

Abstract
Painful experiences are multilayered, composed of sensory, affective, cognitive and behavioral facets. Whereas it is well accepted that the development of chronic pain is due to maladaptive neuronal changes, the underlying molecular mechanisms, their relationship to the different pain modalities, and indeed the localization of these changes are still unknown. Brain-derived neurotrophic factor (BDNF) is an activity-dependent neuromodulator in the adult brain, which enhances neuronal excitability. In the spinal cord, BDNF underlies the development and maintenance of inflammatory and neuropathic pain. Here, we hypothesized that BDNF could be a trigger of some of these plastic changes. Our results demonstrate that BDNF is upregulated in the anterior cingulate cortex (ACC) and the primary sensory cortex (S1) in rats with inflammatory pain. Injections of recombinant BDNF (into the ACC) or a viral vector synthesizing BDNF (into the ACC or S1) triggered both neuronal hyperexcitability, as shown by elevated long-term potentiation, and sustained pain hypersensitivity. Finally, pharmacological blockade of BDNF-tropomyosin receptor kinase B (TrkB) signaling in the ACC, through local injection of cyclotraxin-B (a novel, highly potent, and selective TrkB antagonist) prevented neuronal hyperexcitability, the emergence of cold hypersensitivity, and passive avoidance behavior. These findings show that BDNF-dependent neuronal plasticity in the ACC, a structure known to be involved in the affective-emotional aspect of pain, is a key mechanism in the development and maintenance of the emotional aspect of chronic pain.
Keywords: hyperexcitability, neurotrophic factor, pain, viral vector
Introduction
Chronic pain relies on complex maladaptive changes in neural networks activated by noxious or non-noxious stimuli, and which are clinically expressed as hyperalgesia and allodynia (Apkarian et al., 2009; Sandkühler and Lee, 2013). The molecular mechanisms of this plasticity, as well as the roles of the respective neuronal networks, are still not completely understood.
Brain-derived neurotrophic factor (BDNF) and its high-affinity receptor tropomyosin receptor kinase B (TrkB) are distributed across the CNS, including in nociceptive pathways (Pezet and McMahon, 2006; Merighi et al., 2008), where this factor acts as an activity-dependent neuromodulator. In the brain, BDNF has been reported to enhance neuronal excitability (Thoenen, 1995) by promoting neurotransmitter release (Takei et al., 1997), phosphorylation of specific glutamate receptor subunits (Levine et al., 1995; Suen et al., 1997), exerting direct effects on ion channel conductance (Kafitz et al., 1999), and inducing structural and functional neuronal changes (Marty et al., 1997; Abidin et al., 2008).
Due to its neurotrophic and excitatory neuromodulatory roles, BDNF has a strong physiological role in the hippocampus in learning and memory (Thoenen, 2000), although this activity-dependent modulation also leads to pathophysiological conditions, such as hippocampal hyperexcitability in epilepsy (Binder et al., 2001; Lu et al., 2013). Endogenous BDNF upregulation has been reported following nerve injury or peripheral inflammation in peripheral tissues (Shu et al., 1999; Narita et al., 2000), in the spinal cord (Cho et al., 1997; Geng et al., 2010; Lin et al., 2011), and in supraspinal regions, such as the periaqueductal gray and rostral ventromedial medulla (Guo et al., 2006). This overexpression of BDNF is necessary for synaptic changes and thus the development of chronic pain (Obata and Noguchi, 2006). Numerous studies have described a network of brain areas activated by experimental painful stimulations, forming a neuronal network involved in the different dimensions of pain perception (Treede et al., 1999). These brain areas include the primary sensory, motor, anterior cingulate, and insular cortices, as well as the amygdala and the lateral thalamus.
Several key factors permitted us to hypothesize that BDNF might be overexpressed and induce a functional plasticity underlying sustained thermal and/or mechanical hypersensitivity in a rat model of inflammatory pain, where several brain areas receive information directly or indirectly from tonically active nociceptors: (1) BDNF has a recognized role as an activity-dependent positive modulator in the brain, (2) increased electrophysiological activity is observed in thalamic and cortical areas in animal models of inflammatory pain (Lamour et al., 1983; Guilbaud et al., 1993), and (3) BDNF is capable of inducing long-term synaptic and molecular changes. This study investigated the putative role of BDNF signaling in cortical areas (somatomotor, cingular, and insular cortex) through behavioral, electrophysiological, biochemical, and immunohistochemical approaches in an animal model of inflammatory pain induced by unilateral injection of complete Freund's adjuvant (CFA) in the hindpaw. Our results reveal BDNF-dependent plasticity mostly in the cingulate cortex, a brain area involved in the affective-emotional aspect of pain.
Materials and Methods
Animals
One-hundred and sixty male Sprague-Dawley rats weighing 200–225 g at the beginning of the experiments (Janvier) were used in this study. Experimental procedures were performed in agreement with the European Community Council Directive of 24 November, 1986 (86/609/EEC), and the guidelines for investigation of experimental pain in animals published by the International Association for the Study of Pain were followed. Great care was taken, particularly concerning housing conditions, to avoid or minimize discomfort to the animals. The animals were housed in a solid-floor cage with a deep layer of sawdust to accommodate excess urine, and cages were changed twice per week. The cages were kept at a constant temperature of 22 ± 1°C with an alternating 12 h light/dark cycle. Food and water were available ad libitum.
Note: in all the experiments, the experimenters were blinded to the animal (or slice) treatment as the treatments were performed by another experimenter. The animals were numbered at the base of their tail and the animals included in each experiment were randomized (including per cage).
Habituation.
The animals were subjected to a habituation period before the beginning of the surgical procedure (implantation of the cannula or stereotaxic injection of the viral vector). During this period, animals were habituated to the room and the experimenter, and to different pain-assessing and passive-avoidance environments (the electronic Von Frey testing chamber and the cold-plate chamber).
Induction of peripheral inflammation using CFA.
Animals were anesthetized with Isoflurane (Isoflo, Baxter) and an intraplantar injection of 50 μl of CFA (Sigma-Aldrich) was administered to the right hindpaw. Control animals received 50 μl of saline.
Stereotaxic injections
Stereotaxic injections of GFP, BDNF expression viral vectors, or recombinant BDNF.
Animals were anesthetized by intraperitoneal (i.p.) injection of Metomidine (Domitor, 0.3 mg/kg) and Ketamine (Imalgène, 40 mg/kg), and placed on a stereotaxic frame. Under sterile conditions, a small incision was made in the cranial skin and one to four holes (1 × 1 mm) were performed using a drill kit (Foredom). Two to four injections (depending on the site of injection; Table 1) of either BDNF lentiviral vector (p156sinRRLpptCAG-BDNF, provided by Professor George M. Smith, Temple University; 0.5 μl of solution containing 1.34 × 106 TU/μl) or GFP lentiviral vector (2.67 × 106 TU/μl) were performed per hemispheric side, using a 1 μl Hamilton syringe. The injection coordinates were chosen according to the well accepted rat brain atlas (Paxinos and Watson, 2006). After the slow injection of viral vectors (during 10 min, followed by a 10 min pause before redrawing the needle), the muscles and skin were sutured and the animal was returned to its cage. The animals were then left to recover from anesthesia and surgery, and measurements of thermal/mechanical sensitivity were performed at 1, 2, 3, and 4 weeks after the injection. In a preliminary study using 12 animals, we followed the temporal expression of GFP expression induced by GFP injection in the primary sensory cortex, hindlimb part (S1HL); the animals were then killed (n = 3 per group) at 1, 2, 3, or 4 weeks postinjection and the phenotypic characterization of the transduced cells (see Fig. 2) was performed on these animals.
Table 1.
Information regarding the stereotaxic injections of viral vectors, rBDNF or CTX-B
| Injected molecule | Cingulate cortex |
S1HL |
||
|---|---|---|---|---|
| Coordinates | Concentrations, volumes | Coordinates | Concentrations, volumes | |
| GFP-expressing lentiviral vector | One bilateral injection at AP bregma + 1 mm, lateral: 0.75 mm, depth: 1 mm | 0.5 μl of GFP lentiviral vector solution at 1.3 × 106 TU/μl per injection site | 2 bilateral injections at AP bregma −0.5 mm and −1.5 mm, lateral: 2.5 mm, depth: 1 mm | 0.5 μl of GFP lentiviral vector solution at 1.3 × 106 TU/μl per injection site |
| Mix of GFP- and BDNF-expressing viral vectors | One bilateral injection at AP bregma + 1 mm, lateral: 0.75 mm, depth: 1 mm | 0.5 μl of solution containing 1.3 × 106 TU/μl of BDNF and 1.3 × 106 TU/μLGFP lentiviral vectors per injection site | 2 bilateral injections at AP bregma −0.5 mm and −1.5 mm, lateral: 2.5 mm, depth: 1 mm | 0.5 μl of solution containing 1.3 × 106 TU/μl of BDNF and 1.3 × 106 TU/μLGFP lentiviral vectors per injection site |
| rBDNF | One bilateral injection at AP bregma + 1 mm, lateral: 0.75 mm, depth: 1 mm using a 26-gauge preimplanted double-guide (Plastic One) | 0.5 μl of a solution at 1 μg/μl; controls: 0.5 μl saline | ||
| CTX-B | One bilateral injection at AP bregma + 1 mm, lateral: 0.75 mm, depth: 1 mm using a 26-gauge preimplanted double-guide (Plastic One) | 0.5 μl of a 10 μg/μl solution dissolved in saline; controls: 0.5 μl saline | ||
Volumes and concentrations of vectors or drugs, and also the coordinates of injections are indicated for each type of experiment.
Figure 2.

BDNF expression induced by peripheral inflammation occurred in all types of cells. A–C, Double-immunostaining shows that BDNF immunostaining colocalized with the astrocyte marker GFAP (A, arrows), but also with the microglial marker Iba1 (B, arrows), and NeuN (C, arrows), a neuronal marker. Cells positive for BDNF, but negative for the relevant marker, were also observed and are indicated here with arrowheads. Scale bars: A, B, 20 μm; C, 50 μm.
Stereotaxic injections of either recombinant BDNF or CTX-B before the administration of CFA.
To facilitate administration of rBDNF or cyclotraxin-B (CTX-B) to the cingulate cortex and to follow the behavior shortly after the injection, animals were anesthetized with a mix of Domitol and Vetalar (see above). Implants were performed in the cingulate cortex (coordinates: AP bregma +1 mm lateral: 0.75 mm, depth: 1 mm) using a bilateral cannula system, consisting of a 26 gauge double-guide with two cannulae separated by 1.5 mm (Plastics One), attached to the crane with photopolymerisable dental cement (Henry Schein). Animals were then allowed 1 week of rest, in which baselines for mechanical and thermal sensitivity were measured. Finally, either rBDNF (0.5 μl at 1 μg/μl; Peprotech; Table 1) or CTX-B (0.5 μl of a 2 ng/μl solution dissolved in 0.1% BSA, saline; Bio S&T; Table 1) was injected under isoflurane anesthesia; CTX-B is a highly potent and selective trk-B receptor inhibitor (Tat-cyclotraxin-B; Bio S&T, Montreal; IC50 of ∼70 pm (Cazorla et al., 2010). Control animals received a 0.5 μl saline injection. Induction of inflammation (or saline injection for controls) was performed as described above, immediately after saline/CTX-B injections, while the animal was still under isoflurane anesthesia. Animals in the four experimental groups (saline-saline, saline-CFA, CTX-saline, CTX-CFA; n = 6 animals per group) were then allowed to recover from anesthesia, and measurements of thermal/mechanical sensitivity were performed at 6, 24, 48, and 72 h after the injection.
Targeting viral vector injections in animals.
To measure the extend of diffusion of the viral vectors in the cortex, we performed a preliminary experiment where we injected increasing volume of the GFP-expressing vector to optimize the number and volume of injections to efficiently fill in the structure of interest. Our injections of viral vector were verified by the following procedure: intracardiac perfusion of 4% paraformaldehyde (see “Immunohistochemistry” for details), cryoprotection, sectioning of the brain, measurement of the spread of viral vector injected, and finally analysis of the spread of GFP-positive transduced cells. The assessment of the injection's coordinates was performed using the same procedure. The stereotaxic coordinates at which the final experimental viral injections or where the guides were implanted (injections of rBDNF and CTX in the ACC) were determined using the same procedure. However, in the case of nonfluorescent molecules, only the location of the guide was determined, as it leaves a small lesion in the brain.
Behavioral testing
Assessment of mechanical sensitivity.
An electronic Von Frey hair unit (EVF-3; Bioseb Chaville) was used to measure the sensitivity threshold of the rats, with measurements ranging from 0.1 to 100 g with a 0.2 g accuracy, as previously described (Thibault et al., 2012). Rats were individually placed on an elevated (21 cm from ground) clear plastic chamber (18.5 × 20 × 14 cm) with a wire mesh floor, and were adapted to the testing environment for 15 min. Punctuate stimuli were delivered to the midplantar area of each hindpaw from below the mesh floor, through a plastic spring tip with increasing pressure. Paw sensitivity threshold (i.e., the minimum pressure required to elicit a robust and immediate pedal withdrawal) was recorded as displayed on the EVF-3 screen. Voluntary movement associated with locomotion was not considered to be a withdrawal response. The stimulus was applied to each hindpaw three times, in 15 s intervals. For the CTX-B experiment, the value adopted as the rat's mechanical sensitivity threshold was the average of the three values measured from each hindpaw. For the BDNF-overexpression study, the value adopted as the rat's mechanical sensitivity threshold was the average of the six values (pooling the measurements from both hindpaws). Mechanical allodynia was defined as a significantly lower value in withdrawal thresholds to EVF-3 application when compared with the control groups.
Assessment of cold sensitivity.
Cold thermal sensitivity was measured with a cold-plate analgesia meter (Bioseb), as previously described (Thibault et al., 2012). Briefly, 10 min after placing the animals individually on the plate in a transparent plastic chamber (16.5 × 16.5 × 25 cm), the temperature was gradually decreased (driven by a computer) from 22°C to 0°C at a rate of 5°C/min. Cold-awareness threshold is defined as the temperature at which the animal is startled and/or stops its current activity (e.g., exploring, grooming, scratching, etc.) and begins to stare at/examine the surface of the cold plate. Cold-nociceptive threshold is defined as the temperature at which the animal starts to exhibit a nociceptive response (i.e., licking its hindpaw, escaping, or tapping, such as multiple withdrawals of the hindpaw in a short time period). All behaviors were assessed at room temperature (22 ± 1°C). A significant increase in the cold awareness threshold was interpreted as a sign of cold allodynia, whereas a significant increase in the nociceptive threshold is a marker for cold hyperalgesia.
Assessment of the passive avoidance behavior evoked by an aversive mechanical stimulus.
This test was adapted from (LaBuda and Fuchs, 2000). In a room with the light level set at 240 ± 5 Lux, rats were individually placed on an elevated (21 cm from ground) Plexiglas chamber (18.5 × 40 × 14 cm) with a wire mesh floor, made of two parts of equal dimensions (20 cm width). The first one was dark (4 Lux) and covered with a black cardboard, whereas the second one was brighter (20 Lux) and covered with white cardboard. A first session of habituation was performed, where animals habituated to the dual compartments in presence of the experimenter but animals were not stimulated. Three more habituation sessions were further performed on separate days for 20 min. In these sessions, as on the days of testing after the induction of peripheral inflammation, the animals were mechanically stimulated for 1 s every 15 s for 20 min (LaBuda and Fuchs, 2000). If the animal was located in the dark side, the right paw was then stimulated, whereas the left paw was stimulated when the animal was in the bright compartment. The time spent in the bright compartment was measured in 5 min increments. Animals that spent <20% in the bright compartment at the end of the baseline sessions were excluded from the study, as previously described by LaBuda and Fuchs (2000). Although n = 9 animals per experimental groups were initially included in our study, due to this exclusion factor, the final number of animals included per group was between 6 and 8.
Results are expressed in time (in seconds) spent by the animal in the light chamber during the last 5 min increment, i.e., the 15–20 min period. This period was chosen because we and LaBuda and Fuchs (2000) found that 15–20 min were required for the animals to perceive the stimulations in the dark chamber as aversive and develop a copying strategy (the passive avoidance). An increase in the time spent by the animals in the light chamber was interpreted as a sign of passive avoidance.
Statistical analyses.
Animal treatments were randomized within each cage. For the BDNF lentiviral vectors and rBDNF experiments, we performed a two-way ANOVA followed by a Student-Newman–Keuls post hoc test for multiple comparisons using the Sigmastat software. For the CTX-B experiments, we performed three-way ANOVAs, followed by a Student-Newman–Keuls post hoc test for multiple comparisons. ANOVA results are given as the value of the Fisher distribution Fx,y obtained from the data, where x is the degrees of freedom for groups and y is the total degrees of freedom of the distribution. Data were expressed as mean ± SEM, and the levels of significance were set as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Electrophysiological recordings
Slice preparation
Ten male rats, 2- to 3-months-old (n = 4 GFP, n = 3 BDNF, n = 3 CFA), were anesthetized using an intraperitoneal injection of sodium pentobarbital (100 mg/kg; Ceva Santé) and decapitated. Brains were quickly removed and placed into cold cutting solution (∼4°C) containing the following (in mm): 124 NaCl, 3 KCl, 2.5 CaCl, 1.15 MgSO4, 1.15 KH2PO4, 24 NaHCO3 and 10 d-glucose, pH 7.4, continuously bubbled with 95% O2–5% CO2. To avoid excitotoxicity, 1 mm of kynurenic acid (Sigma-Aldrich) was added to the solution. Coronal brain slices (350 μm thick) comprising the somatosensory cortex were cut with a vibratome (VT1200S; Leica) and transferred to a holding chamber filled with artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 20 glucose, and 1 kynurenic acid, constantly oxygenated (95% O2–5% CO2) and held at room temperature. Individual slices were then submerged in a recording chamber kept at 32°C and perfused (1.5 ml/min) with oxygenated aCSF (without kynurenic acid).
Electrophysiology
Slices were submerged in a thermostat-controlled recording chamber (Luigs and Neuman), placed on the stage of an Axioskop 2FS microscope (Carl Zeiss) equipped with Dodt gradient contrast optics (Luigs and Neuman) and a CoolSnap FX CCD camera (Roper Scientific), and visualized under infrared (IR) illumination. The preparation was continuously perfused (1–2 ml/min) with oxygenated aCSF and the transfection zone in S1 was identified by its fluorescence.
Extracellular field potentials in cortical layer II were recorded with an electrode (2–5 MΩ) filled with aCSF solution. The potentials were evoked by stimulation of axons coming from the thalamus, at the border of the white matter and layer VI, using a bipolar concentric stimulating electrode. Long-term potentiation (LTP) was induced by three trains of theta burst stimulation (TBS; 200 ms interbursts separated by 15 s), with 1 theta burst consisting of four pulses (0.2 ms/pulse) at 100 Hz.
Electrophysiological stimulation protocol
The stimulating electrode was placed at the border between the white matter and layer VI to stimulate vertical pathways projecting to cortical layers II/III and IV (Woodward et al., 1990; Kenan-Vaknin and Teyler, 1994). Field potentials were recorded at 32°C in the current-clamp mode of a MultiClamp 700B amplifier (Molecular Devices). Signals were filtered at 10 kHz and digitized at 50 kHz with an analog signal converter (Digidata 1322A; Molecular Devices), and analyzed using pClamp 9.2 software (Molecular Devices). The stimulation electrode was set at 0.2–1 mA for 200 μs, with a stimulation intensity that yielded a half-maximal response. Baseline responses were obtained every 20 s over 10 min. LTP was induced by TBS: three trains of 10 theta bursts (with 200 ms interbursts) separated by 15 s, with 1 theta burst consisting of four pulses (0.2 ms/pulse) at 100 Hz (He et al., 2009). Post-TBS acquisition was performed over a 35 min period with stimulations every 20 s. Recorded data were analyzed using the Clampfit, Igor pro, and Excel software programs. The amplitude of the maximum negative field potential in layer II was used as a measure of the evoked population excitatory current. The last 5 min of the recordings were averaged and compared with the baseline; all recordings with an average greater than twice the noise of the baseline were considered as LTP. Recordings whose baseline varied by >10% were excluded. Data were then pooled together for each experimental group and expressed as the percentage change from the control baseline ± SEM. Statistical comparisons were performed by applying a Mann–Whitney rank sum test. Differences were considered significant at p < 0.05.
BDNF ELISA titration
Animals were deeply anesthetized with sodium pentobarbital (100 mg/kg; Ceva Santé) and killed by decapitation. Their brains were placed on ice. Slices were performed using a sharp blade and several brain areas were then quickly dissected using the coordinates of the Paxinos and Watson (2006) rat brain atlas. Brain tissues were then individually collected in 2 ml tubes and immediately frozen on dry ice. These brain areas comprised the unilateral primary sensory cortices, pooled bilateral cingulate cortices, pooled bilateral insular cortices, and pooled bilateral motor cortices. Tissues from all animals were homogenized using Ultraturax in lysate NP-40-based buffer, supplemented by protease inhibitor as recommended by the manufacturer (Promega) and centrifuged 20 min at 10,000 × g. Supernatant was collected and BDNF was titrated using the BDNF ELISA kit from Promega. The protein concentrations were determined on the same lysates using a Biorad DC Protein Assay kit (Bio-Rad Laboratories) and results were expressed in picograms BDNF/mg protein. For the S1HL, statistical significance was determined using two-way ANOVA followed by the Student-Newman–Keuls comparison post hoc test. The first parameter was the treatment of the animals (saline vs CFA) and the second parameter was the cortex side counted (ipsilateral vs contralateral side). ANOVA results are given as the value of the Fisher distribution Fx,y obtained from the data, where x is the degrees-of-freedom for groups and y is the total degrees of freedom of the distribution. Mann–Whitney rank sum tests were performed for other structures (cingulate, insular, and motor cortices). The titration was performed on at least four animals per condition [CFA experiment: n = 4 for the saline group and n = 4 for the CFA group for the cingulate, insular and motor cortices, whereas n = 5 for the saline group and n = 5 for the CFA group for the S1HL; BDNF vector experiment: n = 4 for the GFP group and n = 4 for the BDNF group for each time point, observation day (D)7 and D28].
Immunohistochemistry
Animals were deeply anesthetized by 25 mg/ml urethane injection (1.5 g/kg, i.p.) and killed by transcardial perfusion with 200 ml of 0.9‰ NaCl, followed by 500 ml of 4% paraformaldehyde (PFA) plus 15% picric acid in 0.2 m phosphate buffer (PB), pH 7.4. The brain was dissected out and postfixed overnight in 4% PFA plus 15% picric acid at 4°C. The brain was then cryoprotected in 30% sucrose in 0.2 m PB, pH 7.4, overnight at 4°C. Twenty-micrometer-thick coronal brain sections were serially cut using a cryostat (HM550, Microm Microtech). Sets of 10 sections were mounted onto Superfrost slides, so that four series of 10 slides were obtained, corresponding to whole structures of interest including the cingulate cortex and primary somatosensory cortex.
BDNF protein immunostaining: immunohistochemical detection protocol.
Serial sections from the brain were immunostained for BDNF immunoreactivity according to the avidin-biotin-peroxidase method followed by DAB revelation, as previously described (Pezet et al., 1999). Tissue sections were incubated overnight at room temperature in primary antiserum directed against the BDNF protein (sheep anti-BDNF, Abcam, no. ab75040) diluted 1:1000 in PB containing 0.9% NaCl (PBS, 0.02 m), 0.3% triton and 0.02% sodium azide (PBS-T-azide). Incubated sections were washed in three successive baths of PBS and subsequently incubated in 1:300 biotinylated rabbit anti-sheep IgG (no. ab6746, Abcam) in PBS-T for 1 h at room temperature. Sections were then washed three times in PBS and incubated for 1 h in avidin-biotin-peroxidase complex (1:500; Vectastain, Vector Laboratories) diluted in PBS. Finally, sections were washed three times in PBS and revealed with a peroxidase substrate kit (Vector Laboratories, SK-4100) according to the supplier's protocol. The reaction proceeded for 5 min at 20°C; the reaction was then stopped by washing in distilled water. Sections were sequentially differentiated in 70%, 90%, 95%, and absolute alcohol, and finally air dried, xylene-treated, and coverslipped with DPX (Sigma-Aldrich). To account for the possibility that immunochemistry might vary from one experiment to the next, brain sections of rats from all experimental groups were immunostained at the same time.
Quantification of BDNF staining.
To quantify BDNF staining in both groups of animals, black and white 256 gray level images were acquired with a 20× objective on a Zeiss microscope. All acquisitions were performed with the same acquisition setup. To count the number of positive cells, we performed a cell counter analysis using the ImageJ software. The same regions of interest were used for each animal as illustrated in Figure 1D. Cell counting was performed separately for the ipsilateral and contralateral sides of the brain for each animal. Statistical significance was determined by two-way ANOVA followed by Student-Newman–Keuls comparison post hoc test, using the Sigmastat software. The first parameter was the treatment of the animals (saline vs CFA) and the second parameter was the cortex side counted (ipsilateral vs contralateral side). ANOVA results are given as the value of the Fisher distribution Fx,y obtained from the data, where x is the degrees-of-freedom for groups and y is the total degrees of freedom of the distribution. Quantification was performed on four sections per animal (n = 5 animals per group).
Figure 1.

Peripheral inflammation induced an increased BDNF expression in both the cingulate and primary sensory cortices. A–C, ELISA titration showing that BDNF concentration was increased markedly in the cingulate cortex (pool of bilateral sites; A) and to a lesser extent in the primary sensory cortex (B) and unchanged in the insulate and motor cortices (C) 24 h after the induction of peripheral inflammation by unilateral intraplantar injection of CFA. E–J, Immunohistochemical staining of BDNF (G–J) and its quantification (E, F) reveals an increased number of BDNF-positive cells in the right and left cingulate cortex (E) in inflamed animals. There is also an increase in the contralateral somatosensory cortex, hindlimb part (F). D, Schematic showing the area used to quantify the number of BDNF-positive cells in the cingulate cortex (D, top) and the S1HL (D, bottom). G, H, Representative BDNF immunostaining in the ACC of a control (G) and a CFA-injected rat (H). I, J, Representative BDNF immunostaining in the S1HL of a control (I) and a CFA-injected rat (J). Scale bars: G, H, 200 μm; I, J, 100 μm. All data are expressed as mean ± SEM; N = 5 CFA-treated animals, n = 5 control animals. Two-way ANOVA followed by Student-Newman–Keuls comparison post hoc test. B: F(1,18) = 2017, p = 0.176; E: F(1,25) = 16.62, p < 0.001; F: F(1,25) = 17.25, p < 0.001; interaction: F(1,25) = 4.95, p = 0.037; *p < 0.05, **p < 0.01, ***p < 0.001 versus control rats.
Double-staining BDNF: GFAP/Iba1 or NeuN.
Serial brain sections from the animals used for the quantification of BDNF expression in the ACC or S1 (Fig. 2), or from animals used in the temporal expression study (Fig. 3) were incubated overnight at room temperature with the appropriate antibodies diluted in PBS-T-azide: 1:300 BDNF (sheep anti-BDNF, ab75040); 1:1000 GFAP, an astrocyte marker (rabbit anti-GFAP, Abcam, ab48050); 1:1000 Iba1, a microglia and macrophage marker (rabbit anti-Iba1, no. 019-19741, Wako Chemicals); and 1:2000 NeuN, a neuronal marker (mouse anti-NeuN, no. VMA377, AbCys). Following washes, sections were incubated in AlexaFluor 568 anti-rabbit IgG or AlexaFluor 568 anti-mouse IgG (Invitrogen) diluted 1:1000 in PBS containing 0.3% triton and fluorescein anti-sheep IgG (Vector Laboratories, FI-6000). Because the fluorescein anti-sheep secondary antibody was generated in rabbit, as were the anti-GFAP and anti-Iba1 antibodies, these double-immunostainings were performed sequentially, with GFAP or Iba1 immunostaining being performed first. Following incubation in AlexaFluor 568 anti-rabbit IgG, brain sections were incubated overnight at room temperature with anti-BDNF. Following washes, sections were incubated 2 h in fluorescein anti-sheep IgG and mounted in Vectashield medium.
Figure 3.

Time course and cell-specific expression of GFP or BDNF induced by intracerebral injection of recombinant lentiviral vectors. A, Representative examples of GFP lentiviral expression in the hindlimb part of the S1HL at 1, 2, 3, and 4 weeks after local injection. GFP fluorescence was strictly localized in the S1HL and was maintained throughout the 4 weeks. B, ELISA titration performed on cortical samples 7 or 28 d after intracortical injection of GFP- or [GFP + BDNF]-expressing lentiviral vectors, showing that BDNF was significantly upregulated at both times. All data are expressed as mean ± SEM; **p < 0.01, ***p < 0.001 GFP-transfected versus BDNF-transfected rats (n = 4 GFP, n = 4 BDNF). C–E, Double-immunofluorescent staining showed that GFP (C–E, green) colocalized with GFAP (astrocyte cell; C), Iba1 (microglial cell; D, purple), and Cutl1 (pyramidal cortical neuron; E, purple). Scale bars: A, 1 mm; C, D, 10 μm; E, 20 μm.
Double-staining GFP: GFAP/Iba1 or Cutl1.
Serial brain sections from the animals used for the quantification of BDNF expression in the ACC or S1 (Fig. 2), or from animals used in the temporal expression study (Fig. 3), were incubated overnight at room temperature with the appropriate antibodies diluted in PBS-T-azide: 1:1000 GFAP (rabbit anti-GFAP, Abcam, ab48050); 1:1000 Iba1 (rabbit anti-Iba1, no. 019-19741, Wako Chemicals) or 1:1000 Cutl1, a pyramidal marker (mouse anti-Cutl1, no. ab54583, Abcam). Following several washes, sections were incubated in AlexaFluor 568 anti-rabbit IgG or AlexaFluor 568 anti-mouse IgG (Invitrogen) diluted 1:1000 in PBS-T. After several washes, sections were mounted in Vectashield medium.
p-Erk protein immunostaining: immunohistochemical detection protocol.
As described above, serial sections from the brain of the animals used in the behavioral experiment (see Fig. 6) were immunostained for p-Erk immunoreactivity according to the avidin-biotin-peroxidase method, followed by DAB revelation. Tissue sections were incubated overnight at room temperature in primary antiserum directed against the p-Erk protein (rabbit anti-Phospho p44/42 MAPK, Cell Signaling Technology, no. 4370L) diluted 1:1000 in PBS-T-azide. After incubation in 1:300 biotinylated goat anti-rabbit IgG (Vector Laboratories) in PBST for 1 h at room temperature and 1 h in avidin-biotin-peroxidase complex (1:500; Vectastain, Vector Laboratories) diluted in PBS, sections were washed and revealed with a peroxidase substrate kit (Vector Laboratories, SK-4100). Sections were sequentially differentiated in 70%, 90%, 95%, and absolute alcohol, and finally air dried, xylene-treated, and coverslipped with DPX for histology (Sigma-Aldrich). Brain sections of rats from the same experimental group were immunostained at the same time.
Figure 6.

Blockade of BDNF/TrkB signaling by intra-ACC injection of TrkB antagonist prevented the cold hypersensitivity and cortical hyperexcitability induced by peripheral inflammation. A–D, Blockade of the BDNF/TrkB signaling pathway by the antagonist CTX-B prevented the occurrence of cold allodynia in CFA-injected rats (A). It also temporarily prevented the development of cold hyperalgesia for 2 d (B), but had no effect on the mechanical allodynia observed in the hindpaw ipsilateral to CFA injection (C). Neither intraplantar injection of CFA into the right paw nor intra-ACC injection of CTX-B modified the withdrawal threshold for the contralateral hindpaw (D, left). E–F, Representative examples of pERK staining in the cingulate cortex in a control (E) and a CFA-treated (F) rat. Arrows indicate pERK positive neurons. Scale bar, 90 μm. G, Quantification of pERK-positive neurons, showing that the antagonistic effect of CTX-B on BDNF-TrkB signaling prevented a peripheral inflammation-evoked increase in the number of pERK neurons in the ACC. H, Representative photomicrographs of pERK and BDNF immunostainings and colocalisation (arrows) in neurons of the cingulate cortex. Scale bar, 40 μm. All data are expressed as mean ± SEM; N = 6 animals per group, three-way ANOVA, followed by a Student-Newman–Keuls post hoc test. A, Inflammation treatment parameter: F(1,116) = 9.334, p = 0.003; CTX-B treatment parameter: F(1,116) = 4.213, p = 0.043; B, Inflammation treatment parameter: F(1,116) = 106.608, p < 0.001; CTX-B treatment parameter: F(1,116) = 7.245, p = 0.008; C, Inflammation treatment parameter: F(1,116) = 236.029, p < 0.001; CTX-B treatment parameter: F(1,116) = 1.104, p = 0.296; **p < 0.01 CFA-saline versus saline-saline rats; §p < 0.05 CFA-saline versus CFA-CTX-B rats. D, *p < 0.05, **p < 0.01, ***p < 0.001 CFA-saline versus saline-saline rats; ††p < 0.01, †††p < 0.001 CFA-CTX-B versus saline-CTX-B rats; §p < 0.05, §§§p < 0.001 CFA-saline versus CFA-CTX-B rats. G, F(3,20) = 5.875, p = 0.006.
Quantification of p-Erk staining.
Tissue sections were first examined by dark-field microscopy to determine structures of interest. Cells with labeled nuclei were counted using a camera lucida attachment. The number of p-Erk-positive neurons in the cingulate cortex were counted; all p-Erk-IR neurons were counted regardless of staining intensity. The investigator responsible for counting p-Erk-IR neurons was blind to the animal's treatment. As no difference was observed between the ipsilateral and contralateral sides of the cingulate cortex, the number of p-Erk-positive cells in the two sides were pooled and averaged. Statistical significance was determined using one-way ANOVA followed by the Student-Newman–Keuls comparison post hoc test. ANOVA results are given as the value of the Fisher distribution Fx,y obtained from the data, where x is the degrees-of-freedom for groups and y is the total degrees-of-freedom of the distribution. Quantification was performed on seven sections per animal (saline-saline, n = 6; saline-CTX, n = 5; CFA-saline, n = 6; CFA-CTX, n = 4).
Results
Peripheral inflammation increases BDNF expression in both the cingulate and somatosensory cortices
Twenty-four hours after CFA injection into the right hindpaw, ELISA titration of BDNF levels revealed a marked increase in the cingulate cortex (Fig. 1A; p < 0.05) of rats with clear-cut inflammatory pain (Fig. 6A–D). A mild increase was also noted in the whole primary somatosensory cortex although this change did not reach statistical significance (Fig. 1B). In contrast, BDNF levels in the insular and motor cortices did not differ in CFA- versus saline-treated rats (Fig. 1C). CFA-induced peripheral inflammation was associated with an increased density of BDNF-immunoreactive cells in the cingulate cortex, both in the ipsilateral (right) and the contralateral (left) sides, compared with control animals (Fig. 1E; p = 0.001 and p < 0.05, respectively). Moreover, we observed a statistically significant increase in the number of BDNF-immunoreactive cells in the contralateral side of the S1HL (Fig. 1E; p < 0.001). Colocalization with markers of different cell types revealed that this BDNF immunoreactivity was expressed in astrocytes, microglial cells, and neurons (Fig. 2). Quality control testing of the antibody used for Western blots showed that it recognizes both mature BDNF and pro-BDNF (data not shown).
Cortical upregulation of BDNF in naive animals induces a sustained cold hypersensitivity and LTP
To directly investigate the role of BDNF in the induction of hypersensitivity, we measured behavioral changes after injection of a lentiviral vector coexpressing BDNF and GFP in the S1HL or the ACC of naive animals. The control group received a version of the viral vector that only expresses GFP. The spatiotemporal expressions of GFP and BDNF were, respectively, visualized by GFP fluorescence (Fig. 3A) and BDNF ELISA titration showed a long-lasting expression of both transgenes from 7 d to 4 weeks postinjection (Fig. 3B). The colocalization studies revealed that this lentiviral vector was transfected in all cell types: astrocytes, microglial cells, and neurons (Fig. 3C–E, respectively).
For both the S1HL and ACC, rats injected with the BDNF-expressing vector displayed symptoms of cold hypersensitivity (Fig. 4A,B,G,H). Whereas BDNF overexpression in the S1HL was most potent to induce cold hyperalgesia, overexpression in the ACC induced cold allodynia. However, neither S1HL nor ACC injections induced any changes in mechanical withdrawal threshold (Figs. 4C,I, respectively). Finally, to decipher the mechanisms underlying the induction of hypersensitivity, we examined the capacity for LTP induction in S1HL. As illustrated in Figure 4, a TBS induced LTP in S1HL from adult rats injected with the BDNF vector; this was measured as an increase in field potentials (FPs) of 116.00 ± 3.53% (Fig. 4D–F; n = 5, p < 0.001), compared with control rats injected with the GFP vector. Interestingly, TBS stimulation performed on CFA-treated animals also enhanced FPs by 109.52 ± 5.06 (Fig. 4F; n = 6, p < 0.05), suggesting that BDNF induces an increased synaptic efficacy, as previously reported (Kang and Schuman, 1995; Messaoudi et al., 1998). Our results also suggest that the S1HL is a site of increased synaptic efficacy. Note that the numerical value of the p given in the legends of figures is the value of the variance analysis. In Figure 4H, although there are overall statistical differences at the ANOVA, there are no individual significances when tested by post hoc tests.
Figure 4.

Upregulation of BDNF expression by injection of lentiviral vector expressing BDNF in the S1 HL or in ACC induced sustained cold hypersensitivity and neuronal hyperexcitability. A–C, Behavioral studies showed that upregulation of BDNF in S1HL induced a cold thermal allodynia trend (A) and cold thermal hyperalgesia (B), but no modification of mechanical sensitivity (C). D–F, Electrophysiological experiments showed a significant increase in LTP in the S1HL of rats injected locally with BDNF expressing vector or in inflamed animal. Representative FPs recorded before (1) and after (2) TBS (D), mean relative FP amplitude of TBS-induced LTP for each experimental group (E, F). G–I, Behavioral studies reveal that upregulation of BDNF in ACC induced a significant cold allodynia (G) and a trend toward cold hyperalgesia (H), but no modification of mechanical sensitivity (I). All data are expressed as mean ± SEM. Statistics: A–C: n = 8 GFP; n = 8 BDNF, two-way ANOVA followed by a Student-Newman–Keuls post hoc test; A: F(1,62) = 3.089, p = 0.085; B: F(1,62) = 3.059, p = 0.018; C: F(1,54) = 0.49, p = 0.487; F, n = 7 GFP, n = 6 BDNF, n = 6 CFA, Mann–Whitney rank sum test, *p < 0.05, **p < 0.01, versus control rats; G–I, n = 6 GFP, n = 6 BDNF, two-way ANOVA followed by a Student-Newman–Keuls post hoc test; G: F(1,50) = 6140, p = 0.017; H: F(1,50) = 5.941, p = 0.019; I: F(1,50) = 0.809, p = 0.373, *p < 0.05 versus control rats. Note that the numerical value of the p value given in the legends of figures is the value of the variance analysis.
rBDNF injection into the ACC of naive animals rapidly induces sustained cold hypersensitivity
We directly injected recombinant BDNF (rBDNF, 1 ng) into the ACC of naive rats. Strikingly, as early as 6 h after rBDNF injection, rats displayed a sustained increase in the cold awareness threshold compared with the control rats (Fig. 5A; p < 0.001). In addition, the cold nociceptive threshold was increased in rBDNF-injected rats. This change reached statistical significance as soon as 1 d postinjection (Fig. 5B; p < 0.05) and persisted until the last observation day (D13; Fig. 5A,B). In contrast, rats injected with rBDNF into the ACC did not display mechanical allodynia (Fig. 5C). These results are consistent with those obtained with the BDNF vector, and clearly show the emergence of cold hypersensitivity in response to elevated levels of BDNF in the ACC. The slow induction of pain brought about by virally induced BDNF raises the question of whether mechanisms upstream of BDNF (such as production and processing of BDNF) or downstream of BDNF (such as changes in neuronal excitability) might be the limiting factor. The long-lasting cold hypersensitivity observed after injection of rBDNF is consistent with previous observations made by Constandil et al. (2011), showing that a single intrathecal injection of 0.003 ng of BDNF was able to decrease the nociceptive threshold (Randall–Selitto test) in normal rats, for at least a 42 d period. Although the half-life of rBDNF is unknown in vivo, it is possible that such effects may not be due to direct effects of BDNF, but rather due to the activation of secondary mechanisms, such as glial-cell activation.
Figure 5.
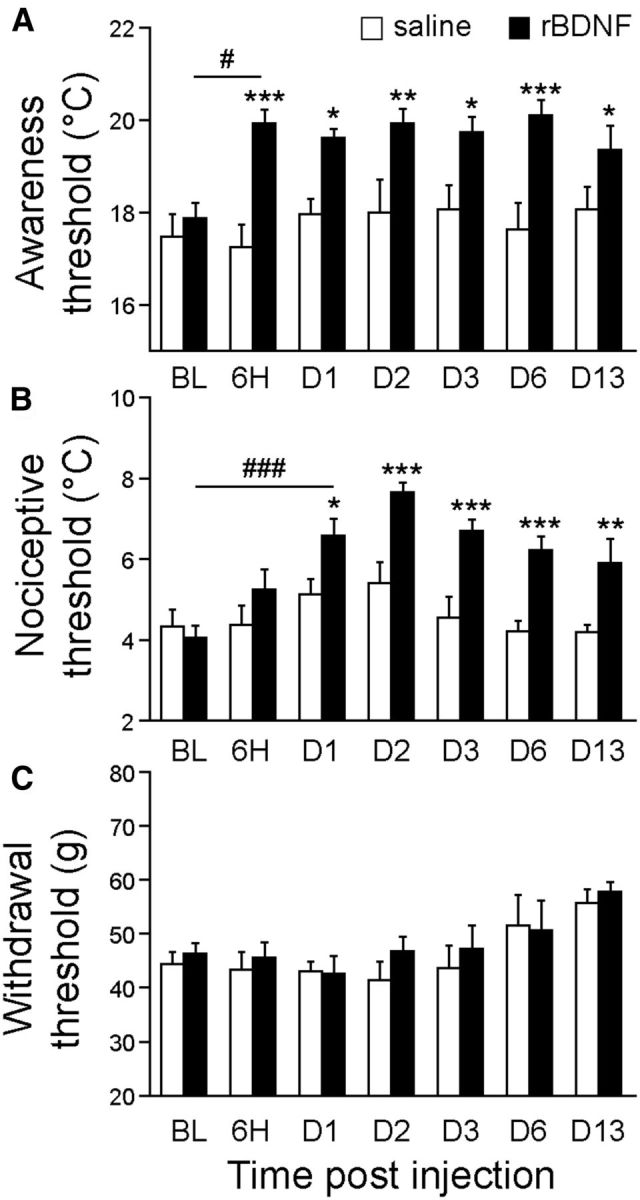
Local injection of recombinant BDNF protein into the cingulate cortex induced sustained cold hypersensitivity. A–C, Behavioral studies indicate that rBDNF injection into ACC induced a significant cold allodynia (A) and a cold hyperalgesia (B) that was maintained for (at least) 13 d postinjection, but no modification of mechanical sensitivity (C); N = 7 GFP, n = 6 rBDNF, two-way ANOVA followed by a Student-Newman–Keuls post hoc test; A: F(1,85) = 53.318, p < 0.001; B: F(1,85) = 44.451, p < 0.001; C: F(1,85) = 1.167, p = 0.282; *p < 0.05, **p < 0.01, ***p < 0.001 versus control rats; #p < 0.05, ###p < 0.001 comparison between different time points.
Intra-ACC injections of CTX-B prevent cold hypersensitivity but not mechanical hypersensitivity in CFA-inflamed rats
As previously reported by other authors (Goff et al., 1998; Allchorne et al., 2005) and herein confirmed, CFA-inflamed rats display cold hypersensitivity (Fig. 6A,B) and mechanical hypersensitivity (Fig. 6C,D). To assess whether the increased expression of BDNF within the ACC could be causally related to the emergence of cold hypersensitivity and mechanical hypersensitivity in CFA-inflamed rats, we blocked the BDNF/TrkB signaling pathway by microinjecting an effective dose (10 μg) of the TrkB antagonist Tat-CTX-B (Cazorla et al., 2010) directly into the ACC. Although this treatment did not affect the thermal and mechanical sensitivity of noninflamed animals (Fig. 6A–D), intra-ACC administration of CTX-B totally prevented the cold allodynia normally observed at 6 h after intraplantar injection of CFA (Fig. 6A; p < 0.05). This protection was maintained for 3 d (Fig. 6A; p < 0.05). In addition, the cold hyperalgesia typically observed 6 h following injection of CFA was also totally prevented by CTX-B administration (Fig. 6B; p < 0.001). However, 2 d after CFA injection, the cold hyperalgesia was only partially blocked (Fig. 6B; p < 0.01 and p < 0.05 for the CFA CTX-B group vs the saline CTX-B group and the CFA saline group, respectively), and 3 d postinjection, the protective effect of CTX-B had disappeared (Fig. 6B; p < 0.01). As soon as 6 h after the intraplantar injection of CFA, rats displayed a noticeable mechanical hypersensitivity of the right hindpaw (ipsilateral side of the injection) that lasted for at least 3 d postinjection (Fig. 6C; p < 0.001). By contrast, no modification of the withdrawal threshold of the contralateral hindpaw was observed (Fig. 6D). Intra-ACC administration of CTX-B did not prevent the mechanical allodynia elicited by CFA-induced inflammation (Fig. 6C; p < 0.001).
Previous studies have shown that the MAP kinase ERK is phosphorylated in several brain areas (such as, the primary and secondary cortices, ACC, insular cortex, rostral ventromedial medulla, and central amygdala) under conditions of noxious stimulation, or inflammatory or neuropathic pain (Carrasquillo and Gereau, 2007; Toyoda et al., 2007; Imbe et al., 2008, 2009; Wei and Zhuo, 2008; Alvarez et al., 2009; Cao et al., 2009a; Thibault et al., 2012). ERK can also be activated by BDNF via TrkB (Klein et al., 1991). In line with these data, we observed a significant increase in the number of pERK-positive cells in the ACC of CFA-inflamed animals compared with the controls (Fig. 6E–G; p < 0.01). This upregulation was totally prevented by an intra-ACC administration of CTX-B (Fig. 6G). Notably, we also observed a colocalization between pERK and BDNF immunolabeling (Fig. 6H), suggesting that the CFA-induced increase in BDNF expression might be responsible for at least some of the induction of ERK phosphorylation in ACC neurons.
Intra-ACC injections of CTX-B prevent the development of passive avoidance behavior
Finally to explore directly our hypothesis of a key role of endogenous BDNF in the ACC in the emotional aspect of pain, we tested the ability of intra-ACC injection of CTX-B to alter the passive avoidance behavior induced by mechanical static stimulation. Using a well described paradigm that was showed to measure the emotional aspect of pain (LaBuda and Fuchs, 2000; LaGraize et al., 2004; Fuchs et al., 2014), we observed that although peripheral stimulation of the inflamed paw (right) induced a passive avoidance of the animals (317% increase in the time spent in the right chamber 24 h after the induction of peripheral inflammation; Fig. 7), this behavior was completely prevented by a single intra-ACC injection of the TrkB antagonist (Fig. 7), confirming that increased BDNF synthesis in the ACC is a key mediator of the emotional aspect of pain.
Figure 7.

Blockade of BDNF/TrkB signaling by intra-ACC injection of TrkB antagonist prevented the passive avoidance behavior induced by an aversive mechanical stimulation in link to peripheral inflammation. Blockade of the BDNF/TrkB signaling pathway by the antagonist CTX-B prevented passive avoidance behavior induced by static mechanical injection of the right paw previously injected with CFA (inflammation treatment parameter: F(1,99) = 14.995, p < 0.001; CTX-B treatment parameter: F(1,99) = 12.463, p < 0.001; interaction between these parameters: F(1,99) = 6.875, p = 0.01). All data are expressed as mean ± SEM; *p < 0.05, **p < 0.01 CFA-saline versus saline-saline rats; §p < 0.05, §§§p < 0.001 CFA-saline versus CFA-CTX-B rats (saline-saline, n = 8; saline-CTX-B, n = 6; CFA-saline, n = 8; CFA-CTX-B, n = 7 animals).
Discussion
This study investigated the putative role of BDNF signaling in cortical plasticity underlying or inducing the perception of pain. Using behavioral, electrophysiological, biochemical, and immunohistochemical approaches in an animal model of inflammatory pain, we demonstrated that BDNF is upregulated in the ACC and the S1 in inflamed rats. Furthermore, while increased BDNF in the ACC of naive rats is sufficient to induce cold hypersensitivity, blockade of BDNF signaling in the ACC of inflamed animals prevents the development of both cold hypersensitivity and avoidance behavior. These findings show that BDNF-dependent neuronal plasticity in the ACC is a key mechanism in the development and maintenance of the affective aspect of pain.
BDNF is an activity-dependent pain modulator in the ACC and the S1HL
Numerous studies have described a network of brain areas activated by experimental painful stimulations, forming a “pain matrix” involved in the different dimensions of pain perception (Treede et al., 1999; Davis, 2000; Peyron et al., 2000; Apkarian et al., 2005; Tracey, 2005; Moisset and Bouhassira, 2007; Hayes and Northoff, 2012). These brain areas include the S1, motor (M), ACC, and insular cortices, as well as the amygdala and the lateral thalamus. Whereas the S1 and M cortical areas and the lateral thalamus are preferentially associated with the sensory-discriminative aspect of pain, the ACC and the anterior insular cortex (which are components of the limbic system) seem to be more involved in the affective-emotional aspect of pain (Davis, 2000; Oertel et al., 2008; Hayes and Northoff, 2012).
The focus of this study was restricted to changes in cortical regions. Among the areas analyzed, we only found increased BDNF expression in the ACC and the S1HL of rats injected with CFA. This compelling result suggests a particular BDNF-related plasticity in these two brain areas. Consistent with these findings, a recent study used a similar inflammatory pain model in mice to demonstrate an enhanced responsiveness of S1 cortical neurons (in layers 2–3), as well as a close link between S1 neuronal activity and AMPA-R-mediated activation of ACC neurons (Eto et al., 2011). Although it still remains to be determined whether this AMPA-R-dependent facilitation of ACC activity by S1 inputs (Eto et al., 2011) involves a direct or an indirect pathway, our results strongly suggest that BDNF could be one possible trigger of this specific plasticity.
Within the S1 cortical area, BDNF-related changes were specifically observed in the S1HL, as previously described in the barrel cortex following whisker stimulation (Rocamora et al., 1996); this observation suggests an activity-dependent change in BDNF that follows cortical somatotopy. This increased BDNF expression was found in both neurons and glial cells of cortical layers 2–3, strengthening the idea that BDNF could be released in an activity-dependent manner in neuronal pathways (Thoenen, 2000), and also synthetized and released locally by glial cells (McNaught and Jenner, 2000; Hutchinson et al., 2009). Indeed, this mechanism was previously described in the spinal cord, where BDNF was shown to affect the synthesis of the potassium chloride cotransporter KCC2, leading to local hyperexcitability and hypersensitivity (Coull et al., 2003, 2005). Very few studies have investigated the functional consequences of increased synaptic plasticity in restricted parts of the S1. It could be postulated that this cortical hyperexcitability is a key element in the long-term development of cortical reorganization and pain chronicization as observed after incomplete spinal cord lesions (Yague et al., 2011) or neuropathic pain (Kim and Nabekura, 2011; Gustin et al., 2012).
The BDNF/TrkB signaling pathway in the ACC enhances the affective aspect of pain
Because the ACC is mainly responsible for the affective component of pain (Rainville et al., 1997; Johansen et al., 2001; LaGraize and Fuchs, 2007), to evaluate the functional role of increased BDNF in the ACC, an appropriate behavior test had to be used to measure the affective aspect of pain. In human studies, the affective (unpleasant) component of persistent pain, such as fear, worry, etc., can easily be dissociated from the sensory component, such as being stabbed, cramped, or pinched (Melzack, 1975). In contrast, it is very difficult in animal studies to precisely define unpleasantness, and the complex paradigms in use often involve cognitive-affective components such as memory and anxiety, which in turn can introduce bias into data interpretation. So far, pain-related escape/avoidance paradigm (Qu et al., 2011; LaBuda and Fuchs, 2000) or a pain-conditioning Pavlovian fear procedure have been used to assess supraspinal areas involved in pain's affect/emotion/aversion (King et al., 2009).
The mechanical hypersensitivity behavioral test that we also used in the present study (electronic Von Frey) is more related to the sensory discriminative component of pain than to its affective component (Sandkühler, 2009). Interestingly, BDNF/TrkB signaling in the ACC did not affect this type of sensitivity.
In contrast, the cold-awareness or cold-nociceptive responses measured in this study (as performed by Allchorne et al., 2005) are not reflex-mediated responses (as performed by Yalcin et al., 2009 using the same apparatus) by scoring a cumulative number of responses at each temperature), but an integrated response. Indeed, we determined two temperature thresholds at which the rat exhibited prototypical behaviors: examination of the floor for the first one (awareness) and paw licking or attempts to escape for the second one (nociceptive). These behaviors are clearly not reflex-mediated responses, but are adaptive coping responses to perceived cold stimuli and we propose that the awareness threshold could be an indirect measure of a component of the emotional aspect of pain. Using this test, we observed a BDNF-induced cold sensitivity, which we propose, could be an increased unpleasantness to a cold stimulation. To further characterize this role of BDNF, a more classical test: the passive avoidance behavior induced by a mechanical aversive stimulus (LaBuda and Fuchs, 2000; LaGraize et al., 2004; Fuchs et al., 2014) was used. It showed that this behavior, that is known to measure the emotional aspect of pain, is prevented when the BDNF signaling pathway was blocked in the ACC, whereas the mechanical sensitivity was not modified. These results therefore strongly support our hypothesis that BDNF upregulation in the ACC potentiates the emotional aspect of cold, although it does not affect mechanical sensitivity. In addition, it suggests that the cold awareness test should be more largely used to measure the emotional adaptation to this cold stimulation (Thibault et al., 2014).
Pain-related plasticity in the cingulate cortex
Our study indicates that BDNF expression is increased in the ACC during inflammation, and is sufficient to induce a series of plastic changes such as LTP and ERK phosphorylation leading to cold hypersensitivity. It is well accepted that the ACC is important in the anticipation of a painful stimulus or feeling the pain applied to another subject (empathy; Ploghaus et al., 1999; Lamm et al., 2011). Several lines of evidence suggest that decreasing ACC activity by meditation or biofeedback reduces pain sensation (deCharms et al., 2005), and has protective effects on changes of brain gray matter and functionality observed in several chronic pain diseases (Grant et al., 2010; Luders et al., 2012). Conversely, animal studies have shown that increased activity in the ACC can increase the emotional aspect of pain (Wei and Zhuo, 2008; Cao et al., 2009b). Measuring LTP, used as an electrophysiological paradigm to assess synaptic efficacy (Toyoda et al., 2007), has allowed demonstrating that peripheral inflammation (Cao et al., 2009a) and nerve damage induce (Wei and Zhuo, 2008) increased synaptic efficacy in the ACC and S1. Finally, several underlying mechanisms that contribute in the ACC to neuronal hyperexcitability and associated sustained pain have been shown to involve astrocytic activation (Chen et al., 2012), adenylyl cyclase 1 (Liauw et al., 2005), calcium calmodulin kinase IV (Wei et al., 2001), the NR2A and 2B subunits of NMDA receptors, the GluA1 type of AMPA receptor (Toyoda et al., 2009), ERK phosphorylation (Toyoda et al., 2007; Cao et al., 2009b), the protein kinase M ζ (Li et al., 2010), and DNA methylation (Tajerian et al., 2013).
To our knowledge, our study is the first to show a facilitatory role of BDNF in the ACC. Using the same inflammatory pain model, Guo et al. (2006) reported that BDNF promotes pain signaling in the rostral ventromedial medulla. This finding is complementary to the present study, as both sets of results suggest a common role for central BDNF as a facilitator of pain in distinct neuroanatomical pathways. In closing, we note that: (1) BDNF is involved in central plasticity linked to memory (Parks et al., 2011), (2) a “memory of pain” contributes to mechanisms underlying chronic pain (Apkarian et al., 2009; Apkarian, 2011; Sandkühler and Lee, 2013), and (3) the anterior cingulate cortex plays a key role in both injury-related unpleasantness in animal models of pain and memory (Tang et al., 2005). We therefore confidently propose that the BDNF-related plasticity herein observed in the ACC could be one such mechanism behind the induction and maintenance of the affective-emotional aspect of pain.
Footnotes
This work was supported by the Centre National pour la Recherche Scientifique (CNRS), the Ecole Supérieure de Physique et Chimie Industrielles de la mairie de Paris, ParisTech, l'Institut UPSA de la Douleur (S.P.) and the Agence Nationale pour la Recherche (M.H.; ANR Contract “TrkBDNFarmod” 2012–2015). We thank Marcel Leopoldie for animal husbandry and Brandon Loveall of Improvence for English proofreading of the paper.
The authors declare no competing financial interests.
References
- Abidin I, Eysel UT, Lessmann V, Mittmann T. Impaired GABAergic inhibition in the visual cortex of brain-derived neurotrophic factor heterozygous knockout mice. J Physiol. 2008;586:1885–1901. doi: 10.1113/jphysiol.2007.148627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allchorne AJ, Broom DC, Woolf CJ. Detection of cold pain, cold allodynia and cold hyperalgesia in freely behaving rats. Mol Pain. 2005;1:36. doi: 10.1186/1744-8069-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Dieb W, Hafidi A, Voisin DL, Dallel R. Insular cortex representation of dynamic mechanical allodynia in trigeminal neuropathic rats. Neurobiol Dis. 2009;33:89–95. doi: 10.1016/j.nbd.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Apkarian AV. The brain in chronic pain: clinical implications. Pain Manag. 2011;1:577–586. doi: 10.2217/pmt.11.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87:81–97. doi: 10.1016/j.pneurobio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 2001;24:47–53. doi: 10.1016/S0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- Cao H, Cui YH, Zhao ZQ, Cao XH, Zhang YQ. Activation of extracellular signal-regulated kinase in the anterior cingulate cortex contributes to the induction of long-term potentiation in rats. Neurosci Bull. 2009a;25:301–308. doi: 10.1007/s12264-009-0904-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Gao YJ, Ren WH, Li TT, Duan KZ, Cui YH, Cao XH, Zhao ZQ, Ji RR, Zhang YQ. Activation of extracellular signal-regulated kinase in the anterior cingulate cortex contributes to the induction and expression of affective pain. J Neurosci. 2009b;29:3307–3321. doi: 10.1523/JNEUROSCI.4300-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Jouvenceau A, Rose C, Guilloux JP, Pilon C, Dranovsky A, Prémont J. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FL, Dong YL, Zhang ZJ, Cao DL, Xu J, Hui J, Zhu L, Gao YJ. Activation of astrocytes in the anterior cingulate cortex contributes to the affective component of pain in an inflammatory pain model 6. Brain Res Bull. 2012;87:60–66. doi: 10.1016/j.brainresbull.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Kim JK, Zhou XF, Rush RA. Increased brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia and spinal cord following peripheral inflammation. Brain Res. 1997;764:269–272. doi: 10.1016/S0006-8993(97)00597-0. [DOI] [PubMed] [Google Scholar]
- Constandil L, Aguilera R, Goich M, Hernández A, Alvarez P, Infante C, Pelissier T. Involvement of spinal cord BDNF in the generation and maintenance of chronic neuropathic pain in rats 5. Brain Res Bull. 2011;86:454–459. doi: 10.1016/j.brainresbull.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De KP, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Davis KD. The neural circuitry of pain as explored with functional MRI. Neurol Res. 2000;22:313–317. doi: 10.1080/01616412.2000.11740676. [DOI] [PubMed] [Google Scholar]
- deCharms RC, Maeda F, Glover GH, Ludlow D, Pauly JM, Soneji D, Gabrieli JD, Mackey SC. Control over brain activation and pain learned by using real-time functional MRI. Proc Natl Acad Sci U S A. 2005;102:18626–18631. doi: 10.1073/pnas.0505210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Wake H, Watanabe M, Ishibashi H, Noda M, Yanagawa Y, Nabekura J. Inter-regional contribution of enhanced activity of the primary somatosensory cortex to the anterior cingulate cortex accelerates chronic pain behavior. J Neurosci. 2011;31:7631–7636. doi: 10.1523/JNEUROSCI.0946-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs PN, Peng YB, Boyette-Davis JA, Uhelski ML. The anterior cingulate cortex and pain processing. Front Integr Neurosci. 2014;8:35. doi: 10.3389/fnint.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng SJ, Liao FF, Dang WH, Ding X, Liu XD, Cai J, Han JS, Wan Y, Xing GG. Contribution of the spinal cord BDNF to the development of neuropathic pain by activation of the NR2B-containing NMDA receptors in rats with spinal nerve ligation. Exp Neurol. 2010;222:256–266. doi: 10.1016/j.expneurol.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Goff JR, Burkey AR, Goff DJ, Jasmin L. Reorganization of the spinal dorsal horn in models of chronic pain: correlation with behaviour. Neuroscience. 1998;82:559–574. doi: 10.1016/S0306-4522(97)00298-4. [DOI] [PubMed] [Google Scholar]
- Grant JA, Courtemanche J, Duerden EG, Duncan GH, Rainville P. Cortical thickness and pain sensitivity in zen meditators. Emotion. 2010;10:43–53. doi: 10.1037/a0018334. [DOI] [PubMed] [Google Scholar]
- Guilbaud G, Benoist JM, Condes-Lara M, Gautron M. Further evidence for the involvement of SmI cortical neurons in nociception: their responsiveness at 24 hr after carrageenin-induced hyperalgesic inflammation in the rat. Somatosens Mot Res. 1993;10:229–244. doi: 10.3109/08990229309028834. [DOI] [PubMed] [Google Scholar]
- Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K. Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. J Neurosci. 2006;26:126–137. doi: 10.1523/JNEUROSCI.3686-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin SM, Peck CC, Cheney LB, Macey PM, Murray GM, Henderson LA. Pain and plasticity: is chronic pain always associated with somatosensory cortex activity and reorganization? J Neurosci. 2012;32:14874–14884. doi: 10.1523/JNEUROSCI.1733-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DJ, Northoff G. Common brain activations for painful and nonpainful aversive stimuli. BMC Neurosci. 2012;13:60. doi: 10.1186/1471-2202-13-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Liu MG, Gong KR, Chen J. Differential effects of long and short train theta burst stimulation on LTP induction in rat anterior cingulate cortex slices: multi-electrode array recordings. Neurosci Bull. 2009;25:309–318. doi: 10.1007/s12264-009-0831-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson AJ, Chou CL, Israel DD, Xu W, Regan JW. Activation of EP2 prostanoid receptors in human glial cell lines stimulates the secretion of BDNF. Neurochem Int. 2009;54:439–446. doi: 10.1016/j.neuint.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbe H, Kimura A, Okamoto K, Donishi T, Aikawa F, Senba E, Tamai Y. Activation of ERK in the rostral ventromedial medulla is involved in hyperalgesia during peripheral inflammation. Brain Res. 2008;1187:103–110. doi: 10.1016/j.brainres.2007.10.075. [DOI] [PubMed] [Google Scholar]
- Imbe H, Okamoto K, Donishi T, Kawai S, Enoki K, Senba E, Kimura A. Activation of ERK in the locus coeruleus following acute noxious stimulation. Brain Res. 2009;1263:50–57. doi: 10.1016/j.brainres.2009.01.052. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Fields HL, Manning BH. The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proc Natl Acad Sci U S A. 2001;98:8077–8082. doi: 10.1073/pnas.141218998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kenan-Vaknin G, Teyler TJ. Laminar pattern of synaptic activity in rat primary visual cortex: comparison of in vivo and in vitro studies employing the current source density analysis. Brain Res. 1994;635:37–48. doi: 10.1016/0006-8993(94)91421-4. [DOI] [PubMed] [Google Scholar]
- Kim SK, Nabekura J. Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J Neurosci. 2011;31:5477–5482. doi: 10.1523/JNEUROSCI.0328-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain 16. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBuda CJ, Fuchs PN. A behavioral test paradigm to measure the aversive quality of inflammatory and neuropathic pain in rats. Exp Neurol. 2000;163:490–494. doi: 10.1006/exnr.2000.7395. [DOI] [PubMed] [Google Scholar]
- LaGraize SC, Fuchs PN. GABAA but not GABAB receptors in the rostral anterior cingulate cortex selectively modulate pain-induced escape/avoidance behavior. Exp Neurol. 2007;204:182–194. doi: 10.1016/j.expneurol.2006.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGraize SC, Labuda CJ, Rutledge MA, Jackson RL, Fuchs PN. Differential effect of anterior cingulate cortex lesion on mechanical hypersensitivity and escape/avoidance behavior in an animal model of neuropathic pain. Exp Neurol. 2004;188:139–148. doi: 10.1016/j.expneurol.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Lamm C, Decety J, Singer T. Meta-analytic evidence for common and distinct neural networks associated with directly experienced pain and empathy for pain. Neuroimage. 2011;54:2492–2502. doi: 10.1016/j.neuroimage.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Lamour Y, Guilbaud G, Willer JC. Altered properties and laminar distribution of neuronal responses to peripheral stimulation in the SmI cortex of the arthritic rat. Brain Res. 1983;273:183–187. doi: 10.1016/0006-8993(83)91111-3. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci U S A. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liauw J, Wu LJ, Zhuo M. Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J Neurophysiol. 2005;94:878–882. doi: 10.1152/jn.01205.2004. [DOI] [PubMed] [Google Scholar]
- Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, Shim J, Lee K, Collingridge GL, Kaang BK, Zhuo M. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science. 2010;330:1400–1404. doi: 10.1126/science.1191792. [DOI] [PubMed] [Google Scholar]
- Lin YT, Ro LS, Wang HL, Chen JC. Up-regulation of dorsal root ganglia BDNF and trkB receptor in inflammatory pain: an in vivo and in vitro study. J Neuroinflammation. 2011;8:126. doi: 10.1186/1742-2094-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- Luders E, Phillips OR, Clark K, Kurth F, Toga AW, Narr KL. Bridging the hemispheres in meditation: thicker callosal regions and enhanced fractional anisotropy (FA) in long-term practitioners. Neuroimage. 2012;61:181–187. doi: 10.1016/j.neuroimage.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty S, Berzaghi M, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci. 1997;20:198–202. doi: 10.1016/S0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Dysfunction of rat forebrain astrocytes in culture alters cytokine and neurotrophic factor release. Neurosci Lett. 2000;285:61–65. doi: 10.1016/S0304-3940(00)00982-4. [DOI] [PubMed] [Google Scholar]
- Melzack R. The McGill pain questionnaire: major properties and scoring methods. Pain. 1975;1:277–299. doi: 10.1016/0304-3959(75)90044-5. [DOI] [PubMed] [Google Scholar]
- Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R. BDNF as a pain modulator. Prog Neurobiol. 2008;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Messaoudi E, Bârdsen K, Srebro B, Bramham CR. Acute intrahippocampal infusion of BDNF induces lasting potentiation of synaptic transmission in the rat dentate gyrus. J Neurophysiol. 1998;79:496–499. doi: 10.1152/jn.1998.79.1.496. [DOI] [PubMed] [Google Scholar]
- Moisset X, Bouhassira D. Brain imaging of neuropathic pain. Neuroimage. 2007;37:S80–S88. doi: 10.1016/j.neuroimage.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Narita M, Yajima Y, Aoki T, Ozaki S, Mizoguchi H, Tseng LF, Suzuki T. Up-regulation of the TrkB receptor in mice injured by the partial ligation of the sciatic nerve. Eur J Pharmacol. 2000;401:187–190. doi: 10.1016/S0014-2999(00)00449-0. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K. BDNF in sensory neurons and chronic pain. Neurosci Res. 2006;55:1–10. doi: 10.1016/j.neures.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Oertel BG, Preibisch C, Wallenhorst T, Hummel T, Geisslinger G, Lanfermann H, Lötsch J. Differential opioid action on sensory and affective cerebral pain processing. Clin Pharmacol Ther. 2008;83:577–588. doi: 10.1038/sj.clpt.6100441. [DOI] [PubMed] [Google Scholar]
- Parks EL, Geha PY, Baliki MN, Katz J, Schnitzer TJ, Apkarian AV. Brain activity for chronic knee osteoarthritis: dissociating evoked pain from spontaneous pain. Eur J Pain. 2011;15:e1–e14. doi: 10.1016/j.ejpain.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in strereotaxic coordinates. San Diego: Academic; 2006. [Google Scholar]
- Peyron R, Laurent B, García-Larrea L. Functional imaging of brain responses to pain: a review and meta-analysis (2000) Neurophysiol Clin. 2000;30:263–288. doi: 10.1016/S0987-7053(00)00227-6. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Pezet S, Onténiente B, Grannec G, Calvino B. Chronic pain is associated with increased TrkA immunoreactivity in spinoreticular neurons. J Neurosci. 1999;19:5482–5492. doi: 10.1523/JNEUROSCI.19-13-05482.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploghaus A, Tracey I, Gati JS, Clare S, Menon RS, Matthews PM, Rawlins JN. Dissociating pain from its anticipation in the human brain. Science. 1999;284:1979–1981. doi: 10.1126/science.284.5422.1979. [DOI] [PubMed] [Google Scholar]
- Qu C, King T, Okun A, Lai J, Fields HL, Porreca F. Lesion of the rostral anterior cingulate cortex eliminates the aversiveness of spontaneous neuropathic pain following partial or complete axotomy. Pain. 2011;152:1641–1648. doi: 10.1016/j.pain.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968–971. doi: 10.1126/science.277.5328.968. [DOI] [PubMed] [Google Scholar]
- Rocamora N, Welker E, Pascual M, Soriano E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci. 1996;16:4411–4419. doi: 10.1523/JNEUROSCI.16-14-04411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Sandkühler J, Lee J. How to erase memory traces of pain and fear. Trends Neurosci. 2013;36:343–352. doi: 10.1016/j.tins.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu XQ, Llinas A, Mendell LM. Effects of trkB and trkC neurotrophin receptor agonists on thermal nociception: a behavioral and electrophysiological study. Pain. 1999;80:463–470. doi: 10.1016/S0304-3959(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc Natl Acad Sci U S A. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, Szyf M, Stone LS. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One. 2013;8:e55259. doi: 10.1371/journal.pone.0055259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Tang J, Ko S, Ding HK, Qiu CS, Calejesan AA, Zhuo M. Pavlovian fear memory induced by activation in the anterior cingulate cortex. Mol Pain. 2005;1:6. doi: 10.1186/1744-8069-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault K, Calvino B, Dubacq S, Roualle-de-Rouville M, Sordoillet V, Rivals I, Pezet S. Cortical effect of oxaliplatin associated with sustained neuropathic pain: exacerbation of cortical activity and down-regulation of potassium channel expression in somatosensory cortex. Pain. 2012;153:1636–1647. doi: 10.1016/j.pain.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Thibault K, Calvino B, Rivals I, Marchand F, Dubacq S, McMahon SB, Pezet S. Molecular mechanisms underlying the enhanced analgesic effect of oxycodone compared to morphine in chemotherapy-induced neuropathic pain 1. PLoS One. 2014;9:e91297. doi: 10.1371/journal.pone.0091297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. doi: 10.1016/S0079-6123(00)28016-3. [DOI] [PubMed] [Google Scholar]
- Toyoda H, Zhao MG, Xu H, Wu LJ, Ren M, Zhuo M. Requirement of extracellular signal-regulated kinase/mitogen-activated protein kinase for long-term potentiation in adult mouse anterior cingulate cortex. Mol Pain. 2007;3:36. doi: 10.1186/1744-8069-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Zhao MG, Ulzhöfer B, Wu LJ, Xu H, Seeburg PH, Sprengel R, Kuner R, Zhuo M. Roles of the AMPA receptor subunit GluA1 but not GluA2 in synaptic potentiation and activation of ERK in the anterior cingulate cortex. Mol Pain. 2009;5:46. doi: 10.1186/1744-8069-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey I. Functional connectivity and pain: how effectively connected is your brain? Pain. 2005;116:173–174. doi: 10.1016/j.pain.2005.05.034. [DOI] [PubMed] [Google Scholar]
- Treede RD, Kenshalo DR, Gracely RH, Jones AK. The cortical representation of pain. Pain. 1999;79:105–111. doi: 10.1016/S0304-3959(98)00184-5. [DOI] [PubMed] [Google Scholar]
- Wei F, Zhuo M. Activation of Erk in the anterior cingulate cortex during the induction and expression of chronic pain. Mol Pain. 2008;4:28. doi: 10.1186/1744-8069-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M. Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci. 2001;4:164–169. doi: 10.1038/83993. [DOI] [PubMed] [Google Scholar]
- Woodward WR, Chiaia N, Teyler TJ, Leong L, Coull BM. Organization of cortical afferent and efferent pathways in the white matter of the rat visual system. Neuroscience. 1990;36:393–401. doi: 10.1016/0306-4522(90)90434-6. [DOI] [PubMed] [Google Scholar]
- Yague JG, Foffani G, Aguilar J. Cortical hyperexcitability in response to preserved spinothalamic inputs immediately after spinal cord hemisection. Exp Neurol. 2011;227:252–263. doi: 10.1016/j.expneurol.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Yalcin I, Charlet A, Freund-Mercier MJ, Barrot M, Poisbeau P. Differentiating thermal allodynia and hyperalgesia using dynamic hot and cold plate in rodents. J Pain. 2009;10:767–773. doi: 10.1016/j.jpain.2009.01.325. [DOI] [PubMed] [Google Scholar]