

Abstract
Synaptic rearrangements during critical periods of postnatal brain development rely on the correct formation, strengthening, and elimination of synapses and associated dendritic spines to form functional networks. The correct balance of these processes is thought to be regulated by synapse-specific changes in the subunit composition of NMDA-type glutamate receptors (NMDARs). Among these, the nonconventional NMDAR subunit GluN3A has been suggested to play a role as a molecular brake in synaptic maturation. We tested here this hypothesis using confocal time-lapse imaging in rat hippocampal organotypic slices and assessed the role of GluN3A-containing NMDARs on spine dynamics. We found that overexpressing GluN3A reduced spine density over time, increased spine elimination, and decreased spine stability. The effect of GluN3A overexpression could be further enhanced by using an endocytosis-deficient GluN3A mutant and reproduced by silencing the adaptor protein PACSIN1, which prevents the endocytosis of endogenous GluN3A. Conversely, silencing of GluN3A reduced spine elimination and favored spine stability. Moreover, reexpression of GluN3A in more mature tissue reinstated an increased spine pruning and a low spine stability. Mechanistically, the decreased stability in GluN3A overexpressing neurons could be linked to a failure of plasticity-inducing protocols to selectively stabilize spines and was dependent on the ability of GluN3A to bind the postsynaptic scaffold GIT1. Together, these data provide strong evidence that GluN3A prevents the activity-dependent stabilization of synapses thereby promoting spine pruning, and suggest that GluN3A expression operates as a molecular signal for controlling the extent and timing of synapse maturation.
Keywords: critical period, hippocampus, NMDA, rat, receptors, synapse
Introduction
Experience-driven activity shapes the development of neural networks during critical periods through mechanisms that maintain a high level of structural plasticity, thereby creating a permissive environment for circuit rewiring. This is notably illustrated by the high rate of excitatory spine synapse formation and elimination that characterizes the developing cortex and hippocampus during the first weeks after birth (Holtmaat and Svoboda, 2009). The mechanisms underlying these structural synaptic rearrangements and the high level of plasticity expressed during early development remain poorly understood. Growing evidence indicates that experience-driven synaptic activity or induction of forms of plasticity, such as LTP or LTD, may significantly affect synaptic network remodeling by promoting spine formation and a selective stabilization or elimination of synapses (Engert and Bonhoeffer, 1999; De Roo et al., 2008b; Caroni et al., 2012). These mechanisms have thus been proposed to contribute to the structural basis of learning and long-term memory storage (Xu et al., 2009; Yang et al., 2009).
Because NMDARs are principal mediators of synaptic plasticity, much attention has been directed to understand their roles on synaptic rearrangements and maturation (Feldman and Knudsen, 1998; Barth and Malenka, 2001; Gambrill and Barria, 2011). NMDARs assemble as heterotetrameric combinations of an obligatory GluN1 subunit, at least one GluN2(A–D), and in some cases GluN3(A,B) subunits. Different subtypes are differentially expressed during development and exhibit ionic conductances with distinct properties, amplitude and duration, which makes them variably permissive for synaptic plasticity (Paoletti et al., 2013). This is particularly true for subtypes that include the nonconventional GluN3A subunit. Inclusion of GluN3A in NMDAR channels reduces their calcium (Ca2+) permeability and sensitivity to magnesium (Mg2+) blockade (Pérez-Otaño et al., 2001; Sasaki et al., 2002), thus modifying the two properties of NMDARs responsible for induction of long-lasting forms of synaptic plasticity. GluN3A expression peaks between P8 and P25 in rodents and the first years of life in humans (Henson et al., 2010) but is largely downregulated afterward, and variations in expression modulate synapse maturation and spine number (Das et al., 1998; Roberts et al., 2009; Henson et al., 2012). Further, continued expression of GluN3A beyond its natural time window was reported to attenuate LTP and interfere with long-term memory storage (Roberts et al., 2009). Importantly, the removal of GluN3A-containing NMDARs from synapses is coupled to activity via a number of trafficking mechanisms (Pérez-Otaño et al., 2006; Chowdhury et al., 2013). These properties of GluN3A and its preferential expression during periods of high structural plasticity suggested that it could work as a brake to prevent an early or nonselective stabilization of neuronal networks.
We investigated this hypothesis and the underlying mechanisms using a combination of genetic approaches to enhance or silence GluN3A expression with time-lapse monitoring of spine dynamics in hippocampal slice cultures. Our data demonstrate that GluN3A promotes spine elimination by limiting the activity-dependent stabilization of spines. They further support a role for endocytic GluN3A removal in regulating these mechanisms and thereby contributing to selectively stabilize active synapses during neuronal network remodeling.
Materials and Methods
Slice cultures and transfection.
Transverse hippocampal organotypic slice cultures (400 μm thickness) were prepared from 6- to 7-day-old-rat pups of either sex (Stoppini et al., 1991) using a protocol approved by the Geneva veterinary office and maintained under culture conditions as described previously (De Roo et al., 2008b). Biolistic transfection was completed at 7 d in vitro (DIV7), unless otherwise stated, using the Gene Gun (Bio-Rad) method with CX-mRFP1 for full visualization of neurons and spines (De Roo et al., 2008b) and one of the following plasmids: GFP-GluN3A in pRK5 vector (Pérez-Otaño et al., 2001), full-length GFP-GluN3A carrying mutations in the YWL endocytic motif (Chowdhury et al., 2013), full-length GFP-GluN3A lacking a C-terminal 1082–1115 amino acid stretch (GFP-GluN3A-ΔGIT1) (Fiuza et al., 2013), shRNA1392 directed to PACSIN1 plus a scrambled control of this shRNA (Marco et al., 2013), and shRNAs 2532 (target sequence: GGACAAAGCCCTTCTGGATTA) and 1185 (target sequence: CTACAGCTGAGTTTAGAAA (Yuan et al., 2013) directed to two separate regions in GluN3A. The efficiency of the shRNAs (shGluN3A2532, shGluN3A1185, shPACSIN11392) has been previously characterized in neurons (Yuan et al., 2013), recombinant cells (Sproul et al., 2011), or HEK293 cells (Marco et al., 2013). As control for mutant constructs, we analyzed cells transfected with either mRFP alone or scrambled shRNA. As these control conditions showed no significant differences in terms of protrusion density across time, protrusion dynamics, and stability, we pooled them together (see Figs. 2, 3B–E, 4, and 5). For all conditions, slices were left 3–4 d after transfection before the first observation. Fluorescence signal for all proteins was usually observed by 1–2 d after transfection and remained stable for a at least 10–15 d. The level of GluN3A overexpression obtained in these experiments was estimated to be 1.8 ± 0.2-fold based on analysis of fluorescence intensity in 6 transfected cells using immunohistochemistry.
Figure 2.
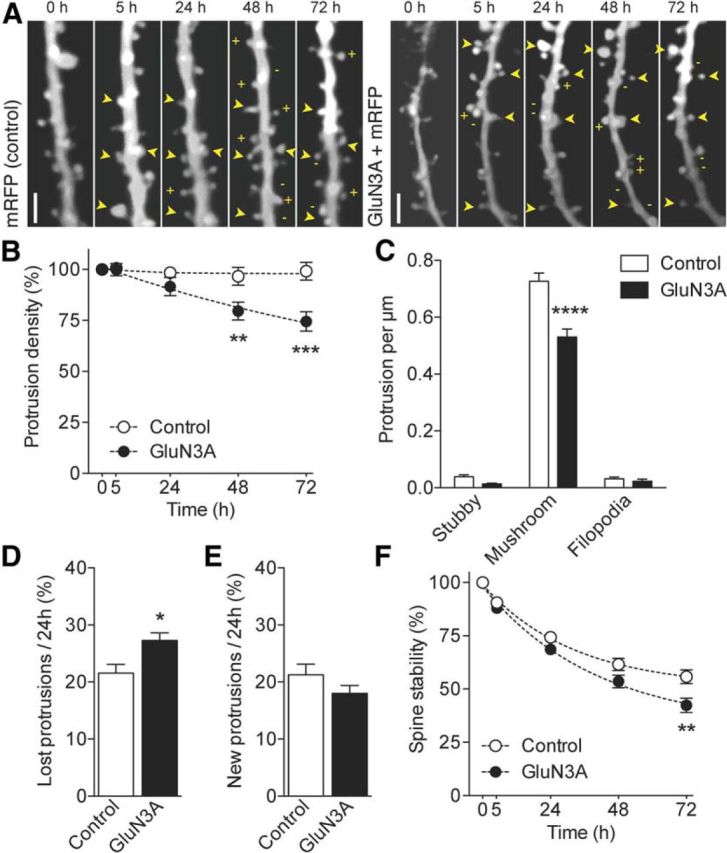
Overexpression of GluN3A promotes spine elimination and decreases spine stability. A, Repetitive imaging of a dendritic segment from mRFP (control) and mRFP/GluN3A-transfected CA1 pyramidal neurons (DIV18) at the indicated time points. Images correspond to the red (mRFP) channel. + and − indicate newly formed and eliminated protrusions. Arrowheads indicate stable spines. Scale bar, 2 μm. B, Protrusion density normalized to the first observation and expressed across time (two-way ANOVA with Bonferroni post test). C, Protrusion density across different morphological categories (control: n = 38 and GluN3A: n = 39; two-way ANOVA with Bonferroni post test). D, Fraction of protrusions eliminated over 24 h time periods in control and GluN3A-transfected cells (control: n = 20 and GluN3A: n = 10). *p = 0.023 (unpaired t test). E, Fraction of newly formed protrusions observed per 24 h periods. F, Preexisting spine stability assessed as the proportion of spines present at time 0 h and still present at the subsequent observations (control: n = 20 and GluN3A: n = 10; two-way ANOVA with Bonferroni post test). *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Figure 3.
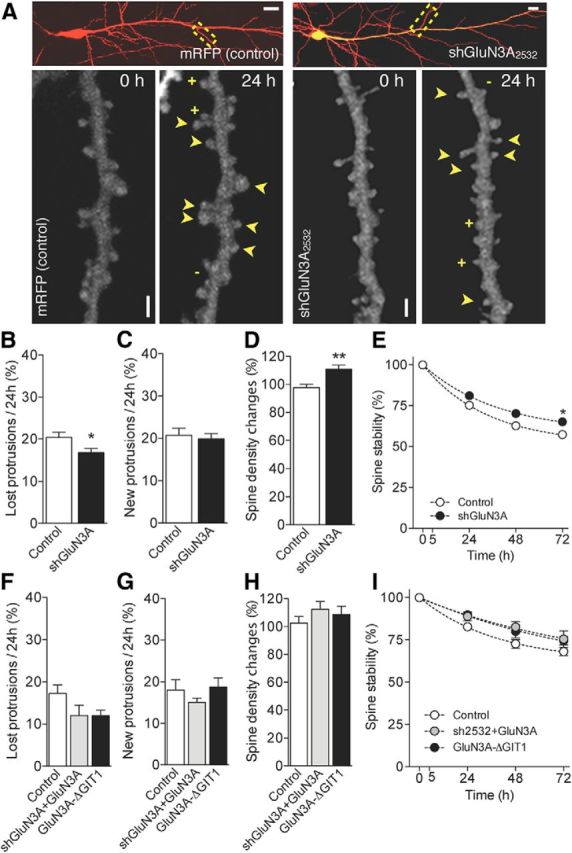
Interference with GluN3A expression or function reduces spine elimination and increases spine stability. A, Illustration of CA1 pyramidal neurons transfected with mRFP or mRFP + shGluN3A2532. Scale bar, 10 μm. The yellow rectangle represents the dendritic segments shown at higher magnification below at 0 and 24 h. Scale bar, 1 μm. + indicates new spines; − indicates lost spines. Arrowheads indicate stable spines. B, Fraction of protrusions eliminated over 24 h time periods in control and shGluN3A-transfected cells (control: n = 24, shGluN3A: n = 12). *p < 0.05 (unpaired t test). C, Fraction of newly formed spines observed per 24 h periods. D, Changes in spine density observed over a 48 h period (control: n = 24, shGluN3A: n = 12). **p < 0.01 (unpaired t test). E, Preexisting spine stability is increased in cells transfected with shGluN3A (control: n = 24, GluN3A: n = 11). *p < 0.05 (two-way ANOVA with Bonferroni post test). Error bars are smaller than symbols. F, Fraction of protrusions eliminated over 24 h time periods in control conditions and in cells transfected with shGluN3A2532+GluN3A and GluN3A-ΔGIT1 mutant (control: n = 4, shGluN3A2532+GluN3A: n = 5, GluN3A-ΔGIT1: n = 4). p > 0.05. G, Fraction of newly formed spines observed per 24 h periods. H, Changes in spine density observed over a 48 h period (control: n = 4, shGluN3A2532+GluN3A: n = 5, GluN3A-ΔGIT1: n = 4). I, Preexisting spine stability in control cells and cells transfected with shGluN3A2532+GluN3A and GluN3A-ΔGIT1 mutant (control: n = 4, shGluN3A2532+GluN3A: n = 5, GluN3A-ΔGIT1: n = 4).
Figure 4.

Alteration in spine dynamics produced by a endocytosis-deficient GluN3A mutant. A, Decreased rectification index of NMDAR-mediated EPSCs in GluN3AYWL/AAA-transfected neurons versus control, neighboring cells (control: n = 8 and GluN3AYWL/AAA: n = 8). *p = 0.048 (unpaired t test). B, Illustration of dendritic segments from a control and GluN3AYWL/AAA-transfected neuron at 0 and 24 h. + indicates new spines; − indicates lost spines. Arrowheads indicate stable spines. C, Fraction of protrusions eliminated over 24 h time periods in control and GluN3AYWL/AAA-transfected cells (control: n = 20 and GluN3AYWL/AAA: n = 5). **p = 0.0029 (unpaired t test). D, Fraction of newly formed protrusions observed per 24 h periods. E, Changes in spine density observed over a 72 h period. F, Decrease in spine stability in GluN3AYWL/AAA-transfected compared with control cells (control: n = 20 and GluN3A: n = 5, two-way ANOVA with Bonferroni post test). **p < 0.01. ***p < 0.001.
Figure 5.

Alterations of spine dynamics produced by silencing of the endocytic adaptor protein PACSIN1. A, Isolated NMDAR-mediated EPSCs of transfected shPACSIN1neurons reveal a significant shift in rectification index (control: n = 8 and shPACSIN1: n = 5). *p = 0.024. B, Illustration of protrusion density in dendritic segments of control and shPACSIN1-transfected neurons at 0 and 24 h. Scale bar, 2 μm. C, Fraction of protrusions eliminated over 24 h time periods in control and shPACSIN1-transfected cells (control: n = 20 and shPACSIN1: n = 12; ***p < 0.001 unpaired t test). D, Fraction of newly formed protrusions observed per 24 h periods. E, Changes in spine density observed over a 72 h period (control: n = 20 and shPACSIN1: n = 12; **p < 0.01 unpaired t test). F, Preexisting spine stability is decreased in shPACSIN1-transfected compared with control cells (control: n = 20 and shPACSIN1: n = 11; two-way ANOVA with Bonferroni post test). **p < 0.01. ***p < 0.001.
Protein extraction and Western blots.
For protein extractions, organotypic slices were plated after dissection and kept under culture conditions as above. Six to eight slices were collected at DIV 6, 12, 18, 24, and 30, frozen on liquid nitrogen, and stored at −80°C. After thawing, slices were sonicated in 150–200 μl of lysis buffer (25 mm Tris-HCl pH 8, 150 mm NaCl, 1 mm DTT, Triton X-100 1%, glycerol 10%, EDTA 2 mm, and 1× proteases inhibitor) and spun at 3400 rpm for 15 min.
Primary cultured corticohippocampal neurons were infected with control lentivirus or lentivirus-expressing GluN3A under the control of a synapsin I promoter. Neurons were collected 5 d later, homogenized in lysis buffer, and centrifuged as above. Supernatants were used for protein quantification using Pierce BCA assay (Thermo Instruments). Proteins were resolved by SDS-PAGE, transferred onto nitrocellulose membrane, and detected by immunoblot using the following antibodies: rabbit anti-GluN3A (1:2000, 07–356, Millipore), mouse anti-PACSIN1 (1:10,000, 611810, BD Transduction Laboratories), mouse anti-β actin (1:40,000 AC-74, Sigma), mouse anti-GluN2B (1:200, 73–097, NeuroMab), rabbit anti-GluA1 (1:1000, AB1504, Millipore), rabbit anti-GluN2A (1:1000, clone A12W, Millipore), mouse anti-PSD95 (1:10000, 05–494, Millipore), and mouse anti-β tubulin (1: 20000, T8660, Sigma).
Electrophysiology.
Electrophysiology was performed on transfected slices using whole-cell patch-clamp techniques (Boda et al., 2004). Connections between CA1 and CA3 were cut 2 h before recording to prevent the formation of epileptiform discharges. Slices were submersed in a recording chamber and continually perfused with extracellular aCSF solution containing (in mm) NaCl 119, KCl 2.5, MgCl 1.3, CaCl2 2.5, Na2HPO4 1.0, NaHCO3 26.2, and glucose 11, bubbled with 95% O2 and 5% CO2 supplemented with 100 μm picrotoxin. CA1 pyramidal neurons positive for plasmids were detected using fluorescence and video microscopy. Control cells were taken in the vicinity of transfected cells and subjected to the same parameters. Whole-cell recordings were performed using patch electrodes filled with the following internal solution (mm): CsCl 130, NaCl 4, MgCl 2, EGTA 1.1, HEPES 5, Na2ATP 2, sodium creatine-phosphate 5, Na3GFP 0.6, and spermine 0.1. Currents were amplified, filtered at 5 kHz, and digitized at 20 kHz. The liquid junction potential was small (−3 mV), and traces were therefore not corrected. Voltage-clamp recordings from 40 to −80 mV were taken to obtain an I/V curve of NMDA EPSCs in the presence of 10 μm NBQX. EPSCs were evoked by stimulating Schaffer collaterals at 0.1 Hz through a glass pipette electrode. Representative example traces are shown as the average of 20 consecutive EPSCs typically obtained at each potential. Experiments were discarded if the access resistance varied by >20%. Controls in each figure are from nontransfected neurons pooled from all conditions as there were no significant differences.
Confocal imaging.
Imaging was performed 3–4 d after transfection. Slices were previewed using either an epifluorescence microscope or an Olympus Fluoview 300 system to identify CA1 pyramidal neuron transfected with mRFP and the plasmid of interest. Laser intensity and acquisition conditions were kept to a minimum and remained stable across the observation period. Cell morphology or viability was not altered across the observation periods. Repetitive time-lapse imaging of dendritic segments was performed using the Visitron spinning disk system, with a two-line excitation laser (488 and 568 nm). Slices were submersed in prewarmed (CO2 controlled) culture medium and short imaging sessions (10–15 min) performed. Z-stacks of CA1 pyramidal neurons were taken of secondary or tertiary dendritic segments of 35–50 μm length using a 40× or 60× water-immersion objective. Images were captured using MetaMorph software.
For θ burst stimulation (TBS), repetitive time-lapse imaging of dendritic segments was performed using the Olympus Fluoview 300 system. Stimulation was performed in an interface chamber under continual presence of aCSF at 32°C, perfused with 95% O2 and 5% CO2. Field EPSPs evoked by stimulation of a group of Schaffer collaterals were recorded by an electrode placed in the CA1 stratum radiatum. LTP was induced by TBS (five trains at 5 Hz composed each of four pulses at 100 Hz, repeated twice at 10 s interval) using stimulation intensities that evoked responses just above the threshold for action potential. These stimulations have been shown to activate ∼30%-40% of synapses (De Roo et al., 2008b). Control slices were placed in the same conditions but were not given any type of stimulation.
Image analysis.
Protrusions refer to all structures extending from the dendrite. Long, thin protrusions without an enlarged head were classified as filopodia; protrusions with large head but without a neck were classified as stubby spines, and protrusions with enlarged head and thin necks were classified as mushroom spines (Harris et al., 1992). For organotypic cultures, dendritic segments were repetitively imaged at 0, 5, 24, 48, and 72 h. Between imaging sessions slices were returned to a 33°C incubator. Analysis also included protrusion density and protrusion head width (measured as the diameter of the largest part of the spine head). All protrusion measurements were made on individual z-stack images of 1–2 dendritic segments per CA1 pyramidal neuron. Analysis was completed using OsiriX software, developed with a plug-in designed for spine quantification. Protrusion turnover was quantified by analyzing all new and lost protrusions (spines and filopodia) that appear or disappear, respectively, between any two observation periods. Turnover was calculated as the sum of the rate of spine formation/24 h plus the rate of spine elimination/24 h divided by 2. Filopodia were counted separately and excluded from spine stability analyses. Protrusions that could not be unambiguously analyzed were excluded, but these did not represent more than ∼1% of cases. Spine stability was calculated as the percentage of spines that were present at 0 h and were still present across subsequent observation periods. Analysis of spine enlargement after TBS was performed before and 5 h after TBS. Spines were considered enlarged if the spine head width increased by >0.1 μm.
Statistical analyses were performed with Prism, using Student unpaired t test unless otherwise indicated. Data are represented as mean ± SEM.
Results
Overexpression of GluN3A decreases spine stability and promotes spine elimination
GluN3A expression is developmentally regulated, with a peak during the first two postnatal weeks followed by a progressive decline into adulthood (Sasaki et al., 2002). A known mechanism for GluN3A synaptic removal involves endocytosis mediated by the adaptor protein PACSIN1/syndapin1, which shows a reciprocal expression pattern to that of GluN3A (Pérez-Otaño et al., 2006). These patterns of expression are essentially preserved in hippocampal slice cultures, with GluN3A being highly expressed during the first 2 weeks after explantation and then strongly declining, whereas PACSIN1 expression increases over the first 2 weeks and then stays high later on (Fig. 1A). This suggests that the developmental regulation of NMDAR subunit composition and underlying mechanisms are maintained under culture conditions. To assess the role of GluN3A on spine dynamics, CA1 pyramidal neurons in hippocampal organotypic slices were transfected with mRFP alone (control) or together with GFP-GluN3A (Fig. 1B) at DIV7 or DIV14, and the behavior of spines was repetitively monitored starting at DIV12 or DIV18, respectively, for a period of 4 d. To confirm the functional overexpression of GluN3A at synapses and its assembly with endogenous NMDAR subunits, we recorded isolated NMDAR-mediated EPSCs. Consistent with reports in transgenic GluN3A overexpressing mice (Roberts et al., 2009), I-V curves of evoked NMDA EPSCs recorded in the presence of 2.5 mm Mg2+ showed that cells expressing GFP-GluN3A had a significantly larger amplitude at hyperpolarized potentials than control (neighboring, nontransfected) neurons, resulting in a decrease in rectification (Fig. 1C) calculated as the ratio of NMDA responses measured at 40 mV and −60 mV (Fig. 1D). These data indicated that NMDARs with decreased Mg2+ sensitivity (i.e., one of the electrophysiological signatures of GluN3A-containing NMDAR subtypes) were present at synapses of GluN3A-transfected neurons. We additionally examined whether GluN3A overexpression affected the expression levels of other glutamate receptor subunits or synaptic proteins, by infecting dissociated hippocampal neurons with increasing concentrations of lentiviral particles expressing GluN3A. Whereas GluN3A expression was significantly increased in a dose-dependent manner compared with control neurons, no changes in GluN2B, GluN2A, GluA1, PSD95 were observed (Fig. 1E).
Figure 1.

Development and synaptic expression of GluN3A. A, Western blot analysis of GluN3A and PACSIN1 expression across several DIV in organotypic hippocampal slice cultures. B, Illustration of an organotypic hippocampal slice culture (left; scale bar, 30 μm) and a CA1 pyramidal neuron transfected using biolistics to express both mRFP (middle; scale bar, 15 μm) and GFP-GluN3A (right) within the same neuron. C, Traces, Representative NMDAR-evoked EPSCs from nontransfected, neighboring (control) and GFP-GluN3A-positive neurons. Scales, 50 ms and 50 pA. Graph represents the current/voltage relationship obtained in the two conditions in the presence of 2.5 mm Mg2+. D, Index of rectification measured in the same experiments by calculating the amplitude ratio of responses recorded at +40 and −60 mV (control, n = 8; and GluN3A, n = 8 neurons; p = 0.0057, unpaired t test). **p < 0.01. E, Expression levels of GluN3A, GluN2A, GluN2B, GluA1, and PSD95 in dissociated hippocampal neurons transfected with increasing concentrations of control lentivirus or lentivirus expressing GluN3A (5, 15, or 30 μg/ml).
We then assessed protrusion density and dynamics in control and GluN3A-transfected cells (Fig. 2A). Protrusion density, including all spines and filopodia, was reduced at the first observation time in GluN3A-overexpressing CA1 pyramidal neurons compared with control neurons (control: n = 20, 0.81 ± 0.04 vs GluN3A: n = 10, 0.65 ± 0.06 protrusions per μm, p = 0.024 unpaired t test). The effect was amplified over the course of the experiments, and by 72 h, GluN3A-transfected neurons showed a further 28 ± 4.8% decrease in density compared with control neurons (p < 0.0001; Fig. 2B). Classification of protrusions into stubby, mushroom and filopodia showed that the decrease in protrusion density specifically affected mushroom spines (Fig. 2C) with no effects on stubby spines or filopodia in agreement with previous work (Roberts et al., 2009). An analysis of spine dynamics demonstrated that the gradual decrease in protrusion density was the result of two underlying mechanisms. First, GluN3A overexpression caused an imbalance in protrusion turnover because of a selective increase in the rate of protrusions eliminated per 24 h (Fig. 2D), without significant changes in rates of protrusion formation (Fig. 2E). Second, the stability of preexisting spines, defined as spines present at the first observation, was significantly reduced over time in GluN3A-overexpressing neurons (Fig. 2F). Thus, GluN3A overexpression led to a decrease in protrusion density, specifically targeting mushroom spines, by increasing protrusion elimination and reducing the fraction of week-long persistent spines.
Silencing GluN3A reduces spine loss
To further verify the role of GluN3A in spine dynamics, we used a loss of function approach and silenced its endogenous expression by using two different short hairpin RNAs that targeted two separate sites of GluN3A, shGluN3A2532 and shGluN3A1185. The silencing efficiency of these shRNAs has been previously demonstrated (Sproul et al., 2011; Yuan et al., 2013) and, as they yielded comparable effects, the data obtained with the two constructs were pooled. Transfection was performed at DIV7 when endogenous GluN3A expression is still high and neurons were imaged between DIV12 and DIV15 (Fig. 3A). Turnover analysis revealed a significant decrease in the rate of spine elimination per 24 h (Fig. 3B) but no changes in the rate of spine growth (Fig. 3C) in shGluN3A-transfected cells compared with control neurons. This resulted in an overall increase in spine density (Fig. 3D). Furthermore, the stability of preexisting spines was significantly increased upon GluN3A knockdown when measured 72 h later (Fig. 3E). As an additional test for the efficiency of our shRNA, we coexpressed GluN3A together with shGluN3A2532. As shown in Figure 3F–I, shGluN3A2532 reversed the spine loss phenotype induced by GluN3A overexpression and tended to promote spine stability.
We further investigated whether interfering with GluN3A signaling mechanisms could affect spine dynamics. A recent study showed that GluN3A binds the G-protein-coupled receptor kinase-interacting protein GIT1 through its intracellular C-terminal domain, inhibiting Rac1/PAK/actin signaling and spine morphogenesis (Fiuza et al., 2013). As illustrated in Figure 3F–I, expression of a GluN3A mutant lacking the GIT1 binding domain in pyramidal neurons did not reproduce the spine loss effects of GluN3A but rather promoted an increase in spine stability. These results show that interfering with GluN3A expression or function promotes spine survival and reduces spine elimination.
Endogenous GluN3A trafficking regulates spine dynamics
Clathrin-dependent endocytosis is a prominent mechanism to achieve downregulation of the functional surface expression of GluN3A-containing NMDARs (Pérez-Otaño et al., 2006). Recent work identified a tyrosine-based YWL motif in the C-terminal domain of GluN3A that is critical for its removal from the neuronal surface by recruiting the clathrin-adaptor protein AP2 (Chowdhury et al., 2013). Direct binding of GluN3A to the multifunctional adaptor protein PACSIN1 further promotes endocytosis (Pérez-Otaño et al., 2006). We therefore tested whether interfering with GluN3A removal by targeting these trafficking mechanisms modified spine dynamics.
First, we overexpressed an endocytosis-deficient GluN3A mutant in which the YWL motif was mutated to AAA (GluN3AYWL/AAA); this mutation has been shown to enhance surface expression of GluN3A-containing NMDARs in cultured hippocampal neurons (Chowdhury et al., 2013). Electrophysiological assays confirmed that the endocytosis-deficient GluN3A mutant was functionally incorporated into synapses, as shown by the decrease in rectification observed in neurons transfected with GluN3AYWL/AAA relative to control neurons (Fig. 4A). Analysis of spine dynamics (Fig. 4B) revealed that CA1 pyramidal neurons transfected with the mutant displayed an increase in the rate of protrusion elimination that exceeded that produced by wild-type GluN3A (compare Fig. 4C with Fig. 2D; p < 0.05). No significant effects on spine growth rates were detected, although there was a tendency toward a compensatory increase in spine formation (Fig. 4D). As a result, spine density also tended to decrease over the next 72 h (Fig. 4E). Finally, the stability of preexisting spines was markedly reduced in cells expressing GluN3AYWL/AAA across all observation time points (Fig. 4F). Thus, combining exogenous overexpression with the shutdown of endocytic mechanisms that normally limit surface expression accentuates the effects of GluN3A on spine dynamics.
As a second approach, we silenced PACSIN1 using an shRNA (shPACSIN1) to prevent ongoing endocytosis of endogenous GluN3A and thus foster functional surface expression (Marco et al., 2013). Expression of shPACSIN1 in CA1 pyramidal neurons also resulted in a marked decrease in the rectification index of NMDAR currents (Fig. 5A), confirming that PACSIN1 silencing increased the number of functional GluN3A-containing NMDARs at synapses. At the morphological level, dendritic segments of cells transfected with this shPACSIN1 displayed fewer protrusions than control cells at the first observation time (Fig. 5B; control: n = 20, 0.91 ± 0.04 vs shPACSIN1: n = 11, 0.65 ± 0.06 protrusions/μm; p = 0.031). shPACSIN1induced a significant increase in protrusion elimination (Fig. 5C), without changes in the rate of spine formation (Fig. 5D), leading to a significant decrease in spine density over the next 72 h (Fig. 5E). In addition, the stability of preexisting spine was significantly decreased in shPACSIN1-transfected cells at all time points analyzed (Fig. 5F). Thus, blocking endogenous mechanisms for GluN3A removal mimicked the effects of exogenous overexpression.
GluN3A expression in mature cells reinstates a low spine stability phenotype
We finally explored whether reactivation of GluN3A expression beyond its natural time window could reinstate a low spine stability phenotype. To do this, we transfected mature organotypic cultures (DIV21-DIV22), which express low levels of GluN3A (Fig. 1A), and analyzed spine dynamics between DIV25 and DIV28. At this age, spine density was higher (DIV25: n = 7, 1.69 ± 0.14 vs DIV12: n = 20, 0.87 ± 0.06 protrusion/μm, p < 0.001) and basal spine turnover was reduced relative to younger slice cultures (DIV25: 6.07 ± 0.59% vs DIV12: 21.40 ± 1.19%, p < 0.0001) consistent with previous findings (De Roo et al., 2008a). Reexpression of GluN3A resulted in striking changes. The major change was a marked increase in rates of spine elimination (Fig. 6A) without detectable modification in spine formation (Fig. 6B). Spine density was significantly decreased already at the first observation (GluN3A, n = 5, 1.1 ± 0.04 vs control, n = 7, 1.7 ± 0.14 protrusion/μm, p < 0.01) and further decreased over the following observation points (Fig. 6C). A significant decrease in spine stability was also observed (Fig. 6D). Overall, these data showed that reexpressing GluN3A in mature tissue interferes with spine stabilization mechanisms and increases rates of spine pruning by twofold close to levels typical of young neurons.
Figure 6.
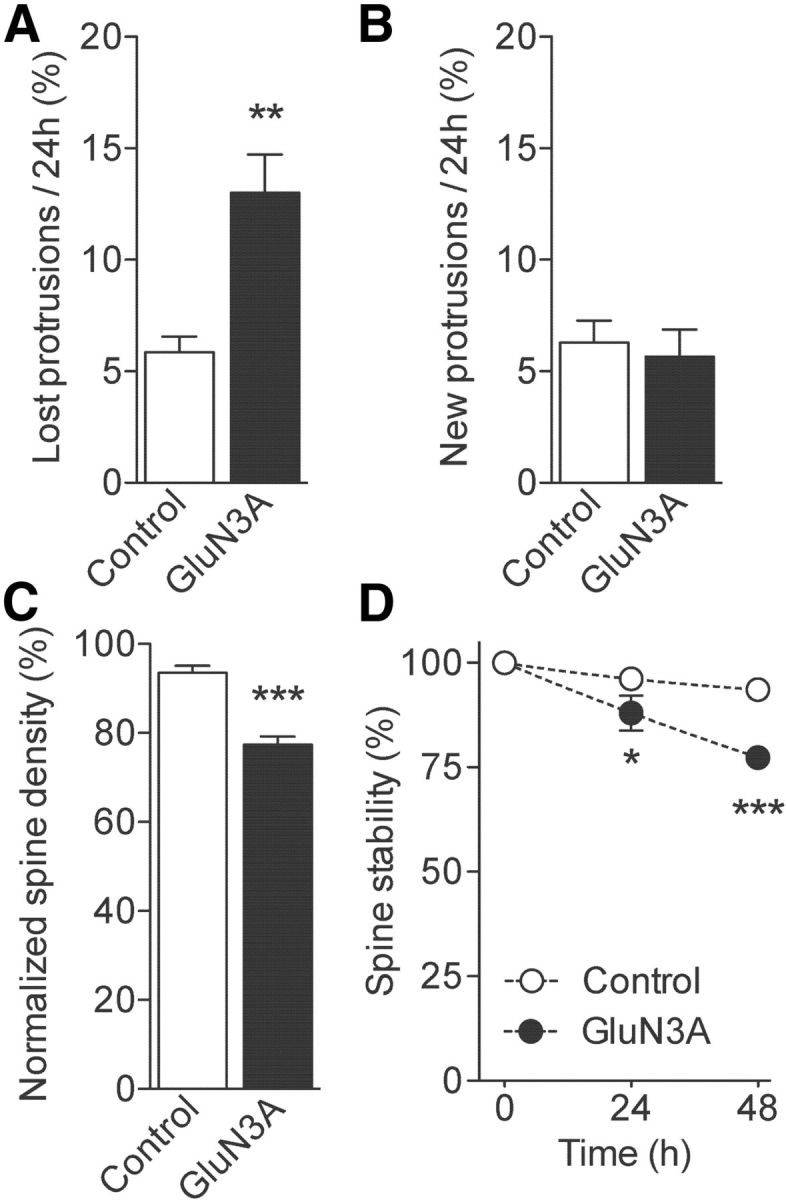
Expression of GluN3A in mature cultures reinstates spine instability. A, Fraction of protrusions eliminated over 24 h time periods in control and GluN3A-transfected cells (control: n = 7 and GluN3A: n = 3; unpaired t test). B, Fraction of newly formed protrusions observed per 24 h periods. C, Changes in spine density observed over a 48 h period (control: n = 5 and GluN3A: n = 3; unpaired t test). D, Preexisting spine stability is decreased in GluN3A-transfected neurons compared with control cells (control: n = 5 and GluN3A: n = 3; two-way ANOVA with Bonferroni post test). *p < 0.05. **p < 0.01. ***p < 0.001.
GluN3A interferes with activity-dependent spine stabilization
We next investigated the mechanisms underlying GluN3A-induced pruning. The results on preexisting spine stability suggested that the increased elimination of spines seen in GluN3A-overexpressing neurons reflected a general decrease in the mean lifetime of spines, affecting predominantly spines with week-long survival rates. However, these experiments did not rule out the possibility that the enhanced pruning could also be the result of a high proportion of transient spines (Holtmaat et al., 2005) that are rapidly eliminated within a few hours or days. To address this issue, we exclusively analyzed the number and stability of the newly formed protrusions observed during a short interval of 5 h (Fig. 7A). Neither the number of newly formed spines (Fig. 7B) nor their stability over the next 3 d (Fig. 7C) differed between control and GluN3A-, shPACSIN1-, and GluN3AYWL/AAA-transfected neurons. Thus, spines seem to form and mature normally during the first days of their life, indicating that the main defect is indeed a decrease in the fraction of persistent spines with week-long lifetimes.
Figure 7.

GluN3A expression does not alter spine formation mechanisms. A, Illustration of newly formed transient spines observed between 0 and 5 h that have disappeared at 72 h. Arrowheads indicate newly formed spines and their survival. B, Proportion of newly formed spines during a 5 h observation period for each condition. C, Stability of newly formed spines over a 72 h observation period for each condition.
As previous work indicated that patterns of activity that induce LTP promote spine stability (De Roo et al., 2008b), we tested whether GluN3A expression interfered with this mechanism. We applied TBS to Schaffer collaterals in hippocampal slice cultures, which induces robust LTP (De Roo et al., 2008b), and monitored spine dynamics over the next 2 d (Fig. 8A). In control neurons, TBS induced a significant increase in basal protrusion turnover rates affecting both protrusion elimination (Fig. 8B) and formation (Fig. 8C). In contrast, no changes in protrusion turnover were observed in GluN3A-overexpressing cells after application of TBS (Fig. 8B,C). Along with promoting protrusion turnover, TBS has also been shown to drive the enlargement of a subset of spines that then display enhanced stability over time (De Roo et al., 2008b). We therefore tested whether TBS was able to promote spine stabilization in conditions of GluN3A overexpression by comparing the stability of spines that enlarged or not 5 h after TBS. Enlarged spines in control neurons exhibited a much higher probability to remain stable over time compared with the nonenlarging spines (Fig. 8D; p < 0.01), consistent with previous reports (De Roo et al., 2008b). In GluN3A-transfected cells, the stability of enlarged spines was initially preserved but dropped dramatically after 24 h to the level of nonenlarged spines (Fig. 8E). The proportion of enlarging spines, however, was not significantly different between the two conditions (32.2 ± 5.9%, GluN3A, vs 41 ± 6.6%, control, n = 4–6). These results indicated that GluN3A overexpression interferes with the regulation of spine dynamics by activity and prevents activity-mediated spine stabilization.
Figure 8.

GluN3A overexpression prevents activity-dependent spine dynamics and stabilization. A, Illustration of dendritic segments from GluN3A (top) and mRFP (control, bottom) transfected neurons before (0 h) and after θ burst stimulation (5 and 48 h, TBS). +, new spines; −, lost spines. Arrowheads indicate stable spines. Scale bar, 2 μm. B, Increase in spine elimination triggered by TBS in control but not GluN3A-transfected cells (n = 6 and 7). *p = 0.0112 (unpaired t test). C, Same but for spine formation (n = 6 and 7). *p = 0.0154 (unpaired t test). D, Long-term stability of spines that enlarge at 5 h after TBS (black circles) compared with nonenlarging spines (open circle; n = 6; 265 spines analyzed; 58 enlarging spines; two-way ANOVA with Bonferroni post test). E, Same but in GluN3A-transfected neurons. Note the drastic drop of stability of enlarging spines observed after 24 h (black circles, n = 4; 126 spines analyzed, 32 enlarged spines; two-way ANOVA with Bonferroni post test). **p < 0.01. ns, Not significant.
Discussion
Previous studies suggested a very specific function of GluN3A subunits during postnatal brain development. Inclusion of GluN3A into NMDAR channels yields nonconventional receptors that are likely to modify synaptic function and plasticity (Pérez-Otaño et al., 2001; Tong et al., 2008). Consistent with these altered properties, continuous expression of GluN3A in a reversible transgenic mouse model resulted in alterations of LTP induction and memory deficits that could be rescued by suppressing transgene expression (Roberts et al., 2009). The functional alterations were associated with structural modifications of synapses, such as a decreased spine densities and a reduction in mature, mushroom type of spines (Roberts et al., 2009). Knocking out GluN3A yielded the opposite phenotype, increasing spine numbers and accelerating the expression of molecular markers of synaptic maturation (Das et al., 1998; Henson et al., 2012). These observations raised the possibility that GluN3A could regulate the functional maturation of synapses and possibly limit the number of synapses able to undergo potentiation and stabilization during critical periods of development. The present data bring strong support to this interpretation by providing direct evidence for the involvement of GluN3A in spine-pruning mechanisms.
During postnatal development, continuous synaptic rearrangements are critical for the formation of functional neuronal networks. Experiments using time-lapse confocal imaging in living mice have shown that excitatory synapses are characterized by a high level of turnover that is developmentally regulated and considerably reduced in adulthood (Holtmaat et al., 2005; Zuo et al., 2005). Synaptic rearrangements are strongly affected by patterns of activity. In hippocampal slice cultures, plasticity-inducing protocols increase spine turnover and promote a selective stabilization of activated spines (De Roo et al., 2008b). In line with this observation, a motor training task in mice promoted spine formation and elimination in the motor cortex as well as stabilization of selective populations of spines (Xu et al., 2009; Yang et al., 2009). These synaptic rearrangements have thus been interpreted as representing a structural basis for learning and memory.
It remains, however, unclear how these structural plasticity properties are regulated at the molecular level and notably what mechanisms sustain the high level of structural plasticity present during early phases of development. Our results indicate that the expression of GluN3A-containing NMDARs at synapses interferes with mechanisms for activity-dependent spine stabilization, decreasing the mean lifetime of spine synapses and promoting their elimination. This conclusion is supported by several observations. First, we used various approaches to increase the surface expression of GluN3A-containing NMDARs and all yielded a similar phenotype: increased spine elimination associated with a decreased stability of spines resulting in a global decrease in spine density. These effects were likely the result of GluN3A-containing NMDARs expressed at synapses because we could measure changes in the rectification properties of synaptic NMDAR currents indicative of lesser Mg2+ blockade, one electrophysiological signature of GluN3A subtypes, in all these conditions. Moreover, the different approaches selectively affected spine elimination and not spine growth mechanisms, although when spine elimination was particularly intense as seen for example with the endocytosis-resistant GluN3A mutant, there was also a compensatory increase in spine formation. It is important to note that these effects were not only observed under conditions of overexpression but also when preventing endocytosis of endogenous GluN3A-containing NMDARs by silencing the GluN3A-selective endocytic adaptor PACSIN1. This finding confirms a physiological role of endocytosis in regulating the synaptic expression of GluN3A subunits and modulating synapse remodeling. Second, converse effects were observed when interfering with GluN3A expression by RNA interference or by preventing association of GluN3A with the adaptor protein GIT1. Together, these data demonstrate bidirectional effects of GluN3A expression on spine elimination and stability, which could account for the synaptic alterations observed in GluN3A-deficient and overexpressing mice (Das et al., 1998; Roberts et al., 2009). Third, reexpression of GluN3A in mature tissue, at times when downregulation was almost complete, reinstated a high level of spine pruning and instability (Roberts et al., 2009). Finally, our stimulation experiments directly demonstrate a role of GluN3A in the regulation of activity-dependent spine dynamics. In these experiments, overexpression of GluN3A occluded both the activity-dependent increase in spine turnover and spine stabilization, consistent with the notion that these two mechanisms depend upon calcium fluxes through NMDARs (De Roo et al., 2008b) and that continuous expression of GluN3A can interfere with LTP induction (Roberts et al., 2009).
The exact mechanisms through which GluN3A prevents spine stabilization remain unclear. Several molecular events have been proposed to contribute to spine stability, including protein synthesis (Caroni et al., 2012), the cytoskeletal regulatory protein β-adducin (Bednarek and Caroni, 2011), the Rac1 downstream effector protein kinase PAK3 (Boda et al., 2004; Dubos et al., 2012), which regulates the actin cytoskeleton, and the adhesion molecule N-cadherin (Mendez et al., 2010), also strongly associated with the spine cytoskeleton. Interestingly, the GluN3A subunit interacts with protein phosphatase 2A (Chan and Sucher, 2001; Ma and Sucher, 2004), which is implicated in LTD (Thiels et al., 1998), with the small GTPase Ras homolog enriched in brain (Rheb) (Sucher et al., 2010), an activator of the mTOR signaling complex, and with GIT1, a postsynaptic scaffold that regulates local Rac1/PAK/actin signaling (Fiuza et al., 2013). Through these interactions, GluN3A could not only reduce the calcium fluxes that are required for spine stabilization but also affect signaling pathways implicated in the regulation of protein synthesis or cytoskeleton reorganization (Fiuza et al., 2013). Our observation that expression of a GluN3A mutant lacking the intracellular binding site for GIT1 does not reproduce the phenotype of GluN3A overexpression suggests that actin signaling mechanisms are implicated in the effects of GluN3A expression on spine dynamics.
The ability of GluN3A to destabilize spines uncovered here could have important clinical implications. In Huntington's disease, disruption of PACSIN1 function drives reexpression of GluN3A-containing NMDARs in striatal neurons (Marco et al., 2013), leading to aberrant synapse loss and likely contributing to the degeneration of neurons. Altered GluN3A expression also occurs in schizophrenia and mood disorders, where defects in pruning are thought to play a major role (Mueller and Meador-Woodruff, 2004). Understanding whether and how GluN3A mediates these effects could therefore be of primary interest.
Together, the present study provides strong evidence that a main consequence of GluN3A expression at synapses is to prevent their stabilization through activity-dependent mechanisms, accounting for GluN3A effects on spine dynamics. This results in reduced spine lifetimes and enhanced pruning, which could underlie the spine loss in overexpressing mouse models or disease conditions linked to elevated GluN3A (Roberts et al., 2009; Marco et al., 2013). The restrictive temporal expression of GluN3A might represent a key mechanism to prevent an unwanted formation of persistent connections during the peak of synapse development and thus contribute to regulate the magnitude and timing of synapse maturation. This could ensure the exquisite specificity in the organization of neural circuits that supports brain functioning.
Footnotes
This work was supported by Spanish Ministry of Science Grants SAF2010–20636 and CSD2008–00005 to I.P.-O., a NARSAD Independent Investigator Award to I.P.-O., the UTE project Centro de Investigacion en Medicina Aplicada, and Swiss National Science Foundation Grant 310030B-144080 to D.M.
The authors declare no competing financial interests.
References
- Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- Bednarek E, Caroni P. β-Adducin is required for stable assembly of new synapses and improved memory upon environmental enrichment. Neuron. 2011;69:1132–1146. doi: 10.1016/j.neuron.2011.02.034. [DOI] [PubMed] [Google Scholar]
- Boda B, Alberi S, Nikonenko I, Node-Langlois R, Jourdain P, Moosmayer M, Parisi-Jourdain L, Muller D. The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J Neurosci. 2004;24:10816–10825. doi: 10.1523/JNEUROSCI.2931-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Donato F, Muller D. Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci. 2012;13:478–490. doi: 10.1038/nrn3258. [DOI] [PubMed] [Google Scholar]
- Chan SF, Sucher NJ. An NMDA receptor signaling complex with protein phosphatase 2A. J Neurosci. 2001;21:7985–7992. doi: 10.1523/JNEUROSCI.21-20-07985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Marco S, Brooks IM, Zandueta A, Rao Y, Haucke V, Wesseling JF, Tavalin SJ, Pérez-Otaño I. Tyrosine phosphorylation regulates the endocytosis and surface expression of GluN3A-containing NMDA receptors. J Neurosci. 2013;33:4151–4164. doi: 10.1523/JNEUROSCI.2721-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Sasaki YF, Rothe T, Premkumar LS, Takasu M, Crandall JE, Dikkes P, Conner DA, Rayudu PV, Cheung W, Chen HS, Lipton SA, Nakanishi N. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998;393:377–381. doi: 10.1038/30748. [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Mendez P, Poglia L, Muller D. Activity-dependent PSD formation and stabilization of newly formed spines in hippocampal slice cultures. Cereb Cortex. 2008a;18:151–161. doi: 10.1093/cercor/bhm041. [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Muller D. LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol. 2008b;6:e219. doi: 10.1371/journal.pbio.0060219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubos A, Combeau G, Bernardinelli Y, Barnier JV, Hartley O, Gaertner H, Boda B, Muller D. Alteration of synaptic network dynamics by the intellectual disability protein PAK3. J Neurosci. 2012;32:519–527. doi: 10.1523/JNEUROSCI.3252-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Knudsen EI. Experience-dependent plasticity and the maturation of glutamatergic synapses. Neuron. 1998;20:1067–1071. doi: 10.1016/S0896-6273(00)80488-2. [DOI] [PubMed] [Google Scholar]
- Fiuza M, González-González I, Pérez-Otaño I. GluN3A expression restricts spine maturation via inhibition of GIT1/Rac1 signaling. Proc Natl Acad Sci U S A. 2013;110:20807–20812. doi: 10.1073/pnas.1312211110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Roberts AC, Pérez-Otaño I, Philpot BD. Influence of the NR3A subunit on NMDA receptor functions. Prog Neurobiol. 2010;91:23–37. doi: 10.1016/j.pneurobio.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Larsen RS, Lawson SN, Pérez-Otaño I, Nakanishi N, Lipton SA, Philpot BD. Genetic deletion of NR3A accelerates glutamatergic synapse maturation. PLoS One. 2012;7:e42327. doi: 10.1371/journal.pone.0042327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Ma OK, Sucher NJ. Molecular interaction of NMDA receptor subunit NR3A with protein phosphatase 2A. Neuroreport. 2004;15:1447–1450. doi: 10.1097/01.wnr.0000132773.41720.2d. [DOI] [PubMed] [Google Scholar]
- Marco S, Giralt A, Petrovic MM, Pouladi MA, Martínez-Turrillas R, Martínez-Hernández J, Kaltenbach LS, Torres-Peraza J, Graham RK, Watanabe M, Luján R, Nakanishi N, Lipton SA, Lo DC, Hayden MR, Alberch J, Wesseling JF, Pérez-Otaño I. Suppressing aberrant GluN3A expression rescues synaptic and behavioral impairments in Huntington's disease models. Nat Med. 2013;19:1030–1038. doi: 10.1038/nm.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez P, De Roo M, Poglia L, Klauser P, Muller D. N-cadherin mediates plasticity-induced long-term spine stabilization. J Cell Biol. 2010;189:589–600. doi: 10.1083/jcb.201003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller HT, Meador-Woodruff JH. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr Res. 2004;71:361–370. doi: 10.1016/j.schres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Pérez-Otaño I, Schulteis CT, Contractor A, Lipton SA, Trimmer JS, Sucher NJ, Heinemann SF. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci. 2001;21:1228–1237. doi: 10.1523/JNEUROSCI.21-04-01228.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Otaño I, Luján R, Tavalin SJ, Plomann M, Modregger J, Liu XB, Jones EG, Heinemann SF, Lo DC, Ehlers MD. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat Neurosci. 2006;9:611–621. doi: 10.1038/nn1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AC, Díez-García J, Rodriguiz RM, López IP, Luján R, Martínez-Turrillas R, Picó E, Henson MA, Bernardo DR, Jarrett TM, Clendeninn DJ, López-Mascaraque L, Feng G, Lo DC, Wesseling JF, Wetsel WC, Philpot BD, Pérez-Otaño I. Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron. 2009;63:342–356. doi: 10.1016/j.neuron.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YF, Rothe T, Premkumar LS, Das S, Cui J, Talantova MV, Wong HK, Gong X, Chan SF, Zhang D, Nakanishi N, Sucher NJ, Lipton SA. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. J Neurophysiol. 2002;87:2052–2063. doi: 10.1152/jn.00531.2001. [DOI] [PubMed] [Google Scholar]
- Sproul A, Steele SL, Thai TL, Yu S, Klein JD, Sands JM, Bell PD. N-Methyl-d-aspartate receptor subunit NR3a expression and function in principal cells of the collecting duct. Am J Physiol Renal Physiol. 2011;301:F44–F54. doi: 10.1152/ajprenal.00666.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-M. [DOI] [PubMed] [Google Scholar]
- Sucher NJ, Yu E, Chan SF, Miri M, Lee BJ, Xiao B, Worley PF, Jensen FE. Association of the small GTPase Rheb with the NMDA receptor subunit NR3A. Neurosignals. 2010;18:203–209. doi: 10.1159/000322206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Norman ED, Barrionuevo G, Klann E. Transient and persistent increases in protein phosphatase activity during long-term depression in the adult hippocampus in vivo. Neuroscience. 1998;86:1023–1029. doi: 10.1016/S0306-4522(98)00135-3. [DOI] [PubMed] [Google Scholar]
- Tong G, Takahashi H, Tu S, Shin Y, Talantova M, Zago W, Xia P, Nie Z, Goetz T, Zhang D, Lipton SA, Nakanishi N. Modulation of NMDA receptor properties and synaptic transmission by the NR3A subunit in mouse hippocampal and cerebrocortical neurons. J Neurophysiol. 2008;99:122–132. doi: 10.1152/jn.01044.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Mameli M, O'Connor EC, Dey PN, Verpelli C, Sala C, Pérez-Otaño I, Lüscher C, Bellone C. Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron. 2013;80:1025–1038. doi: 10.1016/j.neuron.2013.07.050. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005;46:181–189. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]