

Abstract
The vesicular glutamate transporter (VGLUT) plays an essential role in synaptic transmission by filling vesicles with glutamate. At mammalian synapses, VGLUT expression level determines the amount of glutamate packaged into vesicles, and the specific paralog of VGLUT expressed affects the release probability. In this study, we investigate whether there is a link between the number of VGLUTs on vesicles and release probability. We used a combination of electrophysiology and imaging techniques in cultured mouse hippocampal neurons where the VGLUT expression level has been severely altered. We found that vesicles with drastically reduced VGLUT expression were released with a lower probability. This deficit in release could only be rescued by a functional transporter, suggesting that the transport function, and not the molecular interactions, of the protein affects vesicle release. Based on these data, we propose a novel means of presynaptic vesicle release regulation—the intravesicular glutamate fill state of the vesicle.
Keywords: glutamate, hippocampus, release probability, synaptic transmission, vesicular transporter
Introduction
Vesicular glutamate transporters (VGLUTs) play a critical role in synaptic transmission by mediating the transport of glutamate into vesicles. In addition to the essential role for this protein in the vesicle cycle, two additional roles have recently been proposed. First, at glutamatergic mammalian synapses, it has been suggested that the number of VGLUTs influences the amount of glutamate packaged into the synaptic vesicle (Wilson et al., 2005; Moechars et al., 2006; Wojcik et al., 2004). Estimates for the number of VGLUTs on vesicles range from 4 to 14 (Takamori et al., 2006; Mutch et al., 2011).
The second novel role for VGLUTs is a paralog-specific effect on vesicular release probability wherein vesicles containing VGLUT1 have a lower release probability than vesicles containing VGLUT2 or VGLUT3 (Weston et al., 2011). This effect is mediated through an interaction between VGLUT1 and endophilin 1, which buffers the latter and decreases its ability to facilitate vesicle release. This suggests that VGLUT influences the intrinsic release probability of vesicles within the synapse.
Initial characterization of VGLUT1 knock-out (KO) mice revealed several important findings. First, the significant decrease in synaptic responses in hippocampal neurons in both acute slice and autaptic culture suggests that VGLUT1 is the predominant transporter subtype in this region (Fremeau et al., 2004; Wojcik et al., 2004). Second, a decrease in the quantal size, as assayed by miniature excitatory postsyanptic currents (mEPSCs), was observed in VGLUT1 KO neurons compared with wild-type (WT) autaptic neurons, suggesting that the number of transporters affects the amount of transmitter packaged into a vesicle, as this was presumably mediated by a low expression level of the endogenous VGLUT2 paralog (Wojcik et al., 2004). Finally, analysis of the residual response in VGLUT1 KO autaptic neurons revealed that these neurons trended toward a lower vesicular release probability than WT controls (Wojcik et al., 2004). This is surprising, because vesicles containing VGLUT2 should have a higher release probability than those containing VGLUT1 in the WT neurons (Weston et al., 2011). This raises the possibility that the decreased vesicular release probability in VGLUT1 KO neurons is mediated by the decrease in VGLUT copy number.
In this study, we investigated whether the number of VGLUTs on a synaptic vesicle affects its release probability (Pr). Using a synaptophysin-pHluorin (SypHy) technique with sensitivity equivalent to the paired-pulse ratio (PPR) for detecting qualitative differences between synaptic Prs, we determined that the empty vesicles, in addition to the partially full vesicles detected by electrophysiology, in VGLUT1 KO neurons have a lower Pr than those in WT neurons. We show that this is not due to differences in glutamate signaling during development or presynaptic calcium influx between autapses of the two genotypes. By performing our Pr imaging protocol in VGLUT1 KO neurons expressing a WT or transport-deficient VGLUT2 protein tagged with pHluorin, we show that the reduced Pr in KO neurons is not due to the absence of the protein itself, but likely is due to the decreased glutamate fill state of the vesicles.
Materials and Methods
Hippocampal culture preparation.
Microisland cultures were prepared from hippocampal neurons and maintained as previously described (Arancillo et al., 2013). All procedures for experiments involving animals were approved by the animal welfare committee of Charité Medical University and the Berlin state government (permit number T0220/09). Hippocampi were harvested from postnatal day 0 (P0) to P2 mice of either sex of VGLUT1 KO (Wojcik et al., 2004; RRID:MGI_MGI:3718199) or WT (C57BL/6N) genotypes as indicated. Hippocampal neurons from littermates were plated in experiments using both VGLUT1 KO and WT genotypes (see Figs. 2, 3, 4). For electron microscopy (EM) experiments, VGLUT1 KO hippocampal neurons were plated en masse on a layer of astrocytes at a density of ∼1000 cells/cm2 glass-bottom culture dishes (MatTek) or glass coverslips.
Figure 2.
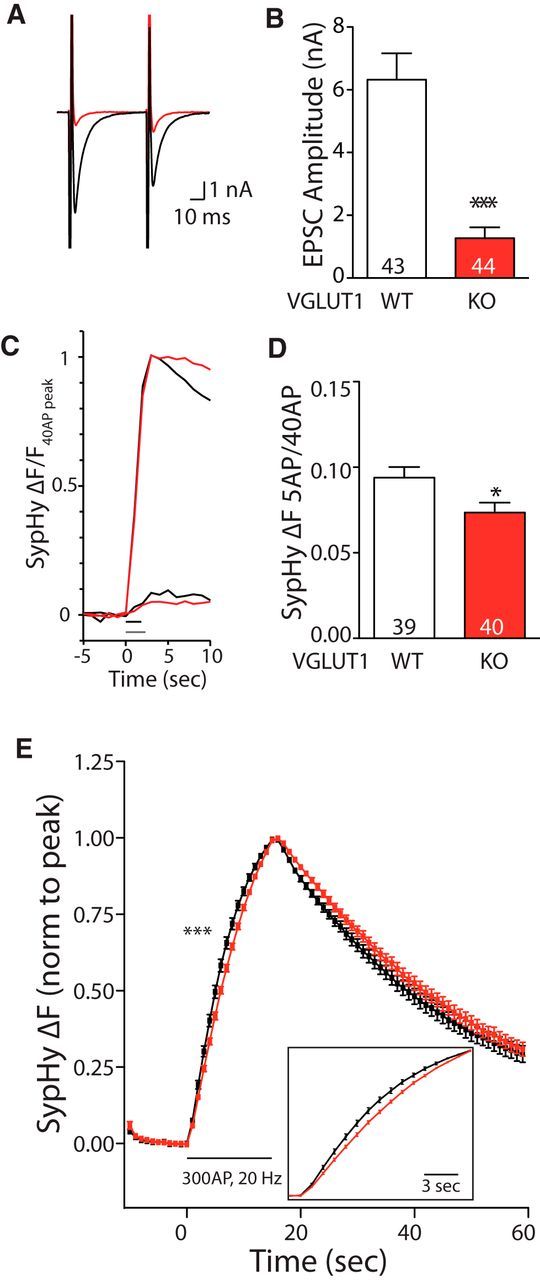
VGLUT1 KO autapses show decreased release probability with SypHy measurements. A, Example traces of EPSCs produced in WT (black) or VGLUT1 KO (red) autapses also expressing SypHy. B, Average EPSC amplitudes produced in WT and VGLUT1 KO autapses expressing SypHy. Signficance determined by Mann–Whitney U test: ***p < 0.0001. C, Example SypHy ΔF signal produced by 5 APs at 2 Hz and 40 APs at 20 Hz averaged per cell and normalized to ΔF 40 APs for a WT (black) and VGLUT1 KO (red) autapses. Bars parallel to the x-axis indicate stimulation with 5 APs (2 Hz; gray) or 40 APs (20 Hz; black). D, Average SypHy ΔF 5 AP/40 AP signal ratio recorded in WT and VGLUT1 KO autapses. For B and D, significance was determined by Student's t test: *p = 0.019. E, Average SypHy ΔF response to 300 APs, 20 Hz, normalized to the peak per cell for WT (black; n = 33) and VGLUT1 KO (red; n = 36) autapses. Significance indicates comparison of 20–80% rise times (Student's t test, ***p = 0.0003). For all panels, n values (number of cells) are indicated in graphs; error bars represent the SEM.
Figure 3.

Release probability deficit in VGLUT1 KO neurons with input from WT synapses. A, Top, Schematic representation of a two-neuron microculture with mixed genotypes (black, WT; red, VGLUT1 KO). Each neuron makes synaptic connections onto itself (autaptic) and onto the partner neuron (heterosynaptic). Bottom, Example traces of excitatory postsynaptic responses (i.e., EPSCs) from a mixed genotype pair. Stimulation of the VGLUT1 KO cell (red) produces an autaptic response in itself and a heterosynaptic response in its partner, as does stimulation of the WT cell (black). Depolarization artifacts in autaptic responses have been blanked for illustrative purposes. B, Bar graph of the average EPSC input (sum of autaptic EPSC of a neuron and heterosynaptic EPSC from the partner neuron) from VGLUT1 KO (red) and WT (black) neurons in mixed pairs. C, Same as B for the average EPSC output (A, dashed boxes; sum of autaptic and heterosynaptic EPSC from a given neuron). D, Comparison of EPSCs produced by VGLUT1 KO (red) and WT (black) neurons in the pair illustrated in A. Responses are scaled to the maximum amplitude of the first pulse to illustrate differences in PPR between genotypes. E, Bar graph of average PPR (interstimulus interval, 25 ms; autaptic and heterosynaptic PPR averaged within cell) for VGLUT1 KO (red) and WT (black) neurons recorded in mixed genotype pairs. Numbers in bar graphs represent n (number of cells). For B–C and E, Significance was determined by Mann–Whitney U test: *p = 0.021; **p = 0.0024; ***p < 0.0001. Error bars represent the SEM.
Figure 4.
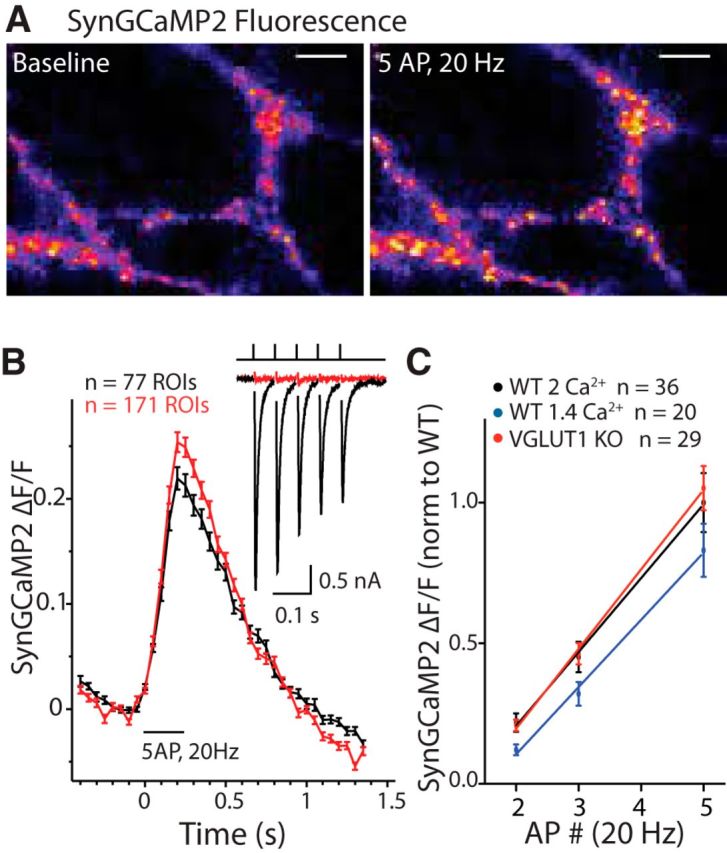
Presynaptic Ca2+ influx is not changed in VGLUT1 KO versus WT autapses. A, Left, Example image of SynGCaMP2 fluorescence in a VGLUT1 KO autapse at baseline (left) and after stimulation with 5 APs at 20 Hz (right). Images are set to the same intensity scale. Scale bars, 10 μm. B, Average SynGCaMP2 ΔF/F0 responses to a 5 AP train at 20 Hz in a single WT (black) or VGLUT1 KO (red) autapse. n value indicates number of ROIs selected per cell. Inset, Accompanying EPSC responses recorded in each neuron. Depolarization artifacts have been blanked for illustrative purposes. C, Average ΔF/F0 response to 2, 3, or 5 APs at 20 Hz compared over several VGLUT1 KO (red) or WT (black) autapses, or WT autapses in reduced [Ca2+]e (1.4 mm; blue). n values indicated in graph equal the number of cells. Solid lines indicate linear fits of data. Significance was tested by two-way ANOVA with Bonferroni post hoc test. For all graphs, error bars represent the SEM.
For WT, VGLUT1 KO pairs, cells from each genotype were labeled with PKH67 Green Fluorescent or PKH26 Red Fluorescent Cell Linker Kit for General Cell Membrane Labeling (catalog #PKH67GL or PKH26GL, respectively; Sigma-Aldrich). Pairs of the appropriate genotype were identified by fluorescence of green or red at the soma.
Plasmid construction and lentivirus production.
For the transport-deficient VGLUTs, mutations in VGLUT2 were made using the Site-Directed Mutagenesis Kit Quick Change II (Agilent Technologies). In tagged-protein constructs, mKate (TagFP635; Evrogen) or miniSOG (Shu et al., 2011) cDNA was fused in frame at the C terminus of VGLUT2 via a PacI restriction site. VGLUT2-pHluorin was generated by inserting pHluorin cDNA into the first luminal loop between residues glycine 108 and lysine 109 separated by a 9 aa glycine-rich flexible linker, a position synonymous with placement of pHluorin in VGLUT1-pHluorin (Voglmaier et al., 2006). SynGCaMP2 (Plasmid 26124; Addgene), SypHy (Plasmid 24478; Addgene), and SypHy2X (Zhu et al., 2009) cDNAs were cloned into lentiviral shuttle vectors under control of the synapsin promoter.
Lentivirus was produced as previously described (Lois et al., 2002). Briefly, an 80% confluent 75 cm2 flask of HEK293T cells was transfected with 10 μg of shuttle vector and mixed helper plasmids (pCMVd8.9 7.5 μg and pVSV-G 5 μg) using XtremeGene 9 DNA Transfection Reagent (Roche Diagnostics). After 48 h, cell culture supernatant was harvested and cell debris was removed by filtration. Aliquots of the filtrate were flash frozen in liquid nitrogen and stored at −80°C until use. Viral titer was estimated by counting cells in mass culture WT hippocampal neurons expressing mKate, pHluorin (in 50 mm NH4Cl) or GFP as a fluorescent reporter. Primary hippocampal cultures were infected with 150–250 μl of the viral solution (0.5–1 × 106 IU/ml) 24–48 h after plating.
shRNA knock-down experiments.
Two VGLUT2-specific siRNA (1: 5′-TCA CTT GGA TAT TGC TCC A-3′; 2: 5′-CAG CAC TAT CCA CCG CGG A-3′) and one FoxP2-specific siRNA as control (5′-GGA ACG CGA ACG TCT TCA A-3′) were obtained using GenScript siRNA Target Finder (GenScript) and cloned as shRNA in a U6 promoter expression cassette within a lentiviral shuttle vector. For viral titering, this vector contained a synapsin promoter-controlled GFP expression cassette. A VGLUT2 shRNA rescue construct was made based on shRNA 1 at the third (“wobble”) position of triplet base coding sequences for L435 and I437, generating an EcoRV site for later discrimination from the WT VGLUT2 cDNA.
Electrophysiology.
Whole-cell voltage-clamp experiments were performed using one or two channels of a MultiClamp 700B amplifier (Molecular Devices) under control of a Digidata 1440A Digitizer (Molecular Devices) and pCLAMP Software (RRID:rid_000085, Molecular Devices). Neurons were recorded at 14–18 d in vitro (DIV). EPSCs were evoked by 2 ms depolarization to 0 mV, resulting in an unclamped action potential (AP). Data were sampled at 10 kHz and Bessel filtered at 3 kHz. Series resistance was typically <10 MΩ, and only cells with <15 MΩ resistance were included. Series resistance was compensated by at least 70%. Extracellular solution for all experiments, unless otherwise indicated, contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 MgCl2, and 2 CaCl2, pH 7.4. For SynGCaMP2 (see Fig. 4) and SypHy characterization experiments (Fig. 1), [MgCl2]e was 1 mm and [CaCl2]e varied as indicated. Intracellular solution contained the following (in mm): 126 KCl, 17.8 HEPES, 1 EGTA, 0.6 MgCl2, 4 MgATP, 0.3 Na2GTP, 12 creatine phosphate, and phosphocreatine kinase (50 U/ml). All reagents were purchased from Carl Roth GmbH with the exception of Na2ATP, sodium Na2GTP, and creatine-phosphokinase (Sigma-Aldrich), and phosphocreatine (EMD Millipore Chemicals).
Figure 1.
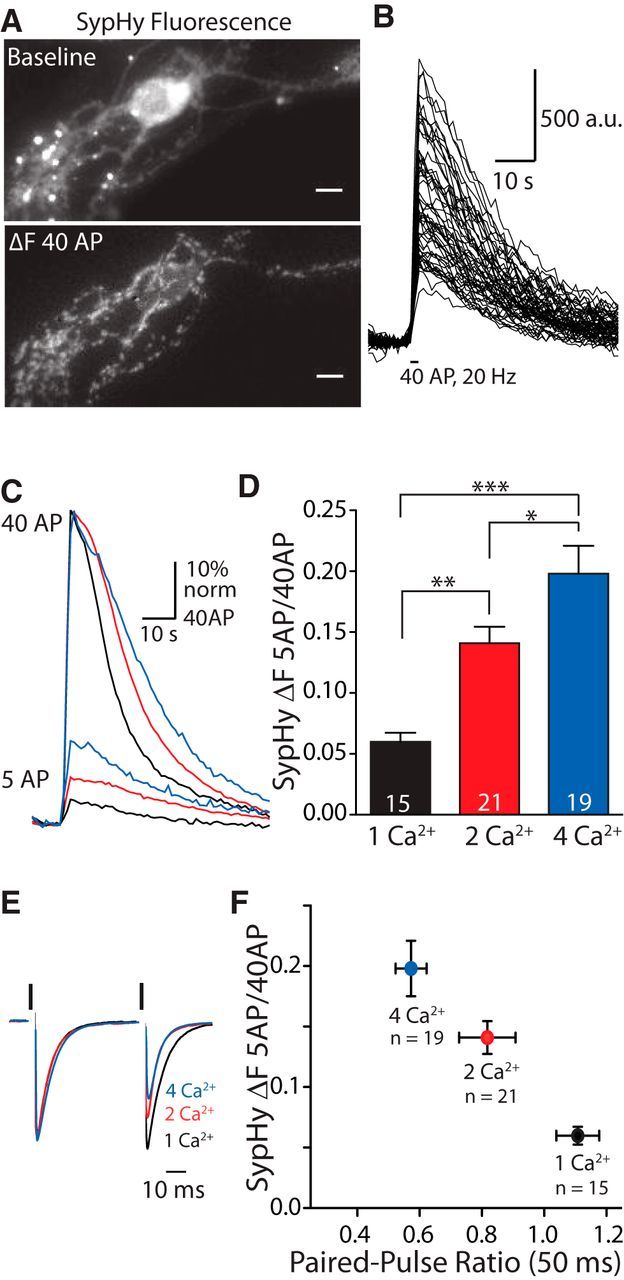
SypHy imaging reveals differences in release probability with high sensitivity. A, Example image of WT hippocampal autapse expressing SypHy at baseline (left) and after stimulation with 40 APs at 20 Hz (right; baseline subtracted) in 2 mm Ca2+. Images were set to the same intensity range. Scale bars, 10 μm. B, Quantification of change in fluorescence over time from ROIs selected from the cell in A. C, Average ΔF responses per cell to 5 APs given at 2 Hz normalized to the peak of the 40 AP (20 Hz) signal in example cells recorded in 1 (black), 2 (red), or 4 (blue) mm [Ca2+]e. D, Bar graph of average ratios of the SypHy ΔF signal in response to 5 APs at 2 Hz over that in response to 40 APs at 20 Hz calculated per cell recorded in [Ca2+]e of 1 (black), 2 (red), or 4 (blue) mm. Measurements with different [Ca2+]e values were compared between cells. n values represent the number of cells. Significance was determined by one-way ANOVA with Tukey post hoc test: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. E, Example EPSC responses to the first two pulses in a 40 AP train at 20 Hz in cells recorded in 1 (black), 2 (red), or 4 (blue) mm [Ca2+]e. Responses are scaled to the peak of the first response. Black bars indicate 70 mV depolarization. The depolarization artifact has been blanked for illustrative purposes. F, Average 5 AP/40 AP SypHy ratio plotted against the average PPR (interstimulus interval, 50 ms) recorded from the same cells in [Ca2+]e, as indicated. n values equal the number of cells. For all graphs, error bars represent the SEM.
Electrophysiological recordings were analyzed using Axograph X (Axograph), Excel (Microsoft), and Prism (GraphPad; RRID:rid_000081). mEPSCs were detected by template function (Axograph; template: rise, 0.5 ms; decay, 3 ms; criteria range: rise, 0.15–1.5 ms; decay, 0.5–5 ms). Nonspecific noise was detected by template function on sweeps in the presence of kynurenic acid (3 mm; Tocris Bioscience) and subtracted from the total number of mEPSCs detected. Cells were excluded from analysis if noise events exceeded one third of the total number of events detected. Statistical significance was determined using a Student's t test (for two groups with normal distribution), Mann–Whitney U test (for two groups without normal distribution), one-way ANOVA with Tukey post hoc test (for more than two groups with normal distribution), or nonparametric Kruskal–Wallis test with Dunn's post-test (for more than two groups without normal distribution). Distributions were tested for normality by D'Agostino–Pearson tests.
pHluorin and SynGCaMP2 imaging.
Images were acquired with an Andor iXon or Hamamatsu EM back-illuminated CCD camera with a Polychrome V Illumination System (Till Photonics; for SypHy experiments) or LED (470 nm; Prizmatix; for VGLUT2-pHluorin and SynGCamp2 experiments) illumination at 60× magnification (numerical aperture 1.2). For pHluorin experiments, 256 × 256 pixel (2 × 2 binning) images of fluorescence responses to stimulus trains were collected at a 1 Hz sampling rate with a 100 ms exposure. Responses to NH4Cl (50 mm) were collected at 5 Hz. For SynCaMP2 experiments, 128 × 128 pixel (2 × 2 binning) images of fluorescence responses to stimulus trains were acquired at 20 Hz with a 49 ms exposure time.
Images were analyzed using ImageJ with custom macros (National Institute of Health; RRID:nif-0000-30467). For pHluorin images, 2 μm2 regions of interest (ROIs) were identified from the peak change in fluorescence (ΔF) in response to a 40 AP train at 20 Hz from baseline subtraction (baseline F = average of three images immediately before stimulation). The same ROI set was applied to all stimulation frequencies/durations. Image sequences were background subtracted (“Rolling Ball” background subtraction; 50 pixel radius; ImageJ), and the ΔF values per image were measured in ImageJ. ΔF measurements over time for each ROI per cell were baseline subtracted and averaged in Axograph X (Axograph). For the 5 AP/40 AP pHluorin ratio, measurements were excluded if the 5 AP measurement was smaller than bleaching (negative ratio value). For all groups, the number of excluded cells were equal (WT SypHy, n = 4 of 40; VGLUT1 KO SypHy, n = 4 of 42; VGLUT2 WT-pHluorin, n = 0 of 54; VGLUT2 TPM-pHluorin, n = 2 of 52). For SynGCaMP2, ROIs were identified in the same manner, but from the peak ΔF image in response to 10 APs at 20 Hz. ΔF measurements over time for each ROI per cell were normalized to baseline (F0) and averaged in Axograph X. ΔF or ΔF/F0 values per cell were averaged and compared using Excel (Microsoft) and Prism (GraphPad). Statistical significance was determined by Student's t test (for two groups with normal distribution), Mann–Whitney U test (for two groups without normal distribution), one-way ANOVA with Tukey's post hoc test (for more than two groups), Kruskal–Wallis test with Dunn's post-test (for more than two groups without normal distribution), or two-way ANOVA (for two independent variables). Distributions were tested for normality by the D'Agostino–Pearson test.
Electron microscopy and immunocytochemistry.
At DIV 16, coverslips containing lentiviral-infected cultures of VGLUT1 KO hippocampal neurons were broken, and prepared for immunocytochemistry (ICC) and EM. For ICC, cells were fixed in 4% paraformaldehyde and processed as previously described (Arancillo et al., 2013). Primary antibodies to VGLUT2 [1:1000; guinea pig; catalog #135 404 (RRID:AB_887884), Synaptic Systems] and synaptophysin 1 [Syp1; 1:1000; rabbit; catalog #101 002 (RRID:AB_2198887), Synaptic Systems] were used. Secondary antibodies were used from Jackson ImmunoResearch Laboratories (1:500). The VGLUT2 signal per image was normalized to Syp1 signal using a thresholding function (85% of maximum; ImageJ). VGLUT2/Syp1 values from multiple images (5–10) were collected per culture, and statistical significance was determined using the Kruskal–Wallis test with Dunn's post-test.
For EM, the experimenter was blinded to culture group from before fixation through analysis. Cells were fixed with 4% paraformaldehyde, 0.25% glutaraldehyde in 0.1 m phosphate buffer for 45 min at room temperature. The fixative was rinsed by washing four times for 15 min with 0.1 m phosphate buffer. Afterward, cells were postfixed with 0.5% osmium tetraoxide in 0.1 m phosphate buffer for 1 h at room temperature. Cells were washed twice with 0.1 m phosphate buffer and four times with water before they were incubated with 2% uranyl acetate in water for 45 min at room temperature. The fixative was rinsed by washing four times with water. Subsequently, cells were dehydrated by incubating in solutions with an increasing percentage of ethanol (1× 10 min 30% ethanol, 1× 10 min 50% ethanol, 2× 10 min 70%, 1× 10 min 90%, 1× 10 min 96%, 2× 10 min 100%). Dehydrated cells were incubated with N-(2-hydroxypropyl)methacrylamide (HPMA; Sigma-Aldrich) for 20 min at room temperature, and the solution was changed once. Infiltration of plastic (Epon, Sigma-Aldrich) was started by incubating the cells with HPMA-Epon (1:1) for 1 h at room temperature. Afterward, the solution was renewed and cells were kept in HPMA-Epon (1:1) overnight. The next day, cells were further infiltrated with pure freshly prepared Epon for 8 h. Epon was changed three times within this 8 h period. The plastic was polymerized at 60°C for 48 h. Seventy nanometer sections were cut using a microtome (Reichert Ultracut S, Reichert/Leica) and collected on 0.5% Formvar grids (G2200C, Plano). The sections were stained with 2.5% uranyl acetate in 70% methanol for 5 min before imaging.
Sections were imaged on a Zeiss EM900 transmission electron microscope equipped with a 1k slow-scan CCD camera (TW 7888, ProScan). Presynaptic structures were identified by morphology. Image analysis was performed using custom macros (ImageJ). Briefly, vesicle structures were outlined with the “Freehand” selection tool, and circularity was measured [macro formula: 4π (area/perimeter2), where the measurement of a perfect circle = 1]. An average circularity measurement was calculated for all vesicles measured per group and was normalized to the average value in the VGLUT2 WT group. Statistical significance was determined using Kruskal–Wallis test with Dunn's post-test.
Results
pHluorin imaging detects qualitative differences in Pr between cells with a similar sensitivity to PPR
The Pr in hippocampal VGLUT1 KO autapses has been assessed by electrophysiological measurement of the postsynaptic response (Wojcik et al., 2004). However, this assay is limited to measuring the Pr of vesicles that are at least partially full of glutamate and provides no information about the Pr of empty vesicles. Empty vesicles may have different release properties. Therefore, we designed an assay to measure presynaptic release probability independent of the postsynaptic response using the pH-sensitive variant of ecliptic green fluorescent protein, pHluorin, targeted to synaptic vesicles (Fig. 1). To test the sensitivity of our assay, we expressed SypHy (Granseth et al., 2006) in WT autapses. Individual cells were under electrical control by whole-cell recordings in voltage-clamp mode. We found that stimulation of SypHy-expressing cells with 40 APs at 20 Hz produced a detectable increase in fluorescence (ΔF) in a punctate-like pattern indicative of presynaptic boutons (Fig. 1A,B). To assess Pr, the average ΔF per cell elicited by 5 APs at 2 Hz was compared with the 40 AP (20 Hz) signal (ΔF 5 AP/40 AP ratio; Fig. 1C,D). To determine the sensitivity of this technique to detect differences in Pr, we compared the averaged ΔF 5 AP/40 AP ratio among WT autapses with 1, 2, and 4 mm [Ca2+]e (1 mm [Mg2+]e; Fig. 1C–E). Averaged ratios with different [Ca2+]e values were compared between cells instead of within a cell to preserve any inherent variability in release probability that was present within the population of hippocampal autapses. Not only could we distinguish significant differences in the ΔF 5 AP/40 AP ratio between cells recorded in each [Ca2+]e, these differences were consistent with the differences observed between groups in average PPR, an indicator of Pr, recorded with electrophysiology from the same cells (Fig. 1E,F; PPR 1 Ca2+ vs 4 Ca2+, ***p ≤ 0.001; PPR 1 Ca2+ vs 2 Ca2+, *p ≤ 0.05; PPR 2 Ca2+ vs 4 Ca2+, NS; significance was determined by Kruskal–Wallis test with Dunn's post-test). Together, these data suggest that the ΔF 5 AP/40 AP ratio can be used to assess differences in Pr with a similar sensitivity to PPR.
To test whether the decreased Pr in VGLUT1 KO autapses is preserved when empty vesicles are included in the assay, we expressed SypHy, or the double pHluorin copy version, Sy2pHy (Zhu et al., 2009), in VGLUT1 KO and WT autapses (data for both probes were combined; Fig. 2). We found that, as expected, the excitatory postsynaptic current (EPSC) amplitude was significantly decreased in VGLUT1 KO compared with WT autapses (Fig. 2A,B). Consistent with previous work (Wojcik et al., 2004), we found a strong trend toward a mean (±SEM) increase in the PPR for the VGLUT1 KO group compared with WT group (PPR VGLUT1 KO, 1.10 ± 0.09, n = 28; PPR WT, 0.90 ± 0.05; p = 0.0501, n = 43); however, nonresponding autapses in the VGLUT1 KO group could not be included in this analysis. Therefore, we used our presynaptic imaging assay to compare release probability differences between the two groups and found that the ΔF 5 AP/40 AP ratio in the VGLUT1 KO autapses was significantly reduced (Fig. 2C,D). This suggests that both empty and partially full vesicles in the VGLUT1 KO autapse have a lower Pr, which can be assayed independently of a postsynaptic response. To confirm this effect, we also examined the ΔF 5 AP response normalized to alkalinization of SypHy in the vesicles with NH4Cl (50 mm). We found that the 5 AP signal normalized to the NH4Cl response per cell was significantly smaller in VGLUT1 KO autapses than WT autapses (as a percentage of NH4Cl: KO, 0. 58 ± 0.1%, n = 43; WT, 1.26 ± 0.16%, n = 42; ***p = 0.0006, Mann–Whitney U test). However, it should be noted that the ΔF NH4Cl was on average significantly larger in VGLUT1 KO autapses (ΔF NH4Cl: KO autapses, 5986 ± 616 a.u., n = 43; WT autapses, 4248 ± 590 a.u., n = 42; *p = 0.022, Mann–Whitney U test), which could indicate a higher number of SypHy molecules on vesicles lacking transporter protein or a role for intracellular glutamate in determining the maximum pH reached in the presence of NH4Cl. As the measurements we used to assess Pr would not be affected by either of these possibilities, we did not further investigate this observation.
In addition to the ΔF response to 5 and 40 APs, we also measured the ΔF response to a 300 AP train at 20 Hz (Fig. 2E). Though there was no decrease in the peak amplitude of the 300 AP ΔF signal for SypHy in VGLUT1 KO autapses (111 ± 14% of WT cells normalized per culture; KO autapses, n = 36; WT autapses, n = 33; p = 0.88), we did see a difference in the kinetics of the rise in the fluorescence signal (Fig. 2E; 20–80% rise time; WT autapses, 6.95 ± 0.20 s, n = 33; KO autapses, 7.77 ± 0.10 s, n = 36; ***p = 0.0006). We suggest that this decrease in rise time may indicate an impairment of vesicles to release, and a general disruption of the vesicle cycle for vesicles with a decreased VGLUT copy number.
VGLUT1 KO autapse Pr decrease persists with glutamatergic input during development
Could the decreased Pr observed in VGLUT1 KO cultured hippocampal autaptic neurons by our pHluorin imaging assay or by electrophysiology (Wojcik et al., 2004) be due to a lack of glutamatergic signaling during development? To address this possibility, we restored glutamatergic input to mutant neurons by coculturing VGLUT1 KO with WT neurons in heterotypic two-neuron pairs grown on a glial microisland, and investigated the synaptic characteristics of each cell with electrophysiology (Fig. 3). Pairs with the appropriate genotypes were identified by a membrane dye applied to dissociated hippocampal cells from each genotype before plating (see Materials and Methods). Stimulation of an unclamped action current by brief depolarization of each cell resulted in an EPSC in the stimulated cell (autaptic) and the unstimulated partner cell (heterosynaptic; Fig. 3A). We found that though the VGLUT1 KO neurons had on average only a small reduction in the amount of glutamatergic input (Fig. 3B), likely due to a preference of glutamatergic neurons to make autaptic connections in two-neuron microisland cultures (Chang et al., 2014), their glutamatergic output was drastically reduced, consistent with the abolishment of their predominant VGLUT subtype (Fig. 3C). However, analysis of the PPR showed that VGLUT1 KO output synapses were more facilitating than those from WT partner neurons (Fig. 3D,E). This suggests that reduced Pr observed in VGLUT1 KO autapses is not an artifact of minimal exposure to glutamatergic input during development.
Decreased Pr in VGLUT1 KO autapse is not due to changes in presynaptic Ca2+ influx
Another explanation for Pr differences between VGLUT1 KO and WT autapses could be reduced presynaptic Ca2+ influx in KO autapses. To address this possibility, we compared the signal of a genetically encoded calcium indicator conjugated to synaptophysin for presynaptic localization (SynGCaMP2) in response to 2, 3, and 5 APs at 20 Hz in VGLUT1 KO and WT autapses (Fig. 2). This method has previously been demonstrated to reveal qualitative differences in presynaptic Ca2+ influx between synapses in different culture conditions (Zhao et al., 2011). Stimulation of the autapses produced detectable increases in SynGCaMP2 fluorescence (Fig. 4A). The average SynGCaMP2 ΔF/F0 per cell was calculated from ROIs selected from the ΔF response to 10 APs at 20 Hz. As indicated from a single VGLUT1 KO (red) and a single WT (black) autapse, the SynGCaMP2 ΔF/F0 produced by a 5 AP train (20 Hz) was similar, even when the EPSC amplitudes from those cells showed a large disparity (0.05 and 3.45 nA, respectively; Fig. 4B). A single ΔF/F0 value for each of 2, 3, and 5 AP trains at 20 Hz was generated per autapse from the average of several ROIs. When averaged across cells, the VGLUT1 KO autapses showed a clear decrease in EPSC amplitude (WT, 10.3 ± 1.3 nA, n = 36; KO, 3.1 ± 1.0 nA, n = 29; p > 0.0001; 2 mm [Ca2+]e, 1 mm [Mg2+]e), but there was no difference in the SynGCaMP2 ΔF/F0 signal with 2, 3, or 5 APs at 20 Hz (Fig. 4C). Because we were interested in qualitative differences in the SynGCaMP2 Ca2+, we normalized each culture to the average signal generated in WT autapses by the 5 AP stimulus (SynGCaMP2 ΔF/F0 = 0.14 ± 0.02; n = 36). Additionally, to assure that our method was sensitive enough to detect qualitative differences in presynaptic Ca2+ influx, we also measured the SynGCaMP2 ΔF/F0 in response to 2, 3, and 5 AP (20 Hz) in WT autapses with reduced [Ca2+]e (1.4 vs 2 mm in control conditions; blue; Fig. 2C). Analysis by two-way ANOVA showed that the average normalized ΔF/F0 values were significantly reduced in 1.4 mm [Ca2+]e compared with 2 mm [Ca2+]e (*p = 0.03), while no significant effect of genotype on these values was detected (WT vs VGLUT1 KO; p = 0.74). Because 2, 3, and 5 APs at 20 Hz is a range at which the SynGCaMP2 signal has been shown to be linear (Dreosti et al., 2009), we performed linear regression analysis on the data. Though linear regression analysis revealed lines with similar slopes for all conditions (VGLUT KO, 0.28 ± 0.01; WT 2 Ca2+, 0.26 ± 0.01; WT 1.4 Ca2+, 0.24 ± 0.01; linear regression fit to values was normalized to 5 APs in WT 2 Ca2+ per culture), the x-intercept for WT 1.4 mm (1.56) was shifted by ∼28% to either VGLUT1 KO (x-intercept, 1.30) or WT 2 mm Ca2+ (x-intercept, 1.22). This suggests that, though there is a lower limit for the sensitivity of the technique, SynGCaMP2 Ca2+ imaging can be used to detect qualitative differences in Ca2+ influx between genotypes in autaptic neurons, and that reduced Ca2+ influx is unlikely to be the cause of the decreased Pr in VGLUT1 KO autapses.
Knockdown of VGLUT2 in VGLUT1 KO autapses results in decreased EPSC amplitude and increased PPR
VGLUT1 KO autapses display a range of response amplitudes from none to WT-like levels, presumably due to the variability in expression levels of a secondary transporter subtype in the hippocampus (Wojcik et al., 2004). To test whether further reducing the transporter copy number on vesicles in VGLUT1 KO autapses decreases Pr, we knocked down expression of the putative residual transporter subtype, VGLUT2, with shRNA (Fig. 5). We found that in VGLUT1 KO autapses, transcription of VGLUT2 shRNA nearly abolished the EPSC amplitude from the entire population of cells compared with VGLUT1 KO neurons without shRNA or with shRNA directed toward FoxP2, a protein not expressed in hippocampus (Ferland et al., 2003; Fig. 5A,B). Additionally, we found that responses from VGLUT1 KO autapses transduced with the VGLUT2 shRNA virus exhibited more facilitation than those with control FoxP2 shRNA (Fig. 5C,D). This confirms the identity of VGLUT2 as the second resident VGLUT transporter in these neurons and supplies supporting evidence that the VGLUT protein level decrease is responsible for the decreased Pr observed in VGLUT1 KO autapses. This is particularly important as vesicles with VGLUT2 have a higher release probability than vesicles with VGLUT1 when the proteins are expressed at the same level (Weston et al., 2011). Together, these data suggest that the relationship between VGLUTs and Pr is a convolution of both the isoform and the copy number effects of the VGLUT protein, with the copy number playing a dominant role at low expression levels, and the isoform playing a dominant role at higher expression levels.
Figure 5.
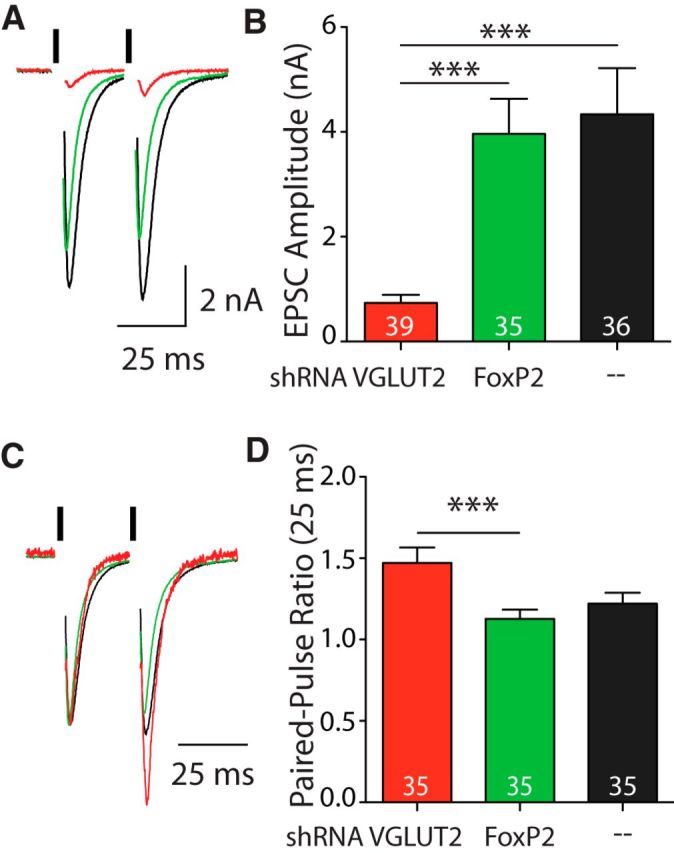
shRNA knockdown of VGLUT2 affects responses in VGLUT1 KO autapses. A, Example traces of EPSCs from VGLUT1 KO autapses expressing shRNA against VGLUT2 (red) or FoxP2 (green), or cytosolic green fluorescent protein (GFP; black). Black bars indicate 2 ms depolarization. Depolarization artifacts have been blanked for illustrative purposes. B, Average EPSC amplitude recorded from two cultures of groups described in A. C, Same traces as A scaled to the peak amplitude of the EPSC from pulse 1. D, Average paired-pulse ratios calculated from two EPSCs at a 25 ms interstimulus interval recorded from VGLUT1 KO autapses groups as described in A. For B and D, significance was determined by Kruskal–Wallis test with Dunn's post-test: ***p ≤ 0.001. For all graphs, error bars represent the SEM. n values (number of cells) are indicated in graphs.
Point mutations in VGLUT2 abolish transport activity
What is the mechanism of the VGLUT expression level effect on Pr? The effects of transporter number on vesicle characteristics can be reduced to the following two components: (1) the presence of transporter proteins in the vesicle membrane (molecular); and (2) the amount of glutamate in the vesicle (functional). This suggests that the decreased Pr in VGLUT1 KO autapses may be due to either a decrease in the number of release-promoting protein–protein interactions or a reduced glutamate fill state. To distinguish between these possibilities, we designed two VGLUT2 proteins with double point mutations (DPMs; R88Q, R184K) and triple point mutations (TPMs; R88Q, R184K, R322Q) in residues putatively involved in the transport of glutamate for VGLUT2 (Juge et al., 2006) and VGLUT1 (Almqvist et al., 2007). We then performed rescue experiments with these mutants in VGLUT1 KO autapses. We chose to use the VGLUT2 transporter subtype for these experiments to avoid complications by the interaction of VGLUT1 with endophilin, which results in a decrease of Pr (Weston et al., 2011). VGLUT1 KO autapses were transduced with lentivirus to induce the expression of WT, DPM, or TPM VGLUT2 mutants. We found that while the WT VGLUT2 protein expression produced large-amplitude EPSCs in VGLUT1 KO autapses, the expression of either DPM or TPM VGLUT2 did not (Fig. 6A,B). Additionally, VGLUT1 KO autapses expressing the transport-deficient VGLUT2 mutants had significantly lower mEPSC frequency and amplitude than those expressing VGLUT2 WT protein (Fig. 6C,D), suggesting a reduced quantal amplitude as a result of a decreased number of functional VGLUTs, as reported by Wojcik et al., 2004.
Figure 6.
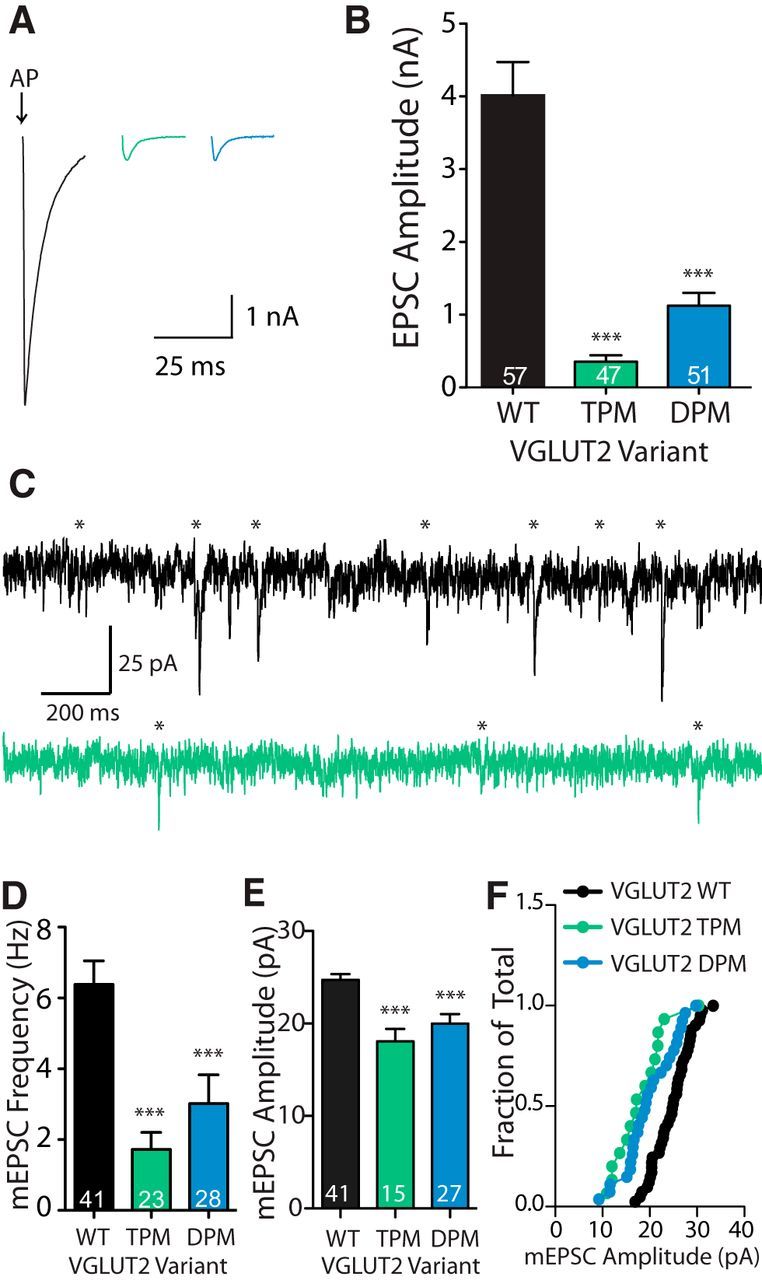
Mutation of putative transport residues renders VGLUT2 transport deficient. A, Example response (average, four traces) of VGLUT1 KO autapses rescued with VGLUT2 WT (black), VGLUT2 TPM (R184K R88Q R322Q; green), or VGLUT2 DPM (R184K R88Q; blue). Arrow indicates 70 mV depolarization. Depolarization artifacts have been blanked for illustrative purposes. B, Quantification of average EPSC amplitude recorded from many autaptic neurons (n values indicated in graph) over three cultures. Significance was determined by Kruskal–Wallis test with Dunn's post-test. ***p ≤ 0.001. C, Example traces of mEPSC recordings from VGLUT1 KO autapses expressing VGLUT2 WT (black) or VGLUT2 TPM (green) proteins. Template-detected events indicated by asterisks. D, E, Quantification of average mEPSC frequency (D) and amplitude (E). F, Cumulative frequency distribution of average mEPSC amplitude for each cell included in the average mEPSC amplitude (E). Significance was determined by Kruskal–Wallis test with Dunn's post-test (D) or one-way ANOVA with Tukey post hoc test (E): ***p ≤ 0.001. For all graphs, error bars represent the SEM. n values, representing the number of cells, are indicated in the graphs.
Decreased Pr in VGLUT1 KO autapses cannot be rescued by nonfunctional transporter protein
We next asked whether transport-deficient VGLUT2 expression in VGLUT1 KO autapses could rescue the Pr deficit to the level of WT VGLUT2 rescue independent of the vesicle fill state. To assess this, we expressed pHluorin-tagged VGLUT2 WT (VGLUT2 WT-pHluorin) or TPM (VGLUT2 TPM-pHluorin) proteins in VGLUT1 KO autapses using a lentivirus expression system. We found that the VGLUT2 TPM-pHluorin protein was trafficked to acidic compartments, which displayed a punctate fluorescence pattern upon application of NH4Cl (50 mm), suggesting appropriate trafficking of the protein to synaptic vesicles (Fig. 7A). However, VGLUT1 KO autapses expressing the VGLUT2 TPM-pHluorin had significantly smaller EPSCs than those expressing VGLUT2 WT-pHluorin (Fig. 7B), consistent with the transport deficiency of the protein. Additionally, analysis of the VGLUT1 KO autapses expressing VGLUT2 TPM-pHluorin that had detectable postsynaptic responses revealed an increase in the PPR compared with those expressing VGLUT2 WT-pHluorin (Fig. 7C). Moreover, the expression levels of both transporters at the synapse were similar, as the average fluorescence signal induced by NH4Cl application was not different between cells expressing VGLUT2 WT-pHluorin and VGLUT2 TPM-pHluorin (Fig. 7E).
Figure 7.

The presence of nonfunctional VGLUT protein cannot rescue release probability deficits in VGLUT1 KO autapses with deficient glutamate release. A, Example image of a VGLUT1 KO expressing VGLUT2 TPM-pHluorin (VGLUT2pH TPM) in the vesicle lumen under baseline conditions (top) and during application of ammonium chloride (50 mm; bottom). B, C, Average EPSC amplitude (B; ***p ≤ 0.0001) and PPR (C; *p = 0.025) of VGLUT1 KO autapses expressing VGLUT2 WT- (VGLUT2pH WT; black) or VGLUT2 TPM-pHluorin (green). Significance determined by Mann–Whitney U test. D, Average ΔF 5 AP/40 AP signal ratio per cell generated by the same VGLUT1 KO autapses expressing VGLUT2 WT-pHluorin or VGLUT2 TPM-pHluorin, as in B and C. Significance was determined using an unpaired Student's t test: **p = 0.006. E, Comparison of average ΔF generated in response to 5 APs at 2 Hz (***p < 0.0001), 40 APs at 20 Hz (***p < 0.0001), 300 APs at 20 Hz (*p = 0.020), and 50 mm NH4Cl (p = 0.815). Significance was determined using the Mann–Whitney U test. F, ΔF response to a 300 AP 20 Hz train normalized to the peak per cell and averaged over the entire population. Significance indicates the comparison of 20–80% rise times (Mann–Whitney U test, ***p < 0.0001). For all graphs, error bars represent the SEM. n values represent the number of cells and are indicated in graphs.
To explore whether VGLUT2 TPM-pHluorin could recover the Pr in VGLUT1 KO autapses to the level of VGLUT2 WT-pHluorin rescue without relying on the significantly different postsynaptic responses (Figs. 6A, 7B), we compared the ΔF 5 AP/40 AP ratio as previously described (Figs. 1, 2). We found that the ΔF 5 AP/40 AP ratio for VGLUT1 KO autapses expressing VGLUT2 TPM-pHluorin was significantly lower than those expressing VGLUT2 WT-pHluorin (Fig. 7D), suggesting that vesicles containing a nonfunctional transporter protein still have a release probability deficit compared with those with functional transporters. In addition, we found that the VGLUT2 TPM-pHluorin ΔF signal produced in response to all stimulus frequencies and durations tested was on average lower than in VGLUT1 KO autapses expressing VGLUT2 WT-pHluorin, even though both proteins had comparable expression levels as assayed by 50 mm NH4Cl (Fig. 7E), which is indicative of an impairment of vesicle release. Furthermore, the signal produced by 5, 40, and 300 APs normalized to the response of the total pool to NH4Cl application within cells also showed a significant reduction in the VGLUT2 TPM-pHluorin protein compared with VGLUT2 WT-pHluorin protein (as a percentage of the NH4Cl signal: 5 APs: TPM-pHluorin, 0.6 ± 0.1%; WT-pHluorin, 1.5 ± 0.1%; ***p < 0.0001, Mann–Whitney test; 40 APs: TPM-pHluorin, 7.1 ± 0.4%; WT-pHluorin, 13.3 ± 0.7%; ***p < 0.0001, Student's t test; 300 APs: TPM-pHluorin, 30.6 ± 1.2%; WT-pHluorin, 42.7 ± 1.6%; ***p < 0.0001, Student's t test; for all stimulations: TPM-pHluorin, n = 52; WT-pHluorin, n = 53). Consistent with these analyses, the rise of the 300 APs at 20 Hz-evoked ΔF was slower on average in VGLUT1 KO autapses expressing VGLUT2 TPM-pHluorin than those expressing VGLUT2 WT-pHluorin (Fig. 6F; 20–80% rise time; VGLUT2 WT-pHluorin, 6.86 ± 0.13 s; VGLUT2 TPM-pHluorin, 7.62 ± 0.11 s; ***p < 0.0001). Together, these data suggest that the presence of a transporter protein alone, without transport function, is not sufficient to rescue the release probability and vesicle cycling deficiencies of subglutamate fill-state vesicles.
Shape of vesicles expressing VGLUT2 TPM consistent with empty vesicles as assessed by EM
Overall, these results favor the hypothesis that the filling of vesicles with glutamate modulates release probability. To gain further support for this, we investigated the morphology of vesicles expressing transport-deficient VGLUTs. We took advantage of a morphological phenotype observed in standard, aldehyde fixation electron micrographs, a deformation of vesicles in VGLUT1 KO synapses, which has been ascribed to the hypo-osmolarity of empty vesicles (Siksou et al., 2013). A similar phenotype has been observed in mouse neuromuscular junction (NMJ) when the vesicular acetylcholine transporter (vAchT) was genetically eliminated or blocked with a pharmacologic agent (Rodrigues et al., 2013). However, previous studies had not investigated the effects of expressing a transport-deficient vesicular transporter on the shape of empty vesicles with aldehyde fixation. Therefore, we expressed shRNA-resistant VGLUT2 WT or VGLUT2 TPM proteins both fused to a variant of GFP, miniSOG (Shu et al., 2011), in VGLUT1 KO cultures in which residual, endogenous VGLUT2 was eliminated by shRNA expression (Fig. 5). Analysis of vesicle morphology in each of these conditions revealed that vesicles expressing VGLUT2 TPM protein had significantly decreased levels of circularity compared with those expressing VGLUT WT protein (Fig. 8A,B,D), demonstrating that only VGLUTs with transport capabilities can rescue the empty vesicle EM phenotype described by Siksou et al. (2013). Vesicles from VGLUT1 KO with shRNA VGLUT2 knock-down synapses without a VGLUT2 rescue protein showed a trend toward decreased circularity compared with the VGLUT2 WT rescue, but did not reach significance (Fig. 8C,D). However, for the purposes of our study, the most important comparison is between the vesicles expressing functional and nonfunctional transporters. The area of vesicles expressing VGLUT2 WT or VGLUT2 TPM proteins, or are without a rescue protein, when averaged per culture were not different (VGLUT2 TPM, 91.7 ± 25.2% of VGLUT2 WT; no rescue, 93.4 ± 0.4%). Ratiometric imaging of VGLUT2 immunofluorescence compared with synaptophysin 1 immunofluorescence (see Materials and Methods) revealed no difference between the expression of VGLUT2 WT and VGLUT2 TPM between cultures examined by EM (Fig. 8E).
Figure 8.
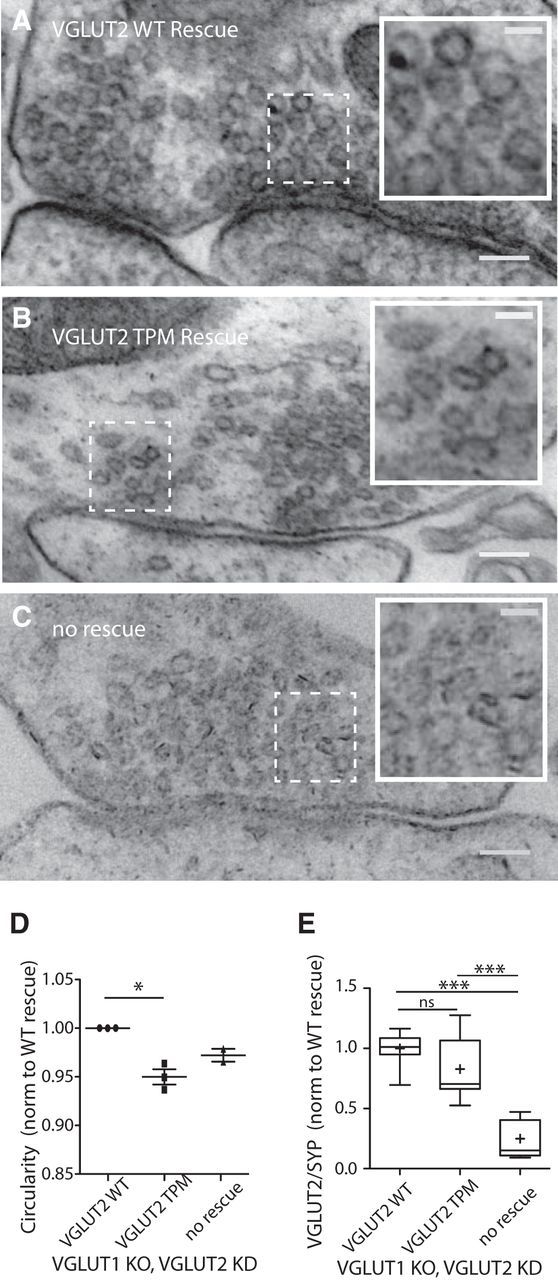
The transport-deficient VGLUT2 protein does not rescue the distorted morphology of empty vesicles in standard EM. A–C, Example electron micrographs of synapses from VGLUT1 KO, VGLUT2 knock-down (KD) hippocampal cultures rescued with shRNA-resistant VGLUT2 WT (A) or VGLUT2 TPM (B) proteins, or cultures without rescue (C). Scale bars: full images, 100 nm; insets, 40 nm (insets are indicated by a dashed box in the full image). D, Quantification of vesicle circularity measurement averaged per culture and normalized to the VGLUT2 rescue condition. Total number of vesicles measured: VGLUT2 WT: n = 1202, three cultures; VGLUT2 TPM: n = 1245, three cultures; no rescue: n = 656, two cultures. Significance was determined by Kruskal–Wallis test with Dunn's post-test: *p ≤ 0.05. Error bars represent the SEM. E, Ratiometric immunofluorescence signals for VGLUT2 over synaptophysin 1 measured for the same cultures as measured in D. VGLUT2/Syp1 signal normalized to the VGLUT2 WT rescue per culture plotted as a box-and-whisker plot with median (horizontal bar), mean (cross), 25th to 75th percentiles (box), and minimum to maximum values (whiskers). For each group, n = 19 images over three cultures. Significance was determined by Kruskal–Wallis test with Dunn's post test: ***p ≤ 0.001.
Discussion
In this study, we have uncovered a novel mechanism affecting Pr: the fill state of a neurotransmitter within a vesicle. We characterized the reduced Pr in VGLUT1 KO autaptic neurons, and found that this phenotype was not a result of autaptic culture conditions, reduced presynaptic Ca2+ influx, or biased sampling of a population of vesicles containing a low-copy number VGLUT2. Additionally, imaging experiments using pHluorin show that the decreased Pr in VGLUT1 KO autapses was independent of the postsynaptic response and also extended toward empty vesicles, which are undetectable with electrophysiology. Finally, directly imaging pHluorin attached to WT or transport-deficient VGLUT2 proteins revealed that the presence of the protein alone could not rescue the Pr deficit of synapses lacking functional VGLUTs. Together, these data suggest that intravesicular glutamate content, which is determined by VGLUT copy number, may directly affect vesicle Pr.
It should be noted that many factors influence the Pr of a vesicle, including presynaptic calcium influx, AP waveform, and components of the release machinery (Schneggenburger et al., 2002; von Gersdorff and Borst, 2002; Rosenmund et al., 2003). For instance, at the calyx of Held synapse, VGLUT1 protein levels increase over development (Billups, 2005). However, this increase does not result in an increased Pr due to a concurrent decrease in AP width (von Gersdorff and Borst, 2002), suggesting in this case that AP width is likely a more dominant factor in determining Pr than fill state.
Neurotransmitter specificity for effect of fill state on Pr
One question raised by our study is whether the effects of intravesicular neurotransmitter content on quantal size (q) and Pr are specific to glutamate. Initial characterization of the vesicular GABA transporter Viaat showed that both q and vesicular Pr are decreased in GABAergic autapses from the Viaat KO mouse (Wojcik et al., 2006). These data suggest that the fill-state effect may extend beyond glutamate. Additionally, a recent study (Wang et al., 2013) suggests that the depletion of cytosolic GABA, and in turn the intravesicular GABA concentration, inhibits vesicle cycling in the terminals of GABAergic neurons. Though this study attributes the inhibition of release to the cytosolic GABA concentration, the possibility remains that empty vesicles also contribute to this deficit. On the other hand, at the mouse NMJ, partially filled vesicles with reduced vAChT expression did not have a reduced Pr (Lima et al., 2010). Nor was there an effect on vesicle cycling of empty vesicles released at the end of a stimulus train (Tabares et al., 2007) or when vesicle refilling was blocked by vAChT inhibition (Parsons et al., 1999). The discrepancy between these results and our study may indicate that the effect of fill state on Pr is neurotransmitter specific.
One argument against a release deficit for empty glutamatergic vesicles stems from the observation that when refilling is blocked by inhibition of the vesicular proton pump, there is no change in the rate stimulation-induced release of FM1–43 compared with control (Zhou et al., 2000). However, we noted in comparative studies that FM1–43 destaining shows less sensitivity to Pr differences in the range we observed between VGLUT1 KO and WT synapses. For instance, our assay with pHluorin could distinguish Pr differences between WT cells in 1, 2, and 4 mm [Ca2+]e (Fig. 1), while similar experiments using FM1–43 found barely significant differences between 1 and 4 mm [Ca2+]e (data not shown).
The comparison between VGLUT2 WT and VGLUT2 TPM tagged with pHluorin indicates that the transporter must be functional to affect Pr. Though it is possible that the point mutations made to the transporter might disrupt protein–protein interactions, we find this unlikely as the mutations were made in the pore of the putative structure (Juge et al., 2006).
Vesicular fill state can be altered for a variety of neurotransmitters
Though little is known about how it affects vesicle Pr per se, vesicular transporter expression level has been shown to affect q. For example, overexpression of the vesicular monoamine transporter VMAT increases the q of dopamine from both synaptic vesicles and large dense core vesicles (LDCVs; Pothos et al., 2000). Overexpression of VGLUTs has been shown to increase q in both cultured neurons and at the Drosophila NMJ (Wilson et al., 2005; Daniels et al., 2004, respectively). Consequentially, changes in VGLUT copy number on vesicles may be a mechanism by which synapses can dynamically alter q, and possibly, vesicle Pr. It should be noted that at the Drosophila NMJ, though overexpression of Drosophila VGLUT (dVGLUT) does cause an increase in q, there is an overall decrease in quantal content, suggesting a decrease in Pr (Daniels et al., 2004). However, as this synapse exhibits homeostatic plasticity, the effect of increased fill state on Pr could be masked by a homeostatic decrease in Pr by another mechanism (Davis, 2013). Activity-dependent homeostatic scaling of VGLUT copy number occurs in cultured neocortical neurons (De Gois et al., 2005). Additionally, the amount of VGLUT1 on vesicles has been shown to vary in a time-of-day-dependent manner (Yelamanchili et al., 2006; Darna et al., 2009). Though in neither of these examples has it been confirmed that a concomitant change in q occurs, in the VGLUT2 heterozygous mouse, where the protein level is halved, a decrease in q at the cellular level and behavioral phenotypes at the whole-animal level were observed (Moechars et al., 2006). Decreased expression of VGLUT has also been reported in human brain samples from patients with Alzheimer's disease (Kashani et al., 2008) and Parkinson's disease (Kashani et al., 2007).
Intravesicular transmitter content can also be altered by the available concentration of transmitter in the cytosol. At the calyx of Held giant synapse, a high concentration of glutamate in the intracellular solution causes an increase in q (Ishikawa et al., 2002; Wu et al., 2007; Yamashita et al., 2009). Consistent with these results, the exposure of PC12 cells to l-DOPA, a dopamine precursor, causes an increase in LDCV content (Colliver et al., 2000), and acetylcholine exposure increases q at frog NMJ (van der Kloot and van der Kloot, 1986). Together, these data indicate that by altering the neurotransmitter available for transport into vesicles, the intravesicular content can be changed, which could affect the Pr of the vesicle. This suggests another mechanism by which a cell could modulate its release efficacy.
Vesicular fill state influences physical properties of vesicles
Whether the intravesicle neurotransmitter content could change the properties of a vesicle is a topic of debate. In LDCVs, an increase or decrease in the intravesicular neurotransmitter content coincides with an increase or decrease of the LDCV volume, respectively (Colliver et al., 2000). It has been hypothesized that this effect is due to differences in osmolarity between the LDCV lumen and the cytosol, which is supported by experiments where changes in osmolarity alone produce similar changes in LDCV volume (Thureson-Klein et al., 1975). One possibility may be that the increased osmolarity caused by filling changes the membrane tension of vesicles, which, in turn, could promote vesicle fusion. However, another intriguing hypothesis from Monte-Carlo simulation suggests that vesicle size itself could produce changes in Pr as a larger vesicle may result in a greater retardation of Ca2+ through voltage-activated channels and a higher local [Ca2+] near the release machinery (Glavinović and Rabie, 2001).
Is there evidence for this phenomenon in synaptic vesicles? In one set of experiments, isolated empty synaptic vesicles show a distinct increase in diameter when exposed to glutamate and ATP (Budzinski et al., 2009). This suggests that the intravesicular glutamate fill state may directly affect synaptic vesicle volume. Additionally, as shown with EM, vesicle diameter is increased at the Drosophila NMJ in a mutant where dVGLUT is overexpressed (Daniels et al., 2004). On the contrary, EM investigation of vesicles in mouse NMJ, where vAChT was reduced to 30%, showed no change in vesicle volume from WT vesicles, even though these mutants showed a reduction in q (Lima et al., 2010); and did not we see a significant change in area for VGLUT1 KO vesicles expressing a transport-deficient VGLUT2, which is consistent with a previous description of empty vesicles in hippocampal synapses (Siksou et al., 2013).
VGLUT copy number as an intrinsic indicator of Pr and q for vesicles
The number of VGLUTs on vesicles in mammalian synapses has long been proposed to affect q (Daniels et al., 2004; Wojcik et al., 2004; Moechars et al., 2006; but see Daniels et al., 2006; Edwards, 2007); however, we show in this study that VGLUT copy number can also affect Pr. This arouses the intriguing possibility that VGLUT copy number may serve as a vesicle-intrinsic link between Pr and q. Additionally, as we show that empty vesicles have a reduced Pr, the synapse displays a bias for releasing vesicles that are filled with neurotransmitter. This suggests that the synapse maintains efficiency by favoring the release of vesicles most likely to cause a postsynaptic response. Whether the decreased release probability of empty vesicles is due to an active “quality control” mechanism or a more passive effect of physical changes in the membrane due to the hypo-osmotic condition remains to be determined.
Footnotes
This work was supported by the Alexander von Humboldt Foundation (Fellowship for Postdoctoral Researchers 1141215, to M.A.H.), the European Research Council (Advanced Grant SYNVGLUT, to C.R.), and the German Excellence Initiative (Grant Exc 257, to C.R.). We thank B. Brokowski, A. Felies, S. Lenz, K. Pötschke, and B. Söhl-Kielczynski for technical assistance; Dr. M. Weston and L. Mamer for unpublished data; Dr. B. Rost for cloning; and members of the Rosenmund Laboratory for helpful discussions.
The authors declare no competing financial interest.
References
- Almqvist J, Huang Y, Laaksonen A, Wang DN, Hovmöller S. Docking and homology modeling explain inhibition of the human vesicular glutamate transporters. Protein Sci. 2007;16:1819–1829. doi: 10.1110/ps.072944707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancillo M, Min SW, Gerber S, Münster-Wandowski A, Wu YJ, Herman M, Trimbuch T, Rah JC, Ahnert-Hilger G, Riedel D, Südhof TC, Rosenmund C. Titration of Syntaxin1 in mammalian synapses reveals multiple roles in vesicle docking, priming, and release probability. J Neurosci. 2013;33:16698–16714. doi: 10.1523/JNEUROSCI.0187-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B. Colocalization of vesicular glutamate transporters in the rat superior olivary complex. Neurosci Lett. 2005;382:66–70. doi: 10.1016/j.neulet.2005.02.071. [DOI] [PubMed] [Google Scholar]
- Budzinski KL, Allen RW, Fujimoto BS, Kensel-Hammes P, Belnap DM, Bajjalieh SM, Chiu DT. Large structural change in isolated synaptic vesicles upon loading with neurotransmitter. Biophys J. 2009;97:2577–2584. doi: 10.1016/j.bpj.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Trimbuch T, Chao HT, Jordan JC, Herman MA, Rosenmund C. Investigation of synapse formation and function in a glutamatergic-GABAergic two-neuron microcircuit. J Neurosci. 2014;34:855–868. doi: 10.1523/JNEUROSCI.0229-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colliver TL, Pyott SJ, Achalabun M, Ewing AG. VMAT-mediated changes in quantal size and vesicular volume. J Neurosci. 2000;20:5276–5282. doi: 10.1523/JNEUROSCI.20-14-05276.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, DiAntonio A. A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle. Neuron. 2006;49:11–16. doi: 10.1016/j.neuron.2005.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darna M, Schmutz I, Richter K, Yelamanchili SV, Pendyala G, Höltje M, Albrecht U, Ahnert-Hilger G. Time of day-dependent sorting of the vesicular glutamate transporter to the plasma membrane. J Biol Chem. 2009;284:4300–4307. doi: 10.1074/jbc.M805480200. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gois S, Schäfer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci. 2005;25:7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreosti E, Odermatt B, Dorostkar MM, Lagnado L. A genetically encoded reporter of synaptic activity in vivo. Nat Methods. 2009;6:883–889. doi: 10.1038/nmeth.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RH. The Neurotransmitter cycle and quantal size. Neuron. 2007;55:835–858. doi: 10.1016/j.neuron.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Glavinović MI, Rabie HR. Monte Carlo evaluation of quantal analysis in the light of Ca2+ dynamics and the geometry of secretion. Pflugers Arch. 2001;443:132–145. doi: 10.1007/s004240100631. [DOI] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y, Takahashi T. A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron. 2002;34:613–621. doi: 10.1016/S0896-6273(02)00692-X. [DOI] [PubMed] [Google Scholar]
- Juge N, Yoshida Y, Yatsushiro S, Omote H, Moriyama Y. Vesicular glutamate transporter contains two independent transport machineries. J Biol Chem. 2006;281:39499–39506. doi: 10.1074/jbc.M607670200. [DOI] [PubMed] [Google Scholar]
- Kashani A, Betancur C, Giros B, Hirsch E, El Mestikawy S. Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson disease. Neurobiol Aging. 2007;28:568–578. doi: 10.1016/j.neurobiolaging.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, Simon A, Delacourte A, Giros B, Epelbaum J, Betancur C, El Mestikawy S. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex in correlated with cognitive decline in Alzheimer disease. Neurobiol Aging. 2008;29:1619–1630. doi: 10.1016/j.neurobiolaging.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Lima Rde F, Prado VF, Prado MA, Kushmerick C. Quantal release of acetylcholine in mice with reduced levels of vesicular acetylcholine transporter. J Neurochem. 2010;113:943–951. doi: 10.1111/j.1471-4159.2010.06657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Moechars D, Weston MC, Leo S, Callaerts-Vegh Z, Goris I, Daneels G, Buist A, Cik M, van der Spek P, Kass S, Meert T, D'Hooge R, Rosenmund C, Hampson RM. Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J Neurosci. 2006;26:12055–12066. doi: 10.1523/JNEUROSCI.2556-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch SA, Kensel-Hammes P, Gadd JC, Fujimoto BS, Allen RW, Schiro PG, Lorenz RM, Kuyper CL, Kuo JS, Bajjalieh SM, Chiu DT. Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J Neurosci. 2011;31:1461–1470. doi: 10.1523/JNEUROSCI.3805-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RL, Calupca MA, Merriam LA, Prior C. Empty synaptic vesicles recycle and undergo exocytosis at vesamicol-treated motor nerve terminals. J Neurophysiol. 1999;81:2696–2700. doi: 10.1152/jn.1999.81.6.2696. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues HA, Fonseca Mde C, Camargo WL, Lima PM, Martinelli PM, Naves LA, Prado VF, Prado MA, Guatimosim C. Reduced expression of the vesicular acetylcholine transporter and neurotransmitter contents affects synaptic vesicle distribution and shape in mouse neuromuscular junction. PLoS One. 2013;8:e78342. doi: 10.1371/journal.pone.0078342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Rettig J, Brose N. Molecular mechanisms of active zone function. Curr Opin Neurobiol. 2003;13:509–519. doi: 10.1016/j.conb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/S0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:E1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siksou L, Silm K, Biesemann C, Nehring RB, Wojcik SM, Triller A, El Mestikawy S, Marty S, Herzog E. A role for vesicular glutamate transporter 1 in synaptic vesicle clustering and mobility. Eur J Neurosci. 2013;37:1631–1642. doi: 10.1111/ejn.12199. [DOI] [PubMed] [Google Scholar]
- Tabares L, Ruiz R, Linares-Clemente P, Gaffield MA, Alvarez de Toledo G, Fernandez-Chacón R, Betz WJ. Monitoring synaptic function at the neuromuscular junction of a mouse expressing synaptopHluorin. J Neurosci. 2007;27:5422–5430. doi: 10.1523/JNEUROSCI.0670-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Thureson-Klein A, Klein RL, Chen Yen SH. Morphological effects of osmolarity on purified noradrenergic vesicles. J Neurocytol. 1975;4:609–627. doi: 10.1007/BF01351540. [DOI] [PubMed] [Google Scholar]
- van der Kloot W, van der Kloot TE. Catecholamines, insulin and ACTH increase quantal size at the frog neuromuscular junction. Brain Res. 1986;376:378–381. doi: 10.1016/0006-8993(86)90203-9. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Borst JG. Short-term plasticity at the calyx of held. Nat Rev Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- Wang L, Tu P, Bonet L, Aubrey KR, Supplisson S. Cytosolic transmitter concentration regulates cycling at hippocampal GABAergic terminals. Neuron. 2013;80:143–158. doi: 10.1016/j.neuron.2013.07.021. [DOI] [PubMed] [Google Scholar]
- Weston MC, Nehring RB, Wojcik SM, Rosenmund C. Interplay between VGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron. 2011;69:1147–1159. doi: 10.1016/j.neuron.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Wu XS, Xue L, Mohan R, Paradiso K, Gillis KD, Wu LG. The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J Neurosci. 2007;27:3046–3056. doi: 10.1523/JNEUROSCI.4415-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Kanda T, Eguchi K, Takahashi T. Vesicular glutamate filling and AMPA receptor occupancy at the calyx of Held synapse of immature rats. J Physiol. 2009;587:2327–2339. doi: 10.1113/jphysiol.2008.167759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamanchili SV, Pendyala G, Brunk I, Darna M, Albrecht U, Ahnert-Hilger G. Differential sorting of the vesicular glutamate transporter 1 into defined vesicular pool is regulated by light signaling involving the clock gene Period2*. J Biol Chem. 2006;281:15671–15679. doi: 10.1074/jbc.M600378200. [DOI] [PubMed] [Google Scholar]
- Zhao C, Dreosti E, Lagnado L. Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J Neurosci. 2011;31:7492–7496. doi: 10.1523/JNEUROSCI.6636-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Petersen CC, Nicoll RA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol. 2000;525:195–206. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Xu J, Heinemann SF. Two pathways of synaptic vesicle retrieval revealed by single vesicle imaging. Neuron. 2009;61:397–411. doi: 10.1016/j.neuron.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]