min), the A1R-induced GluA2 internalization was completely abrogated by the p38α MAPK or JNK1 shRNAs (Fig. 8B). A modest but significant increase in GluA2 surface levels was also observed in neurons transfected with JNK1 shRNA (Fig. 8B, right). Together, these results indicate that stimulation of A1R-induced GluA2 internalization is dependent on p38 MAPK and JNK activities.
The A2AR is not involved in GluA2 trafficking but is involved in GluA1 trafficking
As per the coimmunoprecipitation and colocalization results shown in Figures 1 and 2, A2ARs are not physically associated with GluA1 and GluA2 AMPARs. However, this does not preclude a functional interaction between A2ARs and AMPARs. Indeed, it has been shown that selective agonist activation of A2ARs increased the GluA1 levels in hippocampus (Dias et al., 2012). To determine whether the stimulation of the A2ARs alters GluA2 surface expression, we incubated hippocampal slices with the A2AR agonist CGS 21680 (10 nm) or the A2AR antagonist SCH 58261 (30 nm) for 1 h. The results showed that CGS 21680 (Fig. 9A) and SCH 58261 (Fig. 9B) did not alter the surface level of GluA2, suggesting that the stimulation of A2ARs does not change the surface level of GluA2. To test whether the stimulation of A2ARs alters the surface expression of GluA1, we also quantified the GluA1 surface levels with treatment of CGS 21680 or SCH 58261 in hippocampal slices. The results showed that CGS 21680 (Fig. 9A), but not SCH 58261 (Fig. 9B), increased the surface level of GluA1, indicating that the stimulation of A2ARs increased the surface level of GluA1.
Figure 9.

The A2AR does not affect A1R-induced GluA2 internalization but does affect A1R-induced GluA1 internalization. A, Surface biotinylation of hippocampal slices that were preincubated with DMSO (control) or CGS 21680 (10 nm), an A2AR agonist, for 1 h followed by CPA treatment (500 nm, 45 min). Blots (left) were probed for GluA2 and GluA1 levels, and summary bar charts (right) show that CGS 21680 alone or in combination with CPA did not mimic or prevent the inhibitory effect of CPA on GluA2 surface expression (left bar chart). Conversely, CGS 21680 prevented CPA-induced GluA1 internalization, and CGS 21680 by itself significantly increased GluA1 surface levels without CPA treatment (right bar chart). B, Biotinylation of hippocampal slices preincubated in SCH 58261 (30 nm), an A2AR antagonist followed by CPA treatment (500 nm, 45 min). Summary bar charts show that SCH 58261 did not significantly affect CPA's effect on the surface levels of GluA2 and GluA1. C, Hippocampal biotinylation using the same protocol as in A labeled for A1R and A2AR. Summary bar chart for A1R (left chart) shows that CPA and CGS 21680 with CPA induced a reduction in surface A1Rs and were not affected by CGS 21680 by itself. The A2AR surface levels (right chart) were not affected by CPA treatment alone but were reduced with CGS 21680 treatments. D, Using the same drug treatments as in B, biotinylation shows A1R and A2AR expression levels with CPA, SCH 58261, or CPA and SCH 58261 together. Summary bar chart for A1R (left chart) shows that CPA alone and CPA with SCH 58261 induced reduced surface levels of A1R, but SCH 58261 alone did not affect A1R levels. A2AR surface levels (right chart) show that CPA and SCH 58261 together as well as SCH58261 by itself caused an increase in A2AR surface levels, but CPA alone did not affect surface levels. Values are mean ± SEM. **p < 0.01. ***p < 0.001.
To determine whether A1R stimulation and A2AR stimulation alter surface expression of either adenosine receptor, we preincubated hippocampal slices with CGS 21680 (10 nm) alone or CGS 21680 (10 nm) for 1 h before CPA incubation (500 nm, 45 min). Incubation of the slices with CGS 21680 (Fig. 9C) or SCH 58261 (Fig. 9D) did not prevent the CPA-induced internalization of A1R. However, SCH 58261 alone or in combination with CPA increased A2AR surface levels (Fig. 9D), suggesting that endogenous adenosine tone was sufficient to cause A2AR desensitization in the hippocampal slices. To address the relationship of stimulation and internalization between A1Rs and A2ARs, we incubated the hippocampal slices with the agonists and antagonists of A1Rs and A2ARs. The results show that CPA application alone decreased the surface level of A1Rs, but not A2ARs (Fig. 9C), suggesting that CPA specifically stimulates A1Rs. Application of CGS 21680 decreased the surface level of A2ARs, but not A1Rs (Fig. 9C). Preincubation of CGS 21680 before CPA treatment did not prevent CPA-induced A1R internalization but still caused A2AR internalization (Fig. 9C), suggesting that CGS 21680 specifically activates A2ARs. SCH 58261 treatment increased the surface level of A2ARs but not A1Rs (Fig. 9D), suggesting that SCH 58261 specifically promotes A2AR surface expression. Preincubation of SCH 58261 before CPA did not prevent CPA-induced A1R internalization, suggesting that stimulation of A2ARs is not involved in the CPA-induced A1R internalization. Preincubation of SCH 58261 before CPA treatment still increased the surface level of A2ARs (Fig. 9D), suggesting that A1R stimulation is not involved in SCH 58261-induced increase in surface level of A2ARs. In summary, the A2AR is not involved in A1R-induced internalization of AMPARs, but A2AR stimulation affects the surface level of GluA1, but not GluA2. In addition, stimulation of A1Rs and A2ARs independently alters their surface expression levels.
Hypoxia mediates GluA2 and GluA1 internalization via clathrin-mediated endocytosis
It is widely accepted that hypoxia increases the extracellular levels of adenosine (Van Wylen et al., 1986; Phillis et al., 1987; Fowler, 1993; Dale et al., 2000). Because of high concentrations of adenosine in hypoxia, adenosine A1 receptors are expected to be activated to mediate hypoxia-induced synaptic depression (Fowler, 1989). We also previously reported that a 5 min hypoxic insult caused significant synaptic depression in CA1 region of hippocampus, and this was shown to be dependent on A1R-mediated activation of p38 MAPK and JNK (Brust et al., 2006, 2007). Earlier studies by Sebastião and colleagues provided the first report of an incomplete recovery of synaptic transmission after slightly more prolonged hypoxic insult to hippocampal slices (Lucchi et al., 1996). In the present study, we performed surface biotinylation and membrane fractionation studies using hippocampal brain slices after a 20 min hypoxic insult to test whether prolonged hypoxic insult induces clathrin-mediated internalization of GluA2- and GluA1-containing AMPARs through A1R activation. Hippocampal slices were preincubated with Tat-GluA2–3Y or scrambled Tat-peptide for 1 h before applying hypoxic stimulation for 20 min. After hypoxia, the membrane fractions or the biotinylated proteins were isolated and analyzed by Western blotting. As shown in Figure 10, the hypoxic insult mimicked the effect of selective A1R stimulation with CPA (see Fig. 3), by significantly decreased GluA2 and GluA1 levels in hippocampal membrane fractions (Fig. 10A,B) and in surface biotinylated samples (Fig. 10C,D).
Figure 10.

Hypoxia-induced internalization of both GluA2 and GluA1 requires clathrin-mediated endocytosis and functional A1Rs in rat hippocampus. A, Hippocampal slices were preincubated with Tat-GluA2–3Y (2 μm) or scrambled Tat-GluA2–3Y (2 μm) peptides for 1 h before applying hypoxic insult (ACSF solution saturated with 95% N2/5% CO2). After a 20 min hypoxic stimulation, hippocampal membrane fractions were isolated and GluA2 levels were subsequently determined by Western blotting. GluA2 signals in membrane fractions were normalized to GAPDH values. During hypoxia, membrane expression of GluA2 was significantly decreased and Tat-GluA2–3Y peptide, but not its scrambled version, prevented this GluA2 downregulation. B, Similar to GluA2, the Tat-GluA2–3Y peptide prevented the decrease in GluA1 expression in hippocampal membrane fractions. A, B, Values are mean ± SEM for GluA2 and GluA1 from six independent experiments. *p < 0.05 versus control. **p < 0.01 versus control. ***p < 0.001 versus control. C, D, Rat hippocampal slices preincubated with DPCPX (500 nm) or Tat-GluA2–3Y peptide (2 μm) were exposed to a 20 min hypoxic insult, and surface proteins were subsequently biotinylated and analyzed by Western blotting. Hypoxia-induced decrease in surface GluA2 (C) and GluA1 (D) was prevented by DPCPX and Tat-GluA2–3Y peptide. In contrast, GluA2 and GluA1 levels in whole hippocampal lysates were not altered (C,D, second row of blots), and β actin was only detected in whole lysate blots (fourth row), but not in the biotinylated blots (third row). C, D, Data are mean ± SEM from three independent experiments. *p < 0.05. ***p < 0.001.
However, the Tat-GluA2–3Y peptide, but not the scrambled Tat-peptide, was also effective in blocking hypoxia-mediated GluA2 and GluA1 internalization, as shown in hippocampal membrane fractions (Fig. 10A,B) and in biotinylated hippocampal tissue (Fig. 10C,D). To confirm that the hypoxia-induced reduction in GluA2 and GluA1 surface expression was caused by A1R stimulation, preincubation of hippocampal slices with the A1R antagonist DPCPX blocked these changes in GluA2 and GluA1 surface levels (Fig. 10C,D, summary bar chart). These results indicate a previously unknown mechanism involving excess elevation of adenosine during hypoxia that leads to clathrin-mediated AMPAR internalization and hypoxia-mediated synaptic depression.
Focal cortical ischemia in an in vivo PVD small-vessel stroke model alters hippocampal surface expression of AMPARs and adenosine receptors, contributes to tonic synaptic depression, and increases neurodegeneration in the hippocampus
Many focal cerebral ischemia models involve occlusion of large cerebral blood vessels, such as the middle cerebral artery, which results in damage to the striatum and cortex to varying degrees depending on the duration of vessel occlusion (Traystman, 2003). During hypoxia, transient global ischemia or focal cerebral ischemia, it is well accepted that there is an increase in the extracellular levels of adenosine (Van Wylen et al., 1986; Rudolphi et al., 1992; Valtysson et al., 1998; Dale et al., 2000; Chu et al., 2013). Brain damage in global and focal ischemia models occurs within selectively vulnerable areas, such as the hippocampal CA1 region, neocortex, and striatum (Kirino, 1982; Smith et al., 1984; McBean and Kelly, 1998; Traystman, 2003; Prosser-Loose et al., 2010). Global ischemia has been shown to selectively reduce the expression of GluA2-containing AMPARs in the CA1 region in rats and gerbils (Pellegrini-Giampietro et al., 1992; Pollard et al., 1993; Gorter et al., 1997; Pellegrini-Giampietro et al., 1997). In this study, we have used a modified PVD protocol, which mimics mild, small-vessel strokes. This involves disruption of Class II size pial vessels and has been shown to produce a consistent cone-shaped cortical lesion damage that does not extend to the corpus callosum (Wang and Walz, 2003; Hua and Walz, 2006b; Cayabyab et al., 2013). Because this represents a permanent nonreperfusion injury model, we hypothesized that adenosine surges will be sufficiently prolonged to cause GluA2 and GluA1 downregulation and induce damage in brain regions distant from the site of injury, such as the hippocampus.
As shown in Figure 11A, B, GluA2 and GluA1 surface expressions in the ipsilateral side of the hippocampus were reduced 2 d after performing the PVD lesion surgeries. Surprisingly, these levels were also downregulated in the contralateral side of the hippocampus. Consistent with our results showing that surface levels of GluA2 and GluA1 are reduced by the A1R agonist CPA, and both AMPAR subunits coimmunoprecipitated with A1Rs, we found that A1R surface expression was reduced both in ipsilateral and contralateral sides of the lesion (Fig. 11C). This result agrees with previous findings (Coelho et al., 2006) showing that hypoxia-mediated increase in extracellular adenosine downregulates A1Rs. In contrast, A2AR surface expression increased in the ipsilateral and contralateral side of the cortical lesion damage. It is likely that, during PVD or hypoxia, stimulation of both A1Rs and A2ARs could result in activation of intracellular signaling pathways (e.g., PP2A, p38MAPK, and JNK via A1Rs and PKA via A2ARs) that could contribute to A2AR surface expression, compared with when A2ARs are activated alone with the CGS21680. However, future studies are needed to further define the molecular mechanisms of A2AR surface insertion after cerebral ischemic damage.
Figure 11.

A focal cortical cerebral ischemia model with PVD injury affects expressions of AMPARs and adenosine receptors in hippocampus. Two days after PVD or sham surgeries, hippocampal slices were prepared for biotinylation and subsequent immunoblotting, and some slices were used for electrophysiology (Fig. 12). The resulting focal cortical lesions decreased surface expression of GluA2 (A), GluA1 (B), and A1R (C) but increased A2AR expression (D) in PVD at both ipsilateral and contralateral sides of the hippocampus compared with sham-operated animals. Values in summary bar charts represent mean ± SEM (N = 4 animals each). *p < 0.05. **p < 0.01. ***p < 0.001.
Because it is widely accepted that adenosine is tonically elevated during cerebral ischemia, we tested the hypothesis that adenosine surges in the brain after focal disruption of cortical pial vessels (no reperfusion) may be sufficient to affect vulnerable brain regions, such as the hippocampus, and influence the induction of synaptic depression. Therefore, we evaluated the effects of PVD versus sham surgeries on synaptic transmission 2 d after surgeries. The fEPSP recordings from hippocampal slices were obtained from the ipsilateral side of PVD surgery lesion or sham surgery. Consistent with a downregulation of A1Rs after PVD, we observed less synaptic potentiation and paired-pulse depression when the A1R antagonist DPCPX was applied to the PVD slices compared with sham brains (Fig. 12A,B). These data indicate that persistent synaptic depression in PVD hippocampal slices reflects changes in both presynaptic (decreased probability of transmitter release) and postsynaptic (altered levels of AMPARs and adenosine receptors) loci. These results also indicate that a focal cortical ischemia can potentially affect vulnerable areas of the brain distant from the site of injury.
Figure 12.

PVD model of focal cortical cerebral ischemia leads to increased synaptic depression in hippocampus. A, Hippocampal slices from sham-operated or PVD-lesioned brains were exposed to 500 nm DPCPX for 30 min to assess the level of synaptic depression (reflecting adenosine tone). DPCPX induced greater synaptic transmission in sham versus PVD hippocampal slices. B, Responses to paired pulses (50 ms apart) revealed greater paired-pulse depression in sham versus PVD hippocampal slices. A, B, Values are mean ± SEM from 4 independent experiments (4 animals each). *p < 0.05 (unpaired Student's t test). **p < 0.01 (unpaired Student's t test). Calibration: A, 0.5 mV, 5 ms; B, 10 ms. The numbers “1” and “2” associated with figure traces and time course charts correspond to fEPSPs at baseline and fEPSP in DPCPX, respectively. C, Confocal images of Fluoro-Jade B staining of Sham hippocampus (Ci) and PVD hippocampus (Cii). Images are from the ipsilateral side of sham surgery or PVD lesion and are representative of slices from four different animal experiments. Scale bars: low- and high-magnification images, 500 and 100 μm, respectively. Boxed regions near CA1, CA2, and CA3 pyramidal body layers in low-magnification images are shown below.
Finally, to quantify neurodegenerative processes in the hippocampus in PVD versus sham, poststroke (48 h) hippocampal slices were obtained to perform Fluoro-Jade B staining. Confocal imaging results of Fluoro-Jade B staining show that more neurodegeneration was observed in the hippocampus in PVD brain slices compared with sham animals (Fig. 12C). This suggests that the disruption of Class II size pial vessels in PVD causes the impairment of hippocampal neurons.
Discussion
In hippocampus, adenosine is implicated in neuroprotection through A1Rs (Rudolphi et al., 1992; Wardas, 2002) and neuronal damage through A2ARs (Rudolphi et al., 1992; Cunha, 2005). However, the signal transduction pathways involved in these contrasting actions are not yet well understood. Acknowledging the contributions of glutamate and adenosine to synaptic depression in cerebral ischemia, we investigated the potential roles of AMPARs in A1R-mediated persistent synaptic depression, which we have termed APSD. We discovered a novel functional and biochemical interactions between A1Rs and AMPARs that ultimately contribute to hippocampal APSDs. Our results indicate that hippocampal APSD is mediated by clathrin-mediated, dynamin-dependent internalization of GluA2- and GluA1-containing AMPARs after prolonged A1R stimulation. To further explore the molecular mechanisms of APSD induction and A1R-mediated AMPAR internalization, pharmacological inhibition or genetic knockdown of p38 MAPK and JNK was used. Inhibition of p38 MAPK and/or JNK activity prevented GluA2, but not GluA1, AMPAR downregulation. This study suggests an important regulatory pathway with potential therapeutic targets to mitigate adenosine-induced cerebral hypoxic/ischemic damage.
The actions of p38 MAPK and JNK have diverse biological functions, including regulation of gene expression, synaptic plasticity, and cell survival (Cargnello and Roux, 2011; Denise Martin et al., 2012), and are also implicated in internalization of epidermal growth factor receptor and AMPARs (Xiong et al., 2006; Boudreau et al., 2007; Lambert et al., 2008; Lambert et al., 2010). Because A1Rs and hypoxia lead to the activation of p38 MAPK and JNK in the brain (Brust et al., 2006; Brust et al., 2007; Sanchez et al., 2012), we hypothesized that A1R-induced AMPAR endocytosis is dependent on p38 MAPK and JNK. Activation of p38 MAPK by the protein synthesis inhibitor anisomycin has been shown to induce clathrin-mediated internalization of GluA2 AMPARs (Xiong et al., 2006), which is in accordance with our finding that A1R activation or hypoxic/ischemic insult leads to clathrin-mediated GluA2 internalization dependent on p38 MAPK and JNK activation. However, our results showed that pharmacological inhibition or shRNA knockdown of p38 MAPK and JNK prevented GluA2, but not GluA1 internalization, indicating that p38 MAPK and JNK could selectively target GluA2. In contrast, p38 MAPK, JNK, and extracellular signal-regulated kinase phosphorylation was shown to be inversely proportional to the surface expression of GluA1 in the nucleus accumbens after cocaine challenge (Boudreau et al., 2007).
The C terminus of each of the four AMPAR subunits contains phosphorylation sites for major serine/threonine kinases. Two major phosphorylation sites of GluA1, Ser831 and Ser845, targets of PKC/CaMKII or PKA, respectively, control GluA1 surface expression and synaptic translocation, which are important in synaptic plasticity (Barria et al., 1997; Derkach et al., 1999; Esteban et al., 2003; He et al., 2011). These phosphorylation sites are dynamically regulated by phosphorylation and dephosphorylation events, where dephosphorylation involves protein phosphatase 1 (PP1) and calcineurin (PP2B), which promote GluA1 internalization and long-term depression (Kameyama et al., 1998; Lee et al., 1998, 2000, 2003; Ehlers, 2000).
Because A1R stimulation leads to PP2A activation and translocation to membrane fractions (Brust et al., 2006), we hypothesized that PP2A inhibition prevents GluA1 internalization by altering the levels of phosphorylated Ser845 (pSer845). Pretreatments of hippocampal slices with the PP2A inhibitors okadaic acid and fostriecin prevented CPA-induced GluA1 and GluA2 internalization after CPA treatment. This is in accordance with other studies that show GluA1 phosphorylation states are integral in the regulation of GluA1 surface expression and function (Lee et al., 1998, 2003). Consistent with our hypothesis, we found that high concentrations of CPA reduced GluA1-pSer845.
We found that GluA2 and GluA1 are physically associated with A1Rs, but not with A2ARs. Because the C terminus of AMPAR subunits contains binding sites for a complex array of signaling proteins, it is possible that a direct or indirect protein–protein interaction exists between AMPARs and A1Rs. Signaling proteins known to bind to the C-terminal regions of AMPARs include N-ethylmaleimide-sensitive factor (Henley et al., 1997; Nishimune et al., 1998; Osten et al., 1998; Song et al., 1998; Zou et al., 2005), SAP-97 (Leonard et al., 1998; Sans et al., 2001), proteins 4.1G/4.1N (Shen et al., 2000; Lin et al., 2009), adaptor protein 2 (Lee et al., 2002), glutamate receptor interacting protein (Dong et al., 1997), and protein interacting with C kinase-1 (Staudinger et al., 1995; Xia et al., 1999). However, it remains to be determined whether these interacting proteins are present in the same macromolecular protein complex as A1Rs and AMPARs to regulate hippocampal AMPAR trafficking, synaptic plasticity, and neurotoxicity.
The cellular mechanisms underlying A1R-mediated changes in synaptic plasticity and neurotoxicity are still unclear. Our current findings indicate a dynamic physiopathological adaptation of glutamatergic synapses to insults triggering a massive elevation of cerebral adenosine tone. We determined that prolonged A1R stimulation with CPA or in focal cortical stroke model led to substantial APSD that did not recover after treatments with A1R antagonist DPCPX. Although fEPSP recovery was observed in the presence of Tat-GluA2–3Y peptide, APSD was not abolished in Tat-GluA2–3Y-pretreated hippocampal slices. Nevertheless, these data indicate that GluA2-containing AMPARs are required for restoration of synaptic activity after prolonged exposure to adenosine during focal cortical ischemia. We found in earlier studies (Brust et al., 2006, 2007) that CPA applied for shorter duration (10 min) and at 10-fold lower concentration (40–50 nm) produced lower levels of APSDs (∼25% compared with 50–60% APSDs in present study using 500 nm CPA for 30 min). We also observed that 50 nm CPA did not significantly alter levels of GluA1-pSer845 (Fig. 7D), indicating that stronger and more prolonged A1R stimulation may be required for robust changes in surface AMPARs, such as during cerebral ischemic insults. This prolonged A1R stimulation led to clathrin-mediated internalization of GluA2-containing AMPARs. The resultant decrease in GluA2-containing AMPARs and A1Rs, as well as the more pronounced APSDs, may contribute to the increased susceptibility of hippocampal CA1 pyramidal neurons to ischemic damage after PVD. Interestingly, we found that p38MAPK and JNK inhibitors selectively reduced GluA2 AMPAR internalization, whereas PP2A inhibitors caused similar reductions in clathrin-mediated endocytosis of GluA2 and GluA1 AMPARs. Results also showed that the neuroprotective and inhibitory A1R surface expression decreased, whereas the neurotoxic and excitatory A2AR surface levels increased during PVD. These results indicate that APSD could mediate neuronal damage in subsequent periods after PVD cortical stroke, although it appears to afford neuronal protection during acute neuronal insult, such as acute hypoxia.
Chronic and acute A1R activation is known to exert a different influence on synaptic plasticity. For example, chronic exposure to CPA enhanced learning in mice (Von Lubitz et al., 1993), whereas acute administration of CPA impaired learning capacity (Normile and Barraco, 1991). In middle-aged rats (7–10 months old), LTP was impaired (Rex et al., 2005), which was consistent with higher adenosine tone that likely enhanced disinhibition of synaptic transmission in aged rats (Sebastião et al., 2000). In line with these behavioral findings, acute administration of A1R antagonist enhanced, while endogenous adenosine reduced, LTP, LTD, and depotentiation (de Mendonça and Ribeiro, 1994, 2000; de Mendonça et al., 1997). LTP was also inhibited by the adenosine uptake blocker nitrobenzylthioinosine, presumably by increasing extracellular adenosine (de Mendonça and Ribeiro, 1994). Cunha and colleagues have also reported previously that A2ARs play a prominent role over A1Rs in regulating hippocampal LTP triggered by electrical stimulation in adult and aged rodents (Costenla et al., 2010, 2011). This requires an A2AR-regulated and NMDAR-mediated enhancement of LTP (Rebola et al., 2008). In addition, Dias et al. (2013) recently reported that oxygen glucose deprivation unmasks a novel form of LTP mediated by increased expression of GluA2-lacking AMPARs and requiring the stimulation of A2ARs, which is opposite to the role of A1Rs in APSD during PVD and hypoxia (present study). Our findings obtained from juvenile rats indicate a novel form of long-lasting synaptic depression, known as APSD, which is triggered by prolonged A1R stimulation, involving activation of p38 MAPK, JNK, and PP2A, and clathrin-mediated endocytosis of GluA2-containing AMPARs. We propose that p38 MAPK and JNK inhibitors would be potent inhibitors of the A1R-mediated excitotoxic potential, by decreasing GluA2 internalization and promoting the neuroprotective GluA2-containing AMPARs on neuronal membranes. Therefore, both A2ARs and A1Rs contribute to adenosine neuromodulation of glutamatergic synapses by increasing GluA2-deficient AMPARs and inducing LTP (for A2ARs, see Dias et al., 2013) and increasing GluA2 and GluA1 endocytosis to produce APSD (for A1Rs, present study). Because previous reports suggested that A2ARs are increased while A1Rs are decreased in middle-aged rats (Cunha et al., 1995; Sebastião et al., 2000; Rebola et al., 2003b), it remains to be established whether this novel form of long-lasting synaptic depression is maintained in middle-aged animals.
Our present study supports the hypothesis that prolonged A1R activation during hypoxia or focal cortical ischemia causes clathrin-mediated GluA2 and GluA1 AMPAR endocytosis and persistent synaptic depression, which could contribute significantly to increased neuronal death. The novel signaling complex formed by A1Rs and GluA2-containing AMPARs represents an important mechanism of ischemic damage that may provide effective therapeutic targets in cerebral ischemia.
Footnotes
This work was supported by Heart and Stroke Foundation of Saskatchewan Grant HSFS GIA 000492, the Canadian Foundation for Innovation, and the Saskatchewan Health Research Foundation. We thank Krishnamoorthy Gowribai for technical assistance and support with PVD surgeries.
The authors declare no competing financial interests.
References
- Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, Taghibiglou C, Wang Y, Lu J, Wong TP, Sheng M, Wang YT. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004;23:1040–1050. doi: 10.1038/sj.emboj.7600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/S0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Brust TB, Cayabyab FS, Zhou N, MacVicar BA. p38 mitogen-activated protein kinase contributes to adenosine A1 receptor-mediated synaptic depression in area CA1 of the rat hippocampus. J Neurosci. 2006;26:12427–12438. doi: 10.1523/JNEUROSCI.4052-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust TB, Cayabyab FS, MacVicar BA. C-Jun N-terminal kinase regulates adenosine A1 receptor-mediated synaptic depression in the rat hippocampus. Neuropharmacology. 2007;53:906–917. doi: 10.1016/j.neuropharm.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Lüscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayabyab FS, Gowribai K, Walz W. Involvement of matrix metalloproteinases-2 and -9 in the formation of a lacuna-like cerebral cavity. J Neurosci Res. 2013;91:920–933. doi: 10.1002/jnr.23223. [DOI] [PubMed] [Google Scholar]
- Chu S, Xiong W, Zhang D, Soylu H, Sun C, Albensi BC, Parkinson FE. Regulation of adenosine levels during cerebral ischemia. Acta Pharmacol Sin. 2013;34:60–66. doi: 10.1038/aps.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Ferré S, Casadó V, Cortés A, Cunha RA, Lluis C, Franco R. Heterodimeric adenosine receptors: a device to regulate neurotransmitter release. Cell Mol Life Sci. 2006;63:2427–2431. doi: 10.1007/s00018-006-6216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho JE, Rebola N, Fragata I, Ribeiro JA, de Mendonça A, Cunha RA. Hypoxia-induced desensitization and internalization of adenosine A1 receptors in the rat hippocampus. Neuroscience. 2006;138:1195–1203. doi: 10.1016/j.neuroscience.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Costenla AR, Cunha RA, de Mendonça A. Caffeine, adenosine receptors, and synaptic plasticity. J Alzheimers Dis. 2010;20(Suppl 1):S25–S34. doi: 10.3233/JAD-2010-091384. [DOI] [PubMed] [Google Scholar]
- Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J, Agostinho PM, Ribeiro JA, Cunha RA, de Mendonça A. Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci. 2011;34:12–21. doi: 10.1111/j.1460-9568.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Constantino MC, Sebastião AM, Ribeiro JA. Modification of A1 and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. Neuroreport. 1995;6:1583–1588. doi: 10.1097/00001756-199507310-00029. [DOI] [PubMed] [Google Scholar]
- Dale N, Pearson T, Frenguelli BG. Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice. J Physiol. 2000;526:143–155. doi: 10.1111/j.1469-7793.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendonça A, Ribeiro JA. Endogenous adenosine modulates long-term potentiation in the hippocampus. Neuroscience. 1994;62:385–390. doi: 10.1016/0306-4522(94)90373-5. [DOI] [PubMed] [Google Scholar]
- de Mendonça A, Ribeiro JA. Long-term potentiation observed upon blockade of adenosine A1 receptors in rat hippocampus is N-methyl-d-aspartate receptor-dependent. Neurosci Lett. 2000;291:81–84. doi: 10.1016/S0304-3940(00)01391-4. [DOI] [PubMed] [Google Scholar]
- de Mendonça A, Sebastião AM, Ribeiro JA. Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport. 1995;6:1097–1100. doi: 10.1097/00001756-199505300-00006. [DOI] [PubMed] [Google Scholar]
- de Mendonça A, Almeida T, Bashir ZI, Ribeiro JA. Endogenous adenosine attenuates long-term depression and depotentiation in the CA1 region of the rat hippocampus. Neuropharmacology. 1997;36:161–167. doi: 10.1016/S0028-3908(96)00173-6. [DOI] [PubMed] [Google Scholar]
- Denise Martin E, De Nicola GF, Marber MS. New therapeutic targets in cardiology: p38 alpha mitogen-activated protein kinase for ischemic heart disease. Circulation. 2012;126:357–368. doi: 10.1161/CIRCULATIONAHA.111.071886. [DOI] [PubMed] [Google Scholar]
- Dennis SH, Jaafari N, Cimarosti H, Hanley JG, Henley JM, Mellor JR. Oxygen/glucose deprivation induces a reduction in synaptic AMPA receptors on hippocampal CA3 neurons mediated by mGluR1 and adenosine A3 receptors. J Neurosci. 2011;31:11941–11952. doi: 10.1523/JNEUROSCI.1183-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias RB, Ribeiro JA, Sebastião AM. Enhancement of AMPA currents and GluR1 membrane expression through PKA-coupled adenosine A(2A) receptors. Hippocampus. 2012;22:276–291. doi: 10.1002/hipo.20894. [DOI] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Sebastião AM. Ischemia-induced synaptic plasticity drives sustained expression of calcium-permeable AMPA receptors in the hippocampus. Neuropharmacology. 2013;65:114–122. doi: 10.1016/j.neuropharm.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/S0896-6273(00)00129-X. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Figueiredo BC, Plüss K, Skup M, Otten U, Cuello AC. Acidic FGF induces NGF and its mRNA in the injured neocortex of adult animals. Brain Res Mol Brain Res. 1995;33:1–6. doi: 10.1016/0169-328X(95)00099-E. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Res. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists alter the synaptic response to in vitro ischemia in the rat hippocampus. Brain Res. 1990;509:331–334. doi: 10.1016/0006-8993(90)90560-X. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Purine release and inhibition of synaptic transmission during hypoxia and hypoglycemia in rat hippocampal slices. Neurosci Lett. 1993;157:83–86. doi: 10.1016/0304-3940(93)90648-5. [DOI] [PubMed] [Google Scholar]
- Fowler JC, Gervitz LM, Hamilton ME, Walker JA. Systemic hypoxia and the depression of synaptic transmission in rat hippocampus after carotid artery occlusion. J Physiol. 2003;550:961–972. doi: 10.1113/jphysiol.2003.039594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Gervitz LM, Davies DG, Omidvar K, Fowler JC. The effect of acute hypoxemia and hypotension on adenosine-mediated depression of evoked hippocampal synaptic transmission. Exp Neurol. 2003;182:507–517. doi: 10.1016/S0014-4886(03)00160-2. [DOI] [PubMed] [Google Scholar]
- Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Greene RW. Adenosine enhances afterhyperpolarization and accommodation in hippocampal pyramidal cells. Pflugers Arch. 1984;402:244–247. doi: 10.1007/BF00585506. [DOI] [PubMed] [Google Scholar]
- He K, Lee A, Song L, Kanold PO, Lee HK. AMPA receptor subunit GluR1 (GluA1) serine-845 site is involved in synaptic depression but not in spine shrinkage associated with chemical long-term depression. J Neurophysiol. 2011;105:1897–1907. doi: 10.1152/jn.00913.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM, Nishimune A, Nash SR, Nakanishi S. Use of the two-hybrid system to find novel proteins that interact with alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor subunits. Biochem Soc Trans. 1997;25:838–842. doi: 10.1042/bst0250838. [DOI] [PubMed] [Google Scholar]
- Henley JR, Cao H, McNiven MA. Participation of dynamin in the biogenesis of cytoplasmic vesicles. FASEB J. 1999;13(Suppl 2):S243–S247. doi: 10.1096/fasebj.13.9002.s243. [DOI] [PubMed] [Google Scholar]
- Herrera DG, Robertson HA. N-methyl-d-aspartate receptors mediate activation of the c-fos proto-oncogene in a model of brain injury. Neuroscience. 1990;35:273–281. doi: 10.1016/0306-4522(90)90081-E. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Hua R, Walz W. The need for animal models in small-vessel brain disease. Crit Rev Neurobiol. 2006a;18:5–11. doi: 10.1615/CritRevNeurobiol.v18.i1-2.20. [DOI] [PubMed] [Google Scholar]
- Hua R, Walz W. Minocycline treatment prevents cavitation in rats after a cortical devascularizing lesion. Brain Res. 2006b;1090:172–181. doi: 10.1016/j.brainres.2006.03.072. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/S0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Kumar J, Mayer ML. Functional insights from glutamate receptor ion channel structures. Annu Rev Physiol. 2013;75:313–337. doi: 10.1146/annurev-physiol-030212-183711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laghi Pasini F, Guideri F, Picano E, Parenti G, Petersen C, Varga A, Di Perri T. Increase in plasma adenosine during brain ischemia in man: a study during transient ischemic attacks, and stroke. Brain Res Bull. 2000;51:327–330. doi: 10.1016/S0361-9230(99)00240-3. [DOI] [PubMed] [Google Scholar]
- Lambert S, Ameels H, Gniadecki R, Hérin M, Poumay Y. Internalization of EGF receptor following lipid rafts disruption in keratinocytes is delayed and dependent on p38 MAPK activation. J Cell Physiol. 2008;217:834–845. doi: 10.1002/jcp.21563. [DOI] [PubMed] [Google Scholar]
- Lambert S, Frankart A, Poumay Y. p38 MAPK-regulated EGFR internalization takes place in keratinocyte monolayer during stress conditions. Arch Dermatol Res. 2010;302:229–233. doi: 10.1007/s00403-009-1020-0. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Liu F. Direct interactions between NMDA and D1 receptors: a tale of tails. Biochem Soc Trans. 2004;32:1032–1036. doi: 10.1042/BST0321032. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/S0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/S0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/S0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG, Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lucchi R, Latini S, de Mendonça A, Sebastião AM, Ribeiro JA. Adenosine by activating A1 receptors prevents GABAA-mediated actions during hypoxia in the rat hippocampus. Brain Res. 1996;732:261–266. doi: 10.1016/0006-8993(96)00748-2. [DOI] [PubMed] [Google Scholar]
- Lupica CR, Proctor WR, Dunwiddie TV. Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: analysis of unitary EPSP variance measured by whole-cell recording. J Neurosci. 1992;12:3753–3764. doi: 10.1523/JNEUROSCI.12-10-03753.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/S0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Malinow R. AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:707–714. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/S0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Graf R, Rosner G, Shimada N, Heiss WD. Flow thresholds for extracellular purine catabolite elevation in cat focal ischemia. Brain Res. 1992;579:309–314. doi: 10.1016/0006-8993(92)90066-I. [DOI] [PubMed] [Google Scholar]
- McBean DE, Kelly PA. Rodent models of global cerebral ischemia: a comparison of two-vessel occlusion and four-vessel occlusion. Gen Pharmacol. 1998;30:431–434. doi: 10.1016/S0306-3623(97)00284-X. [DOI] [PubMed] [Google Scholar]
- Mezler M, Müller T, Raming K. Cloning and functional expression of GABA(B) receptors from Drosophila. Eur J Neurosci. 2001;13:477–486. doi: 10.1046/j.1460-9568.2001.01410.x. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Johe KK, Moghadam B, Adams RN. Studies on the ionic mechanism for the neuromodulatory actions of adenosine in the brain. Brain Res. 1988;473:249–260. doi: 10.1016/0006-8993(88)90854-2. [DOI] [PubMed] [Google Scholar]
- Newton AJ, Kirchhausen T, Murthy VN. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc Natl Acad Sci U S A. 2006;103:17955–17960. doi: 10.1073/pnas.0606212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/S0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- Normile HJ, Barraco RA. N6-cyclopentyladenosine impairs passive avoidance retention by selective action at A1 receptors. Brain Res Bull. 1991;27:101–104. doi: 10.1016/0361-9230(91)90288-U. [DOI] [PubMed] [Google Scholar]
- Ochiishi T, Chen L, Yukawa A, Saitoh Y, Sekino Y, Arai T, Nakata H, Miyamoto H. Cellular localization of adenosine A1 receptors in rat forebrain: immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J Comp Neurol. 1999;411:301–316. doi: 10.1002/(SICI)1096-9861(19990823)411:2<301::AID-CNE10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/S0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci U S A. 1992;89:10499–10503. doi: 10.1073/pnas.89.21.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Pulsinelli WA, Zukin RS. NMDA and non-NMDA receptor gene expression following global brain ischemia in rats: effect of NMDA and non-NMDA receptor antagonists. J Neurochem. 1994;62:1067–1073. doi: 10.1046/j.1471-4159.1994.62031067.x. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/S0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Walter GA, O'Regan MH, Stair RE. Increases in cerebral cortical perfusate adenosine and inosine concentrations during hypoxia and ischemia. J Cereb Blood Flow Metab. 1987;7:679–686. doi: 10.1038/jcbfm.1987.122. [DOI] [PubMed] [Google Scholar]
- Pollard H, Héron A, Moreau J, Ben-Ari Y, Khrestchatisky M. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience. 1993;57:545–554. doi: 10.1016/0306-4522(93)90004-Y. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Dunwiddie TV. Adenosine inhibits calcium spikes in hippocampal pyramidal neurons in vitro. Neurosci Lett. 1983;35:197–201. doi: 10.1016/0304-3940(83)90550-5. [DOI] [PubMed] [Google Scholar]
- Prosser-Loose EJ, Verge VM, Cayabyab FS, Paterson PG. Protein-energy malnutrition alters hippocampal plasticity-associated protein expression following global ischemia in the gerbil. Curr Neurovasc Res. 2010;7:341–360. doi: 10.2174/156720210793180792. [DOI] [PubMed] [Google Scholar]
- Ramos AJ, Rubio MD, Defagot C, Hischberg L, Villar MJ, Brusco A. The 5HT1A receptor agonist, 8-OH-DPAT, protects neurons and reduces astroglial reaction after ischemic damage caused by cortical devascularization. Brain Res. 2004;1030:201–220. doi: 10.1016/j.brainres.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Rebola N, Pinheiro PC, Oliveira CR, Malva JO, Cunha RA. Subcellular localization of adenosine A(1) receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003a;987:49–58. doi: 10.1016/S0006-8993(03)03247-5. [DOI] [PubMed] [Google Scholar]
- Rebola N, Sebastião AM, de Mendonça A, Oliveira CR, Ribeiro JA, Cunha RA. Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J Neurophysiol. 2003b;90:1295–1303. doi: 10.1152/jn.00896.2002. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Rex CS, Kramár EA, Colgin LL, Lin B, Gall CM, Lynch G. Long-term potentiation is impaired in middle-aged rats: regional specificity and reversal by adenosine receptor antagonists. J Neurosci. 2005;25:5956–5966. doi: 10.1523/JNEUROSCI.0880-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolphi KA, Schubert P. Adenosine and adenine nucleotides: from molecular biology to integrative physiology. New York: Springer; 1995. Adenosine and brain ischemia; pp. 391–397. [Google Scholar]
- Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-R. [DOI] [PubMed] [Google Scholar]
- Salter MW. D1 and NMDA receptors hook up: expanding on an emerging theme. Trends Neurosci. 2003;26:235–237. doi: 10.1016/S0166-2236(03)00081-X. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Tripathy D, Yin X, Desobry K, Martinez J, Riley J, Gay D, Luo J, Grammas P. p38 MAPK: a mediator of hypoxia-induced cerebrovascular inflammation. J Alzheimers Dis. 2012;32:587–597. doi: 10.3233/JAD-2012-120829. [DOI] [PubMed] [Google Scholar]
- Sans N, Racca C, Petralia RS, Wang YX, McCallum J, Wenthold RJ. Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J Neurosci. 2001;21:7506–7516. doi: 10.1523/JNEUROSCI.21-19-07506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Vissel B, Petralia RS, Wang YX, Chang K, Royle GA, Wang CY, O'Gorman S, Heinemann SF, Wenthold RJ. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J Neurosci. 2003;23:9367–9373. doi: 10.1523/JNEUROSCI.23-28-09367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastião AM, Cunha RA, de Mendonça A, Ribeiro JA. Modification of adenosine modulation of synaptic transmission in the hippocampus of aged rats. Br J Pharmacol. 2000;131:1629–1634. doi: 10.1038/sj.bjp.0703736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Intracellular analysis of a postsynaptic action of adenosine in the rat hippocampus. Eur J Pharmacol. 1982;79:193–199. doi: 10.1016/0014-2999(82)90625-2. [DOI] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Schubert P. Adenosine depression of hippocampal neurons in vitro: an intracellular study of dose-dependent actions on synaptic and membrane potentials. Neurosci Lett. 1981;23:55–60. doi: 10.1016/0304-3940(81)90186-5. [DOI] [PubMed] [Google Scholar]
- Smith ML, Bendek G, Dahlgren N, Rosén I, Wieloch T, Siesjö BK. Models for studying long-term recovery following forebrain ischemia in the rat: 2. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/S0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. PICK1: a perinuclear binding protein and substrate for protein kinase C isolated by the yeast two-hybrid system. J Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Fourreau L, Bloch B, Fredholm BB, Gonon F, Le Moine C. Opposite tonic modulation of dopamine and adenosine on c-fos gene expression in striatopallidal neurons. Neuroscience. 1999;89:827–837. doi: 10.1016/S0306-4522(98)00403-5. [DOI] [PubMed] [Google Scholar]
- Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J. 2003;44:85–95. doi: 10.1093/ilar.44.2.85. [DOI] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtysson J, Persson L, Hillered L. Extracellular ischaemia markers in repeated global ischaemia and secondary hypoxaemia monitored by microdialysis in rat brain. Acta Neurochir (Wien) 1998;140:387–395. doi: 10.1007/s007010050113. [DOI] [PubMed] [Google Scholar]
- Van Wylen DG, Park TS, Rubio R, Berne RM. Increases in cerebral interstitial fluid adenosine concentration during hypoxia, local potassium infusion, and ischemia. J Cereb Blood Flow Metab. 1986;6:522–528. doi: 10.1038/jcbfm.1986.97. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DK, Paul IA, Bartus RT, Jacobson KA. Effects of chronic administration of adenosine A1 receptor agonist and antagonist on spatial learning and memory. Eur J Pharmacol. 1993;249:271–280. doi: 10.1016/0014-2999(93)90522-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Walz W. Unusual topographical pattern of proximal astrogliosis around a cortical devascularizing lesion. J Neurosci Res. 2003;73:497–506. doi: 10.1002/jnr.10683. [DOI] [PubMed] [Google Scholar]
- Wardas J. Neuroprotective role of adenosine in the CNS. Pol J Pharmacol. 2002;54:313–326. [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/S0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Xiong W, Kojic LZ, Zhang L, Prasad SS, Douglas R, Wang Y, Cynader MS. Anisomycin activates p38 MAP kinase to induce LTD in mouse primary visual cortex. Brain Res. 2006;1085:68–76. doi: 10.1016/j.brainres.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/S0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, van Aelst L, Sheng M, Zhu JJ. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]
- Zou S, Li L, Pei L, Vukusic B, Van Tol HH, Lee FJ, Wan Q, Liu F. Protein-protein coupling/uncoupling enables dopamine D2 receptor regulation of AMPA receptor-mediated excitotoxicity. J Neurosci. 2005;25:4385–4395. doi: 10.1523/JNEUROSCI.5099-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]