

Abstract
Laminins are major constituents of the gliovascular basal lamina of the blood–brain barrier (BBB); however, the role of laminins in BBB development remains unclear. Here we report that Lama2−/− mice, lacking expression of the laminin α2 subunit of the laminin-211 heterotrimer expressed by astrocytes and pericytes, have a defective BBB in which systemically circulated tracer leaks into the brain parenchyma. The Lama2−/− vascular endothelium had significant abnormalities, including altered integrity and composition of the endothelial basal lamina, inappropriate expression of embryonic vascular endothelial protein MECA32, substantially reduced pericyte coverage, and tight junction abnormalities. Additionally, astrocytic endfeet were hypertrophic and lacked appropriately polarized aquaporin4 channels. Laminin-211 appears to mediate these effects at least in part by dystroglycan receptor interactions, as preventing dystroglycan expression in neural cells led to a similar set of BBB abnormalities and gliovascular disturbances, which additionally included perturbed vascular endothelial glucose transporter-1 localization. These findings provide insight into the cell and molecular changes that occur in congenital muscular dystrophies caused by Lama2 mutations or inappropriate dystroglycan post-translational modifications, which have accompanying brain abnormalities, including seizures. Our results indicate a novel role for laminin–dystroglycan interactions in the cooperative integration of astrocytes, endothelial cells, and pericytes in regulating the BBB.
Keywords: astrocyte, blood–brain barrier, dystroglycan, gliovascular, laminin, pericyte
Introduction
The blood–brain barrier (BBB) is comprised of physical barriers and transport mechanisms that critically regulate CNS homeostasis (Abbott et al., 2010). Brain endothelial cells (BECs), together with astrocytes, pericytes, and vascular smooth muscle cells (VSMCs), comprise the gliovascular unit. Complex cellular interactions within the gliovascular unit regulate the development of unique BEC properties, including specialized tight junctions, suppressed vesicular transport, and barrier-specific transport proteins (Reese and Karnovsky, 1967; Girardin, 2006; Armulik et al., 2010; Daneman et al., 2010b). In addition to cell–cell interactions, cell–extracellular matrix (ECM) interactions are also proposed to regulate BBB properties, as an extensive ECM network resides between perivascular astrocytic endfeet and BECs. This gliovascular basal lamina (BL) exists either as two distinct BLs, an astrocyte-produced parenchymal BL and a BEC-produced endothelial BL, or, as a fusion of the two BLs, creating a composite BL (Sixt et al., 2001). Pericytes, which also critically regulate barrier properties, reside within this composite gliovascular BL, having an interface with both astrocytes and BECs (Del Zoppo et al., 2006; Armulik et al., 2010; Daneman et al., 2010b). Although the gliovascular BL comprises a key “outer barrier” of the mature BBB, the role of specific cell–ECM interactions in the maturation and function of the BBB remains unclear.
The laminins are major components of the gliovascular BL, serving as ligands for receptors, such as integrins (Sixt et al., 2001; Del Zoppo and Milner, 2006; Abraham et al., 2008) and dystroglycan (Moore et al., 2002). Laminins are heterotrimers, with each laminin having a designation reflecting its subunit composition (e.g., laminini-111 is composed of laminin α1, β1, and γ1 subunits) (Colognato and Yurchenco, 2000; Miner and Yurchenco, 2004). Individual laminins are distributed differentially throughout the composite gliovascular BL, with the parenchymal BL containing laminins-111 and -211 produced by astrocytes, and the endothelial BL containing laminins-411 and -511 produced by endothelial cells (Sixt et al., 2001). Laminins-211, -411, and -511 are similar to laminin-111 but, in place of α1, contain α2, α4, or α5, respectively. Endothelial laminins are essential for brain vascular development, as mice that lack laminin α4 have vascular BL defects, vessel dilation, and decreased vessel integrity (Thyboll et al., 2002). Interestingly, laminin α4-deficient mice upregulate laminin α5, which inhibits T-cell extravasation across the BBB during brain inflammation, thus providing resistance to experimental allergenic encephalomyelitis (Wu et al., 2009).
In contrast to endothelial laminins, the roles of astrocytic laminins are less clear. Initially, ultrastructural analysis in dy/dy mice (in which laminin α2 is deficient but not absent) revealed no obvious defects (Jucker et al., 1996). However, mutations in Lama2, which encodes laminin α2, or in genes that encode enzymes needed for dystroglycan post-translational modifications, cause congenital muscular dystrophies with accompanying brain abnormalities, including seizures, perturbed cortical development, and MRI white matter hypointensities (Miyagoe-Suzuki et al., 2000; Allamand and Guicheney, 2002; Moore et al., 2002; Buteică et al., 2008; Yoshioka et al., 2008; Fujii et al., 2011), with MRI abnormalities hypothesized to reflect, at least in part, BBB defects (Caro et al., 1999). Recently, mice that lack laminin γ1 expression selectively (removing laminins-111 and -211) in astrocytes were reported to have impaired VSMC function and hemorrhagic stroke (Chen et al., 2013). However, the best understood role for astrocytic laminins at the gliovascular interface is as a ligand for dystroglycan, a component of the dystrophin–glycoprotein complex (DGC) localized at perivascular astrocytic endfeet (Zaccaria et al., 2001; Del Zoppo and Milner, 2006). Disruption of components of the DGC (e.g., α-dystrobrevin or dystrophin) results in altered AQP4 distribution at glial endfeet and increased vascular permeability (Nico et al., 2004; Lien et al., 2012). And, in cultured astrocytes, laminin binding to dystroglycan facilitates AQP4 polarization (Blake et al., 1999; Zaccaria et al., 2001; Rurak et al., 2007; Noël et al., 2009). In addition, pericytes themselves reportedly express laminin α2 (Armulik et al., 2010). Intriguingly, in mice with an engineered pericyte deficiency, areas of the vasculature devoid of pericyte coverage coincide with laminin α2 loss at astrocyte endfeet, suggesting that α2-containing laminins may also regulate pericyte–gliovascular interactions (Armulik et al., 2010).
To gain insight into how ECM interactions at the gliovascular interface contribute to BBB development, we: (1) inactivated the Lama2 gene to selectively eliminate laminin-211 from all cells of the developing brain and (2) inactivated the DAG1 gene that encodes dystroglycan in neural lineage cells. We identified laminin-211 and dystroglycan as novel regulators of the selective permeability of the BBB, both contributing to the acquisition of cerebrovascular pericyte coverage in the developing brain and multiple aspects of gliovascular cell–cell and cell-ECM interactions, including glial endfeet architecture. Our findings suggest that laminin–dystroglycan interactions at the gliovascular interface regulate the maturation and function of the BBB and that BBB dysregulation may contribute to CNS abnormalities observed in CMD.
Materials and Methods
Mice.
Laminin α2 subunit-null mice (Lama2−/−) were generated by breeding Lama2 heterozygous pairs as described previously (Miyagoe et al., 1997; Relucio et al., 2012). Genotyping of Lama2 mutants was performed on tail DNA to detect both the Lama2 wild-type (WT) allele (5′-CCAGATTGCCTACGTAATTG-3′ and 5-CCTCTCCATTTTCTAAAG-3′) and the Lama2 null allele (5′-CTTTCAGATTGCAAGC-3′ and 5′-TCGTTTGTTCGGATCCGTCG-3′). Homozygous and heterozygous WT littermates of either sex served as age-matched controls to homozygous null Lama2 mutants. The adhesion receptor DAG was removed from neural stem cells by crossing mice heterozygous for DAG1 lox/lox mice and nestin-cre. Nestin-cre mice and DAG1 loxP/loxP mice were described previously (Tronche et al., 1999; Moore et al., 2002). Genotyping was performed on tail DNA to detect DAG1 loxP and nestin-cre alleles. The primers used to detect the dystroglycan loxP allele were as follows: 5′-GGAGAGGATCAATCATGG-3′ and 5′-CAACTGCTGCATCTCTAC-3′. The primers used to detect nestin-cre alleles were as follows: 5′-GACGGAAATCCATCGCTCACCAG-3′ and 5′-GACATGTTCAGGGATCGCCAGGCG-3′. DAG1 conditional knock-outs were homozygous DAG1 loxP and positive for nestin-cre. Nestin-cre-negative littermates were used as age-matched controls. Mice of either sex were used in experimental and control conditions. All experimental procedures were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the State University of New York at Stony Brook University Institutional Animal Care and Use Committee.
Antibodies.
The following rabbit polyclonal IgG antibodies were used: α-smooth muscle actin (α-SMA) (Abcam); AQP4 (Millipore); CD31 (i.e., PECAM-1) (Abcam); Collagen IV (Sigma); GFAP (DAKO); glucose transporter-1 (Glut-1; Millipore); Iba-1 (Wako); laminin-1 (Sigma); and VE-Cadherin (Abcam). Antibodies against laminin α4-subunit (clone 377b) and laminin α5-subunit (clone 504) were kindly provided by Dr. L. Sorokin (Institute of Physiological Chemistry and Pathobiochemistry; Münster University). The following rat monoclonal IgG antibodies were used: CD45 (BD Biosciences PharMingen); β1-Integrin (Millipore); laminin α2-subunit (clone 4H82, Sigma); laminin γ1-subunit (clone A5, Millipore); laminin α1-subunit (MAB1905, Chemicon) MECA32, also called plasmalemma vesicle-associated protein (BD Biosciences PharMingen and Abcam). The Armenian hamster polyclonal IgG antibody against CD3ε was kindly provided by Dr. Nicholas Carpino (Department of Molecular Genetics and Microbiology, Stony Brook University). The following mouse monoclonal IgG antibodies were used: β-actin (Sigma), β-DAG (Abcam), GFAP (Sigma), and p115 (BD Biosciences). The following additional antibodies were used: chicken anti-cow GFAP polyclonal IgY (Abcam) and goat anti-mouse CD140b (i.e., PDGFR-β polyclonal IgG) (Neuromics). The following monoclonal blocking antibodies were used: IIH6 (i.e., α-dystroglycan) (mouse, Millipore) and Ha2/5 (i.e., β1-Integrin) (hamster, BD Biosciences PharMingen). The following IgG control antigens were used: mouse (κ isotype, BioLegend) and hamster (λ1 isotype, BD Biosciences PharMingen).
Fluorescent immunohistochemistry using frozen sections.
Cerebral cortices from WT, DAG1 CKO, or Lama2−/− littermates at postnatal days 1 and 8 were frozen and embedded in TissueTek OCT. Mice at postnatal day 15 and beyond were anesthetized with Avertin and perfused with saline to remove residual blood from the cerebral vasculature. Frozen tissues were cryosectioned to a thickness of 20–35 μm. Sections were fixed in acetone for 10 min at −20°C and then washed with PBS. Sections were blocked for 1 h in PBS containing 10% donkey serum and then incubated overnight with primary antibodies in block at 4°C. Sections were then washed with PBS, followed by incubation for 1 h at room temperature with either Cy3- or dylight488-conjugated secondary antibodies in blocking solution. Sections were then washed with PBS and counterstained with 10 μg/ml DAPI in PBS and mounted using SlowFade Gold.
Fluorescent immunohistochemistry using PFA-fixed sections.
Cerebral cortices from WT, DAG1 CKO, or Lama2−/− littermates at postnatal days 1 and 8 were fixed with 4% PFA by submersion for 16–24 h. Mice postnatal day 15 and beyond were anesthetized and perfused intracardially with saline, followed by filtered 4% PFA. Brains were removed and postfixed for 2 h in 4% PFA, washed with PBS, then equilibrated in a gradient of 20% followed by 30% sucrose at 4°C. Brains were frozen in TissueTek OCT using a liquid nitrogen bath, and coronal cryostat sections of 20–35 μm were prepared. Sections were blocked in 10% donkey serum with 0.2%–0.5% Triton X-100 for 1 h before incubating overnight with primary antibodies at 4°C. Sections were then washed and incubated with either Cy3- or Dylight488-conjugated secondary antibodies in blocking solution for 1 h, followed by DAPI counterstain, and mounted with SlowFade Gold. To evaluate Glut-1 immunoreactivity, postnatal day 21 sections were postfixed with 2:1 ethanol acetic acid for 20 min at −20°C before immunostaining as described. Evaluation of the vasculature (CD31) and gliovascular interface (GFAP and AQP4) was performed with sections heated in Antigen Retrieval Solution, pH 6 (Dako) for 30 min, followed by PBS washes before blocking.
Analysis of astrocyte development.
Immunohistochemistry with GFAP was performed to quantify astrocytes within the cortex of Lama2−/− mice and WT littermates. PFA-fixed coronal sections were pretreated in PBS for 10 min at room temperature before heating at 95°–99°C for 25 min in 1× Target Retrieval Solution citrate buffer, pH 6.0 (Dako). Sections were then cooled at room temperature for 15 min. Before blocking with 10% donkey serum with 0.5% Triton X-100 for 1 h, sections were washed with PBS to remove residual antigen retrieval solution. Immunohistochemistry for GFAP was performed overnight at 4°C. After primary antibody incubation, slides were washed with PBS and then incubated with Dy-Light 488 secondary antibodies for 1 h, before counterstaining with DAPI and mounting with SlowFade Gold. Sections were imaged using a Zeiss 200M epifluorescence microscope or Zeiss 510 Meta confocal microscope. GFAP+ cell bodies within the cortex were quantified in matching sections of WT and Lama2−/− using Axiovision Rel. 4.8 software (Zeiss). Number of GFAP+ cells per field and area of GFAP immunoreactivity per field in the cortex were analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− and WT pair (n = 3). Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of GFAP coverage. Area of total GFAP immunoreactivity coverage was then normalized to total GFAP+ cells per field of view. Protein levels of GFAP were quantified using whole-cortex lysates obtained from Lama2−/− and WT littermate controls at postnatal day 21.
Analysis of gross vascular morphology.
Blood vessels were visualized by using collagen IV immunohistochemistry and imaged using an LSM Meta 510 microscope (Carl Zeiss). For analysis of vascular density and complexity, five corresponding areas in three pairs of Lama2−/− and WT mice were analyzed at postnatal day 21. Blood vessel area was quantified in maximum image projections and normalized to the total vessel length per field. Vascular complexity was quantified by counting the number of branch points along each vessel per field. The number of branch points was standardized to 100 μm segments of the vasculature.
Analysis of α-SMA vascular coverage.
To quantify the degree of α-SMA coverage along vSMCs, we used α-SMA immunohistochemistry in combination with pan-laminin immunohistochemistry, to visualize vSMCs and vascular BL, respectively. Frozen cortical sections (25 μm) of WT and Lama2−/− mice at postnatal days 21–22 were fixed in acetone at −20°C for 10 min followed by several washes with PBS. Sections were then blocked with 10% donkey serum with 0.2% Triton X-100 at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of α-SMA immunoreactivity relative to the total area of vascular pan-laminin immunoreactivity per field in the cortex and striatum was analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− and WT pair (n = 3). Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of α-SMA coverage. Intensity thresholding of pan-Laminin immunoreactivity was adjusted appropriately to reflect total vascular coverage per field. Area of total α-SMA immunoreactivity coverage was then normalized to total vascular coverage per field of view.
Analysis of Glut-1 vascular coverage.
To quantify the degree of Glut-1 coverage along the vasculature, we used Glut-1 immunohistochemistry in combination with pan-Laminin immunohistochemistry, to visualize Glut-1 transporters and blood vessels, respectively. Frozen cortical sections (25 μm) of WT and Lama2−/− or DAG1 CKO mice at postnatal days 21–22 were fixed in acetone at −20°C for 10 min followed by several washes with PBS. Sections were then blocked with 10% donkey serum at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of Glut-1 immunoreactivity relative to the total area of vascular pan-Laminin immunoreactivity per field in the cortex was analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− or DAG1 CKO and WT pair (n = 3). Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of Glut-1 coverage. Intensity thresholding of pan-Laminin immunoreactivity was adjusted appropriately to reflect total vascular coverage per field. Area of total Glut-1 immunoreactivity coverage was then normalized to total vascular coverage per field of view.
Analysis of MECA32 vascular coverage.
To quantify the degree of MECA32 coverage along the vasculature, we used MECA32 immunohistochemistry in combination with pan-Laminin immunohistochemistry to visualize MECA32 expression and blood vessels, respectively. Frozen cortical sections (25 μm) of WT and Lama2−/− mice at postnatal days 21–22 were fixed in acetone at −20°C for 10 min followed by several washes with PBS. Sections were then blocked with 10% donkey serum with 0.5% Triton X-100 at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of MECA32 immunoreactivity relative to the total area of vascular pan-Laminin immunoreactivity per field in the cortex, striatum, and hippocampus was analyzed on a minimum of three matched sections; five images per were acquired for each Lama2−/− and WT pair (n = 3). MECA32 immunoreactivity was confirmed in both Lama2−/− and WT mice by acquiring a representative field from the circumventricular region of the brainstem that expresses MECA32. Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of MECA32 coverage. Intensity thresholding of pan-laminin immunoreactivity was adjusted appropriately to reflect total vascular coverage per field. Area of total MECA32 immunoreactivity coverage was then normalized to total vascular coverage per field of view.
BBB permeability assay.
Postnatal day 21 mice were either injected with 0.2 μm filtered 2% Evans blue in saline (150 μl/10 g of body weight) intraperitoneally or anesthetized and perfused with Evans blue intracardially. After an acute 30 min dye circulation, mice were anesthetized and perfused with 150 μl of saline. To verify systemic dye distribution, kidney tissue was analyzed to confirm successful circulation. Analysis of cortical extravasation of Evans blue was performed by dissecting and weighing cerebral cortices following dye circulation and snap freezing cortices in liquid nitrogen, storing samples at −80°C. Evans blue dye extraction for all samples was processed in parallel, performed by immersing cortices (n = 4) in 500 μl of formamide in a shaking heat block at 65°C for 24 h. Samples were centrifuged at 13,200 RPM for 30 min, and the supernatant was removed for analysis. Absorbance at 620 nm was measured using a florescent plate reader. Visualization of dye extravasation in cerebral cortices and cerebella of fresh tissue was obtained by mounting dried tissue with DAPI Fluromount (Southern Biotechnology) and imaging immediately using an inverted Zeiss Axioplan epifluorescence microscope.
Brain water content.
Mice ∼3 weeks of age were killed, and their brains were rapidly dissected. Isolated cerebral cortices were weighed to obtain wet mass and then snap frozen in liquid nitrogen and stored. A total of n = 9 mice were processed concurrently in a vacuum oven (Savant Speed Vac SC110) for 16 h at −1000 mbar (Savant VP Two Stage Vacuum Pump) to completely desiccate cortices. The percentage of brain water content was calculated as follows: (wet mass − dry mass) × 100/(wet mass).
Analysis of Iba-1 positive microglia.
Immunohistochemistry with monocyte-specific antigens was performed to quantify microglia within the striatum of Lama2−/− mice and WT littermates. PFA-fixed coronal sections were pretreated in PBS for 10 min at room temperature before heating at 95°C–99°C for 25 min in 1× Target Retrieval Solution citrate buffer, pH 6.0 (Dako). Sections were then cooled at room temperature for 15 min. Before blocking with 10% donkey serum with 0.5% Triton X-100 for 1 h, sections were washed with PBS to remove residual antigen retrieval solution. Immunohistochemistry for Iba-1, a calcium binding protein found in microglia, was performed overnight at 4°C. After primary antibody incubation, slides were washed with PBS and then incubated with Dy-Light 488 secondary antibodies for 1 h, before counterstaining with DAPI and mounting with SlowFade Gold. Sections were imaged using a Zeiss 200M epiflurescence microscope or Zeiss 510 Meta confocal microscope. Iba-1+ cells within the striatum were quantified in matching sections of WT and Lama2−/− using Axiovision Rel. 4.8 software (Carl Zeiss). The number of Iba-1+ cells per field in the striatum were analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− and WT pair. Qualitative analysis of Iba-1+ cells was determined by morphological characteristics of cell bodies and retracted processes consistent with activated microglial phenotypes.
Analysis of β-dystroglycan immunoreactivity on astrocytic endfeet.
To quantify the relationship between laminin α2 and the attachment of astrocytic endfeet via β-dystroglycan, we used β-dystroglycan immunohistochemistry in combination with GFAP immunohistochemistry. GFAP endfeet making contact with β-dystroglycan+ vessels, extending back to an astrocyte cell body, were counted as β-dystroglycan+ astrocytes, whereas GFAP processes that extended from an astrocyte cell body but not in contact a β-dystroglycan-immunoreactive vessel were counted as β-dystroglycan−. Data were obtained from a minimum of three matched sections; three images (20× magnification) per ROI were acquired for each Lama2−/− and WT pair (n = 3).
Analysis of β1-integrin vascular coverage.
To quantify the degree of β1-integrin coverage along the vasculature, we used β1-integrin immunohistochemistry in combination with collagen IV immunohistochemistry, to visualize the adhesion receptor β1-integrin and blood vessels, respectively. Frozen cortical sections (25 μm) of WT and Lama2−/− or DAG1 CKO mice at postnatal days 21–22 were fixed in acetone at −20°C for 10 min followed by several washes with PBS. Sections were then blocked with 10% donkey serum with 0.5% Triton X-100 at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of β1-integrin immunoreactivity relative to the total area of vascular collagen IV immunoreactivity per field in the cortex was analyzed on a minimum of three matched sections; five images were acquired for each Lama2−/− and WT pair (n = 3). Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of β1-integrin coverage. Intensity thresholding of collagen IV immunoreactivity was adjusted appropriately to reflect total vascular coverage per field. Area of total β1-integrin immunoreactivity coverage was then normalized to total vascular coverage per field of view.
Analysis of AQP4 clusters along astrocytic endfeet.
To quantify the degree of AQP4 coverage along astrocytic endfeet, we used AQP4 immunohistochemistry in combination with GFAP immunohistochemistry, to visualize the osmotic regulatory channel AQP4 and astrocyte endfeet, respectively. PFA-fixed coronal sections were pretreated in PBS for 10 min at room temperature before heating at 95°C–99°C for 25 min in 1× Target Retrieval Solution citrate buffer, pH 6.0 (Dako). Sections were then cooled at room temperature for 15 min. Before blocking with 10% donkey serum with 0.5% Triton X-100 for 1 h, sections were washed with PBS to remove residual antigen retrieval solution. Sections were then blocked with 10% donkey serum with 0.5% Triton X-100 at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of AQP4 immunoreactivity relative to the total length vessel length, ensheathed by circumferential astrocytic endfeet per field in the cortex, were analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− or DAG1 CKO and WT pair (n = 3). Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of AQP4 coverage. The total vessel length per field in each region for mutants and WT controls was calculated; area of total AQP4 immunoreactivity coverage was then normalized to total vascular length per field of view.
Analysis of pericyte vascular coverage.
To quantify the degree of pericyte coverage along the vasculature, we used PDGFR-β immunohistochemistry in combination with collagen IV immunohistochemistry, to visualize pericytes and blood vessels, respectively. Frozen cortical sections (25 μm) of WT and Lama2−/− mice at postnatal days 21–22 were fixed in acetone at −20°C for 10 min followed by several washes with PBS. Sections were then blocked with 10% donkey serum with 0.5% Triton X-100 at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of PDGFR-β immunoreactivity relative to the total area of vascular collagen IV immunoreactivity per field in the frontal cortex, striatum, and hippocampus was analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− and WT pair. Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of PDGFR-β coverage. Intensity thresholding of collagen IV immunoreactivity was adjusted appropriately to reflect total vascular coverage per field. Area of total PDGFR-β immunoreactivity coverage was then normalized to total vascular coverage per field of view.
Analysis of laminin vascular coverage.
To quantify the specific endothelial laminin chain coverage along the vasculature, we used laminin α4, α5, and γ1 immunohistochemistry. Frozen cortical sections (25 μm) of WT and Lama2−/− mice at postnatal days 21–22 were fixed in acetone at −20°C for 10 min followed by several washes with PBS. Sections were then blocked with 10% donkey serum with 0.5% Triton X-100 at room temperature for 1 h. Primary antibody incubation was performed at 4°C overnight. Area of laminin α4, α5, or γ1 immunoreactivity relative to the total area of vascular immunoreactivity per field in the cortex were analyzed on a minimum of three matched sections; five images per ROI were acquired for each Lama2−/− and WT pair (n = 3). Equal intensity thresholding was applied to maximum image projections in each region for mutants and WT controls to obtain area of laminin coverage. Then, intensity thresholding was adjusted to determine total vascular (laminin) coverage per field. Area of total endothelial laminin immunoreactivity coverage was then normalized to total vascular coverage per field of view.
Western blot analysis.
Cerebral cortices were removed, dissected free of meninges, and frozen in liquid nitrogen from WT and Lama2−/− littermates at postnatal days 1, 8, 15, and 21. Tissues were pulverized immediately preceding lysis followed by lysis in 1% SDS, 20 mm Tris, pH 7.4, with protease and phosphatase inhibitor mixtures (Calbiochem) at 95°C for 10 min, with occasional trituration. Supernatant was collected after centrifugation at 16,100 relative centrifugal force for 10 min, and protein concentration was determined (Bio-Rad). Laemmli solubilizing buffer with 2% β-mercaptoethanol was added to lysates and incubated at 95°C for 5 min. Proteins were then separated by SDS-PAGE using 7.5%, 10%, or 12% acrylamide mini-gels and transferred to 0.45 μm nitrocellulose. Membranes were blocked in Tris-buffered saline with 0.1% Tween 20 (TBS-T) containing either 1% nonfat milk or 4% BSA for 1 h, followed by primary antibody incubation overnight in blocking buffer at 4°C. Membranes were washed in TBS-T, incubated for 1 h with dye-conjugated antibodies diluted at 1:5000 in blocking buffer, washed with TBS-T, and then visualized using a LICOR Odyssey Classic. Alternatively, membranes were washed with TBS-T, incubated with anti-biotin secondary antibodies diluted at 1:10,000 in blocking buffer followed by dye-conjugated streptavidin diluted at 1:5000. Relative protein level quantifications were determined using the Odyssey Infrared Imaging System (version 3.0) analysis program.
Transmission electron microscopy (TEM).
Postnatal day 22 littermate mice were processed for electron microscopy by intracardial perfusion with 4% PFA/2.5% glutaraldehyde in 0.1 m PBS. Brains were removed and postfixed overnight at 4°C. Brains were cut along the coronal plane on a Leica VT-1000 Vibratome at a thickness of 50–60 μm. Samples were processed as previously described (Relucio et al., 2012). Processed samples were viewed using a Tecnai Spirit BioTwin G2 transmission electron microscope (FEI). Images were acquired with an AMT XR-60 CCD Digital Camera System (Advanced Microscopy Techniques). The BL ultrastructure of the gliovascular interface and tight junction morphology in the hippocampus of Lama2−/− and WT control littermate mice was analyzed and compared on acquired images using Adobe Photoshop, version 5.5. Quantification of discontinuous regions of gliovascular BL in Lama2−/− and WT littermate controls was determined by counting the number of breaches per length of BL in a minimum of 10 vessel fields per genotype (n = 4). Discontinuous BL regions were identified and expressed as the number of total number of breaches over the total length observed.
Astrocyte cultures.
For experiments including dystroglycan (IIH6) and integrin β1 (Ha2/5) blocking antibodies, primary cortical astrocyte cultures were prepared from postnatal day 0–2 rat cortices (Sprague Dawley, Harlan) and cultured at 37°C, 7.5% CO2 in poly-d-lysine (PDL)-coated flasks with high-glucose DMEM containing 10% FCS and penicillin/streptomycin. Media was changed every 3–4 d for 2 weeks to obtain mixed glial cultures containing oligodendrocyte progenitor cells and microglia on astrocyte monolayers. Oligodendrocyte progenitor cells and microglia were removed from the astrocyte monolayer by physical agitation for ∼16 h on an orbital shaker. Remaining astrocytes were removed from the flasks using trypsin-EDTA and resuspended in modified Sato's medium containing 0.5% FCS (differentiation medium) and plated on Permanox 8-well chamber slides (NalgeNunc) coated with 10 μg/ml PDL at a density of 10,000 cells/well. Four days after seeding, the cells were incubated with IIH6, Ha2/5 or isotype control antibodies in differentiation medium for 2 h preceding 7 h treatment with 20 nm murine laminin-111 (Sigma-Aldrich). Cells were then fixed with methanol for 5 min at −20°C.
Primary cortical astrocytes from postnatal day 0–2 WT and Lama2−/− mice were prepared as described above. Genotyping was performed to identify homozygous WT and Lama2−/− cultures derived from individual pups. Astrocytes were resuspended in differentiation media and grown for 4 d in eight-well chamber slides coated with either PDL or with Laminin-211 (purified human merosin; Millipore). Laminin-211 slides were prepared by precoating with PDL at 37°C for 1 h, washing twice with water, and then incubating with laminin-211 at 37°C for 4 h, followed by 3 washes in PBS.
Analysis of aquaporin 4 clusters.
The surface area of aquaporin 4 (AQP4) clusters and the total area of GFAP immunoreactivity were determined on images following intensity thresholding using the National Institutes of Health ImageJ Processing and Analysis Program. Cluster areas were defined as areas of intense AQP4 immunoreactivity as determined by uniformly applied intensity thresholding. The area occupied by these intensely AQP4-immunoreactive regions, relative to total GFAP area coverage, was calculated per field. Within each experiment (5 fields per group, blocking experiments; 2 wells/group, WT/Lama2−/− experiments: minimum 3 wells/group, n = 3), images were acquired using identical parameters on an inverted Zeiss Axioplan epifluorescence microscope fitted with a 10× eyepiece and 20× (0.5 NA) objective. High-magnification AQP4 images were captured with a 63× oil-immersion objective. All images were acquired using a Zeiss Axiocam MRM digital camera controlled by Zeiss Axiovision software.
Statistical analysis.
Statistical significance of brain water content, cortical Evans Blue dye extravasation, GFAP/Iba-1-positive cell density, GFAP area, vascular coverage of pericyte (PDGFR-β)/α-SMA/β1-integrin/MECA32, vascular density complexity, or density and protein quantification by Western immunoblotting was determined using the Student's two-tailed t test. Statistical significance of AQP4 cluster analysis was determined using the one-way Holm–Sidak ANOVA for pairwise comparisons or Student's two-tailed t test where indicated. Statistical significance of TEM analysis was determined using the Mann–Whitney Rank Sum test. All analyses were conducted using SigmaPlot 11.0 (Systat Software). Bars represent the mean values; error bars indicate SEM.
Results
The development of the cerebral vasculature in the absence of laminin α2
The distribution of laminins has been well characterized in the vasculature of the adult cerebral cortex, where a composite BL is derived from endothelial and parenchymal (astrocytes and meninges) sources. The endothelial laminin heterotrimers (laminins-411 and -511) contain laminin α4 and α5, respectively, whereas the parenchymal laminin heterotrimers (laminins-111 and -211) contain laminin α1 and α2, respectively (Yousif et al., 2013). The loss of laminin a2 expression in people causes developmental brain abnormalities of which the underlying cellular disturbances remain unclear (Jimenez-Mallebrera et al., 2005). To determine whether laminin-211 influences the development and function of the cerebral vasculature, we analyzed Lama2−/− mice relative to their WT littermates. Lama2−/− mice die at ∼4 weeks of age, so vascular development was evaluated at 3 weeks of age, reflecting a late juvenile/young adult stage of development.
To confirm the lack of laminin α2 protein in Lama2−/− brains, we conducted laminin α2 immunohistochemistry (Fig. 1a,b). As expected, WT mice had laminin α2 protein most prominently in the BLs of the pia and vasculature (Fig. 1a), with higher magnifications revealing laminin α2 protein enrichment at the interface between astrocytic endfeet and vascular endothelial cells (Fig. 1b). In matched regions obtained from Lama2−/− littermate mice, no laminin α2 protein was observed in any region (Fig. 1a,b).
Figure 1.
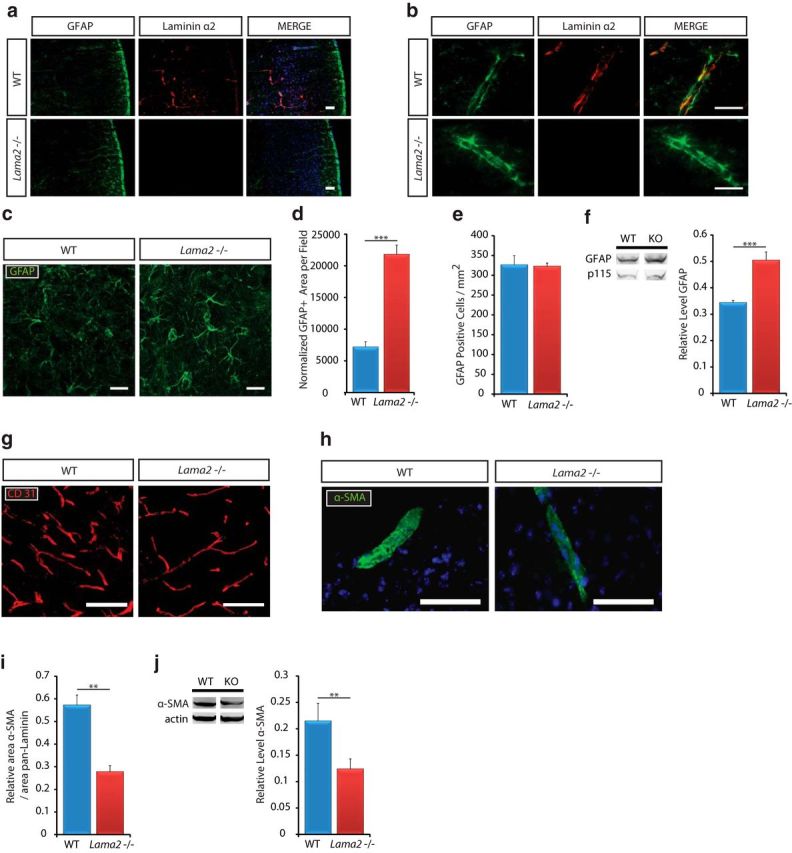
Gross gliovascular development in Lama2−/− mice. a, Laminin α2 (Lm α2) immunoreactivity (red) was observed in the pial BL and in the vasculature BL in WT cerebral cortices but was absent in Lama2−/− cerebral cortices. b, Laminin α2 immunoreactivity (Lm α2; red) was detected along blood vessels at the gliovascular interface of astrocytic endfeet (GFAP, green) of WT mice (WT) at P21, but not in Lama2−/− mice. c, g, h, Immunohistochemistry to detect astrocytes (GFAP), endothelial cells (CD31), and vSMCs (α-SMA) in the cerebral cortices of Lama2−/− mice or WT littermates at postnatal day 21. c, In Lama2−/− mice, astrocytic processes (GFAP, green) appear to be hypertrophic, characteristic of reactive gliosis. d, GFAP-immunoreactive area was increased in Lama2−/− cortices relative to that in WT cortices. e, The density of GFAP+ cells did not significantly differ between Lama2−/− and WT littermate cerebral cortices at postnatal day 21. f, Western immunoblotting for GFAP and p115 (protein-loading control) in lysates from P21 Lama2−/− and WT cerebral cortices. Protein levels of GFAP were significantly elevated in Lama2−/− mice. g, Vascular endothelial cell density (CD31, red) appeared grossly normal, with decreased branching complexity (see Results). h, α-SMA immunoreactivity (green) in Lama2−/− mice appears to be decreased compared with that in WT littermates. i, Quantification of area of α-SMA immunoreactivity coverage relative to laminin-111 (pan-laminin immunoreactivity as vasculature area) in Lama2−/− and WT at postnatal day 21. Coverage of α-SMA was reduced in Lama2−/− mice. j, Quantification of α-SMA by Western blot; α-SMA protein levels in Lama2−/− cerebral cortical lysates are significantly reduced relative to those from WT littermates. **p ≤ 0.01, ***p < 0.001, Student's t test. Error bars indicate SEM. Scale bars, 50 μm.
Next, to determine whether the absence of laminin α2 influenced the development of the cerebral vasculature, several key cell types were assessed. We evaluated the morphology of astrocytes and their processes to elucidate potential changes in the gliovascular interface by conducting GFAP immunohistochemistry (Fig. 1c) at postnatal day 21. Overall, Lama2−/− mice had increased GFAP immunoreactivity suggestive of reactive gliosis, which was particularly apparent in astrocytic processes lining the vasculature of the cerebral cortex and included robust hypertrophied processes enveloping the vasculature. We quantified the area of GFAP immunoreactivity relative to the number of GFAP+ cells. Lama2−/− mice had significantly increased GFAP area relative to the total number of GFAP+ cells (Fig. 1d) in the cortex (21,827.14 ± 1426.28 μm2 in Lama2−/− vs 7192.65 ± 882.17 μm2 in WT; n = 3; p < 0.0001). However, in Lama2−/− mice, the total number of cortical GFAP+ cells (Fig. 1e) was not significantly different relative to WT littermate controls (323.24 ± 7.6 cells/mm2 in Lama2−/− vs 326.56 ± 23.21 cells/mm2 in WT; n = 3; p = 0.91). We further quantified GFAP levels by Western immunoblotting and found that at postnatal day 21 protein levels of GFAP were significantly higher in Lama2−/− cerebral cortices than in WT littermate cerebral cortices (Fig. 1f; 0.505 ± 0.031 in Lama2−/− vs 0.344 ± 0.008 in WT, n = 6, p < 0.001).
We next evaluated the gross morphology of the vasculature in the cerebral cortex by performing immunohistochemistry to detect the endothelial cell protein CD31 followed by morphometric analysis of vascular density and complexity (Fig. 1g). In Lama2−/− mice, cortical vascular density was not significantly different (4.619 ± 0.35 in Lama2−/− vs 5.587 ± 0.20 in WT, n = 3, p = 0.08). However, cortical vascular complexity, measured as branch points per 100 μm segments, was significantly lower in Lama2−/− cerebral cortices relative to that in WT littermate controls (0.929 ± 0.12 in Lama2−/− vs 1.374 ± 0.14 in WT, n = 3, p = 0.006). To visualize VSMCs that are associated with larger-caliber blood vessels, we conducted α-SMA immunohistochemistry (Fig. 1h). The blood vessels that were positive for α-SMA in Lama2−/− mice had a decreased intensity of immunoreactivity, suggestive of reduced levels of α-SMA protein, which would be indicative of decreased VSMC contractility. Next, α-SMA coverage along blood vessels was quantified at postnatal day 21. Lama2−/− mice had significantly reduced (51.5% reduction) α-SMA immunoreactivity relative to blood vessel area (Fig. 1i; 0.278 ± 0.028 in Lama2−/− vs 0.573 ± 0.044 in WT; n = 3; p = 0.005). We further quantified α-SMA at postnatal day 21 by Western immunoblotting and found that α-SMA protein levels in Lama2−/− mice were significantly lower relative to those in WT littermates (Fig. 1j; 0.124 ± 0.019 in Lama2−/− vs 0.215 ± 0.033 in WT; n = 6; p = 0.01). Together, these data confirm that laminin α2 is absent in Lama2−/− mice and that all major cell types in the glial-vascular unit that contribute to the barrier properties are present in Lama2−/− mice. However, Lama2−/− brains have hallmarks of reactive gliosis, altered gliovascular morphology, and reduced α-SMA-positive blood vessels, suggestive of BBB dysfunction and/or immaturity.
Laminin regulates the permeability of the BBB
To investigate the impact of laminin α2 on the functional integrity of the BBB, we assessed several indicators. First, an acute 30 min circulation of the azo-dye Evans Blue was performed at postnatal day 21. Evans Blue dye functions by binding protein components of the circulating blood, such as serum albumin, which can then cross into the brain parenchyma in pathological situations where the BBB is compromised. Using identical acquisition parameters, large amounts of Evans Blue were observed in the parenchymal space in the cortex, striatum, hippocampus, and cerebellum of Lama2−/− mice relative to that in WT littermates (Fig. 2a). Furthermore, extraction of Evans Blue dye from the whole cerebral cortex followed by a reading of optical density at 620 nm (Fig. 2b) indicated a statistically significant difference in the level of Evans Blue in Lama2−/− cortices relative to that in WT littermates (3.013 ± 0.571 in Lama2−/− vs 0.978 ± 0.057 in WT; n = 3; p = 0.024). To further investigate the possibility of a compromised BBB, we assessed the presence of leukocytes, which should not enter the cerebral parenchyma under normal physiological conditions. We performed immunohistochemistry against the common leukocyte antigen CD45 and against laminin-111 to visualize the BL of blood vessels (Fig. 2c, left). In Lama2−/− brains, CD45+ cells can be detected outside of the vasculature in the parenchymal space. Because CD45 can be expressed by activated microglial under some circumstances, we further assessed potential leukocyte infiltration by performing immunohistochemistry against the ε chain of the T-cell receptor-associated CD3 complex, to detect leukocytes within the parenchymal space, in conjunction with laminin-111 immunohistochemistry to visualize the BL of blood vessels. In Lama2−/− brains, CD3+ cells can be detected outside of the vasculature in the parenchymal space (Fig. 2c, right), further confirming leukocyte transmigration. A leaky BBB with accompanying leukocyte infiltration would be predicted to elicit an innate immune response within the brain. We therefore analyzed microglial activation using immunohistochemistry against the monocyte-specific protein Iba-1 in the striatum of postnatal day 21 mice (Fig. 2d). In Lama2−/− brains, Iba-1-positive microglia are more plentiful and have an amoeboid morphology, characteristic of activated microglia, whereas Iba-1-positive microglia in WT mice have long branch-like processes characteristic of resting microglia (Fig. 2d, far right) and are less numerous (Fig. 2e; 445.91 ± 13.57 cells/mm2 in Lama2−/− vs 223.78 ± 9.95 cells/mm2 in WT; n = 3; p < 0.001). Together, these data indicate that the BBB in Lama2−/− mice has compromised selective permeability characteristics, leading to activation of resident immune cells within the CNS.
Figure 2.
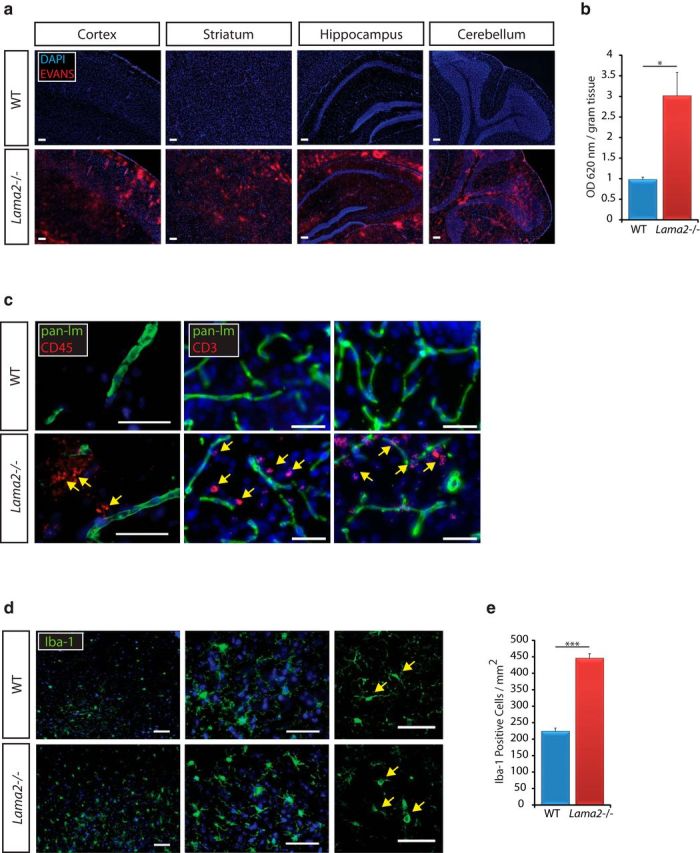
Increased BBB permeability in Lama2−/− mice. a, Evans Blue dye (red) is seen in the parenchymal space in coronal sections taken from the cerebral cortex, striatum, hippocampus, and cerebellum in Lama2−/− and WT littermate mice at P21. Residual signal in WT was restricted to endothelial lumen. b, Quantification of Evans Blue dye extracted from Lama2−/− and WT cerebral cortices at P21. The level of Evans Blue in Lama2−/− cortices was significantly elevated compared with that in WT littermates. c, Immunohistochemistry to detect laminin-111 (“pan-lm” as antibody detects laminin α1, β1, and γ1 subunits, green), CD45 (red, left), and CD3 (red, right) in Lama2−/− and WT mice at postnatal day 21. CD45+ and CD3+ cells were observed in the cerebral parenchyma of Lama2−/− but not WT mice. d, Immunohistochemistry to detect microglia (Iba-1, green) in the striatum of Lama2−/− and WT mice at postnatal day 21. The striatum of Lama2−/− mice had more microglia with the amoeboid morphology characteristic of activated microglia (right) relative to those in WT littermates. e, Quantification of Iba-1+ cells (microglia) in the striatum of Lama2−/− and WT mice at postnatal day 21. Iba-1+ cells per area in the striatum of Lama2−/− mice were significantly elevated compared with those in WT littermates. *p < 0.05, ***p < 0.001, Student's t test. n = 3. Scale bars, 50 μm.
Impaired maturation of the BBB in Lama2−/− mice
To determine whether laminins influenced the maturation of the BBB, we evaluated features of the cerebral vasculature that are known to be regulated by the induction and maturation of barrier features. We conducted immunohistochemistry against Glut-1, a transporter that regulates the metabolic environment of the cerebral parenchyma and whose expression normally coincides with the BBB maturation. Relative to that in WT littermates, however, we observed no significant change in Glut-1 immunoreactivity in Lama2−/− mice at postnatal day 21 (Fig. 3a; 0.561 ± 0.067 in Lama2−/− vs 0.536 ± 0.026 in WT, n = 3, p = 0.74). Protein analysis by Western immunoblotting also indicated no significant changes in Glut-1 levels at postnatal days 1, 8, 15, and 21 (Fig. 3b). Relative Glut-1 protein levels at postnatal day 1 (P1) were 6.878 ± 1.42 in Lama2−/− cerebral cortices vs 8.303 ± 2.54 in WT cortices (n = 3, p = 0.650); at P8 were 9.163 ± 0.81 in Lama2−/− versus 11.077 ± 2.48 in WT (n = 3, p = 0.504); at P15 were 1.501 ± 0.25 in Lama2−/− vs 1.062 ± 0.16 in WT (n = 3, p = 0.211); and at P21 were 1.179 ± 0.12 in Lama2−/− vs 0.912 ± 0.08 in WT (n = 3, p = 0.136). These data suggest that laminin α2 does not significantly affect overall Glut-1 protein levels or localization at the gliovascular interface.
Figure 3.
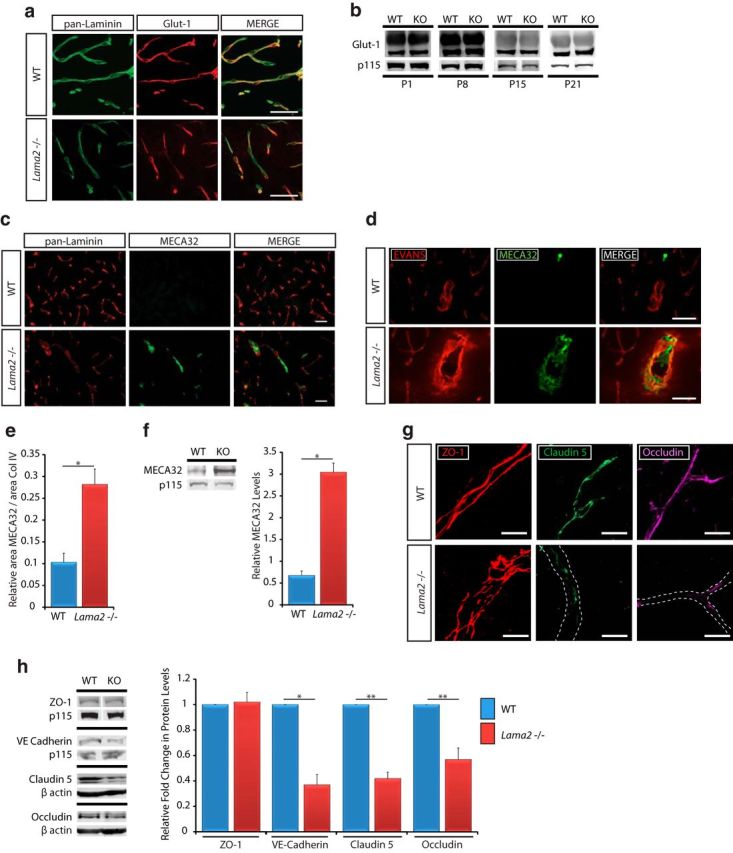
Impaired maturation of the BBB in Lama2−/− mice. a, Immunohistochemistry to detect Glut-1 and any subunit of laminin-111 ((laminin α1, β1, or γ1 subunits; i.e., pan-laminin) in the vascular endothelial cells in coronal sections at postnatal day 21 in Lama2−/− and WT cerebral cortices. Glut-1 immunoreactivity in Lama2−/− mice appears to be comparable with that in WT control littermates. b, Western immunoblotting for Glut-1 at postnatal day 1 (P1), P8, P15, and P21 using lysates from Lama2−/− and WT cerebral cortices. No changes in Glut-1 levels were observed. p115 blots are shown as loading controls. c, Immunohistochemistry to detect any subunit of laminin-111 and MECA-32 in the vascular endothelial cells in coronal sections from postnatal day 21 Lama2−/− and WT mice. MECA-32 immunoreactivity was detected in Lama2−/− brain vascular endothelial cells but not in WT. d, The presence of Evans Blue dye (red) in the parenchymal space is associated with MECA32 immunoreactivity (green) in Lama2−/− cerebral cortices but not in WT. e, Quantification of area of MECA32 immunoreactivity coverage relative to laminin-111 coverage (pan-laminin, to indicate total vasculature area) in Lama2−/− and WT cerebral cortices at postnatal day 21. Coverage of MECA32 was increased in Lama2−/− mice. f, Western immunoblotting for MECA-32 and p115 (protein-loading control) in lysates from P21 Lama2−/− and WT cerebral cortices. MECA-32 levels were significantly elevated in Lama2−/− cerebral cortices compared with those in WT controls. g, Immunohistochemistry to detect the interendothelial tight junction proteins: ZO-1 (red), Claudin-5 (green), and Occludin (magenta) in Lama2−/− and WT cerebral cortices at postnatal day 21. High-magnification confocal imaging reveals ZO-1 morphology in altered in Lama2−/−; however, protein levels were not significantly decreased. In contrast, immunoreactivity for Claudin-5 and Occludin is significantly reduced in Lama2−/− mice. White dashed line indicates the path of the blood vessel. h, Representative Western immunoblots for ZO-1, VE-Cadherin, Claudin-5, and Occludin (p115 and β-actin, protein-loading controls) at P21 in lysates from Lama2−/− and WT cerebral cortices. VE-Cadherin, Claudin-5, and Occludin levels were significantly decreased in Lama2−/− cerebral cortices. *p < 0.05, **p < 0.01, Student's t test. Error bars indicate SEM; n = 6. Scale bars: a, c, e, 50 μm; g, 10 μm.
To further evaluate BBB development, we conducted immunohistochemistry against MECA32 (i.e., plasmalemma vesicle-associated protein, Plvap), which is expressed in the peripheral vasculature throughout development but ceases to be expressed in the cerebrovasculature during embryonic development (corresponding with the establishment of barrier properties). MECA32/Plvap is involved in vesicular transport in endothelial cells and is highly expressed in permeable vessels of peripheral tissues (Ioannidou et al., 2006; Shue et al., 2008; Daneman et al., 2010b). In the postnatal CNS, however, MECA32 expression is limited to circumventricular organs, which reside outside of the confines of the BBB (Schulz and Engelhardt, 2005). However, reexpression of MECA32 has been observed in pathological conditions where CNS inflammation is prominent, such as experimental autoimmune encephalitis (EAE) and other models of neuroinflammation (Cannella et al., 1991; Engelhardt et al., 1994; Yu et al., 2012). Immunoreactivity for MECA32 was observed in a subset of large vessels distributed throughout the cortex, striatum, and hippocampus of postnatal day 21 Lama2−/− mice, whereas no MECA32 immunoreactivity was detected in WT mice outside of the circumventricular organs (Fig. 3c). We further characterized MECA32 vascular coverage at postnatal day 21 and found that Lama2−/− mice had significantly increased levels of MECA32 relative to blood vessel area (Fig. 3d; 0.282 ± 0.035 in Lama2−/− versus 0.103 ± 0.021 in WT; n = 3; p = 0.012). We also performed MECA32 immunohistochemistry on brain sections from mice that had been injected with Evans Blue dye and found that MECA32-immunoreactive large blood vessels were always associated with Evans Blue leakage into the brain parenchyma (Fig. 3e). It should be noted that in Figure 3e, we substantially increased the brightness of the Evans Blue signal to visualize all blood vessels in the field; therefore, the Evans Blue difference between WT and knock-out is no longer apparent in these images. Finally, we analyzed MECA32 by Western immunoblotting and found that MECA32 protein levels were significantly higher in Lama2−/− cerebral cortices at postnatal day 21 than in WT littermate cerebral cortices (Fig. 3f; 3.041 ± 0.21 in Lama2−/− vs 0.677 ± 0.1 in WT, n = 3, p = 0.029).
To evaluate the development of interendothelial tight junctions, which act as physical barriers against passage through vascular fenestrations, we conducted immunohistochemistry against tight junction components zonula occludens 1 (ZO-1), claudin-5, and occludin. Although the formation of tight junctions was apparent in Lama2−/− mice and WT controls at postnatal day 21, differences in morphology were observed in high-magnification confocal images in Lama2−/− mutants (Fig. 3g). ZO-1 immunoreactivity was distributed in a pattern consistent with continuous adherens complexes in both Lama2−/− and WT; however, Lama2−/− mice displayed morphological abnormities, including regions of increased junctional hypertrophy and undulation. In addition, we observed decreased immunoreactivity of claudin-5 and occludin in Lama2−/− mice relative to WT littermates (Fig. 3g). Additionally, we quantified overall protein levels of tight junction components by Western immunoblotting. Quantification of ZO-1 protein levels by Western immunoblotting indicated that ZO-1 levels (Fig. 3h) did not significantly differ between Lama2−/− mice and WT littermate controls (0.971 ± 0.233 in Lama2−/− vs 1.001 ± 0.167 in WT, 1.02 ± 0.3-fold of WT, n = 3, p = 0.92), indicating that ZO-1 morphology, but not production, differs in Lama2−/− mice. We further quantified the protein levels of the adherens junction components, VE-cadherin, claudin-5, and occludin. Western blots indicated that VE-cadherin levels were significantly lower in Lama2−/− mice relative to those in WT littermates at postnatal day 21 (Fig. 3h; 2.198 ± 0.51 in Lama2−/− vs 5.976 ± 0.881 in WT, 0.37 ± 0.08-fold of WT, n = 3, p = 0.021). Claudin-5 levels were also significantly lower in Lama2−/− mice relative to those in WT littermate control mice (Fig. 3h; 0.153 ± 0.025 in Lama2−/− vs 0.379 ± 0.065 in WT, 0.42 ± 0.05-fold of WT, n = 6, p = 0.005). Additionally, Western blots indicated that occludin levels were significantly lower in Lama2−/− mice relative to those in WT littermates (Fig. 3h; 0.576 ± 0.142 in Lama2−/− versus 0.995 ± 0.139 in WT, 0.57 ± 0.09-fold of WT, n = 6, p = 0.01).
Inappropriate MECA32 expression in the postnatal CNS vasculature and significant decreases in the adherens junction components VE-cadherin, claudin-5, and occludin, with additional morphological differences in ZO-1-immunoreactive tight junctions, together indicate that the absence of laminin α2 impacts BBB maturation and further suggests that perturbed vascular endothelial maturation or homeostasis may contribute to increased BBB permeability.
Laminin regulates cerebral vasculature pericyte coverage
The cerebral vasculature is extensively covered by pericytes, which express high levels of the PDGFR-β, a necessary signaling receptor for pericyte recruitment (Lindblom et al., 2003). The absence or loss of vascular pericyte coverage is highly correlated with vascular leakage at the BBB (Armulik et al., 2010). To assess pericyte coverage throughout different regions of the CNS, we used PDGFR-β and collagen IV immunohistochemistry to visualize pericytes and the BL of the cerebral vasculature, respectively. First, we performed laminin α2, PDGFR-β, and collagen IV immunohistochemistry to assess whether laminin α2 is colocalized with pericytes along the vasculature in WT mice (Fig. 4a). We observed a high level of colocalization of laminin α2 and PDGFR-β, which suggests a close association of laminin α2 with pericytes and furthermore supports the possibility that pericytes themselves may express laminin α2 in vivo. Next, we sought to quantify pericyte coverage along the cerebral vasculature in the cerebral cortex, striatum, and hippocampus of Lama2−/− and WT mice. Pericyte coverage along blood vessels, visualized in confocal three dimensional reconstructions of PDGR-β/collagen IV immunohistochemistry, appeared profoundly lower in Lama2−/− cerebral cortices than that in WT littermates (Fig. 4b). Next, pericyte coverage along blood vessels was quantified in mice at postnatal day 21. Lama2−/− mice had significantly reduced pericyte coverage relative to blood vessel area in all regions evaluated (Fig. 4c): in the cortex, a 60.47% reduction (0.17 ± 0.011 in Lama2−/− vs 0.43 ± 0.014 in WT; n = 3; p = 0.007); in the striatum, a 33.34% reduction (0.08 ± 0.007 in Lama2−/− vs 0.25 ± 0.003 in WT; n = 3; p = 0.001); and in the hippocampus, a 56.52% reduction (0.20 ± 0.014 in Lama2−/− vs 0.46 ± 0.023 in WT; n = 3; p = 0.001) relative to WT littermates. We then compared pericyte coverage of the cerebral vasculature in developing littermates at postnatal days 1, 8, 15, and 21 to determine whether changes in pericytes coverage in the vasculature of Lama2−/− mice occurred earlier in postnatal development (Fig. 4d). Throughout postnatal development, the vasculature of Lama2−/− cerebral cortices had decreased pericyte coverage compared with that in WT littermates. At postnatal day 1, Lama2−/− mice already had significantly reduced pericyte coverage in the cortical vasculature relative to WT littermates (0.103 ± 0.016 in Lama2−/− vs 0.278 ± 0.06 in WT; n = 3; p = 0.048). Together, these data indicate that the absence of laminin α2 significantly perturbs the ability of pericytes to interact with the cerebrovasculature during postnatal development. The fact that pericyte coverage appears to be disturbed in numerous brain regions, and is apparent from birth, furthermore supports our observations that BBB leakage is widespread and not confined to particular regions as a localized “damage response” but instead is a developmental abnormality.
Figure 4.
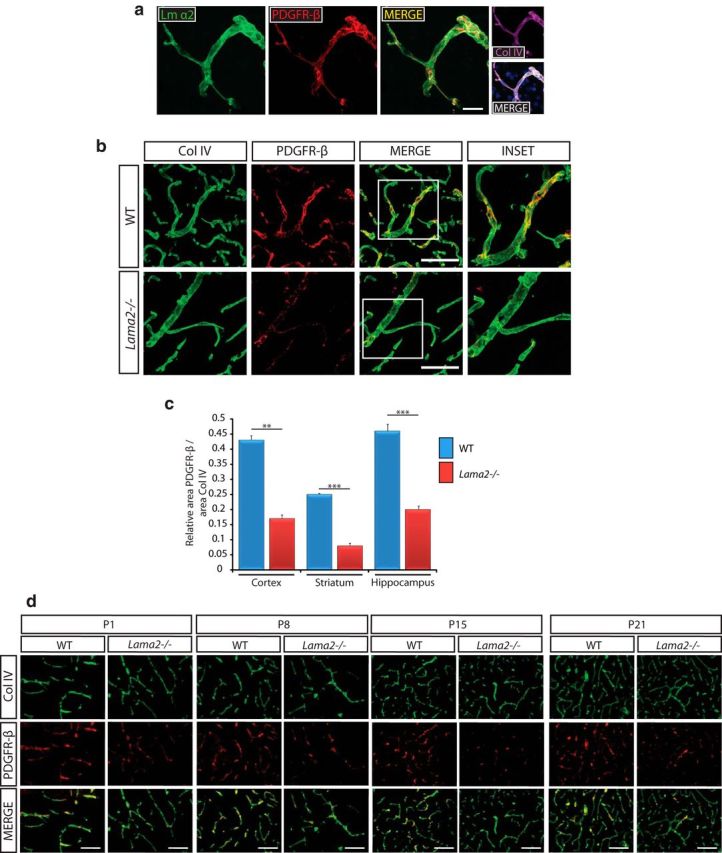
Decreased pericyte coverage of the vasculature in Lama2−/− brains. a, Immunohistochemistry to detect laminin α2 (Lm α2, green), collagen type IV (Col IV, purple), and pericytes (PDGFRβ, red) in WT brains. Lm α2 colocalizes with the pericyte marker PDGFRβ along the BL of brain vascular endothelial cells. b, Immunohistochemistry to detect PDGFRβ (red) and Col IV (green) associated with the vasculature in Lama2−/− and WT brains at postnatal day 21. Lama2−/− mice have decreased PDGFRβ immunoreactivity along the brain vasculature. c, Quantification of area of PDGFRβ coverage relative to Col IV area (vasculature area) in Lama2−/− and WT brains at postnatal day 21. Pericyte coverage was reduced in the cerebral cortex, striatum, and hippocampus of Lama2−/− mice. d, Immunohistochemistry to detect PDGFRβ (red) and Col IV (green) associated with the vasculature in Lama2−/− and WT brains through development (postnatal days 1, 8, 15, and 21). Lama2−/− mice have decreased PDGFRβ immunoreactivity along the vasculature from birth throughout postnatal development. **p < 0.01, ***p < 0.001, Student's t test. Error bars indicate SEM; n = 3. Scale bars: a, 20 μm; b–d, 50 μm.
Channel localization at astrocytic endfeet is altered in Lama2−/− mice
Astrocytic endfeet use the DGC to facilitate the localization of homeostatic channels, such as AQP4 to the gliovascular interface. The laminin receptor dystroglycan acts as a central component of the DGC, physically linking AQP4 through intracellular adaptor proteins, such as α-dystrobrevin. Previous studies have provided evidence that physical interactions with α-dystrobrevin are required to maintain polarized distributions of AQP4 at astrocytic endfeet (Lien et al., 2012). To investigate the role of laminin α2 in localizing channels at astrocytic endfeet, we conducted immunohistochemistry against AQP4 in conjunction with GFAP to visualize circumferential astrocytic processes at the gliovascular interface. We observed decreased immunoreactivity of AQP4 (Fig. 5a) at astrocytic endfeet in Lama2−/− cerebral cortices relative to that in WT littermates at postnatal day 21. Lama2−/− mice had significantly decreased levels of AQP4 immunoreactivity distributed at vascular-associated astrocytic endfeet (Fig. 5b; 3.628 ± 0.97 in Lama2−/− vs 14.213 ± 1.21 in WT; n = 3; p = 0.002). However, relative protein levels of AQP4 were not significantly different between groups at postnatal day 21 (Fig. 5c; 1.847 ± 0.31 in Lama2−/− vs 1.99 ± 0.23 in WT; n = 3; p = 0.730). These data indicate that AQP4 protein levels do not significantly differ, but that AQP4 localization, and perhaps clustering, is perturbed. To address whether AQP4 function was compromised, we evaluated brain water content, which we found to be significantly elevated in Lama2−/− mice compared with that in WT littermates (Fig. 5d; 80.15 ± 0.380 in Lama2−/− vs 78.32 ±. 0.654 in WT; n = 9; p = 0.028). These data suggest that laminin α2 is required for both maintaining the polarization of AQP4 channels at astrocytic endfeet and facilitating the function of AQP4 to regulate cerebral osmotic homeostasis.
Figure 5.

Channel localization at astrocytic endfeet is altered in Lama2−/− mice. a, Immunohistochemistry to detect AQP4 (red) and GFAP (green) in Lama2−/− and WT astrocytic endfeet. AQP4 immunoreactivity was decreased in Lama2−/− mice. b, Quantification of AQP4 immunoreactivity along astrocytic endfeet relative to vessel length. AQP4 coverage along astrocytic endfeet was significantly reduced in Lama2−/− mice relative to littermate controls. **p < 0.01, Student's t test. Error bars indicate SEM; n = 3. c, Western immunobloting for AQP4 and p115 (protein-loading control) in lysates from postnatal day 21 Lama2−/− and WT cerebral cortices. No significant differences in protein levels were observed. d, Quantification of brain water content in cortices isolated from Lama2−/− and WT mice. Brain water content was elevated in Lama2−/− mice relative to littermate controls. *p < 0.05, Student's t test. Error bars indicate SEM; n = 3. e–h, Laminin-binding adhesion receptors located at astrocytic endfeet were evaluated in Lama2−/− and WT cerebral cortices at postnatal day 21. e, Immunohistochemistry to detect β-dystroglycan (red) and GFAP (green) in Lama2−/− and WT astrocytic endfeet. f, Western immunoblotting for β-dystroglycan and p115 (protein-loading control) in lysates from postnatal day 21 Lama2−/− and WT cerebral cortices. No significant differences in protein levels were observed. g, Immunohistochemistry to detect β1-integrin (red) and collagen IV (Col IV, green) in Lama2−/− and WT along astrocytic endfeet and vasculature. h, Quantification of area of β1-integrin immunoreactivity coverage relative to collagen IV immunoreactivity (collagen IV to indicate total vasculature area) in Lama2−/− and WT cerebral cortices at postnatal day 21. Vascular coverage of β1-integrin was reduced in Lama2−/− mice. i, Western immunoblotting for β1-integrin and p115 (protein-loading control) in lysates from postnatal day 21 Lama2−/− and WT cerebral cortices. No significant differences in protein levels were observed. Scale bars: a, 20 μm; e, g, 50 μm.
Next, we sought to characterize the adhesion receptors that are known to interact with laminin α2 at the gliovascular interface by conducting β-dystroglycan and β1-integrin immunohistochemistry (Fig. 5e,g). In Lama2−/− mice at postnatal day 21, there was no change in β-dystroglycan immunoreactivity along the vasculature (Fig. 5e), whereas a decrease in β1-integrin vascular coverage was observed (Fig. 5g). Furthermore, protein lysates prepared from cerebral cortices at postnatal day 21 were evaluated by Western blot, and no significant differences in total β-dystroglycan protein levels were observed in Lama2−/− mice (Fig. 5f; 90.723 ± 11.053 in Lama2−/− vs 92.097 ± 4.704 in WT; n = 3; p = 0.915). Increased levels of cleaved 30 kDa dystroglycan, mediated by MMPs 2 and 9, are observed during periods of disturbed BBB function in EAE (Agrawal et al., 2006), and we noted that there was a slight increase in the relative amount of the cleaved 30 kDa β-dystroglycan fragment in Lama2−/− cerebral cortices, although not significantly so (13.74 ± 0.29% in Lama2−/− vs 9.53 ± 2.23% in WT; n = 3; p = 0.135). To investigate whether the absence of Lama2 influenced the degree to which astrocyte endfeet were associated with β-dystroglycan, we quantified astrocytic endfeet processes in association with β-dystroglycan immunoreactivity at the gliovascular interface. We found a significant decrease in the percentage of β-dystroglycan positive astrocytes in association with blood vessels in Lama2−/− mice relative to those in WT littermates (45.035 ± 3.19% in Lama2−/− vs 53.613 ± 1.104% in WT; n = 4; p = 0.031). These results suggest that disturbed adhesive interactions at astrocytic endfeet may contribute to the dysfunction of the neurovascular unit of Lama2−/− mice.
To further investigate potential changes in β1-integrins in Lama2−/− brains, we quantified β1-integrin vascular coverage. Lama2−/− mice had significantly reduced (a 27.45% reduction) β1-integrin vascular-associated immunoreactivity relative to blood vessel area at postnatal day 21 (Fig. 5h; 0.259 ± 0.018 in Lama2−/− vs 0.357 ± 0.019 in WT; n = 3; p = 0.02). We further quantified overall levels of β1-integrin in the cerebral cortex at postnatal day 21 by Western immunoblotting and found no significant differences (Fig. 5i; 1.39 ± 0.13 in Lama2−/− vs 1.92 ± 0.34 in WT; n = 6; p = 0.128). It should be noted that β1 integrin is expressed in astrocytic endfeet but is also expressed in pericytes, oligodendrocytes, microglia, neural stem cells, and vascular endothelial endothelial cells; thus, changes in β1 integrin localized to the BBB are not likely to be reflected in whole-cortex lysates. These data suggest that laminin α2 is required for the correct localization of AQP4 at astrocyte endfeet and contributes to the ability of β1-integrins to localize along the vasculature, presumably also in astrocytes but perhaps additionally through changes in pericyte and/or endothelial cell integrin distribution. In contrast, β-dystroglycan localization at the gliovascular interface may be compensated for by other ECM components, such as α1-subunit-containing laminins.
Lama2−/− mice have breaches in the gliovascular BL
The gliovascular BL acts to physically separate the resident cells of the perivascular environment while also creating a continuous matrix that links each cell type together. To investigate the role of laminin α2 on the structure and composition of the gliovascular BL, we used two strategies: (1) ultrastructure analysis via TEM and (2) immunohistochemistry against known components of the gliovascular BL. We observed no obvious differences in the BL or gross vascular defects in capillary ultrastructure of Lama2−/− mice (Fig. 6a) compared with WT mice. However, we observed perturbed tight junction morphology in blood vessels of Lama2−/− mice in contrast to those WT littermates (Fig. 6b; tight junctions colorized green). In Lama2−/− mice, many tight junctions appeared to be narrow and undulating, extending throughout the endothelia lumen. Moreover, large-caliber vessels of Lama2−/− mice had more endocytic vesicles than typical, indicative of transport dysfunction (Fig. 6b, red arrows). In addition, detachment and/or discontinuity of both the parenchymal and the endothelial BL were readily observed in large vessels of Lama2−/− mice (Fig. 6c, white asterisks; parenchymal BL, blue; endothelial BL, red). Lama2−/− mice had discontinuous regions in the gliovascular BL (182 breaches were found upon inspection of 109,030.13 nm of BL in Lama2−/−, whereas 0 breaches were found upon inspection of 106,145.12 nm of BL in WT; Mann–Whitney U statistic = 0.00; T = 10.00; n = 4; p = 0.029).
Figure 6.
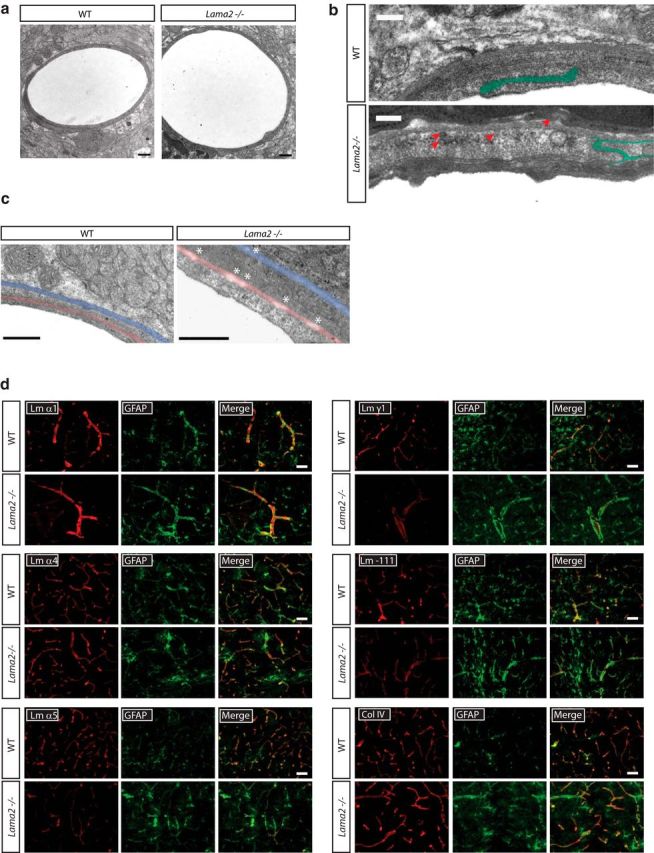
Altered structure and composition of the BBB BL in Lama2−/− mice. a–c, Transmission electron micrographs to evaluate the ultrastructure of the gliovascular interface in Lama2−/− and WT littermate brains at postnatal day 21. a, No obvious defects were apparent in the brain capillary BLs of Lama2−/− mice. b, Tight junction morphology (green) in Additionally, endocytic vesicles (red arrows) were observed in Lama2−/− vascular endothelial cells. c, Regions of the parenchymal BL (blue) and endothelial BL (red) are discontinuous (white asterisk) in large-caliber blood vessels of Lama2−/− mice. d, Immunohistochemistry to detect components of the BL in conjunction with GFAP in the cerebral cortices of Lama2−/− and WT mice at postnatal day 21. Laminin α1, α4, α5, γ1, laminin-111 (a polyclonal antibody that recognizes laminin α1, β1, and γ1 subunits), and collagen IV (col IV) were detected in the gliovascular BL of Lama2−/− and WT brains. Laminin α5 immunoreactivity was reduced in Lama2−/− mice. Scale bars: a, 500 nm; b, 100 nm; c, 500 nm; d, 50 μm.
Next, we conducted immunohistochemistry to detect α1, α4, α5, and γ1 laminin subunits, in conjunction with GFAP immunohistochemistry, to determine whether the levels of other laminins were altered in Lama2−/− brains at postnatal day 21 (Fig. 6d). To characterize endothelial cell laminins, we quantified vascular coverage of α4-, α5-, and γ1-containing laminins. Lama2−/− mice had no significant changes in laminin α4 vascular coverage relative to that in WT littermates (0.94 ± 0.02 in Lama2−/− vs 0.963 ± 0.005 in WT; n = 3; p = 0.443). In contrast, Lama2−/− mice had significantly reduced laminin α5 vascular coverage relative to that in WT littermates (0.664 ± 0.033 in Lama2−/− vs 0.966 ± 0.014 in WT; n = 3; p < 0.001). Because laminin-511 production by vascular endothelial cells usually increases postnatally during BBB maturation (Yousif et al., 2013), a deficit in laminin-511 levels could reflect the lack of BBB maturity in Lama2−/− brains. Lama2−/− mice also had significantly reduced laminin γ1 vascular coverage relative to that in WT littermates (0.442 ± 0.027 in Lama2−/− vs 0.936 ± 0.013 in WT; n = 3; p < 0.001). We also observed a modest increase in laminin α1 immunoreactivity in Lama2−/− mice relative to that in WT littermates (data not shown), suggesting that the loss of laminin-211 led to an increase in laminin-111, possibly as a means to compensate. No change in collagen IV immunoreactivity was observed in Lama2−/− mice relative to controls. These data suggest that laminin α2 is required for the proper formation and/or maintenance of the parenchymal and endothelial BL within the cerebrovasculature of Lama2−/− mice.
Laminin–dystroglycan interactions potentiate the ability of astrocytes to cluster AQP4
To investigate the ability of laminins to facilitate the polarization of homeostatic channels, such as AQP4, we used two astrocyte-based assays. First, we used primary astrocytes isolated from rat neonates to test the effects of blocking dystroglycan and/or β1-containing integrins with blocking antibodies (IIH6 and Ha2/5, respectively) before the acute administration of soluble laminin, which normally induces clusters of AQP4 (GuaDAGno and Moukhles, 2004). We conducted AQP4 and GFAP immunohistochemistry (Fig. 7a) and then uniformly applied intensity thresholding to determine cluster areas (i.e., areas of intense AQP4 immunoreactivity). AQP4 cluster area relative to total GFAP+ cell coverage area was then calculated per field. After 7 h of laminin incubation, we observed a significant increase in AQP4 clusters in control cells, as has been reported previously (Fig. 7b; 0.100 ± 0.009 in control plus laminin-111 vs 0.023 ± 0.003 in control alone, n = 3, p < 0.001). In the presence of dystroglycan-blocking antibodies (IIH6), there was a substantial decrease in laminin-induced AQP4 clusters (0.0233 ± 0.003 IIH6 vs 0.100 ± 0.009 in control plus laminin-111, n = 3, p < 0.001). Blocking the β1-integrin subunit also resulted in a significant decrease in laminin-induced AQP4 clusters, although not as large a decrease as was observed by blocking dystroglycan (0.051 ± 0.007 Ha2/5 vs 0.100 ± 0.009 in control plus laminin-111, n = 3, p < 0.001). The largest decrease in laminin-induced AQP4 clusters, however, resulted from blocking both dystroglycan and β1-integrins concurrently (0.015 ± 0.002 dual antibody blockade vs 0.100 ± 0.009 in control plus laminin-111, n = 3, p < 0.001).
Figure 7.

Lama2−/− vascular endothelial cell is abnormal in contrast to that observed in WT littermates. Lama2−/− tight junctions appear to be elongated with long, spindly regions traversing the endothelium. Laminin–dystroglycan interactions potentiate the ability of astrocytes to cluster AQP4. a, Immunohistochemistry to detect GFAP (red) and AQP4 (green) on primary astrocytes treated either laminin-111 alone or in conjunction with prior addition of blocking antibodies to dystroglycan (IIH6) or β1 integrins (Ha2/5). Colorized AQP4 clusters (bottom, red) depicts regions where AQP4 immunoreactivity intensity was above threshold value (i.e., cluster region). b, Quantification of relative coverage by AQP4 clusters after no treatment, treatment with laminin alone, or with laminin in conjunction blocking antibody pretreatments. Dystroglycan-blocking antibodies, and, to a lesser extent, integrin-blocking antibodies, reduce the ability of laminin to induce AQP4 clusters. **p < 0.01, ***p < 0.001, Holm–Sidak ANOVA. Error bars indicate SEM; n = 3. c, Immunohistochemistry to detect GFAP (red) and AQP4 (green) in astrocytes derived from Lama2−/− and WT mice. Lama2−/− astrocytes were deficient in their ability to assemble AQP4 clusters in long-term cultures; however, growth on purified laminin-211 substrate rescued the ability of Lama2−/− astrocytes to form AQP4 clusters. Colorized AQP4 clusters (bottom, red) depicts regions where AQP4 immunoreactivity intensity was above threshold value. d, Quantification of relative coverage by AQP4 clusters in astrocytes derived from Lama2−/− and WT mice on poly-d-lysine (PDL) or laminin-211 substrates. Relative coverage by AQP4 clusters was significantly reduced in Lama2−/− astrocytes grown on PDL relative to WT astrocytes. **p ≤ 0.01, Student's t test. n.s., Not significant. Error bars indicate SEM; n = 5. The ability of Lama2−/− astrocytes to form AQP4 clusters was rescued by laminin-211 substrate. Scale bars, 25 μm.
We next prepared primary astrocytes from Lama2−/− and WT littermate brains to test the ability of endogenous astrocytic laminin α2 to organize AQP4. When cultured over several days, astrocytes organized AQP4 into mesh-like networks and clusters at the interface between the cell and the plastic; this clustering ability was enhanced by growing the astrocytes on slides coated with laminin (Fig. 7c, left). It should be noted that astrocytes grown on PDL substrates express endogenous laminins-111 and -211, which contribute to the “baseline” of clusters in this long term culture. In contrast, astrocytes from Lama2−/− neonates had a marked deficit in AQP4 clusters when grown on PDL (Fig. 7c, right, PDL). However, the ability of Lama2−/− astrocytes to cluster AQP4 was rescued by growing the cells on laminin-211 (Fig. 7c, right, laminin). To quantify changes in the ability of Lama2−/− astrocytes to cluster AQP4, we conducted AQP4 and GFAP immunohistochemistry and then uniformly applied intensity thresholding to determine cluster areas (i.e., areas of intense AQP4 immunoreactivity). AQP4 cluster area relative to total GFAP+ cell coverage area was then calculated per field. When astrocytes were grown on PDL, we observed significantly decreased relative AQP4 clusters in astrocytes derived from Lama2−/− mice compared with WT astrocytes (Fig. 7d; 0.063 ± 0.025 in Lama2−/− vs 0.126 ± 0.038 in WT, n = 5; p = 0.01). In contrast, the ability of Lama2−/− astrocytes to cluster AQP4 on laminin substrate was similar to WT astrocytes grown on a laminin substrate (Fig. 7d; 0.386 ± 0.075 in Lama2−/− vs 0.491 ± 0.061 in WT, n = 5; p = 0.312). Together, these data suggest that laminin interactions with dystroglycan and, to a lesser extent, β1-integrins, potentiate the ability of astrocytes to aggregate AQP4 channels into clusters. The ability of laminin substrates to rescue AQP4 clustering in Lama2−/− astrocytes furthermore indicates that Lama2−/− have appropriate levels of functional laminin receptors that are responsive to the exogenously applied laminin.
Dystroglycan regulates the function of the BBB
To investigate the role of the adhesion receptor dystroglycan in the formation and function of the BBB in vivo, we used a conditional knock-out strategy to eliminate DAG1 expression from all neural lineage cells, including astrocytes. Permeability of the BBB was assessed using Evans Blue dye circulated through the vasculature. In the absence of neural lineage dystroglycan, extravasation of Evans blue into the brain parenchyma was apparent in sections obtained from the frontal cortex, striatum, and hippocampus (Fig. 8a). Furthermore, a statistically significant increase in the level of Evans Blue in extracts from cerebral cortices was found in DAG1 conditional knock-outs relative to that in WT littermates (Fig. 8b; 2.493 ± 0.109 in DAG1 CKO vs 1.164 ± 0.143 in WT; n = 4; p < 0.001). Considering the altered pericyte coverage observed in Lama2−/− mice, we next sought to elucidate whether laminin–dystroglycan interactions regulate vascular pericyte cell coverage. We performed PDGFRβ and collagen IV immunohistochemistry in matched cortical sections from DAG1 conditional knock-outs and WT littermates at postnatal day 23 (Fig. 8c) and found decreased PDGFRβ immunoreactivity along blood vessels in DAG1 conditional mutants relative to the robust coverage observed in WT mice (Fig. 8c). Quantification revealed significantly reduced (∼50.0% reduction) pericyte coverage along blood vessels in DAG1 conditional mutants relative to that in WT littermates (Fig. 8d; 0.235 ± 0.036 in DAG1 CKO vs 0.47 ± 0.018 in WT; n = 4; p = 0.001). To elucidate whether dystroglycan regulates the induction of transporters associated with barrier development, we next conducted Glut-1 immunohistochemistry (Fig. 8e). We observed a decrease in Glut-1 immunoreactivity in DAG1 conditional knock-out brains relative to that in WT littermates (a 38.83% reduction), suggesting a developmental delay or deficit in BBB maturation (Fig. 8f; 0.323 ± 0.022 in DAG1 CKO vs 0.528 ± 0.055 in WT; n = 3; p = 0.026).
Figure 8.
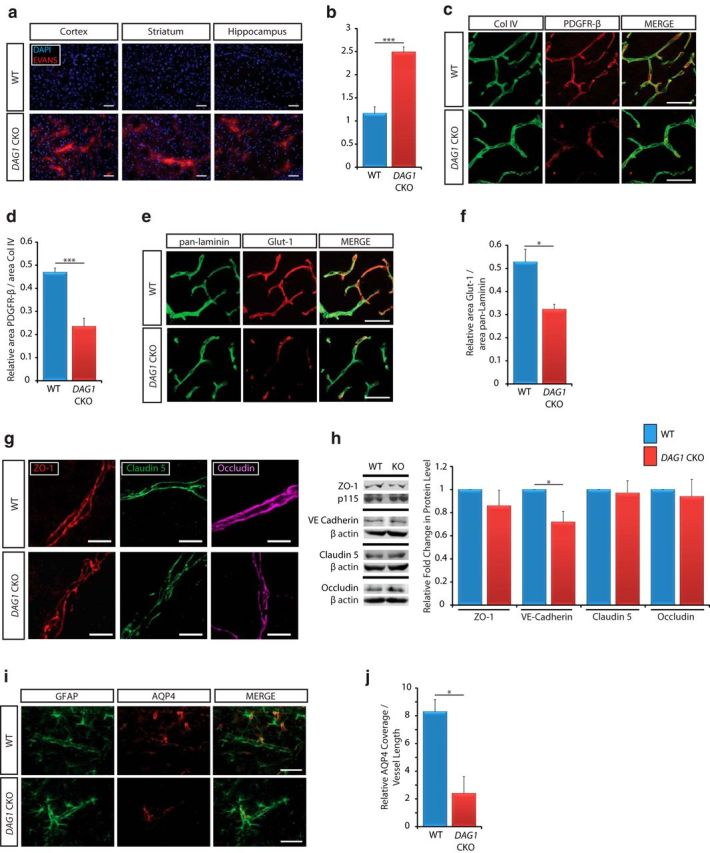
Dystroglycan regulates the function of the BBB. a, Evans Blue dye (red) in the brain parenchyma is apparent in coronal sections taken from cerebral cortex, striatum, and hippocampus in DAG1 CKO mice at postnatal day 23. Residual signal in WT littermate control mice was restricted to the endothelial lumen. b, Quantification of Evans Blue dye extracted from DAG1 CKO and WT cerebral cortices at postnatal day 21. The level of Evans Blue in DAG1 conditional knockout cortices was significantly elevated compared with that in WT littermates (n = 4). c, PDGFRβ immunohistochemistry to detect pericytes (red) and collagen IV immunohistochemistry (col IV, green) to visualize the endothelial BL associated with the vasculature in DAG1 CKO and WT cerebral cortices at postnatal day 23. In DAG1 CKO mice, PDGFRβ immunoreactivity was notably decreased. d, Quantification of area of PDGFRβ coverage relative to collagen IV area (total vasculature area) in DAG1 and WT brains at postnatal day 21. Pericyte coverage was reduced in the cerebral cortex of DAG1 conditional knockout mice (n = 4). e, Immunohistochemistry to detect Glut-1 (red) and laminin-111 (pan-Laminin, green) in vascular endothelial cells in the cerebral cortex from DAG1 CKO and WT littermates. Glut-1 immunoreactivity appeared to be less intense in DAG1 CKO brain vascular endothelial cells than in WT controls. f, Quantification of area of Glut-1 immunoreactivity coverage relative to laminin-111 (pan-Laminin, vasculature area) in DAG1 conditional knockout and WT at postnatal day 21. Coverage of Glut-1 was increased in DAG1 CKO mice (n = 3). g, Immunohistochemistry to detect the interendothelial tight junction proteins: ZO-1 (red), Claudin-5 (green), and Occludin (magenta) in DAG1 CKO and WT cerebral cortices at postnatal day 21. High-magnification confocal images reveal ZO-1, Claudin-5, and Occludin morphology; scale bar, 10 microns. h, Representative Western immunoblots for ZO-1, VE-Cadherin, Claudin-5, and Occludin (p115 and β-actin, protein-loading controls) at postnatal day 21 in lysates from DAG1 CKO and WT cerebral cortices. No significant changes in ZO-1, Claudin-5, or Occludin protein levels were observed; however, VE-Cadherin levels were significantly reduced in DAG1 CKO mice (n = 5). i, Immunohistochemistry to detect GFAP (green) and AQP4 (red) in the cerebral cortices of DAG1 CKO and WT mice. DAG1 CKO mice have decreased AQP4 immunoreactivity and increased GFAP immunoreactivity. j, Quantification of AQP4 coverage along astrocytic endfeet relative to vessel length. AQP4 coverage along astrocytic endfeet was significantly reduced in DAG1 CKO mice relative to WT controls (n = 3). *p < 0.05, ***p < 0.001, Student's t test. Error bars indicate SEM. Scale bars, 50 μm (except in g).
To evaluate the development of interendothelial tight junctions, we conducted immunohistochemistry against ZO-1, claudin-5, and occludin. Although the formation of tight junctions was apparent in DAG1 CKO mice at postnatal day 21, the junctional morphology in DAG1 CKO mutants appeared to be discontinuous with regions of focal hypertrophy and undulation observed in high-magnification confocal images (Fig. 8g). We also quantified the levels of junctional proteins by Western immunoblotting (Fig. 8h). ZO-1 protein levels did not significantly differ between DAG1 knock-out mice and WT littermates (Fig. 8h; 0.641 ± 0.15 in DAG1 CKO vs 0.728 ± 0.07 in WT, 0.86 ± 0.14-fold of WT, n = 3, p = 0.404). Claudin-5 protein levels did not significantly differ between DAG1 knock-out mice and WT littermates (Fig. 8h; 1.503 ± 0.227 in DAG1 CKO vs 1.581 ± 0.22 in WT, 0.97 ± 0.102-fold of WT, n = 5, p = 0.67). Occludin protein levels did not significantly differ between DAG1 knock-out mice and WT littermate controls (Fig. 8h; 2.16 ± 0.057 in DAG1 CKO vs 2.54 ± 0.42 in WT, 0.94 ± 0.15-fold of WT, n = 5, p = 0.43). However, the protein level of VE-cadherin was significantly reduced in DAG1 CKO mutants (Fig. 8h; 0.584 ± 0.089 in DAG1 CKO vs 0.81 ± 0.035 in WT, 0.72 ± 0.09-fold of WT, n = 5, p = 0.04).
To determine whether dystroglycan is necessary to maintain the polarized organization of astrocytic endfeet in vivo, we conducted AQP4 immunohistochemistry in conjunction with GFAP immunohistochemisty (Fig. 8i). We observed decreased immunoreactivity of AQP4 in DAG1 conditional knock-out brains at postnatal day 21, whereas in WT littermates AQP4 channels were highly enriched at GFAP+ processes that line the vasculature (Fig. 8i). It should also be noted that, in DAG1 conditional knock-out brains, perivascular GFAP immunoreactivity was increased, characteristic of reactive gliosis and swollen astrocytic endfeet. To characterize the spatial distribution of AQP4, we quantified AQP4 along astrocyte endfeet at postnatal day 21 and found that DAG1 conditional knock-out mice had significantly decreased AQP4 distributed along astrocytic endfeet relative to that in WT littermates (Fig. 8j; 2.406 ± 1.203 in DAG1 CKO vs 8.295 ± 0.878 in WT; n = 3; p = 0.017). Together, these data indicate that dystroglycan expressed in neural lineage cells (presumably in astrocytes) is required to develop functional barrier properties and to localize homeostatic channel proteins and metabolic transporters at astrocytic endfeet. In addition, although many interendothelial junctional proteins were expressed at normal levels, VE-cadherin levels were decreased and morphological abnormalities in junctional protein localization were observed, which may contribute to of the observed changes in vascular permeability. Our findings furthermore indicate that dystroglycan-mediated interactions, likely with laminin-211, promote pericyte coverage along the cerebral cortical vasculature (Fig. 9).
Figure 9.

Overview of cellular and molecular changes in the BBB that occur in the absence of laminin α2 expression. A, Pericyte (red) coverage of the brain vasculature (green) is decreased in Lama2−/− mice. B, Laminin α2 is produced by astrocytes (purple) and, possibly, pericytes (red) at the gliovascular interface. In the absence of laminin α2 expression (Lama2−/−), both parenchymal (pBL) and endothelial (eBL) are present but contain breaches. Vascular endothelial cells in Lama2−/− brains have elevated levels of endocytic vesicles, indicative of transcytosis and BBB immaturity, and altered tight junction composition and morphology. Astrocytic processes appear hypertrophic in Lama2−/− brains, with astrocytic endfeet lacking the appropriate polarization of aquaporin4 (AQP4) channels (blue ovals).
Discussion
ECM proteins are thought to contribute to BBB function by generating the physical barrier of the gliovascular BL. Here we report that α2-subunit-containing laminins not only have an important contribution to the physical integrity of the gliovascular BL but also promote BBB maturation by increasing pericyte interactions with the vascular endothelium. The failure to produce α2-containing laminins results in a leaky BBB with characteristics of immaturity: expression of embryonic vascular proteins, such as MECA32, limited pericyte coverage, and poorly polarized channel distribution in astrocytes. Although vascular endothelial tight junctions are present in Lama2−/− brains, some observations suggest tight junction immaturity, including reduced levels of VE-cadherin, occludin, and claudin-5, a thinner and undulating tight junction appearance by TEM, differences in junctional protein immunoreactive morphology, and innappropriate appearance of endocytic vesicles. As the removal of dystroglycan expression in neural cells leads to increased BBB permeability in conjunction with a similar set of gliovascular cellular disturbances as in Lama2−/− brains, laminin–dystroglycan interactions are likely to be mediating a significant portion of a requirement for α2-containing laminins. However dystroglycan-deficient brains may even be more profoundly compromised than laminin-deficient brains, as Glut-1 distribution, though normal in Lama2−/− cerebral vasculature, is perturbed in DAG1 conditional knock-outs. Together, these findings provide new insight into the cell and molecular changes in the brains of people with Lama2 mutations or with congenital dystroglycanophathies in which muscular dystrophy is accompanied by seizures, cognitive deficits, and neurodevelopmental disorders, such as autism.
Laminins, pericytes, and BBB maturation
In the current study, we provide evidence that Lama2−/− mice have profound alterations in BBB development and function. For example, Evans Blue dye from the circulatory system quickly crosses into the brain parenchyma in Lama2−/− mice, with all regions of the CNS examined thus far (the frontal cortex, striatum, hippocampus, and cerebellum) exhibiting the “leaky brain” phenotype. However, decreased pericyte coverage along the vasculature may underlie this increased BBB permeability, rather than a failure of vascular endothelial cells to form tight junctions. However, tight junctions in the Lama2−/− vasculature, although present, may be functionally compromised to some degree as they have various morphological abnormalities and decreased levels of VE-cadherin, occludin, and claudin-5. Perturbed pericyte coverage in Lama2−/− mice appears to be a developmental defect, as it was observed from birth onwards until early adulthood. α-SMA levels were also lower in Lama2−/− brains, suggesting that laminins normally contribute to VSMC maturation, which is in agreement with a recent finding that astrocyte-derived laminins promote VSMC maturation (Chen et al., 2013). A conditional knock-out of β1-containing integrins in both pericytes and VSMCs using PDGFRβ-Cre resulted in decreased VSMC differentiation, providing further support for a role for laminin receptor interactions in promoting VSMC maturation (Abraham et al., 2008). However, a separate more recent study, using the mice that lack laminin expression selectively in astrocytes, reported increased levels of α-SMA immunoreactivity associated with the cerebral vasculature, suggesting that laminins can also suppress VSMC maturation (Yao et al., 2014). The mechanism by which laminins could either decrease or increase VSMC maturation remains unclear; however, these studies clearly highlight laminins as key regulators of VSMCs.
In the cerebral cortex of adult Lama2−/− mice, we observed inappropriate expression of MECA32 (i.e., Plvap), which normally associates with caveolae and regulates endothelial vesicular trafficking (Komarova and Malik, 2010). MECA32 is found postnatally in various tissues with fenestrated endothelium, but in the brain MECA32 expression is strictly embryonic, with MECA32 loss coinciding with BBB maturation (Hallmann et al., 1995). Coinciding with inappropriate MECA32 expression, we also detected an overabundance of endocytotic vesicles in the vasculature endothelial cells of Lama2−/− mice. Pericytes have the ability to suppress Plvap expression (Daneman et al., 2010b), indicating that the decreased pericyte coverage in Lama2−/− mice may result in dysregulation of vascular endothelial cell transport mechanisms. Together, these findings indicate a novel role for laminin α2 in the regulation of pericytes. In addition, the loss of pericyte coverage in DAG1 conditional knock-out mice closely mirrors that in Lama2−/− mice, suggesting that pericyte defects observed in Lama2−/− mice, at least in part, are a result of losing key gliovascular laminin–dystroglycan interactions.
Laminin α2, as part of the laminin-211 heterotrimer, contributes to the parenchymal and composite BLs of large-caliber blood vessels and capillaries, respectively (Sixt et al., 2001). Laminin α2 is produced by astrocytes and can be localized to astrocytic endfeet that run along cerebral vessels, and thus is often referred to as an astrocytic laminin. However, in the complete absence of laminin-211 expression from astrocytes in laminin γ1 conditional knock-outs, laminin α2 protein is still observed in protein lysates from the cerebral cortex, albeit at lower levels than normal, suggesting that other cell types in addition to astrocytes contribute laminin α2 (Chen et al., 2013). Indeed, purified pericytes from the CNS have been observed to express several ECM proteins in culture, including laminin α2 (Daneman et al., 2010a); intriguingly, in vivo studies suggest that, in the absence of pericytes, laminin α2 protein levels at the gliovascular interface are substantially lower (Jucker et al., 1996; Armulik et al., 2010). And loss of laminin γ1 gene expression from astrocytes did not significantly impact PDGFR-β immunoreactivity along the vasculature of the CNS (Chen et al., 2013), in contrast to what was observed in our study with a global loss of Lama2 expression from all brain cells. Additionally, an astrocyte-specific conditional β1-integrin knock-out resulted in a profound loss of astrocytic laminin-111 and -211 expression; however, these β1-integrin astrocyte knock-out mice had no change in BBB permeability (although pericyte coverage per se was not addressed) (Robel et al., 2009). Thus, it is worth considering in future studies whether the pericyte abnormalities observed in Lama2−/− brains reflects failed autocrine interactions between pericytes and pericyte-secreted laminins, and whether disturbances in brain vascular endothelial cells, at least in part, result from a loss of laminin from pericytes.
Laminin regulates gliovascular ECM composition
An extensive network of ECM contacts all of the resident cell types of the gliovascular interface facilitating both BBB structural integrity and cellular communication. In the current study, we observed structural defects (i.e., areas of discontinuous or breached BL) in the parenchymal and endothelial BLs of large-caliber blood vessels of Lama2−/− brains. However, it remains unclear whether these breaches reflect a failure in BL development or reflect a failure in BL maintenance. Furthermore, the relative contribution of various laminin subunits appeared to be altered in the Lama2−/− gliovascular interface, indicating that the observed phenotypes could be quite complex and, in some instances, may reflect changes in other laminin subunits that arise as a secondary consequence of changes in laminin α2 expression. Most strikingly in the Lama2−/− cerebral vasculature, we observed decreased immunoreactivity of laminin α5. The laminin α5 subunit is a key component of laminin-511, which along with laminin-411 comprises the brain vascular endothelial laminins (Sixt et al., 2001). Unlike laminin-411 expression, which is present in embryonic brain vascular endothelial cells, laminin-511 expression appears later and can be found concentrated in areas of high pericyte recruitment (Sixt et al., 2001). It remains unclear whether the dysregulation of laminin-511 levels in Lama2−/− brains is related specifically to the poor pericyte coverage or simply may reflect the more global failure of brain vascular endothelial maturity. Overall, these data support a role for α2-containing laminins (i.e., laminin-211) in orchestrating the integrity and composition of BLs, including the endothelial BL, at the gliovascular interface.
Laminin and CNS immune surveillance
The CNS vasculature is a dynamic, yet highly controlled, system that helps regulate the brain parenchyma. Normally, the vasculature BL acts as a physical barrier to transendothelial migration of leukocytes. In Lama2−/− mice, however, we observed leukocytes in the brain parenchyma, indicating that peripheral immune cells were inappropriately crossing the BBB. Laminin α5 has been shown to negatively regulate the ability of T cells to extravasate into the brain parenchyma during EAE (Wu et al., 2009), suggesting that the relatively low level of laminin α5 seen in the Lama2−/− brain vasculature may contribute to inappropriate T-cell infiltration. The proteolytic cleavage of β-dystroglycan by MMPs during EAE has also been shown to facilitate transendothelial migration of leukocytes (Agrawal et al., 2006). In the current study, we observed an increased level of cleaved β-dystroglycan in Lama2−/− brains, suggesting that inappropriate dystroglycan cleavage may contribute to the observed increased T-cell extravasation. On the other hand, an increase in dystroglycan cleavage may also result as a consequence of BBB breakdown and the subsequent innate immune activation, as activated microglia are known to secrete MMPs responsible for dystroglycan cleavage (Agrawal et al., 2006). Additionally, immune cell migration has been observed at sites devoid of pericyte coverage, suggesting that the decreased pericyte coverage in Lama2−/− mice contributes to T-cell migration into the brain parenchyma (Wang et al., 2006; Voisin et al., 2010).
When the BBB is compromised, microglia, the resident immune cells of the CNS, encounter “foreign” substances, which result in microglial process contraction and activation. Lama2−/− brains had more iba1+ microglia, with many more of them displaying an activated morphology indicating that microglia had been activated by the presence of infiltrating immune cells. As stated previously, MECA32, usually confined to embryonic brain vascular endothelial cells, was observed in Lama2−/− brain vasculature at postnatal day 21 and even as late as postnatal day 28 (data not shown). In pathological conditions, such as Alzheimer's disease and EAE, inflammation is known to trigger reexpression of MECA32 in the compromised vasculature of the CNS (Cannella et al., 1991; Engelhardt et al., 1994; Sparks et al., 2000; Yu et al., 2012). Thus, inflammatory changes (e.g., increased microglial cell activation) may contribute to reexpression of MECA32 in Lama2−/− brains.
Laminin–dystroglycan interactions regulate astrocytic endfeet architecture
Components of the DGC contribute to the establishment and maintenance of appropriate astrocytic endfoot architecture with polarized localization of homeostatic channels. In both Lama2 and conditional DAG1 mutants, we observed disturbances in the polarized localization of AQP4, indicating that laminin binding to dystroglycan is required to regulate AQP4 polarization. In support of this, we found that, by blocking dystroglycan, or, to a lesser extent, astrocyte β1-integrin, we significantly attenuated the ability of cultured astrocytes to cluster AQP4 in response to exogenously provided laminin. Additionally, in longer-term cultures, with the absence of endogenously produced laminin α2, astrocytes derived from Lama2−/− brains were defective in their ability to form AQP4 aggregates and clusters (but were rescued by exogenous laminin). Finally, although AQP4 distribution was clearly different in Lama2−/− brains, we did not observe changes in overall AQP4 protein levels, consistent with our hypothesis that laminins influence AQP4 distribution but not AQP4 protein production or stability. These findings support existing reports that other components of the DGC (e.g., dystrophin, syntrophins, and dystrobrevin) also regulate glial endfeet channel polarity, such that osmotic regulation and neuronal metabolism are disturbed in their absence (Vajda et al., 2002; Amiry-Moghaddam et al., 2003a, b; Noël et al., 2009; Lien et al., 2012). In the current study, we also found that brain water content is increased in Lama2−/− mice, providing further support for the hypothesis that loss of AQP4 polarity contributes to osmotic disturbances.
In conclusion, we identified new roles for laminin and dystroglycan at the CNS gliovascular interface, where laminin–dystroglycan interactions regulate BBB development, structure, and function, at least in part by regulating pericyte coverage of brain vascular endothelial cells. We furthermore provide the first report of a specific role for α2-containing laminins (laminin-211) in gliovascular BL structure and function, with implications for the CNS pathologies that result from Lama2 mutations in congenital muscular dystrophies (MDC1A). We conclude that laminin-mediated signals from nonendothelial cells, such as astrocytes and pericytes, underlie successful BBB development/function, a finding that has implications for understanding the connection between BBB breakdown and conditions with neurodegenerative pathology, including stroke, Alzheimer's disease, and multiple sclerosis. Future studies will be of interest to determine whether laminins act as extrinsic organizers of the spatial arrangement of the gliovascular interface or directly modulate cellular phenotypes through signaling mechanisms downstream of adhesion receptors, such as dystroglycan.
Footnotes
This work was supported by the National Multiple Sclerosis Society. We thank Ms. Susan Van Horn of the Stony Brook University Electron Microscopy facility for assistance with TEM, Dr. Guo-Wei Tan of the Multiphoton Confocal Microscope facility for helpful suggestions, Dr. Nicholas Carpino and Dr. Lydia Sorokin for the kind gifts of CD3 antibody and laminin antibodies, respectively, and members of the H.C. laboratory for helpful discussions.
The authors declare no competing financial interests.
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Abraham S, Kogata N, Fässler R, Adams RH. Integrin beta1 subunit controls mural cell adhesion, spreading, and blood vessel wall stability. Circ Res. 2008;102:562–570. doi: 10.1161/CIRCRESAHA.107.167908. [DOI] [PubMed] [Google Scholar]
- Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allamand V, Guicheney P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, Lama2gene coding for alpha2 chain of laminin) Eur J Hum Genet. 2002;10:91–94. doi: 10.1038/sj.ejhg.5200743. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A. 2003a;100:2106–2111. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003b;100:13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol. 1999;147:645–658. doi: 10.1083/jcb.147.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buteică E, Roşulescu E, Burada F, Stãnoiu B, Zãvãleanu M. Merosin-deficient congenital muscular dystrophy type 1A. Rom J Morphol Embryol. 2008;49:229–233. [PubMed] [Google Scholar]
- Cannella B, Cross AH, Raine CS. Adhesion-related molecules in the central nervous system: upregulation correlates with inflammatory cell influx during relapsing experimental autoimmune encephalomyelitis. Lab Invest. 1991;65:23–31. [PubMed] [Google Scholar]
- Caro PA, Scavina M, Hoffman E, Pegoraro E, Marks HG. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. AJNR Am J Neuroradiol. 1999;20:324–326. [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A, Strickland S. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol. 2013;202:381–395. doi: 10.1083/jcb.201212032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood–brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010a;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010b;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Zoppo GJ, Milner R. Integrin–matrix interactions in the cerebral microvasculature. Arterioscler Thromb Vasc Biol. 2006;26:1966–1975. doi: 10.1161/01.ATV.0000232525.65682.a2. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Koziol JA. Vascular matrix adhesion and the blood–brain barrier. Biochem Soc Trans. 2006;34:1261–1266. doi: 10.1042/BST0341261. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Conley FK, Butcher EC. Cell adhesion molecules on vessels during inflammation in the mouse central nervous system. J Neuroimmunol. 1994;51:199–208. doi: 10.1016/0165-5728(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Fujii Y, Sugiura C, Fukuda C, Maegaki Y, Ohno K. Sequential neuroradiological and neurophysiological studies in a Japanese girl with merosin-deficient congenital muscular dystrophy. Brain Dev. 2011;33:140–144. doi: 10.1016/j.braindev.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Girardin F. Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin Neurosci. 2006;8:311–321. doi: 10.31887/DCNS.2006.8.3/fgirardin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadagno E, Moukhles H. Laminin-induced aggregation of the inwardly rectifying potassium channel, Kir4.1, and the water-permeable channel, AQP4, via a dystroglycan-containing complex in astrocytes. Glia. 2004;47:138–149. doi: 10.1002/glia.20039. [DOI] [PubMed] [Google Scholar]
- Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995;202:325–332. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- Ioannidou S, Deinhardt K, Miotla J, Bradley J, Cheung E, Samuelsson S, Ng YS, Shima DT. An in vitro assay reveals a role for the diaphragm protein PV-1 in endothelial fenestra morphogenesis. Proc Natl Acad Sci U S A. 2006;103:16770–16775. doi: 10.1073/pnas.0603501103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F. Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci. 2005;62:809–823. doi: 10.1007/s00018-004-4510-4. [DOI] [PubMed] [Google Scholar]
- Jucker M, Tian M, Norton DD, Sherman C, Kusiak JW. Laminin alpha 2 is a component of brain capillary basement membrane: reduced expression in dystrophic dy mice. Neuroscience. 1996;71:1153–1161. doi: 10.1016/0306-4522(95)00496-3. [DOI] [PubMed] [Google Scholar]
- Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- Lien CF, Mohanta SK, Frontczak-Baniewicz M, Swinny JD, Zablocka B, Górecki DC. Absence of glial alpha-dystrobrevin causes abnormalities of the blood–brain barrier and progressive brain edema. J Biol Chem. 2012;287:41374–41385. doi: 10.1074/jbc.M112.400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Nabeshima Y, Takeda S. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 1997;415:33–39. doi: 10.1016/S0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- Miyagoe-Suzuki Y, Nakagawa M, Takeda S. Merosin and congenital muscular dystrophy. Microsc Res Tech. 2000;48:181–191. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<181::AID-JEMT6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Nico B, Paola Nicchia G, Frigeri A, Corsi P, Mangieri D, Ribatti D, Svelto M, Roncali L. Altered blood–brain barrier development in dystrophic MDX mice. Neuroscience. 2004;125:921–935. doi: 10.1016/j.neuroscience.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Noël G, Tham DK, Moukhles H. Interdependence of laminin-mediated clustering of lipid rafts and the dystrophin complex in astrocytes. J Biol Chem. 2009;284:19694–19704. doi: 10.1074/jbc.M109.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TS, Karnovsky MJ. Fine structural localization of a blood–brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relucio J, Menezes MJ, Miyagoe-Suzuki Y, Takeda S, Colognato H. Laminin regulates postnatal oligodendrocyte production by promoting oligodendrocyte progenitor survival in the subventricular zone. Glia. 2012;60:1451–1467. doi: 10.1002/glia.22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Mori T, Zoubaa S, Schlegel J, Sirko S, Faissner A, Goebbels S, Dimou L, Götz M. Conditional deletion of beta1-integrin in astroglia causes partial reactive gliosis. Glia. 2009;57:1630–1647. doi: 10.1002/glia.20876. [DOI] [PubMed] [Google Scholar]
- Rurak J, Noel G, Lui L, Joshi B, Moukhles H. Distribution of potassium ion and water permeable channels at perivascular glia in brain and retina of the Large(myd) mouse. J Neurochem. 2007;103:1940–1953. doi: 10.1111/j.1471-4159.2007.04886.x. [DOI] [PubMed] [Google Scholar]
- Schulz M, Engelhardt B. The circumventricular organs participate in the immunopathogenesis of experimental autoimmune encephalomyelitis. Cerebrospinal Fluid Res. 2005;2:8. doi: 10.1186/1743-8454-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shue EH, Carson-Walter EB, Liu Y, Winans BN, Ali ZS, Chen J, Walter KA. Plasmalemmal vesicle associated protein-1 (PV-1) is a marker of blood–brain barrier disruption in rodent models. BMC Neurosci. 2008;9:29. doi: 10.1186/1471-2202-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood–brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933–946. doi: 10.1083/jcb.153.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DL, Kuo YM, Roher A, Martin T, Lukas RJ. Alterations of Alzheimer's disease in the cholesterol-fed rabbit, including vascular inflammation: preliminary observations. Ann N Y Acad Sci. 2000;903:335–344. doi: 10.1111/j.1749-6632.2000.tb06384.x. [DOI] [PubMed] [Google Scholar]
- Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, Iivanainen A, Sorokin L, Risling M, Cao Y, Tryggvason K. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22:1194–1202. doi: 10.1128/MCB.22.4.1194-1202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Vajda Z, Pedersen M, Füchtbauer EM, Wertz K, Stødkilde-Jørgensen H, Sulyok E, Dóczi T, Neely JD, Agre P, Frøkiaer J, Nielsen S. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc Natl Acad Sci U S A. 2002;99:13131–13136. doi: 10.1073/pnas.192457099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin MB, Probstl D, Nourshargh S. Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. J Pathol. 2010;176:482–495. doi: 10.2353/ajpath.2010.090510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203:1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, Loser K, Beissert S, Georges-Labouesse E, Sorokin LM. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009;15:519–527. doi: 10.1038/nm.1957. [DOI] [PubMed] [Google Scholar]
- Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. doi: 10.1038/ncomms4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure–genotype relationship in Fukuyama-type congenital muscular dystrophy. Brain Dev. 2008;30:59–67. doi: 10.1016/j.braindev.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Yousif LF, Di Russo J, Sorokin L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh Migr. 2013;7:101–110. doi: 10.4161/cam.22680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Corbett B, Yan Y, Zhang GX, Reinhart P, Cho SJ, Chin J. Early cerebrovascular inflammation in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2012;33:2942–2947. doi: 10.1016/j.neurobiolaging.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104:311–324. doi: 10.1016/S0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]