

Abstract
The hypothalamic suprachiasmatic nucleus (SCN) plays a pivotal role in the mammalian circadian clock system. Bmal1 is a clock gene that drives transcriptional-translational feedback loops (TTFLs) for itself and other genes, and is expressed in nearly all SCN neurons. Despite strong evidence that Bmal1-null mutant mice display arrhythmic behavior under constant darkness, the function of Bmal1 in neuronal activity is unknown. Recently, periodic changes in the levels of intracellular signaling messengers, such as cytosolic Ca2+ and cAMP, were suggested to regulate TTFLs. However, the opposite aspect of how clock gene TTFLs regulate cytosolic signaling remains unclear. To investigate intracellular Ca2+ dynamics under Bmal1 perturbations, we cotransfected some SCN neurons with yellow cameleon together with wild-type or dominant-negative Bmal1 using a gene-gun applied for mouse organotypic cultures. Immunofluorescence staining for a tag protein linked to BMAL1 showed nuclear expression of wild-type BMAL1 and its degradation within 1 week after transfection in SCN neurons. However, dominant-negative BMAL1 did not translocate into the nucleus and the cytosolic signals persisted beyond 1 week. Consistently, circadian Ca2+ rhythms in SCN neurons were inhibited for longer periods by dominant-negative Bmal1 overexpression. Furthermore, SCN neurons transfected with a Bmal1 shRNA lengthened, whereas those overexpressing wild-type Bmal1 shortened, the periods of Ca2+ rhythms, with a significant reduction in their amplitude. BMAL1 expression was intact in the majority of neighboring neurons in organotypic cultures. Therefore, we conclude that proper intrinsic Bmal1 expression, but not passive signaling via cell-to-cell interactions, is the determinant of circadian Ca2+ rhythms in SCN neurons.
Keywords: intracellular calcium, slice culture, yellow cameleon
Introduction
The suprachiasmatic nucleus (SCN) is a central pacemaker in the mammalian circadian clock system (Moore and Eichler, 1972; Stephan and Zucker, 1972). The common molecular elements of circadian rhythms are hypothesized to be transcriptional-translational feedback loops (TTFLs) in clock genes (Reppert and Weaver, 2002). In mice, the BMAL1:CLOCK heterodimer drives the transcription of the mammalian Period (Per) and Cryptochrome (Cry) gene families. PER2 upregulates Bmal1 expression, whereas CRY1/2 blocks the transcriptional activity of Clock and Bmal1 (Honma et al., 1998; Reppert and Weaver, 2002; Sato et al., 2006). Bmal1 is an exclusive gene, and its single deletion (Bmal1−/−) produces arrhythmic behavior under constant darkness (Bunger et al., 2000). Furthermore, brain-specific rescue of Bmal1 in Bmal1−/− mice restores behavioral rhythms (McDearmon et al., 2006). Therefore, Bmal1 is believed to play an essential role in TTFLs and rhythm generation in the SCN pacemaker.
The physiological activity rhythms in SCN neurons are primarily controlled by intracellular machinery, as shown by the presence of circadian action potential firing rhythms in individual SCN neurons in dispersed cell cultures (Welsh et al., 1995). Despite evidence that Bmal1 functions as an essential clock gene, the aspect of how Bmal1 regulates the activity of SCN neurons is currently unclear. Intriguingly, Per2 promoter-driven luciferase rhythms are sustained in the SCN of Bmal1−/− mice (Ko et al., 2010). Because these rhythms are weakened in low-density dispersed cell cultures, the residual rhythms could be nonintrinsic noise-driven oscillations generated via cell-to-cell interactions. In addition, it has been reported that specific Gq-coupled receptor signaling pathways could act as couplers or amplifiers for the majority of SCN rhythms (Maywood et al., 2006; Brancaccio et al., 2013). However, the origin of Gq-mediated oscillations and their relationships with specific intracellular machinery are currently unknown.
We have developed a method to visualize the physiological activities of mouse SCN neurons using the yellow cameleon Ca2+ sensor (Ikeda et al., 2003a). This method enables monitoring of circadian Ca2+ rhythms (CCR) in SCN neurons. CCR are driven by release of Ca2+ from ryanodine-sensitive internal Ca2+ stores, and are resistant to the Na+ channel blocker tetrodotoxin (TTX; Ikeda et al., 2003a). Brancaccio et al. (2013) recently used a GCaMP3 Ca2+ sensor together with a luciferase reporter for the Ca2+/cAMP-responsive element and Per1/2 gene transcription, and detected the most phase-advanced rhythms occurring in CCR. Therefore, determination of the molecular generator(s) of CCR is an important issue. Bmal1−/− mice show reduced expression of ryanodine receptors and caffeine-induced Ca2+ mobilization in SCN neurons (Pfeffer et al., 2009). Therefore, it is reasonable to hypothesize that Bmal1 is a regulator of CCR.
To directly address the involvement of Bmal1 in CCR, we used a particle bombardment (i.e., gene-gun) technique for organotypic SCN cultures and examined the effects of Bmal1 RNAi and wild-type or dominant-negative (DN) Bmal1 expression. This technique allows monitoring of the effects of Bmal1 knockdown or overexpression in randomly selected SCN neurons within intact SCN circuits. Here, we show that Bmal1 is an essential intracellular regulator for the generation of CCR in SCN neurons.
Materials and Methods
Plasmid constructs.
The expression vectors encoding human Bmal1 (hBmal1), human Clock (hClock), and mouse Bmal1 (mBmal1) were described previously (Yu et al., 2002). Deletion mutants were generated based on mBmal1 cDNA sequences using the PCR primers listed in Figure 2a. The resultant PCR products were cloned into the pCR3.1 vector (Invitrogen). The mutant Bmal1 sequences were finally verified using a DNA Sequencer. The firefly reporter plasmid (E54-TK) was kindly provided by Dr M. Noshiro (Hiroshima University, Higashi-Hiroshima Japan). To investigate the intracellular distribution of BMAL1, wild-type, and mutant mBmal1 were individually inserted into the pcDNA3.1/His A vector (Invitrogen), with an Xpress-tag epitope bound to the N-terminus of mBmal1.
Figure 2.
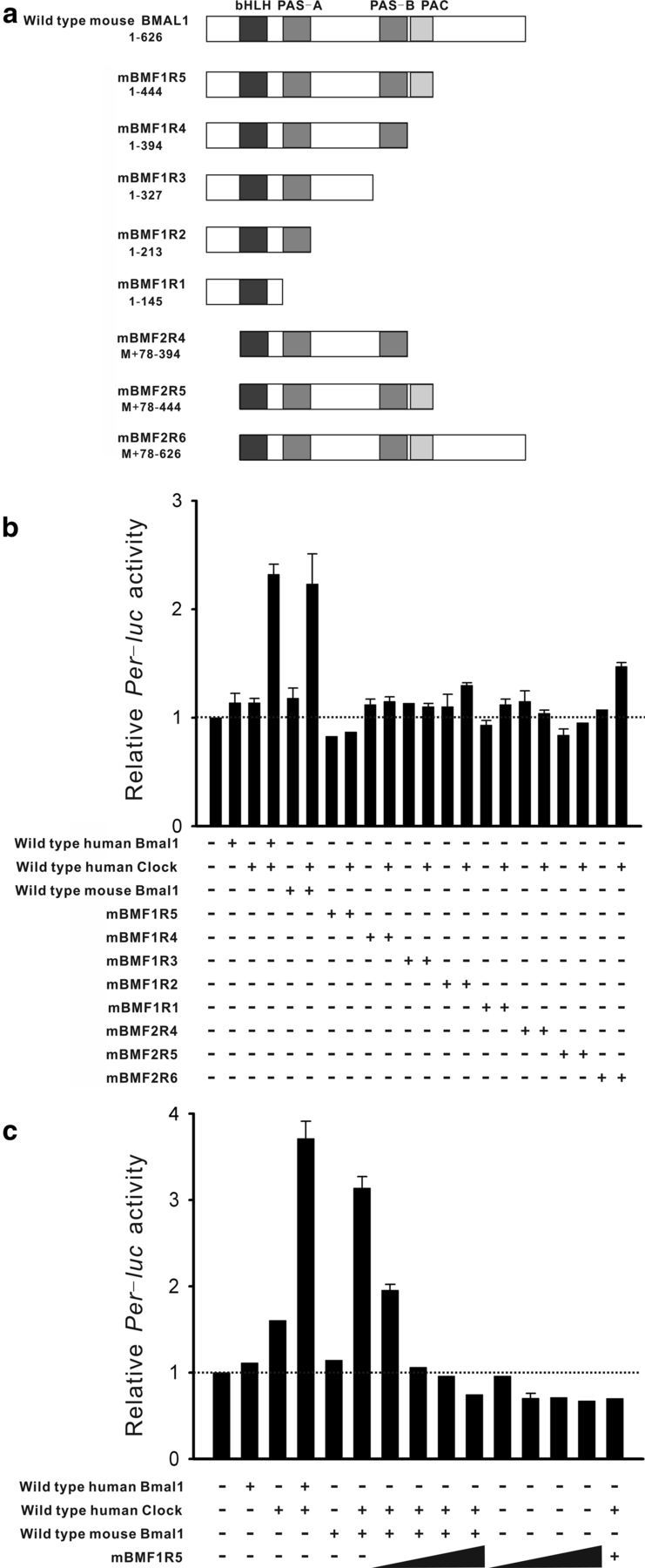
a, Schematic illustrations of the structure of the mouse Bmal1 mutants generated. The sizes and locations of the structural amino acids are shown under the mutant names. bHLH, Basic helix-loop-helix domain; PAS, PAR-ARNT-SIM domain; PAC, C-terminal motif of PAS likely contributing to PAS motifs. b, Effects of deletions (mBMF1R5 to mBMF2R6) on Per1-luc reporter expression in NIH 3T3 fibroblasts. Wild-type human Bmal1 and wild-type human Clock were cotransfected as controls. Plus and minus symbols denote the presence and absence of gene transfection. Cotransfection of human Bmal1 or wild-type mouse Bmal1 and human Clock significantly increased the Per1-luc signals compared with those in untransfected NIH 3T3 cells (dashed line). Cotransfection of Bmal1 mutants lacking the C-terminus (mBMF1R5 or mBMF2R5) and human Clock failed to increase the Per1-luc signals. c, Per1-luc induction by wild-type mouse Bmal1 and human Clock was dose-dependently inhibited by cotransfection of mBMF1R5 (0.01, 0.003, 0.05, and 0.1 μg/dish are shown as a slope). Data points indicate means ± SE. Each trial was repeated four or more times.
The psiRNA-hH1 neo shRNA expression vector was purchased from InvivoGen. An independent 21-bp sequence (5′-AAGGATCAAGAATGCAAGGGA-3′) within the coding region of the mBmal1 gene was selected as the insert for shRNA expression using siDirect (http://sidirect2.rnai.jp/), cloned into the psiRNA-hH1neo G1 vector, and designated psiRNA-mBmal1-204. The psiRNA hH-neo G1-scr vector supplied by the manufacturer was used as a negative control for mBmal1 silencing.
Monoclonal antibody production.
In collaboration with Dr K. Yoshimura (Nihon Institute of Medical Science, Moroyama, Saitama, Japan), a monoclonal antibody against BMAL1 was generated by immunization using peptides with common sequences between murine and human BMAL1. Hybridoma supernatants were screened using an ELISA-based assay and the resulting positive clone was named 2F11. For ascites production, clones were intraperitoneally injected (∼1 × 107 cells per injection) into BALB/c mice primed with Pristane (Wako Pure Chemical Industries) as described previously (Yoshimura et al., 1996). Typing of the 2F11 monoclonal antibody was performed using a mouse monoclonal antibody isotyping kit (GE Healthcare).
Transfection of mBmal1 mutants into NIH 3T3 fibroblasts.
NIH 3T3 fibroblasts were maintained in high-glucose DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (Invitrogen). The cell cultures were maintained in a CO2 incubator at 37 ± 0.5°C under 5% CO2. For dual luciferase assays, cells were grown in 24-well plates and transfected with 0.06 μg of E54-TK luciferase reporter vector and 0.01–0.1 μg of Bmal1 expression vector using Lipofectamine (Invitrogen) according to the manufacturer's instructions. As a control, 6 ng of Renilla luciferase expression vector was transfected into cells in separate wells. The total amount of expression plasmids transfected per well was kept constant (0.16 μg) by adding empty vector. The cells were lysed and monitored for their luciferase activity using a PicaGene Dual Seapansy Luminescence Kit (Nippon Gene) at 24 h after transfection. The E54-TK luciferase activity was normalized by the Renilla luciferase activity in the neighboring well. For immunofluorescence assays, wild-type Bmal1 and DN-Bmal1 (mBMF1R5) were subcloned into an epitope-tagged vector (pcDNA3.1/His A; Invitrogen). The resulting vectors were transfected into cells according to a standard protocol using Lipofectamine 2000 (Invitrogen) at 1 d after seeding on coverslips. The cell density was adjusted to reach 60–70% confluency at the time of transfection, and the cells were cultured in reduced (5%) FBS-containing medium to maintain the cell density until fixation.
Western blotting.
NIH 3T3 cells in 35 mm dishes were transiently transfected with 0.25 μg of mBmal1 expression construct (mBmal1/pcDNA3) and 0.25 μg of shRNA control vector (psiRNA-hH1-neoG1-scr) or shRNA expression construct for mBmal1 (psiRNA-mBmal1-204), using Lipofectamine 2000 as described above. Nuclear extracts were prepared using a Nuclear Extract Kit (Active Motif), mixed with three times SDS sample buffer, heated at 95°C for 3 min, and loaded onto a 10% SDS-polyacrylamide gel. After electrophoresis, the separated proteins were transferred to a polyvinylidene fluoride membrane and incubated in 5% ECL Blocking Reagent (GE Healthcare) at room temperature for 1 h. The membrane was then incubated at room temperature for 1 h with an anti-BMAL1 antibody (2F11) or anti-GAPDH antibody (Sigma-Aldrich) diluted in 0.5% Western blocking reagent. An HRP-conjugated goat anti-mouse IgG antibody (Zymed Laboratories) was used as the secondary antibody and detected with ECL Prime Reagent (GE Healthcare). The chemiluminescent signals were quantified using a bioimage analyzer (LAS 1000 Plus; Fujifilm).
Organotypic SCN culture.
The details for the preparation and maintenance of organotypic slice cultures, yellow cameleon (yc) expression vectors, and Ca2+ imaging were described previously (Ikeda et al., 2003a). Briefly, SCN slice cultures were prepared from 3-d-old male C57BL/6J mice. The animals were deeply anesthetized with pentobarbital, and the brain was quickly removed and dropped into ice-cold artificial CSF bubbled with 95% O2 and 5% CO2. Coronal hypothalamic slices containing the SCN were cut using a vibrating-blade microtome, and placed in 0.40 μm filter cups. The filters were placed in standard 6-well plates and cultured with 1 ml of medium consisting of 50% Eagle's basal medium, 25% Earle's balanced salt solution, and 25% heat-inactivated horse serum, supplemented with glucose and glutamax (Invitrogen). The cultures were maintained in a CO2 incubator at 36.0 ± 0.5°C under 5% CO2. The University of Toyama Animal Care and Use Committee approved all procedures involving animals.
Cotransfection of yc and Bmal1 mutants into cultured SCN neurons.
SCN slice cultures were transfected with YC expression vectors (YC2.1, YC2.6, YC3.1 or YC3.6) using a Helios Gene Gun System (Bio-Rad Laboratories). A HindIII-EcoRI fragment containing the cDNA encoding YC was isolated and subcloned into HindIII- and EcoRI-digested pBluescriptII (Stratagene). For neuron-specific expression, the 5′-flanking sequence (2.7 kb) with exon-1 and intron-1 of rat neuron-specific enolase genomic DNA and SV40 polyadenylation signals from pRc/RSV (Invitrogen) were bound to the HindIII/EcoRI site of pBluescriptII containing the yc gene. For these YC sensors, we initially compared the signal-to-noise ratios of CCR for detection with YC2.1 (primary Kd = 100 nm) > YC3.6 (Kd = 250 nm) > YC2.6 (Kd = 40 nm) > YC3.1 (Kd = 1.5 μm). These reported Kd values (Miyawaki et al., 1999; Nagai et al., 2004) are ∼1.5 times lower in the presence of a competitive cation (1 mm MgCl2) and were thus lower in cells. Although the absolute amplitude of CCR in SCN neurons remains controversial (estimated as 120–440 nm by Ikeda et al., 2003a using YC2.1 and 80–120 nm by Enoki et al., 2012 using YC3.6), we used YC2.1 for the present study because of its better signal-to-noise ratio in our assay system. For biolistics, gold particles were coated with the YC2.1 expression vector together with a vector carrying a Bmal1 mutant or a vector carrying the shRNA encoding Bmal1. The two vectors were suspended in Tris-EDTA buffer containing spermidine (Sigma-Aldrich) and then coated onto gold particles (0.6 μm, 5 mg for 50 bullets) in a droplet of 1 m CaCl2 according to the manufacturer's instructions. The gene-gun bullet was made using a Tubing Prep Station (Bio-Rad Laboratories) and blasted onto 7- to 9-d-old cultures with helium pressure (190–200 psi) using a Helios Gene Gun System (Bio-Rad Laboratories). The efficacy of this double-transfection technique has been evaluated using cell line models (Ikeda et al., 2013).
Immunohistochemistry.
For immunofluorescence staining, SCN slice cultures were fixed with 4% paraformaldehyde in PBS at pH 7.4 for 15 min, rinsed three times with PBS, and incubated in 10% donkey serum (Jackson ImmunoResearch Laboratories) in 0.01% Triton X-100 (Sigma-Aldrich) in PBS overnight at 4°C to block nonspecific antibody binding. The samples were then incubated with 1:2000 diluted 2F11 antibody in 5% donkey serum and 0.01% Triton X-100 in PBS for 48 h at 4°C. Following four 20 min rinses with PBS, the samples were incubated overnight with Cy3-conjugated donkey anti-mouse IgG (1:200 dilution; Jackson ImmunoResearch Laboratories) at 4°C. After another four 20 min rinses with PBS, the slices were embedded in VectaShield mounting reagent containing DAPI (Vector Laboratories).
To examine the intracellular distribution of exogenous BMAL1, NIH 3T3 cells, and organotypic cultures of SCN were transfected with wild-type Bmal1 and DN-Bmal1 linked at the C-terminus with an Xpress-tag (Invitrogen), fixed, rinsed, and incubated in blocking solution as described above. The samples were then incubated with 1:200 diluted monoclonal antibody against Xpress (Invitrogen) dissolved in 5% donkey serum and 0.01% Triton X-100 in PBS for 48 h at 4°C. Following four 20 min rinses with PBS, the samples were incubated overnight with Cy3-conjugated donkey anti-mouse IgG (1:200 dilution; Jackson ImmunoResearch Laboratories) at 4°C. After another four 20 min rinses with PBS, the slices were embedded in VectaShield mounting reagent containing DAPI. The fluorescence images were acquired using a confocal laser-scanning unit (LSM510; Carl Zeiss) mounted on an inverted microscope (Axiovert 200M; Carl Zeiss) with a Plan Apochromat 63×/1.40 oil-immersion objective lens (Carl Zeiss). For quantitative analyses of anti-Xpress immunostaining in NIH 3T3 cells, the intensities of immunofluorescence in the cytosol and nucleus were compared with the intensity of DAPI nuclear staining.
Calcium imaging.
The YC fluorescence levels at 3 d after transfection were observed using an inverted fluorescence microscope (TE-2000s; Nikon) with a 10× objective lens (Plan-Fluor 10×, NA0.3; Nikon) and a GFP filter set. SCN slices that exhibited successful gene transfection were carefully dissected from the filter cups and transferred to collagen-coated glass-bottom dishes (Matsunami Glass Industries) with the transfection surface on the bottom. Half of the culture medium (60–70 μl) was replaced every 1–2 d. On day 5 after gene transfection, the slices on the glass-bottom dishes were transferred to a recording system with an inverted fluorescence microscope (Axiovert 405M; Carl Zeiss) equipped with a custom-built microscope stage, CO2 incubator, mercury arc lamp, excitation filter (435.8 nm DF10; Omega Optical), excitation neutral density filter (ND.5; Omega Optical), dichroic mirror (455DRLP; Omega Optical), and 20× objective lens (Plan-Neofluor 20×, NA0.5; Carl Zeiss). Two emission bandpass filters (480DF30 and 535DF25; Omega Optical) were switched using a filter changer wheel (C4312; Hamamatsu Photonics). Resultant image pairs were acquired through a cooled charge-coupled device camera (C6790; Hamamatsu Photonics) at a sampling rate of one image-pair per 10 min for detecting circadian cytosolic Ca2+ oscillations. The electromagnetic shutter (Copal), filter changer wheel, and image acquisition were controlled using Argus-HiSCA imaging software (Hamamatsu Photonics).
Statistical analyses.
The amplitude and period of circadian cytosolic Ca2+ rhythms were calculated using a four-parameter sine curve fitted using Sigma-Aldrich Plot ver. 7.0 software (SPSS). A two-tailed Student's t test was used for pairwise mean comparisons and ANOVA was used for multiple mean comparisons.
Results
Effects of Bmal1 knockdown on CCR in SCN neurons
To quantify the efficiency of Bmal1 RNAi, a cDNA encoding wild-type mBmal1 was cotransfected with a mBmal1 shRNA or scrambled-sequence shRNA into NIH 3T3 fibroblasts for Western blotting analyses (Fig. 1a). We observed a reduction in the protein expression level by more than half with mBmal1 shRNA (−59.6% of scrambled RNA control), thereby confirming its efficiency. To examine the effect of Bmal1 knockdown on CCR in SCN neurons, we transfected SCN slice cultures using gene-gun bullets (gold particles) that were dual-coated with yellow cameleon and mBmal1 shRNA. At 1 week after transfection, the SCN slice cultures were fixed at light onset time for immunohistochemical quantification. As a result, all of the prepared cultures (n = 28) displayed dense nuclear concentration of BMAL1 protein in SCN neurons (Fig. 1b). At the optimal plasmid concentration ratio (400 ng yc2.1 cDNA/20 pg mBmal1 shRNA per dish), SCN neurons successfully expressed yellow cameleon fluorescence with reduced nuclear BMAL1 expression (Fig. 1c), with no effects on BMAL1 expression in the surrounding yellow cameleon-negative cells. Less than half of the yellow cameleon-positive SCN neurons showed CCR after transfection of mBmal1 shRNA (48.9 ± 4%, 6 slices), whereas the majority of neurons displayed CCR after transfection of the scrambled control shRNA (81.6 ± 6%, 6 slices; Fig. 1d). In addition, a positive correlation was observed between the magnitude of CCR and the intensity of nuclear BMAL1 staining in SCN neurons (r = 0.66, p < 0.01) on day 5 after mBmal1 shRNA transfection (Fig. 1d). Furthermore, SCN neurons displaying CCR on day 5 after mBmal1 shRNA transfection showed progressively damped oscillations thereafter until day 10 (Fig. 1e). During the damped circadian Ca2+ oscillations, the period (τ) of Ca2+ oscillations was significantly prolonged (τ = 28.5 ± 1.4 h, n = 10 in 3 slices) compared with the regular period after scrambled RNA transfection (τ = 23.9 ± 0.3 h, n = 18 in 3 slices; p < 0.01 by Student's t test). These results indicated that (1) CCR were dependent on nuclear BMAL1 expression, and (2) impaired CCR were not rescued by surrounding SCN circuits.
Figure 1.

a, The efficacy of the Bmal1 shRNA was analyzed by Western blotting using NIH 3T3 fibroblasts. The cells cotransfected with wild-type mouse Bmal1 and Bmal1 shRNA (mBmal1 + shRNA) showed less protein synthesis than those cotransfected with the scrambled shRNA control (mBmal1 + Scramble). b, Representative BMAL1 immunofluorescence staining of the SCN (red). 3V, Third ventricle. Before fixation, this culture was cotransfected with Bmal1 shRNA and yellow cameleon. The subregion marked by the red square is enlarged in c. c, Triple-color scanning of yellow cameleon, BMAL1, and DAPI nuclear staining shows that two neurons successfully expressed yellow cameleon and had reduced BMAL1 expression. All other cells in this field show nuclear BMAL1 expression, but the intensity is variable. d, The intensity of nuclear BMAL1 immunoreactivity following Bmal1 shRNA cotransfection (black circles) or scrambled shRNA cotransfection (white circles) was plotted as a function of the circadian amplitude of CCR. The mean amplitude of CCR monitored by single YC2.1 transfection was regarded as 100%. A positive correlation (r = 0.66) was observed between the BMAL1 intensity and the magnitude of CCR. e, Representative CCR in SCN neurons following Bmal1 shRNA cotransfection (4 representative cells are shown as blue, green, red, and black traces). Of these, three cells (blue, green, and red traces) had several sustained circadian oscillations, but the peak Ca2+ levels were decreased and the cycle periods were lengthened after Bmal1 shRNA cotransfection compared with control CCR following scrambled shRNA transfection (gray trace).
Generation and characterization of DN-Bmal1 in SCN neurons
The basic helix-loop-helix (bHLH) PER-ARNT-SIM (PAS) domain of BMAL1 binds to a partner clock gene product, CLOCK (Ikeda and Nomura, 1997). Although preserving the PAS domain, a 43 aa deletion at the C-terminus of Bmal1 reduces Per2 transcription activation and counteracts wild-type Bmal1 actions in Rat1 fibroblasts (Kiyohara et al., 2006), suggesting that this Bmal1 mutant functions as a DN sequence in these cells. In addition, the region upstream of the bHLH-PAS domain contains putative nuclear localization signals, deletion of which inhibits the nuclear localization of BMAL1 in NIH 3T3 fibroblasts (Kwon et al., 2006). The nuclear translocation of BMAL1:CLOCK heterodimers is a critical step for E-box regulation. Therefore, we generated a series of C-terminal and N-terminal deletion mutants from mBmal1 cDNA sequences (mBMF1R to mBMF2R6; Fig. 2a). NIH 3T3 fibroblasts expressing Per1-luciferase (Per1-luc) reporters were transfected with these Bmal1 analogs together with hClock. We found that a 182 aa deletion at the C-terminus with preservation of the PAS domain (mBMF1R5 or mBMF2R5) strongly inhibited Per1-luc induction (Fig. 2b). In addition, the effects of wild-type Bmal1 overexpression were dose-dependently inhibited by cotransfection of mBMF1R5 (Fig. 2c). Therefore, we used mBMF1R5 as DN-Bmal1.
Immunofluorescence staining for the Xpress-tag on the N-terminus of wild-type exogenous BMAL1 was predominantly located in the nucleus of NIH 3T3 fibroblasts, and gradually decreased over 8 d in culture (−25.1%, F(2,73) = 3.46, p < 0.05; Fig. 3a). However, the Xpress-tag immunostaining for DN-BMAL1 showed nuclear and cytoplasmic localizations with greater levels of nuclear expression (Fig. 3b), although both were still significantly decreased over 8 d in culture (−53.2%, F(2,78) = 20.4, p < 0.01 for nuclear expression; −35.3%, F(2,47) = 21.36, p < 0.01 for cytoplasmic expression). Therefore, we examined the function of the C-terminal sequence of Bmal1 for nuclear translocation in hypothalamic slice cultures from mice. Similar to the localization of exogenous BMAL1 in NIH 3T3 cells (Fig. 3) and HeLa cells (Kwon et al., 2006), the wild-type form was localized to the nucleus of astrocytes (Fig. 4a, white asterisk) and SCN neurons (Fig. 4a). Exogenous wild-type BMAL1 degraded faster in SCN neurons than in NIH 3T3 fibroblasts, and the immunostaining signals were almost undetectable at 8 d after transfection (−60.8% on day 8, F(3,25) = 18.24, p < 0.01; Fig. 4b). DN-BMAL1 was only located in the cytoplasm of SCN neurons and did not degrade over 10 d in culture (F(3,36) = 0.56, p > 0.05; Fig. 4c). Therefore, the distribution and lifetime of BMAL1 analogs overexpressed in SCN neurons were similar, but not identical, to those in NIH 3T3 fibroblasts.
Figure 3.

Intracellular localization of wild-type mouse BMAL1 or DN-BMAL1 overexpressed in NIH 3T3 fibroblasts visualized by epitope tag immunostaining (anti-Xpress). a, Top, Exogenous BMAL1 was located in NIH 3T3 fibroblast nuclei. Bottom, The mean exogenous BMAL1 expression declined over 8 d in culture (blue data points indicate means ± SE relative to the intensity of DAPI nuclear staining; n ≥ 6; gray data points indicate individual measurements;*p < 0.05 by one-way ANOVA). b, DN-BMAL1 was distributed in the nucleus and cytoplasm, and also declined over 8 d in culture; **p < 0.01 by one-way ANOVA.
Figure 4.
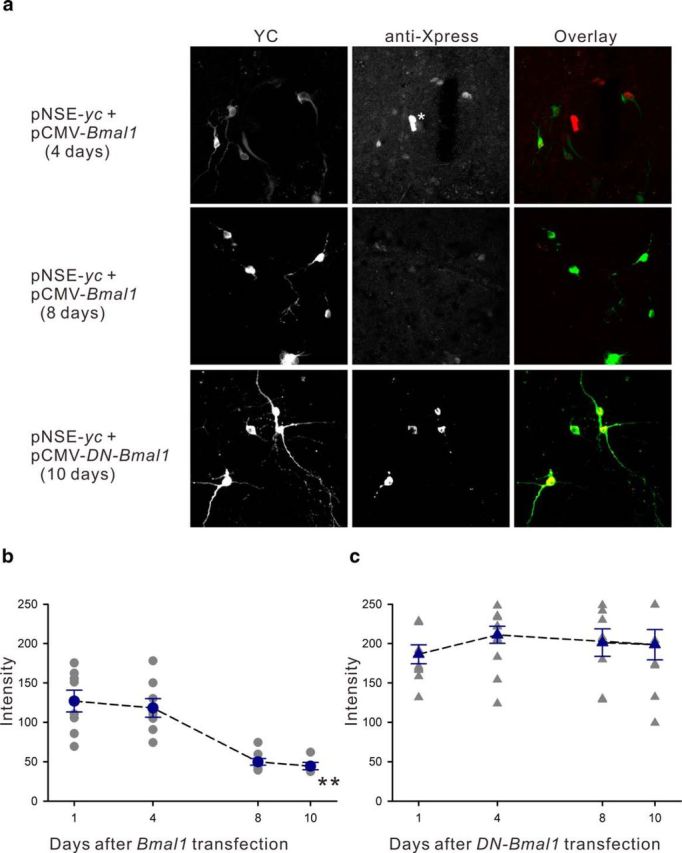
a, Intracellular localization of wild-type mouse BMAL1 or DN-BMAL1 overexpressed in SCN neurons visualized by epitope tag immunostaining (anti-Xpress). Top, Cultured SCN neurons at 4 d after cotransfection with yellow cameleon (pNSE-yc) and mouse Bmal1 (pCMV-Bmal1). YC fluorescence is green in the overlaid panel. BMAL1 was visualized in YC-positive neuronal nuclei using anti-Xpress immunostaining (red). Strong expression can be seen in two nuclei in the middle (white asterisk), indicating BMAL1 expression in astrocytes that failed to express YC with the NSE promoter. Middle, YC signals remained high, whereas BMAL1 expression was significantly decreased at 8 d after transfection. Bottom, After cotransfection with DN-Bmal1, DN-BMAL1 was only expressed in the cytoplasm (visualized by anti-Xpress immunostaining) in SCN neurons throughout the experimental period. b, The mean exogenous BMAL1 expression decreased over 10 d in culture; **p < 0.01 by one-way ANOVA. c, In SCN neurons, DN-BMAL1 was only distributed in the cytoplasm, and the expression level was stable over 10 d in culture.
Effects of Ca2+ channel blockers and Bmal1 overexpression on CCR in SCN neurons
Although we previously reported little effect of TTX and nimodipine on CCR (Ikeda et al., 2003a), the involvement of voltage-dependent Ca2+ channels was further examined using nimodipine (3 μm) together with ω-agatoxin TK (50 nm; P/Q-type Ca2+ channel blocker) and ω-conotoxin GVIA (100 nm; N-type Ca2+ channel blocker), which were previously reported to inhibit depolarization-induced Ca2+ flux in SCN neurons (Ikeda et al., 2003b). We observed a partial reduction in the peak amplitude of the oscillating Ca2+ levels in a subpopulation of SCN neurons (Fig. 5a), although the emergence of the inhibitory actions required 2–3 d after bath application of the blockers. These results indicate indirect involvement of voltage-gated Ca2+ channel activity in the generation of CCR.
Figure 5.
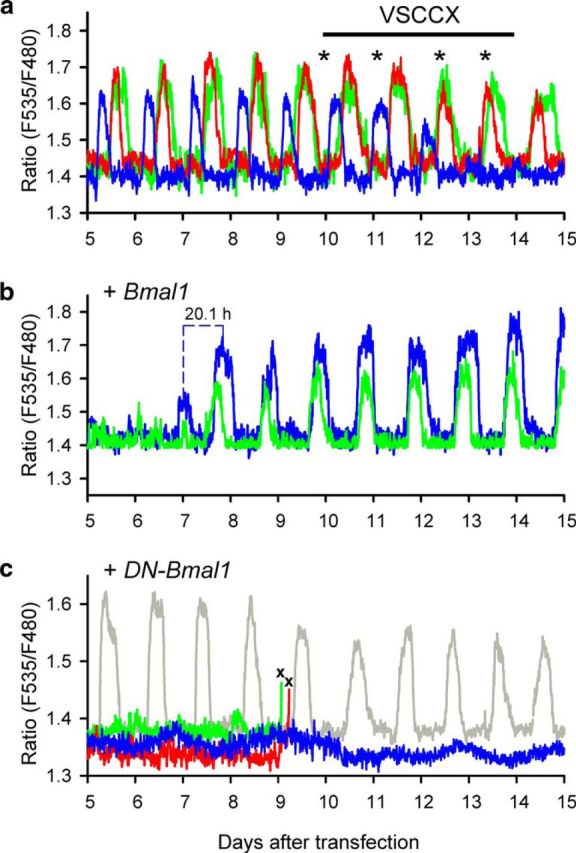
a, Representative CCR in SCN neurons that received single yc transfection. Oscillations in three representative neurons are shown. On the fifth day of recordings, a mixture of voltage-sensitive Ca2+ channel blockers (VSCCX) was bath-applied. Fresh VSCCX was repeatedly applied at the times of medium exchange (asterisks). The subpopulation of SCN neurons showed gradually reduced CCR. b, CCR temporarily ceased after cotransfection with wild-type mouse Bmal1, recovering with a shorter transition cycle period (τ = 20.1 h; blue trace). c, No CCR occurred after cotransfection with DN-Bmal1 in the majority of SCN neurons (blue, green, and red traces). In these neurons, more than half of the neurons did not survive at 9–10 d after transfection (×, cell death). Several cells maintained intact CCR within the same slice (gray trace), but the amplitude was slightly reduced after cell death in the neighboring neurons.
Overexpression of wild-type Bmal1 (40 ng DNA/dish) transiently reduced CCR, but the peak amplitude recovered after ∼8 d in culture (n = 15 in 3 slices; Fig. 5b). The rhythm period was significantly shorter during rhythm recovery (p < 0.01 by Student's t test; τ = 21.3 ± 0.5 h at 7–9 d after transfection, n = 15 in 3 slices) than during the following steady-state (τ = 24.2 ± 0.4 h at 10–15 d after transfection). Although the majority of SCN neurons showed robust CCR under standard conditions (66.3 ± 2.4%; 93/138 neurons in 12 slices; τ = 23.9 ± 0.2 h), overexpression of DN-Bmal1 (40 ng DNA/dish) almost completely abolished CCR (40/42 neurons in 6 slices; Fig. 5c). Notably, more than half of these nonoscillatory cells (22/40 neurons) were dead at 9–10 d after DN-Bmal1 transfection under the optical recording conditions used (Fig. 5c). The morphology of slices viewed under transmitted light following DN-Bmal1 transfection was indistinguishable from that of untransfected slices. In addition, CCR were observed in several neurons within the same DN-Bmal1-transfected slices, and survived until the end of the recordings, whereas the amplitude of CCR was reduced by 25% after cell death in neighboring neurons (2/42 neurons; Fig. 5c). These results indicate the dependence of CCR and cell viability on appropriate BMAL1 expression in SCN neurons.
Discussion
Current issues underlying CCR in SCN neurons
A decade after robust CCR were monitored at the cell body of SCN neurons in mouse organotypic slice cultures (Ikeda et al., 2003a), new evidence has recently been reported. For example, Enoki et al. (2012) transfected YC3.6 into SCN slice cultures using an adeno-associated virus, and monitored CCR from nearly all SCN neurons in single slices. An overview of the SCN revealed propagation of Ca2+ waves from the dorsal to ventral SCN regions. This wave propagation was also reported by Brancaccio et al. (2013), who used GCaMP3 instead of YC3.6.
Inconsistent with three previous studies (Ikeda et al., 2003a; Sugiyama et al., 2004; Hong et al., 2012), however, Enoki et al. (2012) reported a significant reduction in the total range of CCR by TTX treatment. The synaptic influence was further investigated by Brancaccio et al. (2013) using different approaches. They observed that inhibition of Gq-coupled receptor signaling significantly reduced CCR and the uncoupled phase of each cellular oscillation. Conceptually, these observations are more consistent with the TTX study by Enoki et al. (2012), because the Gq-coupled receptor is involved in particular synaptic signaling via action potential firing. The studies by Enoki et al. (2012) and Brancaccio et al. (2013) both used relatively low-affinity Ca2+ sensors. The affinities of YC3.6 for Ca2+ (Kd = 250 nm) and GCaMP3 (Kd = 840 nm) are apparently lower than that of YC2.1 (primary Kd = 110 nm). Because GCaMP3 linearly senses Ca2+ flux caused by action potential firings (Tian et al., 2009), detection of the effect of synaptic influences on CCR (Brancaccio et al., 2013) may be reasonable when considering the dynamic ranges of sensors. Therefore, the underlying issue may be the multiple components of CCR and differential dynamic ranges of Ca2+ sensors used in the experimental setups.
To directly address depolarization-induced Ca2+ influx in the generation of CCR, the present study used inhibitor cocktails to block various voltage-sensitive Ca2+ channels. We observed progressive and irreversible reductions in CCR. However, these inhibitors are capable of blocking voltage-sensitive Ca2+ channels quite rapidly in SCN neurons (Ikeda et al., 2003b). Therefore, the present results suggest that CCR at cell bodies of SCN neurons within the detection limit of YC2.1 were not driven by Ca2+ flux via voltage-sensitive Ca2+ channels. Thus, the present results support our original implication that synaptic interactions among cells are not the principal cause of CCR in SCN neurons.
Relation of TTFLs to CCR
After the discovery of CCR in SCN neurons, the issue was raised regarding the cellular clock as to whether TTFLs are generators of or generated by CCR (Ikeda, 2003, 2004). TTX treatment reversibly and significantly inhibits Per1-luciferase rhythms in SCN neurons (Yamaguchi et al., 2003), suggesting a contribution of synaptic interactions to the production of a particular clock gene rhythm. Because circadian rhythms in cytosolic messenger cAMP have also been reported in SCN neurons, the importance of “cytoscillators” has been suggested for cellular circadian clockworks (Hastings et al., 2008). Brancaccio et al. (2013) compared circadian phases of TTFLs and CCR by referring to the acrophase of Per2-luciferase rhythms. They estimated that the acrophase of circadian cytosolic Ca2+ waves is nearly at the phase of action potential firing rhythms, which is phase-advanced by 5–6 h relative to the Per2 transcriptional rhythms. Together with the recent observations of wave propagation of CCR in a similar fashion to Per1- or Per2-luciferase rhythms, these results lead to speculation that CCR is a driving force of E-box regulatory gene transcriptional rhythms in the SCN. This leads to the issue of which clock gene(s) affect CCR in the SCN.
Despite the generation of a series of clock gene knock-out mice, evidence explaining the link between TTFLs and physiological activity in SCN neurons remains surprisingly limited. The τ in action potential firing rhythms is longer in cultured SCN neurons of Clock mutant mice, yet these rhythms are stable under culture conditions (Nakamura et al., 2002). Studies have also shown that a Per1 antisense oligonucleotide reduces glutamate-induced phase-shifts in action potential firing rhythms of SCN neurons (Akiyama et al., 1999; Tischkau et al., 2003). Per2-deficient mutant mice have reduced mRNA levels of 14-3-3 protein and synapsin I in the SCN (Holzberg and Albrecht, 2003). We analyzed the effects of antisense oligonucleotides for Per1 and Per2, but failed to inhibit CCR in SCN neurons (Sugiyama et al., 2004). Moreover, it was recently shown that Per2-luciferase and action potential firing rhythms are uncoupled, but sustained, in the neonatal SCN dissected from Cry1/Cry2 double-knock-out mice (Ono et al., 2013). This suggests that Cry is involved in cell-to-cell couplings rather than in the intracellular organization of clock function. Although relatively small effects of clock gene deletions on the physiological activity of SCN neurons may be explained by redundant and compensatory functions of multiple clock genes (Miller, 2006), there is a lack of apparent data showing the involvement of TTFLs in the regulation of SCN neuronal activity.
Here, we examined RNAi of Bmal1 and overexpression of Bmal1 with Ca2+ monitoring in SCN neurons in organotypic cultures. The cell death caused by transfection of DN-Bmal1 indicates that the general E-box regulatory functions of BMAL1, such as those via brain-derived neurotrophic factor transcription (Chen et al., 2003), are important for the survival of SCN neurons under particular culture conditions. More importantly, temporal τ shortening following overexpression of wild-type BMAL1 and τ prolongation following BMAL1 knockdown are consistent with the hypothetical clock mechanism in which one circadian cycle is determined by feedback regulation of clock gene transcription by its gene products. Therefore, our study suggests that the Bmal1 TTFL drives CCR, and thus supports the conventional TTFL theory for the generation of cellular physiological rhythms. Furthermore, the present results showed a significant reduction in CCR, even though Bmal1 expression in neighboring neurons was intact. Therefore, our results demonstrate that synaptic interactions with other cells may not rescue the effects of Bmal1 knockdown or overexpression within the cells. These findings provide firm evidence showing that Bmal1 is an intracellular regulator for the generation of CCR in SCN neurons.
SCN-specific regulation of cytosolic Ca2+ and BMAL1
Cytosolic free Ca2+ is a ubiquitous signaling messenger, with plant cells showing strong CCR (Johnson et al., 1995). However, corresponding CCR have not been reported in animal cells, other than in SCN neurons. Endocrine oscillators in Drosophila melanogaster, prothoracic gland cells, fail to display dynamic circadian rhythms at baseline Ca2+ concentrations (Morioka et al., 2012). Moreover, we analyzed a SCN progenitor cell line that stably expresses YC3.6 (SCN2.2YC; Takeuchi et al., 2014), but failed to observe CCR in these cells. Because SCN2.2YC displays Per1-luciferase oscillations and expresses voltage-sensitive Ca2+ channels, other unveiled component(s) that are present in mature SCN neurons may be essential for the generation of CCR.
With regard to SCN-specific machinery, intracellular BMAL1 behaves differently in model cells and SCN neurons. BMAL1 truncated by deletion of 122 aa at the C-terminus is located in the cytoplasm and nucleus of NIH 3T3 cells when coexpressed with Clock (Kwon et al., 2006), similar to the present results in NIH 3T3 cells. In addition, BMAL1 truncated by 178 or 43 aa at the C-terminus is located in the nucleus when overexpressed in COS7 cells (Kiyohara et al., 2006). However, in SCN neurons, DN-BMAL1 is only located in the cytoplasm, suggesting differential intracellular machinery regarding BMAL1 translocation. Mutation of the casein kinase 2α phosphorylation site of BMAL1 at Ser90 results in impaired nuclear BMAL1 accumulation and disruption of clock function in NIH 3T3 fibroblasts (Tamaru et al., 2009). In our study, Ser90 and other phosphorylation residues for casein kinase 1ε (Eide et al., 2002) were preserved in DN-BMAL1. Furthermore, BMAL1 in NIH 3T3 fibroblasts is thought to be phosphorylated after translocation to the nucleus (Tamaru et al., 2009), but this did not occur with DN-BMAL1 in SCN neurons in our study. Therefore, blocking of phosphorylation sites may not be the cause of the cytosolic immobilization of DN-BMAL1 in SCN neurons. An alternative explanation for the function of DN-BMAL1 is impaired interaction with the other clock component CRY, because the C-terminus contains the element responsible for the CRY1 interaction (Sato et al., 2006). This protein-to-protein interaction is hypothesized to facilitate nuclear translocation (Kwon et al., 2006). Therefore, these lines of evidence suggest differential involvement of the C-terminal motif of Bmal1 between SCN neurons and fibroblasts, and that nuclear translocation of BMAL1 is more strictly regulated in SCN neurons.
Finally, the issue of how BMAL1 generates CCR in an SCN-specific manner remains to be resolved. Cyclic ADP ribose-mediated Ca2+ release from endoplasmic reticulum stores is the proposed machinery for CCR in SCN neurons (Ikeda et al., 2003a). In addition, expression of ryanodine receptors is reduced in Bmal1−/− SCN neurons (Pfeffer et al., 2009). Therefore, linking molecules, such as nicotinamide adenine dinucleotide/sirtuins (Kondratov et al., 2006; Asher et al., 2008; Belden and Dunlap, 2008; Nakahata et al., 2008), are of particular interest for complete understanding of CCR in SCN neurons, although the specific molecules involved have not yet been identified.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research (No. 22300108) from the Ministry of Education, Culture, Sports, Science, and Technology Japan to Masayuki Ikeda and Masaaki Ikeda. We thank Dr Michael Hastings (University of Cambridge) for helpful comments on this project, and Tomoya Ozaki, Tomoyoshi Kojiya, Eiichi Yamada, and Megumi Kumagai for their excellent technical assistance.
The authors declare no competing financial interests.
References
- Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci. 1999;19:1115–1121. doi: 10.1523/JNEUROSCI.19-03-01115.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- Belden WJ, Dunlap JC. SIRT1 is a circadian deacetylase for core clock components. Cell. 2008;134:212–214. doi: 10.1016/j.cell.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M, Maywood ES, Chesham JE, Loudon AS, Hastings MH. A Gq-Ca2+ axis controls circuit-level encoding of circadian time in the suprachiasmatic nucleus. Neuron. 2013;78:714–728. doi: 10.1016/j.neuron.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/S0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME. Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci. 2003;23:2572–2581. doi: 10.1523/JNEUROSCI.23-07-02572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide EJ, Vielhaber EL, Hinz WA, Virshup DM. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase lepsilon. J Biol Chem. 2002;277:17248–17254. doi: 10.1074/jbc.M111466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoki R, Kuroda S, Ono D, Hasan MT, Ueda T, Honma S, Honma K. Topological specificity and hierarchical network of the circadian calcium rhythm in the suprachiasmatic nucleus. Proc Natl Acad Sci U S A. 2012;109:21498–21503. doi: 10.1073/pnas.1214415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings MH, Maywood ES, O'Neill JS. Cellular circadian pacemaking and the role of cytosolic rhythms. Curr Biol. 2008;18:R805–R815. doi: 10.1016/j.cub.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Holzberg D, Albrecht U. The circadian clock: a manager of biochemical processes within the organism. J Neuroendocrinol. 2003;15:339–343. doi: 10.1046/j.1365-2826.2003.00992.x. [DOI] [PubMed] [Google Scholar]
- Hong JH, Jeong B, Min CH, Lee KJ. Circadian waves of cytosolic calcium concentration and long-range network connections in rat suprachiasmatic nucleus. Eur J Neurosci. 2012;35:1417–1425. doi: 10.1111/j.1460-9568.2012.08069.x. [DOI] [PubMed] [Google Scholar]
- Honma S, Ikeda M, Abe H, Tanahashi Y, Namihira M, Honma K, Nomura M. Circadian oscillation of BMAL1, a partner of a mammalian clock gene Clock, in rat suprachiasmatic nucleus. Biochem Biophys Res Commun. 1998;250:83–87. doi: 10.1006/bbrc.1998.9275. [DOI] [PubMed] [Google Scholar]
- Ikeda M. Response to Honma and Honma: do circadian rhythms in cytosolic Ca2+ modulate autonomous gene transcription cycles in the SCN? Trends Neurosci. 2003;26:654. doi: 10.1016/j.tins.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Ikeda M. Calcium dynamics and circadian rhythms in suprachiasmatic nucleus neurons. Neuroscientist. 2004;10:315–324. doi: 10.1177/10738584031262149. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Nomura M. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS protein (BMAL1) and identification of alternatively spliced variants with alternative translation initiation site usage. Biochem Biophys Res Commun. 1997;233:258–264. doi: 10.1006/bbrc.1997.6371. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Sugiyama T, Wallace CS, Gompf HS, Yoshioka T, Miyawaki A, Allen CN. Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron. 2003a;38:253–263. doi: 10.1016/S0896-6273(03)00164-8. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Yoshioka T, Allen CN. Developmental and circadian changes in Ca2+ mobilization mediated by GABAA and NMDA receptors in the suprachiasmatic nucleus. Eur J Neurosci. 2003b;17:58–70. doi: 10.1046/j.1460-9568.2003.02427.x. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Tsuno S, Sugiyama T, Yamoto K, Takeuchi K, Hashimoto A, Kishi H, Mizuguchi H, Kohsaka S, Yoshioka T. Ca2+ spiking activity caused by the activation of store-operated Ca2+ channels mediates TNF-α release from microglial cells under chronic purinergic stimulation. Biochim Biophys Acta. 2013;1833:2573–2585. doi: 10.1016/j.bbamcr.2013.06.022. [DOI] [PubMed] [Google Scholar]
- Johnson CH, Knight MR, Kondo T, Masson P, Sedbrook J, Haley A, Trewavas A. Circadian oscillation of cytosolic and chloroplastic free calcium in plants. Science. 1995;269:1863–1865. doi: 10.1126/science.7569925. [DOI] [PubMed] [Google Scholar]
- Kiyohara YB, Tagao S, Tamanini F, Morita A, Sugisawa Y, Yasuda M, Yamanaka I, Ueda HR, van der Horst GT, Kondo T, Yagita K. The BMAL1 C terminus regulates the circadian transcription feedback loop. Proc Natl Acad Sci U S A. 2006;103:10074–10079. doi: 10.1073/pnas.0601416103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko CH, Yamada YR, Welsh DK, Buhr ED, Liu AC, Zhang EE, Ralph MR, Kay SA, Forger DB, Takahashi JS. Emergence of noise-induced oscillations in the central circadian pacemaker. PLoS Biol. 2010;8:e1000513. doi: 10.1371/journal.pbio.1000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Shamanna RK, Kondratova AA, Gorbacheva VY, Antoch MP. Dual role of the CLOCK/BMAL1 circadian complex in transcriptional regulation. FASEB J. 2006;20:530–532. doi: 10.1096/fj.05-5321fje. [DOI] [PubMed] [Google Scholar]
- Kwon I, Lee J, Chang SH, Jung NC, Lee BJ, Son GH, Kim K, Lee KH. BMAL1 shuttling controls transactivation and degeneration of CLOCK/BMAL1 heterodimer. Mol Cell Biol. 2006;26:7318–7330. doi: 10.1128/MCB.00337-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Reddy AB, Wong GK, O'Neill JS, O'Brien JA, McMahon DG, Harmar AJ, Okamura H, Hastings MH. Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr Biol. 2006;16:599–605. doi: 10.1016/j.cub.2006.02.023. [DOI] [PubMed] [Google Scholar]
- McDearmon EL, Patel KN, Ko CH, Walisser JA, Schook AC, Chong JL, Wilsbacher LD, Song EJ, Hong HK, Bradfield CA, Takahashi JS. Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science. 2006;314:1304–1308. doi: 10.1126/science.1132430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. Despite mutated gene, mouse circadian clock keeps on ticking. Science. 2006;312:673. doi: 10.1126/science.312.5774.673. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci U S A. 1999;96:2135–2140. doi: 10.1073/pnas.96.5.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972;42:201–206. doi: 10.1016/0006-8993(72)90054-6. [DOI] [PubMed] [Google Scholar]
- Morioka E, Matsumoto A, Ikeda M. Neuronal influence on peripheral circadian oscillators in pupal Drosophila prothoracic glands. Nat Commun. 2012;3:909. doi: 10.1038/ncomms1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura W, Honma S, Shirakawa T, Honma K. Clock mutation lengthens the circadian period without damping rhythms in individual SCN neurons. Nat Neurosci. 2002;5:399–400. doi: 10.1038/nn843. [DOI] [PubMed] [Google Scholar]
- Ono D, Honma S, Honma K. Cryptochromes are critical for the development of coherent circadian rhythms in the mouse suprachiasmatic nucleus. Nat Commun. 2013;4:1666. doi: 10.1038/ncomms2670. [DOI] [PubMed] [Google Scholar]
- Pfeffer M, Müller CM, Mordel J, Meissl H, Ansari N, Deller T, Korf HW, von Gall C. The mammalian molecular clockwork controls rhythmic expression of its own input pathway components. J Neurosci. 2009;29:6114–6123. doi: 10.1523/JNEUROSCI.0275-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Sato TK, Yamada RG, Ukai H, Baggs JE, Miraglia LJ, Kobayashi TJ, Welsh DK, Kay SA, Ueda HR, Hogenesch JB. Feedback repression is required for mammalian circadian clock function. Nat Genet. 2006;38:312–319. doi: 10.1038/ng1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci U S A. 1972;69:1583–1586. doi: 10.1073/pnas.69.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Yoshioka T, Ikeda M. mPer2 antisense oligonucleotides inhibit mPER2 expression but not circadian rhythms of physiological activity in cultured suprachiasmatic nucleus neurons. Biochem Biophys Res Commun. 2004;323:479–483. doi: 10.1016/j.bbrc.2004.08.126. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Mohammad S, Ozaki T, Morioka E, Kawaguchi K, Kim J, Jeong B, Hong JH, Lee KJ, Ikeda M. Serotonin-2C receptor involved serotonin-induced Ca2+ mobilisations in neuronal progenitors and neurons in rat suprachiasmatic nucleus. Sci Rep. 2014;4:4106. doi: 10.1038/srep04106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru T, Hirayama J, Isojima Y, Nagai K, Norioka S, Takamatsu K, Sassone-Corsi P. CK2alpha phosphorylates BMAL1 to regulate the mammalian clock. Nat Struct Mol Biol. 2009;16:446–448. doi: 10.1038/nsmb.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkau SA, Mitchell JW, Tyan SH, Buchanan GF, Gillette MU. CREB-dependent activation of Per1 is required for light-induced signaling in the suprachiasmatic nucleus circadian clock. J Biol Chem. 2003;278:718–723. doi: 10.1074/jbc.M209241200. [DOI] [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Isejima H, Matsuo T, Okura R, Yagita K, Kobayashi M, Okamura H. Synchronization of cellular clocks in the suprachiasmatic nucleus. Science. 2003;302:1408–1412. doi: 10.1126/science.1089287. [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Negishi T, Kaneko A, Sakamoto Y, Kitamura K, Hosokawa T, Hamaguchi K, Nomura M. Monoclonal antibodies specific to the integral membrane protein P0 of bovine peripheral nerve myelin. Neurosci Res. 1996;25:41–49. doi: 10.1016/0168-0102(96)01026-7. [DOI] [PubMed] [Google Scholar]
- Yu W, Nomura M, Ikeda M. Interactivating feedback loops within the mammalian clock: BMAL1 is negatively autoregulated and upregulated by CRY1, CRY2, and PER2. Biochem Biophys Res Commun. 2002;290:933–941. doi: 10.1006/bbrc.2001.6300. [DOI] [PubMed] [Google Scholar]