

Abstract
Previous studies have shown that the Hippo pathway effector yes-associated protein (YAP) plays an important role in maintaining stem cell proliferation. However, the precise molecular mechanism of YAP in regulating murine embryonic neural stem cells (NSCs) remains largely unknown. Here, we show that bone morphogenetic protein-2 (BMP2) treatment inhibited the proliferation of mouse embryonic NSCs, that YAP was critical for mouse NSC proliferation, and that BMP2 treatment-induced inhibition of mouse NSC proliferation was abrogated by YAP knockdown, indicating that the YAP protein mediates the inhibitory effect of BMP2 signaling. Additionally, we found that BMP2 treatment reduced YAP nuclear translocation, YAP-TEAD interaction, and YAP-mediated transactivation. BMP2 treatment inhibited YAP/TEAD-mediated Cyclin D1 (ccnd1) expression, and knockdown of ccnd1 abrogated the BMP2-mediated inhibition of mouse NSC proliferation. Mechanistically, we found that Smad1/4, effectors of BMP2 signaling, competed with YAP for the interaction with TAED1 and inhibited YAP's cotranscriptional activity. Our data reveal mechanistic cross talk between BMP2 signaling and the Hippo-YAP pathway in murine NSC proliferation, which may be exploited as a therapeutic target in neurodegenerative diseases and aging.
Keywords: BMP2, hippo, neural stem cell, proliferation, transcription, YAP
Introduction
Stem cell fate determination is critical for embryonic development, and tissue homeostasis (Julian et al., 2013). Perturbation of the proliferation or differentiation process results in a reduced or excessive number of neurons, which leads to cortical malfunctions (Fang et al., 2013). Identifying mechanisms of neural stem cell (NSC) regulation will have important clinical implications.
Bone morphogenetic proteins (BMPs) have emerged as critical regulators of NSC self-renewal and maintenance. BMP pathway components are highly expressed in both embryonic and adult neurogenic niches (Morrison and Spradling, 2008; Mira et al., 2010). NSC populations can be expanded by blocking BMP signaling (Bonaguidi et al., 2008). Moreover, accumulating evidence has implicated BMP signaling in the cognitive decline associated with aging and neurodegenerative diseases (Limke and Rao, 2002; Chen et al., 2003). Increased BMP signaling results in cognitive impairments, whereas decreased BMP signaling improves cognition (Doetsch, 2003; Gobeske et al., 2009). For example, the overexpression of BMPs has been associated with decreased neurogenesis in mouse models of AD (Lim et al., 2000; Crews and Masliah, 2010). Additionally, BMP gain of function causes a reduction in neurogenesis that can be blocked by noggin overexpression (Tang et al., 2009).
Yes-associated protein (YAP) is involved in the BMP-suppressed differentiation of embryonic NSCs through a direct interaction with Smad1 (Alarcón et al., 2009). In addition, several lines of evidence suggest that YAP participates in regulating NSC behavior (Cao et al., 2008; Fernandez et al., 2009; Gee et al., 2011). Interestingly, the Drosophila gene Yorkie, the homolog of YAP, maintains neuroepithelial cells in an undifferentiated state (Zhang et al., 2011). TEA domain (TEAD) family members are key target transcription factors that mediate YAP function (Zhao et al., 2008), and YAP-TEAD transcriptional activity is involved in stem cell proliferation (Lian et al., 2010; Ramos and Camargo, 2012). Additionally, Dpp, the Drosophila ortholog of the mammalian BMP family, controls tissue growth by regulating the transcription of Bantam (Zhang et al., 2013). Because Bantam is the downstream target of the Hippo-Yki/Sd pathway, this work established an important link between Dpp and Hippo/Yki signaling, although the ability of this cross talk to regulate the proliferation of NSCs in mammals remains to be explored. Above all, both BMP2 signaling and the Hippo pathway play indispensable roles in governing NSC proliferation and differentiation. However, how these two pathways regulate the behavior of embryonic NSCs remains unclear.
In our study, loss-of-function experiments reveal that YAP mediates the BMP2-induced inhibition of embryonic NSC proliferation. Moreover, we show that BMP2-triggered Smad1 nuclear translocation competes with YAP for the interaction with TEAD, resulting in the downregulation of target genes, and we identify ccnd1 as a critical target that regulates in this process. Our findings suggest a new molecular mechanism for the connection of the Hippo/YAP and BMP2 pathways in the regulation of embryonic NSC proliferation, in which BMP2 stimulation inhibits the transcriptional activity of YAP-TEAD, reducing cell cycle ccnd1 expression and thus decreasing NSC proliferation.
Materials and Methods
Cultures of NSCs.
NSC cultures were generated as described previously (Azari et al., 2011). The female mouse E14 ganglionic eminence was dissected and mechanically dissociated into a single-cell suspension with a fire-polished Pasteur pipette. Cells were seeded in noncoated T-25 culture flasks in DMEM/F12 (Invitrogen) containing 2% B27 (Invitrogen), 20 ng/ml epidermal growth factor (ProSpec), and 20 ng/ml basic fibroblast growth factor (ProSpec) at a density of 100,000 cells/ml. Primary neurospheres were dissociated by incubation with Accutase (Millipore) and reseeded in fresh media at 50,000 cells/ml until secondary spheres were generated. All spheres used for experiments were passaged at least once. BMP2 (R&D), Noggin (ProSpec), MG132 (Sigma), and CHX (Sigma) were used at the indicated concentrations.
Sphere-forming assay.
Mouse NSCs were seeded into 96-well plates at a density of 2–5 cells/μl per well. Fresh medium was added every other day, and spheres were counted after 1 week.
Cell cycle assay.
Mouse NSCs were plated on a nonadherent culture system, grown for 4 d, dissociated into single cells, and fixed with 70% ethanol at 4°C for 24 h. After being washed with PBS, cells were incubated with propidium iodide staining solution (0.1% Triton X-100, 0.2 mg/ml RNase A, and 25 μg/ml propidium iodide) for 30 min. Samples were analyzed on a FACS.
Electroporation.
Mouse NSCs were transfected with the Amaxa Mouse NSC Nucleofector Kit (Lonza; VPG-1004; following the manufacturer's instructions) using 4 × 106 cells and 4 μg of various plasmid DNA for each transfection. Cells were resuspended in 4 ml of growth medium and replated for further experiments 24–48 h after transfection.
Infection.
Neurospheres were gently dissociated by Accutase (Millipore) treatment, and 1 × 106 cells/well were plated in 12-well tissue-culture plates. Virus was then added for 4–6 h. Cells were transferred to 10 cm tissue-culture dishes with 8 ml of fresh medium and cultured for 5 d to medium-sized neurospheres. Neurospheres infected with retrovirus were selected by culturing in medium containing puromycin for 3 d, and surviving cells were used in experiments.
Expression constructs.
3xFlag-tagged Smad1 construct was in the pCMV10 expression vector. YAP1–2δ (Gaffney et al., 2012; Sudol, 2013) coding sequence was subcloned into the pCMV-Myc or pCMV10-3xFlag expression vector. HA-TEAD1 construct was in the pCDNA3.1-HA expression vector. YAP-WT and YAP-WW constructs were in the retroviral pQCXIH expression vector.
Preparation of shRNA retrovirus.
293T packaging cells were cotransfected with gag-pol and VSVG packaging plasmids plus pSIREN-RetroQ (Clontech Laboratories). The medium was changed 6 h after transfection, and the supernatants were harvested at 24 and 48 h, filtered through 0.45 μm syringe filters, and pooled.
Primers used for yap shRNA #1: forward: GATCGTCTTAACAGTGGCACCTATTTCAAGAGAATAGGTGCCACTGTTAAG; reverse: AATTAAAAAAATAGGTGCCACTGTTAAGATCTCTTGAATCTTAACAGTGGCACCTATC. Primers used for yap shRNA #2: forward: GATCGTACGGAATATCAATCCCAGTTCAAGAGACTGGGATTGATATTCCGTATTTTTT; reverse: AATTAAAAAATACGGAATATCAATCCCAGTCTCTTGAACTGGGATTGATATTCCGTAC. Primers used for ccnd1 shRNA #1: forward: GATCGCCACAGATGTGAAGTTCATTTCTCGAGCCACAGATGTGAAGTTCATTTTTTTTT; reverse: AATTAAAAAAAAATGAACTTCACATCTGTGGCTCGAGAAATGAACTTCACATCTGTGGC. Primers used for ccnd1 shRNA #2: forward: GATCGGTTCGTGGCCTCTAAGATGAACTCGAGTTCATCTTAGAGGCCACGAACTTTTTT; reverse: AATTAAAAAAGTTCGTGGCCTCTAAGATGAACTCGAGTTCATCTTAGAGGCCACGAACC. Primers used for smad4 shRNA #1: forward: GATCGCCAGCTACTTACCATCATATTCAAGAGATATGATGGTAAGTAGCTGGTTTTTT; reverse: AATTAAAAAACCAGCTACTTACCATCATATCTCTTGAATATGATGGTAAGTAGCTGGC. Primers used for smad4 shRNA #2: forward: GATCGGCCATAGTGAAGGACTGTTTTCAAGAGAAACAGTCCTTCACTATGGCTTTTTT; reverse: AATTAAAAAAGCCATAGTGAAGGACTGTTTCTCTTGAAAACAGTCCTTCACTATGGCC.
Immunofluorescence staining.
Mouse NSCs were grown on fibronectin polyornithine-coated glass coverslips for 4–6 h. The cells were fixed with 4% PFA for 10 min. Cells were analyzed by immunofluorescence according to standard methods. For BrdU staining, the cells were incubated in 2N HCl for 30 min before blocking. The antibodies used for immunofluorescence recognized BrdU (GE Healthcare), Ki67 (Abcam), SOX2 (R&D), Nestin (Sigma), and YAP1 (Novus Biologicals). Nuclei were counterstained with Hoechst 33258 (Sigma).
Immunoprecipitation and immunoblotting.
Immunoprecipitation and immunoblotting analyses were performed as described previously (Yuan et al., 2010). Briefly, cell lysates were incubated with the indicated antibodies in the presence of Protein A: Protein G (2:1) agarose beads for 2 h at 4°C. After washing four times, the immunoprecipitates were subjected to electrophoresis. Protein expression was examined by probing Western blots of total cell lysates or immunoprecipitates with the appropriate antibodies as noted in the figure legends. Band intensity was detected with the ECL Western Blotting Analysis System. The antibodies used for Western blotting recognized Erk1/2 and p-Smad1/5/8 (Cell Signaling Technology), Flag and GAPDH (Sigma), and HA and Myc antibodies (Santa Cruz Biotechnology).
Luciferase reporter assay.
Cells were harvested 48–72 h post transfection and assayed for reporter gene activity with a Dual-Luciferase Reporter Assay System (Promega). The results are presented as the mean of at least three independent experiments that were performed in triplicate.
qRT-PCR.
Total RNA was extracted using TRIzol (Invitrogen). Reverse transcription was performed with M-MLV (Promega) according to the manufacturer's protocol. qPCR was then performed with the specific primers supplied. The average Ct value for each gene was normalized against GAPDH and calibrated against controls, and the comparative Ct value (fold change) was calculated using 2−▵▵Ct. The following primer sequences were used: ccnd1 5′-GCGTACCCTGACACCAATCTC and 3′-CTCCTCTTCGCACTTCTGCTC, yap 5′-TCACAACGATCAGACAACAAC and 3′-CAGGATGTGGTCTTGTTCTTAT, tead1 5′-CATCGCAGGGTGTCAGTGTGA and 3′-AGGCTGGCGGCTACAAGGTA and smad4 5′-GGGTTGTCTCACCTGGAATTG and 3′-AGTCGTGAACGTACTCATCCTT.
ChIP.
Cells were treated with 1% formaldehyde at room temperature for 15 min and then 2.5 m glycine added to stop the reaction. After rinsing three times with cold PBS, cells were harvested in lysis buffer 1 (50 mm HEPES-KOH, pH 7.5, 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton, Roche protease inhibitor cocktail, and 1 mm PMSF), and then resuspended in lysis buffer 2 (10 mm Tris-HCl, pH 8.0, 200 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, Roche protease inhibitor cocktail, and 1 mm PMSF). After centrifugation, each sample was resuspended and sonicated in lysis buffer 3 (10 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, Roche protease inhibitor cocktail, and 1 mm PMSF). Lysates were incubated with 50 μl of Protein A agarose/salmon sperm DNA (Millipore, 16–157) preloaded with 1 μg of YAP or IgG antibody overnight at 4°C. After washing six times with RIPA buffer (50 mm HEPES-KOH, pH 7.5, 500 mm LiCl, 1 mm EDTA, 1% NP-40, and 0.7% sodium deoxycholate), Protein A agarose/salmon sperm DNA was incubated at 65°C to reverse the covalent histone–DNA bonds. DNA was extracted with phenol: chloroform: isopropanol and used for RT-PCR. The antibodies used for immunoprecipitation were YAP1 (Novus Biologicals) and IgG (negative control; Santa Cruz Biotechnology). DNA was analyzed by qPCR for TEAD-binding sites of the ccnd1 regulatory region, and fold induction over input was calculated using 2−▵▵Ct. The following primer sequences were used: ccnd1 5′-CATTCTGCTCACTGCCATTTC and 3′-AACCACGTAACCCACCACCGACAA.
Statistics.
For each experiment, the data are presented as the mean ± SEM, except for the analyses of the luciferase assays, where the data are presented as the mean ± SD. Each experiment was repeated at least three times. Statistical significance (p < 0.01) was determined using Student's t test or an ANOVA, followed by Bonferroni's post hoc test. Data were analyzed using Origin 7.0 Software.
Results
BMP2 represses embryonic NSC proliferation and self-renewal
BMP2 is a multifunctional growth factor and a highly expressed BMP family member in adult mouse NSCs (Chen et al., 2004), suggesting a significant role for BMP2 in this cell type. Previous studies have suggested that BMP signaling plays an important role in maintaining NSC quiescence in the adult brain (Johnston and Lim, 2010). Noggin, a BMP antagonist, can block its activation.
We first explored whether BMP2 activation impacts the self-renewal and proliferation of mouse embryonic NSCs by performing the in vitro neurosphere assay (Vanderluit et al., 2004). Neurosphere size and the number of multipotent neurospheres generated during passaging reflect the growth and self-renewal activity of mouse NSCs (Ahmed et al., 2009). Mouse NSCs cultured in the proliferating condition in vitro treated with BMP2 produced fewer neurospheres with smaller size than the controls (Fig. 1A,B), suggesting that BMP2 activation inhibited the self-renewal and proliferation of NSCs. To confirm these results, we quantified the number of proliferating NSCs by performing the BrdU incorporation (S phase) analysis. BMP2 treatment resulted in an average 20% decrease in the size of the BrdU-positive population (Fig. 1C,D), and the decrease in proliferation was not due to differentiation because the NSC marker Nestin was expressed in the cultured neurospheres (Fig. 1C). In addition, we found that mouse NSCs exposed to BMP2 exhibited a 15% reduction in the S/G2/M phase and a 15% increase in the G0/G1 phase by flow cytometry analysis (Fig. 1E). Consistent with the increased proportion of cells in G0/G1, the population of Ki67-positive cells was significantly reduced (25% decrease in BMP2-treated NSCs compared with the untreated cells; Fig. 1F). Noggin (the BMP antagonist) treatment partially rescued the proliferation inhibition induced by BMP2 signaling (Fig. 1). BMP2 activation represses mouse embryonic NSC self-renewal and proliferation.
Figure 1.
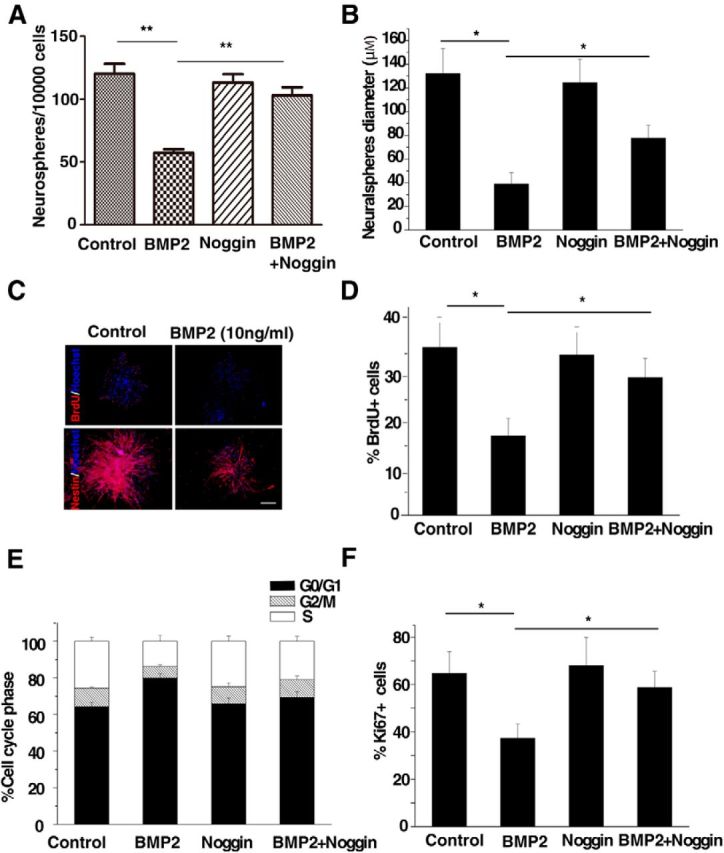
BMP signaling activation inhibits the proliferation of murine embryonic NSCs in vitro. A, B, The self-renewal ability of mouse NSCs isolated from embryonic (E14.5) ganglionic eminences cultured in vitro was analyzed by the neurosphere formation assay. NSCs were treated with BMP2 (10 ng/ml) alone or together with the BMP2 antagonist noggin. Compared with control cells, the number (A) and the diameter (B) of neurosphere-forming cells are decreased in BMP2-treated cultures. Noggin abrogates BMP2-mediated inhibition of NSC proliferation (ANOVA; n ≥ 3, p < 0.01). C, Embryonic NSCs treated with or without BMP2 (10 ng/ml) were stained with an antibody against BrdU (red, top) or Nestin (red, bottom) for the in vitro mouse NSC proliferation analyses. Scale bars: 100 μm. D, Quantification of the number of BrdU+ cells. Murine NSCs were treated as described in A and BrdU (5 μg/ml) was added for 2 h before fixation. Fewer BrdU+ cells are observed after BMP2 treatment, and noggin can inhibit the BMP2-induced proliferation repression (ANOVA; n ≥ 3, p < 0.01). E, Cell cycle analysis of NSCs with the same treatment as described in A. Flow cytometry analysis demonstrated that fewer cells are in the S phase after BMP2 activation, and noggin could restore the BMP2-induced phenotype (ANOVA; n ≥ 3, p < 0.01). F, Ki67 staining was used to measure mouse NSC proliferation. Cells were treated as described in A. Fewer Ki67-positive cells are observed with 10 ng/ml BMP2 treatment, and Noggin blocks the BMP2-inhibitory phenotype (ANOVA; n ≥ 3, p < 0.01).
YAP is required for BMP2-induced proliferation inhibition in embryonic NSCs
Previous studies in chick neural tube and Xenopus laevis embryos revealed that the proliferation of neuroepithelial cells is strongly influenced by YAP activity (Hiemer and Varelas, 2013). To investigate whether YAP was essential for the reduced proliferation of embryonic NSCs induced by BMP2, we performed loss-of-function studies. Mouse NSCs were infected with a bicistronic retrovirus-expressing GFP and short hairpins for YAP (shYAP) or scrambled control sequence (Fig. 2A). Knockdown of YAP reduced the diameters and numbers of neurospheres (Fig. 2B,C), and YAP deficiency abrogated the BMP2-mediated repression of NSC proliferation, indicating that BMP2 represses mouse NSC self-renewal through the YAP protein. Further, BrdU incorporation, Ki67 staining, and cell cycle analysis showed that YAP was the target of BMP2 signaling in regulating proliferation of NSCs (Fig. 2D–F). YAP is required for the BMP2-induced proliferation inhibition of mouse embryonic NSCs.
Figure 2.

BMP2-mediated repression of embryonic NSC proliferation is dependent on YAP. A, NSCs infected with retroviral YAP shRNA or control were lysed and subjected to Western blotting to confirm the knockdown efficiency. Erk was included as the loading control. B, C, Infected mouse NSCs were plated for neurosphere diameter analysis (B) and the neurosphere-forming assay (C), as described in Figure 1A and B. When YAP is knocked down, BMP2 fails to repress the proliferation of mouse NSCs (ANOVA; n ≥ 3, p < 0.01). D, E, Quantification of BrdU incorporation (D) or Ki67 staining (E) in E14.5 ganglionic eminence mouse NSCs infected with retrovirus shYAP or control and treated with or without 10 ng/ml BMP2. Upon knockdown of YAP, BMP2 fails to inhibit the proliferation of mouse NSCs (ANOVA; n ≥ 3, p < 0.01). F, Murine NSCs were treated as in B and stained with propidium iodide for flow cytometry assay. Both BMP2 treatment and YAP knockdown increase the percentage of G0/G1 cells (ANOVA; n ≥ 3, p < 0.01).
BMP2 represses YAP-TEAD transcriptional activity in embryonic NSCs
Previous studies have suggested that YAP-TEAD can induce the expansion of neural progenitors during neural tube development (Hiemer and Varelas, 2013). Here, we performed the 3xSD-luc reporter assay (Wang et al., 2011) to examine the effect of BMP2 on YAP-TEAD-mediated transcriptional activity. We observed that BMP2 treatment decreased YAP-TEAD transcriptional activity, and noggin pretreatment could reverse the inhibition of BMP2 (Fig. 3A). The coimmunoprecipitation assay revealed that YAP formed a physical complex with TEAD1 (Fig. 3B,C), and the interaction between YAP and TEAD1 was reduced by BMP2 treatment (Fig. 3B,C). Consistently, the colocalization of YAP-TEAD1 in nucleus is decreased with BMP2 treatment (Fig. 3D). As a transcriptional cofactor, the nuclear translocation of YAP is tightly regulated by the upstream kinases MST/LAST (Zhang et al., 2008). We then investigated the subcellular localization of YAP during BMP2 treatment. We found that YAP was predominantly found in the nuclei of untreated cells, while BMP2 treatment increased the cytosolic levels of YAP and decreased its nuclear localization (Fig. 3E). These data suggest that BMP2 signaling reduces YAP-TEAD1-mediated transactivation through the regulation of the colocalization of YAP-TEAD1.
Figure 3.

BMP signaling regulates murine embryonic NSC proliferation via inhibiting YAP/TEAD transactivation. A, Murine NSCs were transfected with the 3xSD-luciferase reporter and the Renilla plasmid and treated with 10 ng/ml BMP2 and/or its antagonist noggin. The cell lysates were subjected to the dual luciferase assay. BMP2 reduces YAP/TEAD luciferase activity, and its antagonist noggin abrogates the BMP2-mediated repression (t test; n = 3, p < 0.01). B, Myc-immunoprecipitates of P19 cells transfected with HA-TEAD1 with Myc-YAP or the control vector were immunoblotted with anti-HA antibody treated with or without 10 ng/ml BMP2, respectively. As a control, 2% of the input was blotted with anti-HA or Myc antibody. BMP2 treatment reduces the interaction between exogenous YAP and TEAD1. C, Endogenous interaction between YAP and TEAD1 in embryonic NSCs with or without 10 ng/ml BMP2 treatment. BMP2 inhibits the YAP-TEAD1 interaction. D, Quantification of YAP-TEAD1 colocalization in HT22 with or without 10 ng/ml BMP2 treatment. BMP2 reduced the colocalization of YAP and TEAD1 in nucleus (t test; n = 3, p < 0.01). E, Quantification of the YAP subcellular localization in SH-SY5Y (left) and NSCs (right) with or without 10 ng/ml BMP2 treatment. BMP2 decreases the nuclear retention of YAP (t test; n = 3, p < 0.01).
BMP2 treatment increases the Smad1/4-YAP interaction
In our experiments, we observed that BMP2 treatment induced the activation of the Smad-mediated transcription (BRE-luciferase reporter), while reducing the YAP-TEAD-mediated transactivation (3xSD-luciferase reporter; Fig. 4A). The YAP-TEAD reporter activity was gradually restored after BMP2 washout (Fig. 4B). As the major downstream effectors of BMP2, Smad1, Smad5, and Smad8 become phosphorylated and form a complex with Smad4 upon BMP2 treatment, followed by their nuclear translocation (Chen et al., 2004). Previous studies have shown that YAP interacts with endogenous Smad1 (Alarcón et al., 2009). In our experiments, we confirmed that YAP interacts with Smad1, and also found that the interaction between YAP and Smad1 is reduced when the WW motifs are mutated (Bork and Sudol, 1994; Sudol, 1994; Chen and Sudol, 1995), which indicates that YAP interacts with Smad1 via the WW domain (Fig. 4C). We consistently found that BMP2 treatment increased the endogenous interaction of YAP and Smad1 or Smad4 (Fig. 4D). It has been reported that YAP could interact with the protein, which has the PPxY motif (Chen et al., 1997). We constructed the mutant plasmid Smad1 Y227A, which is speculated to be unable to interact with YAP (Bork and Sudol, 1994; Sudol, 1994; Chen and Sudol, 1995). Western blot analysis confirmed that YAP WW mutant could not interact with Smad1, whereas there is still a weak interaction between Smad1 Y227A and YAP. And there is no interaction between YAP WW and Smad1 Y227A (Fig. 4E). To investigate whether YAP WW was involved in YAP-TEAD transactivation response to BMP2, we performed 3xSD-luc reporter assay. BMP2 treatment reduces WT, but not WW domain-mutated, YAP-mediated transcriptional activation (Fig. 4F). Furthermore, we certified YAP WW function in the neurosphere assay. Mouse embryonic NSCs infected with retroviral YAP WT or YAP WW formed neurospheres at a higher frequency and with a larger size than control, indicating an increase of proliferation. Meanwhile, YAP WW overexpression, not the YAP WT overexpression, could block the BMP2-induced proliferation decrease (Fig. 4G–I), indicating that BMP2's inhibitory function is dependent on the WW domain of YAP. Our findings suggest that BMP2 treatment increases YAP interaction with Smad1/4 via WW domain and reduces the YAP-TEAD1 transactivation in regulating mouse NSC proliferation.
Figure 4.
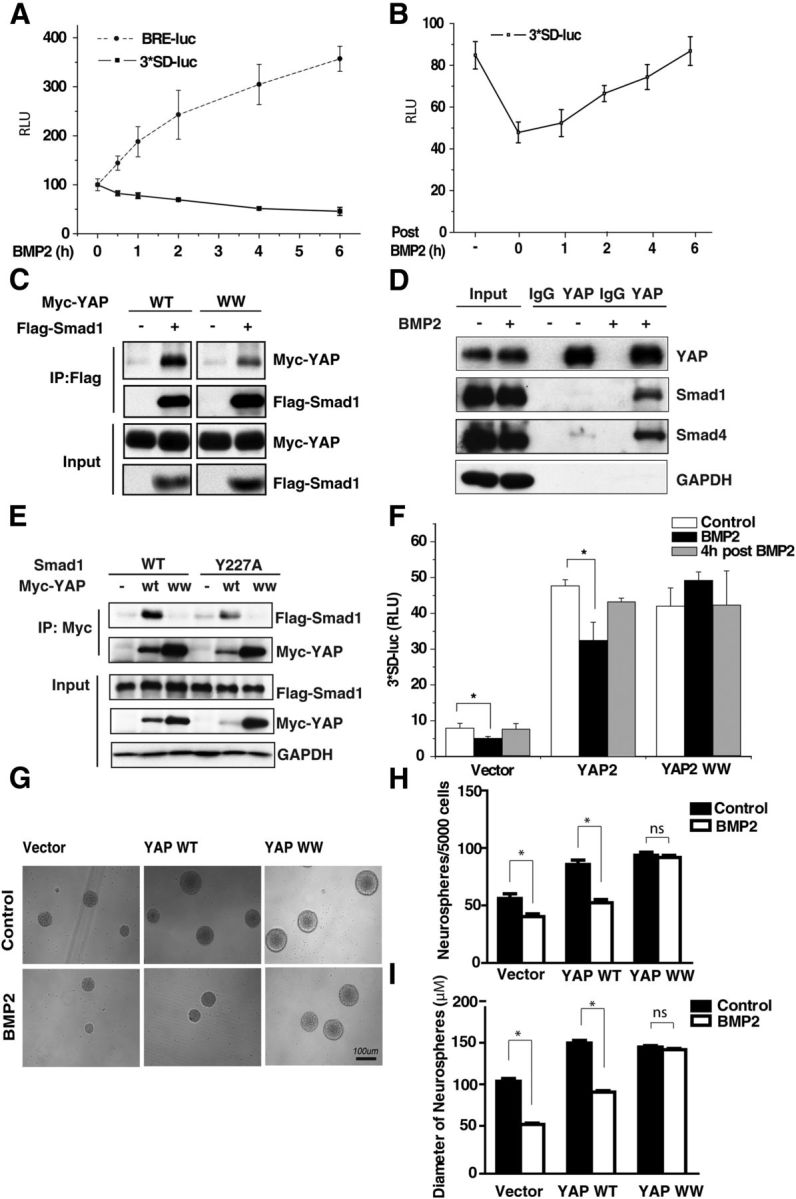
Smad1 is a competitor of the transcription factor TEAD in regulating mouse NSC proliferation. A, Mouse NSCs were transfected with 3xSD-luciferase reporter or BRE-luciferase plasmid and treated with 10 ng/ml BMP2 for different time periods. The luciferase reporter shows that BMP2 reduces YAP/TEAD-mediated transcriptional activity while enhancing Smad-mediated transactivation. B, Mouse NSCs were transfected with 3xSD-luciferase reporter and treated with BMP2. After 2 h, BMP2 was washed out and replaced with the normal medium. YAP/TEAD-mediated transactivity is gradually restored after withdrawal of BMP2 (t test; n = 3, p < 0.01). C, Flag-immunoprecipitates from NSCs transfected with Myc-YAP, WT, or Myc-YAP WW mutant plasmid together with Flag-Smad1 were immunoblotted with anti-Myc antibody and anti-Flag antibody. As a control, 2% of the input was blotted with anti-Flag or Myc antibody. D, Endogenous coimmunoprecipitation between YAP and Smad1/4 in murine embryonic NSCs shows that the YAP and Smad1/4 interaction is increased on BMP2 treatment. E, Myc-immunoprecipitates from 293T cells transfected Myc-YAP WT or Myc-YAP WW together with Flag-Smad1 or Flag-Smad1 Y227A were immunoblotted with anti-Flag antibody. Two percent input was blotted with anti-Flag, Myc, or GAPDH antibody. F, Murine NSCs were transfected with 3xSD-luciferase reporter and Myc-YAP, WT, or WW mutant plasmid. The luciferase assay shows that BMP2 inhibits WT but not WW mutant YAP-mediated transactivation (t test; n = 3, p < 0.01). BMP2 washout restores YAP-mediated transactivation. G, Representative images of neurospheres infected with retroviral YAP WT or YAP WW plasmids. Scale bar, 100 μm. H, I, Murine NSCs were infected with retroviral YAP WT or YAP WW expression vector. The number and size of the neurospheres formed after 1 week were counted and measured. YAP WW overexpression, not the YAP WT, could block the BMP2-induced proliferation inhibition (ANOVA; n ≥ 3, p < 0.01).
Smad4 is involved in the BMP-induced repression of embryonic NSC proliferation
Given that Smad4 forms a physical complex with phosphorylated Smad1 and is responsible for transporting phosphorylated Smad1 into the nucleus upon BMP2 treatment, we asked whether the ablation of Smad4 could result in the increased proliferation of NSCs in the presence of BMP2. RT-PCR analysis confirmed that Smad4 shRNA had a 40% knockdown efficiency (Fig. 5A). Embryonic NSCs infected with Smad4 shRNA retrovirus showed increased proliferation, forming neurospheres at a higher frequency with a larger size (Fig. 5B–D) in the presence of BMP2. Consistent with the above findings, Smad4 knockdown increased YAP-mediated transcription (Fig. 5E) and the interaction of YAP and TEAD1 (Fig. 5F). In addition, we observed that there were more nuclear locations of YAP in Smad4-knockdown murine NSCs (Fig. 5G,H). Thus, the decreased interaction between YAP and TEAD in response to the activated BMP2 signals could be explained by Smad1/4 competitively binding to YAP. This effect reveals a novel mechanism for the regulation of BMP2-induced inhibition of NSC proliferation, possibly through YAP/TEAD, the core components of the Hippo pathway.
Figure 5.

Knockdown of Smad4 increases the proliferation of murine embryonic NSCs by enhancing the YAP/TEAD-mediated transactivation. A, qPCR analysis showed that Smad4 was effectively suppressed by Smad4 shRNA. Primary mouse NSCs were infected with control and Smad4 shRNA (ANOVA; n ≥ 3, p < 0.01). B–D, Murine NSCs infected with retroviral shSmad4 displayed an increase in primary neurosphere formation (B). The number of neurospheres was counted 1 week after mouse NSCs were infected. NSCs infected with shSmad4 retrovirus formed neurospheres at a higher frequency (D) and with a larger size (C) than those from control NSCs (ANOVA; n ≥ 3, p < 0.01). E, Murine NSCs infected with retroviral shSmad4 were transfected with 3xSD-luciferase reporter and Renilla plasmids. The cell lysates were subjected to dual luciferase assay (t test; n = 3, p < 0.01). Smad4 deficiency increases the YAP/TEAD-mediated transactivation (t test; n = 3, p < 0.01). F, Flag-immunoprecipitates from P19 cells transfected with HA-TEAD1 and Flag-YAP in Smad4 stable knockdown cells were immunoblotted with anti-HA or Flag antibody. As a control, 2% of the input was immunoblotted with anti-HA or Flag antibody. The interaction between YAP and TEAD1 increases upon knockdown of Smad4. G, Representative images of the subcellular localization of YAP in control and Smad4-knockdown cells. Scale bars: 100 μm. H, Quantification of the subcellular localization of YAP in control and Smad4-knockdown P19 cells (G) showing that Smad4 knockdown increases nuclear accumulation of YAP (t test; n = 3, p < 0.01).
Ccnd1 is a target of YAP-TEAD signaling in the BMP2-induced inhibition of NSC proliferation
To identify target genes involved in YAP-TEAD-regulated NSC proliferation, we performed RT-PCR on three independent samples. We observed that ccnd1 expression had a 40% reduction by either BMP2 treatment (Fig. 6A) or knockdown of YAP or TEAD1 (Fig. 6B,C), indicating that ccnd1 is a potential target of YAP/TEAD1 in the proliferation of mouse NSCs. Previous studies have shown that ccnd1 affects cell growth and that elevated ccnd1 expression levels accelerate cell cycle progression (Kim and Diehl, 2009). To determine whether YAP directly regulates the transcription of ccnd1 in NSCs, we performed a ChIP assay. We found that the occupancy of YAP on the promoter region of the ccnd1 gene was decreased ∼50% in response to BMP2 treatment (Fig. 6D). To further determine the importance of ccnd1 in murine NSC proliferation, we performed a neurosphere assay in vitro using retroviral ccnd1-shRNA. As expected, ccnd1 knockdown significantly reduced NSC growth by 40% (Fig. 6E) and self-renewal capacity by ∼40% (Fig. 6F). In addition, we observed that BMP2 treatment failed to further reduce the proliferation of NSCs upon ccnd1 knockdown compared with the control (Fig. 6E,F). Our gain-of-function studies showed that YAP WT or YAP WW overexpression could elevate twofold ccnd1 mRNA level, whereas YAP WW, not YAP WT, could block the BMP2-induced inhibition of ccnd1 expression (Fig. 6G). Consistently, we found that the occupancy of overexpressed YAP WT or YAP WW on the promoter region of the ccnd1 gene was increased ∼35% (Fig. 6H). However, the expression of YAP WW mutant blocked the BMP2-induced occupancy reduction in response to BMP2 treatment (Fig. 6H). Our data strongly suggest that ccnd1 is a direct target of the YAP-TEAD1 transcriptional complex in the process of BMP2-inhibited mouse embryonic NSC proliferation.
Figure 6.
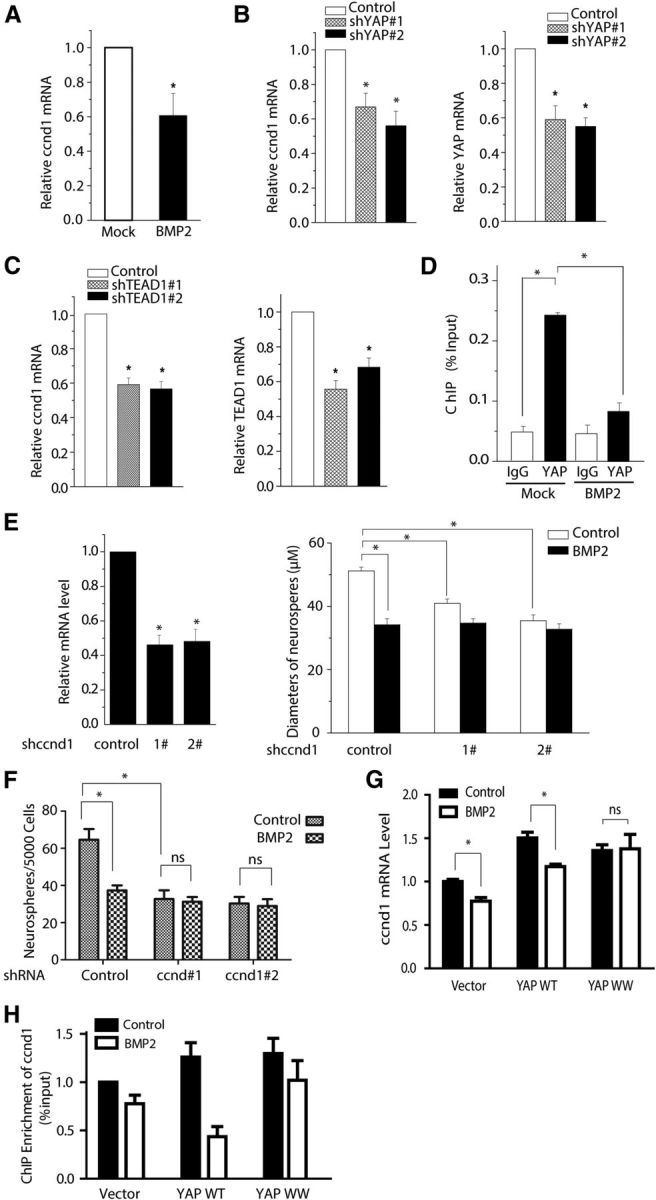
Ccnd1 functions as a downstream target in the BMP2-mediated repression of YAP/TEAD signaling in mouse NSCs. A, qPCR analysis of ccnd1 from the mouse NSCs treated with or without BMP2. BMP2 treatment (10 ng/ml) inhibited ccnd1 mRNA expression in mouse NSCs (t test; n = 3, p < 0.01). B, C, qPCR quantification analysis shows that YAP or TEAD1 were effectively knocked down by shRNA. The ccnd1 expression was reduced in YAP or TEAD1 knockdown mouse NSCs (t test; n = 3, p < 0.01). D, ChIP and qPCR analysis shows that YAP is enriched on the promoter of ccnd1 and BMP2 treatment reduces YAP's binding (t test; n = 3, p < 0.01). Mouse NSCs cultured in the proliferation medium were harvested and sonicated. The cell lysates were incubated with YAP antibody and control IgG (rabbit). DNA fragments were quantified using RT-PCR with primers for the ccnd1 promoter. E, F, qPCR analysis of ccnd1 confirms the knockdown efficiency of retroviral shccnd1 in mouse NSCs (left). BMP2 treatment (10 ng/ml) or knockdown of ccnd1 in NSCs decreases the diameters (E) and the number of neurospheres (F). Mouse NSCs were infected with control, ccnd1, and shRNA retrovirus. Neurospheres were grown for 7 d after infection (ANOVA; n ≥ 3, p < 0.01). G, Mouse NSCs were infected with control, YAP WT, and YAP WW retrovirus. Total RNA was isolated from infected NSCs. The expression of ccnd1 was determined by qPCR and normalized to GAPDH. YAP WW overexpression blocks the ccnd1 decrease induced by BMP2. H, ChIP analysis showed that YAP WW blocked the reduced expression of ccnd1 induced by BMP2 treatment.
Discussion
This study presents three key discoveries. First, we showed that BMP2 signaling represses murine embryonic NSC proliferation through YAP, the core downstream effector of the Hippo pathway. Second, BMP2 inhibits the pro-proliferation function of YAP by increasing the interaction between Smad1/4 and YAP and decreasing YAP-TEAD1 interaction. Third, we revealed a transcriptional mechanism by which YAP/TEAD regulates ccnd1 expression in response to BMP2 signaling in regulating murine embryonic NSC proliferation. The working model is illustrated in Figure 7.
Figure 7.

Working model: BMP2-SMAD signaling represses the proliferation of murine embryonic NSCs through YAP. A, In the absence of BMP2, Smad1 and Smad4 remain in the cytoplasm, and YAP/TEAD1 transcriptionally upregulates ccnd1 expression, leading to the increased proliferation of mouse NSCs. B, Upon BMP2 treatment, the Smad1/4 complex is transported into the nucleus and competes with TEAD1 for binding to YAP, thus repressing ccnd1 expression and resulting in the repression of mouse NSC proliferation. This fine-tuning signaling maintains the proper expansion of the mouse NSC pool.
YAP is required for the BMP2-induced suppression of murine embryonic NSC proliferation
BMPs elicit several different responses in mouse embryonic NSCs, including withdrawal from the cell cycle; differentiation into astrocytes, neurons, or neural crest-like cells; and apoptosis (Lim et al., 2000; Bonaguidi et al., 2008; Tang et al., 2009). These various responses depend on the BMP and BMP receptors involved and the presence of other signaling. We found that embryonic NSCs cultured in vitro with BMP2 treatment arrested more in the G0/G1 phase while retaining NSC marker expression and the capacity to proliferate. This finding is consistent with the fact that BMP2 reduces mouse embryonic NSC proliferation while most of the cells are undifferentiated (Alarcón et al., 2009). As the key downstream mediator of the Hippo pathway, YAP controls the balance of stem cell self-renewal, proliferation, and differentiation (Ramos and Camargo, 2012). However, there is little mechanistic insight into how YAP regulates stem cell properties. A recent study by Lian et al. (2010) suggests that in murine embryonic stem cells, YAP directly binds to the promoters of genes that enhance pluripotency, which suggests that YAP may be a key component of the core pluripotency machinery. We observed that YAP knockdown blocked BMP2-induced inhibition of mouse embryonic NSC proliferation in vitro (Fig. 2). Alarcón et al. (2009) reported that YAP increases Smad1-dependent transcription and is required for BMP suppression of neural differentiation of mouse embryonic stem cells. Our experiments showed that YAP WW could not interact with Smad1 and blocked the proliferation inhibition induced by BMP2 treatment. Our data suggest that YAP is involved in the BMP2-induced suppression of mouse embryonic NSC proliferation.
TAZ is a paralog of YAP and both YAP and TAZ share significant sequence similarity and redundant functions. Moreover, the regulatory mechanism of transcriptional activation is similar between YAP and TAZ (Kanai et al., 2000). However, recent studies suggested that YAP might function differentially from TAZ in stem cell biology. For example, YAP is identified as transcriptional coactivator in stem cell proliferation whereas TAZ has been shown to function as a transcriptional coactivator implicated in modulating mesenchymal stem cell differentiation (Hong and Guan, 2012). We observed that YAP knockdown does not affect the TAZ expression, indicating that the YAP-knockdown phenotype is independent on TAZ. It is still interesting to further investigate the functional interaction of YAP and TAZ in stem cells.
BMP2 enhances the YAP-Smad1/4 interaction and decreases YAP/TEAD transcriptional activity
Based on our findings, we present a model showing how YAP mediates the BMP2-induced modulation of embryonic NSC proliferation (Fig. 7). BMP2 increases the YAP-Smad1/4 interaction that promotes YAP's cytoplasmic translocation and thus inhibits the physical interaction between YAP and TEAD. The dissociation of YAP and TEAD results in the cell cycle exit of mouse embryonic NSCs. YAP-TEAD transcriptional activity was consistently increased, concurrent with the increased interaction of YAP and TEAD upon Smad4 knockdown (Fig. 5F). Meanwhile, we observed that mouse embryonic NSCs produced more neurospheres with larger sizes in the absence of Smad4 under BMP2 treatment (Fig. 5C,D). We also found that YAP accumulated more in the nucleus following Smad4 knockdown in response to BMP2. We propose that the following two possibilities may underlie the BMP2-mediated inhibition of YAP nuclear accumulation and the YAP-TEAD transactivation in mouse NSCs. One possibility is that, upon BMP2 treatment, Smad1/4 proteins transport YAP out of the nucleus, resulting in the reduction of YAP-TEAD binding; another possibility is that the nuclear Smad1/4 competes with TEADs for the binding of YAP, which might release YAP from the YAP/TEAD complex followed by YAP transport out of the nucleus. The exact mechanism whereby Smads regulate YAP translocation and activation needs to be further investigated.
Ccnd1 is the direct target of YAP/TEAD in response to BMP2 activation
We screened candidate YAP-TEAD target genes in the BMP2 modulation of mouse NSC proliferation, especially focusing on genes regulating the G1-S phase transition. p21, one known G1-S phase regulator, is previously reported involved in the cell cycle arrest of mouse NSCs. However, we found that p21 was not the direct target of YAP-TEAD in mouse NSCs. Consistent with our findings, cell cycle withdrawal of mouse NSCs is not fully abolished in p21-deficient mice, suggesting that another signaling pathway is also involved (Mathieu et al., 2008). Another cell cycle factor ccnd1 was reported as a potential YAP-TEAD target gene (Mizuno et al., 2012). We found that both the transactivation of YAP/TEAD and expression of ccnd1 decreased in the presence of BMP2 treatment in mouse NSC, indicating that ccnd1 might be one of the major downstream targets of mouse NSC proliferation. Further, we confirmed ccnd1 involved in BMP2-induced mouse NSC proliferation inhibition (Fig. 6E,F). Finally, our ChIP data (Fig. 6D) indicate that ccnd1 is the direct target gene of YAP/TEAD in response to BMP2.
In summary, we conclude that YAP/TEAD is involved in the BMP2-induced inhibition of embryonic NSC proliferation, and we investigate the molecular mechanism, in which Smad1/4 inhibits YAP transactivation by competing with TEAD binding. We also discovered that ccnd1 is the major transcriptional target of YAP in BMP2-mediated proliferation repression in mouse NSCs. This proposed model for the cross talk of BMP signaling and the Hippo pathway in mouse embryonic NSC proliferation will greatly advance our understanding of mouse NSC physiology. Furthermore, the cross talk between BMP and Hippo-YAP pathway in mouse NSCs could be exploited as a target of drugs that could help to control neurodegenerative diseases.
Footnotes
This work was supported by the Ministry of Science and Technology of China (2013DFA31990 and 973-2012CB910701 to Z.Y.) and the National Science Foundation of China (81200992 to M.Y., 81200993 to P.Z., and 81125010 and 81030025 to Z.Y.).
The authors declare no competing financial interests.
References
- Ahmed S, Gan HT, Lam CS, Poonepalli A, Ramasamy S, Tay Y, Tham M, Yu YH. Transcription factors and neural stem cell self-renewal, growth and differentiation. Cell Adh Migr. 2009;3:412–424. doi: 10.4161/cam.3.4.8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, Sapkota G, Pan D, Massagué J. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azari H, Sharififar S, Rahman M, Ansari S, Reynolds BA. Establishing embryonic mouse neural stem cell culture using the neurosphere assay. J Vis Exp. 2011;pii:2457. doi: 10.3791/2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Peng CY, McGuire T, Falciglia G, Gobeske KT, Czeisler C, Kessler JA. Noggin expands neural stem cells in the adult hippocampus. J Neurosci. 2008;28:9194–9204. doi: 10.1523/JNEUROSCI.3314-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Sudol M. The WW domain: a signalling site in dystrophin? Trends Biochem Sci. 1994;19:531–533. doi: 10.1016/0968-0004(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22:3320–3334. doi: 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- Chen HI, Sudol M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A. 1995;92:7819–7823. doi: 10.1073/pnas.92.17.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HI, Einbond A, Kwak SJ, Linn H, Koepf E, Peterson S, Kelly JW, Sudol M. Characterization of the WW domain of human yes-associated protein and its polyproline-containing ligands. J Biol Chem. 1997;272:17070–17077. doi: 10.1074/jbc.272.27.17070. [DOI] [PubMed] [Google Scholar]
- Chen HL, Lein PJ, Wang JY, Gash D, Hoffer BJ, Chiang YH. Expression of bone morphogenetic proteins in the brain during normal aging and in 6-hydroxydopamine-lesioned animals. Brain Res. 2003;994:81–90. doi: 10.1016/j.brainres.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet. 2010;19:R12–R20. doi: 10.1093/hmg/ddq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F. A niche for adult neural stem cells. Curr Opin Genet Dev. 2003;13:543–550. doi: 10.1016/j.gde.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Fang WQ, Chen WW, Fu AK, Ip NY. Axin directs the amplification and differentiation of intermediate progenitors in the developing cerebral cortex. Neuron. 2013;79:665–679. doi: 10.1016/j.neuron.2013.06.017. [DOI] [PubMed] [Google Scholar]
- Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. 2009;23:2729–2741. doi: 10.1101/gad.1824509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney CJ, Oka T, Mazack V, Hilman D, Gat U, Muramatsu T, Inazawa J, Golden A, Carey DJ, Farooq A, Tromp G, Sudol M. Identification, basic characterization and evolutionary analysis of differentially spliced mRNA isoforms of human YAP1 gene. Gene. 2012;509:215–222. doi: 10.1016/j.gene.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee ST, Milgram SL, Kramer KL, Conlon FL, Moody SA. Yes-associated protein 65 (YAP) expands neural progenitors and regulates Pax3 expression in the neural plate border zone. PLoS One. 2011;6:e20309. doi: 10.1371/journal.pone.0020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeske KT, Das S, Bonaguidi MA, Weiss C, Radulovic J, Disterhoft JF, Kessler JA. BMP signaling mediates effects of exercise on hippocampal neurogenesis and cognition in mice. PLoS One. 2009;4:e7506. doi: 10.1371/journal.pone.0007506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemer SE, Varelas X. Stem cell regulation by the Hippo pathway. Biochim Biophys Acta. 2013;1830:2323–2334. doi: 10.1016/j.bbagen.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23:785–793. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MA, Lim DA. Keeping them quiet: BMPs maintain adult neural stem cell quiescence. Cell Stem Cell. 2010;7:9–10. doi: 10.1016/j.stem.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Julian LM, Vandenbosch R, Pakenham CA, Andrusiak MG, Nguyen AP, McClellan KA, Svoboda DS, Lagace DC, Park DS, Leone G, Blais A, Slack RS. Opposing regulation of Sox2 by cell-cycle effectors E2f3a and E2f3b in neural stem cells. Cell Stem Cell. 2013;12:440–452. doi: 10.1016/j.stem.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC, Yaffe MB. TAZ: a novel transcriptional co-activator regulated by interactions with 14–3-3 and PDZ domain proteins. EMBO J. 2000;19:6778–6791. doi: 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–296. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, Ding S, Guan KL. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106–1118. doi: 10.1101/gad.1903310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, García-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/S0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- Limke TL, Rao MS. Neural stem cells in aging and disease. J Cell Mol Med. 2002;6:475–496. doi: 10.1111/j.1582-4934.2002.tb00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu C, Sii-Felice K, Fouchet P, Etienne O, Haton C, Mabondzo A, Boussin FD, Mouthon MA. Endothelial cell-derived bone morphogenetic proteins control proliferation of neural stem/progenitor cells. Mol Cell Neurosci. 2008;38:569–577. doi: 10.1016/j.mcn.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Mira H, Andreu Z, Suh H, Lie DC, Jessberger S, Consiglio A, San Emeterio J, Hortigüela R, Marqués-Torrejón MA, Nakashima K, Colak D, Götz M, Fariñas I, Gage FH. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell. 2010;7:78–89. doi: 10.1016/j.stem.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, Akatsuka S, Toyokuni S, Yokoi K, Osada H, Sekido Y. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31:5117–5122. doi: 10.1038/onc.2012.5. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A, Camargo FD. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012;22:339–346. doi: 10.1016/j.tcb.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9:2145–2152. [PubMed] [Google Scholar]
- Sudol M. YAP1 oncogene and its eight isoforms. Oncogene. 2013;32:3922. doi: 10.1038/onc.2012.520. [DOI] [PubMed] [Google Scholar]
- Tang J, Song M, Wang Y, Fan X, Xu H, Bai Y. Noggin and BMP4 co-modulate adult hippocampal neurogenesis in the APP(swe)/PS1(DeltaE9) transgenic mouse model of Alzheimer's disease. Biochem Biophys Res Commun. 2009;385:341–345. doi: 10.1016/j.bbrc.2009.05.067. [DOI] [PubMed] [Google Scholar]
- Vanderluit JL, Ferguson KL, Nikoletopoulou V, Parker M, Ruzhynsky V, Alexson T, McNamara SM, Park DS, Rudnicki M, Slack RS. p107 regulates neural precursor cells in the mammalian brain. J Cell Biol. 2004;166:853–863. doi: 10.1083/jcb.200403156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Bai Y, Song B, Wang Y, Liu D, Lai Y, Bi X, Yuan Z. PP1A-mediated dephosphorylation positively regulates YAP2 activity. PLoS One. 2011;6:e24288. doi: 10.1371/journal.pone.0024288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Kim D, Shu S, Wu J, Guo J, Xiao L, Kaneko S, Coppola D, Cheng JQ. Phosphoinositide 3-kinase/Akt inhibits MST1-mediated pro-apoptotic signaling through phosphorylation of threonine 120. J Biol Chem. 2010;285:3815–3824. doi: 10.1074/jbc.M109.059675. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A. 2011;108:2270–2275. doi: 10.1073/pnas.1019603108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Smolen GA, Haber DA. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008;68:2789–2794. doi: 10.1158/0008-5472.CAN-07-6205. [DOI] [PubMed] [Google Scholar]
- Zhang X, Luo D, Pflugfelder GO, Shen J. Dpp signaling inhibits proliferation in the Drosophila wing by Omb-dependent regional control of bantam. Development. 2013;140:2917–2922. doi: 10.1242/dev.094300. [DOI] [PubMed] [Google Scholar]
- Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20:638–646. doi: 10.1016/j.ceb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]