

Abstract
Development of correct topographical connections between peripheral receptors and central somatosensory stations requires activity-dependent synapse refinement, in which the NMDA type of glutamate receptors plays a key role. Here we compared functional roles of GluN2B (GluRε2 or NR2B) and GluN2D (GluRε4 or NR2D), two major regulatory subunits of neonatal NMDA receptors, in development of whisker-related patterning at trigeminal relay stations. Compared with control littermates, both the appearance of whisker-related patterning and the termination of the critical period, as assessed by unilateral infraorbital nerve transection, were delayed by nearly a day in the somatosensory cortex of GluN2B+/− mice but advanced by nearly a day in GluN2D−/− mice. Similar temporal shifts were found at subcortical relay stations in the thalamus and brainstem of GluN2B+/− and GluN2D−/− mice. In comparison, the magnitude of lesion-induced critical period plasticity in the somatosensory cortex, as assessed following row-C whisker removal, was normal in both mutants. Thus, GluN2B and GluN2D play counteractive roles in temporal development and maturation of somatosensory maps without affecting the magnitude of critical period plasticity. To understand the opposing action, we then examined neuronal and synaptic expressions of the two subunits along the trigeminal pathway. At each trigeminal station, GluN2B was predominant at asymmetrical synapses of non-GABAergic neurons, whereas GluN2D was selective to asymmetrical synapses of GABAergic neurons. Together, our findings suggest that GluN2B expressed at glutamatergic synapses on glutamatergic projection neurons facilitates refinement of ascending pathway synapses directly, whereas GluN2D expressed at glutamatergic synapses on GABAergic interneurons delays it indirectly.
Keywords: barrel, critical period, GluN2B, GluN2D, NMDA receptor, somatosensory
Introduction
The precise pattern of neural connectivity depends on both activity-dependent and activity-independent mechanisms (Goodman and Shatz, 1993). The rodent trigeminal system is an excellent model to study molecular mechanisms underlying activity-dependent map formation and plasticity (Erzurumlu and Gaspar, 2012). Whisker-related sensory maps are precisely represented at multiple trigeminal relay stations in rodent brains. Whisker-related central patterning is referred to as barrelettes in the brainstem trigeminal complex, barreloids in the thalamus, and barrels in the cortex (Woolsey and Van der Loos, 1970). During the critical or sensitive period, lesions to whiskers alter the topographical maps, whereas the same lesions after the critical period have little influence on these maps (Van der Loos and Woolsey, 1973; Woolsey et al., 1979; Belford and Killackey, 1980; Durham and Woolsey, 1984; Chiaia et al., 1992). The NMDA type of glutamate receptors plays a key role in activity-dependent refinement of the whisker-related central patterning (Schlaggar et al., 1993; Li et al., 1994; Iwasato et al., 2000).
Postsynaptic Ca2+ influx through NMDA receptors is involved in bidirectional synaptic plasticity known as LTP and LTD (Lisman, 1989; Bliss and Collingridge, 1993; Malenka and Nicoll, 1993; Bear, 1996). In neonates, NMDA receptor-dependent LTP and LTD are readily induced at thalamocortical synapses and are thought to underlie this synapse refinement (Crair and Malenka, 1995; Feldman et al., 1998). Canonical NMDA receptors consist of the obligatory subunit GluN1 (GluRζ1 or NR1) and any of the four regulatory GluN2 subunits (GluRε or NR2) (Seeburg, 1993; Nakanishi and Masu, 1994; Mori and Mishina, 1995). GluN2B (GluRε2 or NR2B) and GluN2D (GluRε4 or NR2D) are the two regulatory subunits expressed in fetal and neonatal brains (Watanabe et al., 1992; Monyer et al., 1994). Whereas GluN2B−/− mice display neonatal lethality, impaired suckling response, and defective whisker-related central patterning, GluN2D−/− mice grow normally and establish the patterning (Ikeda et al., 1995; Kutsuwada et al., 1996; Mori et al., 1998). Furthermore, GluN2B- and GluN2D-containing receptors confer distinct functional properties in terms of single-channel conductance, glutamate affinity, Mg2+ sensitivity, and deactivation kinetics (Stern et al., 1992; Monyer et al., 1994; Kutsuwada et al., 1996; Momiyama et al., 1996; Vicini et al., 1998; Wyllie et al., 1998; Misra et al., 2000a, b). These distinct properties have been thought to contribute to distinct regulation of postsynaptic Ca2+ dynamics, affect the induction of synaptic plasticity and refinement, and lead to contrasting phenotypes of gene-manipulated mice (Lisman, 1989; Artola and Singer, 1993; Hansel et al., 1997). However, it remains unknown whether their functional roles of GluN2B and GluN2D in synaptic refinement in the somatosensory system are synergistic or distinct.
Here we compared the role of GluN2B and GluN2D in whisker-related central patterning and disclosed their opposing action on somatosensory development and maturation. Intriguingly, GluN2B is mainly expressed in glutamatergic neurons constituting the ascending somatosensory pathway and facilitates the formation and maturation of whisker-related patterning, whereas GluN2D is selective to GABAergic neurons constituting local inhibitory circuits and delays the patterning.
Materials and Methods
Animals.
We obtained mouse pups from heterozygous breeding pairs defective in GluN2B or GluN2D, which were backcrossed to C57BL mice for >10 generations (Ikeda et al., 1995; Kutsuwada et al., 1996). Mice were treated according to the guideline for the care and use of laboratory animals of Hokkaido University School of Medicine. Breeding pairs were mated on the same day of the week, and pups born at the 19th day of gestation were used for experiments. The day after overnight mating was counted as embryonic day 0 (E0), and the day of birth within 24 h was designated as postnatal day 0 (P0).
Genotyping.
The genotypes of GluN2B mice were determined by PCR using tail genomic DNA (Takeuchi et al., 2001), whereas genotyping of GluN2D mice was performed using primers E4P1 (5′-ATGAGATTGAGATGCTGGAGCGGCTGTGGC-3′), E4P2 (5′-CTGGAGAAGGCCAGTAGGAAATCCATGCGG-3′), and NeoP1 (Kiyama et al., 1998). The nested PCR products derived from wild-type and mutated GluN2D alleles were 231 and 379 bp long, respectively. Statistical differences in the body weight of mutant and control pups were evaluated by Student's t test.
Fixation.
Mice were fixed by transcardial perfusion with 4% PFA in 0.1 m sodium phosphate buffer (PB), pH 7.2, for light microscopy and 4% PFA/0.1% glutaraldehyde in PB for postembedding immunogold microscopy. Flattened horizontal sections through cortical barrels and coronal brainstem sections were prepared using a microslicer (40 μm) or a cryostat (30 μm), respectively.
Cytochrome oxidase histochemistry.
Whisker-related patterning was examined by cytochrome oxidase (CO) histochemistry. Sections were incubated for 12 h at 37°C in a solution containing cytochrome c (3 mg, Sigma), 3,3′-diaminobenzidine (5 mg, Sigma), and sucrose (450 mg) in 10 ml of 0.1 m sodium phosphate buffer, pH 7.2 (Wong-Riley, 1979). CO-stained patterning was photographed using an AX70 bright-field light microscope (Olympus) equipped with a digital camera DP70 (Olympus).
Nissl staining.
Nissl staining was performed with NeuroTrace 500/525 green fluorescent Nissl stain (Invitrogen) for confocal laser scanning microscopy or with cresyl violet for bright-field microscopy.
Infraorbital nerve transection (ION).
To determine the stage of lesion-induced critical period plasticity termination, transection of the right ION was transected under hypothermia-induced anesthesia by making a vertical slit just behind the mystacial pad, cutting the right ION with a pair of iridectomy scissors, and electrocauterizing the cut edge to prevent nerve regeneration. After 8 d for cortical examination or 5 d for subcortical examination, mice were anesthetized by pentobarbital (100 mg/kg of body weight) and killed for CO histochemistry.
Developmental appearance and critical period plasticity termination.
Developmental appearance and critical period plasticity termination in cortical barrels were assessed by measuring the gray density along two crossing lines through C1, C2, and C3 barrels and through B2, C2, and D2 barrels using an Image Gauge V4.0 software (FUJIFILM), as reported previously (Takasaki et al., 2008). By defining the gray density in barrel septa as a baseline, the relative density was summed for each barrel. Developmental appearance and critical period plasticity termination were judged only when the summed relative density exceeded 20,000 arbitrary units in all measured barrels. In subcortical somatosensory stations, developmental appearance and critical period termination were judged by microscopic presence of segregated whisker-related patterning.
To compare genotypic differences, we estimated the age when barrels appeared in 50% of mice (DA50) and the age when the critical period was terminated in 50% of mice (CPT50). To obtain the optimal DA50 and CPT50 values and their 95% confidence intervals, probit regression analysis was used (Finney, 1971); the fraction of mice that had formed barrels or completed the critical period was converted into probits at each postnatal age. Probit-fitted linear regressis were used for calculation. Differences between control and mutant DA50 or CPT50 values were considered statistically significant if there was no overlap between their confidence intervals. Fisher's exact probability test was used for statistical evaluation.
Lesion-induced critical period plasticity.
To assess the magnitude of lesion-induced critical period plasticity in cortical barrels, hair follicles of right row-C whiskers were cauterized at P2, and barrels were examined by CO histochemistry at P15. The absence of whisker regrowth was checked by stereoscopic and histological observations of whisker pads. The area of a pair of barrels, including their intervening septum, was measured for A2 and A3, B2 and B3, C2 and C3, D2 and D3, or E2 and E3, using IPLab software (Scananalytics). From the scores, map plasticity index was calculated by (B2 + B3 + D2 + D3)/2(C2 + C3) (Lu et al., 2001). Statistical differences in map plasticity index were tested by using Mann–Whitney U test, and p values < 0.05 were considered significant.
In situ hybridization.
We used isotopic in situ hybridization with 33P-dATP-labeled 45-mer antisense oligonucleotide probe for GluN2B or GluN2D mRNA (Watanabe et al., 1993) and nonisotopic in situ hybridization with fluorescein- or digoxygenin (DIG)-labeled cRNA probes for GluN2B, GluN2D, or 67 kDa glutamic acid decarboxylase 67 (GAD67) mRNA. Complementary RNA probes were made by in vitro transcription using Bluescript II plasmid vectors encoding mouse GluN2B cDNA (3901–5112 nucleotide residues; GenBank accession number NM_008171), mouse GluN2D cDNA (151–4086; NM_008172), or mouse GAD67 cDNA (1036–2015; NM_008077).
Fresh frozen sections were treated successively after fixation with 4% PFA–PB, pH 7.2, for 10 min, PBS, pH 7.2, for 10 min, acetylation with 0.25% acetic anhydride in 0.1 m triethanolamine-HCl, pH 8.0, for 10 min, and prehybridization for 1 h in a buffer containing 50% formamide, 50 mm Tris-HCl, pH 7.5, 0.02% Ficoll, 0.02% polyvinylpyrrolidone, 0.02% BSA, 0.6 m NaCl, 200 μg/ml of tRNA, 1 mm EDTA, and 10% dextran sulfate. Isotopic in situ hybridization was performed at 42°C for 12 h in prehybridization buffer supplemented with oligonucleotide (10,000 dpm/μl), followed by washing in 0.1 × SSC containing 0.1% SDS at 55°C for 40 min twice and exposure to x-ray film BioMax (Kodak). FISH with DIG- and fluorescein-labeled cRNA probes was as described previously (Yamasaki et al., 2010). In brief, sections were hybridized with a mixture of DIG- or fluorescein-labeled cRNA probes and followed by stringent posthybridization wash. DIG and fluorescein were detected by the two-step method as follows: the first detection with peroxidase-conjugated anti-fluorescein antibody (Roche Diagnostics; 1:500, 1 h) and the FITC-Tyramide Signal Amplification (TSA) plus amplification kit (PerkinElmer Life and Analytical Sciences), and the second detection with peroxidase-conjugated anti-DIG antibody (Roche Diagnostics, 1:500, 1 h) and the Cy3-TSA plus amplification kit (PerkinElmer Life and Analytical Sciences). Residual activities of peroxidase introduced in the first detection were inactivated by incubation of sections with 1.0% H2O2 for 30 min.
Immunofluorescence.
In the present study, we used primary antibodies against GABA, 65/67 kDa GAD, plasmalemmal glutamate transporter-1 (GLT1), GluN2B, GluN2D, parvalbumin (PV), postsynaptic density protein-95 (PSD-95), and Type 2 vesicular glutamate transporter (VGluT2). Information on the antigen, host species, source, and specificity is summarized in Table 1. Of these primary antibodies, we produced affinity-purified guinea pig antibody against mouse GluN2D; the specificity was confirmed by characteristic immunohistochemical labeling in GluN2D+/+, but not GluN2D−/−, brains (see Fig. 7D,E).
Table 1.
List of primary antibodies used in the present studya
| Molecule | Sequence (NCBI #) | Host | Specificity | Reference |
|---|---|---|---|---|
| GABA | Ms | *1 | Sigma (A0310) | |
| GAD65/67 | 268-593 (A28072) | Rb/Go | IB/PT | (Yamada et al., 2001) |
| GLT1 | 550-525 (NM_001077514) | Rb | IB/KO | (Yamada et al., 1998) |
| (Takasaki et al., 2008) | ||||
| GluN2B | 1379-1458 (D10651) | Rb | IB/KO | (Watanabe et al., 1998) |
| GluN2D | 1221-1300 (D12822) | GP | IB/KO | Current study |
| PSD-95 | 1-64 (D50621) | GP | IB | (Fukaya and Watanabe, 2000) |
| PV | 1-110 (NM_013645) | Rb/GP/Go | IB | (Nakamura et al., 2004) |
| VGluT2 | 559-582 aa (BC038375) | GP/Go | IB | (Miyazaki et al., 2003) |
| (Miura et al., 2006) |
aGo, Goat polyclonal antibody; GP, guinea pig polyclonal antibody; IB, immunoblot with brain homogenates; KO, lack of signals in knockout mouse brains; Ms, mouse monoclonal antibody; PT, preabsorption test.
*1, The specificity of monoclonal mouse GABA antibody was confirmed by intense labeling in GAD(+) GABAergic neuronal elements.
Figure 7.

Immunofluorescence showing distinct distribution pattern of GluN2B and GluN2D immunoreactivities at trigeminal relay stations of adult mice. A, D, Overall staining patterns in wild-type mouse brains for GluN2B (A) and GluN2D (D). B, E, The specificity is indicated by substantial reduction of GluN2B immunolabeling in GluN2B+/− brain compared with GluN2B+/+ brain (B), and by the lack of GluN2D immunolabeling in GluN2D−/− brain (E). C, F, Isotopic in situ hybridization for GluN2B (C) and GluN2D (F) mRNAs in adult mouse brains. G, Triple immunofluorescence for GluN2B (red), GluN2D (green), and VGluT2 (blue) in the S1. Arrows indicate the position of barrels labeled for VGluT2. H, I, High-power views of double immunofluorescence for PSD-95 (green) and GluN2B (red) or GluN2D (red) in the S1. Punctate labeling for both subunits (arrows) well overlaps with PSD-95 signals. J–L, High-power views of double immunofluorescence for GluN2B (red) and GluN2D (green) in the S1 (J), VB and RTN (K), and Pr (L). Intense GluN2B-positive puncta virtually lack GluN2D signals (arrowheads), and GluN2D puncta often possess weak GluN2B signals (arrows). I-VI, Cortical layers I through VI; Cb, cerebellum; CPu, caudate-putamen; Cx, cortex; Hi, hippocampus; HT, hypothalamus; Mb, midbrain; MO, medulla oblongata; OB, olfactory bulb; Po, pons; Th, thalamus. Scale bars: A–F, 1 mm; G, 100 μm; H–J, 2 μm; K, 10 μm; L, 5 μm.
All immunohistochemical incubations were performed at room temperature, and 10% normal donkey serum was used for blocking. In immunofluorescence for NMDA receptor subunits, 4-μm-thick paraffin sections were pretreated to expose antigens with 1 mg/ml of pepsin (DAKO) in 0.2 N HCl at 37°C for 3–5 min for developing brains and for 10 min for adult brains, as reported previously (Watanabe et al., 1998). The TSA kit (TSA-Cy3 and TSA-Fluorescein system, PerkinElmer Life and Analytical Sciences) was used to visualize GluN2B and GluN2D immunolabeling. In double TSA labeling, incubation with guinea pig GluN2D antibody (1 μg/ml) was performed first, followed by successive incubation with biotinylated secondary antibodies for 2 h, avidin-biotin-peroxidase complex for 1 h, and TSA amplification for 10 min. After inactivation of residual peroxidase with 1% H2O2 in PBS for 30 min, the second TSA labeling using rabbit GluN2B antibody (1 μg/ml) was performed. When further combining TSA labeling with conventional immunofluorescence using fluorophore-linked secondary antibodies, conventional immunofluorescence with goat VGluT2 antibody (1 μg/ml) was finally applied to TSA-labeled sections. In conventional immunofluorescence for PV and GAD, microslicer sections (50 μm) were incubated with primary antibodies overnight, and then with Alexa488- or Cy3-conjugated secondary antibodies for 2 h. In some sections, cellular nuclei were counterstained with 0.2 μm of TOTO-3 iodide (Invitrogen).
Photographs were taken with a fluorescent microscope (AX-70) and confocal laser-scanning microscope (FV1000, Olympus) equipped with a HeNe/Ar laser, and PlanApo (10×/0.40) and PlanApoN (60×/1.42, oil-immersion) objective lenses (Olympus). Fluorescence crosstalk was prevented by restricting the width of emission wavelength using spectral slit and by adopting the sequential mode of laser scanning. All images show single optical sections (640 × 640 pixels, pixel size 110 nm).
Postembedding immunogold electron microscopy.
Ultrathin sections on nickel grids were etched with saturated sodium-ethanolate solution for 1–5 s and treated with successive solutions: blocking solution containing 2% normal goat serum (Nichirei) in incubation solution (0.03% Triton X-100 in Tris-buffered saline, pH 7.4) for 20 min, primary antibodies (20 μg/ml for GluN2B and GluN2D labeling; 1:1000 dilution for GABA labeling) diluted with incubation solution overnight, and colloidal gold (10 or 15 nm)-conjugated anti-rabbit, anti-guinea pig, or anti-mouse IgG (1:100, British BioCell International) in blocking solution for 2 h. Finally, grids were washed in 0.03% Triton X-100 in Tris-buffered saline for 30 min, fixed with 2% glutaraldehyde in PBS for 15 min and 1% OsO4 for 20 min, and stained with 2% uranyl acetate for 5 min and Reynold's lead citrate solution for 1 min. Photographs were taken with an H-7100 electron microscope (Hitachi). For quantitative analysis, postsynaptic membrane-associated immunogold particles, defined as those <35 nm apart from the cell membrane, were counted on scanned electron micrographs and analyzed using MetaMorph software (Molecular Devices). Measurements were made from three male mice of each strain. Collected data were pooled together because there was no significant difference in the labeling density among three animals examined.
Results
Development and maturation of whisker-related central patterning
To visualize whisker-related patterning, CO histochemistry was applied to brain samples from GluN2B+/−, GluN2D−/−, and control littermates. We compared the stages of developmental appearance and termination of lesion-induced critical period plasticity at three levels of trigeminal relay stations, and the magnitude of lesion-induced critical period plasticity in the primary somatosensory cortex (S1).
Developmental appearance
In control mice, barrels in the S1 were obscure in all pups at P1–P3, and segregated barrels appeared after P4 (Fig. 1A,E,I,M for GluN2B+/+ mice; Fig. 1C,G,K,N for GluN2D+/+ mice; Table 2, top). In comparison, the appearance of barrels was delayed in GluN2B+/− mice (Fig. 1B,F,J,M) and advanced in GluN2D−/− mice (Fig. 1D,H,L,N). The postnatal day at which barrels appeared in 50% of mice was calculated to be P5.1 (99% confidence interval, P4.7–P5.5) and P4.0 (P3.5–P4.4) for GluN2B+/− and control mice, respectively, and P2.9 (P2.4–P3.5) and P3.9 (P3.5–P4.4) for GluN2D−/− and control mice, respectively, showing significant genotypic differences (p < 0.01 for each, Fisher's exact probability test). Temporal shift in developmental appearance of cortical barrels was replicated by VGluT2 immunofluorescence for thalamocortical afferent patterning (Fig. 1O1–R1) and by Nissl staining (Fig. 1S–V) for cortical neuronal arrangement.
Figure 1.
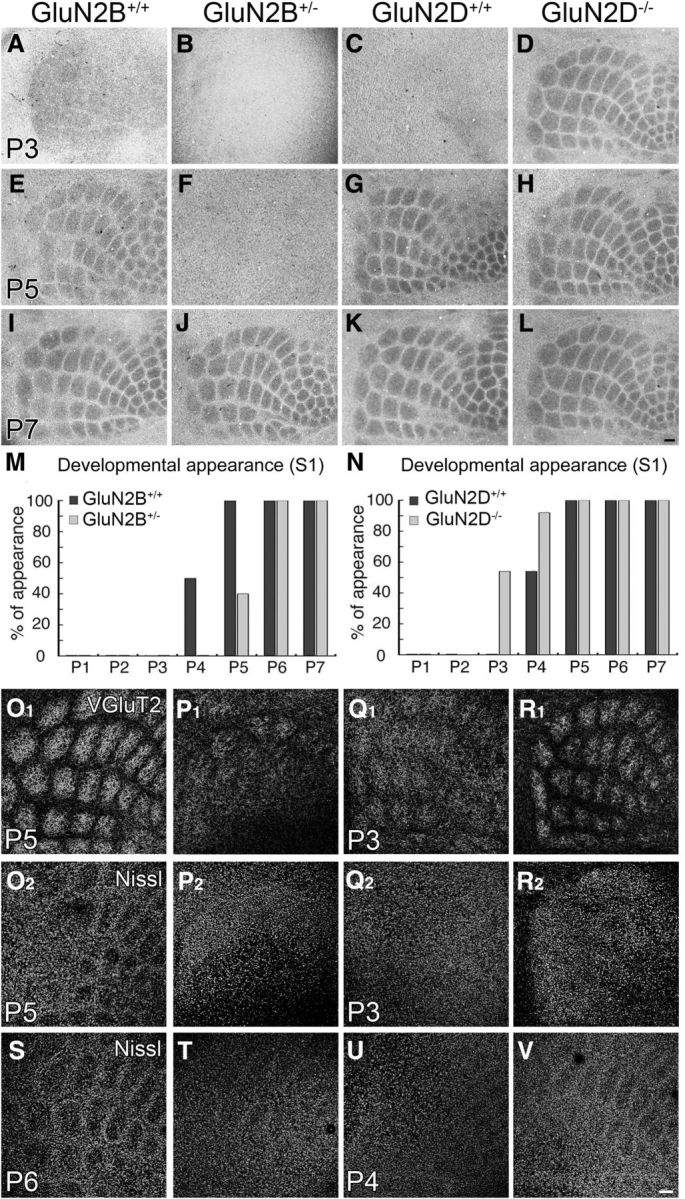
Developmental appearance of cortical barrels is delayed in GluN2B+/− mice and advanced in Glu2D−/− mice. A–L, Cytochrome oxidase histochemistry on flattened cortical sections at P3 (A–D), P5 (E–H), and P7 (I–L). M, N, Bar graphs representing 1 day delay of barrel appearance in GluN2B+/− mice (M) and 1 day advance in GluN2D−/− mice (N). The number of mice examined is listed in Table 2 (top). O–V, Opposing temporal shift between GluN2B+/− and GluN2D−/− mice is reproduced by VGluT2 and Nissl staining. Note the appearance of segregated barrel patterning by VGluT2-positive thalamocortical afferents and Nissl-stained neuronal arrangements in control mice (O), but not GluN2B+/− mice (P), at P5. Also note the appearance of segregated barrel patterning by thalamocortical afferents and neuronal arrangements in GluN2D−/− mice at P3 (R) and P4 (V), respectively, but not in control mice (Q, U). Scale bars, 100 μm.
Table 2.
Percentage of mice in which whisker-related patterning were judged to appear at respective somatosensory stations (developmental appearance)a
| Age | GluN2B+/+ | GluN2B+/− | GluN2D+/+ | GluN2D−/− |
|---|---|---|---|---|
| Cortex | ||||
| P1 | 0 (0/8) | 0 (0/8) | 0 (0/9) | 0 (0/9) |
| P2 | 0 (0/10) | 0 (0/10) | 0 (0/10) | 0 (0/10) |
| P3 | 0 (0/12) | 0 (0/13) | 0 (0/11) | 54 (6/11) |
| P4 | 50 (6/12) | 0 (0/16) | 54 (7/13) | 92 (12/13) |
| P5 | 100 (16/16) | 40 (8/20) | 100 (11/11) | 100 (15/15) |
| P6 | 100 (13/13) | 100 (14/14) | 100 (13/13) | 100 (12/12) |
| P7 | 100 (15/15) | 100 (15/15) | 100 (14/14) | 100 (14/14) |
| Thalamus | ||||
| P0 | 0 (0/3) | 0 (0/2) | 0 (0/2) | 0 (0/2) |
| P1 | 0 (0/5) | 0 (0/4) | 0 (0/4) | 50 (4/8) |
| P2 | 50 (3/6) | 0 (0/6) | 50 (2/4) | 100 (6/6) |
| P3 | 100 (8/8) | 50 (4/8) | 100 (6/6) | 100 (4/4) |
| P4 | 100 (7/7) | 100 (6/6) | 100 (5/5) | 100 (4/4) |
| P5 | 100 (3/3) | 100 (4/4) | 100 (3/3) | 100 (4/4) |
| Brainstem (subnucleus interpolaris) | ||||
| E18 | 0 (0/8) | 0 (0/9) | 0 (0/9) | 100 (0/8) |
| P0 | 100 (8/8) | 0 (0/7) | 100 (7/7) | 100 (9/9) |
| P1 | 100 (6/6) | 100 (7/7) | 100 (7/7) | 100 (8/8) |
aValues in parentheses are % (number of mice judged to have segregated patterning out of the total mice examined).
We also examined the appearance of barreloids in the ventrobasal thalamic nucleus (VB; Fig. 2A–D) and barrelettes in the subnucleus interpolaris of the spinal tract trigeminal nucleus (SpVi; Fig. 2G,H); this subnucleus was selected instead of the principal sensory trigeminal nucleus because of better clarity in the patterning and judgment. Compared with control mice, the appearance of barreloids and barrelettes was also delayed by nearly a day in GluN2B+/− mice (Fig. 2E,I) and advanced by nearly a day in GluN2D−/− mice (Fig. 2F,J) (Table 2, middle and bottom). Therefore, the ablation of GluN2B or GluN2D subunit oppositely shifts the developmental appearance at each trigeminal relay station.
Figure 2.

Opposing temporal shift in developmental appearance of thalamic barreloids and brainstem barrelettes between GluN2B+/− and GluN2D−/− mice. A–D, Cytochrome oxidase-stained thalamic VB. Note the appearance of segregated barreloids in control (A), but not GluN2B+/− (B), mice at P3. Also note the appearance of segregated barreloids in GluN2D−/− (D), but not control (C), mice at P2. E, F, Bar graphs representing 1 day delay of barreloid appearance in GluN2B+/− mice (E) and 1 day advance in GluN2D−/− mice (F). G, H, Cytochrome oxidase-stained subnucleus interpolaris (SpVi) in GluN2B+/− (G) and GluN2D−/− (H) mice at E18 (left), P0 (middle), and P1 (right). Note the first appearance of segregated barrelettes in GluN2B+/− mice at P1, and in GluN2D−/− mice at E18. I, J, Bar graphs representing 1 day delay of barrelette appearance in in GluN2B+/− mice (I) and 1 day advance in GluN2D−/− mice (J). The number of mice examined at each stage is listed in Table 2 (bottom and middle). Scale bars, 200 μm.
Termination of lesion-induced critical period plasticity
The termination stage of critical period plasticity was assessed by CO histochemistry 8 d (S1) or 5 d (VB and SpVi) after transection of the right ION. In control mice, ION transection at P1 and P2 substantially lowered overall CO intensity and blurred patterning in the contralateral hemisphere (Fig. 3A,C). Segregated barrels were maintained in 13%–17% of mice lesioned at P3 and reached 100% at P5–P7 (Fig. 3A,E,I,M,Q for GluN2B+/+ mice; Fig. 3C,G,K,O,R for GluN2D+/+ mice; Table 3, top). By contrast, critical period plasticity termination was delayed in GluN2B+/− mice (Fig. 3B,F,J,N,Q) and advanced in GluN2D−/− mice (Fig. 3D,H,L,P,R). The postnatal day at which the critical period plasticity terminated in 50% of mice was P4.8 (99% confidence interval, P4.4–P5.4) and P3.6 (P3.3–P4.0) for GluN2B+/− and control mice, respectively, and P2.7 (P2.1–P3.2) and P3.6 (P3.1–P4.1) for GluN2D−/− and control mice, respectively, showing significant genotypic differences (p < 0.01 for each, Fisher's exact probability test). Similar temporal shift of critical period plasticity termination was confirmed by VGluT2 immunofluorescence for thalamocortical patterning (Fig. 3S1–V1) and Nissl staining (Fig. 3S2–V2) for neuronal arrangement in the cortex.
Figure 3.
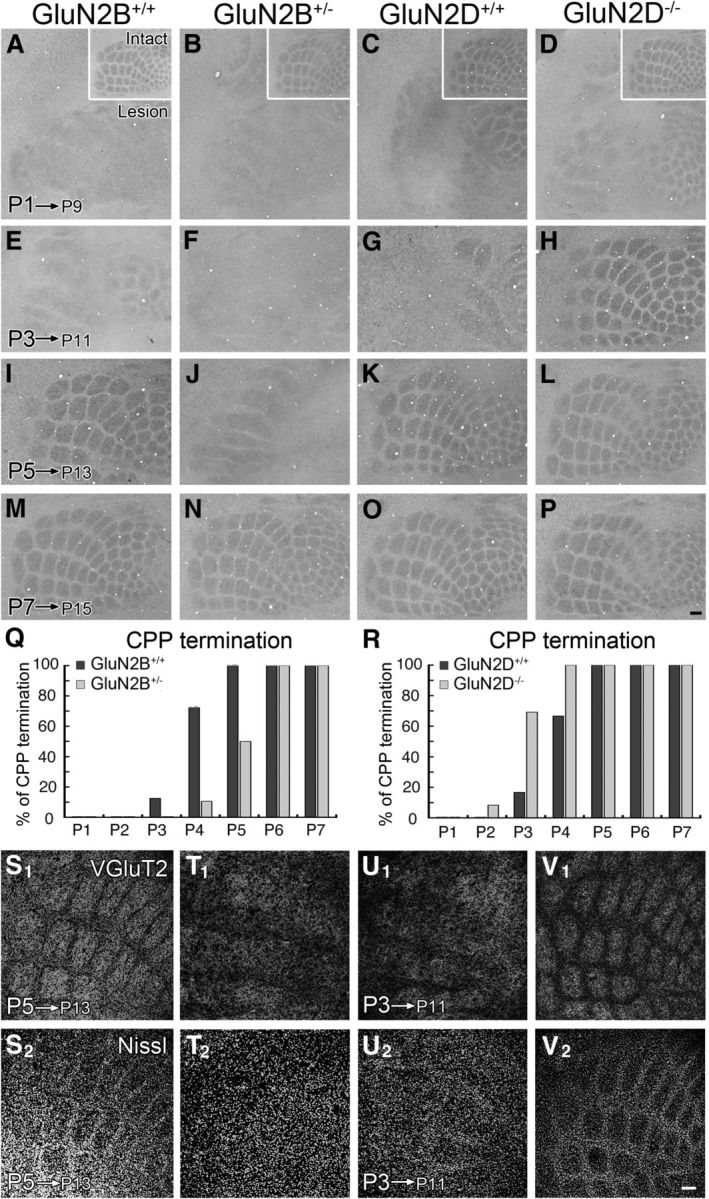
Termination of lesion-induced critical period plasticity in cortical barrels is delayed in GluN2B+/− mice and advanced in GluN2D−/− mice. The right infraorbital nerve was transected at P1 (A–D), P3 (E–H), P5 (I–L), or P7 (M–P), and cytochrome oxidase histochemistry was applied 8 d later. A–P, The contralateral barrel cortices. The ipsilateral cortices are shown in insets (A–D). Q, R, Bar graphs representing 1 day delay of critical period plasticity (CPP) termination in GluN2B+/− mice (Q) and 1 day advance in GluN2D−/− mice (R). The number of mice examined is listed in Table 3 (top). S–V, Opposing temporal shift in CPP termination between GluN2B+/− and GluN2D−/− mice is reproduced by VGluT2 and Nissl staining. The right ION was transected in GluN2B+/− and control mice at P5 (S,T) or GluN2D−/− and control mice at P3 (U, V), and flattened contralateral cortices were subjected to VGluT2 and Nissl staining 8 d later. Critical period plasticity is terminated at P5 in control mice (S), but not in GluN2B+/− mice (T). By contrast, it is terminated as early as P3 in GluN2D−/− mice (V), but not in control (U) mice. Scale bars, 100 μm.
Table 3.
Percentage of mice in which the critical period was complete at respective somatosensory stations (critical period plasticity termination)a
| Age | GluN2B+/+ | GluN2B+/− | GluN2D+/+ | GluN2D−/− |
|---|---|---|---|---|
| Cortex | ||||
| P1 | 0 (0/10) | 0 (0/11) | 0 (0/12) | 0 (0/11) |
| P2 | 0 (0/10) | 0 (0/10) | 0 (0/11) | 8 (1/12) |
| P3 | 13 (2/16) | 0 (0/17) | 17 (2/12) | 69 (9/13) |
| P4 | 72 (13/18) | 10.5 (2/19) | 67 (8/12) | 100 (14/14) |
| P5 | 100 (12/12) | 50 (8/16) | 100 (10/10) | 100 (9/9) |
| P6 | 100 (14/14) | 100 (14/14) | 100 (10/10) | 100 (11/11) |
| P7 | 100 (11/11) | 100 (13/13) | 100 (12/12) | 100 (10/10) |
| Thalamus | ||||
| P0 | 0 (0/3) | 0 (0/3) | 0 (0/3) | 0 (0/4) |
| P1 | 0 (0/5) | 0 (0/4) | 0 (0/4) | 50 (3/6) |
| P2 | 50 (2/4) | 0 (0/5) | 50 (3/6) | 100 (4/4) |
| P3 | 100 (4/4) | 50 (3/6) | 100 (4/4) | 100 (3/3) |
| P4 | 100 (3/3) | 100 (4/4) | 100 (3/3) | 100 (4/4) |
| P5 | 100 (2/2) | 100 (3/3) | 100 (6/6) | 100 (3/3) |
| Brainstem (subnucleus interpolaris) | ||||
| P3 | 0 (0/18) | 0 (0/18) | 0 (5/5) | 0 (5/5) |
| P4 | 0 (0/30) | 0 (0/29) | 0 (0/20) | 25 (12/48) |
| P5 | 25 (7/28) | 0 (0/32) | 25 (4/16) | 56 (9/16) |
| P6 | 67 (16/24) | 22 (8/37) | 69 (9/13) | 90 (/10) |
| P7 | 75 (9/12) | 55 (11/20) | 78 (7/9) | 100 (13/13) |
| P8 | 100 (12/12) | 75 (12/16) | 100 (9/9) | 100 (11/11) |
| P9 | 100 (6/6) | 100 (5/5) | 100 (8/8) | 100 (9/9) |
aValues in parentheses are % (number of mice judged to have segregated patterning out of the total mice examined).
The termination of lesion-induced critical period plasticity in barreloids and barrelettes was also delayed by nearly a day in GluN2B+/− mice (Fig. 4A,B,E for VB; Fig. 4I,J,M for SpVi), and advanced by nearly a day in GluN2D−/− mice (Fig. 4C,D,F for VB; Fig. 4K,L,N for SpVi; Table 3, middle and bottom).
Figure 4.
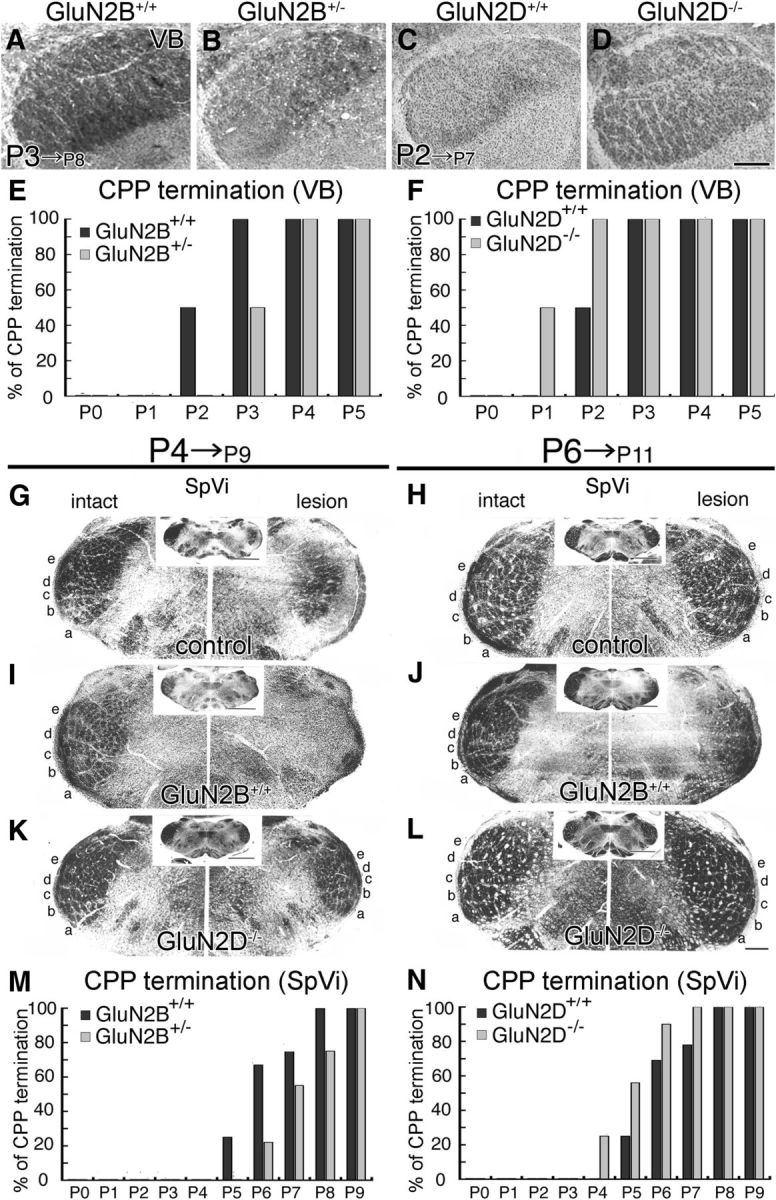
Opposing temporal shift in termination of lesion-induced critical period plasticity in thalamic barreloids and brainstem barrelettes between GluN2B+/− and GluN2D−/− mutants. A–D, Cytochrome oxidase-stained thalamic VB 5 d after right infraorbital nerve lesion. Segregated barreloids are maintained in control (A), but not GluN2B+/− (B), mice lesioned at P3, and in GluN2D−/− (D), but not control (C), mice lesioned at P2. E, F, Bar graphs representing 1 day delay of critical period plasticity (CPP) termination in GluN2B+/− mice (E) and 1 day advance in GluN2D−/− mice (F). G–L, Cytochrome oxidase-stained SpVi 5 d after infraorbital nerve lesion at P4 (G–K) and P6 (H–L) in control (G, H), GluN2B+/− (I, J), and GluN2D−/− (K, L) mice. Intact and lesioned sides of the SpVi are shown to the left or right, respectively, with the whole coronal section images in insets. Segregated barrelettes are maintained after P4 lesion in GluN2D−/− mice only (K), and after P6 lesion in control mice (H). GluN2B+/−mice at P6 are still within the critical period, as barrelettes are obscured in the lesion side (J). M, N, Bar graphs representing 1 day delay of CPP termination in GluN2B+/− mice (M) and 1 day advance in GluN2D−/− mice (N). The number of mice examined at each stage is listed in Table 3 (middle and bottom). Scale bars, 200 μm.
Magnitude of lesion-induced critical period plasticity
The magnitude of lesion-induced critical period plasticity was examined in the S1 by electrocautery of right row-C whisker follicles at P2 and CO histochemistry at P15. In all genotypes, lesioned row-C barrels were robustly shrunk and fused, whereas adjacent row-B and row-D barrels were expanded reciprocally (Fig. 5A,C,E,G), compared with barrels in the intact side (Fig. 5B,D,F,H). The changes were quantitatively evaluated by calculating the map plasticity index: (B2 + B3 + D2 + D3)/2(C2 + C3). In the contralateral hemisphere, the index was 1.89 ± 0.32 (n = 7) and 2.12 ± 0.56 (n = 8) in GluN2B+/− and control mice, respectively, and 2.11 ± 0.42 (n = 7) and 2.36 ± 0.32 (n = 7) in GluN2D−/− and control mice, respectively, showing no significant differences (p > 0.05 for each, Mann–Whitney U test). These data are consistent with previous findings that NMDA receptors are neutral for lesion-induced critical period plasticity (Iwasato et al., 2000; Lu et al., 2001). Because glutamate transporter GLT1 is markedly upregulated in barrel hollows and magnifies lesion-induced plasticity in the neonatal S1 (Takasaki et al., 2008), we compared the expression at neonatal stages. No genotypic differences were discerned for expression patterns and intensities for GLT1 between GluN2B+/− and control mice and between GluN2D−/− and control mice (data not shown).
Figure 5.
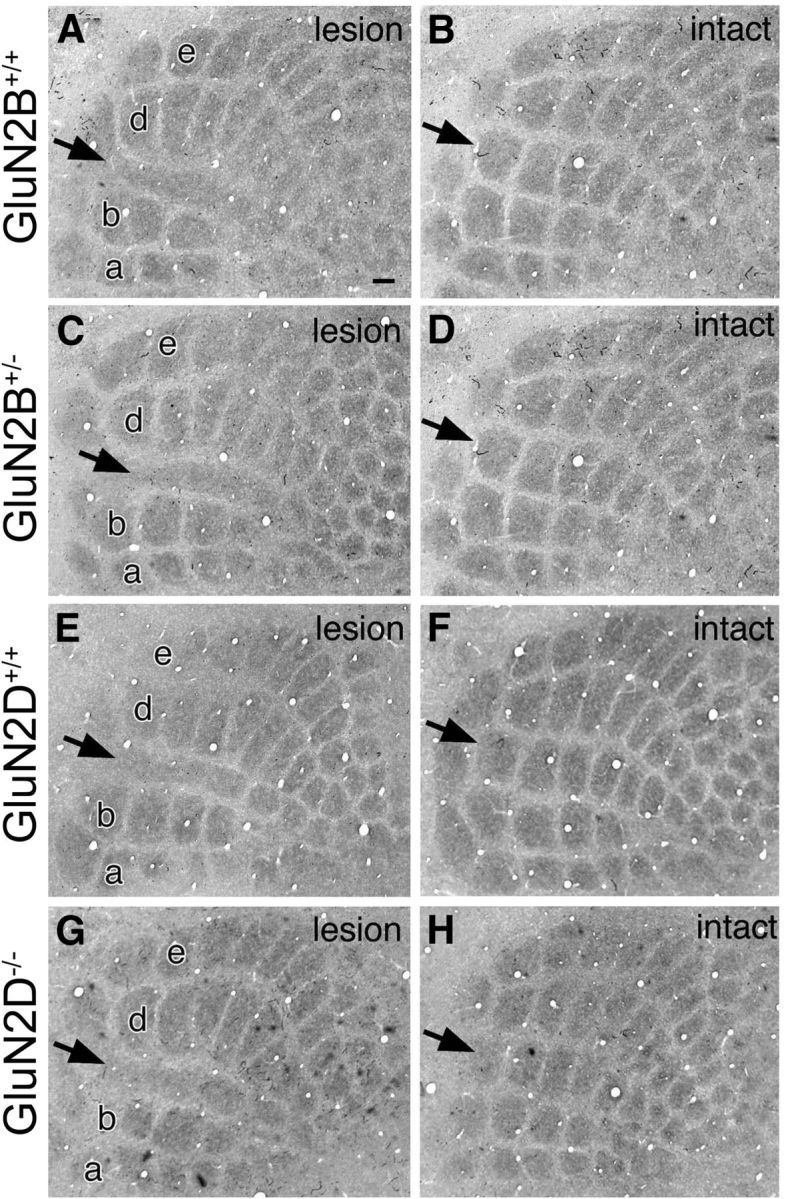
Normal magnitude of critical period plasticity in cortical barrels of GluN2B+/− and GluN2D−/− mutants. Cytochrome oxidase-stained cortical barrels at P15 after electrocautery of right row-C whisker follicles at P2. Lesioned and intact sides of the same animals (A, B, GluN2B+/+; C, D, GluN2B+/−; E, F, GluN2D+/+; G, H, GluN2D−/−) are shown to the left and right, respectively, in each panel. Arrows indicate row-C barrels. Scale bars, 100 μm.
Together, temporal profiles of somatosensory development and maturation are shifted in opposite directions in GluN2B+/− and GluN2D−/− mice throughout the somatosensory axis without affecting the magnitude of lesion-induced critical period plasticity.
Body growth and histoarchitectural differentiation
No significant differences were found in the mean body weight between GluN2B+/− and control mice and between GluN2D−/− and control mice at P0, P4, and P12 (p > 0.05 for each, one-way ANOVA with Bonferroni post hoc, n = 12 for each) (Table 4, top). Moreover, the body weight increased normally after ION lesion given at P3 and P5 (p > 0.05 for each, n = 12 for each) (Table 4, bottom). Furthermore, no apparent differences were discerned in the histoarchitecture of each trigeminal relay station at P5 by Nissl staining with cresyl violet (data not shown). Thus, the shifted temporal development in GluN2B+/− and GluN2D−/− mice is unlikely to result from altered body growth or neuroanatomical differentiation.
Table 4.
Mean body weight (grams) showing normal body growth in GluN2B+/− and GluN2D−/− mutants even after ION transectiona
| Age | GluN2B+/+ | GluN2B+/− | GluN2D+/+ | GluN2D−/− |
|---|---|---|---|---|
| Naive mice | ||||
| P0 | 1.20 ± 0.02 | 1.22 ± 0.02 | 1.23 ± 0.01 | 1.23 ± 0.01 |
| P4 | 2.50 ± 0.07 | 2.50 ± 0.05 | 2.49 ± 0.04 | 2.50 ± 0.04 |
| P12 | 6.09 ± 0.05 | 6.07 ± 0.06 | 6.07 ± 0.05 | 6.08 ± 0.06 |
| ION lesioned mice | ||||
| P11 (P3-lesion) | 5.87 ± 0.11 | 5.83 ± 0.08 | 5.91 ± 0.09 | 5.85 ± 0.11 |
| P11 (sham) | 5.91 ± 0.08 | 5.88 ± 0.11 | 5.84 ± 0.12 | 5.90 ± 0.10 |
| P13 (P5-lesion) | 6.33 ± 0.13 | 6.27 ± 0.18 | 6.25 ± 0.18 | 6.29 ± 0.19 |
| P13 (sham) | 6.31 ± 0.11 | 6.25 ± 0.17 | 6.28 ± 0.15 | 6.31 ± 0.11 |
aIn naive mice, the mean body weight shows no significant differences between control and mutant mice. In ION transection experiments, no significant differences were found between lesioned and sham-operated mice. Data are presented as mean ± SEM. p > 0.05 for each (one-way ANOVA with Bonferroni post hoc comparisons). n = 12 for each group.
Expression in the somatosensory pathway
GluN2B and GluN2D expression at adult trigeminal relay stations
To understand their opposing action, we then examined cellular expression of GluN2B and GluN2D in adult mouse brains by FISH using antisense riboprobes (Fig. 6). Consistent with previous studies by isotopic in situ hybridization (Watanabe et al., 1993; Monyer et al., 1994), GluN2B mRNA was highly expressed throughout the cortex and thalamus (Fig. 6A), whereas GluN2D mRNA was sparsely expressed in these regions (Fig. 6B). To determine their neurochemical phenotypes, we examined expression of GAD67 mRNA as a marker for GABAergic neurons (Fig. 6C). Double-labeling FISH revealed that, in the S1, GluN2B mRNA was expressed intensely in GAD67 mRNA(−) neurons and weakly in GAD67 mRNA(+) neurons (Fig. 6D), whereas GluN2D mRNA was selective to GAD67 mRNA(+) neurons (Fig. 6E). In the thalamus, GAD67 mRNA was exclusively expressed in the reticular thalamic nucleus (RTN) and, accordingly, the thalamic VB was composed of GAD67 mRNA(−) neurons only (Fig. 6C), consistent with a previous study (Liu et al., 1995). GluN2B mRNA was high in VB neurons lacking GAD67 mRNA expression and weak in RTN neurons expressing GAD67 mRNA (Fig. 6F), whereas GluN2D mRNA was selective to RTN neurons expressing GAD67 mRNA (Fig. 6G). This pattern of neuronal expression was also found in the principal sensory nucleus of the trigeminal nucleus (Pr; Fig. 6H–L) and SpVi (Fig. 6M–Q). No significant labeling was found with use of sense riboprobes (data not shown). Thus, it is common to each somatosensory station that non-GABAergic (GAD67 mRNA(−)) neurons expressed GluN2B mRNA exclusively, whereas GABAergic (GAD67 mRNA(+)) neurons expressed GluN2D mRNA at high levels and GluN2B mRNA at low levels.
Figure 6.
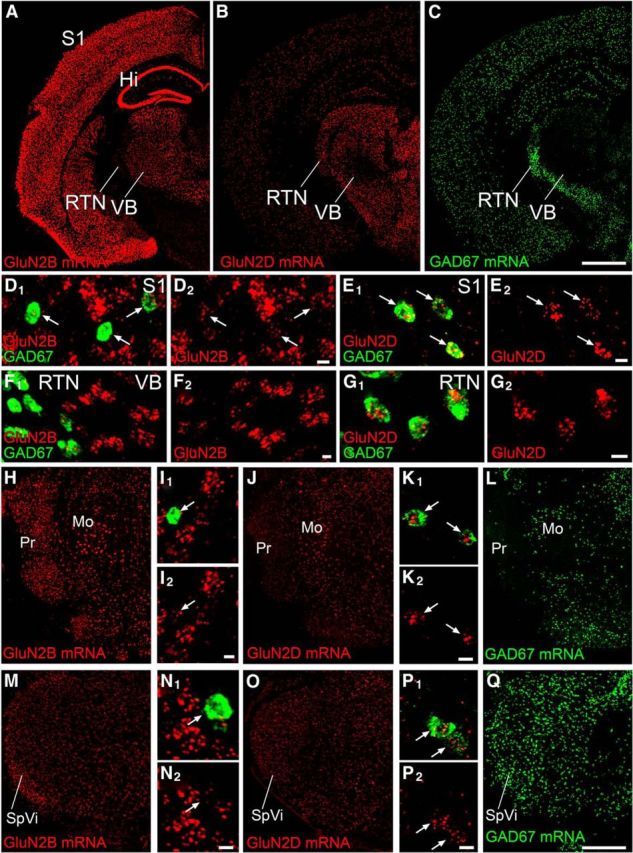
FISH showing distinct neuronal expression of GluN2B and GluN2D mRNAs at trigeminal relay stations in adult mice. A–C, Overall labeling patterns for mRNAs of GluN2B (A), GluN2D (B), and GAD67 (C) in coronal forebrain sections through the S1, VB, and RTN. D–Q, Double-labeling in situ hybridization for GAD67 (green; D–G, I, K, L, N, P, Q) and GluN2B (red; D, F, H, I, M, N) or GluN2D (red; E, G, J, K, O, P) mRNAs in the S1 (D, E), VB and RTN (F, G), trigeminal principal sensory nucleus (Pr; H–L), and subnucleus interpolaris (SpVi; M–Q). Note preferential labeling for GluN2B mRNA in GAD67 mRNA(−) neurons and for GluN2D mRNA in GAD67 mRNA(+) neurons (arrows). Hi, Hippocampus; Mo, trigeminal motor nucleus. Scale bars: A–C (in C), H, J, L, M, O, Q (in Q), 1 mm; D–G, I, K, N, P, 10 μm.
We next examined the distribution of GluN2B and GluN2D immunoreactivities by immunofluorescence using parasagittal paraffin sections (Fig. 7). Pepsin pretreatment of paraffin sections for 10 min, which was optimal for adult specimens, unmasked antigenicity of GluN2B and GluN2D, yielding intense immunolabeling in the forebrain or brainstem, respectively (Fig. 7A,D). The specificity was verified by diminished or blank immunolabeling in GluN2B+/− and GluN2D−/− mouse brains, respectively (Fig. 7B,E), and similar expression patterns to their mRNAs (Fig. 7C,F). In the S1, GluN2B(+) and GluN2D(+) puncta were detected across all layers, including the layer IV where thalamocortical projections labeled by VGluT2 were clustered into barrels (Fig. 7G, arrows). At a higher magnification, these punctate labelings for both subunits are consistently overlapped with PSD-95 immunoreactivity, suggesting postsynaptic localization (Fig. 7H,I, arrows). Double immunofluorescence revealed that bright GluN2B(+) puncta lacked detectable signals for GluN2D, whereas bright GluN2D(+) puncta showed weak signals for GluN2B (Fig. 7J). The VB was crowded with bright GluN2B(+) puncta lacking GluN2D(+) immunoreactivity, whereas bright GluN2D(+) puncta in the RTN had weak immunoreactivity for GluN2B (Fig. 7K). A similar trend was also observed in the Pr (Fig. 7L).
Further postembedding immunogold analyses revealed that GluN2B and GluN2D were specifically localized to asymmetrical synapses in each adult trigeminal relay station (S1, PrV and SpVi in Fig. 8; VB, data not shown). Because GABA is contained in dendrites as well as axon terminals of GABAergic neurons (Bolam et al., 1983), we examined synaptic localization GluN2B and GluN2D on GABA(+) (GABAergic) and GABA(−) (glutamatergic) neurons by double-labeling postembedding immunogold (Fig. 8). Both subunits were preferentially detected on the postsynaptic membrane of asymmetrical synapses in the S1 (Fig. 8A,B). The density of GluN2B labeling per 1 μm of the postsynaptic density was three times higher at synapses on GABA(−) dendrites (mostly on dendritic spines) than synapses on GABA(+) dendrites (mostly on dendritic shafts) (Fig. 8F, left; p < 0.001, U test). By contrast, the density of GluN2D labeling was five times higher at synapses on GABA(+) dendrites than synapses on GABA(−) dendrites, and the density of the latter was almost comparable with the background labeling as determined from the corresponding synapses in GluN2D−/− mice (Fig. 8G, left; p < 0.001, U test). Segregated synaptic distribution was also observed in the Pr and SpVi: preferential synaptic labeling for GluN2B on GABA(−) dendrites (Fig. 8C,D) and for GuN2D on GABA(+) dendrites (Fig. 8E) was seen in both trigeminal nuclei (Fig. 8F,G, middle and right). Therefore, GluN2B is predominantly expressed at asymmetrical synapses on glutamatergic neurons, whereas GluN2D is selective to asymmetrical synapses on GABAergic neurons at each trigeminal station.
Figure 8.
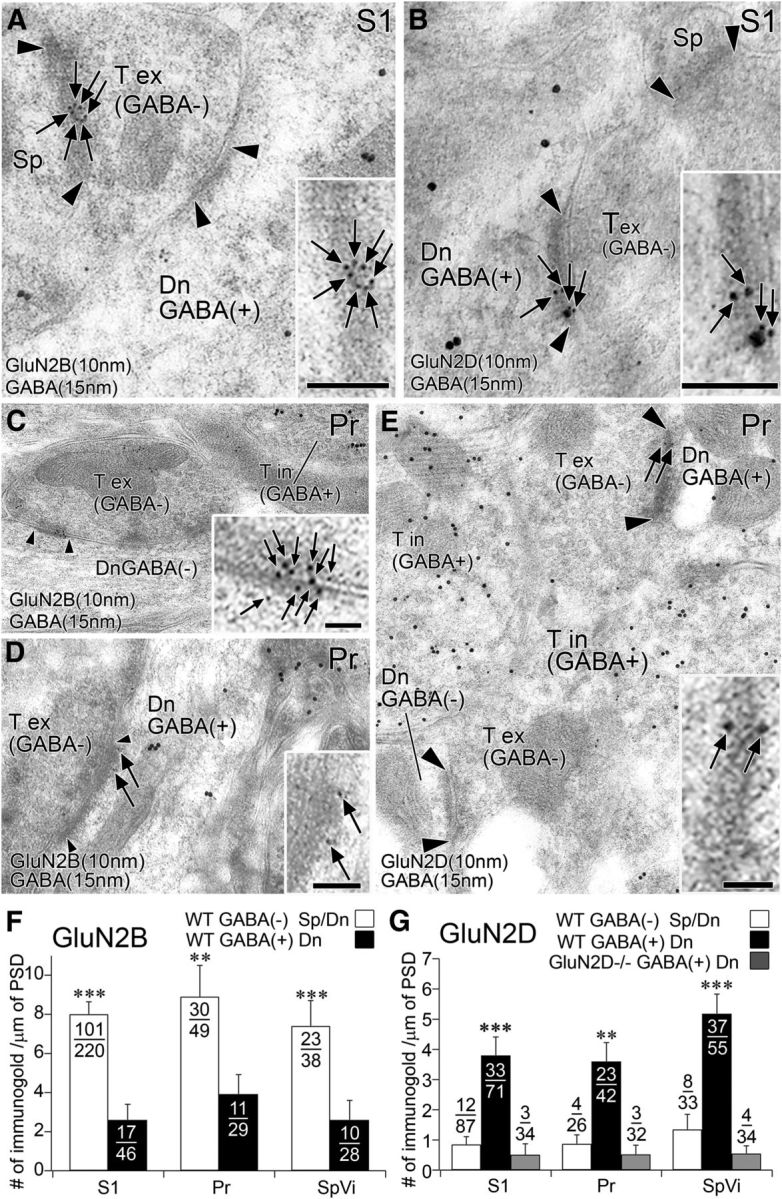
Postembedding immunogold microscopy showing segregated synaptic expression of GluN2B and GluN2D at three relay stations of the adult trigeminal pathway. A–E, Double-labeling postembedding immunogold for GABA (φ = 15 nm) and GluN2B (φ = 10 nm; A, C, D) or GluN2D (φ = 10 nm; B, E) in the S1 (A, B) and Pr (C–E). GluN2B is preferentially expressed at asymmetrical synapses on GABA(−) dendritic shaft and spines, and GluN2D is at GABA(+) dendrites. F, G, Summary bar graphs representing preferential expression of GluN2B (F) and GluN2D (G) at synapses on GABA(−) and GABA(+) postsynaptic compartments in the S1, Pr, and SpVi. The background level of GluN2D labeling was measured at synapses on GABA-positive dendrites in GluN2D−/− mice. Arrowhead indicates the edge of the postsynaptic density. The ratio of labeled synapses to the total number of synapses examined is indicated as a fraction in each column. **p < 0.01 (U test). ***p < 0.001 (U test). Dn, Dendrite; Sp, spine; T ex, GABA-negative (excitatory) terminal; T in, GABA-positive (inhibitory) terminal. Scale bars, 200 nm.
GluN2B and GluN2D expression at neonatal trigeminal relay stations
We further examined neonatal mouse brains at P1 and P5, when whisker-related patterning is differentiating and GluN2B and GluN2D mRNAs are already expressed in the brainstem trigeminal nuclei (Watanabe et al., 1992; Kutsuwada et al., 1996). Immunofluorescence showed that GluN2B was detected widely in neonatal brains, including the brainstem (Fig. 9A,C), whereas GluN2D was mainly expressed in the brainstem and cerebellum (Fig. 9B,D). In the cortex at P5, GluN2B was particularly intense in the layer IV barrels (Fig. 9E), where punctate labeling densely occupied the neuropil, often showing linear arrangements toward the pial surface (Fig. 9F). In comparison, GluN2D was only moderately expressed in the cortex at P5 (Fig. 9G), where it was accumulated in the perikarya of GAD65/67(+) GABAergic neurons and also detected as weak punctate labeling in the neuropil (Fig. 9H,I). The linear arrangements of GluN2B and perikaryal accumulation of GluN2D were unique to neonatal brains, presumably reflecting dynamic differentiation of dendrites and synapses as well as ongoing active synthesis of receptor molecules in neuronal perikarya.
Figure 9.
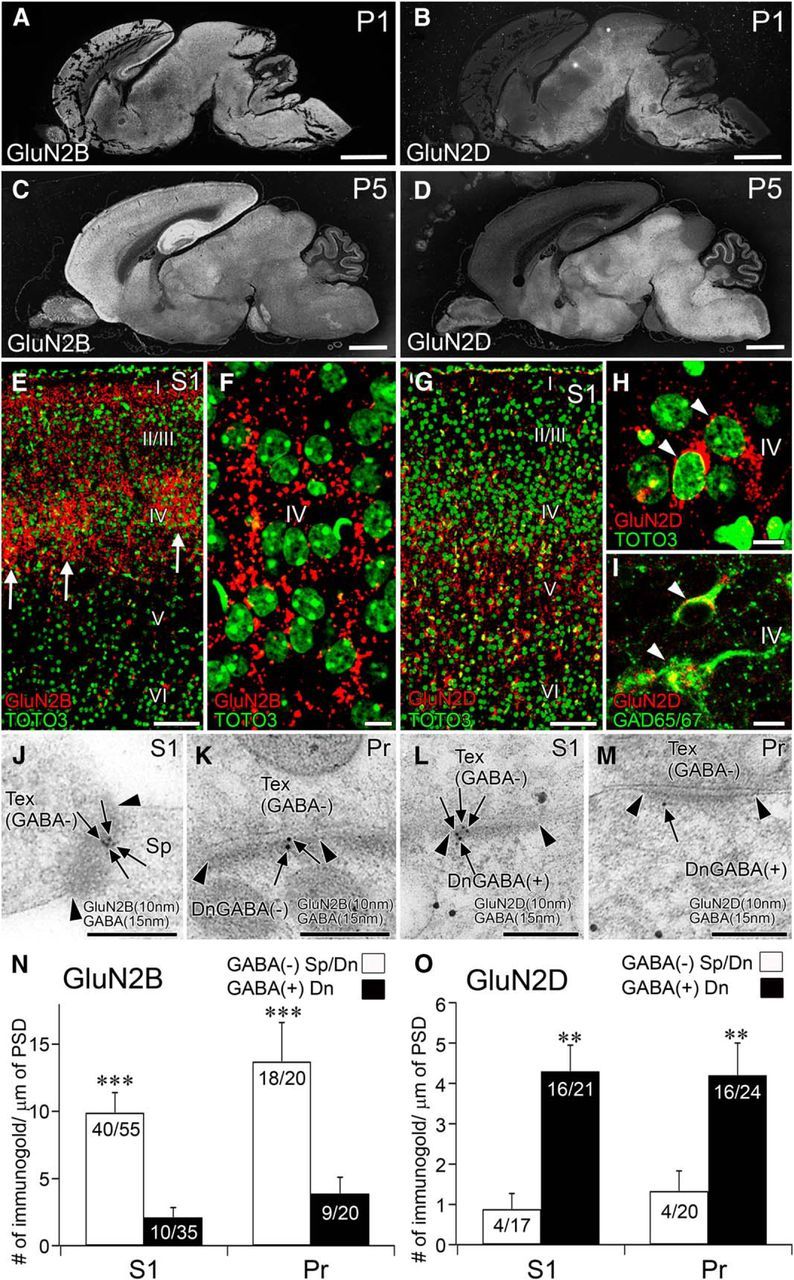
Segregated synaptic expression of GluN2B and GluN2D is preserved at neonatal trigeminal relay stations. A–D, GluN2B (A, C) and GluN2D (B, D) immunolabeling in the brain of wild-type mice at P1 (A, B) and P5 (C, D). E, F, GluN2B immunofluorescence (red) and nuclear counterstaining with TOTO-3 (green) in the S1. Note that barrel hollows (arrows) are filled with intense GluN2B(+) puncta. G–I, GluN2D immunofluorescence (red) with nuclear counterstaining with TOTO-3 (green, G, H) or with GAD65/67 immunofluorescence (green, I) in the S1. Note accumulation of GluN2D in perikarya of GAD65/67(+) neurons (I, arrowheads). J–M, Double-labeling postembedding immunogold for GluN2B (J, K) and GluN2D (L, M) in the S1 (J, L) and Pr (K, M). N, O, Summary bar graphs representing preferential expression of GluN2B (N) and GluN2D (O) at synapses on GABA(−) and GABA(+) postsynaptic compartments, respectively, in the S1 and Pr. **p < 0.01 (U test). ***p < 0.001 (U test). Scale bars: A-D, 1 mm; E, G, 100 μm; F, H, I, 10 μm; J–M, 200 nm.
By double-labeling postembedding immunogold, we further confirmed that GluN2B was preferentially expressed at asymmetrical synapses on GABA(−) dendritic spines in the S1 (Fig. 9J,N, left) and GABA(−) dendritic shafts in the Pr (Fig. 9K,N, right). By contrast, GuN2D was predominantly localized to asymmetrical synapses on GABA(+) dendrites in both regions (Fig. 9L,M,O). Therefore, predominant GluN2B expression at asymmetrical synapses on glutamatergic neurons and selective GluN2D expression at asymmetrical synapses on GABAergic neurons are preserved from the neonatal period.
Whisker-related patterning by PV interneurons
Cortical GABAergic interneurons, particularly PV-positive interneurons, are important in the critical period regulation (Hensch, 2005). Considering the exclusive GluN2D expression in GABAergic interneurons, we lastly investigated whether whisker-related patterning by PV(+) interneurons (Fig. 10), which is known to transiently appear at ∼P10 (del Rio et al., 1994), was affected in GluN2D−/− mice.
Figure 10.
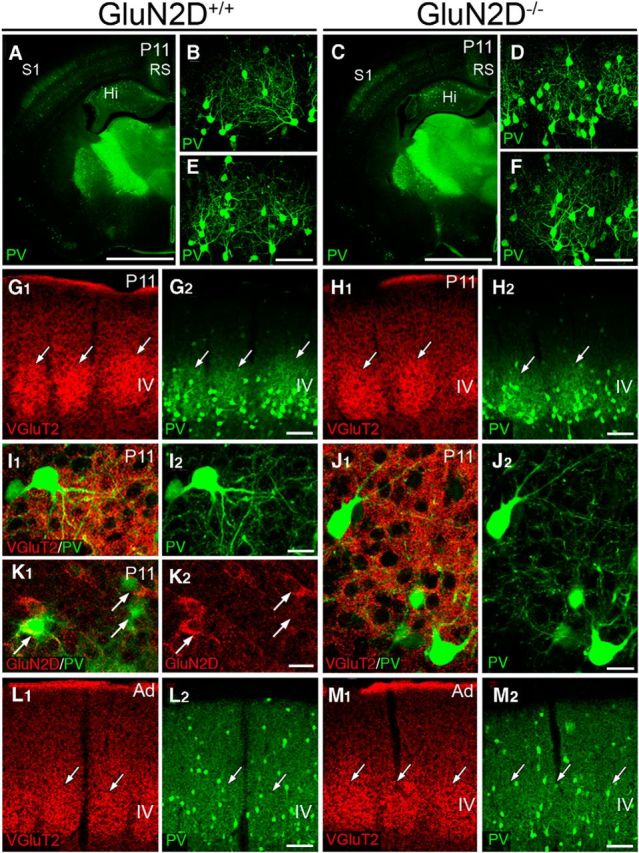
Normal cytodifferentiation of PV-positive interneurons in the S1 of GluN2D−/− mice. A–F, PV immunofluorescence in GluN2D+/+ (A–C) and GluN2D−/− (D–F) mice at P11. PV expression started to increase in the somatosensory cortex (S1) and retrosplenial cortex (RS) (A, D), and whisker-related patterning by PV expression is evident (B, C, E, F) in both mice. G–J, Double immunofluorescence for VGluT2 (red) and PV (green) shows that whisker-related patterning by PV-positive interneurons matches with that by thalamocortical afferents (G, H). In both mice, cell bodies and dendrites of PV-positive interneurons similarly form baskets surrounding barrel hollows, in which VGuT2(+) thalamocortical terminals are densely distributed (I, J). K, Double immunofluorescence for GluN2D (red) and PV (green) in GluN2D+/+ mice at P11. At this stage, PV is upregulated in some interneurons, including those expressing GluN2D (arrows). L, M, Whisker-related patterning by PV-positive interneurons is no longer seen in adult GluN2D+/+ (L) and GluN2D−/− mice (M). Scale bars: A, D, 1 mm; B, C, E, F (in C, F), 200 μm; G, H, L, M, 100 μm; I–K, 20 μm.
In control mice at P11, PV immunoreactivity started to increase in the S1 (Fig. 10A) and exhibited whisker-related patterning in the layer IV (Fig. 10B,C). Whisker-related patterning by PV-positive interneurons was matched with that by VGluT2(+) thalamocortical terminals (Fig. 10G). PV(+) neuronal elements formed baskets surrounding barrel hollows (Fig. 10I) and expressed GluN2D (Fig. 10K, arrows). In GluN2D−/− mice at P11, whisker-related patterning by PV expression was also evident in the S1 (Fig. 10D–F), showing similar basket formation in the barrel field (Fig. 10H,J). In adulthood, whisker-related patterning by PV-positive interneurons was no longer observed in both strains of mice (Fig. 10L,M). Thus, these results suggest normal cytological differentiation and refinement of PV-positive interneurons in GluN2D−/− mice.
Discussion
To test whether GluN2B and GluN2D work synergistically or antagonistically in synaptic circuit development, we compared whisker-related patterning in GluN2B+/− and GluN2D−/− mice with that in their control littermates. Here we demonstrate that temporal profiles of somatosensory development and maturation were shifted in opposite directions in these mutants. Moreover, we examined expression of GluN2B and GluN2D at trigeminal relay stations and clarified their distinct and almost segregated expression.
Positive and negative modulation of somatosensory development and maturation
Developmental appearance and critical period termination were both delayed by nearly a day at each trigeminal relay station in GluN2B+/− mice. The delayed appearance can be taken as a mild phenotype of GluN2B−/− mice, in which barrelettes do not appear even after the survival of the newborn is prolonged for 2 more days by artificial feeding (Kutsuwada et al., 1996). Such a gene dosage effect in GluN2B+/− mice is also manifested as exaggerated nociceptive responses to acute noxious stimuli (Wainai et al., 2001), enhanced acoustic startle response (Takeuchi et al., 2001), and impaired eyeblink conditioning (Takehara et al., 2004). The delayed termination of the critical period further suggests that GluN2B facilitates the maturation into a lesion-insensitive state, rather than prolonging the lesion-sensitive immature state. Conversely, an advance of 1 d was observed at each station in GluN2D−/− mice. Because we analyzed only normal-term delivered pups, it seems unlikely that these temporal shifts are caused by defects associated with preterm or post-term delivery (Toda et al., 2013). Thus, GluN2B positively modulates somatosensory development and maturation, whereas GluN2D counteracts by negative modulation. To our knowledge, the present study has provided the first experimental evidence that GluN2D regulates neural development, with its opposing action to GluN2B. Considering the presence of half the amount of GluN2B protein in GluN2B+/− mice compared with control mice (Kutsuwada et al., 1992; Watanabe et al., 1998), the magnitude of positive modulation by GluN2B exceeds that of negative modulation by GluN2D. Therefore, GluN2B and GluN2D constitute a “push-pull” system on temporal development of the somatosensory system, with higher potency of GluN2B.
Pathway-dependent organization of NMDA receptor subunits
Distinct neuronal and synaptic expression of GluN2B and GluN2D is another important finding in the present study. At trigeminal stations in the cortex and brainstem, glutamatergic (GAD67 mRNA(−)) neurons expressed GluN2B mRNA exclusively, whereas GABAergic (GAD67 mRNA(+)) neurons expressed GluN2D and GluN2B mRNA at high or low levels, respectively. In the thalamus, these two neuronal populations reside separately in the VB and RTN. Considering that the VB and RTN receive glutamatergic projections from ascending afferents, and the RTN sends inhibitory projections to the VB (Jones, 2007), it is common to all trigeminal relay stations that GluN2B mRNA is highly expressed in glutamatergic projection neurons, and GluN2D mRNA is selectively expressed in GABAergic neurons projecting to local or adjacent glutamatergic projection neurons. This was confirmed and further evidenced by postembedding immunogold microscopy that GluN2B protein was predominantly localized at asymmetrical synapses on glutamatergic neurons, and GluN2D protein was selective at asymmetrical synapses on GABAergic neurons. This pathway-dependent organization of NMDA receptor subunits suggests that glutamatergic projection neurons use GluN2B-containing receptors to mediate somatosensory information along the ascending pathway, whereas GABAergic interneurons use GluN2D-containing receptors, presumably, to modulate the excitability and information processing of glutamatergic projection neurons.
Functional considerations
In addition to the opposing action on somatosensory map development, a number of contrasting properties have been reported between GluN2B- and GluN2D-containing NMDA receptors. GluN2B/GluN1 channels require a strong depolarization to overcome Mg2+ blockade and have high conductances, whereas GluN2D/GluN1 channels need only modest depolarization to overcome Mg2+ blockade and show low conductances (Monyer et al., 1994; Momiyama et al., 1996; Misra et al., 2000a, b). The differences in channel conductance and the duration of channel opening might contribute to the distinct regulation of postsynaptic Ca2+ influx in the induction of LTP and LTD (Lisman, 1989; Artola and Singer, 1993; Hansel et al., 1997). GluN2B-containing receptors provide a greater Ca2+ influx per unit of current (Sobczyk et al., 2005) and promote the induction of LTP as exemplified by enhanced hippocampal LTP in GluN2B-overexpressing transgenic mice (Tang et al., 1999). By contrast, GluN2D-containing channels permit modest but prolonged Ca2+ entry (Monyer et al., 1994; Momiyama et al., 1996; Misra et al., 2000a, b), and contribute to LTD induction in the hippocampus (Hrabetova et al., 2000). These contrasting properties and pathway-dependent organization of GluN2B and GluN2D raise the intriguing possibility to account for the opposite modulation of somatosensory development by GluN2B- and GluN2D-containing receptors.
GluN2B-containing receptors expressed along the ascending pathway synapses might confer them with malleable properties and thereby promote activity-dependent synapse refinement directly. GluN2D-containing receptors, on the other hand, are unable to modulate this synapse refinement directly because of the lack of GluN2D expression at the ascending pathway synapses. Normal whisker-related patterning of PV-positive interneurons in GluN2D−/− mice further suggests that advanced somatosensory development in GluN2D−/− mice is unlikely due to altered cytological differentiation and refinement of, at least partly, PV-positive interneurons. Thus, we assume that GluN2D-containing receptors might delay somatosensory development by modulating the activity and function of local interneurons, which construct feedforward and feedback inhibition to regulate the excitability, integration, and plasticity of the ascending projection neurons (Hensch et al., 1998; Klausberger et al., 2002; Freund, 2003; Markram et al., 2004; Katagiri et al., 2007).
In conclusion, GluN2B and GluN2D are organized in a pathway-dependent manner along the trigeminal pathway of the somatosensory system and play opposing roles in temporal development and maturation of whisker-related central patterning.
Notes
Supplemental material for this article is available at http://www.hucc.hokudai.ac.jp/∼e20704/. This material has not been peer reviewed.
Footnotes
This work was supported by Ministry of Education, Culture, Sports, Science and Technology, Japan Grant-in-Aids for Young Scientists (B) 24790187 to M.Y. and Grant-in-Aids for Scientific Research (S) 24220007 to M.W. and the Uehara Memorial Foundation (to M.W.). We thank Dr. Maya Yamazaki (Niigata University) for a kind gift of GluN2D plasmid for riboprobe preparation.
The authors declare no competing financial interests.
References
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-V. [DOI] [PubMed] [Google Scholar]
- Bear MF. A synaptic basis for memory storage in the cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belford GR, Killackey HP. The sensitive period in the development of the trigeminal system of the neonatal rat. J Comp Neurol. 1980;193:335–350. doi: 10.1002/cne.901930203. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Clarke DJ, Smith AD, Somogyi P. A type of aspiny neuron in the rat neostriatum accumulates [3H]gamma-aminobutyric acid: combination of Golgi-staining, autoradiography, and electron microscopy. J Comp Neurol. 1983;213:121–134. doi: 10.1002/cne.902130202. [DOI] [PubMed] [Google Scholar]
- Chiaia NL, Bennett-Clarke CA, Rhoades RW. Differential effects of peripheral damage on vibrissa-related patterns in trigeminal nucleus principalis, subnucleus interpolaris, and subnucleus caudalis. Neuroscience. 1992;49:141–156. doi: 10.1016/0306-4522(92)90082-D. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- del Rio JA, de Lecea L, Ferrer I, Soriano E. The development of parvalbumin-immunoreactivity in the neocortex of the mouse. Dev Brain Res. 1994;81:247–259. doi: 10.1016/0165-3806(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Durham D, Woolsey TA. Effects of neonatal whisker lesions on mouse central trigeminal pathways. J Comp Neurol. 1984;223:424–447. doi: 10.1002/cne.902230308. [DOI] [PubMed] [Google Scholar]
- Erzurumlu RS, Gaspar P. Development and critical period plasticity of the barrel cortex. Eur J Neurosci. 2012;35:1540–1553. doi: 10.1111/j.1460-9568.2012.08075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE, Nicoll RA, Malenka RC, Isaac JT. Long-term depression at thalamocortical synapses in developing rat somatosensory cortex. Neuron. 1998;21:347–357. doi: 10.1016/S0896-6273(00)80544-9. [DOI] [PubMed] [Google Scholar]
- Finney D. Probit analysis. Ed 3. Cambridge, UK: Cambridge UP; 1971. [Google Scholar]
- Freund TF. Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci. 2003;26:489–495. doi: 10.1016/S0166-2236(03)00227-3. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Watanabe M. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: a study in the adult mouse brain. J Comp Neurol. 2000;426:572–586. doi: 10.1002/1096-9861(20001030)426:4<572::AID-CNE6>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell. 1993;72(Suppl):77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. Eur J Neurosci. 1997;9:2309–2322. doi: 10.1111/j.1460-9568.1997.tb01648.x. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabetova S, Serrano P, Blace N, Tse HW, Skifter DA, Jane DE, Monaghan DT, Sacktor TC. Distinct NMDA receptor subpopulations contribute to long-term potentiation and long-term depression induction. J Neurosci. 2000;20:RC81. doi: 10.1523/JNEUROSCI.20-12-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Araki K, Takayama C, Inoue Y, Yagi T, Aizawa S, Mishina M. Reduced spontaneous activity of mice defective in the ε4 subunit of the NMDA receptor channel. Mol Brain Res. 1995;33:61–71. doi: 10.1016/0169-328X(95)00107-4. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knöpfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The thalamus. Ed 2. Cambridge, UK: Cambridge UP; 2007. [Google Scholar]
- Katagiri H, Fagiolini M, Hensch TK. Optimization of somatic inhibition at critical period onset in mouse visual cortex. Neuron. 2007;53:805–812. doi: 10.1016/j.neuron.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M. Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor ε1 subunit. J Neurosci. 1998;18:6704–6712. doi: 10.1523/JNEUROSCI.18-17-06704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Roberts JD, Somogyi P. Cell type- and input-specific differences in the number and subtypes of synaptic GABA(A) receptors in the hippocampus. J Neurosci. 2002;22:2513–2521. doi: 10.1523/JNEUROSCI.22-07-02513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/S0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S. Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cell. 1994;76:427–437. doi: 10.1016/0092-8674(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Warren RA, Jones EG. Synaptic distribution of afferents from reticular nucleus in ventroposterior nucleus of cat thalamus. J Comp Neurol. 1995;352:187–202. doi: 10.1002/cne.903520203. [DOI] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/S0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-T. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Misra C, Brickley SG, Wyllie DJ, Cull-Candy SG. Slow deactivation kinetics of NMDA receptors containing NR1 and NR2D subunits in rat cerebellar Purkinje cells. J Physiol. 2000a;525:299–305. doi: 10.1111/j.1469-7793.2000.t01-1-00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra C, Brickley SG, Farrant M, Cull-Candy SG. Identification of subunits contributing to synaptic and extrasynaptic NMDA receptors in Golgi cells of the rat cerebellum. J Physiol. 2000b;524:147–162. doi: 10.1111/j.1469-7793.2000.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Fukaya M, Sato T, Sugihara K, Asano M, Yoshioka K, Watanabe M. Expression and distribution of JNK/SAPK-associated scaffold protein JSAP1 in developing and adult mouse brain. J Neurochem. 2006;97:1431–1446. doi: 10.1111/j.1471-4159.2006.03835.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci. 2003;17:2563–2572. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
- Momiyama A, Feldmeyer D, Cull-Candy SG. Identification of a native low-conductance NMDA channel with reduced sensitivity to Mg2+ in rat central neurones. J Physiol. 1996;494:479–492. doi: 10.1113/jphysiol.1996.sp021507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995;34:1219–1237. doi: 10.1016/0028-3908(95)00109-J. [DOI] [PubMed] [Google Scholar]
- Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluRε2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/S0896-6273(00)80567-X. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Sato K, Fukaya M, Araishi K, Aiba A, Kano M, Watanabe M. Signaling complex formation of phospholipase Cβ4 with metabotropic glutamate receptor type 1α and 1,4,5-trisphosphate receptor at the perisynapse and endoplasmic reticulum in the mouse brain. Eur J Neurosci. 2004;20:2929–2944. doi: 10.1111/j.1460-9568.2004.03768.x. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Masu M. Molecular diversity and functions of glutamate receptors. Annu Rev Biophys Biomol Struct. 1994;23:319–348. doi: 10.1146/annurev.bb.23.060194.001535. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O'Leary DD. Postsynaptic control of plasticity in developing somatosensory cortex. Nature. 1993;364:623–626. doi: 10.1038/364623a0. [DOI] [PubMed] [Google Scholar]
- Seeburg PH. The Trends Neurosci/TiPS Lecture: the molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern P, Béhé P, Schoepfer R, Colquhoun D. Single-channel conductances of NMDA receptors expressed from cloned cDNAs: comparison with native receptors. Proc Biol Sci. 1992;250:271–277. doi: 10.1098/rspb.1992.0159. [DOI] [PubMed] [Google Scholar]
- Takasaki C, Okada R, Mitani A, Fukaya M, Yamasaki M, Fujihara Y, Shirakawa T, Tanaka K, Watanabe M. Glutamate transporters regulate lesion-induced plasticity in the developing somatosensory cortex. J Neurosci. 2008;28:4995–5006. doi: 10.1523/JNEUROSCI.0861-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehara K, Kawahara S, Munemoto Y, Kuriyama H, Mori H, Mishina M, Kirino Y. The N-methyl-d-aspartate (NMDA)-type glutamate receptor GluRε2 is important for delay and trace eyeblink conditioning in mice. Neurosci Lett. 2004;364:43–47. doi: 10.1016/j.neulet.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Kiyama Y, Nakamura K, Tsujita M, Matsuda I, Mori H, Munemoto Y, Kuriyama H, Natsume R, Sakimura K, Mishina M. Roles of the glutamate receptor ε2 and δ2 subunits in the potentiation and prepulse inhibition of the acoustic startle reflex. Eur J Neurosci. 2001;14:153–160. doi: 10.1046/j.0953-816x.2001.01620.x. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Toda T, Homma D, Tokuoka H, Hayakawa I, Sugimoto Y, Ichinose H, Kawasaki H. Birth regulates the initiation of sensory map formation through serotonin signaling. Dev Cell. 2013;27:32–46. doi: 10.1016/j.devcel.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 1973;179:395–398. doi: 10.1126/science.179.4071.395. [DOI] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Wainai T, Takeuchi T, Seo N, Mishina M. Regulation of acute nociceptive responses by the NMDA receptor GluRε2 subunit. Neuroreport. 2001;12:3169–3172. doi: 10.1097/00001756-200110290-00005. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Distinct distributions of five N-methyl-d-aspartate receptor channel subunit mRNAs in the forebrain. J Comp Neurol. 1993;338:377–390. doi: 10.1002/cne.903380305. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y. Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci. 1998;10:478–487. doi: 10.1046/j.1460-9568.1998.00063.x. [DOI] [PubMed] [Google Scholar]
- Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex: the description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-X. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Anderson JR, Wann JR, Stanfield BB. Effects of early vibrissae damage on neurons in the ventrobasal (VB) thalamus of the mouse. J Comp Neurol. 1979;184:363–380. doi: 10.1002/cne.901840210. [DOI] [PubMed] [Google Scholar]
- Wyllie DJ, Béhé P, Colquhoun D. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J Physiol. 1998;510:1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Watanabe M, Shibata T, Nagashima M, Tanaka K, Inoue Y. Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. J Neurosci. 1998;18:5706–5713. doi: 10.1523/JNEUROSCI.18-15-05706.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Fukaya M, Shimizu H, Sakimura K, Watanabe M. NMDA receptor subunits GluRε1, GluRε3 and GluRζ1 are enriched at the mossy fibre-granule cell synapse in the adult mouse cerebellum. Eur J Neurosci. 2001;13:2025–2036. doi: 10.1046/j.0953-816x.2001.01580.x. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Matsui M, Watanabe M. Preferential localization of muscarinic M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J Neurosci. 2010;30:4408–4418. doi: 10.1523/JNEUROSCI.5719-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]