

Abstract
Terminal Schwann cells (TSCs) are key components of the mammalian neuromuscular junction (NMJ). How the TSCs sense the synaptic activity in physiological conditions remains unclear. We have taken advantage of the distinct localization of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) at the NMJ to bring out the function of different ACh receptors (AChRs). AChE is clustered by the collagen Q in the synaptic cleft and prevents the repetitive activation of muscle nicotinic AChRs. We found that BChE is anchored at the TSC by a proline-rich membrane anchor, the small transmembrane protein anchor of brain AChE. When BChE was specifically inhibited, ACh release was significant depressed through the activation of α7 nAChRs localized on the TSC and activated by the spillover of ACh. When both AChE and BChE were inhibited, the spillover increased and induced a dramatic reduction of ACh release that compromised the muscle twitch triggered by the nerve stimulation. α7 nAChRs at the TSC may act as a sensor for spillover of ACh adjusted by BChE and may represent an extrasynaptic sensor for homeostasis at the NMJ. In myasthenic rats, selective inhibition of AChE is more effective in rescuing muscle function than the simultaneous inhibition of AChE and BChE because the concomitant inhibition of BChE counteracts the positive action of AChE inhibition. These results show that inhibition of BChE should be avoided during the treatment of myasthenia and the pharmacological reversal of residual curarization after anesthesia.
Keywords: acetylcholinesterase, glia, muscle, neuromuscular disease, synaptic homeostasis
Introduction
Acetylcholinesterase (AChE) ends the action of acetylcholine (ACh) in the cholinergic synapses and prevents the repetitive activation of acetylcholine receptors (AChRs). Therefore, AChE inhibitors are used to partially block the degradation of ACh and thus prolong its biological effect in different clinical conditions such as congenital myasthenic syndromes and myasthenia gravis (MG), which are characterized by a decrease of AChR density at the neuromuscular junction (NMJ; Vincent, 2010). In these diseases, the inhibition of AChE increases the half-life of ACh in the synaptic cleft and therefore the number of activated postsynaptic nAChRs. Anesthetists also use AChE inhibitors (e.g., neostigmine) in daily routine practice to reverse the action of nondepolarizing muscle relaxants (Srivastava and Hunter, 2009).
In addition to AChE, butyrylcholinesterase (BChE) is also abundant at the NMJ. AChE and BChE share many properties. BChE hydrolyzes ACh as well as AChE (Nicolet et al., 2003). BChE and AChE are organized in oligomers by the same associated proteins [collagen Q (ColQ) and proline-rich membrane anchor (PRiMA)]. ColQ is a collagen that anchors AChE in basal lamina. PRiMA is a small transmembrane protein that tethers at the plasma membrane (Massoulié et al., 2008). It is generally accepted that AChE plays the major role in the fast hydrolysis of ACh at NMJs, whereas BChE acts simply as a backup enzyme for AChE (Masson and Lockridge, 2010). However, some data support the idea that BChE and AChE have separate roles at the NMJ. AChE and BChE are localized in different sites at the NMJ. AChE is anchored in the synaptic cleft (Bernard et al., 2011), whereas BChE was assumed to be distributed around the terminal Schwann cell (TSC; Davis and Koelle, 1967). AChE and BChE seem to modulate differentially ACh release. It has been shown that the specific inhibition of AChE increases the ACh release through the activation of muscarinic receptors (Minic et al., 2002). On the contrary, the inhibition of both AChE and BChE decreases the probability of ACh release regardless of the muscarinic receptors (Minic et al., 2003).
The initial purpose of this study was to understand the specific function of BChE at the NMJ. Using a specific monoclonal antibody directed against mouse BChE, we found that BChE is anchored by PRiMA at the surface of the TSC. At the NMJ, TSCs detect the nerve activity and modify the release of ACh (Auld and Robitaille, 2003). When BChE was specifically inhibited, we found a significant depression of ACh release that depended on α7 nAChRs localized on the TSC. The reduction of the release was dramatically accentuated when AChE was also inhibited. When a specific AChE inhibitor was used to increase ACh concentration in the synaptic cleft of myasthenic rats, the functional improvement was better than with a nonselective inhibitor of AChE and BChE, during which the inhibition of BChE counteracted the positive action of AChE inhibition. These results show that BChE has a specific function at the NMJ and its inhibition may be counterproductive during the treatment of myasthenia and in anesthesia to reverse residual neuromuscular blockade.
Materials and Methods
Experiments.
All experiments were performed following French guidelines for laboratory animal handling approved by the Animal Committee of Université Paris Descartes in accordance with the European Community Council Directive of February 1, 2013 (82010/63/EEC registration number CEEA34.EK/AGC/LB.111.12).
Endplate electrophysiology and threshold of muscle action potential generation.
Hemidiaphragm nerve–muscle preparations were pinned in translucent chambers and perfused at a rate of 2–3 ml/min with oxygenated (95% O2, 5% CO2) Ringer's–Krebs' solution containing the following (in mm): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 1 NaH2PO4, 15 NaHCO3, and 11 glucose, pH 7.2–7.4. End Plate Potentials (EPPs) and miniature EPPs (mEPPs) were recorded in the synaptic zone using the standard microelectrode technique (3 m KCl, resistance 10–15 MΩ) at 20–22°C. Synaptic responses were recorded using an Axoclamp 900A amplifier and digitized using Digidata 1440A (Molecular Devices) with WinWCP software (John Dempster, University of Strathclyde, United Kingdom). To prevent muscle contractions, μ-conotoxin GIIIB (2 μm; Peptide) was used.
To evaluate the threshold of nerve-evoked action potentials (APs), muscle preparations were incubated in 0.5 μm d-tubocurarine (dTC) for 60 min and then continuously perfused by 3 μm neostigmine and 0.2 μm dTC throughout the recording period (Wood and Slater, 1995). The low concentration of dTC and the inhibition of cholinesterase allowed the recovery of the muscle AP generation in part of the muscle fibers. Contractions of diaphragm in these conditions remained very weak, which made possible intracellular recording by microelectrodes. We analyzed the events in which the rising phase of EPP and the rising phase of the AP triggered by this EPP can be mathematically separated (see Fig. 4C) to determine the threshold (in millivolts) precisely. Motor nerve was stimulated only one time in each muscle fiber. The resting membrane potential was 70–75 mV.
Figure 4.
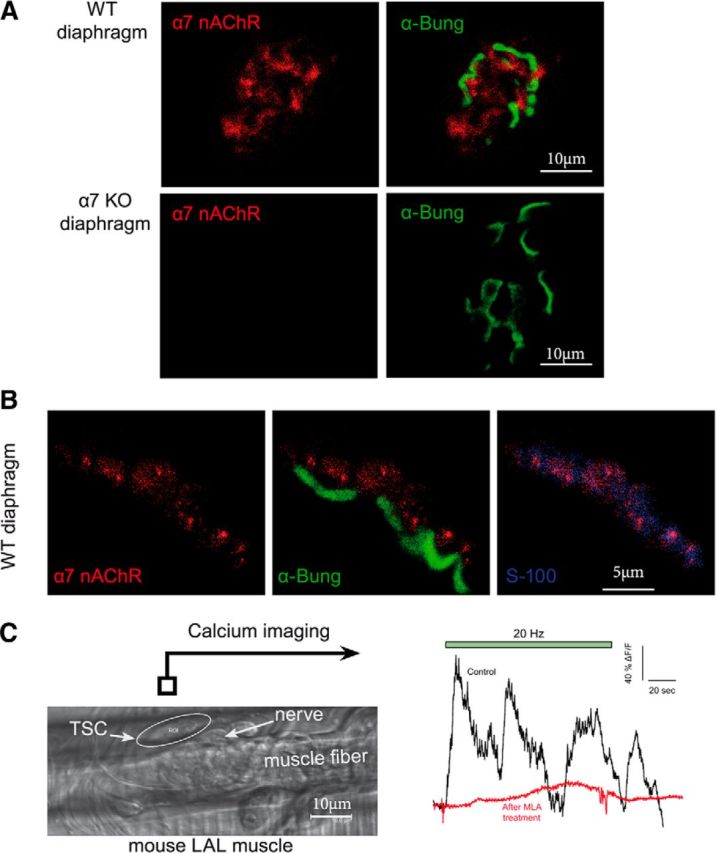
α7 nicotinic receptors are localized at the NMJ on the TSC. A, Immunolocalization of α7 nAChR at the NMJ. WT or α7 KO diaphragm muscles were incubated with a polyclonal antibody directed against α7 nAChR (red). The NMJs were identified with labeled α-bungarotoxin (green). A red signal was observed at the level of WT, but not α7 nAChR KO mice NMJ. B, α7 nAChR is labeled in red, the muscle nAChRs are labeled with α-bungarotoxin (green), and S100 is labeled with a specific antibody (blue). The overlay of α7 nAChR /α-bungarotoxin labeling (middle) and of α7 nAChR /S100 labeling (right) shows that α7 nAChR is mainly localized at the TSC. C, Example of Ca2+ responses elicited in the same TSC of LAL muscle before and after MLA. After a calcium wave had been triggered in the TSC by a first long burst of nerve stimulations, the preparation was left to rest for 30 min with or without MLA (10 nm). A second train of stimulations was then applied and triggered a second calcium wave in all control cases (n = 5) and in none of the muscles incubated wit MLA (n = 5). Left, Image from a transmitted light channel confocal microscope with TSC as a region of interest (ROI). Right, Mean F intensity from ROI represented on transmitted channel image before and after MLA treatment. Green panel is the time of nerve stimulations (20 Hz, 120 s).
Immunolocalization at light microscopy.
Isolated nerve–diaphragm preparations were stretched approximately to their resting length, pinned on Rhodorsil (Rhône-Poulenc)-lined Plexiglas chambers (2 ml volume), perfused with oxygenated Ringer's–Krebs' solution, and fixed with freshly prepared 4% paraformaldeyde (ElectronMicroscopy Sciences) in 0.01 m PBS for 1 h at room temperature. After washing with PBS, the muscles were separated in two groups: (1) hemidiaphragm muscles were immersed in 20–40% sucrose in PBS, frozen in isopentane at −40°C, and transverse sections were obtained with a cryostat at 10 μm and (2) muscle fibers from the other hemidiaphragm muscles were teased apart. Excess aldehyde groups were reduced with 50 mm glycine (Sigma-Aldrich) in PBS solution for 30 min and blocked against nonspecific binding with 5% normal goat serum (Sigma-Aldrich) in PBS for 30 min. BChE was detected in muscle fibers after overnight incubation at 4°C with anti-BChE biotinylated monoclonal antibody 4H1 at 1 μg/ml (1:1000) in PBS supplemented with 1% normal goat serum. α7 nAChRs were detected in muscle fibers after overnight incubation at 4°C with anti-α7 biotinylated polyclonal antibody at 1 μg/ml (1:1000) in PBS supplemented with 1% normal goat serum. S-100 was detected after overnight incubation at 4°C with anti-S-100 biotinylated polyclonal antibody (Abcam) at 1 μg/ml (1:1000) in PBS supplemented with 1% normal goat serum. BChE was revealed by 1 h incubation at room temperature with Alexa Fluor 594-conjugated-streptavidin (Vector Laboratories). α7 nAChR was revealed by 1 h incubation at room temperature with AttoN-647-conjugated streptavidin (1:1000; Invitrogen). S-100 was revealed by 1 h incubation at room temperature with Alexa Fluor-350-conjugated streptavidin (1:1000; Invitrogen). AChRs were stained with Alexa Fluor 488 or Alexa Fluor 647-conjugated α-bungarotoxin (Invitrogen) in PBS and mounted with Vectashield antifade mounting medium (Vector Laboratories). A third group of ex vivo unfixed diaphragm muscles were immunolabeled for BChE by incubation for 1 h with biotinylated 4H1 at 2 μg/ml (1:500), fixed with 4% paraformaldehyde for 1 h, and processed as described at the begining of this paragraph except for glycine incubation.
NMJs were analyzed using a LSM 510 META microscope (Carl Zeiss), mounted on an inverted microscope, and controlled through the manufacturer-supplied software and workstation. Images were collected using an oil-immersion objective [Plan-Apochromat ×63/1.2 numerical aperture (NA)]. The pinhole aperture was set to 1 Airy unit. Images were digitized at 12- or 16-bit resolution into 512 × 512 or 1024 × 1024 pixel arrays. Data were analyzed using Zen 2008 software on a series of “look-through” projections of average intensity.
Immunolocalization by EM.
After perfusion–fixation, as described in Immunolocalization at light microscopy, muscle fibers were incubated in 4% normal horse serum (NHS) for 30 min and then with 4H1 antibody (0.5 μg/ml) supplemented or with 30 nm biotinylated α-BTX (Invitrogen) with 1% NHS at room temperature overnight. After washing, biotin was detected using streptavidin coupled to gold particles (1.4 nm in diameter, 1:100 in PBS/BSA; Nanoprobes) for 2 h. The fibers were then washed and postfixed in 1% glutaraldehyde for 10 min. After washing in acetate buffer (0.1 m, pH 7), immunogold particles were amplified using a silver enhancement kit (HQ silver; Nanoprobes) with a 3 min incubation at room temperature in the dark. After treatment of sections with 1% osmium, dehydration, and embedding in resin, ultrathin sections were cut, stained with lead citrate, and examined in an electron microscope (EM 912 OMEGA; Zeiss) equipped with a LaB6 filament at 80 kV and images were captured with digital camera (SS-CCD, 2kx2k; Veleta).
Biochemical analysis of BChE molecular forms.
Frozen muscles were disrupted through high-speed shaking (TissueLyser; QIAGEN) in 2 ml plastic tubes containing 2 stainless steel beads (5 mm; QIAGEN) and 5 volumes of cold extraction buffer (10 mm HEPES, pH 7.2, 0.8 m NaCl, 1% CHAPS, 10 mm EDTA, 2 mm benzamidine). The extract was mixed thoroughly on a vortex mixer at 4°C for 10 min and then centrifuged at 4°C for 10 min at 20,000 × g. BChE molecular forms were separated in 5–20% sucrose gradients containing 10 mm HEPES, pH 7.4, 0.8 m NaCl, 10 mm EDTA, 0.2% Brij 97, or 1% CHAPS. Brij 97 was added to each sample (final concentration 0.2%) before loading on the gradient. The gradients were centrifuged in a SW41 rotor for 18 h at 37,000 rpm and ∼48 fractions were collected. Positions of internal sedimentation standards (β-galactosidase, 16 S; alkaline phosphatase, 6.1 S) were used to convert fraction numbers to S values. BChE activity was evaluated by the hydrolysis of 1 mm butyrylthiocholine in the presence of 1 mm DTNB and of 10−6 m BW284C51.
Ex vivo twitch tension measurements.
Left hemidiaphragm muscles with their associated phrenic nerves were isolated from mice killed by dislocation of the cervical vertebrae followed by immediate exsanguination. Hemidiaphragms were mounted in a temperature-regulated (32 ± 1°C) chamber and bathed in oxygenated Ringer's-Krebs' solution. For twitch tension measurements, the central tendon of the hemidiaphragm muscle was tied by a stainless steel rod via a linen thread hook to a force sensor (UF1; LCM Systems) and the ribs were blocked with two long hooks. The train-of-four (TOF) responses (2 Hz for 2 s) were evoked by stimulating the phrenic nerve via two microelectrodes with supramaximal current pulses of 0.1 ms duration. For each preparation, the resting tension was adjusted at the beginning of the experiment (to obtain maximal contractile response) and was monitored during the duration of the experiment. Signals from the force sensor were recorded and analyzed digitally, using PowerLab system and LabChart 6 software (ADInstruments).
MG animal model.
An experimental autoimmune model of MG was produced according to the following protocol (Baggi et al., 2003). Female rats 6–8 weeks of age were immunized twice (1 month intervals) by subcutaneous administration of the peptide (DGDFAIVKFTKVLLDYTGHI) mixed in Freund's complete adjuvant (first injection) and incomplete (second injection). The development of muscle weakness was diagnosed in vivo by the marked decrement of surface electromyogram amplitude (i.e., integral AP) of hindlimb muscles. Under urethane anesthesia (1.2 g/kg, i.p.), the sciatic nerve was stimulated (40 Hz, train of 200 stimuli) in the femoral part and the integral muscle action potential from posterior surface of the lower leg was recorded by skin electrodes. The integral muscle action potential was recorded using an FE132 amplifier (ADInstruments) and digitized with the PowerLab4/35 system (ADInstruments). Ambenonium and bambuterol were delivered intraperitoneally dissolved in H2O. Ambenonium was chosen as the AChE inhibitor because it contains quaternary nitrogen atoms and therefore does not cross the blood–brain barrier. It was important to limit the effects of brain AChE inhibition.
Calcium imaging of mammalian TSCs.
Experiments were performed at room temperature on mouse levator auris longus (LAL) nerve–muscle preparations. Muscles were removed under deep anesthesia with ether. LALs were incubated in oxygenated Ringer's–Krebs' solution (see endplate electrophysiology methods) containing 20 mm fluo-3 AM (Invitrogen), 0.02% pluronic acid (Invitrogen), and 0.5% dimethylsulfoxide (Sigma) for 60 min at 20°C temperature. Partial chelation of heavy metal ions was achieved by 20 mm tetrakis (2-pyridylmethyl) ethylenediamine (Invitrogen) to limit the binding of these ions to fluo-3 (Rochon et al., 2001). After this loading procedure, the preparation was washed with Ringer's–Krebs' solution for 15 min. Muscles were then pinned down in a recording chamber coated with Sylgard. Changes in fluorescence (F) intensity were monitored using a confocal microscope (TCS SP5; Leica) equipped with an argon ion laser. Observations were performed using a 20× water-immersion lens (1.00 NA; Leica). The 488 nm laser line was attenuated to 6% intensity and a long-pass filter with cutoff at 515 nm was used to detect the emitted F. Surface NMJs were located using the transmitted light channel of the confocal microscope. F intensity was measured over the area of the TSC cell body and the relative changes in F intensity were evaluated as a percentage as follows: ΔF/F = (F − F0)/F0 × 100. Stimulation of the distal end of the cut motor nerve (20 Hz, 120 s) was achieved using a suction electrode. To prevent muscle contractions evoked by transmitter release, we used μ-conotoxin GIIIB (2 μm; Peptide). If the calcium waves were triggered in a TSC by a burst of nerve stimulations (20 Hz, 120 s), the preparations were allowed to rest for 30 min for recovery with or without methyllycaconitine (MLA) (10 nm) to specifically block α7 nAChR); then, a second burst of nerve stimulations (20 Hz, 120 s) was applied and the F intensity was measured.
Data analysis.
In all cases, data are reported as mean ± SEM and n indicates the number of end plates measured in each group. Two-group comparisons were performed using paired or unpaired Student's t test. Differences between two groups were considered statistically significant at the probability level of p < 0.05.
Chemicals.
Tetraisopropyl pyrophosphoramide (iso-OMPA), atropine monohydrate sulfate, MLA, neostigmine methylsulphate bromide, and bambuterol hydrochloride were all from Sigma-Aldrich. Ambenonium dichloride was from Santa Cruz Biotechnology. μ-conotoxin GIIIB was from Peptide. The solution of fluorocitrate (FC) was prepared as described previously (Bonansco et al., 2011): 8 mg of the barium salt of dl-fluorocitric acid (Sigma) was dissolved in 0.1 m HCl, precipitated by the addition of 0.1 m Na2SO4, buffered with 0.1 m Na2HPO4, and centrifuged at 1000 × g for 5 min. The supernatant containing FC was added to Ringer's buffer at a final concentration of 0.2 mm, pH 7.2–7.4.
Results
BChE is mainly anchored by PRiMA at the plasma membrane at the surface of the TSC
The first experimental step was to determine the accurate localization of BChE at the NMJ. In recent experiments using BChE enzymatic staining in AChE KO mice, BChE was mainly detected in the depth of the NMJ folds that invaginate within the muscle fibers (Blondet et al., 2010), whereas in previous experiments, BChE staining has been localized at the surface of mouse TSCs (Davis and Koelle, 1967). The spatial resolution of the BChE staining based on acetylthiocholine iodine hydrolysis is far from being optimal because it is affected by numerous parameters susceptible to altering the position of the precipitates observed. Indeed, the precipitates obtained can migrate somewhat haphazardly depending upon the experimental conditions. To circumvent this limitation, we used a monoclonal antibody (4H1) directed against mouse BChE that was produced specifically by immunization of BChE KO mice with recombinant mouse BChE. This antibody reveals unequivocally that BChE is abundantly localized at the NMJ (Mrvova et al., 2013). To analyze the distribution of BChE, we looked for colocalization of 4H1 with S100, a specific marker of TSCs (Reynolds and Woolf, 1992). We found a colabeling of BChE with S100, but not with α-bungarotoxin, a well known antagonist of nAChRs (Fig. 1A). To further explore the BChE distribution, we used mice expressing membrane-anchored GFP in motoneurons (De Paola et al., 2003). The triple labeling of BChE with 4H1 (red), muscle nAChR with α-bungarotoxin (blue), and nerve with GFP (green) revealed that BChE is localized mainly on the surface of the TSC at the membrane (Fig. 1A).
Figure 1.
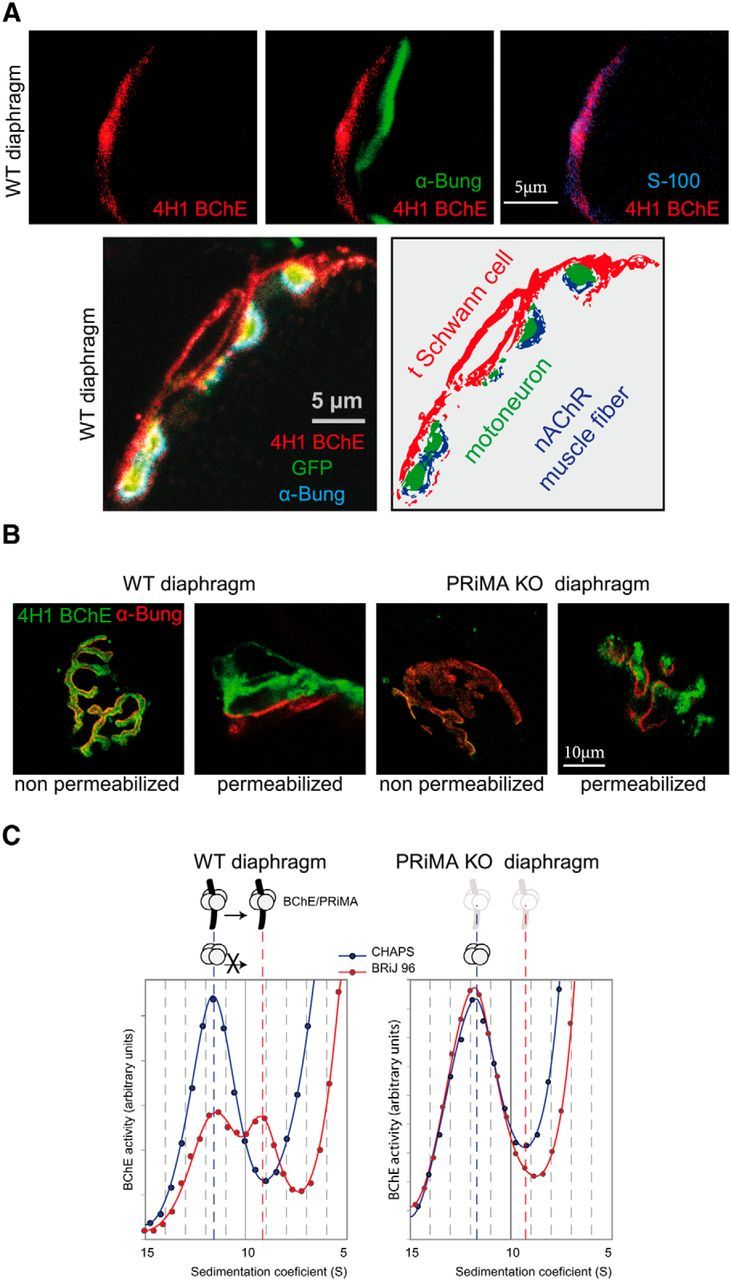
BChE is localized at the TSC with PRiMA. A, Top, BChE was labeled with biotinylated 4H1 (red), the muscle nAChR localized on the top of the fold at the surface were labeled with α-bungarotoxin (green), and S100 a specific marker of the TSCs were labeled with specific antibodies (blue). The overlay of BChE/α-bungarotoxin labeling (middle) and of BChE/S100 labeling (right) shows that BChE is mainly localized at the TSC. Bottom, Immunolocalization with biotinylated 4H1 (red) in a mouse strain in which the motoneuron are labeled by membrane tethered GFP (green). The muscle nAChR are labeled with α-bungarotoxin (blue). The drawing illustrates the localization of BChE on the surface of the TSC. B, Labeling of BChE with biotinylated 4H1 (green) and of muscle nAChR with α-bungarotoxin (red) in muscle diaphragm of WT and PRiMA KO mice. Nonpermeabilized indicates that the muscle was incubated in toto without permeabilization before the application of antibodies and toxin. Note the absence of BChE labeling in PRiMA KO mice. Permeabilized indicates that frozen sections of muscle were incubated with antibodies and toxins. Note the presence of BChE in WT and PRiMA KO mice. The labeling in PRiMA KO mice appears more spotted than in WT mice. Altogether, these images show that BChE is mainly anchored by PRiMA at the surface of the TSC at the NMJ. C, Two BChE tetramers are found in the diaphragm. Enzymes were solubilized and loaded in sucrose gradients containing CHAPS or Brij as the detergent. After centrifugation, BChE activity was determined in all of the fractions. The figure presents a part of the gradient around the peaks of BChE activity that correspond to the tetramer. In the CHAPS gradient, the peak at 11.5 S corresponds to the tetramers. Amphiphilic tetramer (interaction with detergent) shifts in the presence of Brij and is absent in PRiMA KO mice, demonstrating that some of the tetramers of BChE are anchored by PRiMA.
A known anchoring mechanism of AChE and BChE at the cell surface involves the small transmembrane protein PRiMA. In PRiMA KO mice, AChE is not found at the surface of central cholinergic neurons, but rather is retained into the endoplasmic reticulum (Dobbertin et al., 2009). To determine the accurate localization of BChE at the TSC, we incubated in toto nonpermeabilized diaphragms of WT and PRiMA KO mice with the 4H1 monoclonal anti-BChE antibody and found a strong labeling in WT, but not in PRiMA KO mice (Fig. 1B). In contrast, in permeabilized muscle sections, 4H1 labels BChE in both WT and PRiMA KO mice (Fig. 1B). To further prove the anchoring of BChE by PRiMA at the surface of the TSC, we analyzed the distribution of BChE with 4H1 at the NMJ in the tibialis anterior (TA) muscle at the EM level. As illustrated in Figure 2A, 4H1 labeled WT and PRiMA KO NMJ in TA, as in the diaphragm. At the EM level, gold-labeled streptavidin was used to visualize biotinylated 4H1. Immunoparticles were localized along the surface of membrane in extensions close to the nerve terminal and at the cell body close to the NMJ (Fig. 2B). In contrast, in PRiMA KO mice, immunoparticles were absent from the plasma membrane of TSCs (membranes close to the nerve and close to the cell body (Fig. 2C), but were abundant in intracellular compartment close to the nucleus of cells in close proximity of the NMJ. Altogether, the distribution of BChE at the surface of the TSC in WT and its retention inside the cells in PRiMA KO mice reveal that BChE is mainly anchored by PRiMA at the surface of the TSC.
Figure 2.
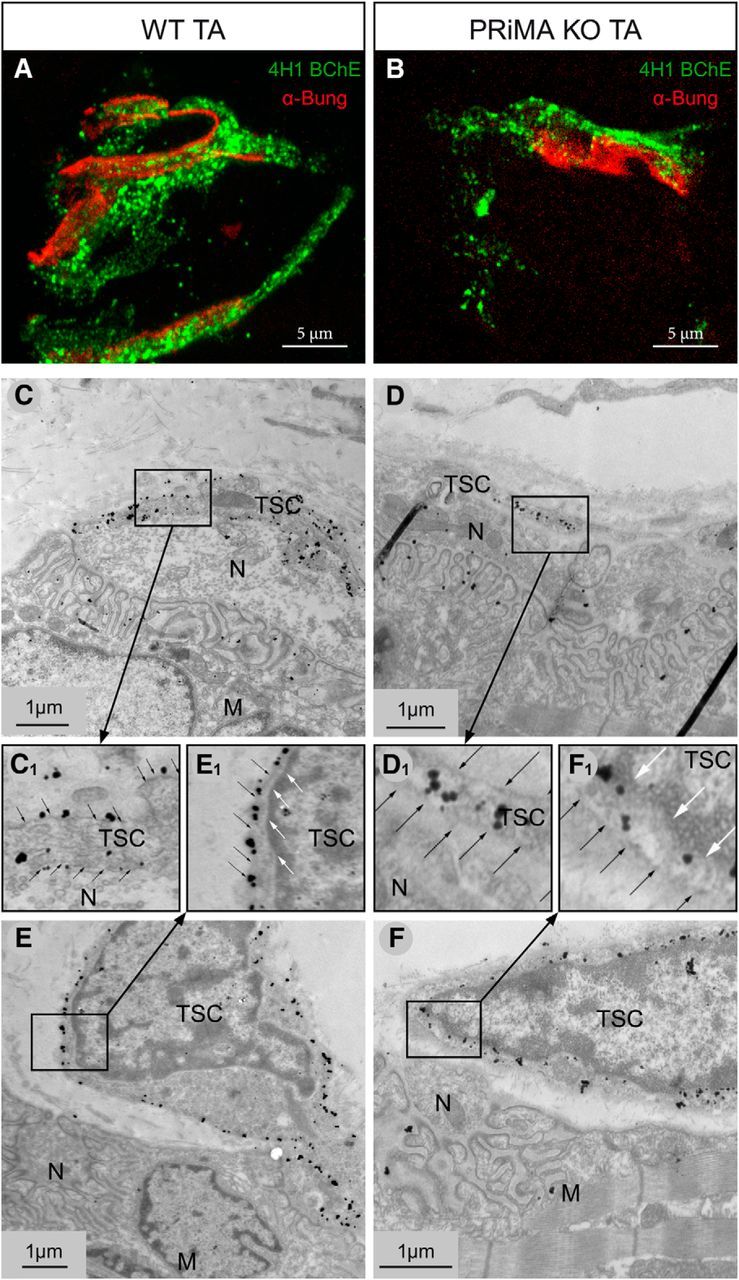
BChE is anchored by PRiMA at the surface of the TSC. A, B, At light microscopy, labeling of BChE with biotinylated 4H1 (green) and of muscle nAChR with α-bungarotoxin (red) in tibialis muscle in WT (A) and PRiMA KO (B) tibialis anterior muscle. Note the apparent weaker signal in PRiMA KO mice versus WT. C–F, At the EM level, labeling of BChE with biotinylated 4H1 was revealed by 1.4 nm gold particle enhanced with silver (C, E). At NMJ of WT mice, the immunoparticles are localized along the plasma membrane. C, Membrane extensions close to the nerve terminal at the NMJ are labeled at the surface. E, The plasma membrane of cell body close to the NMJ is also labeled. C1, E1, Enlargement of the inserts in the C and E. Black and white arrows point to the plasma membrane and the membrane of the nucleus, respectively. D, F, At the NMJ of PRiMA KO mice, immunoparticles are localized only in the cytoplasm, not along the plasma membrane. F, The plasma membrane of cell body close to the NMJ is not labeled; all of the immunoparticles are inside the cells. D1, F1, Enlargements of the inserts illustrating the intracellular localization of BChE in PRiMA KO mice close to the nucleus (white arrows) and the absence at the surface (black arrow). N, Nerve terminal; M, muscle fiber.
We then analyzed the molecular forms of BChE extracted and solubilized from the whole diaphragm. BChE is found mainly as monomers (data not shown), but, as shown in Figure 1C, BChE is also found in tetramers. A peak of BChE activity around 11.5S was observed when the extracts were separated using CHAPS as the detergent. The separation of the tetramers with Brij 97 instead of CHAPS revealed the presence of two types of BChE tetramers: a soluble BChE tetramer that did not shift in presence of BRIJ97 and an amphiphilic tetramer that was shifted with Brij 97. The soluble BChE tetramer is the abundant form of BChE found in the serum and its presence is presumably accounted by the residual serum in the muscle. The amphiphilic BChE tetramer is absent in PRiMA KO mice, demonstrating that the amphiphilic BChE tetramer contains PRiMA.
BChE inhibition decreases the quantal content of EPP when AChE is active
We next addressed the question of the function of BChE anchored at the surface of the TSC. We have previously shown that, when AChE is absent or inhibited, inhibition of BChE reduces the probability of ACh release (Minic et al., 2003). If a similar modulation exists when AChE is active, one expects to observe a reduction the EPP quantal content (QC) after inhibition of BChE. The EPP QC at low frequency of nerve stimulation varies significantly between the NMJs of the diaphragm muscle, but little for a given NMJ during experiments performed ex vivo. Therefore, we analyzed the effect of BChE inhibition on the EPP QC at the level of a single NMJ (Fig. 3A). We found that the EPP QC was reduced by 18% after 30 min application of 50 μm iso-OMPA, a specific, irreversible BChE inhibitor. To validate the selectivity of iso-OMPA toward BChE in our experimental conditions, we measured the EPP QC in BChE KO mice and found that it did not change significantly after application of iso-OMPA (Fig. 3B); then, we compared the mean QC in WT and BChE KO mice and found a reduction of 30% of the mean QC in BChE KO mice compared with WT mice (Fig. 3C). Therefore, the EPP QC is reduced both in BChE KO mice and after the specific pharmacological inhibition of BChE, likely through the action of residual ACh on AChRs.
Figure 3.
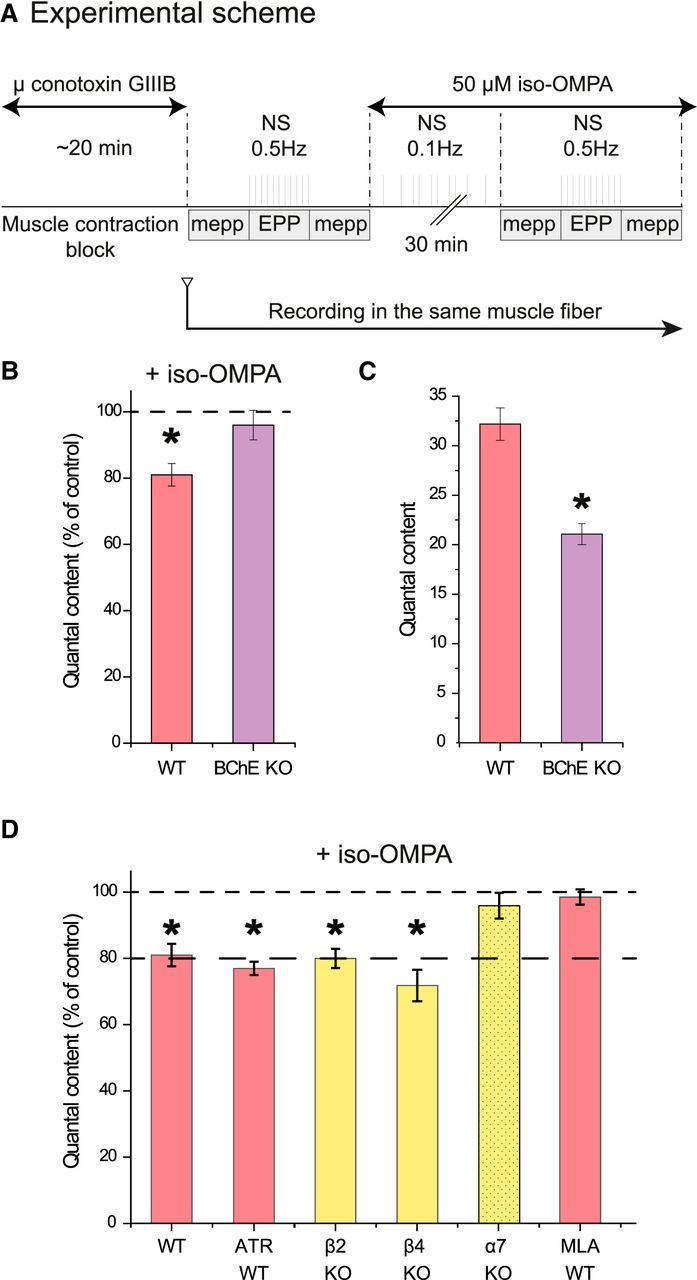
BChE inhibition depresses ACh release at the NMJ. A, Experimental protocol. The muscle contraction was blocked by inhibition of the muscle voltage-gated sodium channels with μ-conotoxin GIIIB. A sharp electrode was inserted close to a NMJ to record focal mEPPs and EPPs. A first file was obtained for the mEPPs (no nerve stimulation), then a second for the EPP (0.5 Hz nerve stimulations), and then a third for the mEPPs (no stimulation). Iso-OMPA (50 μm) was added to the Ringer's buffer and the muscle was stimulated indirectly at 0.1 Hz during the incubation. The electrode was maintained in place. All of the records were obtained in 30–40 min. The QC was obtained by dividing the mean amplitude of the EPPs by the mean amplitude of the mEPPs recorded before and after the EPPs if the resting membrane potential (RMP) did not change >1 mV during mEPP and EPP recording. B, Quantification of the QC in the same muscle fiber after inhibition of BChE with iso-OMPA. The percentage of decrease of the QC in WT mice is higher than in BChE KO mice (n = 5, *p < 0.05, paired Student's t test). C, The mean QC in the NMJs of WT mice is higher than in BChE KO mice (n = 20, *p < 0.05, and n = 5, *p < 0.05, respectively, unpaired Student's t test). D, Quantification of the QC in the same muscle fiber after inhibition of BChE with iso-OMPA when AChRs are absent or blocked. The graph presents the quantification of the QC in the same muscle fiber after iso-OMPA. All of the muscarinic receptors were blocked with 1 μm atropine (n = 5, *p < 0.05, paired Student's t test).
Because BChE is localized on TSCs, we wanted to find out whether TSCs contribute to the reduction of ACh release after BChE inhibition or if they simply act as a scaffold localizing BChE close to, but outside of, the synaptic cleft. To address this question, we lowered specifically the metabolism of the TSC with fluorocitric acid, a Krebs' cycle blocker that affects mainly the glial cells (Bonansco et al., 2011). Incubation in fluorocitric acid (0.2 mm) during 1 h did not alter significantly the EPP QC (92 ± 5%, n = 5), but when fluorocitric acid was washed out and BChE was inhibited by iso-OMPA (50 μm, 30 min), the EPP QC was not significantly reduced (95 ± 6%, n = 5). This result suggests that TSCs may participate actively in the reduction of EPP QC after BChE inhibition.
Blockade of α7 receptors prevents the depression of ACh release after BChE inhibition
The effects of BChE inhibition suggest that the excess of ACh resulting from this inhibition depresses ACh release. To identify the ACh receptors mediating this depression, we estimated the EPP QC after blockade of muscarinic receptors (1 μm atropine) and in the absence of nicotinic β2, β4, or α7 receptors (in β2 KO, β4 KO, or α7 KO mice, respectively). The incubation of the ex vivo nerve–muscle preparations from WT mice with atropine did not prevent the reduction of the EPP QC observed after the specific BChE inhibition by iso-OMPA (Fig. 3C). EPP QC was also decreased after iso-OMPA incubation of ex vivo unitary nerve–muscle preparations from β2 nAChR KO and β4 nAChR KO mice, as in the WT mice (Fig. 3C). In stark contrast, the incubation with iso-OMPA of ex vivo diaphragms obtained from α7 nAChR KO mice did not change the EPP QC (Fig. 3C). To further explore the implication of α7 nAChR, we incubated the WT muscle preparation with 10 nm MLA, which is a specific inhibitor of α7 nAChR, and followed the EPP QC before and after iso-OMPA addition to the bath containing the ex vivo nerve–muscle preparations (Fig. 3C), QC was not changed by iso-OMPA. It is worthwhile to emphasize that MLA did not affect the EPP QC in α7 KO mice.
From these experiments, it can be concluded that the decrease of EPP QC induced by BChE inhibition is mediated by α7 nAChR.
α7 nAChRs are localized and functional at the TSC of the NMJ
To analyze how activation of α7 nAChRs may depress the release of ACh, we attempted to localize them at the NMJ using polyclonal antibodies directed against mouse α7 nAChR (Kalashnyk et al., 2012). As shown in Figure 4A, we obtained a reproducible weak signal at the NMJ identified by α-bungarotoxin labeling. α-Bungarotoxin is an α-neurotoxin known to bind irreversibly and competitively to the nAChRs found at the NMJ and to α7 nAChRs in the brain. The specificity of α7 nAChR labeling observed with the polyclonal antibodies used is substantiated by the absence of any detectable immunolabeling in α7 KO mice with the same polyclonal antibodies. The weak α7 nAChR immunolabeling observed did not overlap with the strong labeling obtained with fluorescent α-bungarotoxin. At the EM level, we also found a dense deposit of gold particles along the postsynaptic membrane labeled with α-bungarotoxin. However, we did not find gold particles along the plasma membranes of the TSC or the nerve terminal. Moreover, immunoparticles that were not associated with the muscle membranes appeared equally distributed in WT and α7 KO muscle preparation. Altogether, these observations failed to reveal α7 nAChR by using labeled α-bungarotoxin, presumably because the density of α7 nAChR on the TSC is much lower than that of the muscle postsynaptic receptors. A low density of α7 nAChR was previously observed in brain astrocytes (Vélez-Fort et al., 2009). To further analyze the distribution of α7 nAChR, we looked for colocalization labeling with S100, a specific marker of TSCs. We found a colabeling of α7 nAChR with S100, but not with α-bungarotoxin (Fig. 3B). These observations suggest that the α7 nAChRs detectable with the polyclonal antibodies are mainly on the TSCs.
TSCs are close to the nerve terminal and sensitive to the synaptic transmission (Auld and Robitaille, 2003). The hallmarks of TSC sensitivity to the synaptic activity are calcium waves recorded specifically in the TSCs after long bursts of nerve stimulations (Todd et al., 2010). We generated calcium waves at the TSC by 120 s nerve stimulations (20 Hz) in presence of μ-conotoxin GIIIB (a specific muscle voltage-gated sodium channel blocker used to prevent muscle contraction). After 30 min of recovery, the calcium waves were again generated by 120 s nerve stimulations (20 Hz). To evaluate whether α7 nAChRs mediate this signaling, the muscle preparation was incubated during the recovery period (30 min) with 10 nm MLA (n = 5 experiments). We found that the second calcium waves were totally abolished after MLA, but not in control (Fig. 4C). This result supports that α7 nAChRs are functional at the TSC, where they are activated by the spillover of ACh. This can actually occur for prolonged stimulations, even when both AChE and BChE are active, but is likely to be much larger in extreme physiological circumstances and/or medical or pathological conditions.
AChE and BChE coinhibition decreases the mean amplitude of EPP
The previous experiments revealed that the selective inhibition of BChE anchored at the surface of TSCs reduces ACh release. This means that, in the presence of AChE, some ACh must escape the cleavage by AChE clustered in the synaptic cleft to reach BChE. This spillover is likely to be moderate, which might explain the modest inhibition of the synaptic transmission observed after BChE specific inhibition at low frequency of nerve stimulation. The inhibition of both AChE and BChE is expected to increase the spillover of ACh and should increase the depression of the EPP QC. Unfortunately, after AChE inhibition, the direct estimation of the QC as the summation of the individual quanta (mEPP) is not possible. In normal conditions, EPP is the simple sum of individual mEPPs. After AChE inhibition, the surfaces of the postsynaptic area ultimately reached by individual quanta overlap, so the number of quanta released cannot be calculated as the quotient of EPP by the mEPP (Hartzell et al., 1975). We thus calculated the mean value of EPP amplitude recorded in the same area of the diaphragm. The amplitude of EPPs is the key parameter for neuromuscular synaptic transmission. The muscle contraction occurs only if the EPPs pass the threshold of AP generation. To record the full EPP, the APs were blocked with μ-conotoxin GIIIB. The resting membrane potential was constant. The mean EPP amplitude was 31.5 ± 0.95 mV in control diaphragm and, after AChE and BChE inhibition (3 μm neostigmine, 20 min), it decreased to 14.2 ± 0.75 mV (Fig. 5D). It is important to note that the amplitude of mEPCs recorded in the interstimuli intervals after ChE inhibition by neostigmine was higher than in the untreated intact preparation (Fig. 5B). Therefore, after the inhibition of AChE and BChE, the reduction of the number of quanta released was likely larger than the reduction of the EPP amplitude.
Figure 5.
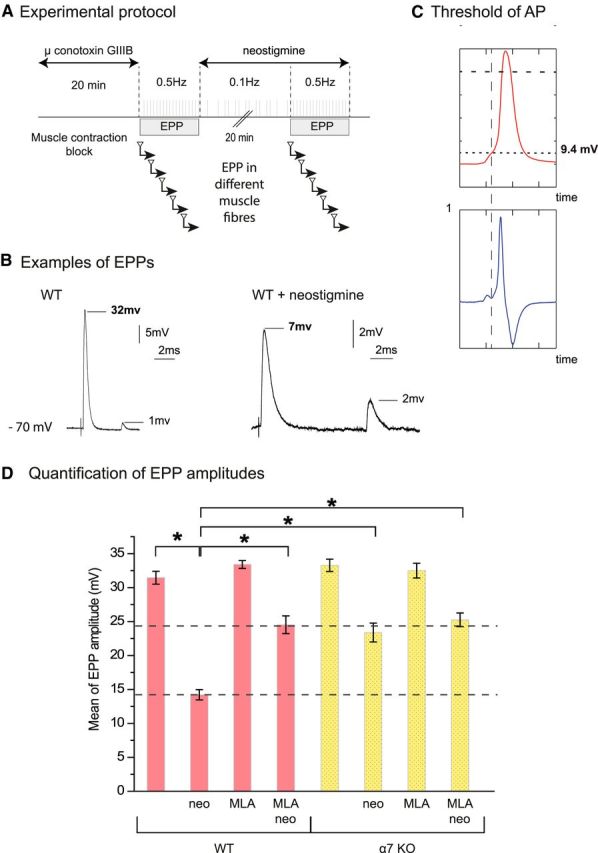
The amplitudes of the EPP are rescued partially by the block of α7 nAChR when both cholinesterases were inhibited. A, Experimental protocol. The muscle contraction in diaphragm was blocked by μ-conotoxin GIIIB. Fifty EPPs were recorded from each muscle fiber. Only data from the fibers in which the resting membrane potential was >70 mV at the end of the recording have been used. Mean amplitude of EPPs was obtained from 20–30 fibers recorded in control conditions and after application of the drugs (neostigmine, MLA). B, Examples of EPPs recorded before and after cholinesterase inhibition with 3 μm neostigmine. Note the difference of scale between the two conditions. In each of the selected traces, one EPP and one mEPP are visible. C, Evaluation of the threshold of nerve-evoked muscle fiber APs. The phrenic nerve was stimulated once for recording an AP in each muscle fiber. The resting membrane potential was −70 to 75 mV. Records of APs were differentiated (lower profile, dV/dt) and the time point on the rising phase of the nerve-evoked AP when the rate of change of voltage with time was closest to zero was determined. The threshold was defined as the voltage at this time. D, Mean amplitude of EPPs recorded in the phrenic-diaphragm preparations of WT mice and α7 nAChR KO mice in control conditions and after application of the drugs (neostigmine 3 μm, MLA 10 nm; n = 20–30, *p < 0.05, unpaired Student's t test).
To test the involvement of α7 nAChR, we used both MLA, a specific blocker of α7 nAChR, and α7 KO mice. After 30 min incubation of the diaphragms of WT mice with MLA (10 nm), the mean EPP was unchanged at 33.4 ± 0.6 mV. Then, after 20 min incubation with neostigmine (3 μm), the mean EPP decreased only to a value of 24.5 ± 1.3 mV. The mean EPP amplitude was 33.3 ± 0.9 mV in the diaphragms of α7 KO mice and 23.4 ± 1.4 mV after 20 min of incubation with neostigmine. We also controlled the specificity of MLA by treating muscles of α7 KO mice. The mean EPP amplitude in α7 KO mice treated by MLA (10 nm) was 32.5 ± 1.1 mV before and 25.2 ± 1.0 mV after neostigmine. These results unambiguously reveal that the activation of α7 nAChR significantly reduces the amplitude of EPP after inhibition of cholinesterase by neostigmine.
The amplitude of EPP after inhibition of AChE and BChE was found to be close to the threshold of AP generation evaluated in extensor digitorum longus and soleus muscles of rat (13 mV and 14 mV, respectively; Wood and Slater, 1995). To perform a direct comparison with the mouse diaphragm, we estimated the threshold of AP generation in mouse diaphragm when both cholinesterases were inhibited by 3 μm neostigmine (Fig. 5C). The average level of threshold of AP generation was 13 ± 3 mV (n = 20), a value close to the mean EPP amplitude (14.2 ± 0.75 mV) obtained after complete inhibition of all cholinesterases with neostigmine. In contrast, the amplitudes of the EPP appear higher after inhibition of cholinesterases when α7 nAChR are absent or inactivated.
AChE and BChE coinhibition and muscle contraction
The decrease of EPP amplitude close or below to the threshold of AP after AChE and BChE inhibition should be translated at the physiological level into a reduction of the muscle contraction triggered by the nerve. The anesthetists routinely assess via neuromuscular monitors the decrease of the EPP amplitude during muscle relaxation. They follow the modification of the muscle contractions triggered by the nerve stimulations. A sensitive method to monitor the neuromuscular blockade is used. The TOF stimulation method consisted of recording the four muscle contractions elicited by four nerve stimulations at 2 Hz (Ali et al., 1975; Srivastava and Hunter, 2009; Plaud et al., 2010). Next, the ratio of the fourth over the first response, known as the TOF ration (TOFR), which reflects the degree of neuromuscular blockade during onset and recovery after nondepolarizing muscle relaxant administration, can be calculated. When this pattern of stimulation was applied to the phrenic nerve diaphragm preparation ex vivo, the amplitudes of the four twitches were identical during hours if TOFs were repeated every 30 s (Fig. 6A). We then examined the effects of neostigmine on the first twitch (T1) and on T2-T4. After 20 min of incubation with 3 μm neostigmine, the mean amplitude of T1 decreased to 60% of the initial amplitude in WT muscle (Fig. 6B,C). A concentration of 0.1 μm atropine (to block all of the muscarinic receptors) did not change the reduction of the T1. In contrast, after blockade of α7 nAChR (MLA treatment, 10 nm) or in α7 KO mice, the mean amplitudes of T1 did not decrease in neostigmine (Fig. 6B,C). Therefore, as expected, the partial recovery of the amplitude of the EPP after α7 nAChR block following AChE and BChE inhibition described in the preceding section is translated in the recovery of T1 amplitude during supramaximal stimulation. These changes required 20 min to be effective and were maintained over time.
Figure 6.
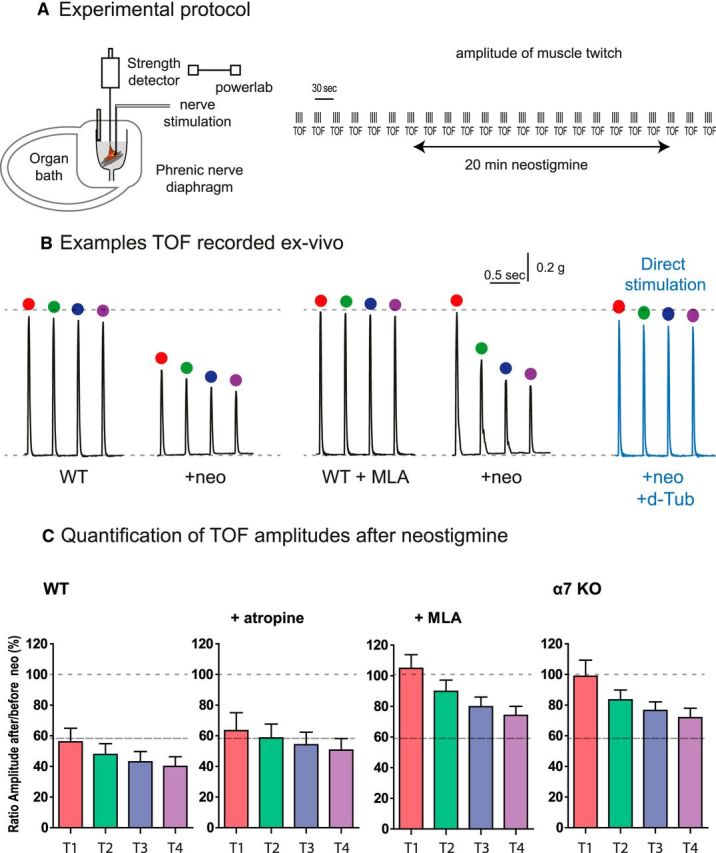
The reduction of the muscle contraction after inhibition of AChE and BChE is partially reversed by the block of α7 nAChR. A, Experimental protocol. The phrenic-diaphragm preparation was mounted in the organ bath. The muscle is indirectly stimulated by the nerve and the twitch was quantified with a tension sensor. The muscle was stimulated each 30 s by 4 stimulations at 2 Hz. B, Example of the representative traces recorded before and 20 min after incubation with neostigmine (3 μm). C, Amplitude of 6 TOFs obtained from in control condition, in atropine (0.1 μm), in MLA (10 nm), and in α7 nAChR KO mice. All values correspond to the mean of six successive TOFs in six different animals.
The analysis of T2-T4 revealed an additional process. When cholinesterases were inhibited in WT, the amplitudes of T4 were smaller than T1 (TOFR ∼0.7). These events occur during the range of few hundred milliseconds to seconds after T1 and are cumulative with T2 and T3. These events are reversible: after 28 s, the amplitude of T1 is returned to the value of T1 of the TOFn-1. A similar ratio was observed after inhibition of muscarinic receptors by atropine. Unexpectedly, the TOFR was also ∼0.6–0.7 when α7 nAChR was not functional, even when T1 mean amplitudes were maintained. To ensure that the decrease of the contraction did not result from the failure of the muscle function, but rather from a decrease of the synaptic transmission, we stimulated the muscles directly. Synaptic transmission was blocked with d-tubocurarine (a nAChR blocker). In these conditions, the four responses after TOF stimulation were identical to the control responses (Fig. 6B,C). Therefore, the reduction of the contraction was due to an alteration in synaptic transmission. The interpretation of this second process needs more investigation and is outside of the scope of this study.
AChE specific versus AChE/BChE nonspecific inhibitors in the treatment of MG
MG is a chronic autoimmune neuromuscular disorder that is characterized by the weakness of voluntary muscle groups. The fluctuating weakness becomes apparent with physical activity and is reversed at rest. Eighty percent of MGs are caused by antibodies directed against muscle nAChRs. These antibodies reduce the number of functional nAChRs at NMJ (Vincent, 2010) and thus the amplitude of the EPP. Pyridostigmine and other cholinesterase inhibitors are commonly used as treatment for MG. When the amplitude of EPPs does not reach the depolarization threshold needed to trigger AP, and thus the contraction of the muscle, AChE inhibition extends the action of ACh and increases the amplitude of EPP. In contrast to this positive action, inhibition of BChE in addition to AChE might have the opposite effect by depressing ACh release and thus reducing the muscle contraction.
To test this hypothesis, we generated an animal model of MG in which rats were immunized with a synthetic peptide (Baggi et al., 2003). After immunization, ∼50% of all animals developed a moderate MG. The evolution of MG was followed by the reduction of the amplitude of the integral muscle APs triggered by the sciatic nerve stimulation by train of 200 pulses applied at 40 Hz. In healthy rats, the amplitude of 200th AP (AP200) was 95 ± 1% of the first one (AP1; Fig. 7B,C). In the affected animals, AP200 was reduced to 81 ± 1% (Fig. 7B,C). Intraperitoneal injection of 5 μg/kg ambenonium (a selective AChE inhibitor that does not cross the blood–brain barrier) maintained the AP200 to control level after 30 min and for several hours (AP200 was 95 ± 1% of AP1). In contrast, intraperitonial injection of 0.2 mg/kg bambuterol (a selective inhibitor of BChE) after ambenonium reduced AP200 to a value of 90 ± 1% (Fig. 7B,C).
Figure 7.
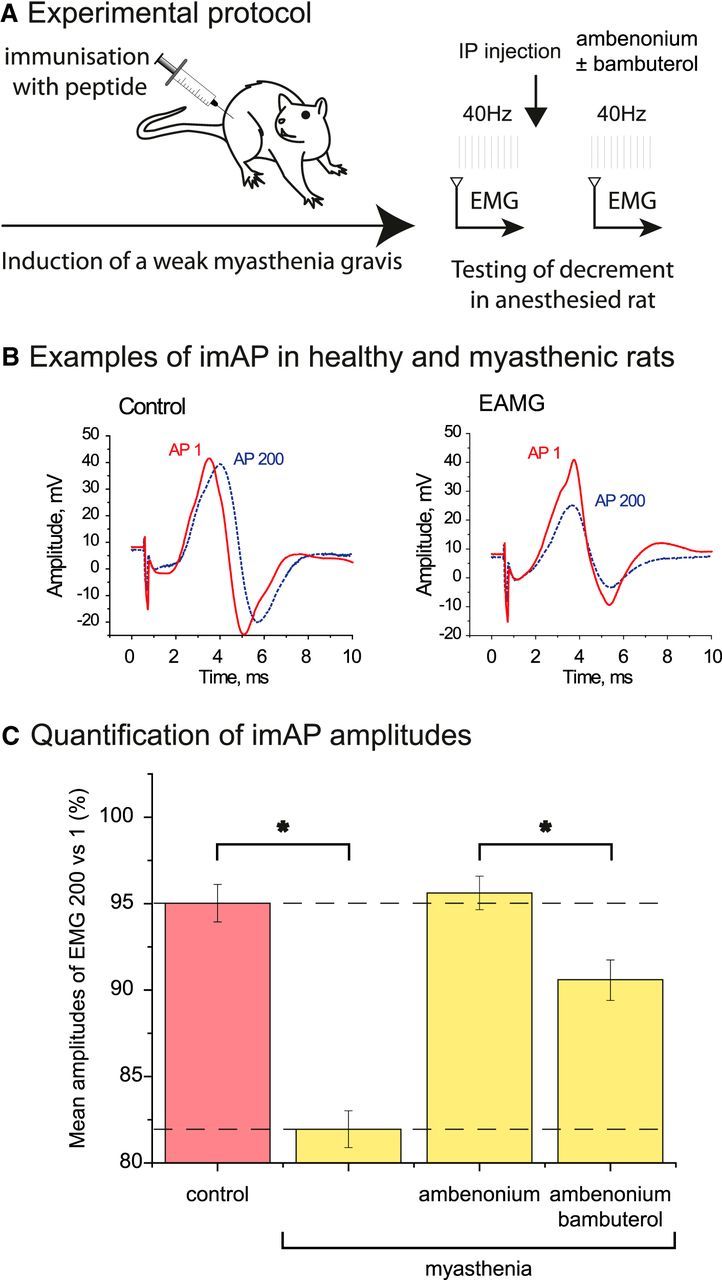
Rescue of MG rat with specific AChE inhibitor and the effect of subsequent BChE inhibition. A, Experimental protocol. Rats were immunized with a peptide (sequence of main immunogenic region of rat muscle type AChR). EAMG was induced in 2 months. MG was tested by electromyography (EMG) recorded on leg of anesthetized rats. B, Examples of integral muscle action potential (imAP) trace in intact (left) and at EAMG (right) muscles. The first nerve stimulation triggered a trace (AP1, red solid line). After 200 nerve stimulations at 40 Hz, the trace (AP200) is shown as a blue dashed line. C, The ratio of the amplitudes of AP200 versus AP1 was used for the quantification. The graph presents the percentage of AP200 versus AP1 in a control rat and in an EAMG rat before and after AChE inhibition (injection of 5 μg/kg ambenonium, i.p.) and subsequent BChE inhibition (injection of 0.2 mg/kg bambuterol, i.p.) (n = 5, *p < 0.05, unpaired Student's t test).
These in vivo experiments suggest that the differential actions of AChE and BChE can be used to improve the treatment of MG.
Discussion
We have identified a negative feedback loop controlling neurotransmitter release at the mammalian NMJ (Fig. 8). When ACh spillover is increased, α7 nAChR activation depresses ACh release. α7 nAChRs work as sensors of ACh outside of the synaptic cleft. Their activation is controlled by BChE anchored by PRiMA on the surface of TSCs at mouse NMJs. The presence of an ACh sensor that reduces release may explain some effects of AChE inhibitors that are commonly used to improve the synaptic transmission at the NMJ in patients affected with MG or to reverse residual blockade at the end of an anesthesia procedure requiring the use of a muscle relaxant. The differential functions of AChE in the synaptic cleft and BChE on the TSC have to be taken into account and suggest that specific AChE inhibitors should be used to ensure the best management in pathological conditions.
Figure 8.

Diagram showing the signaling by ACh at the NMJ. Top, Localization of AChE, BChE, and α2βδε nAChRs and α7 nAChRs in the three domains of the NMJ: the axon terminal, the muscle fiber, and the extension of the TSC. AChE is mainly clustered (by ColQ) in the primary and secondary cleft. It controls the activation of muscle α2βδε nAChRs. BChE is mainly anchored (by PRiMA) at the surface of the TSC and controls the activation of α7 nAChRs located on the TSC. Bottom, Left, When BChE is inhibited, the ACh that has escaped hydrolysis by the cleft AChE and the TSC BChE activates α7 nAChRs on the TSC. This induces a decrease of the QC. Right, When AChE and BChE are both inhibited, ACh spillover dramatically increases and the increased activation of α7 nAChRs produces a large decrease of the QC. This counteracts the increased activation of muscle nAChRs by spilled over ACh.
α7 nAChR at the TSC modulate the QC at the NMJ
Numerous experiments have shown that ACh acts on the nerve terminal and modulates its own release (Ciani and Edwards, 1963; Okamoto and Riker, 1969; Duncan and Publicover, 1979; Dudel, 2007; Kupchik et al., 2011). Many investigators have applied exogenous agonists of AChR to reveal the presence of muscarinic and nicotinic AChRs (Miyamoto and Volle, 1974; Nikolsky et al., 1991, 2004; Vizi and Lendvai, 1997; Slutsky et al., 2001; Minic et al., 2002). In some experiments, muscarinic agonists have inhibited ACh release from motor nerve terminals, whereas in others, ACh release was increased (Abbs and Joseph, 1981; Wessler et al., 1987; Arenson, 1991; Robitaille et al., 1997; Santafé et al., 2007). At the vertebrate NMJ, the activation of m2 AChR reduced the release, whereas the activation of m1/m3 AChR facilitated the release (Slutsky et al., 2001; Minic et al., 2002; Oliveira et al., 2002, 2009; Dudel, 2007).
The regulation of ACh release at NMJs by nAChRs is less documented. The differential effects of nAChR blockers used by the anesthesiologists have been interpreted so far by their potential action on nAChRs. It was assumed that these receptors are localized at the nerve terminal (for review, see Vizi and Lendvai, 1999). Nicotine, a potent agonist of nAChRs, inhibits rhythmic activity of mouse NMJs (Balezina et al., 2006). This effect is prevented by α-cobratoxin (a blocker of muscle and α7 nAChRs) and by apamin (a blocker of Ca2+-activated potassium channels). It was thus proposed that nicotine stimulates presynaptic nAChRs (presumably α7) that activate Ca2+-activated potassium channels. However, previous experiments failed to find α-bungarotoxin (α7 nAChRs ligand in neurons) on the nerve terminal (Jones and Salpeter, 1983) and our results suggest that α7 nAChRs are localized at the TSC. We found a weak labeling with specific antibodies colocalized with S100 (a well established TSC marker). This labeling was absent in α7 KO mice. Calcium waves in the TSC triggered by long bursts of nerve stimulations were abolished by 10 nm MLA (a specific blocker of α7 nAChR). These observations do not exclude that α7 nAChRs are also present at the postsynaptic membrane, where they were found in pathological conditions such as burning or immobilization (Lee et al., 2014).
How the TSCs may affect the release on the nerve terminal is not clear. There are many steps between the activation of α7 nAChR by the spillover of ACh and the reduction of ACh release at the nerve terminal. The initial step is likely to be the entry of calcium in the TSCs and the last one could be a decrease of calcium entry through inhibition of P/Q-type calcium channels of the nerve terminal. Between these two steps, a plausible hypothesis is the release by the TSCs of “gliotransmitters” such as glutamate, ATP, or adenosine because, in the CNS, these compounds have been shown to be released by glial cells, to have receptors on nerve terminals, and to interfere with voltage-dependent calcium channels (Araque et al., 2014), and there is evidence for similar phenomena in the peripheral nervous system (Castonguay et al., 2001). However, our main observations have been made minutes after the activation of α7 nAChRs so one cannot exclude slower cellular mechanisms, including the transcription and/or translation of new proteins or peptides.
The observation that long bursts of nerve stimulations (30 s, 50 Hz) can trigger calcium waves at the TSCs of mammalian NMJ was first made by Rochon et al. (2001). These calcium waves were blocked by 20 μm atropine and were therefore attributed to the activation of muscarinic receptors (mAChRs). However, after 25 min of recovery, long bursts of nerve stimulations were not able to generate new calcium waves (Rochon et al., 2001). In contrast, with a similar pattern of nerve stimulation (120 s, 20 Hz), we triggered repetitive calcium waves in the TSCs when 30 min recovery periods were allowed. Moreover, we found that the second waves were abolished by MLA. The design of the experiments likely explains this discrepancy. Calcium imaging at the NMJ requires that muscle contraction be abolished. To prevent muscle contraction, we used μ-conotoxin GIIIB, a blocker of the muscle voltage-gated sodium channel that does not interfere with α7 nAChRs and muscle nAChRs. In contrast, Rochon et al. (2001) (see also Todd et al., 2007; Rousse et al., 2010) blocked the contraction using d-tubocurarine or α-bungarotoxin, which are known to block α7 nAChRs (Alexander et al., 2013), and applied atropine at 20 μm, a concentration that blocks mAChRs, but also α7 nAChRs (Zwart and Vijverberg, 1997; González-Rubio et al., 2006). α7 nAChRs were thus likely to be blocked in these experiments and thus prevented the generation of a second wave of calcium.
The presence of functional α7 nAChRs was previously shown on astrocytes in cell culture (Sharma and Vijayaraghavan, 2001) and on NG2 cells in brain slices (Vélez-Fort et al., 2009). When α7 nAChRs are activated by ACh they induce calcium entry, but their function remains elusive. We found for the first time here that activation of extrasynaptic α7 nAChRs depresses ACh release. It remains to be determined in which physiological conditions ACh activates these α7 nAChRs. It is tempting to propose that α7 nAChRs play the role of ACh sensor outside of the synaptic cleft at the NMJ. In the context of synaptic homeostasis, it represents the first sensor that detects the excess of neurotransmitter (Davis, 2013).
BChE, a forgotten enzyme
The mammalian cholinesterases include two well known closely related enzymes, AChE (E.C. 3.1.1.7) and BChE (E.C. 3.1.1.8), that hydrolyze ACh efficiently. The participation of BChE in synaptic transmission has been completely ignored until now (Johnson and Moore, 2012). We have now revealed that BChE localized on the TSC regulates the activation of α7 nAChRs, but not the activation of the muscle nAChRs. We found that BChE is anchored by PRiMA, the small transmembrane protein required for the maturation of AChE in the brain (Perrier et al., 2002; Dobbertin et al., 2009). This localization of BChE/PRiMA at the TSC contrasts with the localization of AChE mainly anchored by ColQ in the extracellular matrix of the synaptic cleft (Younkin et al., 1982; Bernard et al., 2011). We can speculate that one of the physiological functions of BChE is to control the spillover of ACh around the TSC and thus the activation of AChRs localized outside of the synaptic cleft. Our results suggest that the spillover of ACh in the CNS described previously (Lamotte d'Incamps et al., 2012) also exists at the NMJ despite the high density of AChE.
Use of specific versus nonspecific AChE inhibitors
The use of specific (AChE) or nonspecific (AChE and BChE) inhibitors to treat myasthenia was described previously, but the rationale to select one or the other was not clear (Komloova et al., 2010). The pharmacokinetics of specific AChE inhibitors is obviously different from the nonspecific inhibitors because BChE is highly abundant in the serum and in several tissues. Indeed, BChE is a scavenger of the nonspecific inhibitors and decreases their active concentration. Moreover, the level of BChE is variable in the human population due to the genetic polymorphism (Benyamin et al., 2011) and to the eventual previous exposure of each individual to drugs and toxins detoxified by BChE (Masson and Lockridge, 2010). Therefore, the nonspecific inhibitors are more difficult to use because they bind to variable levels of BChE, a problem that does not affect the specific AChE inhibitors. We provide here direct evidence for an additional advantage of specific AChE inhibitors over nonspecific inhibitors to improve the muscle function in the treatment of MG. The AChE inhibition increases the lifetime of ACh molecules in the synaptic cleft and thus the number of AChRs opened by ACh, allowing EPP amplitude to reach the threshold of AP and thus to trigger muscle fiber twitch. If BChE is also inhibited, the increase of the EPP is expected to be limited by the negative feedback loop that includes α7 nAChR on the TSC: fewer quanta of ACh are released and the amplitude of the EPP is reduced.
In human surgery, the muscle contraction is blocked with nondepolarizing muscle relaxant (NDMR) agents (competitive antagonists of muscle nAChR). The reversion of the block is a critical period in which NDMRs must unbind the nAChR (Srivastava and Hunter, 2009). The most common method to displace the competitive antagonist is the increase of ACh concentration by inhibition of AChE. Based on empirical practices, neostigmine, the most common used AChE inhibitor (a carbamate-nonspecific AChE and BChE inhibitor), must be applied only when the synaptic transmission has recovered in a part of the fiber, otherwise the muscle enters in a longer block. It is tempting to propose that specific AChE inhibitors would be more efficient because the risk of triggering the depression of the release by the inhibition of BChE would be reduced.
Footnotes
This work was supported by the Russian Government (Program of Competitive Growth of Kazan Federal University among World's Leading Academic Centers Grant to E.N.), the Russian Foundation Basic Research (Grant 12-04-33296 to K.A.P.), and the AFMTelethon (E.K.). We thank Marc Abitbol and Philippe Ascher for critical reading of the paper and helpful discussions; Al-Shekhadat (Biomedical Center, St.Petersburg, Russia) for synthesis of peptide for EAMG induction; M. Skok (Palladin Institute of Biochemistry, Kiev, Ukraine) for the gracious gift of antibodies against α7 nAChR; Géraldine Toutirais for managing the platform Service de Microscopie Électronique of the Institut de Biologie Paris-Seine (Université Pierre et Marie Curie, Paris) and Jean Maurice Petit for managing the platform Service Commun de Microscopie (Université Paris Descartes); and Uwe Maskos for providing the founder KO mice of α7, β2, and β4 nAChRs.
The authors declare no competing financial interests.
References
- Abbs ET, Joseph DN. The effects of atropine and oxotremorine on acetylcholine release in rat phrenic nerve-diaphragm preparations. Br J Pharmacol. 1981;73:481–483. doi: 10.1111/j.1476-5381.1981.tb10446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators The concise guide to pharmacology 2013/14: enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali HH, Wilson RS, Savarese JJ, Kitz RJ. The effect of tubocurarine on indirectly elicited train-of-four muscle response and respiratory measurements in humans. Br J Anaesth. 1975;47:570–574. doi: 10.1093/bja/47.5.570. [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A. Gliotransmitters travel in time and space. Neuron. 2014;81:728–739. doi: 10.1016/j.neuron.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenson MS. The effects of muscarine and atropine reveal that inhibitory autoreceptors are present on frog motor nerve terminals but are not activated during transmission. Naunyn Schmiedebergs Arch Pharmacol. 1991;343:128–133. doi: 10.1007/BF00168599. [DOI] [PubMed] [Google Scholar]
- Auld DS, Robitaille R. Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron. 2003;40:389–400. doi: 10.1016/S0896-6273(03)00607-X. [DOI] [PubMed] [Google Scholar]
- Baggi F, Annoni A, Ubiali F, Longhi R, Mantegazza R, Cornelio F, Antozzi C. Effect on T cell recognition and immunogenicity of alanine-substituted peptides corresponding to 97–116 sequence of the rat AChR α-subunit. Ann N Y Acad Sci. 2003;998:395–398. doi: 10.1196/annals.1254.049. [DOI] [PubMed] [Google Scholar]
- Balezina OP, Fedorin VV, Gaidukov AE. Effect of nicotine on neuromuscular transmission in mouse motor synapses. Bull Exp Biol Med. 2006;142:17–21. doi: 10.1007/s10517-006-0280-3. [DOI] [PubMed] [Google Scholar]
- Benyamin B, Middelberg RP, Lind PA, Valle AM, Gordon S, Nyholt DR, Medland SE, Henders AK, Heath AC, Madden PA, Visscher PM, O'Connor DT, Montgomery GW, Martin NG, Whitfield JB. GWAS of butyrylcholinesterase activity identifies four novel loci, independent effects within BCHE and secondary associations with metabolic risk factors. Hum Mol Genet. 2011;20:4504–4514. doi: 10.1093/hmg/ddr375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Girard E, Hrabovska A, Camp S, Taylor P, Plaud B, Krejci E. Distinct localization of collagen Q and PRiMA forms of acetylcholinesterase at the neuromuscular junction. Mol Cell Neurosci. 2011;46:272–281. doi: 10.1016/j.mcn.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondet B, Carpentier G, Ferry A, Chatonnet A, Courty J. Localization of butyrylcholinesterase at the neuromuscular junction of normal and acetylcholinesterase knockout mice. J Histochem Cytochem. 2010;58:1075–1082. doi: 10.1369/jhc.2010.956623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonansco C, Couve A, Perea G, Ferradas CÁ, Roncagliolo M, Fuenzalida M. Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur J Neurosci. 2011;33:1483–1492. doi: 10.1111/j.1460-9568.2011.07631.x. [DOI] [PubMed] [Google Scholar]
- Castonguay A, Robitaille R. Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci. 2001;21:1911–1922. doi: 10.1523/JNEUROSCI.21-06-01911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani S, Edwards C. The effect of acetylcholine on neuromuscular transmission in the frog. J Pharmacol Exp Ther. 1963;142:21–23. [PubMed] [Google Scholar]
- Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R, Koelle GB. Electron microscopic localization of acetylcholinesterase and nonspecific cholinesterase at the neuromuscular junction by the gold-thiocholine and gold-thiolacetic acid methods. J Cell Biol. 1967;34:157–171. doi: 10.1083/jcb.34.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paola V, Arber S, Caroni P. AMPA receptors regulate dynamic equilibrium of presynaptic terminals in mature hippocampal networks. Nat Neurosci. 2003;6:491–500. doi: 10.1038/nn1046. [DOI] [PubMed] [Google Scholar]
- Dobbertin A, Hrabovska A, Dembele K, Camp S, Taylor P, Krejci E, Bernard V. Targeting of acetylcholinesterase in neurons in vivo: a dual processing function for the proline-rich membrane anchor subunit and the attachment domain on the catalytic subunit. J Neurosci. 2009;29:4519–4530. doi: 10.1523/JNEUROSCI.3863-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudel J. The time course of transmitter release in mouse motor nerve terminals is differentially affected by activation of muscarinic M1 or M2 receptors. Eur J Neurosci. 2007;26:2160–2168. doi: 10.1111/j.1460-9568.2007.05770.x. [DOI] [PubMed] [Google Scholar]
- Duncan CJ, Publicover SJ. Inhibitory effects of cholinergic agents on the release of transmitter at the frog neuromuscular junction. J Physiol. 1979;294:91–103. doi: 10.1113/jphysiol.1979.sp012917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rubio JM, García de Diego AM, Egea J, Olivares R, Rojo J, Gandía L, García AG, Hernández-Guijo JM. Blockade of nicotinic receptors of bovine adrenal chromaffin cells by nanomolar concentrations of atropine. Eur J Pharmacol. 2006;535:13–24. doi: 10.1016/j.ejphar.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Kuffler SW, Yoshikami D. Post-synaptic potentiation: interaction between quanta of acetylcholine at the skeletal neuromuscular synapse. J Physiol. 1975;251:427–463. doi: 10.1113/jphysiol.1975.sp011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson G, Moore SW. Why has butyrylcholinesterase been retained? Structural and functional diversification in a duplicated gene. Neurochem Int. 2012;61:783–797. doi: 10.1016/j.neuint.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Jones SW, Salpeter MM. Absence of [125I] alpha-bungarotoxin binding to motor nerve terminals of frog, lizard and mouse muscle. J Neurosci. 1983;3:326–331. doi: 10.1523/JNEUROSCI.03-02-00326.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnyk OM, Gergalova GL, Komisarenko SV, Skok MV. Intracellular localization of nicotinic acetylcholine receptors in human cell lines. Life Sci. 2012;91:1033–1037. doi: 10.1016/j.lfs.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Komloova M, Musilek K, Dolezal M, Gunn-Moore F, Kuca K. Structure-activity relationship of quaternary acetylcholinesterase inhibitors–outlook for early myasthenia gravis treatment. Curr Med Chem. 2010;17:1810–1824. doi: 10.2174/092986710791111198. [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Barchad-Avitzur O, Wess J, Ben-Chaim Y, Parnas I, Parnas H. A novel fast mechanism for GPCR-mediated signal transduction–control of neurotransmitter release. J Cell Biol. 2011;192:137–151. doi: 10.1083/jcb.201007053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamotte d'Incamps B, Krejci E, Ascher P. Mechanisms shaping the slow nicotinic synaptic current at the motoneuron–Rrenshaw cell synapse. J Neurosci. 2012;32:8413–8423. doi: 10.1523/JNEUROSCI.0181-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yang HS, Sasakawa T, Khan MA, Khatri A, Kaneki M, Martyn JA. Immobilization with atrophy induces de novo expression of neuronal nicotinic α7 acetylcholine receptors in muscle contributing to neurotransmission. Anesthesiology. 2014;120:76–85. doi: 10.1097/ALN.0000000000000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson P, Lockridge O. Butyrylcholinesterase for protection from organophosphorus poisons: catalytic complexities and hysteretic behavior. Arch Biochem Biophys. 2010;494:107–120. doi: 10.1016/j.abb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoulié J, Perrier N, Noureddine H, Liang D, Bon S. Old and new questions about cholinesterases. Chem Biol Interact. 2008;175:30–44. doi: 10.1016/j.cbi.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Minic J, Molgó J, Karlsson E, Krejci E. Regulation of acetylcholine release by muscarinic receptors at the mouse neuromuscular junction depends on the activity of acetylcholinesterase. Eur J Neurosci. 2002;15:439–448. doi: 10.1046/j.0953-816x.2001.01875.x. [DOI] [PubMed] [Google Scholar]
- Minic J, Chatonnet A, Krejci E, Molgó J. Butyrylcholinesterase and acetylcholinesterase activity and quantal transmitter release at normal and acetylcholinesterase knockout mouse neuromuscular junctions. Br J Pharmacol. 2003;138:177–187. doi: 10.1038/sj.bjp.0705010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto MD, Volle RL. Enhancement by carbachol of transmitter release from motor nerve terminals. Proc Natl Acad Sci U S A. 1974;71:1489–1492. doi: 10.1073/pnas.71.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrvova K, Obzerova L, Girard E, Krejci E, Hrabovska A. Monoclonal antibodies to mouse butyrylcholinesterase. Chem Biol Interact. 2013;203:348–353. doi: 10.1016/j.cbi.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- Nikolsky EE, Bukharaeva EA, Strunsky EG, Vyskocil F. Depression of miniature endplate potential frequency by acetylcholine and its analogues in frog. Br J Pharmacol. 1991;104:1024–1032. doi: 10.1111/j.1476-5381.1991.tb12544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolsky EE, Vyskocil F, Bukharaeva EA, Samigullin D, Magazanik LG. Cholinergic regulation of the evoked quantal release at frog neuromuscular junction. J Physiol. 2004;560:77–88. doi: 10.1113/jphysiol.2004.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Riker WF., Jr Motor nerve terminals as the site of initial functional changes after denervation. J Gen Physiol. 1969;53:70–80. doi: 10.1085/jgp.53.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira L, Timóteo MA, Correia-de-Sá P. Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur J Neurosci. 2002;15:1728–1736. doi: 10.1046/j.1460-9568.2002.02020.x. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Timóteo MA, Correia-de-Sá P. Negative crosstalk between M1 and M2 muscarinic autoreceptors involves endogenous adenosine activating A1 receptors at the rat motor endplate. Neurosci Lett. 2009;459:127–131. doi: 10.1016/j.neulet.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Perrier AL, Massoulié J, Krejci E. PRiMA: the membrane anchor of acetylcholinesterase in the brain. Neuron. 2002;33:275–285. doi: 10.1016/S0896-6273(01)00584-0. [DOI] [PubMed] [Google Scholar]
- Plaud B, Debaene B, Donati F, Marty J. Residual paralysis after emergence from anesthesia. Anesthesiology. 2010;112:1013–1022. doi: 10.1097/ALN.0b013e3181cded07. [DOI] [PubMed] [Google Scholar]
- Reynolds ML, Woolf CJ. Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol. 1992;21:50–66. doi: 10.1007/BF01206897. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Jahromi BS, Charlton MP. Muscarinic Ca2+ responses resistant to muscarinic antagonists at perisynaptic Schwann cells of the frog neuromuscular junction. J Physiol. 1997;504:337–347. doi: 10.1111/j.1469-7793.1997.337be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochon D, Rousse I, Robitaille R. Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci. 2001;21:3819–3829. doi: 10.1523/JNEUROSCI.21-11-03819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousse I, St-Amour A, Darabid H, Robitaille R. Synapse-glia interactions are governed by synaptic and intrinsic glial properties. Neuroscience. 2010;167:621–632. doi: 10.1016/j.neuroscience.2010.02.036. [DOI] [PubMed] [Google Scholar]
- Santafé MM, Lanuza MA, Garcia N, Tomàs M, Tomàs J. Coupling of presynaptic muscarinic autoreceptors to serine kinases in low and high release conditions on the rat motor nerve terminal. Neuroscience. 2007;148:432–440. doi: 10.1016/j.neuroscience.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci U S A. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky I, Silman I, Parnas I, Parnas H. Presynaptic M(2) muscarinic receptors are involved in controlling the kinetics of ACh release at the frog neuromuscular junction. J Physiol. 2001;536:717–725. doi: 10.1111/j.1469-7793.2001.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Hunter JM. Reversal of neuromuscular block. Br J Anaesth. 2009;103:115–129. doi: 10.1093/bja/aep093. [DOI] [PubMed] [Google Scholar]
- Todd KJ, Auld DS, Robitaille R. Neurotrophins modulate neuron–glia interactions at a vertebrate synapse. Eur J Neurosci. 2007;25:1287–1296. doi: 10.1111/j.1460-9568.2007.05385.x. [DOI] [PubMed] [Google Scholar]
- Todd KJ, Darabid H, Robitaille R. Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci. 2010;30:11870–11882. doi: 10.1523/JNEUROSCI.3165-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vélez-Fort M, Audinat E, Angulo MC. Functional α7-containing nicotinic receptors of NG2-expressing cells in the hippocampus. Glia. 2009;57:1104–1114. doi: 10.1002/glia.20834. [DOI] [PubMed] [Google Scholar]
- Vincent A. Autoimmune channelopathies: well-established and emerging immunotherapy-responsive diseases of the peripheral and central nervous systems. J Clin Immunol. 2010;30:S97–S102. doi: 10.1007/s10875-010-9401-x. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Lendvai B. Side effects of nondepolarizing muscle relaxants: relationship to their antinicotinic and antimuscarinic actions. Pharmacol Ther. 1997;73:75–89. doi: 10.1016/S0163-7258(96)00139-8. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Lendvai B. Modulatory role of presynaptic nicotinic receptors in synaptic and non-synaptic chemical communication in the central nervous system. Brain Res Rev. 1999;30:219–235. doi: 10.1016/S0165-0173(99)00016-8. [DOI] [PubMed] [Google Scholar]
- Wessler I, Karl M, Mai M, Diener A. Muscarine receptors on the rat phrenic nerve, evidence for positive and negative muscarinic feedback mechanisms. Naunyn Schmiedebergs Arch Pharmacol. 1987;335:605–612. doi: 10.1007/BF00166975. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Action potential generation in rat slow- and fast-twitch muscles. J Physiol. 1995;486:401–410. doi: 10.1113/jphysiol.1995.sp020821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younkin SG, Rosenstein C, Collins PL, Rosenberry TL. Cellular localization of the molecular forms of acetylcholinesterase in rat diaphragm. J Biol Chem. 1982;257:13630–13637. [PubMed] [Google Scholar]
- Zwart R, Vijverberg HP. Potentiation and inhibition of neuronal nicotinic receptors by atropine: competitive and noncompetitive effects. Mol Pharmacol. 1997;52:886–895. doi: 10.1124/mol.52.5.886. [DOI] [PubMed] [Google Scholar]