

Abstract
Phasic increases in dopamine (DA) are involved in the detection and selection of relevant sensory stimuli. The DAergic and cholinergic system dynamically interact to gate and potentiate sensory inputs to striatum. Striatal cholinergic interneurons (CINs) respond to relevant sensory stimuli with an initial burst, a firing pause, or a late burst, or a combination of these three components. CIN responses coincide with phasic firing of DAergic neurons in vivo. In particular, the late burst of CINs codes for the anticipated reward. To examine whether DAergic midbrain afferents can evoke the different CIN responses, we recorded from adult olfactory tubercle slices in the mouse ventral striatum. Olfactory inputs to striatal projection neurons were gated by the cholinergic tone. Phasic optogenetic activation of DAergic terminals evoked combinations of initial bursts, pauses, and late bursts in subsets of CINs by distinct receptor pathways. Glutamate release from midbrain afferents evoked an NMDAR-dependent initial burst followed by an afterhyperpolarization-induced pause. Phasic release of DA itself evoked acute changes in CIN firing. In particular, in CINs without an initial burst, phasic DA release evoked a pause through D2-type DA receptor activation. Independently, phasic DA activated a slow depolarizing conductance and the late burst through a D1-type DA receptor pathway. In summary, DAergic neurons elicit transient subsecond firing responses in CINs by sequential activation of NMDA, D2-type, and D1-type receptors. This fast control of striatal cholinergic tone by phasic DA provides a novel dynamic link of two transmitter systems central to the detection and selection of relevant stimuli.
Keywords: cholinergic interneurons, glutamate corelease, olfactory tubercle, pause response, phasic dopamine, striatum
Introduction
In humans, one of the brain regions that becomes preferentially activated by attended as opposed to unattended odors is the olfactory tubercle (OT) of the striatum (Zelano et al., 2005). The OT has been implicated in multisensory processing and reward learning (Ikemoto, 2007; Wesson and Wilson, 2010). The three-layered OT shares its ontogenetic origin and expression of neurochemical markers with other striatal areas (Price, 1973; Haberly and Price, 1978; Schwob and Price, 1984; Wahle and Meyer, 1986; Wichterle et al., 2001; Nagayama et al., 2010). Olfactory bulb projections target dendrites of odor-responsive spiny projection neurons (SPNs) that are densely packed in layer 2 of the OT (Millhouse and Heimer, 1984; McNamara et al., 2004; Wesson and Wilson, 2010; Sosulski et al., 2011). As part of the striatum, the OT also receives massive innervation from dopaminergic midbrain neurons.
The two neuromodulators, dopamine (DA) and acetylcholine (ACh), have been linked to the selection of relevant and reward-predicting sensory stimuli (Schultz, 1986; Aosaki et al., 1994a; Berridge and Robinson, 1998). A better understanding of the interaction of these two neuromodulators will be critical to eventually dissect the cellular processes underlying olfactory stimulus selection.
Local cholinergic interneurons (CINs) are the main source of ACh in the striatum (Kreitzer, 2009). In the dorsal striatum, inputs to SPNs are gated by CINs (Ding et al., 2010) and may thereby be involved in selection of sensory inputs. CINs respond to relevant sensory stimuli with changes in their firing rate, i.e., an initial burst, a pause, or a delayed burst, or a combination of these three components (Kimura et al., 1984; Aosaki et al., 1994a,1994b; Benhamou et al., 2014; Doig et al., 2014). These CIN responses and the phasic firing of midbrain DAergic neurons frequently coincide (Morris et al., 2004; Schulz and Reynolds, 2013), and CINs receive synaptic afferents from DAergic midbrain neurons (Dimova et al., 1993). Similar to phasic firing of DAergic midbrain neurons, the late burst of CINs encodes the probability and magnitude of the anticipated reward (Apicella et al., 2011), and DA receptor (DAR) antagonists interfere with expression of the CIN response during learning (Aosaki et al., 1994a).
It is, therefore, possible that DAergic midbrain neurons can directly evoke the different firing responses of CINs. CINs respond within hundreds of milliseconds after stimulus presentation (Aosaki et al., 1994a). If DAergic neurons control CIN firing, DA needs to act on a fast timescale in addition to its slow neuromodulatory actions (Kreitzer, 2009). DAergic neurons could also use their corelease of the fast transmitter glutamate (Stuber et al., 2010; Tecuapetla et al., 2010). To begin to address the cellular processes of olfactory stimulus selection in the OT, we examined the mechanisms by which phasic firing of DAergic neurons may directly evoke different firing responses in CINs in an acute slice preparation from adult mice. We observed that optogenetic phasic activation of DAergic midbrain neurons evoked initial bursts, pauses, and late bursts through distinct pathways involving postsynaptic glutamate and unexpectedly fast actions of DA through D1-/D2-type DARs.
Materials and Methods
Animals and husbandry.
We used heterozygous transgenic mice, Tg(DATCre)9075Gsc (Parlato et al., 2006), expressing the Cre recombinase under control of the dopamine transporter (DAT) gene contained in a bacterial artificial chromosome, DAT-Cre. The genetic background was C57BL/6NCrl (Charles River). All experimental subjects have been backcrossed at least 10 generations. Unless specified otherwise, 2–4 mice were housed per cage and were kept in a standard 12 h light/dark cycle and given food and water ad libitum. All animal procedures were approved by the local Animal Welfare Committee and in accordance with National Institutes of Health guidelines. Throughout the study, mice of either sex were used.
Virus preparation.
To induce cell-specific expression of channelrhodopsin2 (ChR2), we injected a Cre-inducible recombinant rAAV1/2-double floxed inverted open reading frame (DIOA)-ChR2:mCherry (Cardin et al., 2009) into the ventral midbrain of DAT-Cre mice. The Cre-inducible recombinant adeno-associated virus (AAV) vectors were produced with AAV1/2 coat proteins and purified with heparin columns to a final viral concentration of ∼1016 genome copies/ml (Monory et al., 2006).
Stereotactic AAV injection.
Mice aged >8 weeks were anesthetized with isoflurane. Purified rAAV1/2-DIOA-ChR2:mCherry virus (0.75 μl) was injected bilaterally into the ventral midbrain (from bregma: posterior, 3.0 mm; lateral, 0.8 mm; ventral, 4.4 mm). All mice recovered for at least 21 d before electrophysiological experiments.
Optical stimulation.
Laser pulses (5 ms duration, 5–10 mW/mm2 at slice) were delivered from 100 μm multimode optical fibers (Thorlabs) coupled to a 25 mW, 473 nm, diode-pumped, solid-state laser (CrystaLaser). The fiber optic was positioned at a distance of >300 μm from the recording site. We chose laser light pulses of 5 ms duration in accordance with studies originally revealing glutamate corelease from DAergic terminals (Stuber et al., 2010; Tecuapetla et al., 2010). Under our conditions, the failure rate of evoking EPSC was slightly lower at 5 versus 1 ms pulse duration (4.7 ± 1.3% vs 10.6 ± 3.2%, respectively, n = 8 cells, p < 0.05, t test). The frequency of the laser pulses was chosen at 40 Hz, a frequency that closely parallels burst firing of DAergic neurons and maximally activated the D1-mediated inward current (see Fig. 4D).
Figure 4.
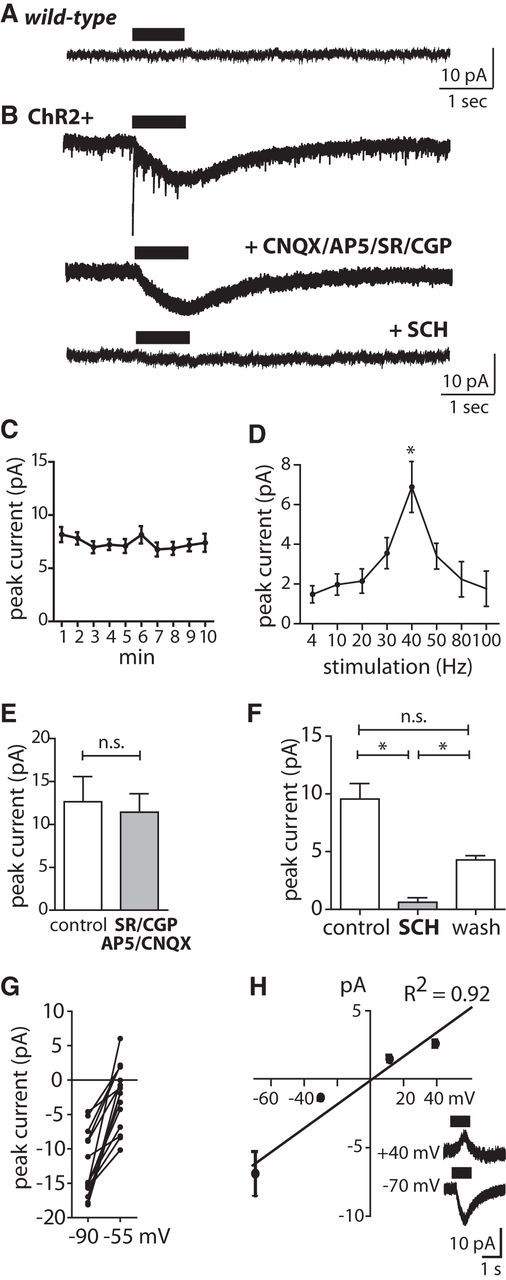
Phasic DA release elicits a D1-type inward current in CINs. A, Whole-cell recording of membrane current response to PLS (40 Hz for 1 s repeated every 20 s) in wild-type brain slice at Vh = −80 mV. B, Same conditions as for A but in brain slice from a ChR2+ DAT-Cre mouse (top trace). Application of d-AP5 (50 μm), CNQX (10 μm), SR95531 (10 μm), and CGP 55845 (5 μm) eliminated fast inward currents (middle trace), and additional application of D1-type DAR antagonist SCH 23390 (10 μm) blocked the slow inward current (bottom trace). C, The amplitude of the slow inward current evoked by PLS (40 Hz, 1 s) at Vh = −80 mV was stable over time (n.s., ANOVA, n = 15). Recordings in D and F–H were performed in the presence of d-AP5 (50 μm), CNQX (10 μm), SR95531 (10 μm), and CGP 55845 (5 μm). D, Dependence of the slow inward current amplitude (Vh = −80 mV) on PLS frequency [p < 0.05, ANOVA with post-test (*p < 0.05), n = 15]. E, The amplitude of the slow inward current evoked by PLS (40 Hz, 1 s) did not change upon application of the NMDAR/AMPAR and GABAA/BR blockers d-AP5 (50 μm), CNQX (10 μm), SR95531 (10 μm), and CGP 55845 (5 μm; n.s., t test, n = 5). F, Additional application of the D1-type DAR blocker SCH 23390 (10 μm) eliminated the PLS-evoked inward current [p < 0.05, ANOVA with post-test (*p < 0.05), n = 8]. G, The amplitude of the slow inward current evoked by PLS (40 Hz, 1 s) was reduced at Vh = −55 mV compared with Vh = −90 mV in K+-based intracellular solution (p < 0.05, t test, n = 13). H, Voltage dependence of PLS-evoked slow current in Cs+-based intracellular solution (R2, correlation coefficient of linear fit, n = 5). Inset, Representative traces at Vh = −70 mV and +40 mV.
Voltammetry.
Fast-scan cyclic voltammetry (FSCV) recordings were made with an EVA8 voltammetric amplifier (HEKA) using 7-μm-diameter and 100- to 200-μm-long carbon fiber microelectrodes scanning from −0.4 V to +1.4 V (vs Ag/AgCl) at a rate of 900 V/s, and sampling at 100 ms intervals. Identification of released DA was based on voltammograms displaying the single oxidation and reduction peak currents that define the voltammetric signature of DA (Robinson et al., 2003). Oxidation peak currents were normalized to baseline [1.5 s before phasic laser stimulation (PLS) onset]. Electrodes were calibrated in a bath chamber with 1 μm DA after the experiment.
Electrophysiology.
Animals were given an overdose of isoflurane, followed by transcardiac perfusion with cold (+4°C) slicing solution containing the following (in mm): 212 sucrose, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 7 MgCl2, 10 glucose (95% O2/5% CO2, 308 mOsm, and pH 7.3). Coronal or sagittal sections (300 μm) were cut in cold slicing solution with a Microm vibratome. Coronal slices with an angle of ∼15° were used, except where otherwise indicated, because the quality of cells in the OT was superior to other cutting angles. We initially also tested whether coronal sectioning would affect the ability to optogenetically evoke postsynaptic responses upon optogenetic stimulation of DAergic terminals. We observed no difference between sagittal and coronal slice preparations. For recovery and recording, slices were incubated at 32°C for 30 min in artificial CSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 2 CaCl2, 25 glucose (95% O2/5% CO2, 312 mOsm, and pH 7.3). IR-differential interference contrast patch-clamp recordings were performed with an EPC-10USB amplifier (HEKA). If not otherwise mentioned, the pipette solution for whole-cell recordings contained the following (in mm): 120 K-gluconate, 10 HEPES, 15 KCl, 4 Mg-ATP, 7 phosphocreatine, 4 ATP-Mg, 0.3 GTP, 0.1 EGTA, pH 7.2, 291 mOsm. Pipette resistance ranged from 4 to 7 MΩ, and cells were only accepted for recording if pipette access resistance was <25 MΩ. The liquid junction potential was not corrected throughout the study. All recordings were collected at 33°C. Data were acquired with Patchmaster software (HEKA) and analyzed with IgorPro software (Wavemetrics). SCH 23390 (7-chloro-3-methyl-1-phenyl-1,2,4,5-tetrahydro-3-benzazepin-8-ol), CGP 35348 [3-aminopropyl(diethoxymethyl)phosphinic acid], sulpiride, and atropine were purchased from Sigma-Aldrich; dihydro-β-erythroidine (DHβE) was purchased from Tocris Bioscience. CNQX, d-AP5, and SR 95531 were purchased from Biotrend.
Whole-cell measurements.
In recordings shown in Figure 1, we first recorded neurons in layer 2. We then performed recordings from layers 1b and 3, selecting cells with larger cell bodies to obtain a larger fraction of neurons with properties of CINs. For the remainder of the study, SPNs were targeted by their small cell bodies in layer 2 and by their active and passive membrane properties. CINs were selected by their large, multipolar somata and by their active and passive membrane properties in whole-cell recordings or by their spontaneous regular firing in cell-attached recordings. The pipette solution for recordings of glutamatergic EPSCs (see Figure 5) contained the following (in mm): 120 Cs-gluconate, 10 HEPES, 15 KCl, 4 Mg-ATP, 7 phosphocreatine, 4 ATP-Mg, 0.3 GTP, 0.1 EGTA, 5 QX314, pH 7.2, 291 mOsm. Resting membrane potential was determined shortly after establishing the whole-cell configuration. Membrane capacitance and resistance were determined by application of a hyperpolarizing pulse (10 mV for 10 ms). Action potential firing was recorded in current-clamp mode at Im = 0 pA by application of rectangular 1 s current steps starting at Im = −200 pA up (step size = +50 pA). The maximum instantaneous firing frequency is defined as the maximum frequency of the first two action potentials (APs). A double exponential fit was used to determine EPSP decay times. For paired-pulse ratio (PPR) analysis, the amplitude of the second EPSP was divided by the first EPSP. Fibers in the lateral olfactory tract were stimulated with a unipolar glass electrode that was positioned in the superficial layer 1a of the OT.
Figure 1.
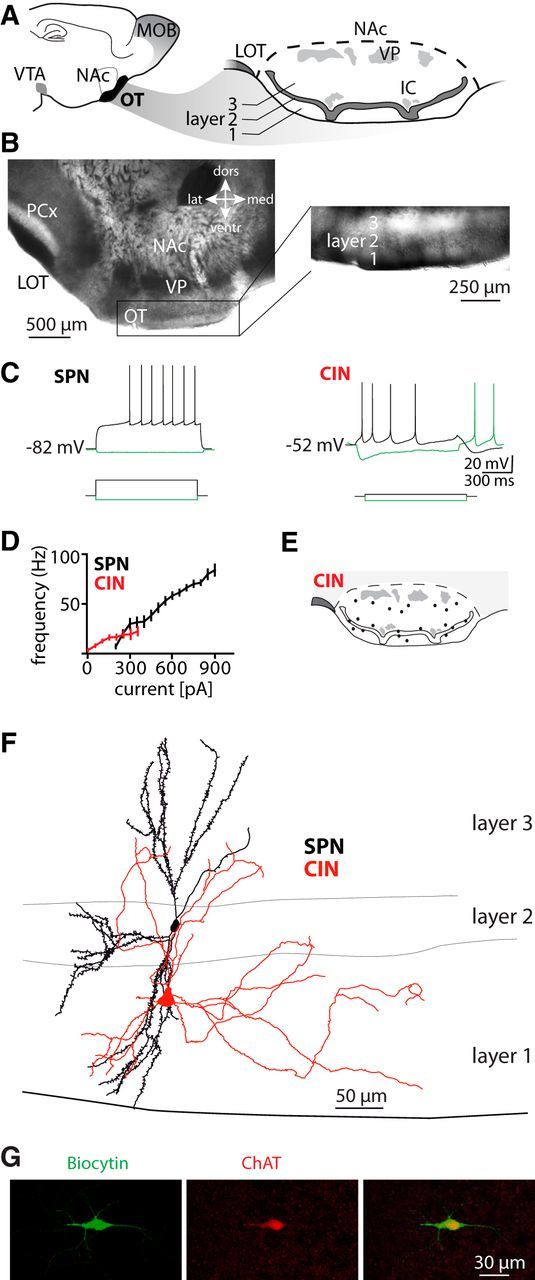
Characterization of OT neurons. A, Sagittal view of the mouse forebrain showing the location of the OT in the ventral striatum (left) and coronal view of the OT (right). MOB, Main olfactory bulb; VTA, ventral tegmental area; IC, islands of Calleja; VP, ventral pallidum. B, Image of the acute coronal slice preparation at lower (left) and higher magnification (differential interference contrast, right) of the adult OT. PCx, Piriform cortex. C, Membrane potential responses to current injection (1 s) in a layer 2 SPN (from Iinj = 0 pA to −50 and +200 pA) and a CIN with long afterhyperpolarization (from Iinj = 0 pA to −50 and +25 pA). D, Relationship of injected step current and evoked instantaneous firing frequency in SPNs (n = 83) and CINs (n = 24). E, Distribution of recorded CINs. F, Reconstruction of a biocytin-filled SPN (black) and an aspiny CIN (red). G, Confocal image of immunoreactivity for biocytin (green) and ChAT (in red) in a CIN. All data in this and subsequent figures are given as mean ± SEM except where otherwise indicated.
Figure 5.

Midbrain afferents evoke glutamate synaptic currents. A, The EPSC evoked by single laser pulses at Vh = +40 mV (in the presence of CNQX 10 μm/SR95531 10 μm) was blocked by NMDAR antagonist d-AP5 (50 μm). EPSCs evoked by laser pulses at Vh = −70 mV (in the presence of SR95531 10 μm) were blocked by the AMPAR antagonist CNQX (10 μm). B, Summary of EPSC amplitude responses at Vh = −70 mV (in the presence of SR95531 10 μm) and on additional application of CNQX [10 μm; p < 0.05, ANOVA with post-test (*p < 0.05), n = 5]. C, Summary of EPSC amplitude responses at Vh = +40 mV (in the presence of CNQX 10 μm and SR95531 10 μm) and on additional application of d-AP5 [50 μm; p < 0.05, ANOVA with post-test (*p < 0.05), n = 5]. D, Membrane current response to PLS in a CIN at Vh = −80 mV (in the presence of SR95531 10 μm) and on application of d-AP5 (50 μm) and CNQX (10 μm). Inset, Relationship of EPSCs to each laser pulse.
Cell-attached recordings.
For voltage-clamp cell-attached recordings, CINs were identified by their morphology and their spontaneous firing pattern. The pipette solution for cell-attached recordings contained 120 mm NaCl. Pipette resistance ranged from 6 to 8 MΩ. Tonically active neurons with firing frequencies >10 Hz and <0.33 Hz or with burst firing pattern were excluded. Very long interspike intervals at baseline resulted in unreliable quantification of the PLS-evoked pause; we therefore excluded CINs with firing frequencies <0.33 Hz. Neurons with baseline firing >10 Hz were excluded to reduce the chances of inclusion of other interneurons that are tonically firing at high frequencies (Beatty et al., 2012). In our voltage-clamp cell-attached recordings, Vh was adjusted to Im = 0 pA. Except where otherwise indicated, the PLS contained 12 laser pulses of 5 ms duration at 40 Hz. We binned the AP firing frequency (bin size = 100 ms) and defined the baseline as the average frequency within the 2 s before PLS.
Immunohistochemistry.
Mice were given an overdose of isoflurane followed by transcardiac perfusion with PBS at 37°C before being perfused with 4% PFA and postfixed in 4% PFA for 12 h at 4°C. Coronal slices (50 μm thick) were cut with a vibratome. Sections were incubated in blocking solution containing primary antibodies at 4°C overnight. Choline acetyltransferase (ChAT) was localized using a goat polyclonal antibody, AB144 (1:250), against ChAT (EMD Millipore, catalog #AB144P-1ML, Research Resource Identifier (RRID): AB_262156), and tyrosine hydroxylase (TH) was localized using sheep polyclonal antibody AB1542 (1:1000) against tyrosine hydroxylase (EMD Millipore, catalog #AB1542, RRID: AB_90755). Subsequently, Alexa Fluor 488/555-conjugated secondary antibodies (1:1000, Invitrogen) were applied in blocking solution at room temperature for 2 h. Blocking solution contained 0.25% Triton X-100 and 1% bovine serum albumin. Sections were mounted with Fluoromount (Sigma) and analyzed using a confocal laser scanning microscope (SPM5, Leica). Confocal images for colocalization were acquired with a 63× oil-immersion objective. The overview image (see Fig. 3A) was acquired as a montage with an epifluorescence microscope (Axio Imager 2, Zeiss) with a Neurolucida system (MicroBrightField).
Figure 3.

Optogenetic activation of DAergic axons in the OT. A, Left, Sagittal overview of ChR2:mCherry expression and its colocalization with immunoreactivity for TH (green) in the OT, NAc, ventral tegmental area (VTA), and substantia nigra pars compacta (SNc; scale bar, 1 mm) 3 weeks after injection of rAAV1/2-DIOA-ChR2:mCherry in DAT-Cre mice. Right, Colocalization of ChR2:mCherry and TH immunoreactivity at higher magnification in the VTA (scale bar, 20 μm) and the OT (scale bar, 10 μm). B, FSCV recording configuration of DA release upon PLS (40 Hz, 5 ms pulses) of ChR2+ DAergic terminals. C, Measurement of [DA] with FSCV at different PLS durations in layer 2 of the OT. Inset, Background-subtracted voltammogram.
Biocytin-filling of neurons and reconstruction.
Brain slices of adult mice were prepared as described for electrophysiology. Neurons were recorded with 2 μg/ml biocytin in the pipette for 10–30 min. After the pipette was withdrawn, slices were kept in the recording chamber for an additional 10 min and subsequently immersion fixed in 4% PFA at 4°C overnight. Biocytin-filled cells were incubated with peroxidase-avidin-biotin complex (ABC, Vector Laboratories) and visualized with DAB-nickel sulfate (Sigma). Subsequently, slices were mounted onto microscope slides and coverslipped with Mowiol. The reconstruction of biocytin-filled neurons was performed with a Neurolucida system (MicroBrightField) with a 100× oil-immersion objective (Axio Imager 2, Zeiss). In a second set of experiments, fixed tissue was resliced with a vibratome to obtain 50 μm sections before immunofluorescent staining. Immunofluorescent staining was performed as described above. Biocytin-filled neurons were visualized by Cy2-conjugated streptavidin (1:500, respectively; Jackson ImmunoResearch Laboratories). Double-labeled sections were processed and analyzed by confocal microscopy as described above.
Statistical analysis.
Except where otherwise indicated, data were normally distributed and are reported as mean ± SEM. Data were analyzed with the two-tailed Student's t test or ANOVA followed by a post-test with Bonferroni correction for multiple comparisons.
Results
Cholinergic interneurons in the olfactory tubercle
To characterize the properties and distribution of putative SPNs and CINs in the OT, we initially performed whole-cell patch-clamp recordings in acute coronal slices of adult mice (Fig. 1A,B). In layer 2 of the OT, most neurons (83 of 92 recorded cells) had functional properties of SPNs (Kawaguchi, 1993). They had a resting membrane potential of −82.2 ± 0.4 mV and an input resistance of 72.9 ± 2.1 MΩ (n = 83 cells). Upon current injection, these neurons showed a regular firing pattern with a slow ramp depolarization and a long latency to spike (Fig. 1C,D). Reconstructions of biocytin-filled neurons with these properties revealed spiny dendrites (spine density = 0.32 ± 0.02/μm, n = 6, Fig. 1F). CINs were found in all three layers (Fig. 1E). They had large somata and the membrane potential at zero current injections fluctuated between −49.3 ± 0.8 mV and −58.2 ± 0.9 mV (n = 24). Their input resistance was 359.5 ± 49.2 MΩ (n = 24 cells). CINs had a rebound discharge upon hyperpolarizing current injections (Fig. 1C). APs were followed by a pronounced afterhyperpolarization with cell type-specific temporal dynamics (time-to-trough 45.1 ± 5.4 ms vs 1.6 ± 0.04 ms, amplitude 14.6 ± 1.2 mV vs 13.7 ± 0.6 mV in 24 CINs vs 83 SPNs, respectively). Reconstructed biocytin-filled cells with these properties were aspiny (n = 7, Fig. 1F) and expressed choline acetyltransferase (8 of 8 cells; Fig. 1G).
In the OT, sensory inputs arrive from the main olfactory bulb via the lateral olfactory tract (LOT) in the superficial parts of layer 1. We recorded from SPNs with electrical stimulation of the LOT (Fig. 2A) and evoked EPSPs that were blocked by application of the AMPA/NMDAR antagonists CNQX (10 μm) and d-AP5 (50 μm, n = 4; Fig. 2B). Paired-pulse stimulation in LOT elicited synaptic facilitation of this input in SPNs (PPR 2.6 ± 0.2, n = 18). To test whether SPNs and CINs are both innervated by LOT projections from the olfactory bulb, we sequentially recorded neighboring SPNs and CINs (18 cells, respectively). We first recorded layer 2 SPNs and electrically stimulated the LOT. This LOT stimulation evoked EPSPs without failures (latency to EPSP peak 4.9 ± 0.3 ms, n = 18 SPNs). We kept the stimulation electrode in the same spot and recorded a neighboring CIN. In these neighboring CINs, LOT stimulation did not evoke EPSPs (n = 18 CINs). We then increased the stimulation intensity up to 50-fold; however, no short-latency EPSPs were evoked (data not shown). After unsuccessful stimulations of three CINs, we evoked EPSPs in another neighboring SPN that showed robust short-latency EPSPs at the original stimulation intensities (data not shown). We observed similar responses for LOT stimulations in sagittal and coronal slices and throughout the mediolateral and rostrocaudal extent of the OT. However, the same CINs responded with an EPSP (latency to EPSP peak = 3.9 ± 0.3 ms, n = 18) when we repositioned the stimulation electrode to layer 3 (Fig. 2C), where association fibers from other olfactory cortices terminate. The EPSP evoked in layer 3 was blocked by application of AMPA/NMDAR antagonists (n = 4; Fig. 2C). Paired-pulse stimulation in layer 3 elicited synaptic facilitation of this input (PPR 1.5 ± 0.1, n = 10). Thus, LOT axons predominantly project to SPNs in layer 2. In contrast, CINs receive excitatory synaptic input from other sources, but not LOT.
Figure 2.

Lateral olfactory tract inputs target SPNs but not CINs. A, Scheme of the recording configuration. B, Short-latency EPSPs in a SPN evoked by electrical stimulation of the LOT were blocked by NMDAR/AMPAR antagonists d-AP5 (50 μm) and CNQX (10 μm) (Vm = −80 mV). C, No EPSP was evoked in a CIN upon LOT stimulation. The same cell, however, responded with a short-latency EPSP upon stimulation in layer 3. The EPSP was blocked by d-AP5 (50 μm) and CNQX (10 μm) (Vm = −80 mV). Experiments in B and C were performed in the presence of SR95531 (10 μm) and with the same stimulation intensity. Stimulation artifacts were truncated. D, Blockade of AChRs with DHβE (2 μm) and atropine (2 μm) increased the amplitude and reduced the PPR of EPSPs in SPNs evoked by LOT stimulation (50 ms interstimulus interval, repeated at 0.1 Hz, Vm = −82 mV). Amplitudes of the first EPSP and the PPR were both normalized to the control condition for statistical analysis [both p < 0.05, ANOVA with post-test (*p < 0.05), n = 7, respectively].
CINs provide a cholinergic tone to the striatum. The cholinergic tone drops during firing pauses of CINs. Such sudden ACh drops gate synaptic inputs to SPNs in dorsal striatum (Pakhotin and Bracci, 2007; Ding et al., 2010) We tested whether blocking the cholinergic tone would enhance the EPSP amplitude of LOT projections to SPNs in the OT (Fig. 2D). We stimulated LOT axons with paired pulses (interpulse interval 50 ms) and recorded EPSPs in layer 2 SPNs. Application of nicotinergic and muscarinergic ACh receptor (AChR) blockers atropine (2 μm) and DHβE (2 μm) reversibly potentiated the amplitude of the EPSPs and reduced the PPR of EPSPs evoked by LOT stimulation (Fig. 2D). These observations support the notion that the cholinergic tone gates sensory input of the LOT to SPNs.
Activation of DAergic terminals evokes postsynaptic currents
We then focused on the actions of phasic activation of DAergic midbrain projections on CINs in the OT and conditionally expressed ChR2 in DAergic midbrain neurons (Fig. 3). We injected a Cre-inducible AAV with a DIOA containing ChR2 fused to mCherry (rAAV1/2-DIOA-ChR2:mCherry) into the ventral midbrain of 8-week-old DAT-Cre mice. Three weeks after injection, we histologically examined the ventral midbrain and OT of injected mice. In the ventral tegmental area, all ChR2:mCherry neurons (n = 1380 cells in 5 mice) expressed TH and 54.3% of TH+ neurons expressed ChR2:mCherry in these mice (Fig. 3A), indicating that ChR2 expression was restricted to DAergic neurons. Also, ChR2:mCherry+ axons in the OT expressed TH. We then tested whether trains of laser stimuli to these ChR2+ afferents evoke DA release in the OT. We stimulated DAergic terminals in OT with PLS (5 ms laser pulses at 40 Hz) and measured [DA] transients with FSCV (Fig. 3B). The [DA] transients increased with the number of laser pulses (n = 9, Fig. 3C). The PLS-evoked [DA] transients had a half-decay time (2.4 ± 0.9 s; n = 9) comparable to that of phasic DA peaks in vivo (Grace, 1991).
We first examined the activation of postsynaptic conductances in CINs during photostimulation of DAergic terminals (Fig. 4). We performed whole-cell recordings in CINs and applied trains of laser pulses. The PLS lasted 1 s and was repeated every 20 s. The PLS evoked a slowly rising inward current in CINs with maximum amplitude of 14.9 ± 3.4 pA at Vh = −80 mV in a K+-based internal solution in ChR2+ mice (n = 37), but not in wild-type mice (n = 8; Fig. 4A,B). Repetitive delivery of a 1 s PLS every 20 s for 10 min evoked an inward current with stable amplitudes (Fig. 4C). We also tested other frequencies of laser stimuli and observed maximal amplitude at 40 Hz (Fig. 4D). The slow current persisted when AMPA, NMDA, or GABAA/BR antagonists were applied (Fig. 4B,E). We then tested DAR antagonists. The D1-type DAR antagonist SCH 23390 (10 μm) blocks both D1Rs and D5Rs. CINs express mainly D5Rs (Yan and Surmeier, 1997). The PLS-evoked inward current was reversibly blocked by SCH 23390 (10 μm; Fig. 4B,F). CINs also express D2Rs (Yan and Surmeier, 1997). Application of the D2-type DAR antagonist sulpiride (10 μm), however, did not reduce the amplitude of the slow inward current recorded under the same conditions (control 9.8 ± 1.5 mV, sulpiride 12.9 ± 2.2 mV, n.s., t test, n = 8). The PLS-activated current decreased in amplitude when the Vh was depolarized from −90 to −55 mV (Fig. 4G). This slow current was not blocked by a Cs+-based internal solution and reversed with this internal solution near Vh = 0 mV (n = 5; Fig. 4H). Thus, phasic activation of endogenous DA release evoked a D1-type DAR-mediated cation conductance.
To determine whether DAergic neurons also release glutamate onto CINs in the OT, we recorded from CINs and applied single 5 ms laser pulses to ChR2+ terminals (Fig. 5A). A total of 86.7% of CINs responded with short-latency EPSCs at Vh = −70 mV (latency = 5.2 ± 0.4 ms, n = 34) upon laser stimulation in the presence of the GABAAR antagonist SR95531 (10 μm; Fig. 5A). These optogenetically evoked EPSCs exhibited CNQX-sensitive AMPAR components at Vh = −70 mV (Fig. 5A,B) with decay time constants of 7.8 ± 1.1 ms (n = 19) and a d-AP5-sensitive NMDAR component at Vh = +40 mV (Fig. 5A,C) with decay time constants of 133 ± 10 ms (n = 19). The AMPA/NMDAR EPSC ratio was 1.8 ± 0.6 (n = 19). A 10 Hz laser stimulation elicited a short-term-depression of EPSCs (PPR 0.69 ± 0.03 at Vh = −70 mV, n = 21). Together, DAergic axons projecting to the OT act dually on CINs by activation of glutamate synaptic currents and a slow D1-type DAR-mediated cation conductance (Fig. 5D).
Firing responses of CINs to phasic activation of DAergic terminals
Based on these glutamatergic and DAergic responses upon activation of endogenous release from DAergic terminals, we examined whether we can also evoke firing responses in CIN in a cell-attached configuration that preserves tonic firing (Fig. 6A,B). Whereas PLS (12 pulses, 40 Hz) did not affect the spontaneous firing of CINs in ChR2− mice (n = 10; Fig. 6C), PLS of ChR2+ DAergic axons (12 pulses, 40 Hz) evoked an initial burst, a firing pause, and a late burst in CINs (Fig. 6D). Thus, phasic activation of DAergic neurons can evoke all components of the CIN response.
Figure 6.
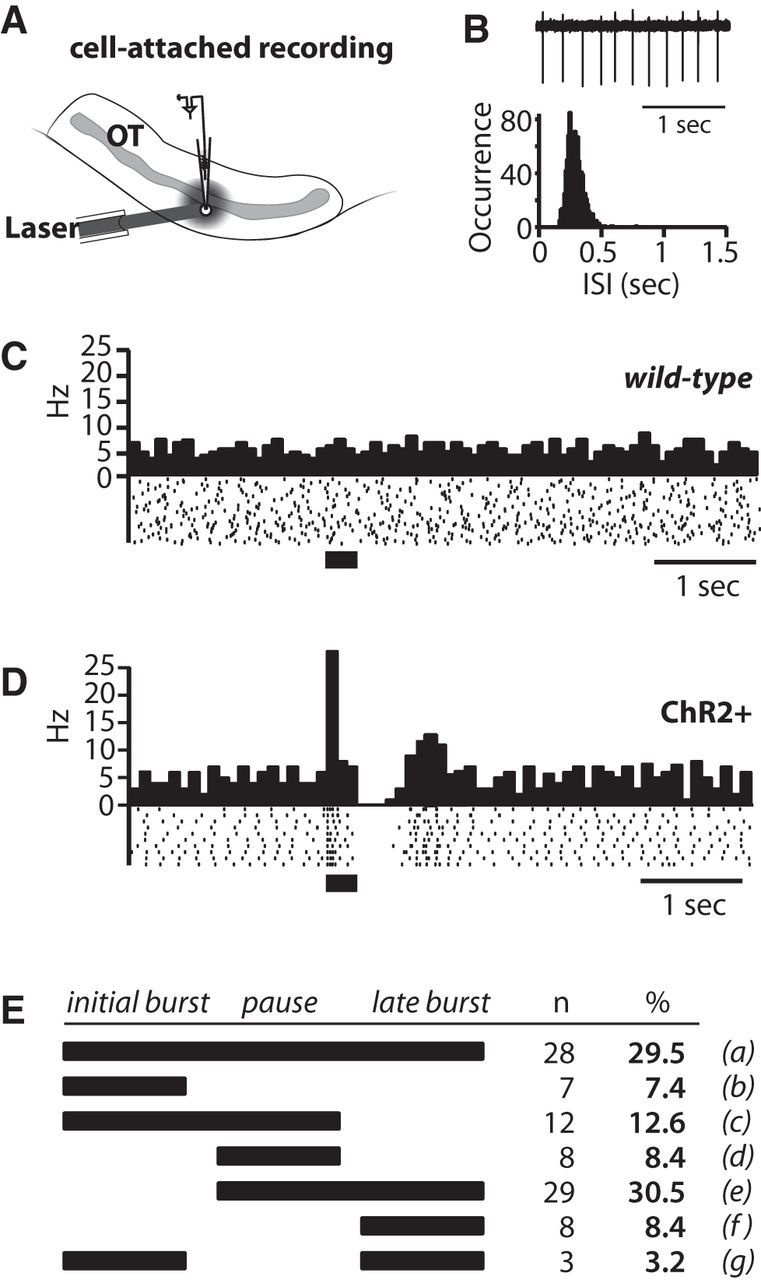
Phasic activation of DAergic terminals evokes a multiphasic firing response in CINs. A, Scheme of recording configuration. B, Spontaneous CIN firing in cell-attached configuration (top) and distribution of interspike intervals (ISI). C, Peristimulus histogram [PSTH; with 0.1 s bins (top) and raster plot of spike activity (bottom)] in a CIN. Application of PLS (40 Hz for 300 ms) in a brain slice from a wild-type (ChR2−) animal had no effect on spontaneous CIN firing. D, PSTH and raster plot showing an initial burst–pause–late burst firing response to PLS in a CIN recorded in a brain slice from a ChR2+ mouse. E, The scheme displays the co-occurrence of the different components of the firing responses at the normal threshold defined in Results. A higher threshold was also applied to the data (for definition, see Results); at this high threshold the co-occurrence was for a = 3.2% (n = 3); b = 18.9% (n = 18); c = 22.1% (n = 21); d = 18.9% (n = 18); e = 18.9% (n = 18); f = 17.9% (n = 17); g = not observed.
For these experiments we defined thresholds for bursts and pauses. The initial burst was defined as a twofold increase in the firing frequency (averages from 40 sweeps) in one bin (100 ms per bin) within 0–200 ms after the PLS onset. The firing pause was defined as a drop in the firing frequency of at least 50% in two consecutive bins within 500 ms after the PLS onset. The late burst was defined as a 50% increase of the firing frequency in two consecutive bins between 300 and 800 ms after the PLS onset. According to these criteria, 95 of 110 recorded CINs in the OT had at least one component of the firing responses. Among CINs with a firing response, 50 cells had an initial burst, 77 cells expressed a pause, and 68 cells responded with a late burst. The co-occurrence of the different components in single CINs is illustrated in Figure 6E. The durations of the initial burst, the pause, and the late burst were 135 ± 8 ms (n = 50), 313 ± 11 ms (n = 77), and 338 ± 9 ms (n = 68), respectively. At this threshold, PLS evoked none of these components in 10 CINs recorded in ChR2− mice (Fig. 6C). To test for robustness of the responses, we also applied a higher threshold for the burst that required a threefold increase for the initial burst and a 100% increase for the late burst. At the high threshold, the majority (95 of 110 recorded) of CINs in the OT still had at least one component of the firing responses (see Fig. 6E legend). Among them, 40 cells had an initial burst, 65 cells expressed a pause, and 42 cells responded with a late burst. Of note, the fraction of CINs with all three components dropped from 29.5% to 3.2% when the threshold was increased. At the same time, the fraction of cells with either an initial burst–pause or a pause–late burst combination increased when the higher threshold was applied. The durations of the initial burst, the pause, and the late burst were 143 ± 27 (n = 40), 393 ± 29 (n = 65), and 354 ± 33 ms (n = 42), respectively. Thus, the duration of the responses did not differ between the two thresholds (all n.s., t test, respectively).
We examined the different components of the PLS-evoked CIN firing response. We first examined the PLS-evoked initial burst–pause sequence (Fig. 7). The initial burst–pause sequence persisted when GABAA/BR antagonists SR95531 (10 μm) and CGP 55845 (5 μm) were applied (pause/baseline ratio in control, 0.18 ± 0.05, vs GABAA/BR antagonists, 0.20 ± 0.04; n.s., t test; n = 6), indicating that the pause was not mediated by polysynaptic inhibition. Thus, all subsequent recordings were performed in the presence of GABAA/BR antagonists (Figs. 7–9). Even though single laser pulses triggered an EPSC in CINs (Fig. 5), this stimulation was not sufficient to evoke a pause response. On average, >3 laser pulses were required to evoke a significant pause (Fig. 7A,B). Consistent with summation of EPSPs being required to evoke a pause following burst, both the initial burst and the following pause were reversibly blocked by the NMDAR antagonist (Fig. 7C,D). It is possible that the initial burst evokes ACh release that then modulates the subsequent pause. Application of nicotinergic and muscarinergic AChR antagonists DHβE (2 μm) and atropine (2 μm) did not significantly change the duration of the pause (control 335.0 ± 87.5 ms vs AChR blockers 385.0 ± 65.7 ms, n.s., t test, n = 6 CINs). Thus, AChRs were not required for the generation of the NMDAR-activated pause. In whole-cell recordings (Fig. 7E), PLS-evoked EPSPs summed and triggered bursts of APs (Fig. 7F) followed by an afterhyperpolarization during which the firing paused (Fig. 7G). The initial burst-induced hyperpolarization had a higher amplitude (13.6 ± 0.8 mV) than the AP-induced afterhyperpolarization during tonic firing (9.9 ± 0.8 mV, p < 0.05, t test; n = 4). We concluded that glutamate release from midbrain DAergic neurons triggers a NMDAR-dependent burst–pause sequence in CINs.
Figure 7.

Phasic activation of DAergic terminals evokes a NMDAR-dependent burst–pause sequence. A, Peristimulus histogram and raster plot of initial burst–pause response to 3 and 6 laser pulses per PLS (40 Hz). These and subsequent recordings were performed in the presence of SR95531 (10 μm). B, Box-whisker-plots (mean and 90–10 percentile) for the number of APs during a PLS with different numbers of laser pulses (left) and the relative increase of the interspike interval (ISI) following the PLS normalized to the mean ISI at baseline [pause factor, right; p < 0.05, ANOVA with post-test (*p < 0.05), n = 7 for both groups, respectively]. C, Raster plot of initial burst–pause response to PLS (40 Hz, 300 ms) showing reversible elimination of this response on application of the NMDAR blocker d-AP5 (50 μm). D, Quantitative analysis of the effect of NMDAR antagonism on CINs with an initial burst–pause response for changes in (left) the ratio of the peak to baseline firing rate during the initial burst, and (right) the pause to baseline firing rate ratio (p < 0.05, ANOVA with post-test (*p < 0.05), n = 11 for both groups, respectively). E, The membrane potential in whole-cell recording of a CIN during the initial burst–pause response revealed an afterhyperpolarization following the burst of APs (Vm = −52 mV). F, Higher temporal resolution of the PLS-triggered initial burst of the cell shown in E. G, Overlay of membrane potential in whole-cell recordings of a CIN during the initial burst–pause response (black, average of 30 sweeps; gray, single sweeps).
Figure 8.
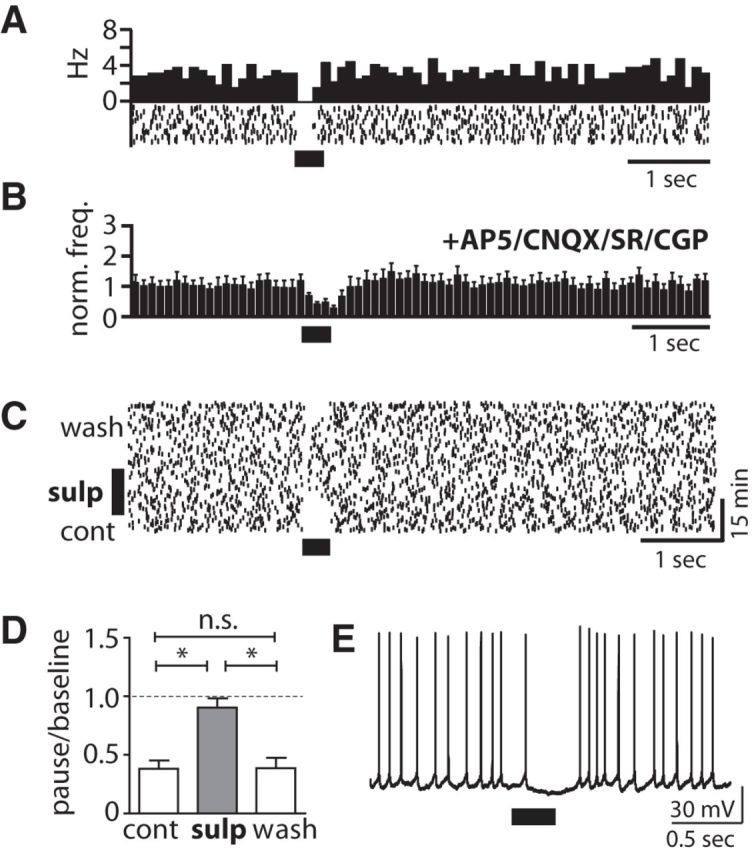
Phasic DA release evokes a D2-type DAR-dependent firing pause. A, Peristimulus histogram (PSTH) and raster plot of a pause response without a preceding burst evoked by PLS (40 Hz for 300 ms). B, The pause (population PSTH, n = 8) without preceding burst persisted in the presence of d-AP5 (50 μm), CNQX (10 μm), SR95531 (10 μm), or CGP 55845 (5 μm). Blockers were also present in C–E. C, Raster plot of isolated pause response to PLS and the reversible elimination of the pause during application of the D2-type DAR blocker sulpiride (10 μm). D, Quantitative assessment of the effect of D2-type DAR antagonism on the change in the firing rate during the pause compared with baseline firing in CINs expressing a pause response without initial burst [p < 0.05, ANOVA with post-test (*p < 0.05), n = 6]. E, Whole-cell recording of example CIN displaying a membrane hyperpolarization during the PLS-evoked pause response (Vm = −53 mV).
Figure 9.
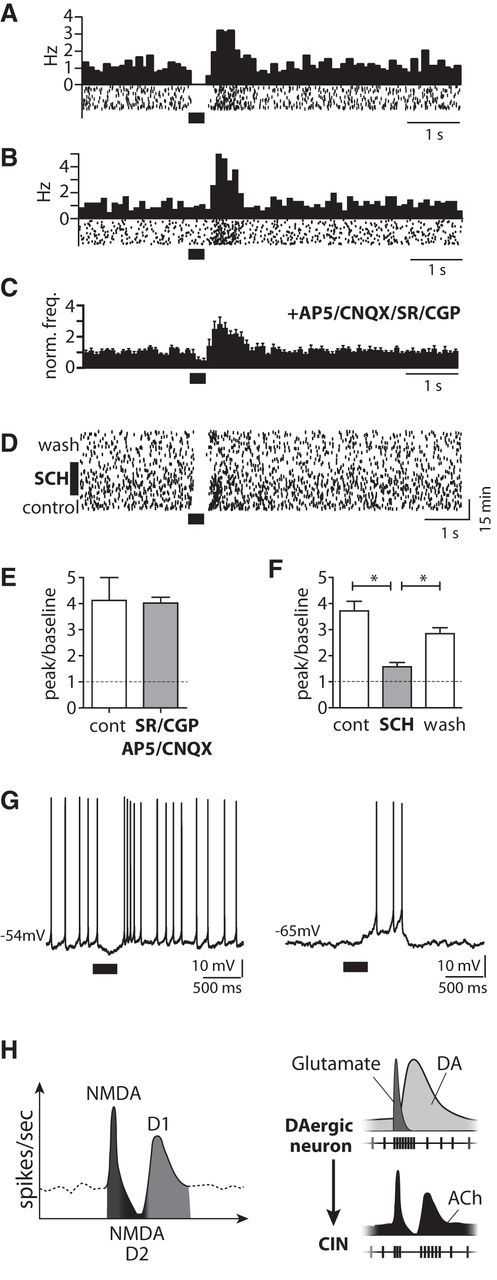
Phasic DA release evokes a D1-type DAR-mediated late burst. A, Peristimulus histogram (PSTH) and raster plot of a pause–late burst sequence evoked by PLS (40 Hz for 300 ms). B, PSTH and raster plot of a PLS-evoked late burst without a preceding pause. C, Late bursts (population PSTH, n = 9 cells) were detected in the presence of d-AP5 (50 μm), CNQX (10 μm), SR95531 (10 μm), and CGP 55845 (5 μm)]. (Blockers were also present in D and F.) D, Raster plot of a pause–late burst response to PLS and the reversible elimination of the late burst on application of the D1-type DAR blocker SCH 23390 (10 μm). E, The firing rate increase of the late burst compared with baseline firing did not change after application of d-AP5 (50 μm), CNQX (10 μm), SR95531 (10 μm), or CGP 55845 (5 μm; n.s., t test; n = 10 cells). F, Change in the firing rate during the late burst compared with baseline firing following application of the D1-type DAR antagonist SCH 23390 [10 μm; p < 0.05, ANOVA with post-test (*p < 0.05), n = 7]. G, Whole-cell recording of example CIN displaying a PLS-evoked late burst after a pause without current injection (left). In the same cell, negative current injection hyperpolarized the cell to Vm = −65 mV (right) so that spontaneous spiking ceased. At this membrane potential, PLS evoked a slow depolarization with a late burst. H, Summary of findings of receptors (left) and transmitters (right) mediating the different components, initial burst–pause–late burst, upon phasic activation of DA terminals.
CINs also expressed a pause without an initial burst (Fig. 8A). This type of pause response was not affected by glutamate or GABAA/BR antagonists (Fig. 8B), but was reversibly abolished by the D2-type DAR antagonist sulpiride (10 μm; Fig. 8C,D). In contrast, a D1-type DAR antagonist, SCH 23390 (10 μm), did not affect expression of the pause (pause/baseline ratio: control, 0.17 ± 0.09, vs SCH 23390, 0.26 ± 0.07; n.s., t test; n = 5). Whole-cell recordings revealed a membrane hyperpolarization during the pause. The pause-related hyperpolarization had a higher amplitude (11.4 ± 1.4 mV) than the AP-induced afterhyperpolarization during tonic firing (8.9 ± 1.4 mV, p < 0.05, t test; n = 5; Fig. 8E). Together, these observations reveal a second type of CIN pause that was independent of glutamate corelease, but required D2-type DAR activation.
Based on these observations, we then tested whether the firing pause of the initial burst–pause sequence required coactivation of D2-type DARs. We applied the D2-type DAR antagonist sulpiride in CINs. In CINs with neurons that expressed an initial burst–pause response, sulpiride (10 μm) neither significantly changed the pause length (pause duration in sulpiride 97.4 ± 14.5% of that before the drug: 427.5 ± 154.5 ms; n.s., t test, n = 4 cells), nor changed the firing rate during the pause (pause/baseline ratio: control, 0.28 ± 0.12, vs sulpiride, 0.23 ± 0.07; n.s., t test, n = 4 cells).
Finally, we examined CINs with a late burst (Fig. 9). The late burst was observed between 300 and 800 ms after PLS onset, whereas the NMDAR-mediated initial burst occurred during the first 200 ms of the PLS (Fig. 7). The late burst was not preceded by a pause in all CINs (Fig. 9A,B; see also Fig. 6E). The late burst without a preceding pause suggests that the late burst was not due to rebound firing after the pause and may thus be mediated by a mechanism that differs from those responsible for the pause. The late burst could still be evoked in the presence of glutamate and GABAA/BR antagonists (Fig. 9C). Also, the increase of the firing response during the late burst did not change compared with control conditions when glutamate and GABAA/BR antagonists were applied (Fig. 9 E). The late burst was, however, reversibly blocked by D1-type DAR antagonist SCH 23390 (10 μm; Fig. 9D,F). In whole-cell recordings (Fig. 9G), PLS evoked a late burst following the pause in CINs with spontaneous firing (Vm = −50 to −53 mV; no intracellular current was injected; n = 3). Recordings were performed in the presence of AMPA/NMDAR and GABAA/BR antagonists. The late burst occurred in a time window of 300–800 ms after PLS onset. We then hyperpolarized the CINs to Vm = −65 mV by negative current injection, so that spontaneous spiking ceased. Under this condition PLS evoked a depolarization generating multiple APs. The time courses of the depolarization and the bursts were comparable with and without negative current injection (Fig. 9G). In contrast to cell-attached recordings, the late burst disappeared within the first few minutes after establishing the whole-cell configuration, possibly because of intracellular dialysis of G-protein-coupled molecules that mediate the D1-type DAR responses. Together, our observations indicated a D1-type DAR-mediated late burst in CINs of the OT upon phasic activation of DAergic neurons that was relatively independent of the occurrence of a preceding pause.
Discussion
DA is generally considered a slow neuromodulator (Kreitzer, 2009). However, phasic firing of DAergic midbrain neurons is also expected to transmit information at fast time scales (Schultz, 1986; Berridge and Robinson, 1998). The synaptic and cellular mechanisms of fast information transfer by DA are not clear. We observed in this study that optogenetically activated phasic DA release controls firing of striatal neurons at a subsecond timescale. This phasic release from DAergic neurons could evoke all three components of the firing response in CINs of the OT by coordinated actions of glutamate and DA release (Fig. 9H). Glutamate receptor activation evoked an initial burst followed by an afterhyperpolarization with a firing pause, whereas DA elicited both a D2-type DAR-dependent firing pause and a late D1-type DAR-dependent burst. Activation of the cholinergic pause response by phasic DA provides a direct connection that links the global midbrain salience signal and the local striatal significance marker. Via this direct connection, phasic bursts of DAergic neurons are able to control the cholinergic tone in the striatum. This transient modulation of the cholinergic tone gates, in turn, sensory inputs to projection neurons.
Glutamate release from midbrain afferents
Phasic release of glutamate from midbrain afferents evoked a burst–pause response in CINs. Several previous studies demonstrated that corelease from DAergic midbrain neurons evoke glutamatergic synaptic currents in SPNs in nucleus accumbens (NAc) and the dorsal striatum (Chuhma et al., 2004; Descarries et al., 2008; Tecuapetla et al., 2010; Tritsch et al., 2012). Together with a recent study in nucleus accumbens of the ventral striatum (Chuhma et al., 2014), our study reveals a first function for glutamate release from ventral midbrain terminals. The release of glutamate from ventral midbrain afferents has been attributed to corelease from DAergic terminals to NAc that express the glutamate transporter vGluT2 (Hnasko et al., 2010). Deletion of vGluT2 abolishes induced glutamate release from these midbrain terminals (Hnasko et al., 2010). Recent studies reported that a small fraction of ventral midbrain neurons express glutamatergic over DAergic markers (Hnasko et al., 2012; Been et al., 2013; Yamaguchi et al., 2013). Compatible with corelease of glutamate, we observed that ChR2+:mCherry expression in our transgenic mice was restricted to TH+ ventral midbrain DAergic neurons. Photostimulation of these ChR2+ ventral midbrain projections evoked robust short-latency synaptic currents in the majority of CINs in the OT, suggesting that glutamate originates most likely from corelease from DAergic terminals.
In the OT, the activation of an initial burst-induced pause required repetitive activation of DAergic afferents. On average, more than three repetitive presynaptic stimuli were necessary to trigger multiple APs and a firing pause. The pause following the burst was NMDAR dependent, compatible with the requirement of EPSP summation to elicit multiple APs and a long afterhyperpolarization. In addition to burst-activated hyperpolarizing conductances, a partial depolarization block of action-potential initiation may contribute to the drop in firing probability following the initial phase of bursting. Interestingly, the pause following a NMDAR-dependent initial burst did not require coactivation of D2-type DARs. Together, DAergic midbrain neurons add to thalamic and cortical afferents (Lapper and Bolam, 1992; Ding et al., 2010; Doig et al., 2014) that provide glutamatergic inputs to directly control CIN firing. This glutamate corelease provides a pathway that allows ventral midbrain neurons to transmit information to target circuits at a fast timescale. Dopaminergic terminals in the dorsal striatum can also corelease the inhibitory transmitter GABA (Tritsch et al., 2012; Nelson et al., 2014). GABAergic transmission did not, however, contribute to the different components of the CIN firing response in the OT elicited by phasic activation of DAergic terminals as revealed here by application of GABAA/BR blockers.
Direct actions of phasic DA release
DA has different actions on postsynaptic computation depending on the local [DA] (Grace, 1991; Kreitzer, 2009). Changes in local [DA] differentially activate low-affinity D1-type and high-affinity D2-type DARs (Grace, 1991). Yet, the cellular effects of phasic DA release are still poorly understood. We observed that evoked phasic endogenous DA release can activate a long hyperpolarization and a firing pause in CINs by activation of D2-type DARs. In the dorsal striatum, D2-type DAR activation can hyperpolarize CINs by inhibition of persistent sodium and HCN channels (Yan et al., 1997; Maurice et al., 2004; Deng et al., 2007). Evoked endogenous DA release also evokes a D2-type DAR-dependent pause in CINs of the dorsal striatum (Chuhma et al., 2014). This report also observed heterogeneity of glutamate corelease- and DA-mediated pause responses in the nucleus accumbens. Interestingly, this study did not describe the third component of the CIN response. The study used only one photostimulus or very few photostimuli at lower frequencies (20 Hz) and did not measure the released [DA]. It is therefore possible that [DA] levels were too low to activate low-affinity D1-type DARs that are required for the late burst.
The late burst is observed in vivo in some, but not all CINs of the dorsal striatum (Aosaki et al., 1994b; Morris et al., 2004; Apicella et al., 2011; Doig et al., 2014). In the OT, we also found a late burst in approximately half of the CINs in response to phasic activation of DAergic projections. In a subset of CINs, the late burst did not depend on the occurrence of a preceding pause. This is compatible with independent receptor pathways by which DA evokes the pause and the late burst. In contrast to the pause, the late burst was blocked by D1-type DAR antagonists. We observed that induced endogenous phasic DA release evoked a D1-type DAR-mediated cation conductance in CINs of the OT. Similarly, in dorsal striatum, puff application of DA elicits a D1-type DAR-mediated cation conductance followed by action-potential firing in CINs (Aosaki et al., 1998; Centonze et al., 2003). Particularly, D1-type DARs activate voltage-gated sodium and calcium channels by protein kinase A (Neve et al., 2004; Kreitzer, 2009). DAergic midbrain neurons operate at two modes, i.e., tonic and burst firing. While tonic firing at ∼4–10 Hz maintains the DA tone, brief periods of bursts at ∼40 Hz by large fractions of DAergic neurons result in peaks of (phasic) DA release (Schultz, 1986). Activation of DAergic terminals at frequencies of ∼40 Hz also optimally activated D1-type-mediated cation conductance under our conditions, as would be predicted for low-affinity DARs. Together, these results provide us the first evidence that evoked endogenous phasic release of DA can elicit a D1-type DAR-dependent, slow-depolarizing conductance and late burst in CINs. Reward probability correlates to phasic midbrain firing in primates (Ljungberg et al., 1992) and, importantly, also to the strength of late burst of the CIN response in the dorsal striatum (Apicella et al., 2011). Assuming related cellular functions in dorsal striatum and the OT, the D1-type DAR-mediated late burst may thus provide a mechanism by which midbrain neurons transmit reward information to the striatal CIN network. These observations also highlight the notion that phasic release of DA itself can transmit information at the timescales of hundreds of milliseconds.
Functional implications of phasic DA-induced CIN responses
The top-down control of striatal cholinergic tone by midbrain DAergic neurons has several functional implications. DAergic midbrain afferents generate firing responses in CINs by the use of distinct transmitter and receptor pathways. These distinct CIN firing responses add to the heterogeneity that has been recently observed in DAergic neurons innervating striatum. For example, subsets of midbrain DAergic neurons either burst or stop firing in response to sensory stimuli or reward (Bromberg-Martin et al., 2010; Cohen et al., 2012; Roeper, 2013). Furthermore, axon terminals of midbrain DAergic neurons arborize only in a relatively small area of the striatum (Matsuda et al., 2009) and are heterogeneous in the expression of their DA, glutamate, and GABA release machinery (Hnasko et al., 2012; Tritsch et al., 2012; Been et al., 2013; Yamaguchi et al., 2013). The heterogeneity in the innervation of the striatum raises the possibility that CINs do not receive DAergic inputs with homogenous properties.
CINs use different DAR pathways to generate distinct response patterns. In particular, activation of D1- and D2-type DARs allows for differential expression of the pause and the late burst. In the ventromedial striatum, CINs display burst firing without a concomitant pause in response to cues and reward (Atallah et al., 2014). Firing pauses and bursts of CINs will have opposing actions. The burst firing of CINs increases ACh release, whereas firing pauses result in drops of the ACh tone provided by spontaneously active CINs. In the dorsal striatum, both increases and decreases in the cholinergic tone differentially gate sensory inputs to striatal projection neurons (Pakhotin and Bracci, 2007; Ding et al., 2010; Witten et al., 2010; English et al., 2012). In the OT, reduction of the cholinergic tone reversibly increased the amplitude of olfactory inputs in SPNs. This indirect gating through CINs may allow midbrain DAergic neurons to exert acute effects on sensory processing in SPNs. The opposing actions of D1-type bursts and D2-type pauses in the bidirectional gating of olfactory inputs described here may be cellular mechanisms relevant to processes involved in the selection of relevant sensory inputs in the OT. These experiments require realistic timing of the ACh release because, in particular, nAChRs desensitize quickly. Dynamic increases and drops in released ACh during the multiphasic CIN response could create a defined time-window for DA-induced plasticity of sensory inputs to projection neurons.
Together, our results show that phasic firing of DAergic neurons directly controls striatal cholinergic neurons by distinct receptor pathways and provides a novel link of two transmitter systems central to neuropsychiatric disorders. The direct control of CIN firing demonstrates that DAergic neurons have the capacity to act at an unexpectedly fast timescale. These cellular mechanisms may be relevant to the selection of sensory inputs and aberrant salience in schizophrenia (Kapur, 2003).
Footnotes
The project was funded by Deutsche Forschungsgemeinschaft Emmy-Noether-Grant KE1661/1-1 to W.K., R.P. was supported by Bundesministerium für Bildung und Frauen Grant 01GS08141 and the Alfred Krupp Prize. We thank Dr. Andreas Meyer-Lindenberg for his support, Dr. Genela Morris for discussions, and Cathy Huber for technical assistance.
The authors declare no competing financial interests.
References
- Aosaki T, Graybiel AM, Kimura M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science. 1994a;265:412–415. doi: 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. 1994b;14:3969–3984. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Kiuchi K, Kawaguchi Y. Dopamine D1-like receptor activation excites rat striatal large aspiny neurons in vitro. J Neurosci. 1998;18:5180–5190. doi: 10.1523/JNEUROSCI.18-14-05180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicella P, Ravel S, Deffains M, Legallet E. The role of striatal tonically active neurons in reward prediction error signaling during instrumental task performance. J Neurosci. 2011;31:1507–1515. doi: 10.1523/JNEUROSCI.4880-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atallah HE, McCool AD, Howe MW, Graybiel AM. Neurons in the ventral striatum exhibit cell-type-specific representations of outcome during learning. Neuron. 2014;82:1145–1156. doi: 10.1016/j.neuron.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty JA, Sullivan MA, Morikawa H, Wilson CJ. Complex autonomous firing patterns of striatal low-threshold spike interneurons. J Neurophysiol. 2012;108:771–781. doi: 10.1152/jn.00283.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Been LE, Staffend NA, Tucker A, Meisel RL. Vesicular glutamate transporter 2 and tyrosine hydroxylase are not co-localized in Syrian hamster nucleus accumbens afferents. Neurosci Lett. 2013;550:41–45. doi: 10.1016/j.neulet.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamou L, Kehat O, Cohen D. Firing pattern characteristics of tonically active neurons in rat striatum: context dependent or species divergent? J Neurosci. 2014;34:2299–2304. doi: 10.1523/JNEUROSCI.1798-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–369. doi: 10.1016/S0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron. 2010;68:815–834. doi: 10.1016/j.neuron.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Grande C, Usiello A, Gubellini P, Erbs E, Martin AB, Pisani A, Tognazzi N, Bernardi G, Moratalla R, Borrelli E, Calabresi P. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J Neurosci. 2003;23:6245–6254. doi: 10.1523/JNEUROSCI.23-15-06245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, Rayport S. Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci. 2004;24:972–981. doi: 10.1523/JNEUROSCI.4317-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron. 2014;81:901–912. doi: 10.1016/j.neuron.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JY, Haesler S, Vong L, Lowell BB, Uchida N. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature. 2012;482:85–88. doi: 10.1038/nature10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P, Zhang Y, Xu ZC. Involvement of I(h) in dopamine modulation of tonic firing in striatal cholinergic interneurons. J Neurosci. 2007;27:3148–3156. doi: 10.1523/JNEUROSCI.5535-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Bérubé-Carrière N, Riad M, Bo GD, Mendez JA, Trudeau LE. Glutamate in dopamine neurons: synaptic versus diffuse transmission. Brain Res Rev. 2008;58:290–302. doi: 10.1016/j.brainresrev.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Dimova R, Vuillet J, Nieoullon A, Kerkerian-Le Goff L. Ultrastructural features of the choline acetyltransferase-containing neurons and relationships with nigral dopaminergic and cortical afferent pathways in the rat striatum. Neuroscience. 1993;53:1059–1071. doi: 10.1016/0306-4522(93)90489-3. [DOI] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Magill PJ, Apicella P, Bolam JP, Sharott A. Cortical and thalamic excitation mediate the multiphasic responses of striatal cholinergic interneurons to motivationally salient stimuli. J Neurosci. 2014;34:3101–3117. doi: 10.1523/JNEUROSCI.4627-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsáki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-U. [DOI] [PubMed] [Google Scholar]
- Haberly LB, Price JL. Association and commissural fiber systems of the olfactory cortex of the rat. J Comp Neurol. 1978;178:711–740. doi: 10.1002/cne.901780408. [DOI] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, Edwards RH. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron. 2010;65:643–656. doi: 10.1016/j.neuron.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Hjelmstad GO, Fields HL, Edwards RH. Ventral tegmental area glutamate neurons: electrophysiological properties and projections. J Neurosci. 2012;32:15076–15085. doi: 10.1523/JNEUROSCI.3128-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S. Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Rajkowski J, Evarts E. Tonically discharging putamen neurons exhibit set-dependent responses. Proc Natl Acad Sci U S A. 1984;81:4998–5001. doi: 10.1073/pnas.81.15.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Lapper SR, Bolam JP. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience. 1992;51:533–545. doi: 10.1016/0306-4522(92)90293-B. [DOI] [PubMed] [Google Scholar]
- Ljungberg T, Apicella P, Schultz W. Responses of monkey dopamine neurons during learning of behavioral reactions. J Neurophysiol. 1992;67:145–163. doi: 10.1152/jn.1992.67.1.145. [DOI] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara AM, Cleland TA, Linster C. Characterization of the synaptic properties of olfactory bulb projections. Chem Senses. 2004;29:225–233. doi: 10.1093/chemse/bjh027. [DOI] [PubMed] [Google Scholar]
- Millhouse OE, Heimer L. Cell configurations in the olfactory tubercle of the rat. J Comp Neurol. 1984;228:571–597. doi: 10.1002/cne.902280409. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Nagayama S, Enerva A, Fletcher ML, Masurkar AV, Igarashi KM, Mori K, Chen WR. Differential axonal projection of mitral and tufted cells in the mouse main olfactory system. Front Neural Circuits. 2010;4:120. doi: 10.3389/fncir.2010.00120. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, Kreitzer AC. Striatal cholinergic interneurons drive GABA release from dopamine terminals. Neuron. 2014;82:63–70. doi: 10.1016/j.neuron.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/RRS-200029981. [DOI] [PubMed] [Google Scholar]
- Pakhotin P, Bracci E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato R, Rieker C, Turiault M, Tronche F, Schütz G. Survival of DA neurons is independent of CREM upregulation in absence of CREB. Genesis. 2006;44:454–464. doi: 10.1002/dvg.20236. [DOI] [PubMed] [Google Scholar]
- Price JL. An autoradiographic study of complementary laminar patterns of termination of afferent fibers to the olfactory cortex. J Comp Neurol. 1973;150:87–108. doi: 10.1002/cne.901500105. [DOI] [PubMed] [Google Scholar]
- Robinson DL, Venton BJ, Heien ML, Wightman RM. Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin Chem. 2003;49:1763–1773. doi: 10.1373/49.10.1763. [DOI] [PubMed] [Google Scholar]
- Roeper J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 2013;36:336–342. doi: 10.1016/j.tins.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Schultz W. Responses of midbrain dopamine neurons to behavioral trigger stimuli in the monkey. J Neurophysiol. 1986;56:1439–1461. doi: 10.1152/jn.1986.56.5.1439. [DOI] [PubMed] [Google Scholar]
- Schulz JM, Reynolds JN. Pause and rebound: sensory control of cholinergic signaling in the striatum. Trends Neurosci. 2013;36:41–50. doi: 10.1016/j.tins.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Price JL. The development of lamination of afferent fibers to the olfactory cortex in rats, with additional observations in the adult. J Comp Neurol. 1984;223:203–222. doi: 10.1002/cne.902230205. [DOI] [PubMed] [Google Scholar]
- Sosulski DL, Bloom ML, Cutforth T, Axel R, Datta SR. Distinct representations of olfactory information in different cortical centres. Nature. 2011;472:213–216. doi: 10.1038/nature09868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, Koos T. Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J Neurosci. 2010;30:7105–7110. doi: 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–266. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahle P, Meyer G. The olfactory tubercle of the cat. II. Immunohistochemical compartmentation. Exp Brain Res. 1986;62:528–540. doi: 10.1007/BF00236031. [DOI] [PubMed] [Google Scholar]
- Wesson DW, Wilson DA. Smelling sounds: olfactory-auditory sensory convergence in the olfactory tubercle. J Neurosci. 2010;30:3013–3021. doi: 10.1523/JNEUROSCI.6003-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Wang HL, Morales M. Glutamate neurons in the substantia nigra compacta and retrorubral field. Eur J Neurosci. 2013;38:3602–3610. doi: 10.1111/ejn.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/S0896-6273(00)80402-X. [DOI] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- Zelano C, Bensafi M, Porter J, Mainland J, Johnson B, Bremner E, Telles C, Khan R, Sobel N. Attentional modulation in human primary olfactory cortex. Nat Neurosci. 2005;8:114–120. doi: 10.1038/nn1368. [DOI] [PubMed] [Google Scholar]