

Abstract
Alzheimer's disease (AD) is characterized by extracellular amyloid-β (Aβ) deposits and microglia-dominated inflammatory activation. Innate immune signaling controls microglial inflammatory activities and Aβ clearance. However, studies examining innate immunity in Aβ pathology and neuronal degeneration have produced conflicting results. In this study, we investigated the pathogenic role of innate immunity in AD by ablating a key signaling molecule, IKKβ, specifically in the myeloid cells of TgCRND8 APP-transgenic mice. Deficiency of IKKβ in myeloid cells, especially microglia, simultaneously reduced inflammatory activation and Aβ load in the brain and these effects were associated with reduction of cognitive deficits and preservation of synaptic structure proteins. IKKβ deficiency enhanced microglial recruitment to Aβ deposits and facilitated Aβ internalization, perhaps by inhibiting TGF-β-SMAD2/3 signaling, but did not affect Aβ production and efflux. Therefore, inhibition of IKKβ signaling in myeloid cells improves cognitive functions in AD mice by reducing inflammatory activation and enhancing Aβ clearance. These results contribute to a better understanding of AD pathogenesis and could offer a new therapeutic option for delaying AD progression.
Keywords: Alzheimer's disease, endocytosis, inflammation, microglia, neurodegeneration, NF-κB
Introduction
Microglia-dominated neuroinflammation is a hallmark of Alzheimer's disease (AD) (Akiyama et al., 2000; Wyss-Coray and Rogers, 2012). Positron emission tomography studies have shown that microglial activation correlates with disease progression in AD patients (Cagnin et al., 2001; Edison et al., 2008; Okello et al., 2009). In AD mouse models, which overexpress Alzheimer's APP in neurons, microglia are observed to be activated and recruited to deposits of pathogenic amyloid β peptides (Aβ) (Bolmont et al., 2008; Meyer-Luehmann et al., 2008).
Increasing evidence suggests that the innate immune system drives chronic neuroinflammation, controls cerebral Aβ load, and subsequently affects neuronal degeneration in AD. In APP-transgenic mice, a deficiency in certain innate immune molecules, such as CD14 (Reed-Geaghan et al., 2010), Toll-like receptor 2 (TLR2) (Liu et al., 2012), TLR4 (Song et al., 2011), myeloid differentiation factor 88 (MyD88) (Hao et al., 2011), and interleukin receptor-associated kinase 4 (IRAK4) (Cameron et al., 2012), attenuates the degree of neuroinflammation, shifts microglial activation from proinflammatory to anti-inflammatory pathways, or both. However, the results of studies of the effects of innate immunity on Aβ pathology and neuronal degeneration in AD mice are inconclusive and sometimes contradictory. For example, one study showed that TLR4 deficiency exaggerates Aβ deposition (Tahara et al., 2006), whereas another study showed that a deficiency in the TLR4 coreceptor CD14 exerts the opposite effect by reducing the cerebral Aβ load (Reed-Geaghan et al., 2010). Several studies have found that a deficiency in MyD88 or its downstream signaling molecule, IRAK4, attenuates Aβ pathology and neuronal damage in APP-transgenic mice (Hao et al., 2011; Lim et al., 2011; Cameron et al., 2012), but other reports state that wild-type MyD88 is necessary for clearing Aβ from the brain and protecting cognitive function (Michaud et al., 2011 and 2012). Although it should be noted that these studies differ in methods and animal models, the apparently conflicting results remain puzzling; therefore, neuroscientists are actively investigating whether innate immunity is “friend or foe” in AD pathogenesis.
To explore this question, we examined the role of inhibitor of NF-κB (IκB) kinase-β (IKKβ), one of the key signaling molecules downstream of TLRs-MyD88-IRAK4 in microglia (Kawai and Akira, 2011). Activated IKKβ phosphorylates IκB and triggers its degradation by proteasomes and this process results in translocation of NF-κB to the nucleus and the induction of NF-κB-dependent transcription of a wide range of immune and inflammatory genes (Lawrence, 2009). We used the Cre-Lox technique to knock out the ikbkb gene (encoding IKKβ) specifically in the myeloid cell lineage (including microglia, macrophages, and monocytes) in APP-transgenic mice under physiological conditions. As shown by Mildner et al. (2011), engraftment of bone marrow (BM)-derived cells in the brain parenchyma of AD mice does not occur normally during disease progression without preconditioning the brain (e.g., by irradiation). In our APP-transgenic mice, no peripheral myeloid cells should infiltrate the brain under physiological conditions, and the effects of the deficiency in IKKβ in endogenous microglia in these mice should be apparent. Here, we report the results of the study.
Materials and Methods
Mice.
TgCRND8 APP-transgenic mice (apptg) on a 129 background (continuously interbred for >9 generations to reach the genetic cognate) expressing a transgene incorporating both the Indiana mutation (V717F) and the Swedish mutations (K670N/M671L) in the human app gene under the control of hamster prion protein (PrP) promoter were kindly provided by D. Westaway (University of Toronto). In this mouse strain, the Aβ load does not differ between male and female mice (Chishti et al., 2001). Ikbkbfl/fl mice carrying loxP site-flanked ikbkb alleles on a C57BL/6N genetic background were kindly provided by M. Pasparakis (University of Cologne; Pasparakis et al., 2002). Ikbkbfl/fl mice express normal levels of IKKβ. LysM-Cre knock-in mice (LysM-Cre+/+) expressing Cre from the endogenous lysozyme 2 gene locus were obtained from The Jackson Laboratory (stock number 004781; Clausen et al., 1999) and were back-crossed to C57BL/6J mice for >6 generations. Therefore, myeloid cell type (e.g., microglia and macrophages)-specific IKKβ-deficient (ikbkbfl/flCre+/−) mice were generated by breeding ikbkbfl/fl mice and LysM-Cre knock-in mice. APP-transgenic mice deficient in IKKβ specifically in myeloid cells (apptgikbkbfl/flCre+/−) were then established by cross-breeding APP-transgenic mice with ikbkbfl/fl and LysM-Cre mice on a constant C57BL/6129 (1:1) genetic background. All mice from the same litter were used for the study without any exclusion so that the phenotype of APP-transgenic mice with or without IKKβ ablation in myeloid cells was compared only between siblings. To demonstrate the cell-specific expression of Cre recombinase, we cross-bred LysM-Cre mice to ROSAmT/mG Cre report mice (The Jackson Laboratory; stock number 007676), which express cell membrane-targeted tomato fluorescent protein (TFP) before Cre exposure and express cell-membrane-targeted GFP in Cre-expressing cells. To track microglia and brain macrophages with deficiency of IKKβ in the AD mice, apptgikbkbfl/flCre+/− mice were further mated to ROSAmT/mG mice to obtain apptgikbkbfl/flROSAmT/mGCre+/− mice. To demonstrate the chemokine (C-C motif) receptor 2 (CCR2)-positive cells in the AD mice, we cross-bred TgCRND8 APP-transgenic mice to CCR2-RFP mice (The Jackson Laboratory; stock number 017586), in which the CCR2 coding sequence has been replaced with an RFP-encoding sequence. All animal experiments were approved by the regional ethical committee in Saarland, Germany.
Cell isolation and primary microglial culture.
Primary microglia were isolated from brains of newborn mice resulting from crosses of ikbkbfl/fl and LysM-Cre knock-in mice (ikbkbfl/flCre−/− and ikbkbfl/flCre+/−). Microglia were cultured in DMEM (Life Technologies) supplemented with 10% FCS (PAN Biotech) in 25 cm2 flasks (BD Biosciences) according to a published protocol (Liu et al., 2005). As shown by flow cytometry, >98% of microglia were CD11b+.
Preparation of Aβ peptides.
The 42 aa form of human Aβ (Aβ42) was kindly provided by Dr. L. Fülöp (Albert Szent Gyorgyi Medical University, Szeged, Hungary). The oligomeric and fibrillar Aβs were prepared according to a published protocol (Dahlgren et al., 2002). Fluorescent Aβ was prepared by mixing HiLyte Fluor 488-labeled human Aβ42 (AnaSpec) and unlabeled Aβ at a ratio of 1:10. Endotoxin concentrations of peptide samples were <0.01 EU/ml, as determined by the Limulus amebocyte lysate assay (Lonza). Western blot analysis confirmed that HiLyte Fluor 488-conjugated and nonconjugated forms of Aβ42 peptides had similar oligomeric conformations.
Flow cytometric analysis of HiLyte Fluor 488-conjugated Aβ42 internalization in primary microglia.
To investigate effects of IKKβ on microglial internalization of Aβ, IKKβ-deficient and wild-type primary microglia cultured in a 24-well plate (BD Biosciences) at a density of 3 × 105 cells per well were treated with 0.2 or 1.0 μm HiLyte Fluor 488-conjugated Aβ42 aggregates for various time periods, as indicated in the Results. To investigate effects of blocking TGF-β-SMAD2/3 signaling on Aβ internalization, wild-type microglia were pretreated with the activin-like kinase-5 (ALK5) inhibitors SB-505124 and SB-431542 at 0, 0.004, 0.02, 0.1, 0.5, or 1.0 μm for 1 h, followed by incubation with 1.0 μm fluorescent oligomeric Aβ42 aggregates for 6 h in the presence of pretreated inhibitors. Thereafter, microglia were washed with a 1× solution of PBS and detached from the plate with trypsin-EDTA (Life Technologies). The mean fluorescence intensity (mFI) of internalized fluorophore-conjugated Aβ42 and the percentages of positive cells with intracellular proteins were immediately determined with a FACSCanto II flow cytometer (BD Biosciences). To examine the surface binding of Aβ, we incubated cells with 1.0 μm HiLyte Fluor 488-conjugated Aβ42 aggregates for 2 h on ice and then analyzed them for mFI.
Western blot detection of phosphorylated p65 and SMAD proteins in microglia.
Primary microglia were cultured in the 6-well plate (BD Biosciences) at a density of 1 × 106 cells/well For detection of phosphorylated and total amount of the p65 component of the NF-κB complex, IKKβ-deficient and wild-type cells were activated with 10 μm oligomeric Aβ42 for 12 min. For detection of phosphorylated and total amount of SMAD2 and SMAD5, IKKβ-deficient and wild-type cells were activated with recombinant mouse TGF-β1 (R&D Systems) at 0, 1, and 10 ng/ml for 30 min. To test the inhibitory effects of ALK5 inhibitors, wild-type primary microglia were pretreated with SB-505124 or SB-431542 at 0, 0.02, and 0.1 μm for 1 h and then treated with 1 ng/ml TGF-β1 for 30 min in the presence of inhibitors. After treatments, cells were immediately lysed in buffer (50 mm Tris/HCl, pH 7.4, 2 mm EDTA, 50 nm okadaic acid, 5 mm sodium pyrophosphate, 50 mm NaF, 1 mm DTT, 1% Triton X-100, and protease inhibitor mixture; Roche Applied Science). For Western blot, rabbit monoclonal antibodies against phosphorylated and total p65, SMAD2, and SMAD5, and β-actin (clone 93H1, D14E12, 138D4, D43B4, D5B10, D4G2, and 13E5, respectively; Cell Signaling Technology) were used. Western blots were visualized via the ECL method (PerkinElmer). Densitometric analysis of band densities was performed with ImageJ software version 1.4.3. For each sample, the protein level was calculated as a ratio of target protein/β-actin from that sample.
Barnes maze test.
The Barnes maze test was used to assess the cognitive function of various APP-transgenic and wild-type littermate mice according to an established protocol (Hao et al., 2011; Liu et al., 2012). The test involved 5 d of acquisition training with 2 trials per day. For each trial, latency to enter the escape chamber and distance traveled were recorded by EthoVision XT version 7.0 tracking software (Noldus Information Technology), and an average time for latency and total distance was obtained by averaging the results of the 2 daily trials. Twenty-four hours after the last training day, a probe trial was performed, in which the escape chamber was removed, the mice were placed in the center of the maze just as during acquisition training, and each mouse was given 5 min to explore the maze. During the probe phase, we recorded the time mice spent in the target zone, which is adjacent to the escape hole and its two neighboring holes, and the frequency with which mice visited the target zone and the nontargeting zone, which surround other holes far from the escape hole (see Fig. 4A). The experimenter was blinded to the genotypes of the mice during the entire test.
Figure 4.

Deficiency in IKKβ in myeloid cells improves cognitive function in APP-transgenic mice. A, Schematic of the Barnes maze used. During the training phase, 6-month-old APP-transgenic mice (APPtg) spent more time in the maze and traveled longer distances to reach the escape hole than did their non-APP-transgenic littermates (APPwt). Ablation of IKKβ in myeloid cells (IKKβko) significantly reduced the traveling time and distance of APPtg mice but not of APPwt mice (B, C; two-way ANOVA; n ≥ 9 per group). In the probe trial, APPtg/IKKβko mice remained in the target zone significantly longer and visited the escape hole more frequently than the APPtg/IKKβwt mice (D, E; one-way ANOVA; n ≥ 9 per group). The amount of PSD-95 and Munc18-1 in the brain homogenate was quantified with Western blotting (F, G). Deficiency in myeloid IKKβ was associated with a higher level of PSD-95 and Munc18–1 in the APPtg mouse, but not in the APPwt mouse (F, G; one-way ANOVA; n ≥ 6 per group).
Positive selection of CD11b+ microglia/brain macrophages in the adult mouse brain.
To determine the gene expression and cell surface antigen expression in microglia/brain macrophages, we carefully dissected the entire cerebrum from 3- or 6-month-old APP-transgenic mice with or without a deficiency in myeloid IKKβ (ikbkbfl/flCre−/− or ikbkbfl/flCre+/−). We prepared a single-cell suspension with the Neural Tissue Dissociation Kit (papain-based) and removed myelin by using Myelin Removal Beads II (both from Miltenyi Biotec) according to the manufacturer's protocols. After pelleting cells by centrifugation, we added 80 μl of blocking buffer containing 25 μg/ml rat anti-mouse CD16/CD32 antibody (2.4G2; BD Biosciences) and 10% FCS to prevent nonspecific binding. Thirty minutes after blocking at 4°C, we added 20 μl of MicroBeads-conjugated CD11b antibody (Miltenyi Biotec) directly to the cells. After 1 additional hour of incubation at 4°C, cells were washed with buffer and loaded onto a MACS LS Column (Miltenyi Biotec) for separation. Lysis buffer was immediately added to both CD11b+ and CD11b− cells for isolation of total RNA with the RNeasy Plus Mini Kit (Qiagen); alternatively, CD11b+ cells were used for flow cytometric analysis after cells were stained with fluorescence-labeled anti-CD45 antibody (clone 30-F11′ eBioscience) or CD11b+ cells were used for Western blot detection of IKKβ after cells were lysed in buffer (50 mm Tris/HCl, pH 7.4, 145 mm NaCl, 1% Triton-100, and protease inhibitor mixture; Roche Applied Science) and the rabbit monoclonal antibody against IKKβ (clone Y466; Abcam) was used to blot the membrane.
Tissue collection for pathological analysis.
Animals were put to death at 6 months of age by inhalation of isoflurane (Abbott Laboratories). Whole blood was collected by intracardial puncture and placed into EDTA-containing Eppendorf tubes. Mice were then rapidly perfused transcardially with ice-cold PBS, and the brain was removed and divided. The left hemibrain was immediately fixed in 4% PFA (Sigma-Aldrich) for histological analysis. A 0.5-mm-thick piece of tissue was sagittally cut from the right hemibrain. The cortex and hippocampus were carefully separated and homogenized in TRIzol (Life Technologies) for RNA isolation. The remainder of the right hemibrain was snap-frozen in liquid nitrogen for biochemical analysis.
Brain homogenates and Aβ and TNF-α ELISA.
The brain was homogenized as described previously (Liu et al., 2012). Briefly, frozen hemispheres were bounce-homogenized in TBS containing a protease inhibitor mixture (Roche Applied Science) and centrifuged at 16,000 × g for 30 min at 4°C. The supernatant (TBS-soluble fraction) was collected and stored at −80°C. The pellets were resuspended in TBS plus 1% Triton X-100 (TBS-T), sonicated for 5 min in a 4°C water bath, and centrifuged at 16,000 × g for another 30 min at 4°C. The supernatant was collected and stored at −80°C as the TBS-T-soluble fraction. The pellets were extracted for a third time with an ice-cold guanidine buffer (5 m guanidine HCl/50 mm Tris, pH 8.0, herein referred to as the guanidine-soluble fraction). The protein concentration of all samples was measured with the Bio-Rad Protein Assay. The Aβ concentrations in three separate fractions of brain homogenates were determined by Aβ42 and Aβ40 ELISA kits (both from Life Technologies). The concentration of aggregated Aβ in TBS-T-soluble fraction was also assayed with Oligomeric Amyloid-β ELISA Kit (Biosensis). The TNF-α concentration in the TBS-soluble brain fraction was measured by ELISA (eBioscience). Results were normalized on the basis of the sample's protein concentration.
Western blot analysis of synaptic proteins.
To detect synaptic proteins in the brain, we used 10% Tris-glycine PAGE to separate TBS-T-soluble brain homogenate derived from APP-transgenic and non-APP-transgenic littermate mice differing in IKKβ expression. Western blots and analysis were performed as described in the section “Western blot detection of proteins in microglia” using the following antibodies: mouse monoclonal anti-PSD-95 (clone 6G6-1C9; Abcam) and rabbit polyclonal anti-the mammalian homolog of unc-18 protein (Munc18)-1 (Cell Signaling Technology). The mouse monoclonal anti-α-tubulin (clone DM1A; Abcam) was used as a protein loading control.
Immunohistochemistry.
Four percent PFA-fixed left hemispheres were embedded in paraffin and serial 40-μm-thick sagittal sections were cut and mounted on glass slides. Immunohistochemical staining with the primary antibody, rabbit anti-ionized calcium-binding adapter molecule 1 (Iba-1) (1:500; Wako Chemicals), or mouse monoclonal anti-human Aβ antibody (clone 6F/3D; Dako), was performed on these sections with the VectaStain Elite ABC kit (Vector Laboratories). Diaminobenzidine (Sigma-Aldrich) was used as the chromogen and counterstaining was performed with hematoxylin for Iba-1 staining. To detect proliferating microglia, we added the rabbit monoclonal antibody (clone SP6; Abcam) directed against Ki67 to sections that had been already stained with Iba-1 antibodies but before hematoxylin counterstaining. Thereafter, we used the VectaStain ABC-AP kit and the VECTOR Blue Alkaline Phosphatase Substrate kit without hematoxylin counterstaining. Ki67 staining was visualized in blue. To investigate potentially brain-infiltrating peripheral leukocytes, we performed immunohistochemical staining with rat anti-mouse CD45 (clone 30-F11; BD Biosciences), rabbit anti-CD3 polyclonal antibody (catalog #ab5690; Abcam), rat anti-mouse neutrophil (clone RM0028-3G23; Novus Biologicals), and mouse anti-CD68 (clone KP1; Abcam) just as we did for Iba-1 staining except that the hematoxylin counterstaining was skipped to improve the sensitivity for observing the positive staining.
Immunofluorescent staining.
To demonstrate the colocalization of IKKβ or GFP and various cellular markers (e.g., Iba-1, S100, and NeuN), we used paraffin-embedded sections. The primary antibodies rabbit polyclonal anti-IKKβ (catalog #NB600-477; Novus Biologicals) and rabbit or chicken polyclonal anti-GFP (catalog #ab290 and catalog #ab13970; Abcam) were first incubated with deparaffinized brain sections and thereafter with Alexa Fluor 488-conjugated goat anti-rabbit IgG or chicken IgY. After thorough washing, additional antibodies against various cellular markers (mouse monoclonal anti-Iba-1, clone 20A12.1; Millipore; mouse monoclonal anti-S100, clone 4C4.9; Abcam; or mouse monoclonal anti-NeuN, clone A60; Millipore) were added and were visualized with relevant Alexa Fluor 546-conjugated second antibodies (all second antibodies were from Life Technologies). To detect CCR2-RFP+ cells in the brain, we used brain sections from 6-month-old BM chimera APP-transgenic mice, which were constructed as in our published study (Hao et al., 2011) with ROSAmT/mG mice as the BM donor, as positive controls. A rabbit polyclonal anti-RFP and its variants (e.g., tdTomato; Rockland Immunochemicals) were used as the first antibody and Alexa Fluor 546-conjugated anti-rabbit IgG was used as the second antibody (Life Technologies).
Congo red staining.
To identify compacted amyloid in a β-sheet secondary structure in the brain tissue, we stained paraffin-embedded sections as described in the section of “Immunohistochemistry” with Congo red according to the standard laboratory procedures (Wilcock et al., 2006). To demonstrate the amyloid Aβ deposits in the blood vessel, we cooked Congo red-stained sections in citrate buffer and then treated them with pepsin (Life Technologies) for the antigen retrieval. After being blocked in goat serum, the sections were incubated with a rabbit polyclonal against collagen type IV (catalog #ab6586; Abcam) as the first antibody and Alexa Fluor 488-conjugated goat anti-rabbit IgG as the second antibody.
Histological image acquisition and analysis.
All images were acquired with a Zeiss AxioImager.Z2 microscope equipped with a Stereo Investigator system (MicroBrightField). We used the stereological technique for all histological analyses and were kept blinded to the genotype of mice during the whole experiment. Briefly, after systematic random sampling of every 10th section throughout the entire hippocampus and the cortex, we used the Optical Fractionator (MicroBrightField) as a stereological probe to quantify Iba-1-labeled cells with 120 × 120 × 18 μm of a dissector and 400 × 400 μm of a sampling grid. In each dissector, only Iba-1+ cells with clear hematoxylin nucleus staining were counted. The estimated coefficient of error was <0.05. Volumes of Aβ, Congo red staining, and brain tissues (hippocampus and cortex) were estimated with the Cavalieri probe (Gundersen and Jensen, 1987) with 20 μm of a grid size, which provided coefficient of error estimates of <0.04. The Aβ load was demonstrated as the ratio of Aβ volume to relevant brain tissue volume.
The proliferating microglia, which appeared with typical brown microglial processes and dark blue nuclei, were counted in the total hippocampus area in the randomly selected sections as described in the preceding paragraph. CD45+, CD3+, neutrophil+, and CD68+ cells were counted in randomly selected sections of the entire hemisphere. Because the cell numbers were low, we did not use stereological analysis, which requires a larger number of countable cells so that the random paradigm can be applied. Data were recorded as the number of antibody-labeled cells divided by the full area (in square millimeters) of interest.
To analyze the distribution of Aβ plaque size, we acquired images of hippocampus after Congo red staining with a 10× objective (Carl Zeiss) using the Virtual Tissue Module (MicroBrightField). The hippocampus was delineated and the area of individual plaques was determined after performing histogram-based segmentation with Image-Pro Plus 6.0 software (Media Cybernetics).
Confocal microscopy.
To investigate the relationship between microglia and Aβ deposits, we costained four equidistant (120 μm interval) serial sections from each mouse (see the section of “Immunohistochemistry”) with rabbit Iba-1 (Wako Chemicals) and monoclonal anti-Aβ (clone 6F/3D; Dako) and then with Alexa Fluor 488-conjugated or Cy3-conjugated second antibodies. Under a Zeiss LSM 510 Meta Confocal Microscope with a 40× objective, Aβ deposits labeled with Cy3 were imaged after excitation with a 543 nm laser. Thereafter, we performed Z-stack serial scanning at 1 μm intervals from −15 to +15 μm under the excitation of 488 and 543 nm lasers. Five randomly chosen areas from each section were analyzed. To count the number of Iba-1+ cells that colocalized with Aβ deposits, we color-coded the images of Alexa Fluor 488 (Iba-1) and Cy3 (Aβ) and reconstructed a 3D structure with the Imaris 7.2.3 software (Bitplane). At the same time, the area of Aβ from each layer of serial scanning was determined with ImageJ software. Finally, the total number of microglia in each section was normalized by the total area of Aβ deposits.
To demonstrate the morphology of microglia with and without IKKβ ablation around the same Aβ plaque in apptgikbkbfl/flROSAmT/mGCre+/− mice, we costained GFP, which represented Cre-mediated gene recombination and IKKβ ablation, and Iba-1 in Alexa Fluor 488 and Cy3 fluorescence, respectively, and Aβ with the blue alkaline phosphatase substrate (the detailed staining procedure is described in the section of “Immunohistochemistry”). Under confocal microscopy, we first identified the blue Aβ deposits using the bright field and then imaged microglia under the excitation of 488 and 543 nm lasers.
Purification of membrane components and β- and γ-secretase activity assays.
Membrane components were purified from 6-month-old APP-transgenic and nontransgenic littermate mouse brains and β- and γ-secretase activities were measured by incubating the crude membrane fraction with secretase-specific FRET substrates according to our established methods (Hao et al., 2011; Xie et al., 2013).
Reverse transcription and quantitative PCR for analysis of gene transcripts.
For transcription analysis, total RNA was extracted from the brain homogenate in TRIzol and from brain cells in the lysis buffer. First-strand cDNA was synthesized by priming total RNA with hexamer random primers (Life Technologies) and using Superscript III reverse transcriptase (Life Technologies). For quantification of gene transcripts, real-time PCR was performed with SYBR green (Roche Applied Science) or TaqMan probes (Life Technologies) with the 7500 Fast Real-Time PCR System (Life Technologies). The primer sequences for detecting transcripts of tnf-α, interleukin (il)-1β, inducible nitric oxide synthase (inos), chemokine (C-C motif) ligand 2 (ccl2), il-10, and gapdh were the same as those used in our earlier study (Liu et al., 2006). TaqMan gene expression assays from Life Technologies were used to measure transcripts of the following genes: mouse tnf-α, il-1β, mannose receptor C type 1 (mrc1), arginase (arg)1, chitinase-3-like protein 3 (chi3l3), cd36, scavenger receptor A (sra), insulin-degrading enzyme (ide), neprilysin (nep), the receptor for advanced glycation end products (rage), low-density lipoprotein receptor-related protein 1 (lrp1), cd40, transforming growth factor β1 (tgf-β1), tgf-β receptor type I and II (tgfβ-r1 and r2), gapdh, and 18s RNA. For the ikbkb transcript detection, the Taqman gene expression assay (Mm01222249_m1) was used with the amplified PCR product overlapping 6–7 exon boundary of ikbkb.
The amount of double-stranded PCR product synthesized in each cycle was measured by detecting the FAM dye freed from the TaqMan probes or SYBR green, which binds to double-stranded DNA. Threshold cycle (Ct) values for each test gene from the replicate PCRs were normalized to the Ct values for 18s RNA or gapdh control from the same cDNA preparations. Transcription ratios were calculated as 2(ΔCt), where ΔCt is the difference between Ct (18s or gapdh) and Ct (test gene).
Statistics.
Data shown in the figures are presented as mean ± SEM. For multiple comparisons, we used one-way or two-way ANOVA followed by Bonferroni's, Tukey's honestly significant difference, or Tamhane's T2 post hoc test (the choice of post hoc test depended on the result of Levene's test for homogeneity for determining the equality of variances). Two independent-samples t tests were used to compare means for two groups. All statistical analyses were performed with SPSS version 19.0 for Windows (IBM). Statistical significance was set at the level of p < 0.05.
Results
Establishment of APP-transgenic mice with IKKβ deficiency in myeloid cells (endogenous microglia in the brain)
To ablate IKKβ specifically in the myeloid cell lineage, especially in microglia, we cross-bred TgCRND8 APP-transgenic mice (apptg) to ikbkb-floxed mice (ikbkbfl/fl) and LysM-Cre knock-in mice (LysM-Cre+/+). In the brains of 3-month-old apptgikbkbfl/flCre+/− mice, immunofluorescence staining confirmed that IKKβ protein levels were greatly reduced by Cre-mediated gene recombination in Iba-1+ cells in the parenchyma, which represent endogenous microglia and potentially infiltrating brain macrophages, but not in NeuN+ cells (neurons) (Fig. 1A,B). IKKβ was undetectable in S100-stained cells (astrocytes) (Fig. 1C). Accordingly, Western blotting determined that the amount of IKKβ in CD11b+ cells isolated from apptgikbkbfl/flCre+/− mice was significantly less than that in control cells isolated from apptgikbkbfl/flCre−/− littermates (IKKβ/actin: 0.037 ± 0.004 vs 0.051 ± 0.003; t test, p = 0.022; Fig. 1D,E). Similarly, quantitative PCR analysis showed that the levels of ikbkb transcripts in CD11b+ brain cells from apptgikbkbfl/flCre+/− mice were 34.53% ± 1.61% of the levels of ikbkb transcripts from apptgikbkbfl/flCre−/− littermate mice (Fig. 1F). In contrast, in CD11b− brain cells, including neurons, astrocytes, and oligodendrocytes, the levels of general ikbkb transcripts were unaffected by Cre expression (Fig. 1F). In additional experiments, we also measured IKKβ protein levels in CD11b+ circulating monocytes and cultured BM-derived macrophages, which had been prepared according to our established protocol (Hao et al., 2011). LysM-Cre could more efficiently recombine the floxed genes in these peripheral myeloid cells than in microglia (IKKβ/actin: 0.095 ± 0.014 vs 0.332 ± 0.034, t test, p < 0.001, in monocytes; 0.013 ± 0.002 vs 0.266 ± 0.063, t test, p = 0.013, in cultured macrophages derived from apptgikbkbfl/flCre+/− and apptgikbkbfl/flCre−/− littermates, respectively). Our results were consistent with those of a previous published report stating that the rate of LysM-Cre-mediated gene recombination is ∼40% in microglia and ∼60% in monocytes (Goldmann et al., 2013).
Figure 1.
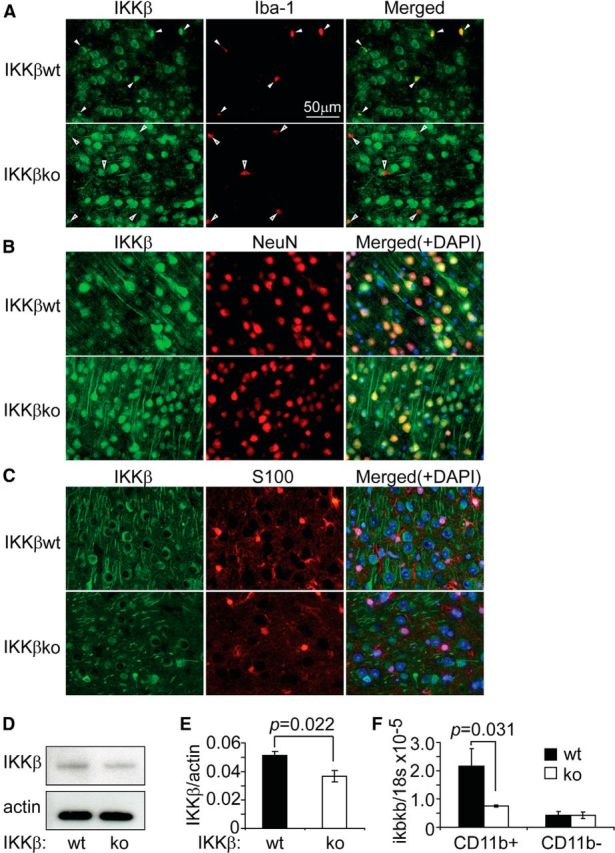
LysM-Cre efficiently excises the floxed ikbkb gene in microglia and brain macrophages. A–C, Brain sections derived from 3-month-old ikbkbfl/flCre+/− (IKKβko) and ikbkbfl/flCre−/− (IKKβwt) mice were stained for IKKβ (in green) and various cellular markers: Iba-1, NeuN, and S100 (in red). In IKKβwt brains, IKKβ was strongly stained in Iba-1+ cells (closed arrowhead), whereas in the IKKβko tissue, IKKβ staining was absent in most Iba-1+ cells (open arrowhead). IKKβ was labeled in all NeuN+, but in no S100+ cells in both IKKβko and IKKβwt tissues. To determine the ablation efficiency of the ikbkb gene by Cre recombinase, CD11b+ and CD11b− cells were isolated from the brains of these two groups of mice. IKKβ protein was detected and quantified by Western blot (D–E; t test; n = 4 per group) and the ikbkb gene transcripts were measured by quantitative PCR (F; t test; n = 4 per group).
To further confirm the cell selectivity of Cre, we cross-bred LysM-Cre mice to ROSAmT/mG Cre reporter mice (Muzumdar et al., 2007). This report system confirmed that Cre recombinase was active only in Iba-1+ cells, not in S100+ cells (data not shown).
It is known that LysM-Cre recombines floxed genes in myeloid cells outside of the brain, including monocytes, macrophages, and neutrophils (Clausen et al., 1999; Goldmann et al., 2013). We wondered whether peripheral myeloid cells had migrated into the brain and affected brain pathology in our APP mice, although one published study found a negative answer to this question (Mildner et al., 2011). We found no positive immunohistochemical staining with neutrophil-specific or CD3-specific antibodies in the brains of 6-month-old APP-transgenic mice (data not shown). Therefore, we excluded the possibility of brain infiltration of neutrophils and T-lymphocytes, both of which are CD45+. However, we did observe a few CD45+ cells in the same brain; these cells were diffused or clustered within a region of the brain parenchyma, but were not distributed throughout the entire brain area where Aβ was deposited. The density of CD45+ cells was 2.01 ± 0.42/mm2 in the hippocampus and cortex of apptgikbkbfl/flCre+/− mice and 1.88 ± 0.38/mm2 in the hippocampus and cortex of apptgikbkbfl/flCre−/− mice (Fig. 2A,B; t test between two groups, p > 0.05). In addition, we used flow cytometry to count fluorescence-labeled cells and found that, in the CD11b+ brain cell populations derived from apptgikbkbfl/flCre+/−, apptgikbkbfl/flCre−/−, and even appwtikbkbfl/flCre−/− mice, ∼2% of cells were CD45+. This ratio did not differ between the three groups of mice (Fig. 2C,D; one-way ANOVA, p > 0.05), but was significantly higher in the experimental autoimmune encephalitis mice than in appwtikbkbfl/flCre−/− mice (10.30% ± 0.54%, Bonferroni's post hoc test, p < 0.001). Experimental autoimmune encephalitis is a mouse model of multiple sclerosis with infiltration of peripheral leukocytes in the CNS. We considered these CD45/CD11b+ cells with limited numbers to be potentially infiltrating brain macrophages.
Figure 2.

Infiltration of CD45+ or CCR2+ cells in APP-transgenic mouse brain. A, Brain sections derived from 6-month-old APP-transgenic mice with (ko) and without (wt) ablation of IKKβ in myeloid cells were stained for CD45 (in brown, arrowhead). IKKβ ablation did not affect the density of CD45+ cells in the hippocampus and cortex (B; t test; n = 6 per group). C, CD11b+ cells were isolated from 6-month-old IKKβko and wt APP-transgenic mice and fluorescently labeled with CD45 antibody for flow cytometry. Histograms show CD45+ cells and comparisons between two groups with control in gray color. D, Columns summarize percentages of CD45+ cells in the CD11b+ brain cell populations (one-way ANOVA; n>7 per group). E, Brain sections derived from 6-month-old APP-transgenic mice that were mated to CCR2-RFP knock-in mice were stained for RFP. A few RFP+ cells (arrowhead) are in close proximity to cerebral blood vessels, but there are no RFP+ cells in the parenchyma. A brain section from a 6-month-old tdTomato-transgenic BM chimera APP-transgenic mouse was used as a positive control, because the chosen antibody recognized both RFP and tdTomato (F). TdTomato+ cells are distributed in the brain parenchyma with typical microglial morphology.
As an additional confirmation of the existence of brain macrophages in AD mice, we performed immunohistochemical staining with a popularly used macrophage-recognizing antibody, anti-CD68. We did not observe CD68+ cells in the brain parenchyma of either apptgikbkbfl/flCre+/− or apptgikbkbfl/flCre−/− mice (data not shown). We cross-bred TgCRND8 APP-transgenic mice to CCR2-RFP mice (Saederup et al., 2010). In 6-month-old APP-transgenic mice heterozygous for the ccr2-rfp gene, we found no RFP+ cells in the brain parenchyma and only a few cells in close proximity to cerebral blood vessels (Fig. 2E; n = 3). As a positive control we used brains from 6-month-old BM chimera APP-transgenic mice, which were constructed as described previously (Hao et al., 2011) with ROSAmT/mG mice as the BM donor. The BM-derived cells were distributed in the brain parenchyma and expressed TFP, which reacts with the same antibody against RFP in CCR2-RFP mice. The results of this experiment provide evidence for the limited infiltration of brain macrophages in AD mice, given that brain macrophages are derived from Ly6ChighCCR2+ monocytes (Mildner et al., 2007; Mizutani et al., 2012; Varvel et al., 2012).
We also investigated the effects of IKKβ deficiency in myeloid cells on peripheral inflammatory status by measuring plasma TNF-α levels. We observed no difference in plasma TNF-α levels between the four mouse groups (appwtikbkbfl/flCre+/−, 35.38 ± 10.56 pg/ml; appwtikbkbfl/flCre−/−, 34.84 ± 11.39 pg/ml; apptgikbkbfl/flCre+/−, 32.68 ± 13.88 pg/ml; and apptgikbkbfl/flCre−/−, 35.88 ± 9.09 pg/ml; one-way ANOVA, p > 0.05; n = 9 per group.
Finally, we investigated the effects of IKKβ deficiency on microglial NF-κB activation. Western blot analysis showed that the amount of IKKβ protein detected in cultured primary microglia derived from ikbkbfl/flCre+/− mice was 49.18% ± 6.90% of the amount detected in cultured microglia derived from ikbkbfl/flCre−/− mice (Fig. 3A,B). The ∼50% reduction in IKKβ protein suppressed the phosphorylation of p65 in the NF-κB complex in the microglia both at the basal level and after activation with 10 μm oligomeric Aβ42 (the ratio of phosphorylated p65 to total p65 was 0.40 ± 0.09 in ikbkbfl/flCre−/− cells and 0.11 ± 0.01 in ikbkbfl/flCre+/− cells at the basal level, and 1.29 ± 0.20 in ikbkbfl/flCre−/− cells and 0.26 ± 0.05 in ikbkbfl/flCre+/− cells after Aβ activation; one-way ANOVA, p < 0.05; Fig. 3C,D). However, IKKβ deficiency did not completely block NF-κB activation by Aβ42 oligomers because Aβ significantly increased the level of phosphorylated p65 in microglia derived from ikbkbfl/flCre+/− mice (t test, p < 0.05; Fig. 3C,D).
Figure 3.

IKKβ ablation inhibits NF-κB activation in primary cultured microglia. IKKβ protein (A, B; t test; n = 3 per group) and phosphorylated and total NF-κB p65 (C, D; t test; n = 3 per group) in the primary cultured microglial cell lysate derived from ikbkbfl/flCre+/− (IKKβko) and ikbkbfl/flCre−/− (IKKβwt) mice were detected and quantified with Western blotting. For NF-κB activation assays, IKKβ wt and ko microglia were activated with 10 μm oligomeric Aβ42 for 12 min.
Deficiency of IKKβ in myeloid cells (microglia) rescues cognitive deficits in APP-transgenic mice
Because deficiencies in TLR2, MyD88, and IRAK4 in myeloid cells, especially in microglia, have been shown to ameliorate AD-like pathology and improve neuronal functions in AD mice (Hao et al., 2011; Cameron et al., 2012; Liu et al., 2012), we investigated whether deficiency of IKKβ in myeloid cells (microglia) improves the cognitive function of APP-transgenic mice. In the Barnes maze (Hao et al., 2011; Liu et al., 2012), the traveling time and distance traveled was significantly shorter for all tested mice when training time increased (Fig. 4A–C; one-way ANOVA, p < 0.05). During the test, there were no significant differences in running speed between various groups of mice or for the same mice on different training dates (two-way ANOVA, p > 0.05). Therefore, both APP-transgenic (apptg) and non-APP-transgenic (appwt) littermate mice with different myeloid expressions of IKKβ retained the ability to use spatial reference points to learn the location of an escape hole (Fig. 4A–C).
There were no differences in traveling time and distance traveled between non-APP-transgenic littermate mice with deficiencies in IKKβ or with wild-type IKKβ in myeloid cells (appwtikbkbfl/flCre−/− and appwtikbkbfl/flCre+/−; Fig. 4B,C; two-way ANOVA, p > 0.05). However, compared with their appwtikbkbfl/flCre−/− and appwtikbkbfl/flCre+/− littermates, 6-month-old APP-transgenic mice with wild-type IKKβ expression in myeloid cells (apptgikbkbfl/flCre−/−) spent significantly more time (Fig. 4B; two-way ANOVA, p < 0.05) and traveled longer distances (Fig. 4C; two-way ANOVA, p < 0.05) before reaching the escape hole. Interestingly, ablation of IKKβ specifically in myeloid cells, especially microglia (apptgikbkbfl/flCre+/− mice), completely rescued these cognitive deficits in 6-month-old APP-transgenic mice as assessed by the Barnes maze test (Fig. 4B,C). Moreover, in the probe trials, appwtikbkbfl/flCre−/− and appwtikbkbfl/flCre+/− mice visited the escape hole similarly, and apptgikbkbfl/flCre−/− mice spent less time around the escape hole and visited the hole less frequently than did non-APP-transgenic mice, although the difference did not reach statistical significance (Fig. 4D,E). Interestingly, compared with apptgikbkbfl/flCre−/− mice, apptgikbkbfl/flCre+/− mice with IKKβ deficiency in myeloid cells visited the escaping area significantly more frequently and spent significantly more time there (Fig. 4D,E).
We also used Western blot analysis to quantify the protein levels of PSD-95 (also known as disks large homolog 4) and presynaptic Munc18–1 in brain homogenates from 6-month-old APP-transgenic and wild-type littermate control mice. Both PSD-95 and Munc18–1 levels were markedly lower in apptgikbkbfl/flCre−/− mice than in their appwtikbkbfl/flCre−/− and appwtikbkbfl/flCre+/− littermates (Fig. 4F,G; one-way ANOVA, p < 0.05). Similarly, the amounts of PSD-95 and Munc18–1 proteins did not differ significantly between appwtikbkbfl/flCre−/− and appwtikbkbfl/flCre+/− mice (Fig. 4F,G; one-way ANOVA, p > 0.05). Interestingly, the reduction in PSD-95 and Munc18–1 proteins due to APP-transgenic expression was attenuated by the deficiency of IKKβ in myeloid cells. The levels of PSD-95 and Munc18–1 proteins were significantly higher in brains from apptgikbkbfl/flCre+/− mice than in brains from apptgikbkbfl/flCre−/− control mice (Fig. 4F,G; one-way ANOVA, p < 0.05).
Deficiency of IKKβ in myeloid cells (microglia) reduces inflammatory activation in aged APP-transgenic mouse brains
Because proinflammatory activation contributes to AD pathogenesis, we investigated whether a deficiency of IKKβ in myeloid cells (microglia) might reduce inflammatory activity in the brain. We used the stereological technique to estimate the total number of Iba-1+ cells, including microglia and potentially infiltrating brain macrophages, in the hippocampus and cortex of 6-month-old APP-transgenic and non-APP-transgenic mice with or without IKKβ expression in myeloid cells (apptgikbkbfl/flCre+/−, apptgikbkbfl/flCre−/−, appwtikbkbfl/flCre+/−, and appwtikbkbfl/flCre−/− mice). The total number of Iba-1+ cells was significantly higher in APP-transgenic mice than in non-APP-transgenic mice (Fig. 5A–C; one-way ANOVA, p < 0.05). The two groups of transgenic mice differed significantly in the total number of Iba-1+ cells: apptgikbkbfl/flCre+/− mice, 20.46 ± 0.70 × 103 cells in the hippocampus and 23.74 ± 0.86 × 103 cells in the cortex dorsal to the hippocampus; apptgikbkbfl/flCre−/− mice, 25.49 ± 1.17 × 103 cells in the hippocampus and 29.54 ± 0.89 × 103 cells in the cortex dorsal to the hippocampus (Fig. 5B,C; one-way ANOVA, p < 0.05). However, there was no significant difference in the number of Iba-1+ cells between the two groups of non-APP-transgenic mice (appwtikbkbfl/flCre+/− mice and appwtikbkbfl/flCre−/− mice; Fig. 5B,C; one-way ANOVA, p > 0.05).
Figure 5.

Deficiency of IKKβ in myeloid cells reduces neuroinflammatory activation in APP-transgenic mice. Six-month-old APP-transgenic mice (APPtg) and their non-APP-transgenic littermates (APPwt) were tested for inflammatory activation. Microglial cell numbers were estimated with stereological methods after immunohistochemical staining of Iba-1 (A; in brown). Proliferating microglia were identified by double staining of Iba-1 and Ki67, which appear in blue nucleus and brown cytoplasm (D; marked with closed arrowheads; pure Iba-1+ cells are marked with open arrowheads). The numbers of Iba-1+ cells were significantly smaller in APPtg mice with ablation of myeloid IKKβ (IKKβko) than in those with normal IKKβ expression (IKKβwt). However, IKKβ ablation did not affect the number of Iba-1+ cells in APPwt mice (B, C; one-way ANOVA; n = 6 per group). The number of Iba-1/Ki67 double-positive cells was significantly smaller in APPtg/IKKβko than in APPtg/IKKwt mice (E; t test; n = 6 per group). TNF-α protein concentration in brain homogenates derived from APPtg and APPwt mice was determined by ELISA (F; one-way ANOVA; n ≥ 6 per group). Inflammatory gene transcripts in the brain (G–N) and in isolated microglia from 6-month-old APPtg mice (O) were measured by quantitative RT-PCR. Both TNF-α protein expression and the number of tnf-α and il-1β transcripts in APPtg brains, but not in the APPwt control brains, were significantly reduced by IKKβ ablation in myeloid cells (F-H; one-way ANOVA; n ≥ 9 per group). Accordingly, the number of tnf-α transcripts in APPtg microglia was also significantly reduced by IKKβ ablation (O; t test; n = 3 per group).
We next investigated the mechanisms by which IKKβ deficiency decreases the number of Iba-1+ cells. Because we had observed no significant difference between groups in the recruitment of monocyte-derived brain macrophages (Fig. 2), we evaluated endogenous microglial proliferation by costaining Iba-1 and Ki67, a cell-proliferation marker (Liu et al., 2013). Because there were too few cells, we could not use the stereological technique. As shown in Figure 5, D and E, there were indeed significantly fewer double-positive cells in the hippocampus of apptgikbkbfl/flCre+/− mice (3.67 ± 0.50 cells/mm2) than in that of apptgikbkbfl/flCre−/− mice (5.53 ± 0.77 cells/mm2; t test, p < 0.05).
We then used ELISA to quantify TNF-α protein levels in the TBS-soluble brain homogenate derived from 6-month-old APP-transgenic and nontransgenic mice and found that TNF-α production was significantly higher in APP mice than in their non-APP-transgenic littermates (Fig. 5F; one-way ANOVA, p < 0.001). A deficiency in IKKβ in myeloid cells did not affect the levels of cerebral TNF-α protein in non-APP-transgenic mice (Fig. 5F), but did significantly decrease levels of TNF-α protein in the brain of APP-transgenic mice compared with littermate APP mice with wild-type IKKβ expression in myeloid cells (Fig. 5F; one-way ANOVA, p = 0.002).
We also quantified transcripts of M1-inflammatory gene markers (tnf-α, il-1β, inos, and ccl2) and M2-inflammatory gene markers (il-10, mrc1, arg1, and chi3l3) (Colton et al., 2006) in the brains of four separate groups of 6-month-old littermate mice [myeloid IKKβ: deficient (ikbkbfl/flCre+/−) and wild-type (ikbkbfl/flCre−/−) mice; APP-transgenic expression: positive (apptg) and negative (appwt) mice]. As shown in Figure 5, G and H, levels of tnf-α and il-1β transcripts were significantly higher in APP mice than in non-APP mice (one-way ANOVA, p < 0.05). A deficiency in IKKβ in myeloid cells (microglia) completely abolished the transcriptional upregulation of tnf-α and il-1β by APP-transgenic expression (one-way ANOVA, p < 0.05). In non-APP-transgenic mice, an IKKβ deficiency in myeloid cells did not change the transcription of tnf-α and il-1β genes (Fig. 5G,H; one-way ANOVA, p > 0.05). Neither the transcription of the other M1 genes (inos and ccl2) that we tested nor the transcription of the M2 activation markers differed between myeloid IKKβ-deficient and wild-type APP-transgenic or nontransgenic mice (Fig. 5I–N).
In an additional experiment, we isolated microglia and potential brain macrophages from 6-month-old APP-transgenic mice. We observed that transcription of the inflammatory gene tnf-α, but not that of other proinflammatory and antiinflammatory genes, was significantly lower in CD11b+ cells isolated from apptgikbkbfl/flCre+/− mice than in cells from apptgikbkbfl/flCre−/− mice (Fig. 5O; t test, p < 0.05).
Deficiency of IKKβ in myeloid cells (microglia) reduces Aβ load in aged APP-transgenic mouse brains
Because Aβ pathology is considered to be the key mechanism mediating neuronal death in AD (Mucke and Selkoe, 2012), we continued to investigate the effects on the cerebral Aβ load of IKKβ deficiency in myeloid cells. We separated brain homogenate from 6-month-old apptgikbkbfl/flCre+/− and apptgikbkbfl/flCre−/− mice into 3× TBS-soluble, TBS-T-soluble, and guanidine chloride-soluble fractions according to our established protocols (Hao et al., 2011; Liu et al., 2012). Compared with apptgikbkbfl/flCre−/− mice, apptgikbkbfl/flCre+/− mice exhibited significantly lower concentrations of Aβ42 and Aβ40 in both TBS-T-soluble fractions and guanidine chloride-soluble fractions, which were enriched in oligomeric and high-molecular-weight aggregated Aβ species (Hao et al., 2011; Fig. 6A; t test, p < 0.05). Using the commercially available oligomeric Aβ ELISA kit, we confirmed that the aggregated level of Aβ (106.58 ± 6.20 pg/mg of wet brain tissue) in TBS-T-soluble brain homogenate from apptgikbkbfl/flCre+/− mice was significantly lower than that (130.52 ± 6.20 pg/mg of wet brain tissue) in homogenate from apptgikbkbfl/flCre−/− mice (t test, p = 0.036). In the TBS-soluble brain homogenate fraction, which contained most of the monomeric Aβ species (Hao et al., 2011), a deficiency in IKKβ in myeloid cells did not significantly affect Aβ concentrations (Fig. 6A; t test, p > 0.05).
Figure 6.
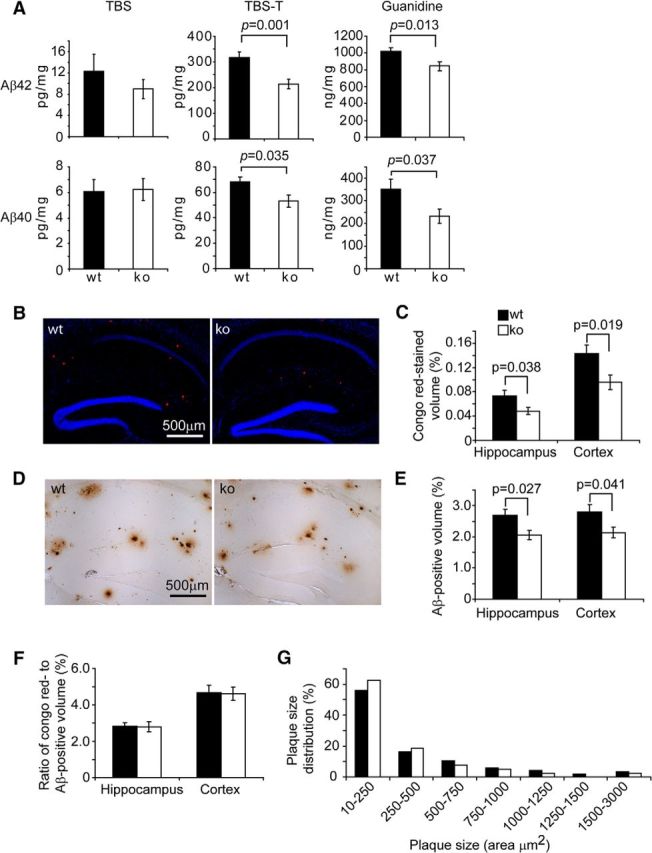
Deficiency of IKKβ in myeloid cells reduces Aβ load in the APP-transgenic mouse brain. The brains of 6-month-old APP-transgenic (APPtg) mice were analyzed for Aβ load. The brain was homogenized and separated into TBS-, TBS-T-, and guanidine-soluble fractions. Amounts of Aβ40 and Aβ42 were measured by ELISA and normalized to the homogenate protein concentration. The concentrations of both Aβ42 and Aβ40 in both TBS-T- and guanidine-soluble fractions were significantly lower in myeloid IKKβ-ablated (ko) APP mice than in IKKβ-wild-type (wt) control mice (A; t test; n = 11 per group). The Aβ volume in the whole hippocampus and cortex was estimated after both Congo red staining (B, C) and immunohistochemistry with human Aβ-specific antibody (D, E) and adjusted by volumes of the relevant brain tissues. Myeloid IKKβ deficiency significantly reduces cerebral Aβ load (t test; n = 8 per group for Congo red staining and n = 13 per group for immunohistochemistry). F, The ratio of Congo red-stained volume to Aβ-immunohistochemically stained volume is calculated (t test; n = 8 per group). G, The size of Aβ plaque was measured and the frequency of Aβ plaques with a certain size is shown as a percentage of the total number of plaques (two-way ANOVA, p > 0.05; n = 8 per wt or ko group).
We used Congo red staining and immunohistochemical analysis to determine the Aβ load in the brain. As shown in Figure 6, B and D, the extent and morphology of the Congophilic amyloid load or of antibody-labeled diffused Aβ or Aβ plaques with dense cores were not changed by a deficiency in IKKβ in the myeloid cell lineage. When we measured Aβ volume (adjusted by the volume of analyzed tissues) with the stereological Cavalieri method (Gundersen and Jensen, 1987), we observed that, after ablation of IKKβ in myeloid cells, the volume of Congophilic Aβ load (0.074% ± 0.009% in the hippocampus and 0.143% ± 0.015% in the cortex) was significantly reduced (to 0.048% ± 0.006% in the hippocampus, p = 0.038, and 0.095% ± 0.012% in the cortex, p = 0.019; Fig. 6B,C; t test for relevant comparisons). Similarly, the volume of immunoreactive Aβ load was markedly higher in apptgikbkbfl/flCre−/− mice (2.687% ± 0.020% in the hippocampus and 2.801% ± 0.234% in the cortex) than in apptgikbkbfl/flCre+/− mice (2.062% ± 0.152% in the hippocampus, p = 0.027, and 2.139% ± 0.178% in the cortex, p = 0.041; Fig. 6D,E; t test for relevant comparisons). Because Congo red typically binds to the β sheet structure of Aβ plaques (Lorenzo and Yankner, 1994), we calculated the ratio of volumes of Congo red staining to volumes of immunohistochemical staining, which were not altered by the deficiency in myeloid IKKβ (Fig. 6F; t test, p > 0.05), a fact suggesting that the ablation of myeloid IKKβ does not affect Aβ aggregation. Furthermore, we quantified Aβ deposits in the hippocampus and analyzed the distribution of their size. As shown in Figure 6G, IKKβ ablation in the myeloid cell lineage tended to shift Aβ plaques from large to small, although the difference in the distribution of plaque size between IKKβ-deficient and wild-type mice was not statistically significant (two-way ANOVA, p > 0.05).
Deficiency in IKKβ enhances microglial and macrophage recruitment toward Aβ deposits and Aβ internalization
Given that a deficiency in IKKβ in microglia reduced both inflammatory activation and Aβ load in the APP-transgenic mouse brain, we hypothesized that IKKβ deficiency might enhance Aβ clearance by internalization into microglia or macrophages. Because it is difficult to measure intracellular Aβ directly in the brain, we first counted microglia and macrophages interacting with Aβ deposits and found that significantly more Iba-1+ cells were recruited to the Aβ deposits in apptgikbkbfl/flCre+/− mice than to the Aβ deposits in their apptgikbkbfl/flCre−/− littermates (Fig. 7A,B; t test, p = 0.008), a finding consistent with our previous findings in AD mice with a deficiency in MyD88 in myeloid cells (Hao et al., 2011). However, the amoeboid morphology of microglia surrounding Aβ appeared not to be changed by myeloid IKKβ deficiency when the IKKβ-deficient cells were tracked by Cre-mediated GFP expression (Fig. 7C). IKKβ deficiency significantly upregulated the transcription of Aβ-cleaning scavenger receptor A (Frenkel et al., 2013) in the brains of APP-transgenic mice, but not in the brains of their non-APP-transgenic littermates (Fig. 7D; one-way ANOVA, p < 0.05). CD36 transcription was lower in apptgikbkbfl/flCre−/− mice than in appwtikbkbfl/flCre−/− littermate controls (Fig. 7E; one-way ANOVA, p < 0.05), a finding consistent with the observations of El Khoury's group (Hickman et al., 2008). Interestingly, a deficiency in IKKβ in microglia was associated with the complete recovery of CD36 expression in the brains of APP-transgenic mice (Fig. 7E; one-way ANOVA, p < 0.05), but had no effects on CD36 transcription in non-APP-transgenic mice (Fig. 7E; one-way ANOVA, p > 0.05).
Figure 7.

IKKβ deficiency increases microglial/brain macrophage recruitment toward Aβ deposits in the APP-transgenic mouse brain. The 6-month-old APP-transgenic (APPtg) mouse brain was analyzed for the interaction between Aβ and microglia and brain macrophages under confocal microscopy after immunofluorescent staining with antibodies against Aβ and Iba-1 (A; Aβ in red and Iba-1 in green). The numbers of microglia and macrophages that colocalized with Aβ were counted and adjusted by Aβ volume. More IKKβ-ablated (ko) than wild-type (wt) microglia and macrophages were recruited to Aβ deposits (B; t test; n = 6 per group). C, IKKβ-ablated microglia and macrophages were tracked by GFP expression. After costaining GFP in green and Iba-1 in red fluorescence, microglia and macrophages expressing both proteins appeared in yellow (closed arrowheads) and cells expressing Iba-1 alone are shown in red (open arrowhead). Aβ plaques were stained in blue color in the bright field and appeared as black holes under confocal microscopy (marked with asterisks). Therefore, IKKβ ablation does not change the amoeboid morphology of microglia and macrophages contacting Aβ plaque. The transcription of scavenger receptor A (sra) and cd36 was measured with quantitative RT-PCR and was upregulated by myeloid IKKβ deficiency in APPtg mouse brains, but not in APP-wild-type (APPwt) mouse brains (D, E; one-way ANOVA; n ≥ 9 per group).
In further experiments, we challenged primary cultured microglia with HiLyte Fluor 488-labeled Aβ42 enriched in oligomeric and fibrillar species (Fig. 8A) to model the Aβ species in TBS-T-soluble and guanidine-soluble brain homogenates. When microglia were treated with aggregated Aβ at a concentration of 1 μm, internalization was detectable within 1 h and levels of internalized Aβ increased as the incubation time increased (Fig. 8B,C). Significantly more oligomeric Aβ42 than fibrillar Aβ42 was internalized by microglia (with or without IKKβ expression), as measured by mFI (Fig. 8B; two-way ANOVA, p < 0.001). After cells were incubated with Aβ42 for 3 h, the mFI of cells treated with Aβ oligomers decreased more quickly than did that of microglia treated with Aβ fibrils. This difference could be due to the difference in the efficiency of degradation and extracellular release between Aβ oligomers and fibrils in microglia (Chung et al., 1999; Yamamoto et al., 2008).
Figure 8.

IKKβ deficiency blocks TGF-β-SMAD2/3 signaling and increases microglial internalization of oligomeric Aβ42. A, Aggregating patterns (o, oligomeric; f, fibrillar) of HiLyte Fluor 488-labeled Aβ42. B, C, Line curves showing the internalization of 1 or 0.2 μm oligomeric Aβ42 (oAβ) and 1 μm fibrillar Aβ42 (fAβ) by primary cultured microglia derived from IKKβ-ablated (IKKβko) and wild-type (IKKβwt) mice for various time periods (two-way ANOVA; n = 3 per group). D, E, Histograms showing the inflammatory gene transcripts in primary microglia 6 h after the Aβ internalization (t test; n ≥ 6 per group). F–H, Transcripts of tgf-β1, tgfβ-r1, and r2 genes in the entire hippocampus and cortex in adult microglia isolated from APP-transgenic mice and in cultured primary microglia with and without treatment of 1 μm oAβ were measured using real-time PCR (t test; n ≥ 5 per group). Primary cultured IKKβwt and IKKβko microglia were activated with TGF-β1 at 0, 1, and 10 ng/ml for 30 min (I–K); IKKβwt microglia were also treated with SB-505124 and SB-431542 at 0, 0.02, and 0.1 μm and then with 1 ng/ml TGF-β1 for 30 min (L). After stimulation, the phosphorylated and total SMAD2 and SMAD5 proteins were detected by Western blotting. The ratios of phospho-/total SMAD2 and SMAD5 reflect SMAD2/3 and SMAD1/5/8 signaling. Therefore, the IKKβ ablation specifically blocks SMAD2/3 signaling without affecting SMAD1/5/8 signaling (J, K; two-way ANOVA, n = 4 per group). SB-505124 and SB-431542 at 0.02 and 0.1 μm inhibit SMAD2/3 signaling and exert less effect on SMAD1/5/8 signaling (L). M, N, IKKβwt microglia were pretreated with SB-505124 and SB-431542 at 0, 0.02, and 0.1 μm for 1 h and then incubated with 1 μm fluorescent oAβ for 6 h in the presence of inhibitors. The internalization of oAβ was assayed with flow cytometry (two-way ANOVA followed by post hoc tests, *p < 0.05 and **p < 0.01 vs the Aβ internalization without inhibitor treatment; n = 4 per group).
Comparison of the internalizing capability of IKKβ-deficient and wild-type microglia showed that IKKβ deficiency significantly increased the uptake of Aβ42 oligomers at a concentration of 1 μm, but not of Aβ fibrils. This difference remained when Aβ42 treatment was extended to 24 h (Fig. 8B,C; two-way ANOVA, p < 0.001). Interestingly, when microglia were treated with oligomeric Aβ42 at a concentration of 0.2 μm, IKKβ deficiency did not enhance Aβ internalization (Fig. 8B,C; two-way ANOVA, p > 0.05). Similar results were seen in internalization assays with BM-derived macrophages, in which IKKβ-deficient macrophages showed a significantly higher uptake of Aβ than IKKβ-wild-type macrophages when cells were treated with oligomeric Aβ42 at concentrations of 1 or 10 μm. This Aβ internalization enhanced by IKKβ deficiency disappeared when oligomeric Aβ42 was administered at a concentration of 0.5 μm or when fibrillar Aβ42 was administered at a concentration of 1 μm (data not shown).
To determine whether elevated inflammatory activation might be inversely related to the internalization of Aβ into microglia (Hao et al., 2011; Liu et al., 2012), we performed two additional experiments. First, we analyzed the pathological changes in 3-month-old APP-transgenic mice with or without IKKβ ablation in myeloid cells as controls for the 6-month-old AD mice that we described in the sections “Deficiency of IKKβ in myeloid cells (microglia) reduces inflammatory activation in aged APP-transgenic mouse brains” and “Deficiency of IKKβ in myeloid cells (microglia) reduces Aβ load in aged APP-transgenic mouse brains.” A deficiency in myeloid IKKβ neither changed cerebral transcripts of tnf-α, il-1β, and inos genes nor altered Aβ concentrations in all TBS-soluble, TBS-T-soluble, and guanidine chloride-soluble brain homogenate fractions (data not shown). Second, we measured the inflammatory activation of cultured microglia during internalization of Aβ over a 6 h interval. As shown in Figure 8, D and E, IKKβ deficiency significantly suppressed inflammatory gene (e.g., tnf-α and il-1β) transcription triggered by oligomeric Aβ at a concentration of 1 μm (two-way ANOVA, p < 0.05). Microglial inflammatory gene transcription was not significantly induced by fibrillar Aβ42 at a concentration of 1 μm or by oligomeric Aβ42 at a concentration of 0.2 μm (Fig. 8D,E; two-way ANOVA, p > 0.05).
Inhibition of TGF-β-SMAD2/3 signaling could be involved in microglial Aβ internalization enhanced by IKKβ deficiency
Because it has been reported that blocking either CD40 or the TGF-β signaling cascade in AD mice leads to a reduction in both inflammatory activation and Aβ load in the brain (Tan et al., 1999 and 2002; Town et al., 2008), the same phenomenon we observed in myeloid IKKβ-deficient AD mice, we decided to investigate whether IKKβ modulates CD40 and TGF-β signaling. Indeed, a deficiency in myeloid IKKβ significantly decreased transcription levels of TGF-β receptor types 1 and 2 (TGFβ-R1 and -R2) in microglia isolated from APP-transgenic mice (Fig. 8G; t test, p < 0.05), although IKKβ deficiency did not change the transcription level of these two receptors or of TGF-β1 in the entire brain (Fig. 8F; t test, p > 0.05). In cultured microglia, IKKβ deficiency reduced tgf-β1 transcription at the basal level and reduced both tgf-β1 and tgfβ-r1 transcription after challenge with oligomeric Aβ42 at a concentration of 1 μm (Fig. 8H; t test, p < 0.05). Notably, we observed that IKKβ deficiency inhibited SMAD2 phosphorylation but did not change SMAD5 phosphorylation in microglia in response to stimulation with TGF-β1 at a concentration of 1 or 10 ng/ml (Fig. 8I–K; two-way ANOVA). However, CD40 gene transcription was not changed by IKKβ deficiency in either adult or cultured microglia (data not shown).
Thereafter, we tested whether Aβ internalization in microglia was increased by blockade of TGF-β-SMAD2/3 signaling, as described previously (Town et al., 2008). Two ALK-5 inhibitors, SB-505124 and SB-431542, at concentrations of 1 and 10 μm, had been shown to inhibit both SMAD2/3 and SMAD1/5/8 signaling in response to exogenous TGF-β1 activation (Town et al., 2008). We observed that both SB-505124 and SB-431542 at concentrations of 0.1 and 0.02 μm reduced SMAD2 phosphorylation, but did not markedly affect SMAD5 phosphorylation in microglia after the same challenge with TGF-β1 (Fig. 8L). This partial inhibition was comparable to the inhibition caused by IKKβ deficiency, as described in the preceding paragraph. Interestingly, the inhibitor-treated cultured microglia internalized significantly more oligomeric Aβ42 aggregates, in a dose-dependent manner, than did the microglia without special treatment (Fig. 8M,N; two-way ANOVA, p < 0.05). The concentrations that promoted Aβ internalization were between 0.5 μm and 20 nm; the maximal effect was induced by a concentration of 20 nm (Fig. 8M,N; two-way ANOVA followed by post hoc tests, p < 0.05). At higher and lower concentrations (1 μm and 4 nm, respectively), the inhibitors failed to promote Aβ internalization.
Deficiency in IKKβ in myeloid cells neither increases Aβ clearance through brain-to-blood transportation and endogenous degradation nor reduces amyloidogenic APP metabolism
Deane et al. (2004) reported that altered communication of Aβ between the brain interstitial fluid and the peripheral bloodstream affects cerebral Aβ load. Peripheral myeloid cells could clear Aβ in the perivascular area so as to reduce Aβ deposition in the parenchyma (Hawkes and McLaurin, 2009; Mildner et al., 2011). We observed that neither the volume of Aβ that was deposited in the blood vessels nor the number of vessels that tested positive for Aβ deposits differed between myeloid IKKβ-deficient and wild-type APP-transgenic mice (Fig. 9A–C; t test, p > 0.05). We also measured Aβ levels in the plasma and the expression of RAGE and LRP1 in the brain, two receptors that act as shuttles to transport Aβ across the blood–brain barrier (Shibata et al., 2000; Deane et al., 2003 and 2004). As shown in Figure 9, neither plasma Aβ40 and Aβ42 concentrations in APP-transgenic mice (Fig. 9F) nor receptor expression in APP-transgenic and nontransgenic mouse brains (Fig. 9D,E) was affected by IKKβ deficiency in myeloid cells (t test, p > 0.05).
Figure 9.

Deficiency of IKKβ in myeloid cells does not increase Aβ clearance through brain-to-blood transportation. The 6-month-old APP-transgenic (APPtg) and non-APP-transgenic (APPwt) littermate mice with (IKKβko) and without IKKβ ablation (IKKβwt) in myeloid cells were analyzed for cerebral vascular Aβ deposits after tissues were costained with Congo red and collagen type IV antibodies (A–C) and for transcription of lrp1 and rage in the brain with quantitative RT-PCR (D, E). Concentrations of Aβ40 and Aβ42 in the plasma from IKKβko and IKKβwt APPtg mice were quantified by ELISA (F). Neither volumes of Congo red staining nor numbers of Congo red-positive blood vessels differed between IKKβko and IKKβwt APPtg mice (t test, p > 0.05; n = 8 per group). Similarly, neither gene transcription (D, E) nor plasma Aβ concentrations (F) was significantly changed by the IKKβ deficiency (one-way ANOVA for gene transcripts, p > 0.05; t test for plasma Aβ concentrations between IKKβko and IKKβwt mice, p > 0.05; n ≥ 9 per group).
To determine whether IKKβ deficiency in myeloid cells affects Aβ production in APP-transgenic mice, we measured the activities of β-secretase and γ-secretase, the two main enzymes that cleave APP to produce Aβ (Mucke and Selkoe, 2012). Surprisingly, neither β-secretase activity nor γ-secretase activity was changed by IKKβ deficiency (data not shown). Quantitative RT-PCR investigation of the Aβ-degrading enzyme neprilysin and the insulin-degrading enzyme (Leissring et al., 2003; Miners et al., 2008) showed no significant increase in the transcription of nep and ide genes in apptgikbkbfl/flCre+/− mice compared with apptgikbkbfl/flCre−/− littermate controls (data not shown), a finding suggesting that microglial IKKβ deficiency has no effect on Aβ catabolism.
Discussion
Microglial activation exerts both toxic and protective effects on neurons in AD pathogenesis, and TLRs and their downstream signaling molecules have been observed to control microglial behaviors that lead to these controversial effects (Tahara et al., 2006; Reed-Geaghan et al., 2010; Hao et al., 2011; Lim et al., 2011; Michaud et al., 2011 and 2012; Song et al., 2011; Cameron et al., 2012; Liu et al., 2012). In this study, we demonstrated that a deficiency in IKKβ in myeloid cells, especially in endogenous microglia, simultaneously reduces inflammatory activation and Aβ load in the brain and improves cognitive function in AD mice. These findings corroborate the results of our earlier studies of the deficiency of myeloid TLR2 or MyD88 in APP-transgenic mice (Hao et al., 2011; Liu et al., 2012).
To the best of our knowledge, we were the first to use the Cre-Lox technique in cross-breeding experiments to conditionally delete the protein of interest in APP-transgenic mice. This deletion allows us to investigate in detail the functions of this protein in AD pathogenesis in a specific tissue and within a certain time frame. We investigated IKKβ in microglia by specifically ablating IKKβ in myeloid cells of APP-transgenic mice, thereby excluding any confounding effects from neuronal IKKβ. Neuronal IKKβ has been reported to activate neuronal NF-κB, thus modifying Aβ generation and the degeneration and plasticity of neurons (Kaltschmidt et al., 1997; Fridmacher et al., 2003; He et al., 2007; Kaltschmidt and Kaltschmidt, 2009; Gutierrez and Davies, 2011).
However, in our AD animal model, the ikbkb gene was deleted not only in endogenous microglia, but also in peripheral myeloid cells (Clausen et al., 1999; Goldmann et al., 2013). One subset of myeloid cells potentially circulates to the brain parenchyma and serves as brain macrophages (Hao et al., 2011). In our APP-transgenic mouse brains we observed a small population of CD45+ cells the distribution of which was localized within one brain region instead of around most Aβ deposits, a finding indicating that these cells are not activated endogenous microglia. These CD45+ cells are also neither neutrophils nor T lymphocytes because they do not express neutrophil or CD3 markers. Therefore, this cell population may be brain macrophages. Surprisingly, the number of CD45+ cells in this population is very limited (<2% of Iba-1+ cells, as estimated on the basis of our observations; Hao et al., 2011; Liu et al., 2012; Xie et al., 2013). Moreover, flow cytometry showed that ∼2% of CD11b+ brain cells are CD45+, not only in IKKβ-deficient and wild-type APP-transgenic mice, but also in non-APP-transgenic mice. Therefore, the recruitment of potential brain macrophages appears to be independent of neuroinflammatory status and of IKKβ expression in AD mice.
Brain macrophages have been shown to originate from CCR2+ monocytes (Mildner et al., 2007; Varvel et al., 2012). In another APP-transgenic mouse model, one allele of the ccr2 gene was replaced by the RFP-encoding sequence, whereas the other allele expressed CCR2 to exert full cellular physiological function (Saederup et al., 2010). In the brains of these animals (apptg/ccr2-rfp+/− mice), no CCR2-RFP+ cells were observed to infiltrate the brain parenchyma, a finding further corroborating the hypothesis that the recruitment of brain macrophages in AD mice is limited. APP-transgenic mouse brains that have not been preconditioned by irradiation are devoid of parenchymal peripherally infiltrated macrophages (Mildner et al., 2011; Kierdorf et al., 2013), a condition that makes it unlikely that IKKβ-ablated brain macrophages can affect Aβ pathology in the brain parenchyma.
In the perivascular area, peripheral myeloid cells can interact with Aβ and modify the cerebral Aβ load (Hawkes and McLaurin, 2009; Mildner et al., 2011). In our AD mouse models, neither the concentration of plasma Aβ (especially Aβ42) and TNF-α nor the volume of Aβ deposited in the blood vessels was changed by the ablation of IKKβ in the myeloid cells. Therefore, although the effects of peripheral myeloid cells on Aβ pathology cannot be completely excluded, the anti-AD phenotype observed in our study appears to result primarily from a deficiency in IKKβ in the endogenous microglia.
As we reviewed in the Introduction, whether microglial activation serves beneficial or detrimental effects in AD pathogenesis is still strongly under debate. The most contentious issue is whether the inhibition of innate immunity simultaneously attenuates Aβ pathology and proinflammatory activation. Our previous studies focusing on TLR2 (Liu et al., 2012) and MyD88 (Hao et al., 2011) and the present study addressing IKKβ have consistently given a positive answer to this question. Indeed, other published observations show that a decrease in the cerebral Aβ load accompanies neuroinflammatory reduction, whether caused by a deficiency in CD40 ligand (Tan et al., 1999 and 2002), IRAK4 (Cameron et al., 2012), NLRP3 (Heneka et al., 2013), or Mrp14 (a neuroinflammation-amplifying protein; Kummer et al., 2012), by blocking TGF-β-SMAD2/3 signaling (Town et al., 2008), or by stimulating PPARγ and retinoid X receptors (Yamanaka et al., 2012). The enhancement of Aβ clearance results from reduced inflammatory activation rather than from direct effects of the innate immune molecules per se. In the brains of 3-month-old APP-transgenic mice and in the microglial/macrophage internalization assays with Aβ at low concentrations (e.g., 0.2 μm), the inflammatory activation level is low. Therefore, the presence or absence of MyD88 or IKKβ does not affect Aβ internalization (Hao et al., 2011).
However, it has been shown that inflammatory activation by the systemic administration of TLR4 ligands can facilitate microglial internalization of Aβ, which decreases the Aβ burden in APP-transgenic mice (Michaud et al., 2013). These results raise an apparent discrepancy with our findings. In our study, we observed that IKKβ deficiency specifically blocks TGF-β-SMAD2/3 signaling but does not affect the SMAD1/5/8 pathway in microglia. Microglial SMAD1/5/8 signaling is constitutively active. Town et al. (2008) reported that SMAD2/3 signaling inhibits macrophage uptake of Aβ, whereas SMAD1/5/8 signaling promotes it; however, an IKKβ deficiency in the microglia of our AD mice may have attenuated the inhibitory effects of SMAD2/3 signaling, thereby enhancing microglial clearance of Aβ in the brain. Using cultured microglia, we have confirmed that the pharmacological inhibition of SMAD2/3 signaling increases Aβ internalization. In Michaud's AD mice, microglial SMAD1/5/8 signaling could be further induced by TLR4-mediated inflammatory activation, so that the Aβ internalization-promoting effect is relatively stronger than the inhibitory effect contributed by SMAD2/3. Therefore, activation of the SMAD1/5/8 signaling cascade by exogenous stimulators may resolve this apparent discrepancy. The hypothesis that the signaling balance between SMAD2/3 and SMAD1/5/8 modulates microglial Aβ internalization could function also in other AD mice deficient in NLRP3 (Heneka et al., 2013) or with PPARγ activation (Yamanaka et al., 2012), because a deficiency in NLRP3 attenuates (Wang et al., 2013) SMAD2/3 phosphorylation in the kidney epithelium, whereas a deficiency in PPARγ increases SMAD2/3 phosphorylation in embryo fibroblasts (Ghosh et al., 2008). Additional studies are needed to further investigate whether this TGF-β hypothesis is a general rule in enhanced microglial Aβ clearance after the inhibition of innate immune signaling and exactly how TGF-β regulates microglial endocytosis.
It is clear that microglial proinflammatory activation contributes to AD pathology, and both M1 and M2 inflammatory activation markers have been observed in AD human and mouse brains (Colton et al., 2006). M1 inflammatory activation contributes to neuronal degeneration (Akiyama et al., 2000; Wyss-Coray and Rogers, 2012). Inhibiting the typical proinflammatory signaling by IL-12/IL-23 (Vom Berg et al., 2012) and IL-1β (Heneka et al., 2013) has been reported to attenuate AD pathology and cognitive deficits. In another study, adoptive transfer of interferon-γ-producing Aβ-specific T lymphocytes into AD mice severely exacerbated AD pathogenesis (Browne et al., 2013). In contrast, M2 inflammatory activation appears to produce beneficial effects in AD by facilitating neurotoxic inflammatory resolution and promoting neuronal protection and regeneration (Colton et al., 2006; Qian et al., 2006; Martinez et al., 2009; Ma et al., 2010; Sharma et al., 2011; Liu et al., 2012). In our studies, ablation of microglial MyD88 and IKKβ reduced only M1 inflammatory activation without affecting M2 activation in the APP-transgenic mouse brain (Hao et al., 2011). In AD mice with a deficiency in CD14 (Reed-Geaghan et al., 2010), TLR2 (Liu et al., 2012), or IRAK4 (Cameron et al., 2012), microglia were skewed from M1 to M2 inflammatory activation, although the underlying mechanisms were not explained.
In this study, a deficiency in IKKβ in myeloid cells did not appear to affect Aβ generation and Aβ efflux from the brain parenchyma. This observation was surprising in light of the fact that neuroinflammation has been reported to change β-secretase and γ-secretase expression and activity (Sheng et al., 2003; He et al., 2007; Xie et al., 2013) and to alter the Aβ transporter function in the blood–brain barrier (Erickson et al., 2012).
In summary, our study shows that the signaling cascade of TLRs-MyD88-IRAK4-IKKβ mediates Aβ-triggered microglial inflammatory activation. Ablation of IKKβ inhibits detrimental neuroinflammation and facilitates beneficial Aβ clearance, thereby improving neuronal function in AD mice. These results contribute to a better understanding of AD pathogenic mechanisms that may eventually translate to therapeutic options for the prevention or treatment of AD progression.
Footnotes
This work was supported by Alzheimer Forschung Initiative e.V. (to Y.L.); Fritz Thyssen Stiftung (to Y.L. and K.F.); Medical Faculty of University of the Saarland through HOMFOR2013 (to Y.L.); and Forschungspreis 2011 der Freunde des Universitätsklinikums des Saarlandes (to W.H.). We thank D. Westaway (University of Toronto, Canada) for providing TgCRND8 APP transgenic mice; Junfa Li (Capital University of Medical Sciences, Beijing, China) for help with the technical equipment; and Mirjam Müller, Rebecca Lancaster, and Andrea Schottek for excellent technical assistance.
The authors declare no competing financial interests.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. 2013;190:2241–2251. doi: 10.4049/jimmunol.1200947. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Cameron B, Tse W, Lamb R, Li X, Lamb BT, Landreth GE. Loss of interleukin receptor-associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer's disease. J Neurosci. 2012;32:15112–15123. doi: 10.1523/JNEUROSCI.1729-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/A:1008942828960. [DOI] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid beta efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, Farfara D, Kingery ND, Weiner HL, El Khoury J. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat Commun. 2013;4:2030. doi: 10.1038/ncomms3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, Pfeiffer J, Kaltschmidt C, Israël A, Mémet S. Forebrain-specific neuronal inhibition of nuclear factor-kappaB activity leads to loss of neuroprotection. J Neurosci. 2003;23:9403–9408. doi: 10.1523/JNEUROSCI.23-28-09403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Wei J, Wu M, Varga J. Constitutive Smad signaling and Smad-dependent collagen gene expression in mouse embryonic fibroblasts lacking peroxisome proliferator-activated receptor-gamma. Biochem Biophys Res Commun. 2008;374:231–236. doi: 10.1016/j.bbrc.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, Luedde T, Heikenwalder M, Jung S, Prinz M. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. 2013;16:1618–1626. doi: 10.1038/nn.3531. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-kappaB. Trends Neurosci. 2011;34:316–325. doi: 10.1016/j.tins.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Liu Y, Liu S, Walter S, Grimm MO, Kiliaan AJ, Penke B, Hartmann T, Rübe CE, Menger MD, Fassbender K. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer's disease-related symptoms and pathology. Brain. 2011;134:278–292. doi: 10.1093/brain/awq325. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol. 2009;1:a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Katzmarski N, Haas CA, Prinz M. Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PLoS One. 2013;8:e58544. doi: 10.1371/journal.pone.0058544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer MP, Vogl T, Axt D, Griep A, Vieira-Saecker A, Jessen F, Gelpi E, Roth J, Heneka MT. Mrp14 deficiency ameliorates amyloid beta burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J Neurosci. 2012;32:17824–17829. doi: 10.1523/JNEUROSCI.1504-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/S0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Lim JE, Kou J, Song M, Pattanayak A, Jin J, Lalonde R, Fukuchi K. MyD88 deficiency ameliorates beta-amyloidosis in an animal model of Alzheimer's disease. Am J Pathol. 2011;179:1095–1103. doi: 10.1016/j.ajpath.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rübe CE, Walter J, Heneka MT, Hartmann T, Menger MD, Fassbender K. TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- Liu Y, Hao W, Letiembre M, Walter S, Kulanga M, Neumann H, Fassbender K. Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J Neurosci. 2006;26:12904–12913. doi: 10.1523/JNEUROSCI.2531-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang M, Hao W, Mihaljevic I, Liu X, Xie K, Walter S, Fassbender K. Matrix metalloproteinase-12 contributes to neuroinflammation in the aged brain. Neurobiol Aging. 2013;34:1231–1239. doi: 10.1016/j.neurobiolaging.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TC, Campana A, Lange PS, Lee HH, Banerjee K, Bryson JB, Mahishi L, Alam S, Giger RJ, Barnes S, Morris SM, Jr, Willis DE, Twiss JL, Filbin MT, Ratan RR. A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a cAMP-independent pathway. J Neurosci. 2010;30:739–748. doi: 10.1523/JNEUROSCI.5266-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JP, Richard KL, Rivest S. MyD88-adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer's disease. Mol Neurodegener. 2011;6:5. doi: 10.1186/1750-1326-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JP, Richard KL, Rivest S. Hematopoietic MyD88-adaptor protein acts as a natural defense mechanism for cognitive deficits in Alzheimer's disease. Stem Cell Rev. 2012;8:898–904. doi: 10.1007/s12015-012-9356-9. [DOI] [PubMed] [Google Scholar]
- Michaud JP, Hallé M, Lampron A, Thériault P, Préfontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, Brichard V, Palmantier R, Pilorget A, Larocque D, Rivest S. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proc Natl Acad Sci U S A. 2013;110:1941–1946. doi: 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, Quinn M, Brück W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol. 2012;188:29–36. doi: 10.4049/jimmunol.1100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, Walker Z, Kennedy A, Fox N, Rossor M, Brooks DJ. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, Krieg T, Rajewsky K, Haase I. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, Wilson B, Hong JS, Flood PM. Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther. 2006;319:44–52. doi: 10.1124/jpet.106.106351. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Reed QW, Cramer PE, Landreth GE. Deletion of CD14 attenuates Alzheimer's disease pathology by influencing the brain's inflammatory milieu. J Neurosci. 2010;30:15369–15373. doi: 10.1523/JNEUROSCI.2637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res. 2011;1373:189–194. doi: 10.1016/j.brainres.2010.11.096. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–145. doi: 10.1016/S0969-9961(03)00069-X. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. J Neuroinflammation. 2011;8:92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Thal DR, Charo IF, Heppner FL, Aguzzi A, Garaschuk O, Ransohoff RM, Jucker M. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A. 2012;109:18150–18155. doi: 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. 2012;18:1812–1819. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, Beck PL, Muruve DA. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. 2013;190:1239–1249. doi: 10.4049/jimmunol.1201959. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Morgan D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat Protoc. 2006;1:1591–1595. doi: 10.1038/nprot.2006.277. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Liu Y, Hao W, Walter S, Penke B, Hartmann T, Schachner M, Fassbender K. Tenascin-C deficiency ameliorates Alzheimer's disease-related pathology in mice. Neurobiol Aging. 2013;34:2389–2398. doi: 10.1016/j.neurobiolaging.2013.04.013. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Walsh SM, Liu J, Kipnis J, Ikezu T. Cytokine-mediated inhibition of fibrillar amyloid-beta peptide degradation by human mononuclear phagocytes. J Immunol. 2008;181:3877–3886. doi: 10.4049/jimmunol.181.6.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]