

Abstract
Synaptic plasticity in hippocampal neurons has been thought to represent a variety of memories. Although accumulating evidence indicates a crucial role of BDNF/TrkB/Akt signaling in the synaptic plasticity of the hippocampus, the mechanism by which Akt, a serine/threonine kinase, controls activity-dependent neuronal plasticity remains unclear. Girdin (also known as APE, GIV, and HkRP1), an actin-binding protein involved both in the remodeling of the actin cytoskeleton and in cell migration, has been identified as a substrate of Akt. Previous studies have demonstrated that deficit of neuronal migration in the hippocampus of Girdin-deficient (Girdin−/−) mice is independent on serine phosphorylation of Girdin at S1416 (Girdin S1416) by Akt. In the present study, we focused on the role of Girdin S1416 phosphorylation in BDNF/TrkB/Akt signaling associated with synaptic plasticity. We found that Girdin in the hippocampus was phosphorylated at S1416 in an activity-dependent manner. Phosphorylation-deficient knock-in mice (GirdinSA/SA mice), in which S1416 is replaced with alanine, exhibited shrinkage of spines, deficit of hippocampal long-term potentiation, and memory impairment. These phenotypes of GirdinSA/SA mice resembled those of Girdin+/− mice, which have 50% loss of Girdin expression. Furthermore, Girdin interacted with Src kinase and NR2B subunit of NMDA receptor, leading to phosphorylation of the NR2B subunit and NMDA receptor activation. Our findings suggest that Girdin has two different functions in the hippocampus: Akt-independent neuronal migration and Akt-dependent NR2B phosphorylation through the interaction with Src, which is associated with synaptic plasticity in the hippocampus underlying memory formation.
Keywords: Girdin, long-term potentiation, memory formation, mutant mice, NMDA receptor, phosphorylation
Introduction
It has been widely believed that the synaptic plasticity in hippocampal neurons is related to the formation of various memories (Burgess et al., 2002; Squire et al., 2004). Synaptic plasticity refers to the ability of synapses to vary their efficacy of chemical or electrical transmission over time in response to activities (Pitman, 1984). Regulation of synaptic transmission and strength may be mediated by several events including changes in expression of synaptic surface receptors and neurotransmitters (Duguid and Sjöström, 2006; Minichiello, 2009), receptor subunit phosphorylation (Soderling and Derkach, 2000), postsynaptic calcium release (Turrigiano and Nelson, 2000), and cytoskeletal remodeling (Dillon and Goda, 2005).
Neurotrophins are considered powerful molecular mediators in synaptic plasticity. For example, BDNF, as well as its receptor TrkB, have emerged as key molecules in the neurobiological mechanisms related to learning and memory (Minichiello, 2009). TrkB is known to activate Ras/ERK, PLC-γ pathways, and phosphatidylinositol 3-kinase (PI3-K)/Akt (Yoshii and Constantine-Paton, 2010). Serine/threonine kinase Akt has been linked to brain development, aging, and neurodegenerative and psychiatric disorders via the phosphorylation of a wide variety of its substrates (Brunet et al., 2001; Manning and Cantley, 2007). Accumulating evidence indicates a crucial role of Akt in the neuronal plasticity of the hippocampus (Sanna et al., 2002; Cammalleri et al., 2003; Man et al., 2003; Balu et al., 2012), but little is known about the mechanism by which Akt controls activity-dependent neuronal plasticity.
Girdin has been identified as an actin-binding Akt substrate that has been reported to regulate migration of various cells (Enomoto et al., 2005). In the CNS, Girdin is highly expressed in the hippocampus, and Girdin-deficient (Girdin−/−) mice have defects in the migration and positioning of the subgranular zone in the DG (Enomoto et al., 2009) and neuroblast chain migration in the subventricular zone toward the olfactory bulb (Wang et al., 2011).
To understand the physiological role of the serine phosphorylation of Girdin by Akt, phosphorylation-deficient knock-in mice (GirdinSA/SA mice), in which Girdin S1416 is replaced with alanine, have been developed (Wang et al., 2011). Phosphorylation level of Girdin S1416 was undetectable in GirdinSA/SA mice while the expression level of Girdin is normal (Wang et al., 2011). Notably, GirdinSA/SA mice show no defects in the migration and positioning of the subgranular zone in the DG (Wang et al., 2011), indicating that Girdin has a crucial role in neuronal migration and positioning, but the effect is independent on its phosphorylation by Akt. Accordingly, a physiological role for Girdin S1416 phosphorylation by Akt in the hippocampus remains obscure.
Here, we report that Girdin S1416 was phosphorylated in an activity-dependent manner, and this phosphorylation was essential for spine morphogenesis, maintenance of hippocampal LTP, and memory formation. Furthermore, Girdin interacts with Src and the NR2B subunit of NMDA receptors, leading to phosphorylation of the NR2B subunit and NMDA receptor activation. Our findings suggest that activity-dependent Girdin phosphorylation at S1416 induced by Akt is crucial for NMDA receptor activation associated with neuronal plasticity underlying hippocampal memory formation.
Materials and Methods
Animals
Construction of the Girdin gene-targeting vector and generation of Girdin-deficient (Girdin−/−) mice were as described previously (Kitamura et al., 2008). Girdin+/− mice were generated by the breeding of Girdin+/− and wild-type (Girdin+/+) mice. The generation of GirdinSA/SA mice has been described previously (Wang et al., 2011). GirdinSA/SA mice were produced at the expected Mendelian ratio by intercrossing GirdinSA/+ mice. C57BL/6J mice were obtained from Charles River Laboratory. Mice were housed under a standard 12 h light/dark cycle (light phase 9:00–21:00) at a constant temperature of 23 ± 1°C, with food and water ad libitum throughout the experiments. All of the animal protocols were approved by the Animal Care and Use Committee of Nagoya University Graduate School of Medicine.
Cell culture
Primary cultured hippocampal neurons were prepared from Girdin+/+ or GirdinSA/SA mice on gestational days 15–16, as described previously with minor modification (Ibi et al., 2013). Embryo hippocampi were digested with 0.25% trypsin and 0.01% DNase for 10 min at 37°C and mechanically dissociated by gentle pipetting. Neurons were seeded on poly-d-lysine-coated coverslips (Sekiya Rika), and cultured in nerve culture medium (Sumitomo Bakelite) on DIV 0. Culture medium was exchanged for new medium every 3–4 d. One micrometer of cytosine β-d-arabinofuranoside was added to the culture medium on DIV 2. Our preparations were 99% pure for neurons, as demonstrated by immunocytochemistry for tau (axonal marker) or microtubule-associated protein 2 (dendritic marker).
BDNF-induced Girdin phosphorylation in cultured hippocampal neurons
On DIV 14, hippocampal primary cultured neurons at a high density (1.0 × 106 cells/well in poly-d-lysine-coated coverslips) were treated with PBS (control) or 50 ng/ml BDNF (PeproTech) for 5, 15, and 30 min. Cells were lysed, briefly sonicated in 100 μl of buffer A [1% SDS, complete protease inhibitor cocktail (Roche), and PhosSTOP phosphatase inhibitor cocktail (Roche)] and used for Western blotting. Alternatively, cultured neurons were pretreated with DMSO (1%), Trk inhibitor K252a (0.2 μm, 0.6 μm, or 1.2 μm; Alomone Labs), PI3-K inhibitor LY294002 (10 μm, 30 μm, or 50 μm; Cell Signaling Technology), or Akt inhibitor IV (0.1 μm, 0.5 μm, or 1.0 μm; Millipore) for 30 min, and then stimulated with PBS (control) or BDNF (50 ng/ml) for 15 min in the presence of each inhibitor. The cells were lysed using buffer A and used for Western blotting.
Girdin phosphorylation in the hippocampus of mouse after fear conditioning
Eight- to 9-week-old male Girdin+/+ mice were habituated in a training chamber for 10 min once daily for at least 6 d before the experiment. The mice were then decapitated and the brain was immediately removed 5, 15, and 30 min after fear conditioning. The detailed procedure is described below (see Fear-conditioning test). Control mice were placed in a training chamber without delivering footshock and tone. The DG was rapidly dissected out on ice, sonicated in 100 μl of buffer A, and used for Western blotting.
Western blotting analysis
Lysates from hippocampal neurons and the DG were briefly boiled for 10 min at 99°C, and cleared by centrifugation at 15,000 × g for 20 min. The protein concentration was determined using a DC Protein Assay Kit (Bio-Rad), and protein was boiled in sample buffer (0.0625 m Tris-HCl, pH 6.8, 2% SDS, 5% glycerol, 0.002% bromophenol blue, and 5% 2-mercaptoethanol); applied to a 6–10% SDS-polyacrylamide gel; and subsequently transferred to a polyvinylidene difluoride membrane (Millipore). The membranes were blocked with Blocking One-P (Nacalai Tesque) at room temperature for 1 h and incubated with a primary antibody at 4°C overnight. Then, membranes were washed three times every 30 min with Tris-buffered saline and Tween 20 (10 mm Tris-HCl, pH 7.4, 100 mm NaCl, and 1% Tween 20). After incubation with a horseradish peroxidase-conjugated secondary antibody for 1 h, the immune complex was detected using ECL Prime Western blotting detection reagents (GE Healthcare). The intensity of the bands on the membranes was analyzed by densitometry (Atto Instruments). To calculate the relative amount of phosphorylated proteins compared with total proteins, the same membranes were stripped with WB Stripping Solution Strong (Nacalai Tesque) at room temperature for 1 h and treated as described above. Because there was no change in the levels of total proteins, values of phosphorylation were normalized to the values of total proteins. All the data from Western blotting are expressed as relative fold change in expression with respect to the control. The primary and secondary antibodies were diluted with Can-Get-Signal Solutions 1 and 2 (Toyobo), respectively, to enhance antibody–antigen binding. The mouse anti-phospho Girdin monoclonal antibody (1:5000) was provided by Dr. M. Asai and K. Omori (Nagoya University). Other primary antibodies were used as follows: a sheep anti-Girdin (1:10,000; R&D Systems), a rabbit anti-NMDA2A subunit (NR2A, 1:4000; Millipore), a mouse anti-NMDAR 2B subunit (NR2B, 1:5000; Millipore), a rabbit anti-Akt (1:50,000; Cell Signaling Technology), a rabbit anti-Src (1:8000; Cell Signaling Technology), a rabbit anti-Fyn (1:6000; Santa Cruz Biotechnology), a mouse anti-β-tubulin (1:5000; Cell Signaling Technology), a goat anti-β-actin (1:4000; Santa Cruz Biotechnology), a rabbit anti-phospho NR2A (1:4000; Abcam), a rabbit anti-phospho NR2B (1:2000; Millipore), a rabbit anti-phospho Akt (1:20,000; Cell Signaling Technology), a rabbit anti-phospho Src (1:8000; Millipore), and anti-V5 (1:8000; Invitrogen). The secondary antibodies were horseradish peroxidase-conjugated anti-rabbit (1:4000–100,000; KPL), anti-mouse (1:8000–50,000; KPL), anti-goat (1:8000–10,000; Santa Cruz Biotechnology), or anti-sheep IgG (1:20,000; Invitrogen).
Analyses of dendritic length and spine density in the dentate granule cells of the hippocampus
Eight- to 12-week-old male Girdin+/+ and GirdinSA/SA mice were anesthetized with 0.02 mg/ml tribromoethanol and transcardially perfused with ice-cold 1.5% paraformaldehyde in 0.1 m PBS. Their brains were removed and postfixed in the same fixative for 1 h. Then, brains were washed with PBS and sliced into 200 μm coronal sections using a vibratome in ice-cold PBS. The sections were collected between –1.34 and –2.06 from the bregma according to the brain atlas (Franklin and Paxinos, 1997). For dendritic and spine staining in the dentate granule cells (DGCs) of the hippocampus, DiOlistic labeling was performed as described previously (Gan et al., 2000). All images (2048 × 2048 pixels) were obtained using a Zeiss LSM 700 laser-scanning confocal microscope (Carl Zeiss Microscopy) with ZEN software (Carl Zeiss Microscopy). The acquisition parameters were kept the same for all images. All dendrites and spines in the DGCs were traced using Neurolucida software (MicroBrightField) and analyzed using NeuroExplorer (MicroBrightField). For all neurons, dendritic spine density, volume, head diameter, and length were measured on the same dendritic branches used for measurements of total dendrite length. These parameters were quantified on at least dendritic segments of secondary and tertiary branches located at least 50 μm away from the cell body (Ayhan et al., 2011; Jurgens et al., 2012). An investigator blinded to the group's status performed neuron selection and tracing.
Hippocampal slice preparation and electrophysiology
Eight- to 12-week-old male mice were decapitated under deep anesthesia with ethyl ether. Brains were quickly removed, and transverse 400-μm-thick slices were cut from the hippocampus using a vibratome in ice-cold modified artificial ACSF containing 206 mm sucrose, 5 mm KCl, 8 mm MgCl2, 1.25 mm KH2PO4, 1 mm CaCl2, 26 mm NaHCO3, and 10 mm d-glucose. ACSF was gassed with 95% O2/5% CO2 and the pH was adjusted to 7.4. Slices were maintained for at least 1 h at room temperature (26–28°C) in an incubation chamber with gassed standard ACSF containing 128 mm NaCl, 5 mm KCl, 1.3 mm MgSO4, 1.25 mm KH2PO4, 2.41 mm CaCl2, 26 mm NaHCO3, and 10 mm d-glucose. A single hippocampal slice was transferred to and maintained for 1 h in the recording chamber where it was superfused continuously with gassed standard ACSF at a rate of 2.0–2.5 ml/min at room temperature. A stimulating electrode (monopolar stimulation) was positioned in the molecular layer 30–50 μm from the GC layer in the DG of the hippocampus to activate the medial perforant path (MPP). Constant-current pulses (100 μs) were supplied by a stimulator (SEN-3301; Nihon Kohden) every 20 s. The intensity of the test stimuli was adjusted to evoke ∼50% of the maximum response. Using the conventional extracellular recording technique, fEPSPs were recorded from the middle molecular layer in the DG with a glass pipette filled with 2 m NaCl (2–3 MΩ). The baseline recording was established by delivering test stimuli for >60 min before experimental recordings. LTP was induced by tetanic stimulation (TET; four trains, 500 ms each, 100 Hz within the train, repeated every 20 s) in the presence of the GABA receptor antagonist picrotoxin (50 μm). For whole-cell patch-clamp recording, a patch electrode was filled with a pipette solution containing the following: 140 mm cesium gluconate, 10 mm NaCl, 2 mm MgCl2, 1 mm EGTA, 10 mm HEPES, 3 mm Mg-ATP, 0.3 mm Na-GTP, and 5 mm QX-314, with 6–8 MΩ of resistance. Whole-cell patch-clamp recordings were made from GCs near the outer border of the GC layer in the DG. DGCs were imaged with IR-DIC optics (BX51WI with 20× water-immersion objective lens; Olympus). For selective recording of the mature GCs, GCs with an input resistance of <350 MΩ were chosen (Spigelman et al., 1992; Liu et al., 1996). Holding potentials were compensated for the junction potential between the pipette solution and the ACSF. To measure NMDA receptor-mediated EPSCs (NMDAR-EPSCs), cells were held at +40 mV to relieve the voltage-dependent Mg2+ block. NMDAR-EPSCs were isolated by blocking AMPA receptors (10 μm NBQX) and GABAA receptors (10 μm bicuculline). Access resistance was monitored continuously during the experiment, and the obtained data were discarded if the access resistance fluctuated >20%. Signals were amplified and filtered at 5 kHz with an amplifier (Axopatch 200B; Molecular Devices). Data acquisition and analysis were performed using pClamp 9.0 software (Molecular Devices). Synaptic transmission in the extracellular recordings was estimated from the rising phase of the fEPSP slope using linear regression during the first 0.6 ms after the presynaptic fiber volley. The paired pulse ratio (PPR) of fEPSPs was calculated with the following formula: PPR = 100 × (S2EPSP/S1EPSP). The magnitude of LTP is expressed as the mean value of percentage changes in 55–60 min after TET with respect to the final 5 min of the baseline period.
Plasmid and transfection
The construction of a human Girdin-V5 cDNA expression vector was as previously described (Enomoto et al., 2009). The mouse NR2B, Src-GFP, and Fyn-GFP expression vectors were purchased from OriGene Technologies. HEK293T cells were plated at 5.0 × 106 cells/well in 100 mm plastic culture plates. The following day, each well was transfected with cDNA using the Lipofectamine LTX (Invitrogen) and Opti-MEM (Invitrogen). Forty-eight hours after transfection, the cells were lysed using buffer B (50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 150 mm NaCl, 1% Nonidet P-40, protease inhibitor cocktail, and PhosSTOP phosphatase inhibitor cocktail).
Immunoprecipitation
Immunoprecipitation (IP) of NR2B was performed using an IP Kit Dynabeads Protein G (Invitrogen), according to the manufacturer's instructions. Briefly, HEK293T cells expressing Girdin-V5/NR2B/Src-GFP or Girdin-V5/NR2B/Fyn-GFP were lysed using buffer B. The lysates of HEK293T cells were briefly sonicated and cleared by centrifugation at 15,000 × g for 20 min. Lysate protein (2 mg) of HEK293T cells was added to 50 μl of Dynabeads/protein G cross-linked to rabbit anti-NR2B antibody (Cell Signaling Technology) overnight at 4°C. Normal rabbit IgG (Santa Cruz Biotechnology) was used as a negative control at a concentration equal to that of anti-NR2B antibody. The immunoprecipitates were washed three times in buffer B and eluted in 30 μl of sample buffer containing SDS. The eluted samples were analyzed by Western blotting as described above.
Behavioral analyses
Behavioral analyses of Girdin+/+, GirdinSA/SA, and Girdin+/− mice were conducted at the age of 7–20 weeks (adult). Animals were used for each behavioral test only once in the adult period in the following order: one group of animals was subjected to locomotor activity test, Y-maze test, open field test, prepulse inhibition (PPI) test, novel object recognition test, fear-conditioning test, and Morris water maze test, while the other group of animals was used in the elevated-plus maze test, forced-swim test, sucrose-preference test, and fear-conditioning test. Each behavioral test was conducted repeatedly with two different batches of animals.
Locomotor activity test in a novel environment.
Each mouse was placed in a standard transparent rectangular rodent cage (25 × 30 × 18 cm) under moderate light (15 lux). Locomotor activity was then monitored for 120 min using an infrared sensor (NS-AS01; Brain Science Idea) placed over the cage (Ibi et al., 2009).
Y-maze test.
The Y-maze test was performed as described previously (Ibi et al., 2010). Each arm was 40 cm long, 12 cm high, 3 cm wide at the bottom, and 10 cm wide at the top under moderate light (30 lux). The arms converge in an equilateral triangular central area that is 4 cm at its longest axis. Each mouse was placed individually at the center of the apparatus and allowed to move freely through the maze during an 8 min session. The number of arm entries was recorded visually. Alternation is defined as successive entries into the three arms on overlapping triplet sets. The percentage alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus 2) multiplied by 100.
Open field test.
Mice were placed in the center of the arena and were allowed to explore the open field (diameter: 60 cm, height: 35 cm) for 5 min under moderate light (80 lux); activity was automatically measured using the EthoVision automated tracking program (Noldus Information Technology). The open field was divided into an inner circle (diameter: 40 cm) and an outer area surrounding the inner circle. Mouse movement was measured via a camera mounted above the open field. Measurements included distance and time spent in the inner and outer sections (Ibi et al., 2009).
PPI test.
The PPI test was performed as described previously (Ibi et al., 2009). Animals were placed in the chamber under moderately bright conditions (180 lux; San Diego Instruments) and were allowed to habituate for 10 min with 65 dB background white noise. Animals then received 10 startle trials, 10 no-stimulus trials, and 40 PPI trials. The intertrial interval was between 10 and 20 s, and the total session lasted 17 min. The startle trial consisted of a single 120 dB white noise burst lasting 40 ms. PPI trials consisted of a prepulse (20 ms burst of white noise at 69 dB, 73 dB, 77 dB, or 81 dB intensity) followed by the startle stimulus (120 dB, 40 ms white noise) 100 ms later. Each of the four prepulse trials (69 dB, 73 dB, 77 dB, or 81 dB) was presented 10 times. Sixty different trials were presented pseudorandomly, ensuring that each trial was presented 10 times and that no two consecutive trials were identical. Animal movements in the startle chamber were measured for 100 ms after the onset of the startle stimulus (sampling frequency 1 kHz), and the maximal response was calculated during the 100 ms period. The basal startle amplitude was determined from the mean amplitude of the 10 startle trials. PPI was calculated according to the following formula: 100 × [1 − (PP×/P120)] %, in which PP× was the mean of the 10 PPI trials (PP69, PP73, PP75, or PP80), and P120 was the basal startle amplitude.
Fear-conditioning test.
The fear-conditioning test was conducted as previously described (Ibi et al., 2010). During the conditioning phase, each mouse was placed in a training chamber (30 × 30 × 40 cm) equipped with a metal floor, and a 15 s white noise tone (85 dB) was delivered (conditioned stimulus). During the last 5 s of the tone stimulus, a footshock of 0.8 mA was delivered through a shock generator as an unconditioned stimulus (Brain Science Idea). This procedure was repeated four times at 15 s intervals. One hour or 24 h after conditioning, the context-dependent test was performed. For this test, each mouse was placed in the training chamber, and the freezing response was measured for 2 min in the absence of the conditioned stimulus. Tone-dependent testing was performed 4 h after the context-dependent test. For the tone-dependent test, the freezing response was measured in a standard transparent rectangular rodent cage (25 × 30 × 18 cm) for 1 min in the presence of a continuous-tone stimulus identical to the conditioned stimulus using mice that had been subjected to the context-dependent test.
Novel object recognition test.
A novel object recognition test was performed as described previously (Nagai et al., 2007). Mice were individually habituated to an open box (30 × 30 × 35 cm height) for 3 d. During the training session on the fourth day, two novel objects were placed in the open box, and the animals were allowed to explore for 10 min under moderate light (10 lux). The exploration time for each object was recorded. During the retention session on the fifth day, the animals were placed into the same box (24 h after the training session) and one of the familiar objects that had been used during training was replaced with a novel object. The mice were then allowed to explore freely for 5 min. The preference index during the retention session and the ratio of the amount of time spent exploring the novel object over the total time spent exploring both objects was used to measure cognitive function. During the training session, the preference index was calculated as the ratio of time spent exploring the object that had been replaced by a novel object in the retention session to the total exploration time.
Morris water maze test.
The Morris water maze test was performed according to a previous report (Ibi et al., 2008) with minor modifications. A pool (120 cm in diameter) was prepared, and the water temperature was maintained at 21–23°C under moderate light (45 lux). Swimming paths were automatically measured using the EthoVision automated tracking program (Noldus Information Technology). In the training trials, the platform (7 cm in diameter) was submerged 1 cm below the water surface. After reaching the platform, the mouse was allowed to remain on it for 20 s. If the mouse failed to reach the platform within 60 s, the trial was terminated and the animal was placed on the platform by the experimenter for 20 s. After training trials for 6 d, mice were subjected to the probe test on day 7, in which the platform was removed and they swam for 60 s in the pool. We measured the time spent in each quadrant of the pool as a measure of spatial memory. At 1 h after the probe test, to measure swimming ability or motivation, mice were subjected to the visible test in which the platform was marked with a flag that protruded 12 cm above the water surface to be highly visible, but in a new location. Three starting positions were used pseudorandomly and each mouse was subjected to three trials per day with a 30 min intertrial interval in the training trials (days 1–6) and visible test (day 7). During training trials and the visible test, we measured both path length (swim distance) and escape latency as measures of performance.
Elevated-plus maze test.
The elevated-plus maze consisted of two open (25 × 8 × 0.5 cm) and two closed (25 × 8 × 20 cm) arms emanating from a common central platform (8 × 8 cm) to form a plus shape (Koike et al., 2009). The entire apparatus was elevated 50 cm above floor level under moderately bright conditions (170 lux). The test began by placing a mouse on the central platform of the maze facing an open arm. An arm entry was defined as all four paws in the arm. The amount of time spent in an arm and the number of arm entries were measured for 5 min.
Forced-swim test.
The forced-swim test was performed as described previously with minor modification (Koike et al., 2009). Mice were placed in a glass cylinder (20 cm high × 13.5 cm diameter) filled with water (24 ± 1°C) to a depth of 12 cm. They were allowed to swim for 6 min, and the amount of time spent immobile, defined as the absence of struggling and floating motionless in water making only movements necessary to keep their heads above water, was recorded during the last 5 min of the test.
Sucrose-preference test.
The sucrose-preference test was conducted according to a method described in previous reports (Catches et al., 2012; Mouri et al., 2012) with minor modification. Total intakes were automatically measured using a recording system (MDC-WR1 and MDC-SOF software; Brain Science Idea). Mice were individually housed during this behavioral test, and water bottles were replaced with two bottles (bottle A and bottle B). Both bottles A and B were first filled with drinking water (w/w) for 24 h. Second, both A and B were filled with a solution of 1% sucrose (s/s) for 48 h. Then, bottle A contained 1% sucrose and bottle B contained drinking water (s/w) for 4 d. The positions of bottles A and B were switched every day to avoid a side bias. Total fluid consumed was calculated as [sucrose + water] and averaged across each day for a given condition (w/w, s/s, or s/w). Sucrose preference was calculated as 100 × (sucrose/[sucrose + water]).
Statistical analysis
Data are presented as means ± SEM. The differences between two groups were analyzed using two-tailed Student's t test. Differences in locomotor activity, PPI, and training in Morris water maze tests were analyzed by repeated-measures ANOVA. Multiple group comparisons were made with one-way ANOVA or two-way ANOVA, followed by the Tukey–Kramer post hoc test when the F ratios were significant (p < 0.05).
Results
BDNF increased Girdin S1416 phosphorylation through the TrkB/PI3-K/Akt pathway
To address the role of Girdin S1416 phosphorylation in the BDNF signaling pathway, we first examined the changes in the levels of phosphorylated Akt and Girdin S1416 following BDNF application in primary cultured hippocampal neurons. Primary cultured hippocampal neurons were prepared from C57BL/6J mice on gestational days 15–16. On DIV 14, hippocampal primary cultured neurons were treated with PBS (control) or 50 ng/ml BDNF for 5, 15, and 30 min. BDNF treatment significantly increased the phosphorylated level of Akt 5 min after the treatment, and the phosphorylation persisted up to 30 min (p < 0.01; Fig. 1A). A significant increase in the level of Girdin S1416 phosphorylation was also evident 15 min after the BDNF treatment (p < 0.05; Fig. 1B). BDNF-induced phosphorylation of Girdin S1416 was inhibited by pretreatment with Trk inhibitor K252a (Fig. 1C), PI3-K inhibitor LY294002 (Fig. 1D), or Akt inhibitor IV (Fig. 1E), indicating that BDNF increases Girdin S1416 phosphorylation through the TrkB/PI3-K/Akt pathway in hippocampal neurons.
Figure 1.
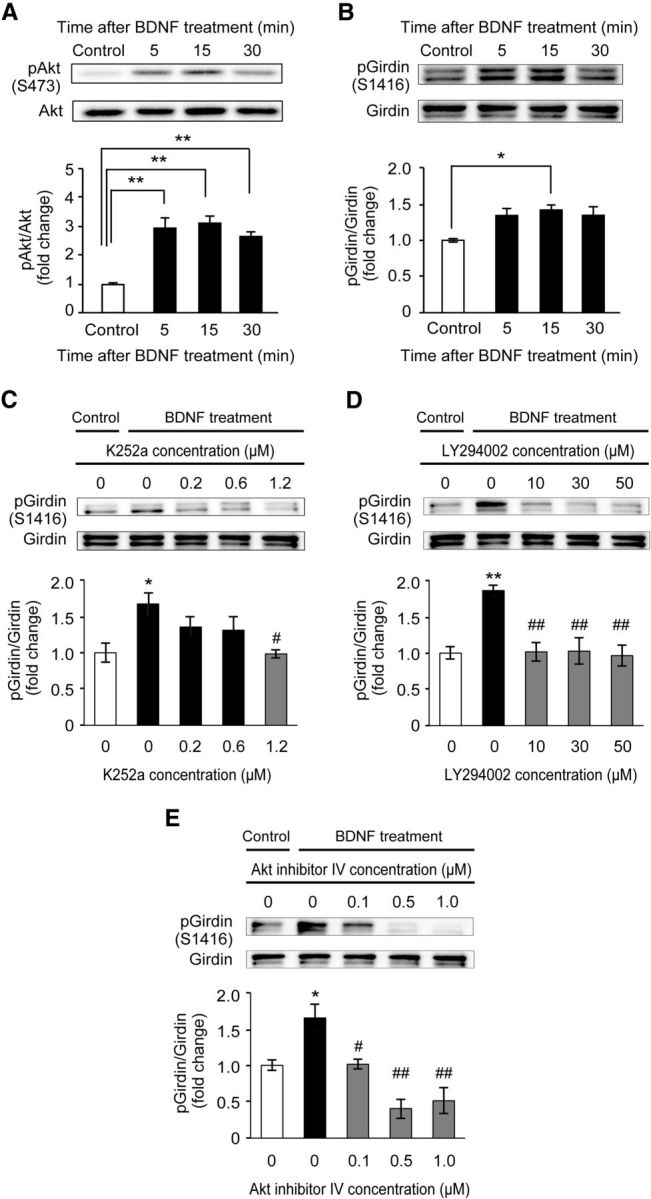
BDNF increased Girdin S1416 phosphorylation through the TrkB/PI3-K/Akt pathway in cultured mouse hippocampal neurons (DIV 14). A, B, Time-course changes in the levels of phosphorylated Akt (A) and Girdin (B) after BDNF (50 ng/ml) application in cultured E15–E16 mouse hippocampal neurons (DIV 14). Data are expressed as mean ± SEM (n = 5 at each time point). One-way ANOVA, F(3,16) = 16.107, p < 0.01 for A; F(3,16) = 4.412, p < 0.05 for B. *p < 0.05 and **p < 0.01 compared with the control (Tukey–Kramer post hoc test). C–E, Effects of Trk (C), PI3-K (D), and Akt (E) inhibitors on BDNF-induced Girdin phosphorylation in cultured E15–E16 mouse hippocampal neurons (DIV 14). Neurons were stimulated with BDNF (50 ng/ml) for 15 min in the presence of DMSO (1%), K252a (0.2 μm, 0.6 μm, or 1.2 μm), LY294002 (10 μm, 30 μm, or 50 μm), and Akt inhibitor IV (0.1 μm, 0.5 μm, or 1.0 μm). Data are expressed as mean ± SEM (n = 4 in each group). DMSO was treated as a control of each inhibitor (shown as 0 μm). One-way ANOVA, F(4,15) = 3.885, p < 0.05 for C; F(4,15) = 8.967, p < 0.01 for D; F(3,16) = 12.837, p < 0.01 for E. *p < 0.05 and **p < 0.01 compared with the control (Tukey–Kramer post hoc test). #p < 0.05 and ##p < 0.01 compared with the BDNF-treated control (Tukey–Kramer post hoc test).
Synaptic morphology and plasticity in the hippocampus of GirdinSA/SA mice
BDNF-TrkB signaling has been implicated in various aspects of neural development including dendritic outgrowth and spine formation (Yoshii and Constantine-Paton, 2010). To explore the role of phosphorylated Girdin in the morphological alteration of neuronal cells, DGCs of the hippocampus in 8- to 12-week-old Girdin+/+ and GirdinSA/SA mice were visualized by DiOlistic labeling. Total dendrite length was slightly, but not significantly, increased in GirdinSA/SA mice (Fig. 2A,B). Morphological analysis of spines revealed that spine density in GirdinSA/SA mice was normal (Fig. 2C,D). However, cumulative frequency and mean spine volume were significantly reduced in GirdinSA/SA mice (p < 0.01 for cumulative frequency, p < 0.05 for mean value of total spines; Fig. 2C,E). The reduction of spine volume in GirdinSA/SA mice was due to shortened spine head diameter (p < 0.01 for cumulative frequency, p < 0.05 for mean value of total spines; Fig. 2C,F), but not the spine length (Fig. 2C,G). These results suggest that phosphorylation of Girdin S1416 may be involved in shortening of thin spine volume, but not spine length in DGCs.
Figure 2.

Phosphorylation of Girdin S1416 regulates spine morphology in DGCs of mice. A, Representative image of DGCs in 8- to 12-week-old Girdin+/+ and GirdinSA/SA mice. Scale bar, 20 μm. B, Cumulative frequency and mean value of total dendrite length of Girdin+/+ and GirdinSA/SA mice. Data are expressed as mean ± SEM (n = 14–18 neurons from 3–4 mice). C, Representative images of the spines of DGCs in 8- to 12-week-old Girdin+/+ and GirdinSA/SA mice. Scale bar, 1 μm. D, Quantitative analysis of spine density. E–G, Cumulative frequency and mean value of spine volume (E), spine head diameter (F), and spine length (G) of DGCs in Girdin+/+ and GirdinSA/SA mice. Data are expressed as mean ± SEM (n = 3386–5536 spines from 3–4 mice). *p < 0.05 (Student's t test) and **p < 0.01 (Kolmogorov–Smirnov test) compared with Girdin+/+ mice.
We next examined whether phosphorylated Girdin S1416 is involved in the synaptic properties. For this purpose, electrophysiological recording was performed in the DG of hippocampal slices prepared from 8- to 12-week-old mice. In addition to GirdinSA/SA mice, we also analyzed heterozygous Girdin-deficient (Girdin+/−) mice, which have 50% loss of Girdin expression because homozygous (Girdin−/−) mice died within a month after the birth (Kitamura et al., 2008). The strength of basal synaptic transmission was analyzed at the MPP-DGC synapses between Girdin+/+ and GirdinSA/SA mice, or between Girdin+/+ and Girdin+/− mice. The basal synaptic transmission was estimated by measuring the amplitude of a presynaptic fiber volley (PSFV) and the initial slope of the fEPSP over a range of stimulus intensities, and input–output relationships in the synaptic transmission could be established by plotting the fEPSP slope against the PSFV amplitude (Fig. 3A,B). The mean value of the fEPSP slope in GirdinSA/SA mice was smaller than in Girdin+/+ mice over the stimulus range, but the difference was not significant (Fig. 3A). There were no marked differences in the PSFV-fEPSP curves between Girdin+/+ and Girdin+/− mice (Fig. 3B). We further examined the paired-pulse ratio, which depends on presynaptic properties in Girdin+/+, GirdinSA/SA, and Girdin+/− mice. The MPP-DGC synapses exhibit paired-pulse depression (PPD; McNaughton, 1980; Brown and Reymann, 1995). All genotypes of mice showed similar levels of PPD without any significant difference between the interevent interval (Fig. 3C,D). These results indicate that synaptic transmission at the MPP-DGC synapses in GirdinSA/SA and Girdin+/− mice is normal.
Figure 3.

Impairments of NMDA receptor-dependent hippocampal synaptic plasticity in DGCs in GirdinSA/SA and Girdin+/− mice. A, B, Normal input–output curve at hippocampal MPP-DG synapses in GirdinSA/SA (A) and Girdin+/− (B) mice. Data are expressed as mean ± SEM (n = 10 for Girdin+/+, n = 10 for GirdinSA/SA in A; n = 10 for Girdin+/+, n = 10 for Girdin+/− in B). C, D, Normal paired-pulse ratio in GirdinSA/SA (C) and Girdin+/− (D) mice. Data are expressed as mean ± SEM (n = 9 for Girdin+/+, n = 9 for GirdinSA/SA in C; n = 9 for Girdin+/+, n = 9 for Girdin+/− in D). E–H, Impaired LTP in GirdinSA/SA (E, F) and Girdin+/− (G, H) mice. LTP was induced by TET (four trains of 100 Hz), and the fEPSP slope was recorded for 60 min after the TET. E, G, The LTP in all genotypes of mice was completely abolished by the NMDA receptor antagonist AP5 (100 μm). F, H, Quantification of the fold changes in the average fEPSP slope 0–5, 25–30, and 55–60 min after TET. Data are reported as mean ± SEM (n = 4–5 for Girdin+/+, n = 4–5 for GirdinSA/SA in E, F; n = 4–6 for Girdin+/+, n = 4–6 for Girdin+/− in G, H. *p < 0.05, **p < 0.01 compared with Girdin+/+ mice (Student's t test). I, K, Typical recordings of the AMPA and NMDA receptor-mediated EPSCs in GirdinSA/SA (I) and Girdin+/− (K) mice. J, L, Reduced NMDA/AMPA ratio in the DGCs in GirdinSA/SA (J) and Girdin+/− (L) mice. Data are expressed as mean ± SEM (n = 8 for Girdin+/+, n = 7 for GirdinSA/SA in J; n = 8 for Girdin+/+, n = 7 for Girdin+/− in L). **p < 0.01 compared with Girdin+/+ mice (Student's t test). M, N, Normal decay time constant of NMDA receptor-mediated EPSCs in the DGCs in GirdinSA/SA (M) and Girdin+/− (N) mice. Data are expressed as mean ± SEM (n = 8 for Girdin+/+, n = 7 for GirdinSA/SA in M; n = 8 for Girdin+/+, n = 7 for Girdin+/− in N). Electrophysiological recording was performed in the DG of 8- to 12-week-old mouse hippocampal slices. n indicates the number of slices tested.
Next, we examined synaptic responses to TET (four trains of 100 Hz) in Girdin+/+, GirdinSA/SA, and Girdin+/− mice at the age of 8–12 weeks. In the slices from Girdin+/+ mice, the TET stimulus resulted in enhancement of the fEPSP slope that persisted for >60 min, indicative of LTP (Fig. 3E–H). The same experimental conditions reliably induced LTP in the DG from GirdinSA/SA mice, but the magnitude of LTP in GirdinSA/SA mice was smaller than that in Girdin+/+ mice (Girdin+/+, 182.0 ± 2.4% of baseline at 55–60 min after the TET; GirdinSA/SA, 146.8 ± 2.6% of baseline at 55–60 min after the TET, p < 0.01; Fig. 3E,F). A significant reduction of LTP magnitude was also observed in Girdin+/− mice (Girdin+/+, 194.5 ± 6.4% of baseline at 55–60 min after the TET; Girdin+/−, 164.7 ± 5.2% of baseline at 55–60 min after the TET, p < 0.01; Fig. 3G,H). Moreover, the LTP in all genotypes of mice was completely abolished by the pretreatment with the NMDA receptor antagonist AP5 at a concentration of 100 μm (Fig. 3E,G), indicating that the NMDA receptors play a crucial role in the induction of LTP under our experimental conditions. We therefore analyzed the ratio of EPSCs mediated by NMDA receptor to AMPA receptor (NMDA/AMPA ratio) in DGCs of the hippocampal slices from Girdin+/+, GirdinSA/SA, and Girdin+/− mice. The NMDA/AMPA ratio in GirdinSA/SA mice was significantly lower than that in Girdin+/+ mice (p < 0.01; Fig. 3I,J). Girdin+/− mice also display marked reduction of NMDA/AMPA ratio compared with Girdin+/+ mice (p = 0.05; Fig. 3K,L). However, both GirdinSA/SA and Girdin+/− mice showed a normal decay time constant of NMDA receptor-mediated EPSCs, which is correlated with the gating kinetics of the channel (Fig. 3M,N). Electrophysiological analysis revealed that GirdinSA/SA and Girdin+/− mice have reduction of the LTP magnitude and NMDA/AMPA ratio in the DG, but synaptic transmission at the MPP-DGC synapses and decay time constant of NMDA receptor-mediated EPSCs is normal. These results suggest that the reduction of the functional NMDA receptors, but not gating kinetics, in DGCs of GirdinSA/SA and Girdin+/− mice causes impairment of the LTP in the DG. Furthermore, GirdinSA/SA mice represented the synaptic properties of Girdin+/− mice, indicating phosphorylation of Girdin S1416 may be responsible for synaptic plasticity associated with LTP induction.
Phosphorylation of Girdin S1416 is required for NR2B phosphorylation
Our next concern is the causal relationship between Girdin S1416 phosphorylation and NMDA receptors. Several studies indicated that tyrosine phosphorylation of NMDA receptor 2 subunit (NR2), especially NR2A and NR2B, can regulate the activity of NMDA receptors, and that the phosphorylation is involved in LTP induction, memory formation, and synaptic plasticity (Wang and Salter, 1994; Köhr and Seeburg, 1996; Rosenblum et al., 1996; Rostas et al., 1996; Salter and Kalia, 2004). It is also well known that the BDNF-TrkB signaling pathway is involved in LTP, hippocampus-dependent learning, spatial memory, and fear memory formation (Minichiello, 2009). First, we examined the effect of BDNF on the phosphorylation level of NR2A and NR2B in primary cultured hippocampal neurons. Primary cultured hippocampal neurons were prepared from Girdin+/+ mice on gestational days 15–16. On DIV 14, hippocampal primary cultured neurons were treated with PBS (control) or 50 ng/ml BDNF for 5 min, 15 min, or 30 min. Time-course analysis revealed that BDNF had little effect on NR2A phosphorylation (Fig. 4A), whereas the same treatment induced phosphorylation of NR2B at 15 min or 30 min after the treatment (p < 0.05; Fig. 4B). These results are consistent with previous findings (Lin et al., 1998; Xu et al., 2006), and suggest that phosphorylation of NR2B, but not NR2A, is regulated by the BDNF-TrkB signaling pathway. Next, to test the involvement of Girdin phosphorylation in this process, we examined the effect of BDNF on NR2B phosphorylation in primary cultured hippocampal neurons of GirdinSA/SA mice. The application of BDNF significantly increased the level of phosphorylated Akt in both Girdin+/+ and GirdinSA/SA neurons (p < 0.01; Fig. 4C). BDNF treatment significantly increased the level of phosphorylated Girdin in Girdin+/+ neurons whereas BDNF-induced Girdin phosphorylation was at an undetectable level in GirdinSA/SA neurons (p < 0.01; Fig. 4D). Furthermore, BDNF treatment significantly increased the phosphorylated NR2B level in Girdin+/+ neurons, but had no effects in GirdinSA/SA neurons (p < 0.01; Fig. 4E). There were no significant differences in total Akt, Girdin, and NR2B between the two groups (data not shown). These results indicated that Akt-mediated phosphorylation of Girdin is required for BDNF-induced phosphorylation of NR2B in the hippocampus.
Figure 4.

Akt-mediated Girdin S1416 phosphorylation regulates BDNF-induced NR2B phosphorylation of NMDA receptor through Src kinase. A, B, Time-course changes in the levels of phosphorylated NR2A (A) and NR2B (B) after BDNF (50 ng/ml) application in cultured E15–E16 mouse hippocampal neurons (DIV 14). Data are expressed as mean ± SEM (n = 4 in each time point for A; n = 5 in each time point for B). One-way ANOVA, F(3,16) = 6.021, p < 0.01 for B. *p < 0.05 compared with control (Tukey–Kramer post hoc test). C–E, Western blot analyses showing the levels of phosphorylated Akt, Girdin, and NR2B after BDNF application. Cultured E15–E16 mouse hippocampal neurons (DIV 14) were treated with PBS (control) or BDNF (50 ng/ml) for 15 min. C, No changes in BDNF-induced Akt phosphorylation in Girdin+/+ and GirdinSA/SA neurons. D, E, BDNF-induced phosphorylation of Girdin S1416 and NR2B was abolished in GirdinSA/SA neurons. Data are expressed as mean ± SEM (n = 5 for Girdin+/+; n = 5 for GirdinSA/SA). Two-way ANOVA; C, genotype, F(1,16) = 0.799, p = 0.3846; BDNF, F(1,16) = 16.981, p < 0.01; genotypes × BDNF, F(1,16) = 0.041, p = 0.8420; D, genotype, F(1,16) = 812.318, p < 0.01; BDNF, F(1,16) = 27.516, p < 0.01; genotypes × BDNF, F(1,16) = 27.516, p < 0.01; E, genotype, F(1,16) = 29.733, p < 0.01; BDNF, F(1,16) = 5.069, p < 0.05; genotypes × BDNF, F(1,16) = 14.556, p < 0.01. **p < 0.01 compared with corresponding control. N.D., not detectable. F, Interaction of Girdin with NR2B and Src, but not Fyn. Coimmunoprecipitation among Girdin, NR2B, and Src was performed in HEK293T cells that overexpressed Girdin-V5 and NR2B together with either Src-GFP or Fyn-GFP.
Interactions of Girdin with Src kinase and NR2B
A previous study has shown that Girdin itself lacks kinase activity and that tyrosine phosphorylation of Girdin at Y1764 and Y1798 by the nonreceptor tyrosine kinase Src promotes activation of PI3-K signaling during cell migration (Lin et al., 2011). Accordingly, we assumed that Girdin might interact with NMDA receptors through Src or Fyn kinases, which have been reported as major kinases to phosphorylate the NR2B subunit (Köhr and Seeburg, 1996; Rostas et al., 1996; Nakazawa et al., 2001; Mizuno et al., 2003; Xu et al., 2006). Therefore, NR2B was immunoprecipitated from HEK293T cells that coexpress Girdin-V5, NR2B, and Src-GFP (Fig. 4F). As expected, Girdin and Src were coimmunoprecipitated with NR2B. We further investigated possible interaction among Girdin, NR2B, and Fyn kinase (Fig. 4F). Girdin was not coimmunoprecipitated with NR2B although Fyn was detected in immunoprecipitant, suggesting that Girdin interacts with NR2B through Src kinase, but not Fyn kinase.
Behavioral phenotypes in GirdinSA/SA mice and Girdin+/− mice
To explore the physiological significance of the phosphorylation of Girdin S1416 in vivo, we analyzed the phosphorylation of Akt, Girdin S1416, and NR2B in the DG of the hippocampus of 8- to 9-week-old Girdin+/+ mice after training for the fear-conditioning test. The level of phosphorylated Akt in the DG of the hippocampus was significantly increased 5 min after fear conditioning (p < 0.05; Fig. 5A). The phosphorylation levels of Girdin S1416 and NR2B Y1472 were significantly increased 15 and 30 min after training, respectively, after fear conditioning (p < 0.05; Fig. 5B,C). Since fear conditioning increased the phosphorylation of Girdin as well as Akt and NR2B in the hippocampus, we examined the performance of Girdin+/+, GirdinSA/SA, and Girdin+/− mice at the age of 7–20 weeks in the fear-conditioning test. GirdinSA/SA and Girdin+/− mice showed context-dependent fear memory impairment 24 h, but not 1 h, after fear conditioning (p < 0.01; Fig. 6A,B), although there was no significant difference in freezing time in the conditioning phase (Table 1). GirdinSA/SA mice also showed impairment of tone-dependent freezing 24 h after the conditioning (p < 0.01), whereas Girdin+/− mice exhibited normal tone-dependent freezing as did Girdin+/+ mice (Table 1). The stimulus thresholds of electric footshock for vocalization were indistinguishable among Girdin+/+, GirdinSA/SA, and Girdin+/− mice (data not shown). The ability of nonspatial memory was also analyzed by the novel object recognition test. In the retention session, which was performed 24 h after the training session, a marked decrease in the exploratory preference for novel objects was evident in GirdinSA/SA and Girdin+/− mice compared with that in Girdin+/+ mice (p < 0.01; Fig. 6C,D). There was no significant difference in the total time taken to explore two objects during the training and retention sessions in Girdin+/+, GirdinSA/SA, and Girdin+/− mice (Table 1). Spatial memory was assessed by the Morris water maze test. GirdinSA/SA and Girdin+/− mice took a longer path length to find the hidden platform than Girdin+/+ mice during training trials (p < 0.05; Fig. 6E,F), although there was no significant difference in the visible test on day 7 and in the swimming velocity during the training trials (Table 1). In addition, Girdin+/− mice showed impairments of time spent and the number of platform crossings in the target quadrant compared with Girdin+/+ mice in the probe test of the Morris water maze test (p < 0.01; Table 1). When short-term memory was assessed by the Y-maze test, no deficits were observed in GirdinSA/SA and Girdin+/− mice (Table 1). These results suggest that phosphorylation of Girdin S1416 is involved in long-term memory, but not short-term memory.
Figure 5.

Increased phosphorylation of Akt, Girdin S1416, and NR2B of NMDA receptor in the hippocampus of mouse after fear conditioning. A–C, Western blot analyses showing the time-course changes in the phosphorylated levels of Akt S473 (A), Girdin S1416 (B), and NR2B Y1472 (C) in the DG. The DG of 8- to 9-week-old mice were removed 5 min, 15 min, or 30 min after fear conditioning. Control mice were placed in a conditioning chamber without shock and tone. Data are expressed as mean ± SEM (n = 5 mice per time point). One-way ANOVA; F(3,16) = 4.795, p < 0.05 for A; F(3,16) = 4.909, p < 0.05 for B; F(3,16) = 4.120, p < 0.05 for C. *p < 0.05 compared with control mice (Tukey–Kramer post hoc test).
Figure 6.

Behavioral abnormalities in adult GirdinSA/SA and Girdin+/− mice. A, B, Performance in fear-conditioning test in GirdinSA/SA (A) and Girdin+/− (B) mice. Context-dependent freezing was measured 1 and 24 h after fear conditioning. Data are expressed as mean ± SEM (n = 8 for 1 h after the conditioning in Girdin+/+, n = 15 for 24 h after the conditioning in Girdin+/+, n = 10 for 1 h after the conditioning in GirdinSA/SA, n = 15 for 24 h after the conditioning in GirdinSA/SA in A; n = 6 for 1 h after the conditioning in Girdin+/+, n = 16 for 24 h after the conditioning in Girdin+/+, n = 6 for 1 h after the conditioning in Girdin+/−, n = 15 for 24 h after the conditioning in Girdin+/− in B). **p < 0.01 compared with Girdin+/+ mice (Student's t test). C, D, Performance in novel object recognition test in GirdinSA/SA (C) and Girdin+/− (D) mice. Data are expressed as mean ± SEM (n = 16 for Girdin+/+, n = 15 for GirdinSA/SA in C; n = 16 for Girdin+/+, n = 16 for Girdin+/− in D). **p < 0.01 compared with Girdin+/+ mice (Student's t test). E, F, Performance in Morris water maze test in GirdinSA/SA (E) and Girdin+/− (F) mice. Path length (swim distance) to find the hidden platform was measured during training trials (days 1–6). Data are expressed as mean ± SEM (n = 14 for Girdin+/+, n = 14 for GirdinSA/SA in E; n = 15 for Girdin+/+, n = 14 for Girdin+/− in F). Repeated-measures ANOVA for E, genotypes, F(1,26) = 6.284, p < 0.05; days, F(5,130) = 47.858, p < 0.01; genotypes × days, F(5,130) = 1.001, p = 0.4198. Repeated-measures ANOVA for F, genotypes, F(1,27) = 5.969, p < 0.05; days, F(5,135) = 51.334, p < 0.01; genotypes × days, F(5,135) = 1.712, p = 0.1360. *p < 0.05 compared with Girdin+/+ mice (Tukey–Kramer post hoc test). G, Locomotor activity in a novel environment in GirdinSA/SA mice. Data are expressed as mean ± SEM (n = 16 for Girdin+/+; n = 15 for GirdinSA/SA). Repeated-measures ANOVA, genotypes, F(1,29) = 6.519, p < 0.05; times, F(35,1015) = 99.176, p < 0.01; genotypes × times, F(35,1015) = 2.011, p < 0.01. *p < 0.05 compared with Girdin+/+ mice (Tukey–Kramer post hoc test). H, Total locomotor activity during 90–120 min in GirdinSA/SA mice. **p < 0.01 compared with Girdin+/+ mice (Student's t test). I, Performance in PPI test in GirdinSA/SA mice. PPI (%) at four different prepulse intensities (69, 73, 77, and 81 dB) in PPI test. Data are expressed as mean ± SEM (n = 15 for Girdin+/+; n = 14 for GirdinSA/SA). Repeated-measures ANOVA, genotypes, F(1,28) = 5.267, p < 0.05; prepulse intensities, F(3,84) = 46.386, p < 0.01; genotypes × prepulse intensities, F(3,84) = 0.237, p = 0.8705. *p < 0.05 compared with Girdin+/+ mice (Tukey–Kramer post hoc test). Behavioral analyses were performed in 7- to 20-week-old mice.
Table 1.
Summary for behavioral phenotypes in GirdinSA/SA and Girdin+/− mice
| Domain of function | Test | Parameter | Number of animals |
Phenotype in GirdinSA/SA | Number of animals |
Phenotype in Girdin+/− | ||
|---|---|---|---|---|---|---|---|---|
| Girdin+/+ | GirdinSA/SA | Girdin+/+ | Girdin+/− | |||||
| Motor activity | Locomotor | Locomotor activity | 16 | 15 | ↑ (p < 0.05) | 16 | 16 | = |
| Affective behavior | Open field | Total movement | 16 | 15 | = | 16 | 16 | = |
| Inner movement | 16 | 15 | = | 16 | 16 | = | ||
| Elevated-plus maze | Open-arm entries | 15 | 15 | = | 16 | 16 | = | |
| Closed-arm entries | 15 | 15 | = | 16 | 16 | = | ||
| Time in open arms | 15 | 15 | = | 16 | 16 | = | ||
| Time in closed arms | 15 | 15 | = | 16 | 16 | = | ||
| Forced swim | Immobility time | 14 | 13 | = | 13 | 14 | = | |
| Sucrose-preference | Sucrose preference | 7 | 8 | = | 7 | 8 | = | |
| Cognition/learning and memory | PPI | Prepulse (69, 73, 77, and 81 dB) | 15 | 14 | ↓ (p < 0.05) | 16 | 16 | = |
| Y-maze | Alternation | 16 | 15 | = | 16 | 16 | = | |
| Fear conditioning (1 h after training) | Freezing time in conditioning phase | 8 | 10 | = | 6 | 5 | = | |
| Context-dependent freezing | 8 | 10 | = | 6 | 5 | = | ||
| Fear conditioning (24 h after training) | Freezing time in conditioning phase | 15 | 15 | = | 16 | 15 | = | |
| Context-dependent freezing | 15 | 15 | ↓ (p < 0.01) | 16 | 15 | ↓ (p < 0.01) | ||
| Tone-dependent freezing | 15 | 15 | ↓ (p < 0.01) | 16 | 15 | = | ||
| Novel object recognition | Exploratory preference | 16 | 15 | ↓ (p < 0.01) | 16 | 16 | ↓ (p < 0.01) | |
| Exploration time | 16 | 15 | = | 16 | 16 | = | ||
| Morris water maze | Escape latency | 14 | 14 | ↓ (p < 0.05) | 14 | 15 | = | |
| Path length | 14 | 14 | ↓ (p < 0.05) | 14 | 15 | ↓ (p < 0.05) | ||
| Visible test | 14 | 14 | = | 14 | 15 | = | ||
| Post-probe test | 14 | 14 | = | 14 | 15 | ↓ (p < 0.01) | ||
↑, higher than +/+; ↓, lower than +/+; =, no difference.
When locomotor activity in a novel environment was measured for 120 min, Girdin+/+ mice displayed an increase in exploratory activity during the first 0–15 min, and then the activity was gradually reduced as they became habituated to the novel environment. GirdinSA/SA mice displayed an increase of total counts during the last 30 min of the test, suggesting impaired habituation to the novel environment (p < 0.05; Fig. 6G,H). GirdinSA/SA mice also exhibited an impairment of sensorimotor gating as indicated by PPI deficits without changes in the acoustic startle amplitude (p < 0.05; Fig. 6I). Girdin+/− mice exhibited a normal locomotor pattern in a novel environment and normal sensorimotor gating (Table 1). No significant changes were observed in GirdinSA/SA and Girdin+/− mice compared with Girdin+/+ mice in open field, elevated-plus maze, forced-swimming, and sucrose-preference tests (Table 1).Together, behavioral phenotypes of GirdinSA/SA and Girdin+/− mice suggest that the phosphorylation of Girdin S1416 may play a role in the habituation to a novel environment and sensorimotor gating response as well as long-term memory.
Discussion
We demonstrated in the present study that BDNF-induced phosphorylation of Girdin S1416 follows the stimulation of TrkB/Akt signaling. Defect of Girdin S1416 phosphorylation decreased spine volume and head diameter, LTP magnitude, NMDA/AMPA ratio, and NR2B Y1472 phosphorylation in the hippocampal neurons, and impaired long-term memory in GirdinSA/SA mice. Girdin+/− mice were remarkably similar to GirdinSA/SA mice in the points of electrophysiological properties and behavioral phenotypes. Furthermore, we found that Src kinase, but not Fyn kinase, is required for interactions of Girdin with NR2B in HEK293T cells. Accordingly, we propose the following scenario for the role of Girdin S1416 phosphorylation in BDNF/TrkB and NR2B-mediated plasticity changes in the hippocampus. BDNF-triggered Akt phosphorylation can lead through Girdin S1416 phosphorylation to altered Src-mediated Y1472 phosphorylation of NR2B, leading to larger LTP responses through increased head volume and postsynaptic densities (PSDs) with an increased NMDA/AMPA ratio of thin spines in the hippocampus. These structural and functional modifications of synapse contribute to long-term memory formation (Fig. 7).
Figure 7.

Working model of the role of Girdin S1416 phosphorylation in BDNF/TrkB and NR2B-mediated plasticity changes in the hippocampus. BDNF-triggered PI3-K/Akt activation through TrkB can lead phosphorylation of Girdin S1416. Phosphorylated Girdin S1416 makes complex with Src and NR2B to increase Src-mediated Y1472 phosphorylation of NR2B. Activation of NMDA receptors containing NR2B leads to larger LTP responses through increased head volume and PSDs with an increased NMDA/AMPA ratio of thin spines in the hippocampus. The structural and functional modifications of synapse associated with Girdin S1416 phosphorylation contribute to long-term memory formation.
In this study, bath application of BDNF increased Girdin S1416 phosphorylation in cultured mouse E15–E16 hippocampal neurons at DIV 14. The BDNF-induced phosphorylation of Girdin S1416 was abolished by pretreatment with inhibitors for Trk, PI3-K, or Akt. These results suggest that BDNF increases Girdin S1416 phosphorylation through the TrkB/PI3-K/Akt pathway. It should be noted, however, that there may be some differences in BDNF responsiveness between immature and mature hippocampal neurons. BDNF mRNA is detected at E19, peaked at 2 weeks of age, and then gradually decreased in the adult brain (Ernfors et al., 1990). A recent study demonstrated that the effects of BDNF on the dendritic architecture of hippocampal neurons were dependent on the neuron's maturation stage (Kellner et al., 2014). It was also indicated that acute and gradual increases in BDNF concentration elicited distinct signaling and functions in neurons (Ji et al., 2010). To verify the role of Girdn S1416 phosphorylation in BDNF signaling cascade, its phosphorylation should be demonstrated in mature hippocampal slice or tissue.
Although impairment of Girdin has been shown to induce severe defects in neurogenesis, GirdinSA/SA mice are normal in terms of their growth, survival rate, and brain development (Wang et al., 2011). Furthermore, GirdinSA/SA mice showed no apparent defects in migration and positioning of the subgranular zone in the DG (Wang et al., 2011). Our results showed that there were no significant differences in total dendrite length between Girdin+/+ and GirdinSA/SA mice, indicating that Girdn S1416 phosphorylation may have little effect on dendritic elongation.
In adult hippocampus, spine shapes differ categorically, with thin and mushroom types (Bourne and Harris, 2007). GirdinSA/SA mice were characterized by thinner spines accompanied by a small volume and shortened spine head diameter. Thin spines are formed and disappear relatively rapidly in response to different levels of synaptic activity (Bourne and Harris, 2007). Thin spines have smaller PSDs that contain NMDA receptors but few AMPA receptors, making them ready for strengthening by the addition of AMPA receptors (Nimchinsky et al., 2004). Tyrosine phosphorylation of NMDA receptors has been implicated in the LTP (Rostas et al., 1996; Nakazawa et al., 2001). In particular, phosphorylation of NR2B Y1472 has been shown to play a crucial role in NMDA receptor activation (Nakazawa et al., 2001; Nakazawa et al., 2006). In the present study, GirdinSA/SA mice exhibited reduction of NMDA receptor function, which causes the impairment of LTP in DGCs of the hippocampus, while synaptic transmission at the MPP-DGC synapses and synaptic AMPA receptor responses in GirdinSA/SA mice were normal. Additionally, hippocampal neurons derived from GirdinSA/SA mice had a deficit of the phosphorylation of NR2B Y1472 induced by BDNF treatment. The total expression level of NR2B did not differ between Girdin+/+ and GirdinSA/SA hippocampal neurons. These results suggest that phosphorylation of Girdin S1416 is involved in NMDA receptor activation, especially through the phosphorylation of NR2B.
TET stimulus induced LTP in the DG in GirdinSA/SA mice, but the magnitude of LTP was smaller than that in Girdin+/+ mice. The maintenance of LTP depends on both local dendritic protein synthesis (Sutton and Schuman, 2006) and nuclear transcription (Zhou et al., 2006). Therefore, Girdin S1416 phosphorylation may be involved in local dendritic protein synthesis and nuclear transcription in the maintenance of LTP. Another possibility is the structural remodeling of synapses mediated by the actin cytoskeleton (Dillon and Goda, 2005). Since the size of spines was reduced in GirdinSA/SA mice, Akt-mediated Girdin phosphorylation may be associated with activity-dependent structural remodeling of synapses after the induction of LTP.
It has been shown that both Src and Fyn kinases directly phosphorylate NR2B Y1472, which has been implicated in LTP and NMDA receptor activation (Köhr and Seeburg, 1996; Lu et al., 1998; Salter and Kalia, 2004). Consistent with previous reports, Src and Fyn were coimmunoprecipitated with NR2B, but Girdin was detected in Src-transfected, but not Fyn-transfected, HEK 293T cells. It has been reported that Src phosphorylates tyrosine of Girdin at Y1764 and Y1798 (Lin et al., 2011). Therefore, Girdin S1416 phosphorylation seems to regulate interaction between Girdin/Src complex and NR2B. The underlying mechanism by which phosphorylated Girdin S1416 regulates NR2B phosphorylation should be elucidated in neurons because we demonstrated interaction of Girdin/Src complex and NR2B under non-neuronal physiological conditions in HEK293T cells. As future direction, we speculate a possible explanation that Girdin S1416 phosphorylation may modulate the localization of Girdin/Src in the cell. It has been reported that Girdin binds to membrane-bound phosphoinositides PI(4)P and PI(3)P, which reside in the plasma membrane and Golgi apparatus, and Akt-mediated phosphorylation allows Girdin to localize at the leading edge and contribute to the formation of short-branched actin filaments in the lamellipodium (Enomoto et al., 2005). Therefore, phosphorylation of Girdin S1416 by Akt may regulate trafficking of the Girdin/Src complex from the plasma membrane to postsynaptic specializations such as PSDs containing a high concentration of NMDA receptors and associated cytoskeletal and signaling proteins.
GirdinSA/SA mice have impairments of long-term memory in fear-conditioning, Morris water maze, and novel object recognition tests. In contrast, short-term memory was normal in GirdinSA/SA mice, as analyzed by Y-maze test and context-dependent test 1 h after fear conditioning. Similar behavioral phenotypes were manifested in Girdin+/− mice. These results are consistent with a previous report demonstrating that Akt-1−/− mice show impairments of context-dependent fear memory and recall of spatial learning (Balu et al., 2012). Furthermore, we demonstrated that phosphorylated levels of Akt and Girdin S1416 in the DG of the hippocampus increase after fear conditioning. These results indicate that the Akt-mediated phosphorylation of Girdin S1416 is involved in hippocampus-dependent long-term memory formation. In addition to memory formation, we found that the phosphorylation of Girdin S1416 may play a role in the habituation to a novel environment and sensorimotor gating response, as indicated by locomotor activity and PPI tests. These impairments are hallmarks of current animal models for mental disorders (Mouri et al., 2013). A growing body of evidence from human and mouse studies suggests that Akt signaling may be important as a risk factor for schizophrenia (Arguello and Gogos, 2008). In addition to memory impairments, Akt-1−/− mice also showed impairments of PPI of the acoustic response (Balu et al., 2012). Therefore, it is possible that interaction of Girdin and Akt may contribute to cognitive dysfunction and sensorimotor gating abnormality in mental disorders.
Footnotes
This work was supported by the following funding sources: Japan Society for the Promotion of Science (JSPS) KAKENHI (243493) from JST; Exploratory Research from JSPS; “Integrated Research on Neuropsychiatric Disorders” and “Bioinformatics for Brain Sciences” performed under the SRPBS from Ministry of Education, Culture, Sports, Science, and Technology (MEXT); Core Research for Evolutional Science and Technology; Grants-in-Aid for Scientific Research (24111518, 25116515, 25460093, 26670121, and 26293053) from MEXT; Health and Labour Sciences Research Grant (H25-Iyaku-Ippan-020) from MHLW; Intramural Research Grant (24-12) for Neurological and Psychiatric Disorders of National Center of Neurology and Psychiatry; and SRF Grant for Biomedical Research. We thank the Division for Research of Laboratory Animals, Center for Research of Laboratory Animals, and Medical Research Engineering (technical staff: Yasutaka Ohya and Kumiko Yano) for animal care and use.
The authors declare no competing financial interests.
References
- Arguello PA, Gogos JA. A signaling pathway AKTing up in schizophrenia. J Clin Invest. 2008;118:2018–2021. doi: 10.1172/JCI35931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan Y, Abazyan B, Nomura J, Kim R, Ladenheim B, Krasnova IN, Sawa A, Margolis RL, Cadet JL, Mori S, Vogel MW, Ross CA, Pletnikov MV. Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol Psychiatry. 2011;16:293–306. doi: 10.1038/mp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Carlson GC, Talbot K, Kazi H, Hill-Smith TE, Easton RM, Birnbaum MJ, Lucki I. Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus. 2012;22:230–240. doi: 10.1002/hipo.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Brown RE, Reymann KG. Metabotropic glutamate receptor agonists reduce paired-pulse depression in the dentate gyrus of the rat in vitro. Neurosci Lett. 1995;196:17–20. doi: 10.1016/0304-3940(95)11825-H. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/S0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Burgess N, Maguire EA, O'Keefe J. The human hippocampus and spatial and episodic memory. Neuron. 2002;35:625–641. doi: 10.1016/S0896-6273(02)00830-9. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Lütjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catches JS, Xu J, Contractor A. Genetic ablation of the GluK4 kainate receptor subunit causes anxiolytic and antidepressant-like behavior in mice. Behav Brain Res. 2012;228:406–414. doi: 10.1016/j.bbr.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Duguid I, Sjöström PJ. Novel presynaptic mechanisms for coincidence detection in synaptic plasticity. Curr Opin Neurobiol. 2006;16:312–322. doi: 10.1016/j.conb.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9:389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Asai N, Namba T, Wang Y, Kato T, Tanaka M, Tatsumi H, Taya S, Tsuboi D, Kuroda K, Kaneko N, Sawamoto K, Miyamoto R, Jijiwa M, Murakumo Y, Sokabe M, Seki T, Kaibuchi K, Takahashi M. Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron. 2009;63:774–787. doi: 10.1016/j.neuron.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Ibáñez CF, Ebendal T, Olson L, Persson H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Natl Acad Sci U S A. 1990;87:5454–5458. doi: 10.1073/pnas.87.14.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego, CA: Academic; 1997. [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/S0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Ibi D, Takuma K, Koike H, Mizoguchi H, Tsuritani K, Kuwahara Y, Kamei H, Nagai T, Yoneda Y, Nabeshima T, Yamada K. Social isolation rearing-induced impairment of the hippocampal neurogenesis is associated with deficits in spatial memory and emotion-related behaviors in juvenile mice. J Neurochem. 2008;105:921–932. doi: 10.1111/j.1471-4159.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Kitahara Y, Mizoguchi H, Koike H, Shiraki A, Takuma K, Kamei H, Noda Y, Nitta A, Nabeshima T, Yoneda Y, Yamada K. Neonatal polyI:C treatment in mice results in schizophrenia-like behavioral and neurochemical abnormalities in adulthood. Neurosci Res. 2009;64:297–305. doi: 10.1016/j.neures.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Koike H, Kitahara Y, Mizoguchi H, Niwa M, Jaaro-Peled H, Nitta A, Yoneda Y, Nabeshima T, Sawa A, Yamada K. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res. 2010;206:32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Nakajima A, Mizoguchi H, Kawase T, Tsuboi D, Kano S, Sato Y, Hayakawa M, Lange UC, Adams DJ, Surani MA, Satoh T, Sawa A, Kaibuchi K, Nabeshima T, Yamada K. Astroglial IFITM3 mediates neuronal impairments following neonatal immune challenge in mice. Glia. 2013;61:679–693. doi: 10.1002/glia.22461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Lu Y, Yang F, Shen W, Tang TT, Feng L, Duan S, Lu B. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci. 2010;13:302–309. doi: 10.1038/nn.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens HA, Amancherla K, Johnson RW. Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci. 2012;32:3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner Y, Gödecke N, Dierkes T, Thieme N, Zagrebelsky M, Korte M. The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front Synaptic Neurosci. 2014;6:5. doi: 10.3389/fnsyn.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, Jiang P, Watanabe T, Usukura J, Kondo T, Costantini F, Murohara T, Takahashi M. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- Köhr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol. 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Ibi D, Mizoguchi H, Nagai T, Nitta A, Takuma K, Nabeshima T, Yoneda Y, Yamada K. Behavioral abnormality and pharmacologic response in social isolation-reared mice. Behav Brain Res. 2009;202:114–121. doi: 10.1016/j.bbr.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Lin C, Ear J, Pavlova Y, Mittal Y, Kufareva I, Ghassemian M, Abagyan R, Garcia-Marcos M, Ghosh P. Tyrosine phosphorylation of the Galpha-interacting protein GIV promotes activation of phosphoinositide 3-kinase during cell migration. Sci Signal. 2011;4:ra64. doi: 10.1126/scisignal.2002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res. 1998;55:20–27. doi: 10.1016/S0169-328X(97)00349-5. [DOI] [PubMed] [Google Scholar]
- Liu YB, Lio PA, Pasternak JF, Trommer BL. Developmental changes in membrane properties and postsynaptic currents of granule cells in rat dentate gyrus. J Neurophysiol. 1996;76:1074–1088. doi: 10.1152/jn.1996.76.2.1074. [DOI] [PubMed] [Google Scholar]
- Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/S0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton BL. Evidence for two physiologically distinct perforant pathways to the fascia dentata. Brain Res. 1980;199:1–19. doi: 10.1016/0006-8993(80)90226-7. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, He J, Nakajima A, Nabeshima T. Involvement of BDNF receptor TrkB in spatial memory formation. Learn Mem. 2003;10:108–115. doi: 10.1101/lm.56003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouri A, Sasaki A, Watanabe K, Sogawa C, Kitayama S, Mamiya T, Miyamoto Y, Yamada K, Noda Y, Nabeshima T. MAGE-D1 regulates expression of depression-like behavior through serotonin transporter ubiquitylation. J Neurosci. 2012;32:4562–4580. doi: 10.1523/JNEUROSCI.6458-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouri A, Nagai T, Ibi D, Yamada K. Animal models of schizophrenia for molecular and pharmacological intervention and potential candidate molecules. Neurobiol Dis. 2013;53:61–74. doi: 10.1016/j.nbd.2012.10.025. [DOI] [PubMed] [Google Scholar]
- Nagai T, Takuma K, Kamei H, Ito Y, Nakamichi N, Ibi D, Nakanishi Y, Murai M, Mizoguchi H, Nabeshima T, Yamada K. Dopamine D1 receptors regulate protein synthesis-dependent long-term recognition memory via extracellular signal-regulated kinase 1/2 in the prefrontal cortex. Learn Mem. 2007;14:117–125. doi: 10.1101/lm.461407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO J. 2006;25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K. The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci. 2004;24:2054–2064. doi: 10.1523/JNEUROSCI.5066-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RM. The versatile synapse. J Exp Biol. 1984;112:199–224. doi: 10.1242/jeb.112.1.199. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Dudai Y, Richter-Levin G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc Natl Acad Sci U S A. 1996;93:10457–10460. doi: 10.1073/pnas.93.19.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostas JA, Brent VA, Voss K, Errington ML, Bliss TV, Gurd JW. Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-D-aspartate receptor in long-term potentiation. Proc Natl Acad Sci U S A. 1996;93:10452–10456. doi: 10.1073/pnas.93.19.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/S0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- Spigelman I, Zhang L, Carlen PL. Patch-clamp study of postnatal development of CA1 neurons in rat hippocampal slices: membrane excitability and K+ currents. J Neurophysiol. 1992;68:55–69. doi: 10.1152/jn.1992.68.1.55. [DOI] [PubMed] [Google Scholar]
- Squire LR, Stark CE, Clark RE. The medial temporal lobe. Annu Rev Neurosci. 2004;27:279–306. doi: 10.1146/annurev.neuro.27.070203.144130. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/S0959-4388(00)00091-X. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kaneko N, Asai N, Enomoto A, Isotani-Sakakibara M, Kato T, Asai M, Murakumo Y, Ota H, Hikita T, Namba T, Kuroda K, Kaibuchi K, Ming GL, Song H, Sawamoto K, Takahashi M. Girdin is an intrinsic regulator of neuroblast chain migration in the rostral migratory stream of the postnatal brain. J Neurosci. 2011;31:8109–8122. doi: 10.1523/JNEUROSCI.1130-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Plummer MR, Len GW, Nakazawa T, Yamamoto T, Black IB, Wu K. Brain-derived neurotrophic factor rapidly increases NMDA receptor channel activity through Fyn-mediated phosphorylation. Brain Res. 2006;1121:22–34. doi: 10.1016/j.brainres.2006.08.129. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70:304–322. doi: 10.1002/dneu.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, Takemori H, Xiong ZQ. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]