

Abstract
Synapsins (Syns) are synaptic vesicle (SV)-associated proteins involved in the regulation of synaptic transmission and plasticity, which display a highly conserved ATP binding site in the central C-domain, whose functional role is unknown.
Using molecular dynamics simulations, we demonstrated that ATP binding to SynI is mediated by a conformational transition of a flexible loop that opens to make the binding site accessible; such transition, prevented in the K269Q mutant, is not significantly affected in the absence of Ca2+ or by the E373K mutation that abolishes Ca2+-binding. Indeed, the ATP binding to SynI also occurred under Ca2+-free conditions and increased its association with purified rat SVs regardless of the presence of Ca2+ and promoted SynI oligomerization. However, although under Ca2+-free conditions, SynI dimerization and SV clustering were enhanced, Ca2+ favored the formation of tetramers at the expense of dimers and did not affect SV clustering, indicating a role of Ca2+-dependent dimer/tetramer transitions in the regulation of ATP-dependent SV clustering.
To elucidate the role of ATP/SynI binding in synaptic physiology, mouse SynI knock-out hippocampal neurons were transduced with either wild-type or K269Q mutant SynI and inhibitory transmission was studied by patch-clamp and electron microscopy. K269Q-SynI expressing inhibitory synapses showed increased synaptic strength due to an increase in the release probability, an increased vulnerability to synaptic depression and a dysregulation of SV trafficking, when compared with wild-type SynI-expressing terminals. The results suggest that the ATP-SynI binding plays predocking and postdocking roles in the modulation of SV clustering and plasticity of inhibitory synapses.
Keywords: ATP binding, inhibitory transmission, molecular dynamics simulations, synapsins, synaptic ultrastructure, synaptic vesicles
Introduction
Synapsins (Syns) are synaptic vesicle (SV)-associated phosphoproteins implicated in the regulation of neurotransmitter release that play important roles in the control of SV trafficking through phosphorylation-dependent interactions with actin and SVs (Chi et al., 2001, 2003; Cesca et al., 2010; Rizzoli, 2014). The best-described function of Syns is the control of neurotransmitter release by clustering SVs and tethering them to the actin cytoskeleton, thus maintaining the integrity of SV pools (Cesca et al., 2010). Moreover, Syns appear to control postdocking events of SV exocytosis, such as priming and fusion, and implicitly, the size of the readily releasable pool (RRP; Hilfiker et al., 1998; Gitler et al., 2004; Fassio et al., 2006; Baldelli et al., 2007; Coleman and Bykhovskaia, 2010; Lignani et al., 2013; Medrihan et al., 2013). Mutant mice in which one or more Syn isoforms are deleted are all prone to epileptic seizures, with the exception of Syn III knock-out (KO) mice, and display an autistic like phenotype (Cesca et al., 2010; Fassio et al., 2011a; Greco et al., 2013). The epilepsy propensity stems from excitation/inhibition imbalances triggered by a primary impairment of inhibitory transmission that is particularly vulnerable to the absence of Syns (Baldelli et al., 2007; Farisello et al., 2013) and several mutations involving the SYN1 and SYN2 genes were found to be associated with epilepsy and autism in humans (Garcia et al., 2004; Fassio et al., 2011b; Corradi et al., 2014).
Presynaptic terminals are believed to be major sites of ATP production and consumption by the activity-driven SV cycling and the resting intraterminal ATP concentration is in the micromolar range (Rangaraju et al., 2014). All Syns isoforms display a major and highly conserved ATP binding site in the central C-domain. The crystal structure of domain C (residues 110–420) of bovine SynI (SynI C) has been resolved (Esser et al., 1998) revealing that SynC monomers are structurally similar to ATP-using enzymes such as glutathione synthase. Although the ATP site is highly conserved, the binding of ATP was reported to be differentially regulated by Ca2+ions in the three main Syn isoforms, being respectively stimulated, unaffected or inhibited by Ca2+ in recombinant Syns I, II, and III expressed in prokaryotic cells (Hosaka and Südhof, 1998). However, the functional role of ATP binding to Syns has not been investigated thus far, except for an increase in SynI tetramer formation upon binding of ATP and Ca2+ (Brautigam et al., 2004).
Here, we studied the effects of ATP in the regulation of SynI binding to SVs, SynI oligomerization and SV clustering, as well as the effects of a SynI mutant constitutively unable to bind ATP on the ultrastructure and function of inhibitory synapses. We found that ATP does not require Ca2+ to bind to SynI, although its binding is enhanced by Ca2+. Moreover, ATP strengthens SynI association with SVs, favors the formation of high order oligomers, and increases SV clustering. Genetic inactivation of the ATP binding site in SynI increased the strength of evoked inhibitory transmission, and the release probability (Pr), associated with an accelerated synaptic depression and a dysregulation of SV trafficking.
Materials and Methods
Materials
Male Sprague-Dawley rats and C57BL/6J mice of either sex were from Charles River. SynI KO mice were generated by homologous recombination (Chin et al., 1995) and extensively backcrossed on the C57BL/6J background for >10 generations. All experiments were performed in accordance with the guidelines established by the European Communities Council (Directive 2010/63/EU of September 22, 2010) and were approved by the Italian Ministry of Health. SynI was purified from bovine brain as described previously (Bähler and Greengard, 1987). The antibodies used were as follows: anti-VGAT (Rabbit, 1:500, no. 131013), anti-VGLUT1 (Guinea Pig, 1:1000, no. 135304) from Synaptic Systems; anti-mCherry (Mouse, 1:500, no. 632543) from Diatech Lab Line, Clontech; peroxidase-conjugated goat anti-rabbit and anti-mouse antibodies from Bio-Rad; polyclonal anti-SynI and anti-synaptophysin antibodies (raised in our laboratory); phosphorylation-state-specific antibodies recognizing phosphorylated sites 1, 3, and 4,5 of SynI (generated in Dr Greengard's laboratory, The Rockefeller University, New York, NY); phosphorylation-state-specific antibodies recognizing phosphorylated site 7 of SynI from Invitrogen. ECL Plus Western blotting reagent and Kodak X-Omat films were from GE Healthcare. The cross-linking agent disuccinimidyl suberate (DSS) was from Pierce Biotechnology and was dissolved in dimethylsulfoxide (DMSO). The protease inhibitor cocktail used throughout included phenyl-methyl-sulfonyl fluoride (0.1 mm), pepstatin (1 μg/ml), and leupeptin (1 μg/ml) was from Sigma-Aldrich. All other chemicals were obtained from Sigma-Aldrich.
MD simulations
All molecular dynamics (MD) simulations were performed using the program NAMD (Phillips et al., 2005) and the CHARMM27 force field with the CMAP correction for protein backbone energetics (Mackerell et al., 2004). Protein atomic coordinates were obtained from the crystal structure of rat SynI C-domain complexed with ATP and Ca2+ (PDB code 1pk8; Brautigam et al., 2004). The different simulated systems were built by immersing the protein monomer in a box of TIP3P model water molecules (Jorgensen et al., 1983) and counter-ions to neutralize the total charge, for a total of 63,000 atoms in all cases. ATP or Ca2+ were removed where necessary by deleting the corresponding atoms and the mutations were introduced by replacing the side-chains of the involved residues. In all simulations, periodic boundary conditions were used to replicate the system and remove box surface effects (Frenkel and Smit, 2001). Short-range nonbonded interactions were cutoff at 12 Å, whereas long-range electrostatic interactions were computed using the particle mesh Ewald method (Darden et al., 1993). Chemical bonds connecting hydrogen atoms to heavy atoms were kept fixed using SHAKE (Ryckaert et al., 1977). The integration time step was 1 fs to ensure stability of the dynamics algorithm. A proper size for the simulation box corresponding to a pressure of 1 atm was obtained by simulating the system in the constant pressure and temperature ensemble using the Nosé-Hoover Langevin piston method for 0.5 ns (Martyna et al., 1994). The simulation ensemble was then switched to constant volume and temperature (or canonical) ensemble for the rest of the simulation by keeping the temperature stationary ∼300 K using Langevin dynamics. Each full simulation run lasted 150 ns. Free Energy landscapes were obtained by calculating the two-dimensional function as follows:
 |
where d1, d2 are two variables functions of the atomic coordinates of the system and usually called reaction coordinates, kB is the Boltzmann constant, T the temperature (300 K), and h indicates the histogram of the two variables computed from the MD simulations and approximating their probability distributions. In the present case, we used as variables the distance between the backbone centers of mass of the multifunctional loop (MFL) residues 336–338 and residue 315, and the root mean square distance (RMSD) of the MFL with respect to its conformation in the crystal structure.
ATP-γ35S binding assays
Purified SynI (500 nm) was incubated with 20 nm ATP-γ35S (PerkinElmer) in 50 mm HEPES, 25 mm NaCl, 0.1 mm EGTA, pH 7.4, for 1 h at room temperature in the absence or presence of 2.1 mm Ca2+ (Hosaka and Südhof, 1998) and of increasing concentrations (0.1–30 μm) of cold ATP as described previously (De Franchi et al., 2010). Samples were spotted onto squares of nitrocellulose membrane (2.0 × 2.0 cm; 0.2 μm pores; Whatman GE Healthcare Life Science) that were extensively washed with deionized water for 30 min, air-dried, and analyzed for radioactivity by using the PerkinElmer Cyclone Plus Phosphor Imager. After subtraction of the background values (samples incubated with radioactive ATP in the absence of SynI), data from individual inhibition curves were fitted with the following dose–response four-parameter inhibition function:
where x is the logarithm of the cold ATP concentration, y0 the amount of ATP-γ35S bound in the absence of cold ATP, ymin the nonspecifically bound ATP-γ35S at the highest cold ATP concentration, IC50 is concentration yielding 50% inhibition of specific binding and HS is the Hill slope using Prism 6.0 (GraphPad Software). Data in the plots are the means ± SEM of at least five independent experiments.
SV purification and binding assays
Purification of SVs from rat brain was performed through the step of controlled-pore glass chromatography (Huttner et al., 1983). Untreated SVs (USVs) containing endogenous SynI were quantitatively depleted of SynI by dilution (10 μg of protein/ml) in 0.15 m glycine, 0.2 m NaCl, 2.5 mm HEPES, pH 7.4, immediately after elution from the column and incubation for 2 h in ice. After incubation, salt-treated SVs (SSVs) were recovered by high-speed centrifugation and resuspended in 0.3 m glycine/5 mm HEPES, pH 7.4 (glycine buffer) at a protein concentration of 1 mg/ml.
Binding of purified SynI.
SSV (10 μg/sample in a final volume of 100 μl) depleted of endogenous SynI were incubated for 1 h at 0°C with purified SynI (50 nm) in the absence or presence of ATP (0.25 mm), GTP (0.25 mm), and/or Ca2+ (2.1 mm) in a buffer containing 0.3 m glycine, 40 mm NaCl, 5 mm HEPES, 0.1 mm EGTA, pH 7.4, and 100 μg/ml bovine serum albumin. After incubation, SV-bound SynI was separated by high-speed centrifugation (400,000 × g for 45 min) through a 10% (w/w) sucrose cushion.
Dissociation of endogenous SynI.
USVs (10 μg/sample in a final volume of 100 μl) containing endogenous SynI were incubated for 1 h at 0°C in the absence or presence of ATP (0.25 mm), GTP (0.25 mm), and/or Ca2+ (2.1 mm) under high ionic strength conditions (0.3 m glycine, 200 mm NaCl, 5 mm HEPES, 0.1 mm EGTA, pH 7.4) in the presence of protease inhibitors, to trigger dissociation of endogenous SynI. After incubation, USVs were recovered by high-speed centrifugation (400,000 × g for 45 min). Aliquots of the resuspended pellets were subjected to SDS-PAGE and the amounts of SynI were determined by immunoblotting with anti-SynI antibodies. The recovery of SVs in the pellets, used to correct the amounts of bound SynI, was evaluated by immunoblotting with anti-synaptophysin antibodies.
Crosslinking of SynI oligomers
The formation of SynI oligomers was assessed by crosslinking experiments using DSS. Briefly, 300 nm of purified SynI dialyzed in 200 mm NaCl, 20 mm NaPO4, 0.1 mm EGTA, pH 7.4, was incubated with 0.5 mm DSS dissolved in DMSO or with DMSO alone in the absence or presence of ATP (0.25 mm), GTP (0.25 mm), and/or Ca2+ (2.1 mm) for 1 h at room temperature. The reaction was stopped by the addition of 10 mm glycine, pH 7.4, and the cross-linked complexes were separated by SDS-PAGE on 6% (w/v) polyacrylamide gels, transferred to nitrocellulose membranes, and detected by immunoblotting with polyclonal anti-SynI antibodies.
Primary cultures of hippocampal neurons
Primary neuronal cultures were prepared from the hippocampi of day 18 embryos from SynI KO mice as described previously (Lignani et al., 2013). The pregnant mice were killed with CO2 and cervical dislocation; embryos were removed after cesarean section and decapitated. Skulls were opened and brains were dissected out and placed into Hank's Balanced Salt Solution (HBSS). Hippocampi were removed under a dissecting microscope and collected. After 15 min of incubation with 0.25% trypsin in HBSS at 37°C, whole hippocampi were washed with HBSS to remove trypsin and then mechanically dissociated. Neurons stained with a vital dye (Trypan blue; Sigma-Aldrich) were counted by using a Burker chamber. Neurons were plated on poly-l-lysine (0.1 mg/ ml; Sigma-Aldrich)-treated 25 mm glass coverslips at a density of 80,000 cells per coverslip (low-density cultures). Cells were plated and maintained in culture medium consisting of Neurobasal, B-27 (1:50 v/v), glutamax (1% w/v), and penicillin-streptomycin (1%) at 37°C in a 5% CO2 humidified atmosphere. All experiments were performed on mature neurons >14 d in vitro (DIV).
Viral transduction procedures
The mCherry-tagged WT- and K269Q-SynI were digested from the GFP backbone and ligated into PGK no. 743 (pCCL-sin-PPT-prom-EGFP-Wpre) lentiviral backbone, using AgeI and SalI sites. All constructs were sequenced. The plasmid pCCL-sin-PPT-prom-EGFP-Wpre was a kind gift from M. Amendola (Tiget, Milano, Italy). The production of VSV-pseudotyped third-generation lentiviruses was performed as previously described (De Palma and Naldini, 2002). Primary hippocampal neurons were infected at 7–11 DIV at 10 multiplicity-of-infection. After 24 h, half of the medium was replaced with fresh medium. All experiments were performed 7 d postinfection (between 14 and 18 DIV). Viral titers (infectious particles) were determined by transduction of HEK293T cells with serial dilution of the viral stocks and evaluation of transduction efficiency by fluorescence intensity.
Immunocytochemistry
Primary SynI KO hippocampal neurons infected at 7 DIV with lentiviral vectors expressing WT-SynI or K269Q-SynI were fixed at 14 DIV with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, for 20 min at room temperature. After several washes in PBS, they were permeabilized and blocked for 30 min in 5% normal goat serum (NGS), 0.1% saponin in PBS and then incubated with primary antibodies diluted in 5% NGS and 0.1% saponin in PBS up to 2 h. Coverslips were then washed twice in 0.1% saponin in PBS and blocked for 10 min in 5% NGS and 0.1% saponin in PBS before being incubated in the same buffer with AlexaFluor-conjugated secondary antibodies (1:500; Invitrogen). After several washes in PBS, coverslips were mounted using Prolong Gold antifade reagent with DAPI staining (Invitrogen). Primary antibodies were omitted to control for the specificity of the staining and of the acquisition procedure. Images were acquired using a 63× objective in a Leica SP5 confocal. Acquired images were analyzed using ImageJ software.
Patch-clamp recordings
Patch electrodes, fabricated from thick borosilicate glass (Kimble Chase), were pulled and fire-polished to a final resistance of 5–7 MΩ. Whole-cell patch-clamp recordings from postsynaptic neurons were performed using an Axon Multiclamp 700B/Digidata1440A system (Molecular Devices) and an upright BX51WI microscope (Olympus) equipped with Nomarski optics. Neurons at 7–11 DIV were infected with lentiviral vectors expressing WT-SynI, K269N-SynI or the empty vector and experiments were performed 7 d postinfection on inhibitory neurons. An extracellular electrode connected to an isolated pulse stimulator (A-M Systems) was used to stimulate release from the presynaptic compartment. Recordings with either leak currents >100 pA or series resistance >15 MΩ were discarded. Experiments were performed at 22–24°C. Evoked IPSCs (eIPSCs) were recorded in extracellular solution (Tyrode solution) containing the following (in mm): 140 NaCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 4 KCl, 10 glucose, pH 7.3. To selectively analyze inhibitory transmission, D-AP5 (50 μm), CNQX (10 μm), and CGP58845 (5 μm) were added to block NMDA, non-NMDA, and GABAB receptors, respectively. A minimal stimulation approach was applied using a 1 μm tip pipette filled with Tyrode solution close to an interneuron body in loose-patch configuration. Current pulses of 0.1/0.3 ms and variable amplitude between 10 and 50 μA were delivered to find the minimal current required to record a postsynaptic response without failures and with an invariant latency and shape during the 0.1 Hz stimulation protocol. To study the response to paired-pulse protocols, we applied two consecutive stimuli at increasing interpulse intervals (25–2000 ms). To analyze fast-depression kinetics, we applied a high-frequency short train (1 s at 40 Hz) and normalized the eIPSC amplitude to the average eIPSC amplitude obtained by stimulating afferent fibers at 0.1 Hz (baseline). Asynchronous release was calculated as the 1 s integral of the release after the end of the 1 s at 40 Hz short train and was normalized to the first eIPSC area of the train. The Pr was calculated with cumulative analysis as previously described (Schneggenburger et al., 1999; Lignani et al., 2013). Synaptic depression was induced by stimulating neurons with a train at 10 Hz for 30 s and by normalizing the eIPSC amplitude to the baseline.
Electron microscopy
Negative staining of purified SVs.
Purified SVs, incubated under the various experimental conditions, were diluted to 0.02 μg/μl and spotted on glow discharged carbon film-coated copper EM grids (EMS). Grids with SVs were incubated for 2 min in a solution of 1% uranyl acetate, dried, and observed with a JEM-1011 transmission electron microscope (Jeol) operating at 100 kV and equipped with an ORIUS SC1000 CCD camera (Gatan). For each experimental condition, at least eight images of SVs were acquired at 10,000× magnification (sampled area per experimental condition: 36 μm2). The SV position was manually determined using ImageJ software and the coordinates were used to evaluate the degree of SV clustering using the software CrimeStat (A Spatial Statistics Program for the Analysis of Crime Incident Locations v3.3 Ned Levine and Associates, Houston, TX, and the National Institute of Justice, Washington, DC). The mean nearest neighbor distance between SVs was calculated and a nearest neighbor hierarchical spatial clustering analysis was performed by entering two predetermined parameters for length of the searching radius to scan the dataset area in search of clusters (0.2 μm) and minimum number of points to build a cluster (Lignani et al., 2013).
Conventional transmission electron microscopy.
Primary cultured hippocampal neurons derived from SynI KO embryos infected at 7–11 DIV with either WT-SynI or K269Q-SynI were fixed at 14–18 DIV with 1.2% glutaraldehyde in 66 mm sodium cacodylate buffer, postfixed in 1% OsO4, 1.5% K4Fe(CN)6, 0.1 m sodium cacodylate, en bloc stained with 1% uranyl acetate, dehydrated, and flat embedded in epoxy resin (Epon 812, TAAB). After baking for 48 h, the glass coverslip was removed from the Epon block by thermal shock. Neurons were identified by means of a stereomicroscope, excised from the block, and mounted on a cured Epon block for sectioning using an EM UC6 ultramicrotome (Leica Microsystem). Ultrathin sections (60–70 nm thick) were collected on copper mesh grids and observed with a JEM-1011 electron microscope as described above. Inhibitory synapses (symmetric, gray type 2) were recognized based on their ultrastructural features including parallel pre-postsynaptic membranes and absence of a postsynaptic density or postsynaptic spine. Synaptic profile area, SV number, and distribution relative to the active zone (AZ) were determined using ImageJ software. The analysis of synaptic ultrastructure at the end of the train stimulation protocol (30 s at 10 Hz) or after recovery (120 s at 0.1 Hz) was performed by fixing the samples with glutaraldehyde at 37°C. Under these conditions, we estimated a complete fixation of synapses within 1–3 s from fixative addition (Leung, 1994).
Ultrastructural determination of Pr.
Primary hippocampal neurons derived from SynI KO embryos infected at 7–11 DIV with either WT-SynI or K269Q-SynI were field stimulated at 14–18 DIV (7 d postinfection) with a train of 30 current pulses (1 ms; 20 μA) at 1 Hz with an insulated pulse stimulator (A-M Systems). Experiments were performed in Tyrode's buffer containing 50 μm APV, 10 μm CNQX, 30 μm bicuculline, and 10 mg/ml soluble horseradish peroxidase (HRP). After the stimulus, neurons were fixed for electron microscopy in 1.2% glutaraldehyde in 66 mm sodium cacodylate buffer. After fixation neurons were washed in 0.1 m sodium cacodylate and incubated for 10 min in a solution containing 0.3 mg/ml of diaminobenzedine (DAB) in 0.1 m sodium cacodylate buffer and then developed in a solution containing 0.3 mg/ml DAB and 0.003% H2O2 in 0.1 m sodium cacodylate buffer until a brown substrate was formed. Neurons were then rinsed in cold dH2O, washed in 0.1 m sodium cacodylate, and then postfixed in 1% OsO4, 1.5% K4Fe(CN)6, 0.1 m sodium cacodylate. After lipid fixation, the standard TEM sample preparation protocol was followed and samples were embedded in Epon resin. Serial 60-nm-thick sections were collected on carbon-coated copper slot formvar and carbon-coated grids, and serial synaptic profiles acquired. Serial sections were aligned using the Midas of IMOD. Inhibitory synapses with one single AZ, at least one HRP-positive SV and >100 but <800 total SVs were reconstructed with the software IMOD, and the total number of HRP-positive and docked SVs were calculated.
Pre-embedding immunogold SynI localization.
Primary SynI KO hippocampal neurons were infected at 7–11 DIV with the two lentiviral vectors and fixed at 14–18 DIV with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, for 30 min at room temperature, permeabilized and incubated with anti-SynI primary antibodies (mouse, 1:500 Synaptic Systems). Neurons were then incubated with a secondary antibody (goat anti mouse, 1:100, Nanoprobes) conjugated to a colloidal nanogold particle (1.4 nm). Neurons were fixed for 1 h in 1.2% glutaraldehyde in PBS and then the nanogold particles size was enhanced using a gold enhancement kit (GoldEnhance, Nanoprobes) to allow signal detection. Neurons were postfixed in 1% osmium tetroxide in 0.1 m sodium cacodylate, pH 7.4, en block stained in uranyl acetate, dehydrated in a graded series of ethanol and embedded in Epon. Bona fide symmetric inhibitory synapses were considered and distances between gold particles and AZ was measured with ImageJ software.
Miscellaneous techniques
Protein concentrations were determined by the Bradford or BCA assays. SDS-PAGE was performed according to Laemmli (1970). Immunoblotting was performed using peroxidase-conjugated secondary antibodies coupled with the ECL chemiluminescence detection system. Immunoblots were quantified by densitometric analysis of the fluorograms using ImageJ software. Data were analyzed by unpaired Student's t test or, in case of more than two experimental groups, by one-way ANOVA followed by multiple-comparison tests using Prism 6.0 (GraphPad Software). Significance level was preset to p < 0.05. Data are expressed as means ± SEM throughout.
Results
The presence of Ca2+ potentiates, but it is not necessary for, the binding of ATP to SynI
The available crystal structure of bovine and rat SynI C complexed with Ca2+ and ATP (Esser et al., 1998; Brautigam et al., 2004) reveals that the protein can form dimers and tetramers, and that the so-called MFL (residues 330–343) is essential for binding ATP. In the crystal structure, the MFL establishes contacts with ATP and shields the ligand and its binding pocket from the solvent. Since such conformation blocks ATP access to the active site, it has been previously reasoned that it should be acquired only after binding (Brautigam et al., 2004). To further investigate the mechanism of ATP binding to SynI and its sensitivity to the external environment and the mutation of critical residues, we performed MD simulations of the native protein (WT-SynI), as well as of the two mutants K269Q-SynI and E373K-SynI in the presence (holo) or absence (apo) of Ca2+/ATP or ATP alone (Fig. 1). In accordance with the structural observations, our simulations proved that binding of ATP to the SynI C-domain is mediated by a conformational transition of the MFL that has to open to let the ligand reach its binding site. The conformational freedom of the MFL under the various conditions was characterized by calculating the Free Energy landscape (or potential of mean force) as a function of: (1) the loop opening measured by its distance from the protein core; and (2) the loop distortion, measured by the RMSD from its conformation in the crystal structure (Brautigam et al., 2004). The holo-SynI Free Energy landscape when both ATP and Ca2+ are bound (Fig. 1A) showed a single narrow basin indicating limited MFL conformational divergence from the crystal structure (Fig. 1A, inset). Conversely, in apo-SynI (absence of Ca2+/ATP; Fig. 1B), a much broader landscape with two distinct basins separated by a low free-energy barrier was observed. The new basin corresponds to MFL conformations that are quite different from those in the crystal structure and in the holo-simulation, in which the loop extended away from the core of the protein toward the solvent, thus making the binding pocket accessible (Fig. 1B, inset). Notably, we observed a large conformational flexibility of the MFL also in the K269Q-SynI (Fig. 1F). This is a consequence of the perturbation introduced by the mutated residue that weakens the interaction between SynI and ATP, eventually causing the binding pocket to open (Fig. 1F, inset). Conversely, a reduced mobility of the MFL was observed in WT-SynI in absence of Ca2+ (Fig. 1C) and in the E373K-SynI mutant (Fig. 1D,E) in which the acidic residue necessary for Ca2+ coordination is replaced by a basic residue, a condition similar to what found in the SynII isoform. These simulated conditions and the observed perturbations of protein-ligand interactions through destabilizing/stabilizing contacts predict that the binding of ATP to SynI is poorly Ca2+-dependent and that the amount of ATP bound to the protein primarily depends on the ligand concentration and the integrity of the ATP binding pocket.
Figure 1.
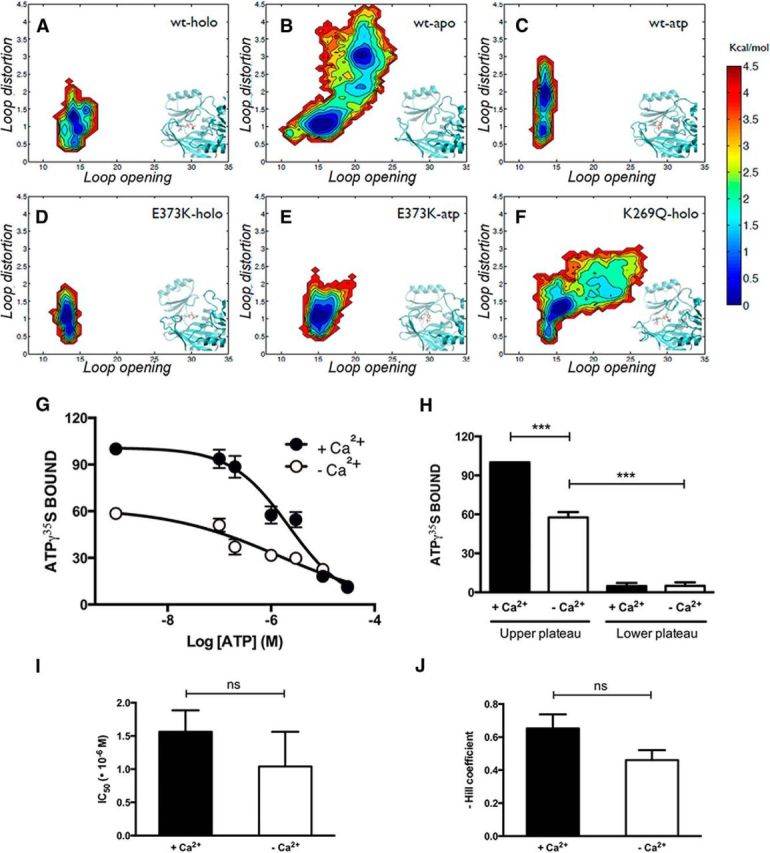
The presence of Ca2+ potentiates, but it is not necessary for, the binding of ATP to SynI. A–F, Free energy surfaces describing the conformations of the MFL calculated from MD simulations are shown. The reaction coordinates used are the distance between the centers of mass of the MFL residues 336–338 and residue 315 (x-axis), and the RMSD of the MFL with respect to its conformation in the crystal structure (y-axis). Results for the following systems are shown: WT-SynI with ATP and Ca2+ (holo; A), WT-SynI without ATP or Ca2+(apo; B), WT-SynI with ATP but no Ca2+ (atp; C),E373K mutant with ATP and Ca2+ (D), E373K mutant with ATP and no Ca2+(E), and K269Q mutant with ATP and Ca2+ (F). Insets, Representative conformations of the protein region in the vicinity of the MFL and the ATP binding site are shown. G, Inhibition of ATP-γ35S (20 nm) binding to purified bovine SynI (500 nm) by increasing concentrations of cold ATP in the absence (open symbols) or presence (closed symbols) of Ca2+ (2.1 mm). The amount of bound ATP-γ35S is expressed in percentage of the binding in the absence of cold ATP and presence of Ca2+. Points in the plot are mean ± SEM of five independent experiments. H–J, Individual inhibition curves were fitted as described in Materials and Methods. Percentage of the amount of bound ATP-γ35S at the upper and lower plateau (H), IC50 values of cold ATP (I), and Hill slope (J) of the fitted functions in the absence (open bars) or presence (solid bars) of Ca2+ are shown as mean ± SEM. Statistical analysis was performed using the Student's t test or one-way ANOVA followed by the Bonferroni's multiple-comparison test; ***p < 0.001; ns, not significant.
To confirm the predictions made by MD simulations, we directly investigated the Ca2+-dependence of ATP binding to bovine SynI purified under nondenaturing conditions. To this aim, we set up an ATP-γ35S binding assay using nitrocellulose membranes to immobilize purified SynI and determined the dose-dependent ability of cold ATP to competitively inhibit the binding of the radioactive ligand (Fig. 1G). In agreement with the predictions, we found that ATP is able to bind SynI under Ca2+-free conditions, but that the presence of Ca2+ positively modulates the ATP binding. Indeed, the Ca2+-independent binding of ATP was ∼60% of the maximal binding measured in the presence of 2.1 mm Ca2+ (Fig. 1H). These data suggest that Ca2+ increases the accessibility of the ATP binding site in SynI, as no significant changes in either IC50 values for cold ATP inhibition (a measure of the binding affinity) or in the Hill slope of inhibition were observed (Fig. 1I,J).
ATP binding increases the association of SynI with SVs
The binding of SynI to SVs is variably modulated by site-specific phosphorylation of SynI (for review, see Cesca et al., 2010). As ATP can bind SynI in either the absence or presence of Ca2+, we studied the effects of such interactions on the binding to purified SVs in vitro. We monitored both the dissociation of endogenous SynI from USVs upon exposure to high ionic strength (Fig. 2A) and the binding of purified SynI to SynI-depleted SVs (SSVs; Fig. 2B) in the presence or absence of ATP, GTP, and/or Ca2+. The presence of ATP significantly decreased the dissociation of SynI from USVs, and consistently, significantly increased the binding of purified SynI to SSVs compared with the control conditions, whereas GTP was completely ineffective. Notably, the enhancement of SynI binding to SVs was closely similar in the absence or presence of Ca2+, suggesting that the amounts of ATP bound under Ca2+-free conditions are sufficient to elicit the effect.
Figure 2.

ATP binding to SynI increases the association with SVs in a Ca2+-independent fashion and promotes SV clustering. A, Purified native USVs were incubated in the absence or presence of ATP, GTP, and/or Ca2+ under high ionic strength conditions as described in Materials and Methods and assayed for the dissociation of endogenously bound SynI. After high-speed centrifugation, the amounts of bound SynI and the SV recovery in the pellets were determined by immunoblotting with anti-SynI and anti-synaptophysin antibodies. Top, Immunoblot of a representative experiment. Bottom, Quantification of the ATP dependence of SynI recovery in the SV pellet after high salt-induced dissociation was performed by densitometric analysis of the fluorograms and is shown as mean ± SEM of n = 6 independent experiments. Statistical analysis was performed using one-way ANOVA followed by the Bonferroni's multiple-comparison test versus the respective control; **p < 0.01. B, Purified bovine SynI was incubated with SSVs in the absence or presence of ATP, GTP, and/or Ca2+ as described in the Materials and Methods. Top, Immunoblot of a representative experiment. Bottom, The extent of SynI binding to SVs is expressed as mean ± SEM of n = 6 independent experiments; **p < 0.01; one-way ANOVA followed by Dunnett's multiple-comparison test versus the respective control. C, Purified native SVs (USV) and purified bovine SynI (SYN) were incubated under the conditions used for the SV binding assays in the absence or presence of ATP and/or Ca2+. After incubation, samples were analyzed by immunoblotting for the level of total and site-specific phosphorylated SynI (1P-SynI: PKA site 1; 3P-SynI: CaMKII site 3; 4,5P-SynI: MAPK/ERK sites 4,5; 7P-SynI: Cdk5 site 7). No significant changes in the phosphorylation state of SynI were detected with purified SVs or purified SynI under the conditions tested. D, Left, Representative immunoblot of equal amounts of purified USVs and SSVs analyzed for the presence of SynIa/b, SynIIa/b, and synaptophysin. The dilution in high salt medium of USVs led to almost quantitative dissociation of SynI from the SVs, whereas SynII and the integral membrane protein synaptophysin were unaffected. Right, Representative transmission electron micrographs of negatively stained native USVs or SynI-depleted SSVs incubated under physiological conditions in the absence or presence of ATP and/or Ca2+ as indicated. Scale bar, 200 nm. E, The number of SV clusters (left) and the percentage of SVs in clusters (right) from n = 5 (USVs) and n = 3 (SSVs) independent SV preparations are reported as mean ± SEM; **p < 0.01; one-way ANOVA followed by Dunnett's multiple-comparison test versus the control sample (no ATP, no Ca2+).
As many SynI functions are modulated by phosphorylation, and since some of the protein kinases phosphorylating SynI have been reported to associate with SVs, we verified that the observed effects on SynI binding to SVs were not attributable to an indirect effect of SynI phosphorylation activated by the presence of ATP and/or Ca2+. Thus, we checked the samples used for binding studies by performing immunoblotting with phosphorylation state-specific antibodies recognizing SynI phosphorylated by PKA/CaM kinase I, CaM kinase II, MAPK/ERK, and Cdk5. No significant differences in the degree of SynI phosphorylation was detectable in the various experimental conditions (Fig. 2C), demonstrating that the observed effects on SV binding were solely attributable to the binding of ATP to SynI.
SynI, by binding to the SV membrane, is known to mediate SV clustering, likely by the activity of Syn oligomers (Shupliakov et al., 2011). Thus, we tested whether the effect of ATP on SV binding was associated with an increase in SV clustering by analyzing aliquots of native and SynI-depleted SVs (USVs and SSVs, respectively) used in the binding assays by negative staining and electron microscopy (Fig. 2D). We observed that, when USVs containing endogenous SynI were incubated in the presence of ATP, the clustering of SVs was significantly higher than in those incubated under control conditions both in terms of number of clusters and percentage of clustered SVs with respect to the total SV counts (Fig. 2E). Unexpectedly, the presence of Ca2+ was not associated with changes in clustering with respect to control conditions (Fig. 2E).
To ascertain that the observed effects were attributable to the ATP-binding activity of SynI, we repeated the experiments with SynI-depleted SSVs (Fig. 2D,E). SSVs were dramatically less clustered than their parent USVs in terms of both number of clusters and percentage of clustered SVs with respect to the total SV counts (Fig. 2E), consistent with the previously reported role of SynI in SV clustering (Benfenati et al., 1993; Monaldi et al., 2010). Interestingly, the quantitative depletion of the sole SynI in SSVs totally abolished the effect of ATP on SV clustering, excluding SynI-independent effects of ATP on SV clustering (Fig. 2D,E).
ATP binding regulates the formation of SynI high order oligomers
Synaptic vesicle clustering and maintenance of SV pools is believed to occur, at least partly, through oligomerization of SynI (Benfenati et al., 1993; Hosaka and Südhof, 1999; Monaldi et al., 2010). Thus, using chemical cross-linking, we asked whether binding of ATP to SynI was able to differentially affect its oligomerization state in the presence or absence of Ca2+. We found that binding of ATP induced a significant increase in the formation of SynI oligomers (i.e., dimers and tetramers) and a corresponding decrease in SynI monomers (Fig. 3A). The effect was specific for ATP, as GTP was completely ineffective. Interestingly, binding of ATP under Ca2+-free conditions favored the formation of SynI dimers from monomers. On the other hand, when ATP was bound in the presence of Ca2+, a significant shift toward higher order oligomers (tetramers) was observed, consistent with previous results (Brautigam et al., 2004) and suggesting a direct effect of both ATP and Ca2+ binding to SynI on its oligomerization properties.
Figure 3.
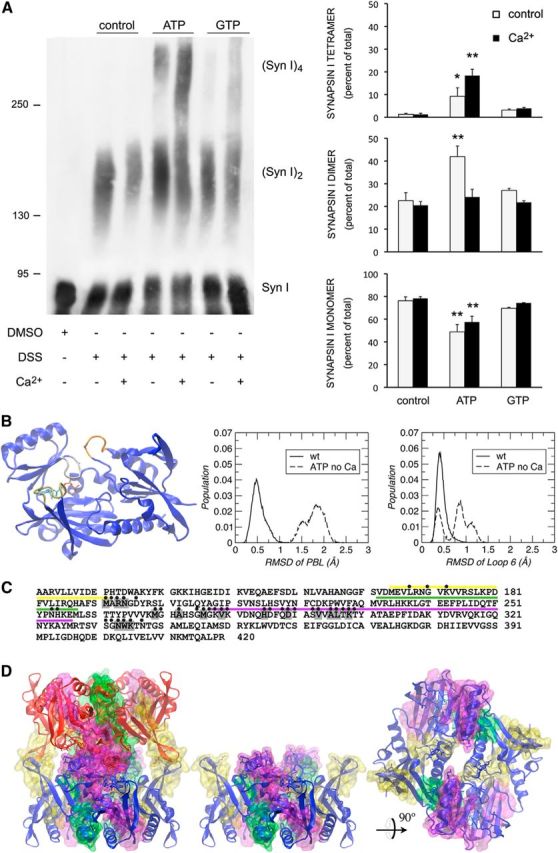
ATP and Ca2+ differentially regulate the oligomerization of SynI. A, Purified bovine SynI, incubated in the absence or presence of ATP, GTP and/or Ca2+, was subjected to chemical cross-linking with DSS dissolved in DMSO. Samples were resolved by SDS-PAGE on 6% (w/v) polyacrylamide gels and developed by immunoblotting with anti-SynI antibodies. Left, Immunoblot of a representative experiment. Right, Quantification of the stoichiometric ratios between SynI monomer (Syn I), dimer [(Syn I)2], and tetramer [(Syn I)4] in the absence or presence of ATP, GTP, and/or Ca2+. The relative intensities of the immunoreactivity of SynI species are shown in percentage of total sample immunoreactivity and are expressed as mean ± SEM of n = 5 independent experiments; *p < 0.05; **p < 0.01; one-way ANOVA followed by Dunnett's multiple-comparison test versus the respective control. B, Left, Pictorial representation of SynI C domain monomer. The MFL is colored in gold, the PBL in gray and the loop 6 in orange. ATP and Ca2+ are represented as sticks and white sphere, respectively. Middle, Histograms of RMSD values for the PBL in wt and ATP/no-Ca2+. Right, Histograms of RMSD values for the loop 6 in wt and ATP/no-Ca2+. C, SynI C domain primary sequence highlighting the overlaps between tetramer interface amino acids and SV membrane-interacting domains (Cheetham et al., 2001). Residues identified with a black circle are involved in the tetramer interface between dimers. Residues highlighted in gray establish direct contacts between the dimers. The membrane-interacting regions are capped with yellow, green, and purple lines. D, Left, Pictorial representation of the SynI C domain tetramer. Middle and Right, Pictorial representation of the SynI C domain dimer. The two main dimers described previously (Esser et al., 1998) are colored blue and red and depicted as ribbons. The membrane-interacting domains are drawn as transparent surfaces and colored according to C. The residues that are part of the tetramer buried interface are represented as sticks. The rotated view of the dimer on the right shows the surface buried upon tetramerization. It can be seen that a large portion of the membrane-interacting domains is part of the tetramer interface, particularly in the purple surface region (residues 278–327).
To further investigate the functional consequences of the transitions between monomer, dimer and tetramer, we analyzed the behavior of the tetramer interface residues in our MD simulations. In particular, we compared the WT protein trajectory of the system with ATP and no Ca2+. The tetramer interface residues belong to the MFL, the phosphate binding loop (PBL), the helix α4 and the loop 6 right before helix α1, all regions that are close to the ATP binding site (for helices and loops numeration, see Esser et al., 1998; Brautigam et al., 2004; Fig. 3B, left). From the comparisons shown in Figure 1(A vs C), we observed that, although the MFL remains closed also when only ATP is bound, its conformation is more distorted than in the WT. Similarly, we observed that the absence of Ca2+ induces conformational distortions in other regions of the tetramer interface, the PBL (Fig. 3B, middle) and the loop 6 before helix α1 (Fig. 3B, right). Because these regions are part of the tetramer interface, their distortion might result in the perturbation of the intramonomer contacts that stabilize the oligomer. Upon tetramerization, the SynI C domain buries a total surface area of ∼11,200 Å2, where 4010 Å2 come from forming the tetramer out of dimers (Brautigam et al., 2004). The tetramer assemblage shows an extensive packing of the regions close to the ATP binding site in each monomer, with the different MFLs also participating in the interface. By comparing the molecular surface area of the tetramer and the isolated dimers, we identified 37 residues per monomer as part of the buried interface (these amino acids are indicated with black circles along the protein sequence in Fig. 3C and represented as sticks in Fig. 3D). Among the 37 residues, 22 belong to the three protein domains that were previously proved to interact with the SV membrane (Fig. 3C, yellow, green, and purple capped residue; D, consistently colored surfaces; Cheetham et al., 2001). This amounts to ∼60% of all buried residues and ∼21% of the SV interacting regions. Among the three segments, the one that contains the highest number of tetramer interface residues is the stretch 278–327 (Fig. 3D, purple surface), where 32% of the amino acids get buried (Fig. 3D). Finally, the tetramer interface shows 16 residues per monomer that establish direct contacts with residues from the facing dimer; eight of these are in the membrane-interacting domains and seven belong to the 278–327 segment.
In conclusion, we observe that a relevant portion of membrane-interacting residues gets buried in the tetramer surface, consistent with a decreased capability of ATP/Ca2+-induced tetramers to bind SV with respect to ATP alone-induced dimers. The deduced lower efficacy of the tetramer in cross-linking SVs with respect to the dimer explains the increased SV clustering observed with ATP in the absence of Ca2+ and its normalization upon exposure to ATP/Ca2+ (Fig. 2D).
The ATP binding mutant K269Q-SynI alters SynI distribution within nerve terminals without affecting its phosphorylation or the density of excitatory and inhibitory synapses
Because ATP binding was found to modulate the association of SynI with SVs and their clustering status, we wondered whether a loss-of-function SynI mutant (K269Q-SynI) lacking the ATP binding site expressed in SynI KO neurons could impact SV trafficking, synaptic transmission, and short-term plasticity. Previous data and our MD simulations demonstrated that the point K269Q mutant of SynI (Hosaka and Südhof, 1999; Fig. 1F) is unable to bind ATP. Thus, we used this mutant to functionally investigate the role of SynI ATP binding site in intact neurons. First we used Western blotting with anti-SynI antibodies to verify the expression of WT-Syn and K269Q-SynI in primary SynI KO hippocampal neurons at 14–18 DIV (7 d after infection) and found that the expression levels of WT and mutant SynI were closely similar, confirming the validity of the viral constructs and the infection efficiency (Fig. 4A).
Figure 4.

The ATP binding mutant alters SynI distribution within nerve terminals without affecting its phosphorylation or the density of excitatory and inhibitory synapses. A, Expression of WT-SynI and K269Q-SynI. Primary hippocampal neurons from SynI KO mice were transduced at 7 DIV with lentiviral vectors encoding either WT-SynI or K269Q-SynI and lysed at 14 DIV. Top, Cell extracts were analyzed by SDS-PAGE, transferred to nitrocellulose membranes and developed by immunoblotting with anti-SynI antibodies. An immunoblot from a representative experiment is shown. Tubulin immunoreactivity was used to control equal loading. Quantification of the SynI immunoreactivity was performed by densitometric analysis of the fluorograms and is shown as mean ± SEM of n = 3 independent experiments in the bottom. B, Basal and activity-dependent phosphorylation of WT-hSynI and K269Q-SynI in SynI KO hippocampal neurons. Neurons, transduced as in A, were solubilized in SDS with phosphatase inhibitors under either basal conditions or immediately after electrical field stimulation at 10 Hz for 30 s. Left, Samples were analyzed by immunoblotting for the level of total and site-specific phosphorylated SynI (1P-SynI: PKA site 1; 3P-SynI: CaMKII site 3; 4,5P-SynI: MAPK/ERK sites 4,5; 7P-SynI: Cdk5 site 7). Tubulin immunoreactivity was used as control for equal loading. Right, Immunoreactivity for phosphorylated and total SynI was quantified by densitometric analysis of the fluorograms. Site-specific phosphorylation of SynI, expressed as the ratio between the phospho-specific immunoreactivity and the total SynI immunoreactivity, is shown as mean ± SEM of n = 3 independent experiments. No significant changes in the phosphorylation state of K269Q-SynI at all phospho-sites were observed with respect to WT-SynI both under basal conditions and in response to electrical stimulation; *p < 0.05; **p < 0.01, one-way ANOVA followed by the Bonferroni's multiple-comparison test versus the respective control. C, Expression of mCherry-labeled WT- or K269Q-SynI and their distribution in excitatory and inhibitory terminals labeled with vGLUT1 and vGAT antibodies, respectively (left). In the bar graphs on the right, the quantification of the density of vGLUT and vGAT-positive puncta is shown as means ± SEM (n = 6 and 19 for WT- and K269Q-SynI, respectively). Scale bar, 20 μm. D, Left, Representative transmission electron micrographs of inhibitory presynaptic terminals from SynI KO neurons expressing an empty vector (SynI KO), WT-SynI or K269Q-SynI on which a pre-embedding immunogold localization of SynI was performed. Right, Gold particle distance from AZ was decreased in the K269Q-SynI expressing synapse, suggesting a mislocalization of mutant-SynI toward the AZ, as shown in the bar plot and in the XY SynI distribution plot. Data are shown as means ± SEM; **p < 0.01 unpaired Student's t test (the number of independent coverslips analyzed was 18 and 39 for WT-SynI and K269Q-SynI, respectively). Scale bar, 200 nm.
Although the point mutation is somewhat far from the major activity-dependent phosphorylation sites, the lack of ATP binding could potentially affect the interactions of mutant Syn I with kinases and thereby alter its association with SVs. To ascertain this possibility, we grew hippocampal neurons under the same conditions used for the electrophysiology/EM studies, subjected them to the same protocol of high-frequency electrical stimulation (30 s at 10 Hz) and harvested them under basal conditions and immediately after the train. Neuronal extracts were then probed with phosphorylation state-specific antibodies to investigate SynI phosphorylation at site 1 (phosphorylated by PKA and CaM kinases I/IV), sites 2/3 (phosphorylated by CaM kinase II), sites 4, 5 (phosphorylated by MAPK/ERK), and site 7 (phosphorylated by Cdk5). As expected, the phosphorylation state of wild-type SynI was low at sites 1, 2, 3 and constitutively high at sites 4, 5, 7 under basal conditions, whereas it increased at sites 1 and 2/3 and decreased at sites 4, 5, 7 upon high-frequency stimulation, in agreement with the activation of CaM kinases phosphorylating sites 1, 2, 3 and of protein phosphatase 2B (calcineurin) dephosphorylating sites 4, 5, 7 upon Ca2+ build-up (Cesca et al., 2010). Notably, the phosphorylation of the ATP binding mutant K269Q-SynI was indistinguishable from that of WT-SynI both under basal conditions and in response to electrical stimulation, ruling out off-target effects of the mutation on presynaptic function (Fig. 4B).
We also performed immunocytochemistry to analyze the patterns of expression of the mutant protein in excitatory and inhibitory synapses. Both WT-SynI and mutant SynI were correctly translated and targeted to both excitatory (vGLUT1-positive) and inhibitory (vGAT-positive) presynaptic terminals with a comparable expression pattern (Fig. 4C, left). Also, the density of excitatory (vGLUT1-positive) and inhibitory (vGAT-positive) terminals was similar in WT-SynI and K269Q-SynI expressing neurons, demonstrating that the mutant does not significantly affect neuronal development and synaptogenesis (Fig. 4C, right).
To assess whether the lack of ATP binding to K269Q-SynI had any effect on the distribution of mutant SynI in nerve terminals, we performed an ultrastructural analysis by pre-embedding immunogold electron microscopy with anti-SynI antibodies in resting inhibitory synapses (Fig. 4D, left). As expected, WT-SynI was specifically enriched at presynaptic terminals with a distribution comparable to that reported for the endogenous protein (Tao-Cheng, 2006) and was characterized by fewer metal particles located near the plasma membrane, as compared with those located in the more central region of the synapse. Interestingly, we found that SynI immunoreactivity was partially mislocalized in K269Q-SynI expressing inhibitory terminals, being closer to the AZ (Fig. 4D, right) than WT-SynI. This result might be interpreted in light of the in vitro observations of a reduced ability of SynI to associate with SV clusters in the absence of ATP that may favor a partial translocation toward the AZ.
The ATP binding mutant K269Q-SynI increases release probability in inhibitory synapses
Since the main phenotype of SynI KO mice is a primary impairment of synaptic inhibition (Baldelli et al., 2007), and because inhibitory modulation of network activity can be finely modulated by ATP (Bhattacharya et al., 2013), we investigated the functional role of ATP-binding to SynI in inhibitory synapses from low-density cultures of SynI KO hippocampal neurons that were transduced with mCherry alone, WT-SynI or K269Q-SynI by performing electrophysiological experiments to analyze synaptic transmission and short-term plasticity.
Infection of SynI KO neurons with WT-SynI rescued the decrease of the eIPSCs amplitude observed in SynI KO cells. Surprisingly, the eIPSC amplitude in K269Q-SynI expressing inhibitory synapses was significantly larger than in neurons infected with WT-SynI (Fig. 5A). Such effect was accompanied by a significant decrease in the coefficient-of-variation (Fig. 5C), suggesting a likely increase in the Pr of inhibitory synapses expressing mutant SynI. No changes in miniature IPSC (mIPSC) frequency and amplitude were observed (frequency: 0.98 ± 0.15, 1.00 ± 0.18, 1.02 ± 0.12; amplitude: 1.04 ± 0.11, 1.00 ± 0.09, 0.96 ± 0.08; n = 9, 11, and 12 neurons; for synapses expressing mCherry, WT-SynI, or K269Q-SynI, respectively; data were normalized to the mean values of frequency, 0.8 Hz, and amplitude, 34 pA, observed in the WT-SynI group), excluding postsynaptic effects or changes in synaptic connectivity (Fig. 4).
Figure 5.
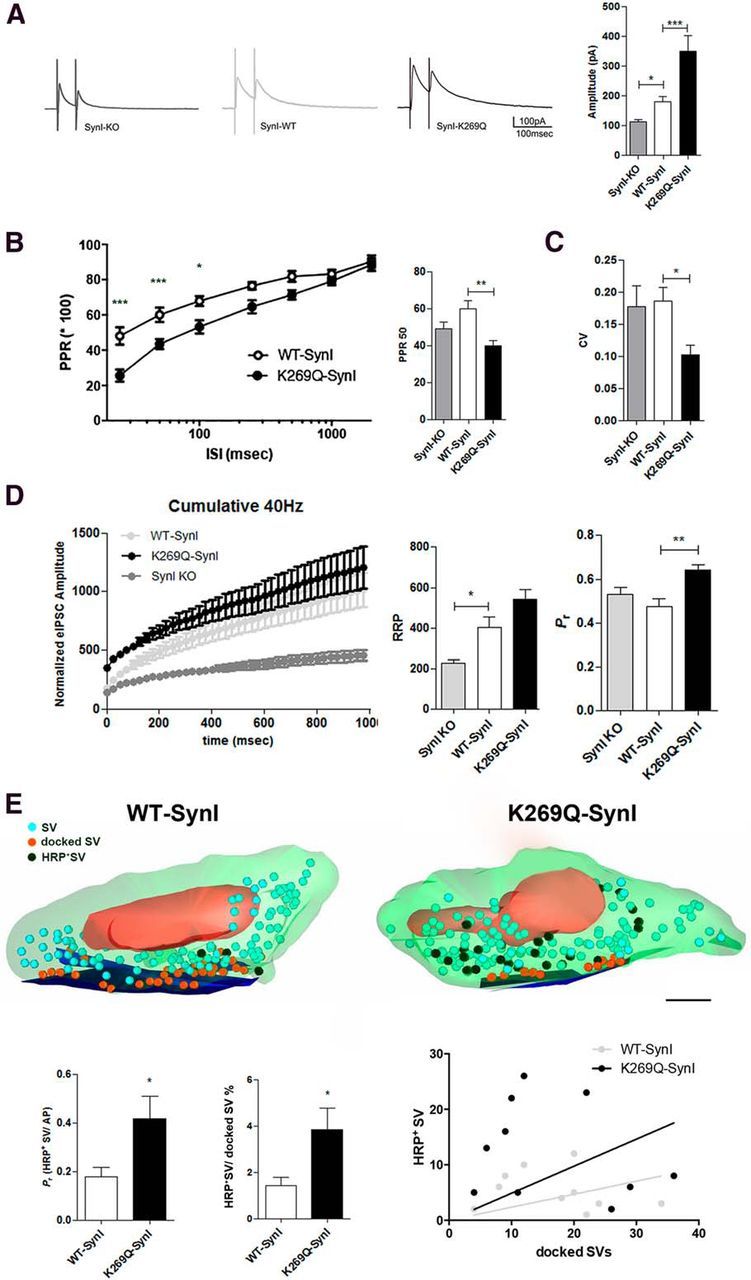
Expression of mutant K269Q-SynI increases inhibitory release probability. A, Patch-clamp recordings of eIPCSs. Averaged traces of eIPSCs are shown for SynI KO (dark gray trace), WT-SynI (light gray trace), and K269Q-SynI (black trace) synapses; bars on the right indicate the mean (± SEM) eIPSC amplitude (n = 16, 25, and 12 for SynI KO, WT-SynI, and K269Q-SynI, respectively); *p < 0.05, ***p < 0.001; one-way ANOVA followed by Fischer's LSD test. B, Paired pulse protocols were applied to inhibitory synapses transduced with either WT-SynI (open symbols) or K269Q-SynI (closed symbols) at ISIs ranging from 25 to 2000 ms. The mean (± SEM) PPR was calculated and plotted against ISI. Expression of K269Q-SynI caused a strong reduction of paired pulse depression in inhibitory synapses at short ISIs (n = 14 and 10 for WT- and K269Q-SynI, respectively); *p < 0.05; ***p < 0.001; two-way ANOVA followed by the Bonferroni's multiple-comparison test versus WT-SynI. The bar plot on the right reports the mean (± SEM) PPR at 50 ms ISI for the three experimental groups (n = 13, n = 14, and n = 10 for SynI KO, WT-SynI, and K269Q-SynI, respectively); **p < 0.01 one-way ANOVA followed by Dunnett's multiple-comparison test versus WT-SynI. C, Bars indicate the mean (± SEM) coefficient of variation of eIPSCs recorded from SynI KO (gray bars), WT-SynI (white bars), or K269Q-SynI (black bars; n = 12, 20, and 11 for SynI KO, WT-SynI, and K269Q-SynI, respectively); *p < 0.05; one-way ANOVA followed by Dunnett's multiple-comparison test versus WT-SynI. D, A brief stimulation train (1 s at 40 Hz) was used to study the quantal properties of synchronous release in inhibitory synapses transduced with mCherry (SynI KO; dark gray; n = 8), WT-SynI (light gray; n = 12), or K269Q-SynI (black; n = 10). The cumulative curves of eIPSC amplitude (in nA; left) were built, and the linear part was fit as described in Materials and Methods. In the bar plots on the right, the RRP for synchronous release and the Pr estimated from the cumulative curves are shown as mean ± SEM; *p < 0.05; **p < 0.01, one-way ANOVA followed by Dunnett's multiple-comparison test versus WT-SynI. E, The SV Pr was estimated by electron microscopy analysis of HRP uptake in serial sections of primary hippocampal neurons after stimulation at 1 Hz for 30 s in the presence of extracellular HRP. Representative three-dimensional reconstructions from 60-nm-thick serial sections obtained from WT-SynI (left) and K269Q-SynI (right) expressing inhibitory synapses. Recycled SVs are easily recognizable in the images due to their electron-dense lumen. In the three-dimensional reconstructions total SVs, HRP-positive SVs, and physically docked SVs are depicted as light blue, black, and orange spheres, respectively. The AZ is given in blue. Scale bar, 200 nm. The number of total SVs, physically docked SVs, and HRP-labeled SVs that underwent exo-endocytosis was quantified. The bar plots in the bottom represent the number of HRP-positive SVs per synapse divided by the number of stimuli (AP), which gives an estimation of Pr and the number of HRP-positive SVs per synapse divided by the number of physically docked SVs. In the correlation plot on the right, the number of HRP-labeled SVs are plotted against the number of physically docked SVs for terminals expressing either WT-SynI (open bars/light gray symbols) or K269Q-SynI (black bars/symbols), and fitted by linear regression (n = 10 for both WT- and K269Q-SynI); *p < 0.05, unpaired Student's t test.
To study the response to paired-pulse stimulation, synapses were subjected to two stimuli applied at interstimulus intervals (ISI) ranging from 25 ms to 2 s. At short ISIs (25–100 ms), all K269Q-SynI expressing synapses displayed a significant decrease of the paired-pulse ratio (PPR; Fig. 5B) consistent with an increase in Pr.
To demonstrate the effects of the ATP site mutation on the quantal parameters of release of inhibitory synapses, we performed cumulative analysis of release during a stimulation train for 1 s at 40 Hz, as previously described (Schneggenburger et al., 1999; Baldelli et al., 2007; Lignani et al., 2013). This analysis showed that both WT-SynI and K269Q-SynI significantly rescued the depressed size of RRP in SynI KO neurons and that, according to the above suggestions, the K269Q-SynI mutant, but not the WT-SynI, specifically and significantly increased the initial Pr (Fig. 5D).
As the conditions assumed by the cumulative analysis (i.e., that the tetanus should completely deplete the RRP bringing the Pr toward one and that recycling rate should remain constant for the whole duration of the train) may not be fully satisfied, we wanted to obtain a fully independent evaluation of Pr. An ultrastructural readout of the average Pr during a fixed number of APs was previously proposed (Fernández-Alfonso and Ryan, 2004; Branco et al., 2008, 2010; Lignani et al., 2013). The method is based on the assumption that the ratio between exocytosis and endocytosis ≤1 Hz stimulation is close to one, making it possible to determine the number of SVs that fuse in response to each stimulus by using extracellular markers, such as soluble HRP, which is internalized upon SV endocytosis and catalyzes the formation of a chromogen substrate. By counting the number of HRP-containing SVs in serial sections of the synaptic terminal, it was possible to estimate the number of SVs that underwent a cycle of exo-endocytosis during a fixed number of action potentials, yielding an evaluation of the average Pr. Thus, we stimulated infected neurons for 30 s at 1 Hz in the presence of HRP, cut serial sections to obtain full reconstructions of presynaptic terminals and counted the number of HRP-positive and docked SVs (Fig. 5E, top). Inhibitory synapses expressing K269Q-SynI were characterized by a significantly higher number of SVs released per action potential compared with WT-SynI-expressing inhibitory terminals (Fig. 5E, bottom). These results suggest a fundamental role of ATP binding to SynI in the postdocking phases of synaptic release by exerting a negative modulation of Pr.
The ATP binding mutant K269Q-SynI increases synaptic depression and slows down the recovery from depression in inhibitory synapses
Synaptic depression and synchronous/asynchronous release balance are key processes regulated by Syns whose dysregulation can affect network dynamics (Baldelli et al., 2007; Lignani et al., 2013; Medrihan et al., 2013). First, we investigated the role of ATP binding to SynI during short high-frequency trains. During 1 s train at 40 Hz, SynI KO inhibitory synapses exhibited a pronounced depression that was significantly rescued by expression of WT-SynI (Fig. 6A). When the ATP mutant K269Q-SynI was expressed in SynI KO neurons it was substantially unable to rescue the depression of the KO neurons and depressed more intensely than neurons expressing WT-SynI (Fig. 6A), a result that is consistent with the increased initial Pr in response to Ca2+ entry (Fig. 5).
Figure 6.

The ATP binding mutant K269Q-SynI increases synaptic depression during both short and sustained high-frequency stimulations in inhibitory synapses. A, The progressive decay of eIPSCs amplitude during the short stimulation train (1 s at 40 Hz) is plotted (means ± SEM) as a function of time. Depression curves in inhibitory SynI KO synapses expressing mCherry (SynI KO; dark gray trace), WT-SynI (light gray trace), and K269Q-SynI (black trace) were individually fitted with mono-exponential functions to yield time constants (τ) shown as means ± SEM in the bar plot on the right. Mutant K269Q-SynI showed a faster depression during short train respect to WT-SynI (n = 7, 12, and 10 for SynI KO, WT-SynI, and K269Q-SynI, respectively); **p < 0.01, one-way ANOVA followed by Dunnett's comparison test versus WT-SynI. B, C, To evaluate synaptic depression during sustained high-frequency stimulation, inhibitory SynI KO neurons transduced with mCherry (SynI KO; dark gray trace/bars), WT-SynI (light gray trace/white bars), or K269Q-SynI (black trace/bars) were stimulated with trains lasting 30 s at 10 Hz. The progressive decay of eIPSCs amplitude during the train (means ± SEM) is plotted as a function of time (B, left). Depression curves were individually fitted with biexponential functions to yield time constants (τ) and steady-state currents (SSCs) after depression. The resulting parameters τslow and SSCs are shown as means ± SEM in the bar plots of B (right) and C, respectively. The performance of K269Q-SynI synapses was specifically impaired during the train (n = 7, 13, and 12 for SynI KO, WT-SynI, and K269Q-SynI, respectively); *p < 0.05, ***p < 0.001, one-way ANOVA followed by Dunnett's comparison test versus WT-SynI. D, To evaluate the recovery from synaptic depression, inhibitory neurons transduced with mCherry (SynI KO; dark gray trace/bars), WT-SynI (light gray trace/white bars), or K269Q-SynI (black trace/bars) were stimulated 0.1 Hz for 120 s after the end of the high-frequency train, starting at t = 30 s. The progressive recovery of the eIPSCs amplitude is plotted (mean ± SEM) as a function of time from the end of the depression-inducing train (left). The first points of the plot represent the SSC of depression (Fig. 6). Recovery curves were individually fitted with a mono-exponential function to yield time constant (τ) and SSCs after recovery. In the bar plots on the right, the normalized value of the first eIPSC elicited 10 s after the end of the train, the time constant (τ) and the SSCs of recovery are shown as means ± SEM. The performance of K269Q-SynI synapses specifically slowed down the IPSC recovery (n = 7, 13, and 12 for SynI KO, WT-SynI, and K269Q-SynI, respectively); *p < 0.05, one-way ANOVA followed by Dunnett's multiple-comparison test versus WT-SynI. E, F, The changes in the absolute values of IPSC amplitude (E) and PPR (F) before train stimulation (Pre), at the last stimulus in the train (Last in train), at the first stimulus of the recovery period (First after train), and after 120 s of recovery (End recovery) are shown for the three genotypes (SynI KO neuron expressing either mCherry, WT-SynI, or K269Q-SynI, respectively). Shaded areas represent the 30 s of stimulation at 10 Hz. No PPR values were obtained during the train; #p < 0.05, ##p < 0.01, ###p < 0.001, one-way ANOVA across genotype followed by Dunnett's multiple-comparison test versus respective WT-SynI values; *p < 0.05, ***p < 0.001, one-way ANOVA for repeated measures within genotype followed by Tukey's multiple-comparison test versus basal values (Pre).
Synapsin I has a major role in the regulation of the reserve pool (RP) of SVs and in the SVs mobilization from the RP to the RRP and published data indicate that deletion of SynI enhances synaptic depression during sustained high-frequency stimulation (Baldelli et al., 2007; Cesca et al., 2010). To evaluate whether ATP binding is also involved in this process, we performed electrophysiology coupled with electron microscopy (Fig. 7) during sustained high-frequency stimulation (30 s at 10 Hz), a protocol known to mobilize SVs from the RP to the RRP (Fig. 6B,C; Baldelli et al., 2007; Lignani et al., 2013). SynI KO neurons transduced with mCherry, WT-SynI, or K269Q-SynI were stimulated with the above protocol and the eIPSC amplitude was followed during the train and in the subsequent recovery phase. Whereas the expression of WT-SynI fully rescued the SynI KO phenotype inducing a significant slow-down in the rate of depression (Fig. 6B) and in the steady-state current (Fig. 6C), the expression of K269Q-SynI was totally unable to rescue the KO phenotype.
Figure 7.

The physiological changes of the ATP binding mutant K269Q-SynI are accompanied by changes in synaptic vesicle trafficking. A, Representative transmission electron micrographs of inhibitory presynaptic terminals from SynI KO neurons expressing only mCherry (SynI KO), WT-SynI, or K269Q-SynI (left, middle, and right columns, respectively). Synaptic ultrastructure was evaluated by fixing neurons under basal conditions (top row), after the train at 10 Hz for 30 s (middle row), and after 120 s of recovery (bottom row). Scale bar, 200 nm. B, Results of the ultrastructural analysis. The total SV density (top row) and the number of docked SVs (bottom row), calculated in the samples subjected to the depression/recovery protocol, are shown as means ± SEM. Shaded areas represent the 30 s of stimulation at 10 Hz. No significant changes in the nerve terminal area and in the length of the AZ were found among the experimental groups. The number of presynaptic terminals analyzed for SynI KO, WT-SynI, and K269Q-SynI, respectively was as follows: basal: 16, 38, and 33; after depression: 31, 14, and 17; after recovery: 11, 15, and 18); #p < 0.05, one-way ANOVA across genotype followed by Dunnett's multiple-comparison test versus respective WT-SynI values; *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA for repeated measures within genotype followed by Dunnett's multiple-comparison test versus basal values (Pre).
After administration of the sustained stimulation protocol (30 s at 10 Hz) that induced depression in inhibitory terminals, the stimulation frequency was brought back to the resting frequency (0.1 Hz), allowing recovery of synaptic strength from depression. Although at the end of the recovery period (120 s after the end of the first train) the rescue of eIPSC amplitude was comparable between WT-SynI and K269Q-SynI expressing inhibitory terminals, the time constant (τ) of recovery was significantly increased in terminals expressing the K269-SynI mutant Fig. 6D).
To have a better insight into the release mechanisms altered by the mutation in the three neuronal genotypes, we plotted the absolute IPSC amplitude along the stimulation/recovery protocol (Fig. 6E) and the PPR measured during the low-frequency stimulation of the recovery period for an indirect evaluation of Pr (Fig. 6F). Interestingly, although all three genotypes fully recovered the basal amplitude 150 s after the end of the train, the recovery “load” was much more intense for K269Q-SynI expressing inhibitory terminals. This was associated with a relative increase of PPR that became similar to those of WT-SynI expressing terminals, accounting for the slower recovery rate of K269Q-SynI expressing inhibitory terminals.
The ATP binding mutant K269Q-SynI affects synaptic vesicle trafficking during depression and recovery
Under basal conditions, transmission electron microscopy followed by ultrastructural analysis revealed that: (1) SynI KO inhibitory terminals were relatively depleted of SVs, (2) the infection with either WT-SynI or K269Q-SynI was able to efficiently rescue the SV depletion phenotype of SynI KO neurons, and (3) inhibitory terminals expressing K269Q-SynI exhibited a general architecture and number of docked SVs similar to Syn KO neurons expressing either mCherry alone or WT-SynI (Fig. 7A, top row, B).
In the temporal window between the end of the 30 s at 10 Hz stimulation protocol and the first response to the 0.1 Hz recovery protocol, all three genotypes displayed a transient increase in total SV density with respect to resting conditions, although the effect was significant only in K269Q-SynI expressing neurons. This was accompanied by a selective and significant increase in the number of docked SVs in K269Q-SynI synapses with respect to SynI KO expressing either mCherry or WT-SynI (Fig. 7A, middle row, B).
After completion of the recovery period (120 s after the end of the train; Fig. 7A, bottom row, B), the total SV density of SynI KO inhibitory terminals expressing either mCherry of WT-SynI returned to the basal levels, while K269Q-SynI expressing terminals became significantly depleted in total SVs. Notably, the number of docked SVs increased at the end of the recovery period in SynI KO inhibitory terminals expressing either mCherry of WT-SynI, indicating an efficient refilling of the RRP, while the number of docked SVs in K269Q-SynI expressing terminals, that was transiently increased at the beginning of the recovery period, returned to basal levels.
Together, these results show that in the absence of ATP binding to SynI, the SynI-mediated trafficking of SVs is dysregulated when terminals are challenged with sustained high-frequency stimulation.
Discussion
In neurons, mitochondria produce the majority of ATP (90%) via aerobic metabolism: this energy supply is critical at synapses, which are the primary site of ATP consumption in the brain (Harris et al., 2012). In fact, proper neuronal function requires ATP to fuel ion pumps, to organize cytoskeletal components, support phosphorylation reactions, and above all sustain the SV cycle (Vos et al., 2010). Positioning mitochondria within nerve terminals is therefore highly important to support neurotransmission: motile mitochondria can be recruited to axonal boutons in an activity-dependent manner, and an elaborate cytoskeletal superstructure has been observed that anchors mitochondria to the presynaptic membrane near the AZ for high-rate metabolism (Ma et al., 2009; Perkins et al., 2010; Sheng, 2014). Moreover, it has been recently demonstrated that mitochondria motility contributes to the variability of presynaptic strength and that terminals that do not contain a mitochondria have a smaller size of total releasable SVs due to an impaired ability to mobilize reserve SVs (Verstreken et al., 2005; Ma et al., 2009; Sun et al., 2013). Accordingly, SV exocytosis in hippocampal synaptosomes was found to correlate directly with total mitochondrial volume (Ivannikov et al., 2013).
It has been recently demonstrated, using a quantitative, optical reporter of presynaptic ATP concentration, that activity drives ATP synthesis and that nerve terminal function, and in particular, SV cycling, rely heavily on activity-stimulated ATP synthesis (Rangaraju et al., 2014). In the absence of electrical activity, glycolysis is sufficient to support maintenance of ATP levels, but both glycolysis and mitochondrial function become necessary to satisfy the activity-dependent ATP needs when primary neurons are stimulated with high-frequency trains (600 AP at10 Hz; Rangaraju et al., 2014). All of these observations suggest a central role of ATP in neuronal transmission, but the exact molecular mechanisms involved in this regulation are only beginning to be deciphered.
In 1998, the crystal structure of the C-domain of bovine SynI was resolved and found to share homology with “ATP-grasp” proteins such as glutathione synthase, that contain a flexible loop contacting bound ATP (Esser et al., 1998). The ATP site in SynI is shared by virtually all Syn isoforms and is phylogenetically highly conserved (Kao et al., 1999). The presence of a Ca2+ binding domain in SynI in which Ca2+ is coordinated by two glutamate residues (Glu373,Glu386) brought to the hypothesis that ATP binding is regulated by Ca2+. Indeed, it was found that ATP binding to SynI is strongly Ca2+-dependent, whereas the binding is Ca2+-insensitive in SynII that has a Lys374 replacing glutamate in the site of Ca2+ coordination (Hosaka and Südhof, 1998). The binding of ATP to SynI was subsequently reported to have an absolute requirement for Ca2+ ions (Hosaka and Südhof, 1998), suggesting that it could be regulated during synaptic activity. The only functional effect that has been reported thus far is the stabilization of a tetramer formed by rat SynI ABC domains in complex with Ca2+ and ATP (Brautigam et al., 2004).
MD simulations indicated that changes at the ATP binding site are connected with conformational fluctuations at the intermonomer surface and the membrane-interacting regions of the protein, providing a structural link between ATP binding, oligomerization, and SynI function. We found that, in the absence of ATP/Ca2+, the MFL is highly flexible and hinders tetramer formation. When ATP alone is bound, a condition experimentally associated with a prevalence of dimers over tetramers, the MFL remains closed over the binding site. This behavior can be explained by the observation that other regions surrounding the ATP binding site and participating in the tetramer interface, become distorted under Ca2+-free conditions, resulting in the perturbation of the intermonomer contacts that stabilize the tetramer and thereby promoting the transition to dimer. SynI dimers are constitutively more apt to cross-link adjacent SVs because of wide exposure of the membrane interacting stretches (Cheetham et al., 2001) on the dimer surface, whereas in tetramers a considerable portion of these surfaces become buried in the tetramer interface. This suggests that the tetramer is less capable of cross-linking adjacent SVs in clusters than the dimer. This prediction was indeed verified by comparing SynI binding to SVs (enhanced by ATP both in the presence and in the absence of Ca2+) with SV clustering (enhanced by ATP only under Ca2+-free conditions). This indicates that the lower ATP binding site occupancy in the absence of Ca2+ is sufficient to increase the association with SVs, whereas SV clustering is enhanced in the absence of Ca2+ by the abundance of SynI dimers and decreased in the presence of ATP/Ca2+ due to the stabilization of SynI tetramers. Consistent with MD predictions, we found that Ca2+ is dispensable for ATP-SynI binding, although it significantly enhances it. These results only partially agree with previous observations (Hosaka and Südhof, 1998) reporting an absolute Ca2+-dependency of ATP binding. However, in this study SynI was purified from bovine brain under nondenaturing conditions (and not from prokaryotes) and tested over a wide range of ATP concentrations.
Although ATP production has been demonstrated to cope with ATP consumption even during high-frequency neural activity, our data using the K269Q-SynI mutant lacking the ATP binding site unambiguously characterize the functional role of the ATP binding to SynI independently of potential changes in the intraterminal ATP concentrations. Although this mutant was targeted to nerve terminals, had a normal phosphorylation behavior and did not alter the synaptic density, it was less associated with the distal pool of SVs, likely due to the decreased SV binding. The functional effects of the deletion of ATP binding were studied in inhibitory synapses that are primarily affected by SynI malfunctioning (Gitler et al., 2004; Baldelli et al., 2007). The main effect of the mutant's expression was an increase of basal synaptic strength due to a selective increase of Pr at rest. Such increase, demonstrated with independent approaches, profoundly affects short-term plasticity by enhancing paired-pulse depression and increasing the dynamics and extent of synaptic depression induced by tetanic stimulation. The effect on the probability of release adds to the postdocking actions of SynI (Hilfiker et al., 1998; Fassio et al., 2006; Baldelli et al., 2007) and indicates that nucleotide binding and immobility of the MFL in SynI represent a brake to the Ca2+-dependent fusion of primed SVs. Such a negative regulation of Pr might occur by the enhanced formation of SynI oligomers at the level of primed SVs exposed to the Ca2+ influx.
Not only release dynamics during high-frequency stimulation and the subsequent recovery were affected by K269Q-SynI, but the SV distribution was also profoundly altered. In particular: (1) the recovery to basal current levels was much more demanding in the mutant because of the much larger baseline amplitude and transient decrease of Pr (PPR was increased to the WT-SynI level after the train); (2) the mutant showed a precocious increase in docked SVs after the train that the other genotypes showed only later on; (3) the mutant was able to fully recover the IPSC amplitude, but at the expense of relative depletion of total SVs; and (4) the decreased SynI binding/clustering of SVs implies a greater SV availability for release in mutant terminals, an effect that potentiates the physiological rise in SV availability during Ca2+ buildup mediated by CaM kinase phosphorylation of SynI, but may also impair SV reclustering during periods of intense recycling.
Under basal conditions, a balance is present in K269Q-SynI synapses between a larger amount of released SVs per action potential and an increased availability of SVs for release (Fig. 8). After train stimulation, the precocious increase in docked SVs may result from the increased availability of SVs for release (due to the additive effects of the mutation and Ca2+-dependent SynI phosphorylation) and a transient Pr decrease (in the presence of a twofold higher IPSC amplitude versus WT-SynI in response to the first stimulus after the train; Fig. 8). During recovery, the number of docked SVs normalizes and the evoked release recovers, albeit with a slower kinetics, to the high basal levels thanks to the usage of the reserve SVs. This process, together with an impaired ability to reconstitute the SV clusters after endocytosis, may lead to the relative and transient depletion of total SVs (Fig. 8). In this respect, SynI has been proposed to act at endocytic sites to recruit SVs back to the nerve terminal clusters (Bloom et al., 2003; Evergren et al., 2007) and ATP binding may be necessary for this activity.
Figure 8.

Mechanistic diagram of the SV dysregulation in the absence of ATP binding to SynI. An inhibitory nerve terminal expressing either WT-SynI (blue symbols; top row) or K269Q-SynI (red symbols; bottom row) is shown under basal conditions (left), at the end of a sustained high-frequency stimulation (HFS; 30 s at 10 Hz; middle) and over recovery from depression (right). The oligomerization state of SynI has been hypothesized on the basis of the data of Figure 3. Under basal conditions, Pr is higher in mutant terminals (Fig. 5) and SVs are more dispersed due to the decreased SV binding of SynI (Fig. 2) and the relative decrease of high-order SynI oligomers (Fig. 3). Accordingly, the mutant SynI accumulates in proximity to the presynaptic membrane (Fig. 4), possibly contributing to the increase in Pr. After sustained HFS, mutant terminals display a precocious increase in nonprimed docked SVs possibly resulting from the increased SV availability and a concomitant decrease in Pr (Figs. 6, 7). After depression, mutant terminals recover more slowly but completely at the expense of a transient depletion of total SVs (Figs. 6, 7). The increase in the formation of SynI tetramers versus dimers upon ATP and Ca2+ binding in WT terminals and the parallel reduction in SV clustering may represent a mechanism to increase SV availability under conditions of sustained stimulation associated with Ca2+ build-up.
In conclusion, we have shown that ATP binding to SynI under Ca2+-free conditions enhances SV binding and stabilizes SynI dimers, whereas Ca2+ has a modulatory role, enhancing ATP binding to SynI and stabilizing SynI tetramers. This Ca2+-dependent switch between SynI dimers/tetramers could represent a SV dynamic clustering/unclustering mechanism regulated by electrical activity and Ca2+ influx into the terminal. This may represent an interesting activity-dependent mechanism, particularly during sustained activity, when intraterminal Ca2+ concentrations increase and SVs have to be released from the clusters to sustain release. In addition to this predocking role, ATP binding has also a postdocking activity in inhibitory synapses by negatively modulating the release probability and reducing depression during high-frequency activity. This may modulate the filtering properties and efficacy of inhibitory synapses and render them less vulnerable to fatigue.
Footnotes
This work was supported by research grants from the Italian Ministry of University and Research (PRIN 2010-11 to F.B. and F.O., and FIRB 2010 “Futuro in Ricerca” to S.G.), EU FP7 Integrating Project “Desire” (Grant no. 602531) and Cure Epilepsy “Advanced Innovator Award” (F.B.). The support of Telethon-Italy (Grant GGP13033 to F.B.) and the IIT Platform “Computation” are also acknowledged. We thank Drs Hung-Teh Kao (Brown University, Providence, RI) and Paul Greengard (The Rockefeller University, New York City, NY) for providing us with the synapsin I mutant mice, Luigi Naldini and Mario Amendola (Tiget, Milano, Italy) for invaluable help in lentiviral production protocols, Marina Nanni and Claudia Chiabrera for the preparation of primary cultures of hippocampal neurons, Federico Pecoraro Bisogni for help with viral transduction, and Alberto Diaspro, Paolo Bianchini, Silvia Galiani, and Chiara Peres for the observation of our samples at the STED fluorescence microscope.
The authors declare no competing financial interests.
References
- Bähler M, Greengard P. Synapsin I bundles F-actin in a phosphorylation-dependent manner. Nature. 1987;326:704–707. doi: 10.1038/326704a0. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Fassio A, Valtorta F, Benfenati F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci. 2007;27:13520–13531. doi: 10.1523/JNEUROSCI.3151-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Rossi MC, Onofri F, Sihra T, Greengard P. Interactions of synapsin I with phospholipids: possible role in synaptic vesicle clustering and in the maintenance of bilayer structures. J Cell Biol. 1993;123:1845–1855. doi: 10.1083/jcb.123.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Vavra V, Svobodova I, Bendova Z, Vereb G, Zemkova H. Potentiation of inhibitory synaptic transmission by extracellular ATP in rat suprachiasmatic nuclei. J Neurosci. 2013;33:8035–8044. doi: 10.1523/JNEUROSCI.4682-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Löw P, Brodin L, Pieribone VA, Greengard P, Shupliakov O. Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol. 2003;161:737–747. doi: 10.1083/jcb.200212140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco T, Staras K, Darcy KJ, Goda Y. Local dendritic activity sets release probability at hippocampal synapses. Neuron. 2008;59:475–485. doi: 10.1016/j.neuron.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco T, Marra V, Staras K. Examining size-strength relationship at hippocampal synapses using an ultrastructural measurements of synaptic release probability. J Struct Biol. 2010;172:203–210. doi: 10.1016/j.jsb.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA, Chelliah Y, Deisenhofer J. Tetramerization and ATP binding by a protein comprising the A–C domains of rat synapsin I. J Biol Chem. 2004;279:11948–11956. doi: 10.1074/jbc.M312015200. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Cheetham JJ, Hilfiker S, Benfenati F, Weber T, Greengard P, Czernik AJ. Identification of synapsin I peptides that insert into lipid membranes. Biochem J. 2001;354:57–66. doi: 10.1042/0264-6021:3540057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin i phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/S0896-6273(03)00151-X. [DOI] [PubMed] [Google Scholar]
- Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9230–9234. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman WL, Bykhovskaia M. Cooperative regulation of neurotransmitter release by Rab3a and synapsin II. Mol Cell Neurosci. 2010;44:190–200. doi: 10.1016/j.mcn.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi A, Fadda M, Piton A, Patry L, Marte A, Rossi P, Cadieux-Dion M, Gauthier J, Lapointe L, Mottron L, Valtorta F, Rouleau GA, Fassio A, Benfenati F, Cossette P. SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum Mol Genet. 2014;23:90–103. doi: 10.1093/hmg/ddt401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- De Franchi E, Schalon C, Messa M, Onofri F, Benfenati F, Rognan D. Binding of protein kinase inhibitors to synapsin I inferred from pair-wise binding site similarity measurements. PLoS One. 2010;5:e12214. doi: 10.1371/journal.pone.0012214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Naldini L. Transduction of a gene expression cassette using advanced generation lentiviral vectors. Methods Enzymol. 2002;346:514–529. doi: 10.1016/S0076-6879(02)46074-0. [DOI] [PubMed] [Google Scholar]
- Esser L, Wang CR, Hosaka M, Smagula CS, Südhof TC, Deisenhofer J. Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J. 1998;17:977–984. doi: 10.1093/emboj/17.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evergren E, Benfenati F, Shupliakov O. The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res. 2007;85:2648–2656. doi: 10.1002/jnr.21176. [DOI] [PubMed] [Google Scholar]
- Farisello P, Boido D, Nieus T, Medrihan L, Cesca F, Valtorta F, Baldelli P, Benfenati F. Synaptic and extrasynaptic origin of the excitation/inhibition imbalance in the hippocampus of synapsin I/II/III knockout mice. Cereb Cortex. 2013;23:581–593. doi: 10.1093/cercor/bhs041. [DOI] [PubMed] [Google Scholar]
- Fassio A, Merlo D, Mapelli J, Menegon A, Corradi A, Mete M, Zappettini S, Bonanno G, Valtorta F, D'Angelo E, Benfenati F. The synapsin domain E accelerates the exo-endocytotic cycle of synaptic vesicles in cerebellar Purkinje cells. J Cell Sci. 2006;119:4257–4268. doi: 10.1242/jcs.03194. [DOI] [PubMed] [Google Scholar]
- Fassio A, Raimondi A, Lignani G, Benfenati F, Baldelli P. Synapsins: from synapse to network hyperexcitability and epilepsy. Semin Cell Dev Biol. 2011a;22:408–415. doi: 10.1016/j.semcdb.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, Pozzi D, Messa M, De Franchi E, Fadda M, Corradi A, Baldelli P, Lapointe L, St-Onge J, Meloche C, Mottron L, Valtorta F, Khoa Nguyen D, Rouleau GA, Benfenati F, Cossette P. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. 2011b;20:2297–2307. doi: 10.1093/hmg/ddr122. [DOI] [PubMed] [Google Scholar]
- Fernández-Alfonso T, Ryan TA. The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron. 2004;41:943–953. doi: 10.1016/S0896-6273(04)00113-8. [DOI] [PubMed] [Google Scholar]
- Frenkel D, Smit B. Understanding molecular simulation: from algorithms to applications. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, Curtis A, Goodship JA. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet. 2004;41:183–186. doi: 10.1136/jmg.2003.013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsin at excitatory and inhibitory synapses. J Neurosci. 2004;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco B, Managò F, Tucci V, Kao HT, Valtorta F, Benfenati F. Autism-related behavioral abnormalities in synapsin knockout mice. Behav Brain Res. 2013;251:65–74. doi: 10.1016/j.bbr.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC. Synapsin I and II are ATP-binding proteins with differential Ca2+ regulation. J Biol Chem. 1998;273:1425–1429. doi: 10.1074/jbc.273.3.1425. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC. Homo- and heterodimerization of synapsins. J Biol Chem. 1999;274:16747–16753. doi: 10.1074/jbc.274.24.16747. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein: III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivannikov MV, Sugimori M, Llinás RR. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J Mol Neurosci. 2013;49:223–230. doi: 10.1007/s12031-012-9848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- Kao HT, Porton B, Hilfiker S, Stefani G, Pieribone VA, DeSalle R, Greengard P. Molecular evolution of the synapsin gene family. J Exp Zool. 1999;285:360–377. doi: 10.1002/(SICI)1097-010X(19991215)285:4<360::AID-JEZ4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leung A. Fixation. In: Woods AE, Ellis RC, editors. Laboratory histopathology: a complete reference. New York: Churchill Livingstone; 1994. [Google Scholar]
- Lignani G, Raimondi A, Ferrea E, Rocchi A, Paonessa F, Cesca F, Orlando M, Tkatch T, Valtorta F, Cossette P, Baldelli P, Benfenati F. Epileptogenic Q555X SYN1 mutant triggers imbalances in release dynamics and short-term plasticity. Hum Mol Genet. 2013;22:2186–2199. doi: 10.1093/hmg/ddt071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Cai Q, Lu W, Sheng ZH, Mochida S. KIF5B motor adaptor syntabulin maintains synaptic transmission in sympathetic neurons. J Neurosci. 2009;29:13019–13029. doi: 10.1523/JNEUROSCI.2517-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. doi: 10.1063/1.467468. [DOI] [Google Scholar]
- Medrihan L, Cesca F, Raimondi A, Lignani G, Baldelli P, Benfenati F. Synapsin II desynchronizes neurotransmitter release at inhibitory synapses by interacting with presynaptic calcium channels. Nat Commun. 2013;4:1512. doi: 10.1038/ncomms2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaldi I, Vassalli M, Bachi A, Giovedì S, Millo E, Valtorta F, Raiteri R, Benfenati F, Fassio A. The highly conserved synapsin domain E mediates synapsin dimerization and phospholipid vesicle clustering. Biochem J. 2010;426:55–64. doi: 10.1042/BJ20090762. [DOI] [PubMed] [Google Scholar]
- Perkins GA, Tjong J, Brown JM, Poquiz PH, Scott RT, Kolson DR, Ellisman MH, Spirou GA. The micro-architecture of mitochondria at active zones: electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J Neurosci. 2010;30:1015–1026. doi: 10.1523/JNEUROSCI.1517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156:825–835. doi: 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO. Synaptic vesicle recycling: steps and principles. EMBO J. 2014;33:788–822. doi: 10.1002/embj.201386357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints and molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/S0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Sheng ZH. Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol. 2014;204:1087–1098. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Haucke V, Pechstein A. How synapsin I may cluster synaptic vesicles. Semin Cell Dev Biol. 2011;22:393–399. doi: 10.1016/j.semcdb.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 2013;4:413–419. doi: 10.1016/j.celrep.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH. Activity-related redistribution of presynaptic proteins at the active zone. Neuroscience. 2006;141:1217–1224. doi: 10.1016/j.neuroscience.2006.04.061. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicles pools in health and disease. Front Synaptic Neurosci. 2010;2:139. doi: 10.3389/fnsyn.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]