

Abstract
Relatively few studies have examined plasticity of inhibitory neuronal networks following stroke in vivo, primarily due to the inability to selectively monitor inhibition. We assessed the structure of parvalbumin (PV) interneurons during a 5 min period of global ischemia and reperfusion in mice, which mimicked cerebral ischemia during cardiac arrest or forms of transient ischemic attack. The dendritic structure of PV-neurons in cortical superficial layers was rapidly swollen and beaded during global ischemia, but recovered within 5–10 min following reperfusion. Using optogenetics and a multichannel optrode, we investigated the function of PV-neurons in mouse forelimb somatosensory cortex. We demonstrated pharmacologically that PV-channelrhodopsin-2 (ChR2) stimulation evoked activation in layer IV/V, which resulted in rapid current sinks mediated by photocurrent and action potentials (a measure of PV-neuron excitability), which was then followed by current sources mediated by network GABAergic synaptic activity. During ischemic depolarization, the PV-ChR2-evoked current sinks (excitability) were suppressed, but recovered rapidly following reperfusion concurrent with repolarization of the DC-EEG. In contrast, the current sources reflecting GABAergic synaptic network activity recovered slowly and incompletely, and was coincident with the partial recovery of the forepaw stimulation-evoked current sinks in layer IV/V 30 min post reperfusion. Our in vivo data suggest that the excitability of PV inhibitory neurons was suppressed during global ischemia and rapidly recovered during reperfusion. In contrast, PV-ChR2 stimulation-evoked GABAergic synaptic network activity exhibited a prolonged suppression even ∼1 h after reperfusion, which could contribute to the dysfunction of sensation and cognition following transient global ischemia.
Keywords: inhibitory synaptic transmission, optogenetics, parvalbumin neuron, reperfusion, stroke, two-photon imaging
Introduction
Global ischemia occurs during cardiac arrest and has been implicated as a complication that can occur following cardiac surgery (Block, 1999). It induces delayed neuronal death in human and animal models, particularly in the hippocampus (Zola-Morgan et al., 1986), but also can affect the cortex (Hossmann and Sato, 1970; Koh et al., 1996; Block, 1999; Chavez and LaManna, 2002; Murphy et al., 2008; S. Chen et al., 2012; Xie et al., 2013). Inhibitory interneurons only constitute ∼20% of cortical neurons, yet these interneurons are crucial in regulating the balance of excitation and inhibition (E/I), plasticity, and functional architecture of cortical circuits (Markram et al., 2004). Among interneuron subtypes, parvalbumin (PV) neurons have been recently shown to modify sensory perception (Cardin et al., 2009; Lee et al., 2012). Moreover, accumulating evidence shows that abnormality of GABA systems, particularly in altered PV expression, is common in many brain disorders, including schizophrenia, bipolar disorder, and major depression (Torrey et al., 2005), and could putatively occur in human patients following cardiac arrest- or cardiac surgery-induced global ischemia (McKhann et al., 1997; Lim et al., 2004).
Postmortem studies have shown that hippocampal PV-neurons are more resistant to global ischemia-induced neuronal cell death. This resistance was attributed to enhanced Ca2+ buffering via PV (Nitsch et al., 1989; Freund et al., 1990; Johansen et al., 1990). However, how the structure and function of PV-neurons change during and following a reversible ischemic event is not clear. Conventional electrophysiological approaches to directly characterize PV-neurons in this setting via patch-clamp electrophysiology are not technically feasible due to the recording duration required and complexities involved with using the ischemic model in vivo. In this study, we used optogenetic tools (Gradinaru et al., 2010; F. Zhang et al., 2010) and Cre-dependent transgenic mouse lines (Madisen et al., 2010; Taniguchi et al., 2011) to investigate cortical PV-neuronal dendritic structure, intrinsic excitability, and network synaptic function during global ischemia and reperfusion. We demonstrate that PV inhibitory neurons are not spared from the damaging effects of global ischemia with respect to their structure and function.
Materials and Methods
Animal and surgical procedures.
Adult male parvalbumin-Cre (PV-Cre) knockin mice (B6;129P2-Pvalbtm1(cre)Arbr/J) and Ai9 Cre reporter strain (B6;Cg-Gt(ROSA)26Dortm9(CAG-tdTomato)Hze/J) aged 2–3 months were used. Experimental protocols were approved by the University of British Columbia Animal Care Committee and conformed to the Canadian Council on Animal Care and Use Guidelines. A 3 × 3 mm cranial window was made over the somatosensory cortex. Body temperature was maintained throughout the surgical and experimental procedures at ∼37°C. In all animals the cortical surface temperature was maintained at ∼36.5°C by attaching a custom-made stainless steel head plate connected to a temperature-controlled water pump (Xie et al., 2012). Intraperitoneal injection of urethane (0.12% w/w) was used for anesthesia except for viral injection.
Global ischemia model.
Five minute common carotid arterial occlusion (CCAO) was performed as in previous studies to induce a reversible global forebrain ischemia under urethane anesthesia (S. Chen et al., 2012; Xie et al., 2013). We only considered ischemia that generated a >6 mV DC-EEG depolarization as being successful for further analysis. The PV-Cre transgenic mouse line used in this study is made within C56BL/6 background that typically lacks posterior communicating arteries, allowing bilateral CCAO to create significant forebrain ischemia (Fujii et al., 1997; Sheng et al., 1999). Reduction of blood flow (>80% of baseline level) in the somatosensory cortex during CCAO was previously confirmed by measuring red blood cell velocity in small vessels (Murphy et al., 2008), and also by assessing regional blood flow in motor and somatosensory cortices using laser speckle imaging (Xie et al., 2013) and laser Doppler flowmetry (Panahian et al., 1996). Reperfusion after 5 min of CCAO can rapidly restore flow to >75% of baseline level within 3–5 min (Panahian et al., 1996; Xie et al., 2013).
Two-photon imaging and analysis.
Two-photon (2P) excitation was performed with a Mai Tai Ti-sapphire laser and tuned to 970 nm to excite tdTomato. A median filter (radius, 1 pixel) was applied to reduce photon and photomultiplier tube noise. Detection and quantification of the fraction of damaged dendrites was performed using BlebQuant, a MATLAB-based software program that examined the first 40–60 μm maximal intensity z-projections of the cortex (S. Chen et al., 2011).
Forepaw stimulation.
To stimulate the forepaw, thin acupuncture needles were inserted into the contralateral forepaw and a 1 ms, 1 mA electrical pulse was delivered by a constant current stimulus isolator (A385; World Precision Instruments) triggered by Clampex-controlled 1322A Digidata system (Molecular Devices).
Viral injection.
A 0.5–0.8 μl AAV1.EF1a.DIO.hChR2(H134R)-EYFP.WPRE.hGH or AAV1.CAG.FLEX.tdTomato.WPRE.bGH (Penn Vector Core) was injected by a syringe infusion pump (model UMC4; World Precision Instruments) as previously reported (Xie et al., 2010) into the forelimb somatosensory cortex (∼2.3 mm lateral to the bregma, depth at 350–400 μm, speed at ∼1 nl/s) of PV-Cre mice 3–4 weeks before imaging and optrode recording (Fig. 1B). Isoflurane (1.5%) mixed with oxygen was used for anesthesia.
Figure 1.

PV-Cre-dependent expression within PV interneurons and experimental design with global ischemia and reperfusion model. A, Confocal fluorescence images of coronal sections through layer I to VI in the forepaw somatosensory cortex of PV-Cre line crossed with the ROSA-tdTomato reporter line (PV-Cre × Ai9) show overlay between PV-Cre expression and PV-antibody immunohistochemical (IHC) staining. Layer borders are indicated on the left. Ratios of PV IHC+ cells distributed through layer I to VI and tdTomato expressed cells/PV IHC+ cells are shown on the right (mean ± SEM, n = 2). B, AAV1.DIO.ChR2-eYFP or AAV1.flex.tdTomato was injected into the somatosensory cortex (350–400 μm depth) of PV-Cre mice at ∼1 nl/s using glass pipettes (tips are <25 μm in diameter) at least 3 weeks before experiments. ChR2-eYFP or tdTomato expressing cells and PV IHC are shown. Stars indicate ChR2-eYFP (and tdTomato) and PV IHC+ colocalized cells. Ratio of viral-expressing cells to PV IHC+ cells is shown on the right. C, Experimental design: one group of mice with AAV1.flex.tdTomato injection was used to perform 2P imaging of PV-neuron structure; the other group of mice with AAV1.DIO.ChR2-eYFP injection is used to perform functional study of PV-neurons with optogenetic stimulation. Both groups of mice receive a 5 min CCAO-induced global ischemia followed by reperfusion. Isc, ischemia.
EEG recording and analysis.
The methods have been described previously (S. Chen et al., 2012). Briefly, a single surface electrode was placed within 1 mm of the forelimb somatosensory cortex for recording DC-EEG. EEG signals were sampled at 1 kHz. AC-EEG was filtered into 0.3–3 Hz from DC-EEG using a Chebyshev filter.
Optrode stimulation, recording, and analysis.
To obtain information from different cortical depths, a silicon optrode (A1x16–3 mm-50–413-Op16; NeuroNexus Technologies) was used for recording LFP and multi-unit activity (MUA), and also for laser-inducing ChR2 stimulation (S. Chen et al., 2012). The optrode contained 16 recording sites (413 μm2, 0.7–1.2 MΩ impedance), arranged linearly with 50 μm spacing between each site. A 473 nm laser (IKE-473–100 OP; IkeCool) was coupled to the optrode using a fiber-optic patch cord (FC-x.x-NNC; NeuroNexus Technologies). Using coordinates obtained from an atlas of mouse cortex (Allen Institute), the optrode probe was inserted into the forelimb somatosensory representation (posterior 0.3 mm, lateral 2.0 mm to the bregma) to a depth of 800 μm to target layers I–VI. ChR2 stimulation was performed with an optrode-integrated laser fiber (105 μm diameter). The fiber tip was positioned ∼50 μm above the recording site nearest to the cortical surface. Laser power was adjusted to ∼5 mW at the fiber tip and 10 ms laser stimulation induced consistent and stable LFP responses in all recording sites. LFP signals were digitized using a portable 16-channel data acquisition system (USB-ME16-FAI-System; Multi Channel Systems). The 16-channel LFP signals were recorded and amplified (×1000), and all 16-channel LFP signals were sampled at 25 kHz. To quantify the PV-ChR2 stimulation-evoked LFP responses, the differences between baseline (averaged from 100 ms before the ChR2 stimulation) and downward peak (minimum LFP within 10–50 ms after the onset of ChR2 stimulation) were quantified as the putative depolarization of PV-neurons; the differences between the baseline and upward peak (maximum LFP within 10–50 ms after the onset of ChR2 stimulation) were quantified as the putative network hyperpolarization mediated by inhibitory synaptic transmission. To resolve MUA from LFP recording, we applied a similar analysis as shown in our previous report (S. Chen et al., 2012), using an open sources MATLAB-based program WaveClus (Quiroga et al., 2004). Briefly, an off-line bandpass filtering (600–6000 Hz, elliptic filter) was performed (Kahn et al., 2011). Automated amplitude thresholding (4–7 times SD of background noise, negative peak only) was used to detect spikes from the filtered data. Relatively large laser stimulation artifacts were removed by setting an upper bound threshold (40 times SD of background noise). Each spike was saved as 35 time point data. The spikes detected from baseline recording were averaged (reference spike) for sorting MUA (correlation coefficient to reference spike >0.9). We did not perform single-unit sorting because we found the optrode was not sensitive enough to resolve single unit, and also global ischemia changed the waveform. Only LFP signals from 400–600 μm depth (layer IV/V) were quantified for group data.
Current sources density analysis.
Recording from a 16-channel optrode was used for functional analysis. For pharmacological data, current source density (CSD) analysis was averaged from at least 30 repetitions of PV-ChR2 or forepaw electrical stimulation trials. For ischemic data, CSD analysis was averaged from 10 repetitions from baseline recording (before global ischemia), and 3 repetitions following global ischemia and reperfusion for optimized temporal resolution. Baseline correction was applied using the 50–80 ms prestimulus period as baseline. CSD analysis was performed using the MATLAB-based program CSDplotter (Pettersen et al., 2006). We applied the cubic spline iCSD method, where cortical parameters were set as follows: SD of the Gaussian filter = 0.1 mm, extracellular cortical conductivity = 0.3 S/m, top conductivity = 0.3 s/m, activity diameter = 0.5 mm. The peak amplitude of current sinks or current sources was sorted within 50 ms following PV-ChR2 or forepaw stimulation relative to the baseline recording (50–80 ms prestimulus). CSD data from 400 to 600 μm depth (layer IV/V) were averaged and quantified for group data.
Pharmacology.
Ten PV-ChR2-expressed mice were used for pharmacological experiments. For experiments involving GABAA receptor antagonist, gabazine (1 μm; Sigma, n = 5) or TTX (30 μm; Sigma, n = 5) was dissolved in physiological saline solution and applied to the craniotomy (Harrison et al., 2012). The compounds were allowed to incubate for ∼20 min before starting optrode stimulation and recording.
Transcardial perfusion and immunohistochemistry.
Mice were transcardially perfused with PBS followed by chilled 4% PFA in PBS. Coronal brain sections (50 μm thickness) were cut on a vibratome (Leica VT1000S), and were incubated with mouse anti-parvalbumin antibody (1:500; Sigma) overnight, and subsequently incubated with Alexa 488 or 594 anti-mouse (1:1000; Invitrogen) for 2 h. Images were acquired using confocal microscopy (Zeiss LSM510).
Statistical analysis.
All values were normalized to the percentage of pre-CCAO value (100%) except DC-EEG. A one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc tests was used to compare differences in dendritic damage, LFP peaks, and CSD results at each time point to pre-CCAO baselines in experimental groups. The other comparisons were analyzed with repeated measures (RM)-ANOVA. Significance was set at p < 0.05.
Results
AAV viral transduced selective expression of tdTomato and ChR2-EYFP in PV-Cre mice
Our initial work involved confirming that the ChR2 expression pattern was associated with PV inhibitory neurons. We visualized the Cre expression pattern of PV-Cre mice by crossing them with a Rosa26 tdTomato reporter line, leading to expression patterns expected for Cre-dependent AAV vectors (Kuhlman and Huang, 2008). Immunoreactivity with PV antibody almost exclusively labeled PV-Cre cells (Fig. 1A). PV-neurons were relatively enriched in layer IV and V of the forepaw somatosensory cortex (Fig. 1A). To allow a selective investigation of the structure and function of PV-neurons, we injected Cre-inducible AAV vector containing tdTomato or ChR2-eYFP into the somatosensory cortex of PV-Cre mice (Fig. 1B). The virally transduced expression of tdTomato or ChR2-eYFP spanned from layer I to VI and colocalizes with PV immunostaining (Fig. 1B).
Reversible loss of PV-neuron dendritic structure following global ischemia and reperfusion
We investigated the impact of reversible global ischemia on the dendritic structure of PV-neuron dendrites located within the first 100 μm of somatosensory cortex (Fig. 1C). In contrast to layer 5 excitatory neurons (Brown et al., 2007), PV-neurons exhibited few or no clear spines on their dendrites (Fig. 2Aiia). Therefore, we focused on potential structural alterations apparent within the dendrites. All dendrites were imaged using ∼40 to 60 μm z-stacks within layer 1 taken at ∼2 min intervals. Cortical surface temperature was maintained at ∼36.5°C using a temperature-controlled thermoregulation system (Tran et al., 2012; Xie et al., 2012). Six of 10 animals exhibited >6 mV DC-EEG depolarization (Fig. 2B) following induction of global ischemia; and this DC shift was associated with >90% suppression of spontaneous EEG power (filtered into 0.3–3 Hz; Fig. 2B). Damage to dendritic structure was assessed using an automated software procedure we previously developed termed BlebQuant (S. Chen et al., 2011). CCAO induced dendritic damage to PV-neurons within 4 min (77.4 ± 7.6% blebbed dendrites), which correlated with the appearance of ischemic depolarization (DC-EEG). The damage consisted of regular dendritic swellings or blebs similar to reports from pyramidal neurons (S. Zhang et al., 2005; Andrew et al., 2007; Takano et al., 2007; Murphy et al., 2008; Mostany et al., 2010; Risher et al., 2010; Mostany and Portera-Cailliau, 2011). Reperfusion rapidly recovered the dendritic structure to near the baseline levels within 10 min (1.6 ± 0.9% blebbed dendrites) and remained stable for the following hour (0.2 ± 0.1% blebbed dendrites at 1 h). These results indicated that the dendritic structure of PV-neurons is vulnerable to ischemic injury, but this apparent injury is reversible following reperfusion.
Figure 2.

Global ischemia induces dendritic damage to PV-neurons (inhibitory interneurons) which rapidly recovers following reperfusion. Ai, 2P imaging (0–60 μm in depth) following 5 min global ischemia and reperfusion. The white-boxed area on the top left corresponds to a region in higher magnification illustrations below (Aii). B, Changes of average DC potential and spontaneous EEG power (0.3–3 Hz) are shown (mean ± SEM, n = 6). C, Average percentage of damaged dendrites during global ischemia and reperfusion are shown (mean ± SEM, *p < 0.05, ***p < 0.001, one-way ANOVA followed by Bonferroni's post-test, compared with pre-CCAO group, n = 6). Isc, ischemia; Rep, reperfusion.
Pharmacological characterization of PV-ChR2 stimulation-evoked electrical responses in the somatosensory cortex
In pilot studies, we were unable to reliably detect light-induced PV-ChR2 stimulation-evoked responses as EEG signals using a surface silver wire (data not shown). However, to achieve more sensitive recordings and to also provide cortical laminar information regarding PV-ChR2 stimulation-evoked responses, we used a 16-channel optrode. The optrode permitted stimulation of ChR2-expressed PV-neurons from a laser fiber positioned ∼50 μm above the cortical surface and simultaneously recording of the light-evoked LFP responses throughout the layers of the somatosensory cortex. At alternate time intervals we recorded the contralateral forepaw electrical stimulation-evoked LFP (Fig. 3A). Noticeably, there were larger PV-ChR2 stimulation-evoked LFP signals in layer IV/V compared with shallower layers, even with much less laser exposure at deeper layers (Fig. 3B).
Figure 3.
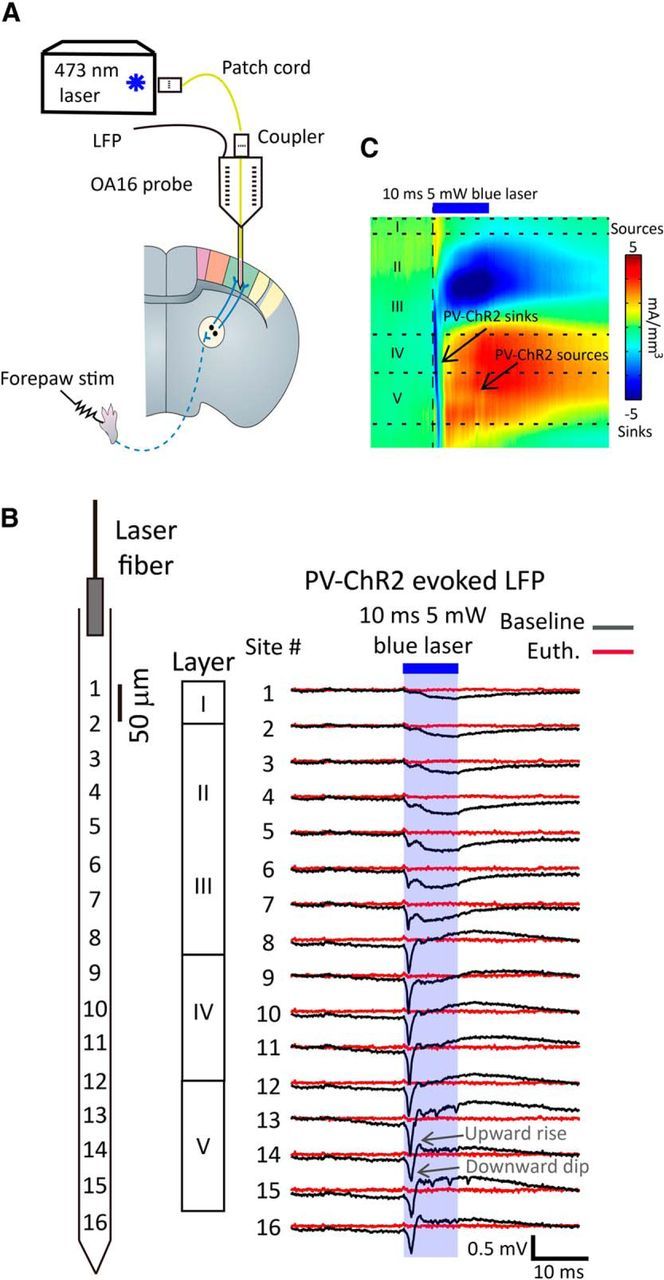
Sixteen-channel optrode laminar recording of PV-ChR2-mediated local field potential signals and CSD analysis in the cortex. A, A 16-channel optrode (OA16; NeuroNexus Technologies) with an optical fiber attached at ∼50 μm above the first recording site was placed into the somatosensory cortex of AAV.DIO.ChR2-injected PV-Cre mice for local field potential recording and ChR2 stimulation. The contralateral forepaw was electrically stimulated to induce sensory cortical responses for comparison. B, Left, A diagram shows the distribution of each recording site of the optrode in the somatosensory cortex. Right, 10 ms, 5 mW of blue laser PV-ChR2 stimulation-evoked LFP signals corresponding to laminar layers before (black) and after being killed (red). C, CSD analysis is performed with cubic spline iCSD method based on 16-channel LFP signals using the MATLAB-based program CSDplotter. Euth, euthanasia.
To further confirm the localization of PV-ChR2 and forepaw stimulation-evoked responses and eliminate far-field signal contribution, we performed CSD analysis on 16-channel LFP data. CSD analysis showed that a 10 ms, 5 mW blue laser stimulation induced rapid current sinks followed by current sources in layer IV/V (Fig. 3C), appearing as a downward dip followed by a rapid upward rise in LFP signals (Fig. 3B). In theory, these were typical responses generated from photocurrent and action potential-induced current influx (sinks) followed by an inhibitory synaptic activation-mediated current outflow (sources) in the local network (Mitzdorf, 1985). A 1 mA, 1 ms forepaw electrical stimulation induced typical current sinks in layer IV that rapidly extended toward layers II/III and upper layer V (Fig. 4C), similar to previously reported findings in the somatosensory cortex (Mégevand et al., 2009).
Figure 4.
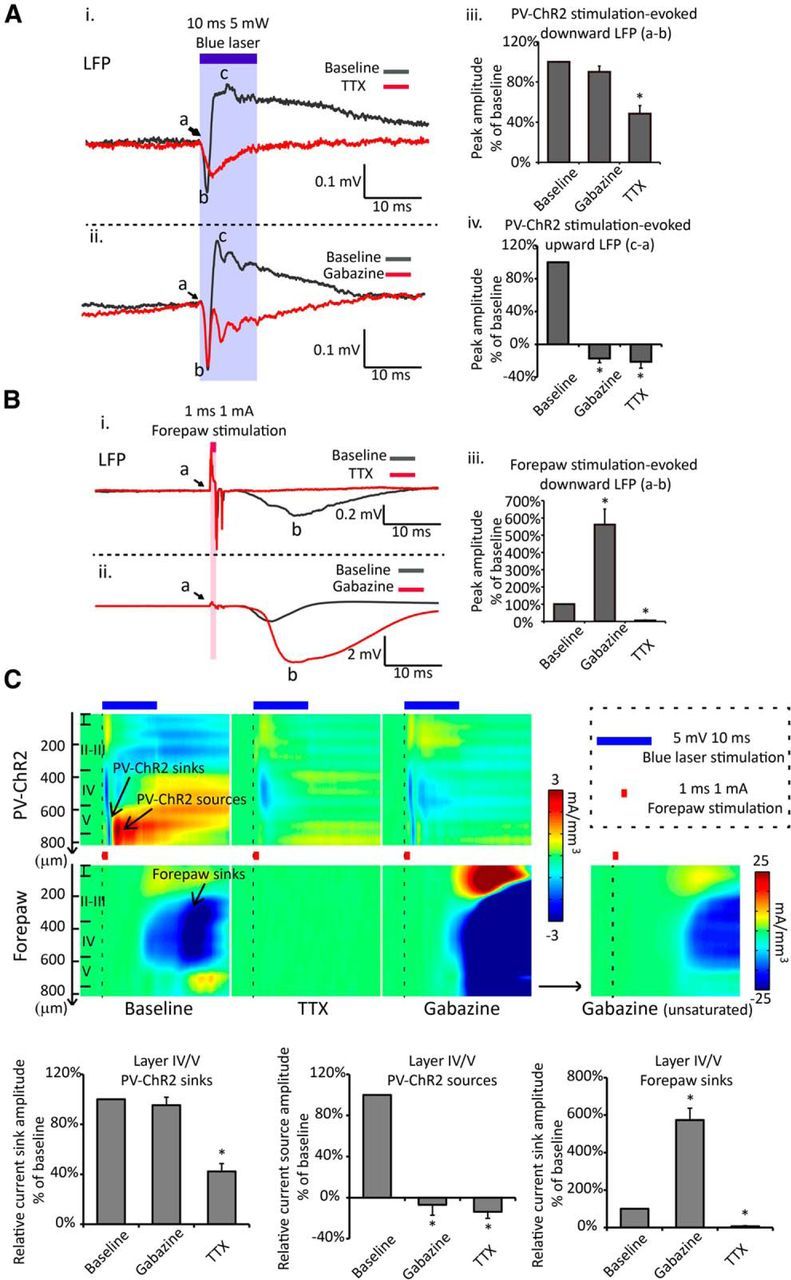
Pharmacological definition of PV-ChR2 stimulation-evoked LFP signals. A, Cortical application of 30 μm TTX partially suppresses the peak value of 10 ms, 5 mW light-evoked PV-ChR2 downward LFP signals [Ai, Aiii; peak value is calculated as the difference between the downward peak (b) and baseline (a)] and abolishes the light-evoked upward LFP signals (Ai, Aiv, c−a; c, upward peak; a, baseline value) in layer IV/V. Cortical application of 1 μm gabazine depresses the PV-ChR2 stimulation-evoked upward LFP signals (Aii, Aiv), while it does not significantly change the peak value of the downward LFP signals (Aii, Aiii) in layer IV/V. B, The same application of TTX or gabazine abolishes (Ai, Aiii) or amplifies (Aii, Aiii) the peak value of 1 ms, 1–1.5 mA forepaw stimulation-evoked LFP signals (b−a) at the same recording site, respectively, confirming the drug action deep input layers. C, CSD analysis of the PV-ChR2 stimulation-evoked and the forepaw stimulation-evoked responses through layer I to V confirms the conclusion from LFP measurements without potential far-field signal contamination. TTX partially suppresses the PV-ChR2 stimulation-evoked current sinks and abolishes the PV-ChR2 stimulation-evoked current sources in layer IV/V; gabazine abolishes the PV-ChR2 stimulation-evoked current sources in layer IV/V. All numbers are normalized to the value before drug application, mean ± SEM, *p < 0.001, RM-ANOVA, n = 5 for each drug. All traces in A–C are averaged from 20 to 30 repetitions. Some sweeps with occasional seizure-like depolarization in gabazine experiment were removed from the analysis.
Cortical surface bath application of 30 μm TTX suppressed the peak amplitude of layer IV/V PV-ChR2 stimulation-evoked current sinks to 42.3 ± 14.2% of baseline level (downward LFP to 48.5 ± 17.9% of baseline level) and abolished the current sources/upward LFP (Fig. 4Ai,Aiii,Aiv,C). Bathing with 1 μm GABAA receptor antagonist gabazine abolished the peak value of the current sources/upward LFP without affecting the peak value of the current sinks/downward LFP (Fig. 4Aii–iv,C). The actions of TTX and gabazine within layer IV/V were confirmed by the blockade and amplification of forepaw stimulation-evoked current sinks/LFP signals, respectively (Fig. 4Bi–iii,C). We suggest that the current sinks/downward LFP reflected the excitability of PV-neurons (ChR2 channel photocurrent and PV-neuron action potential), while the current sources/upward LFP reflected a GABAergic inhibitory synaptic network activity upon PV-neurons stimulation.
ChR2-based functional assessment of PV-neurons in the cortex following a 5 min transient global ischemia and reperfusion
Pharmacologically defined excitability (current sinks/downward LFP) and GABAergic synaptic network activity upon PV-ChR2 stimulation (current sources/upward LFP) were used to characterize the function of PV-neurons in layer IV/V following a 5 min global ischemia and reperfusion in the forelimb somatosensory cortex in mice. We considered an animal that has >6 mV DC-EEG shift following the onset of global ischemia as successful global ischemia (Fig. 5A, n = 5). AC-EEG power (0.3–3 Hz) exhibited faster suppression during global ischemia and delayed recovery compared with DC-EEG shift following reperfusion (Fig. 5A), which was consistent with our previous reports (Murphy et al., 2008; S. Chen et al., 2012; Xie et al., 2013) and structural experiments (Fig. 2B). We recorded 10 ms, 5 mW blue laser-stimulated PV-ChR2-evoked LFP responses as well as 1 ms, 1–1.5 mA electrical stimulation of contralateral forepaw-evoked LFP responses throughout cortical layers (0–800 μm) before, during a 5 min global ischemia, and following reperfusion (Fig. 1C) and then performed CSD analysis (Fig. 5B,D). MUA was extracted from raw LFP signals, where spiking activity was counted over a 150 ms period following the onset of electrical forepaw stimulation and over a 70 ms period following the onset of blue laser stimulation (Fig. 5Ci–iii). We considered the short-latency PV-ChR2 stimulation-evoked MUA as mostly PV-neuron single-unit activity, given that the stimulation of the inhibitory neurons should have suppressed spontaneous MUA. This proposal is supported by a silent period following the laser stimulation where few spikes were observed (Fig. 5Cii, baseline). Peak values of current sinks (downward LFP) and current sources (upward LFP) from the prestimulation values were measured. Five minute global ischemia greatly suppressed all forms of activity in layer IV/V evoked by PV-ChR2 stimulation or forepaw stimulation. Activity gradually recovered within ∼1 h following reperfusion (Fig. 5E), suggesting that the function of PV inhibitory interneurons was not spared from the effect of ischemia, but can be partially restored by introducing reperfusion.
Figure 5.
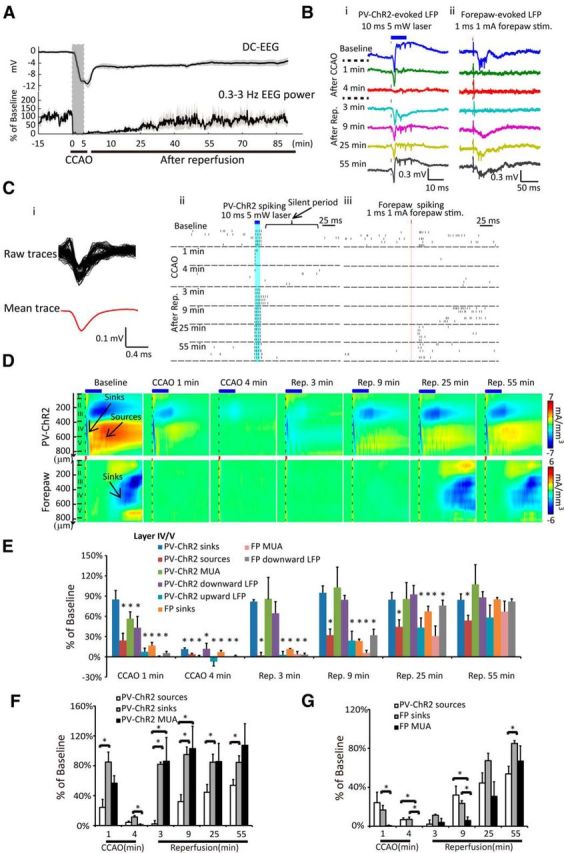
Reperfusion recovers layer IV/V PV-neuron excitability rapidly, but recovers GABAergic synaptic network activity more slowly and incompletely from a 5 min global ischemia-induced suppression. A, Changes of average DC potential, spontaneous EEG power (0.3–3 Hz) are shown (mean ± SEM, n = 5). Bi, 10 ms, 5 mW PV-ChR2 stimulation-evoked LFP responses. Bii, 1 ms, 1 mA forepaw electrical stimulation-evoked LFP responses in layer IV/V before and after global ischemia and reperfusion. Traces represent single LFP signals at various indicated time points. Ci, Averaging spike waveform for multi-unit spike sorting. Cii, Ciii, Raster plots show the PV-ChR2 stimulation-evoked spiking activities and the forepaw stimulation-evoked spiking activities. Five trials are shown at each time point. D, CSD shows the PV-ChR2 stimulation and the forepaw stimulation-evoked current sinks and sources through layer I to V at various indicated time points. E, Changes of all the forms of the PV-ChR2 stimulation and the forepaw stimulation-evoked responses in layer IV/V at various indicated time points corresponding to the 5 min global ischemia and reperfusion, compared with the baseline level. All numbers are normalized to the value before global ischemia (100%), mean ± SEM, n = 5, *p < 0.05, RM-ANOVA. F, Group comparison of PV-ChR2 simulation-evoked current sinks, current sources, and time-locked spiking number (MUA) in layer IV/V at various time points is shown. All numbers are normalized to the value before global ischemia (100%), mean ± SEM, n = 5, *p < 0.05, RM-ANOVA. G, Group comparison of PV-ChR2 simulation-evoked current sources, forepaw stimulation-evoked current sinks, and forepaw stimulation-evoked spiking number (MUA) in layer IV/V. All numbers are normalized to the value before global ischemia (100%), mean ± SEM, n = 5, *p < 0.05, RM-ANOVA. Stim, stimulation.
We then compared the PV-ChR2 stimulation-evoked current sources, sinks, and MUA to further determine the relationship between the changes of putative excitability of PV-neurons (current sinks, time-locked MUA) and GABAergic synaptic network activity upon PV-neuron stimulation (current sources, upward LFP). One minute after the onset of global ischemia, the PV-ChR2 stimulation-evoked current sources were suppressed more rapidly than the current sinks (24.2 ± 10.7 vs 84.9 ± 13.4% of baseline, p < 0.01). Global ischemia further depressed both the current sources and sinks (putative GABAergic synaptic network activity and PV-neurons excitability measure respectively) to <10% of baseline level at 4 min. Approximately 3 min after the induction of reperfusion, the excitability of PV-neurons (current sinks) started to recover, whereas the inhibitory synaptic transmission-mediated network activity (current sources) exhibited prolonged suppression (84.9 ± 13.4% of baseline vs 2.1 ± 4.3%, p < 0.001). The mismatch between the recovery of current sinks and sources was present during the first hour after reperfusion (Fig. 5F). Interestingly, the stimulation time-locked MUA of PV-neurons showed a slower suppression and faster recovery compared with the current sources during global ischemia and immediately following reperfusion. This suggested a period that PV-neurons can still generate action potentials, yet cannot fully synaptically signal surrounding neurons to generate the GABAergic synaptic network activity. Similar to PV-ChR2 simulation-evoked current sources, the forepaw stimulation-evoked current sinks and MUA were depressed rapidly following the induction of global ischemia and recovered more slowly and incompletely following reperfusion (Fig. 5G), consistent with our previous work with LFP analysis (S. Chen et al., 2012). Noticeably, the forepaw stimulation-evoked MUA in layer IV/V depressed more rapidly during global ischemia, and recovered more slowly within 9 min following reperfusion, compared with the current sinks (Fig. 5G). It indicated a period that the cells in layer IV/V had current influx, but were less likely to generate action potentials following sensory stimulation. There were no differences between the suppression and recovery of PV-ChR2 stimulation-evoked GABAergic synaptic activity and the forepaw stimulation-evoked responses within the first 0.5 h following reperfusion (p > 0.05; Fig. 5G). The differences between PV-ChR2 stimulation-evoked current sources and forepaw stimulation-evoked current sinks at 55 min following reperfusion (Fig. 5G) may suggest a mismatch of layer IV/V network activity that is dependent on GABAergic synaptic transmission (Fig. 4C) and glutamatergic synaptic transmission (S. Chen et al., 2012).
Discussion
In the present study, we have selectively investigated the structure and network function of PV-neurons in mouse forelimb somatosensory cortex during acute global ischemia and reperfusion using optogenetic tools, in vivo imaging, and optrode recording. Despite having structurally intact dendrites and excitable membranes, GABAergic synaptic transmission-dependent network activity was not functionally recovered within the first hour after reperfusion (Fig. 6).
Figure 6.

Summary figure: coincident sensitivity of pyramidal and interneurons to the acute effects of global ischemia and reperfusion. Five minute global ischemia induces the loss of apical dendritic integrity and suppression of membrane excitability of both excitatory neuron and PV inhibitory neuron in the cortex, and suppresses the GABAergic synaptic network activity in the cortex and forepaw stimulation-evoked cortical response. Reperfusion rapidly recovers the dendritic structure and membrane excitability of excitatory and PV inhibitory neuron, whereas the GABAergic synaptic network activity and forepaw stimulation-evoked response recover slowly and incompletely. Gray drawing shows neuronal structure. Black traces show electrophysiological signals.
Inhibitory neurons are not spared from dendritic damage during global ischemia
Previous studies in both humans and rodents suggest that global ischemia-induced neuronal loss is enriched in the hippocampal CA1 region, and can result in impaired learning and memory (Zola-Morgan et al., 1986; Block, 1999). However, it has been shown that the cortex can also be impaired by short periods of global ischemia despite lacking widespread neuronal death; and this could contribute to cognitive deficits (McKhann et al., 1997; Lim et al., 2004). We have previously shown that a transient global ischemia can induce a reversible damage of the apical dendritic structure of layer V excitatory neurons during ischemic depolarization (Murphy et al., 2008). Our current findings suggest that PV-neurons are as vulnerable as pyramidal neurons to the acute insult of ischemia on dendritic structure. This vulnerability is apparent despite previous reports indicating enhanced resistance of PV-neurons to ischemia-induced neuronal death, which has been attributed to their enhanced Ca2+ buffering via parvalbumin (Nitsch et al., 1989; Freund et al., 1990; Johansen et al., 1990; Tortosa and Ferrer, 1993). Conceivably, this enhanced Ca2+ buffering is insufficient against the initial ischemic depolarization-triggered massive Ca2+ elevation that can be independent of direct NMDA receptor-mediated Ca2+ influx (Jarvis et al., 2001; Anderson et al., 2005; Thompson et al., 2006; Murphy et al., 2008). It is also possible that some changes to PV-neuron structure may be indirectly caused by ischemic depolarization-induced compression due to swollen components of adjacent cells (Andrew et al., 2007) such as glia (Risher et al., 2012).
Dissecting the cortical laminar electrical signals upon PV-ChR2 stimulation
Compared with the conventional techniques used to measure excitability and synaptic function of select neurons, such as patch-clamp electrophysiology (Pang et al., 2002; Runyan et al., 2010), optogenetic-based measurements have the advantage of potentially being less invasive and able to sample potentially more locations. However, we acknowledge that our approach using extracellular electrophysiological recording of optogenetic stimulation-evoked responses may not be a specific measurement restricted to light-activated neurons, as other synaptically active cells are within the network. In support of the method, we pharmacologically show that the PV-ChR2 stimulation induces rapid current sinks followed by current sources in layer IV/V that were dependent on PV-neuron depolarization and GABAergic inhibitory synaptic transmission-mediated network activity, respectively. It is unclear why the PV-ChR2 simulation-evoked current sinks and sources were localized in relatively deep layers, despite these layers receiving much less illumination from the laser localized above the cortical surface. This might be due to a relatively higher density of PV-neurons in layer IV/V (Fig. 1A; Ren et al., 1992; Pfeffer et al., 2013). It is also possible that the anesthesia suppresses neuronal activity in superficial layers (Petersen et al., 2003; Constantinople and Bruno, 2011; Q. Chen et al., 2012).
Mechanisms of prolonged loss of GABAergic synaptic network activity following reperfusion
In our study, the reduction of PV-neuron excitability during and immediately following ischemic depolarization is likely related to a loss of membrane potential and depolarization block of PV-neurons (Hossmann, 1971). The observed persistent suppression of GABAergic network activity (evoked by PV-ChR2 stimulation) following reperfusion is consistent with previous reports from rat hippocampal brain slices (Zhan et al., 2006), and may in part be mediated by a depression of presynaptic release caused by adenosine receptor activation (Pang et al., 2002; Ilie et al., 2006; Z. Chen et al., 2014). Alternatively, ischemia-induced internalization or downregulation of postsynaptic GABAergic receptors (Alicke and Schwartz-Bloom, 1995; Qü et al., 1998) is a postsynaptic mechanism for loss of inhibitory synaptic activity. Moreover, reversal of GABA transporter following ischemia (Allen et al., 2004) could deplete the sources of presynaptic exocytosis and saturate the postsynaptic receptors to suppress generation of phasic GABA activity.
Implications of prolonged deficits in PV-neuron stimulation-dependent GABAergic synaptic transmission following reperfusion on cognition and functional recovery
Previous evidence has shown that focal stroke can cause sustained alterations in the E/I balance in peri-infarct cortex (Buchkremer-Ratzmann and Witte, 1997; Clarkson et al., 2010, 2011; Carmichael, 2012). Restoring the E/I balance by blocking tonic GABA currents (Clarkson et al., 2010) or stimulating AMPA-type glutamate receptors (Clarkson et al., 2011) promotes functional recovery after focal stroke. Despite the fact that inhibitory neuron-mediated phasic GABA current is not the predominant inhibitory conductance source in the cortex (tonic currents can be greater; Carmichael, 2012), it could still be critical for stroke recovery since it can regulate brain states (Cardin et al., 2009) and sensory processing that leads to perception (Lee et al., 2012; Wilson et al., 2012; Pfeffer et al., 2013). There is no direct in vivo evidence about whether global ischemia and reperfusion could result in E/I imbalance. Previous work using brain slice electrophysiology has shown that 4 min of in vitro ischemia and reperfusion, which mimics in vivo global ischemia and reperfusion model, permanently impaired the excitability and GABAergic synaptic transmission of cortical inhibitory neurons. Interestingly, this manipulation made principal neurons more excitable during reperfusion (Wang, 2003). Our in vivo results have shown that the structure and excitability of both cortical pyramidal neurons and PV inhibitory neurons recovered concurrently with the repolarization of the recorded EEG during reperfusion after stroke (Murphy et al., 2008; S. Chen et al., 2012; Xie et al., 2013), whereas PV-neuron stimulation-dependent GABAergic synaptic network activity exhibited a prolonged suppression at least over the first hour that was assessed. Our work assessing sensory responses suggests excitatory circuits also experience a persistent suppression, but were less impaired than the GABAergic synaptic network activity upon PV-ChR2 stimulation 1 h after reperfusion. We propose that the incomplete recovery of GABAergic synaptic transmission could contribute to E/I imbalance and lead to the deficits of sensory and motor processing in the cortex (Lesnick et al., 1984; S. Chen et al., 2012; Xie et al., 2013). While we mention changes in E/I balance, it is also important to remember that these changes are likely to occur in the background of profoundly suppressed activity that occurs in the days to weeks after stroke (Krakauer et al., 2012). These mismatches may contribute to cognitive dysfunction following a transient global ischemia (Nunn and Hodges, 1994; McKhann et al., 1997; Lim et al., 2004). Optogenetic-based chronic recordings in combination with behavioral assessments are needed to further explore these hypotheses in the future.
In this study, we have developed a novel methodology to assess the structure and network function of a particular inhibitory neuron population in vivo, following a transient ischemic episode. This method can be applied to chronically study changes within inhibitory neuronal networks during other brain diseases with a chronic imaging window (Holtmaat et al., 2009; Drew et al., 2010; Yang et al., 2010) or optrode implantation (Gradinaru et al., 2010; F. Zhang et al., 2010). With the development of multicolor optogenetic techniques (Tye and Deisseroth, 2012; Lin et al., 2013; Klapoetke et al., 2014), it would be possible to monitor the function of different neuronal subtypes in the same mouse following stroke. Moreover, our approach can also be used to test potential stimulation paradigms that target inhibitory neuron networks as potential treatments for stroke patients (Hummel and Cohen, 2006; Kravitz et al., 2010; Cheng et al., 2014).
Footnotes
This work was supported by a Canadian Institutes of Health Research (CIHR) Operating Grant MOP-111009 to T.H.M. and Heart and Stroke Foundation of Canada Grant in Aid. Y.X. holds a CIHR Vanier Canada Graduate Scholarship and a Four Year Fellowship from the University of British Columbia. T.H.M. is supported by a Canadian Partnership for Stroke Recovery expansion Grant. We thank the Allen Institute for Ai9 Cre reporter mouse and AAV1.CAG.FLEX.tdTomato.WPRE.bGH and Karl Deisseroth for pAAV-Ef1a-DIO hChR2 (C128S/D156A)-EYFP. We thank Cindy Jiang and Pumin Wang for assistance with surgical preparations, Jeff LeDue and Jamie Boyd for technical assistance, and Allen W. Chan for helpful discussions and comments on this manuscript.
The authors declare no competing financial interests.
References
- Alicke B, Schwartz-Bloom RD. Rapid down-regulation of GABA(A) receptors in the gerbil hippocampus following transient cerebral ischemia. J Neurochem. 1995;65:2808–2811. doi: 10.1046/j.1471-4159.1995.65062808.x. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Rossi DJ, Attwell D. Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J Neurosci. 2004;24:3837–3849. doi: 10.1523/JNEUROSCI.5539-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD. Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. J Neurophysiol. 2005;93:963–979. doi: 10.1152/jn.00654.2004. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex. 2007;17:787–802. doi: 10.1093/cercor/bhk032. [DOI] [PubMed] [Google Scholar]
- Block F. Global ischemia and behavioural deficits. Prog Neurobiol. 1999;58:279–295. doi: 10.1016/S0301-0082(98)00085-9. [DOI] [PubMed] [Google Scholar]
- Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci. 2007;27:4101–4109. doi: 10.1523/JNEUROSCI.4295-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkremer-Ratzmann I, Witte OW. Extended brain disinhibition following small photothrombotic lesions in rat frontal cortex. Neuroreport. 1997;8:519–522. doi: 10.1097/00001756-199701200-00028. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST. Brain excitability in stroke: the yin and yang of stroke progression. Arch Neurol. 2012;69:161–167. doi: 10.1001/archneurol.2011.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JC, LaManna JC. Activation of hypoxia-inducible factor-1 in the rat cerebral cortex after transient global ischemia: potential role of insulin-like growth factor-1. J Neurosci. 2002;22:8922–8931. doi: 10.1523/JNEUROSCI.22-20-08922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Cichon J, Wang W, Qiu L, Lee SJ, Campbell NR, Destefino N, Goard MJ, Fu Z, Yasuda R, Looger LL, Arenkiel BR, Gan WB, Feng G. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron. 2012;76:297–308. doi: 10.1016/j.neuron.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Tran S, Sigler A, Murphy TH. Automated and quantitative image analysis of ischemic dendritic blebbing using in vivo 2-photon microscopy data. J Neurosci Methods. 2011;195:222–231. doi: 10.1016/j.jneumeth.2010.12.018. [DOI] [PubMed] [Google Scholar]
- Chen S, Mohajerani MH, Xie Y, Murphy TH. Optogenetic analysis of neuronal excitability during global ischemia reveals selective deficits in sensory processing following reperfusion in mouse cortex. J Neurosci. 2012;32:13510–13519. doi: 10.1523/JNEUROSCI.1439-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Xiong C, Pancyr C, Stockwell J, Walz W, Cayabyab FS. Prolonged adenosine A1 receptor activation in hypoxia and pial vessel disruption focal cortical ischemia facilitates clathrin-mediated AMPA receptor endocytosis and long-lasting synaptic inhibition in rat hippocampal CA3-CA1 synapses: differential regulation of GluA2 and GluA1 subunits by p38 MAPK and JNK. J Neurosci. 2014;34:9621–9643. doi: 10.1523/JNEUROSCI.3991-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MY, Wang EH, Woodson WJ, Wang S, Sun G, Lee AG, Arac A, Fenno LE, Deisseroth K, Steinberg GK. Optogenetic neuronal stimulation promotes functional recovery after stroke. Proc Natl Acad Sci U S A. 2014;111:12913–12918. doi: 10.1073/pnas.1404109111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G, Carmichael ST. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci. 2011;31:3766–3775. doi: 10.1523/JNEUROSCI.5780-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinople CM, Bruno RM. Effects and mechanisms of wakefulness on local cortical networks. Neuron. 2011;69:1061–1068. doi: 10.1016/j.neuron.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PJ, Shih AY, Driscoll JD, Knutsen PM, Blinder P, Davalos D, Akassoglou K, Tsai PS, Kleinfeld D. Chronic optical access through a polished and reinforced thinned skull. Nat Methods. 2010;7:981–984. doi: 10.1038/nmeth.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G, Leon A, Baimbridge KG, Somogyi P. Relationship of neuronal vulnerability and calcium binding protein immunoreactivity in ischemia. Exp Brain Res. 1990;83:55–66. doi: 10.1007/BF00232193. [DOI] [PubMed] [Google Scholar]
- Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke. 1997;28:1805–1810. doi: 10.1161/01.STR.28.9.1805. discussion 1811. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison TC, Ayling OG, Murphy TH. Distinct cortical circuit mechanisms for complex forelimb movement and motor map topography. Neuron. 2012;74:397–409. doi: 10.1016/j.neuron.2012.02.028. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA. Cortical steady potential, impedance and excitability changes during and after total ischemia of cat brain. Exp Neurol. 1971;32:163–175. doi: 10.1016/0014-4886(71)90060-4. [DOI] [PubMed] [Google Scholar]
- Hossmann KA, Sato K. Recovery of neuronal function after prolonged cerebral ischemia. Science. 1970;168:375–376. doi: 10.1126/science.168.3929.375. [DOI] [PubMed] [Google Scholar]
- Hummel FC, Cohen LG. Non-invasive brain stimulation: a new strategy to improve neurorehabilitation after stroke? Lancet Neurol. 2006;5:708–712. doi: 10.1016/S1474-4422(06)70525-7. [DOI] [PubMed] [Google Scholar]
- Ilie A, Ciocan D, Zagrean AM, Nita DA, Zagrean L, Moldovan M. Endogenous activation of adenosine A(1) receptors accelerates ischemic suppression of spontaneous electrocortical activity. J Neurophysiol. 2006;96:2809–2814. doi: 10.1152/jn.00466.2006. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex. 2001;11:249–259. doi: 10.1093/cercor/11.3.249. [DOI] [PubMed] [Google Scholar]
- Johansen FF, Tønder N, Zimmer J, Baimbridge KG, Diemer NH. Short-term changes of parvalbumin and calbindin immunoreactivity in the rat hippocampus following cerebral ischemia. Neurosci Lett. 1990;120:171–174. doi: 10.1016/0304-3940(90)90030-D. [DOI] [PubMed] [Google Scholar]
- Kahn I, Desai M, Knoblich U, Bernstein J, Henninger M, Graybiel AM, Boyden ES, Buckner RL, Moore CI. Characterization of the functional MRI response temporal linearity via optical control of neocortical pyramidal neurons. J Neurosci. 2011;31:15086–15091. doi: 10.1523/JNEUROSCI.0007-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, Wang J, Xie Y, Yan Z, Zhang Y, Chow BY, Surek B, Melkonian M, Jayaraman V, Constantine-Paton M, Wong GK, et al. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11:338–346. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Krakauer JW, Carmichael ST, Corbett D, Wittenberg GF. Getting neurorehabilitation right: what can be learned from animal models? Neurorehabil Neural Repair. 2012;26:923–931. doi: 10.1177/1545968312440745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman SJ, Huang ZJ. High-resolution labeling and functional manipulation of specific neuron types in mouse brain by Cre-activated viral gene expression. PLoS One. 2008;3:e2005. doi: 10.1371/journal.pone.0002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kwan AC, Zhang S, Phoumthipphavong V, Flannery JG, Masmanidis SC, Taniguchi H, Huang ZJ, Zhang F, Boyden ES, Deisseroth K, Dan Y. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnick JE, Michele JJ, Simeone FA, DeFeo S, Welsh FA. Alteration of somatosensory evoked potentials in response to global ischemia. J Neurosurg. 1984;60:490–494. doi: 10.3171/jns.1984.60.3.0490. [DOI] [PubMed] [Google Scholar]
- Lim C, Alexander MP, LaFleche G, Schnyer DM, Verfaellie M. The neurological and cognitive sequelae of cardiac arrest. Neurology. 2004;63:1774–1778. doi: 10.1212/01.WNL.0000144189.83077.8E. [DOI] [PubMed] [Google Scholar]
- Lin JY, Knutsen PM, Muller A, Kleinfeld D, Tsien RY. ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci. 2013;16:1499–1508. doi: 10.1038/nn.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Borowicz LM, Goldsborough MA, Enger C, Selnes OA. Depression and cognitive decline after coronary artery bypass grafting. Lancet. 1997;349:1282–1284. doi: 10.1016/S0140-6736(96)09466-4. [DOI] [PubMed] [Google Scholar]
- Mégevand P, Troncoso E, Quairiaux C, Muller D, Michel CM, Kiss JZ. Long-term plasticity in mouse sensorimotor circuits after rhythmic whisker stimulation. J Neurosci. 2009;29:5326–5335. doi: 10.1523/JNEUROSCI.5965-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitzdorf U. Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiol Rev. 1985;65:37–100. doi: 10.1152/physrev.1985.65.1.37. [DOI] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C. Absence of large-scale dendritic plasticity of layer 5 pyramidal neurons in peri-infarct cortex. J Neurosci. 2011;31:1734–1738. doi: 10.1523/JNEUROSCI.4386-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci. 2010;30:14116–14126. doi: 10.1523/JNEUROSCI.3908-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci. 2008;28:1756–1772. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch C, Scotti A, Sommacal A, Kalt G. GABAergic hippocampal neurons resistant to ischemia-induced neuronal death contain the Ca2(+)-binding protein parvalbumin. Neurosci Lett. 1989;105:263–268. doi: 10.1016/0304-3940(89)90631-9. [DOI] [PubMed] [Google Scholar]
- Nunn J, Hodges H. Cognitive deficits induced by global cerebral ischaemia: relationship to brain damage and reversal by transplants. Behav Brain Res. 1994;65:1–31. doi: 10.1016/0166-4328(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Panahian N, Yoshida T, Huang PL, Hedley-Whyte ET, Dalkara T, Fishman MC, Moskowitz MA. Attenuated hippocampal damage after global cerebral ischemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Deng P, Ruan YW, Xu ZC. Depression of fast excitatory synaptic transmission in large aspiny neurons of the neostriatum after transient forebrain ischemia. J Neurosci. 2002;22:10948–10957. doi: 10.1523/JNEUROSCI.22-24-10948.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC, Hahn TTG, Mehta M, Grinvald A, Sakmann B. Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc Natl Acad Sci U S A. 2003;100:13638–13643. doi: 10.1073/pnas.2235811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen KH, Devor A, Ulbert I, Dale AM, Einevoll GT. Current-source density estimation based on inversion of electrostatic forward solution: effects of finite extent of neuronal activity and conductivity discontinuities. J Neurosci Methods. 2006;154:116–133. doi: 10.1016/j.jneumeth.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 2013;16:1068–1076. doi: 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qü M, Mittmann T, Luhmann HJ, Schleicher A, Zilles K. Long-term changes of ionotropic glutamate and GABA receptors after unilateral permanent focal cerebral ischemia in the mouse brain. Neuroscience. 1998;85:29–43. doi: 10.1016/S0306-4522(97)00656-8. [DOI] [PubMed] [Google Scholar]
- Quiroga RQ, Nadasdy Z, Ben-Shaul Y. Unsupervised spike detection and sorting with wavelets and superparamagnetic clustering. Neural Comput. 2004;16:1661–1687. doi: 10.1162/089976604774201631. [DOI] [PubMed] [Google Scholar]
- Ren JQ, Aika Y, Heizmann CW, Kosaka T. Quantitative analysis of neurons and glial cells in the rat somatosensory cortex, with special reference to GABAergic neurons and parvalbumin-containing neurons. Exp Brain Res. 1992;92:1–14. doi: 10.1016/S0079-6123(08)61160-7. [DOI] [PubMed] [Google Scholar]
- Risher WC, Ard D, Yuan J, Kirov SA. Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J Neurosci. 2010;30:9859–9868. doi: 10.1523/JNEUROSCI.1917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Croom D, Kirov SA. Persistent astroglial swelling accompanies rapid reversible dendritic injury during stroke-induced spreading depolarizations. Glia. 2012;60:1709–1720. doi: 10.1002/glia.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan CA, Schummers J, Van Wart A, Kuhlman SJ, Wilson NR, Huang ZJ, Sur M. Response features of parvalbumin-expressing interneurons suggest precise roles for subtypes of inhibition in visual cortex. Neuron. 2010;67:847–857. doi: 10.1016/j.neuron.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Characterization of a recovery global cerebral ischemia model in the mouse. J Neurosci Methods. 1999;88:103–109. doi: 10.1016/S0165-0270(99)00018-7. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 2005;57:252–260. doi: 10.1016/j.biopsych.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Tortosa A, Ferrer I. Parvalbumin immunoreactivity in the hippocampus of the gerbil after transient forebrain ischaemia: a qualitative and quantitative sequential study. Neuroscience. 1993;55:33–43. doi: 10.1016/0306-4522(93)90452-L. [DOI] [PubMed] [Google Scholar]
- Tran S, Chen S, Liu RR, Xie Y, Murphy TH. Moderate or deep local hypothermia does not prevent the onset of ischemia-induced dendritic damage. J Cereb Blood Flow Metab. 2012;32:437–442. doi: 10.1038/jcbfm.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13:251–266. doi: 10.1038/nrn3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH. Short-term cerebral ischemia causes the dysfunction of interneurons and more excitation of pyramidal neurons in rats. Brain Res Bull. 2003;60:53–58. doi: 10.1016/S0361-9230(03)00026-1. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Runyan CA, Wang FL, Sur M. Division and subtraction by distinct cortical inhibitory networks in vivo. Nature. 2012;488:343–348. doi: 10.1038/nature11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Wang T, Sun GY, Ding S. Specific disruption of astrocytic Ca2+ signaling pathway in vivo by adeno-associated viral transduction. Neuroscience. 2010;170:992–1003. doi: 10.1016/j.neuroscience.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Chen S, Murphy T. Dendritic spines and pre-synaptic boutons are stable despite local deep hypothermic challenge and re-warming in vivo. PLoS One. 2012;7:e36305. doi: 10.1371/journal.pone.0036305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Chen S, Anenberg E, Murphy TH. Resistance of optogenetically evoked motor function to global ischemia and reperfusion in mouse in vivo. J Cereb Blood Flow Metab. 2013;33:1148–1152. doi: 10.1038/jcbfm.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc. 2010;5:201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan RZ, Nadler JV, Schwartz-Bloom RD. Depressed responses to applied and synaptically-released GABA in CA1 pyramidal cells, but not in CA1 interneurons, after transient forebrain ischemia. J Cereb Blood Flow Metab. 2006;26:112–124. doi: 10.1038/sj.jcbfm.9600171. [DOI] [PubMed] [Google Scholar]
- Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, Deisseroth K. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Boyd J, Delaney K, Murphy TH. Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci. 2005;25:5333–5338. doi: 10.1523/JNEUROSCI.1085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]