

Abstract
Excessive pathophysiological activity of the nuclear enzyme poly(ADP-ribose) polymerase-1 (PARP1) causes neuron death in brain hypoxia/ischemia by inducing mitochondrial permeability transition and nuclear translocation of apoptosis-inducing factor (AIF). Bcl-2/adenovirus E1B 19 kDa-interacting protein (Bnip3) is a prodeath BH3-only Bcl-2 protein family member that is induced in hypoxia, and has effects on mitochondrial permeability and neuronal survival similar to those caused by PARP1 activation. We hypothesized that Bnip3 is a critical mediator of PARP1-induced mitochondrial dysfunction and neuron death. Hypoxic death of mouse cortical neuron cultures was mitigated by deletion of either PARP1 or Bnip3, indicating that both factors are involved. Direct normoxic PARP1 activation by a DNA alkylating agent enhanced Bnip3 expression, and caused Bnip3-dependent mitochondrial membrane permeability, AIF translocation, and neuron death. Hypoxia produced PARP1-dependent depletion of nicotinamide adenine dinucleotide (NAD+) and inhibition of the NAD+-dependent class III histone deactelyase (HDAC) sirtuin-1 (SIRT1). This, in turn, led to hyperacetylation and nuclear localization of the forkhead box (Fox) protein FoxO3a, followed by enhanced association of FoxO3a with the Bnip3 upstream promoter region, increased levels of Bnip3 transcript, and elevated mitochondrial Bnip3 immunoreactivity. Finally, FoxO3a silencing using a lentiviral short hairpin RNA approach significantly reduced hypoxic Bnip3 expression, mitochondrial damage, and neuron death. Together, these data illustrate a direct PARP1-mediated hypoxic signaling pathway involving NAD+ depletion, SIRT1 inhibition, FoxO3a-driven Bnip3 generation, and mitochondrial AIF release.
Keywords: apoptosis-inducing factor, Bnip3, ischemia, mitochondrial permeability, PARP, sirtuin
Introduction
Poly(ADP-ribose) polymerase-1 (PARP1) is a genomic stability enzyme that responds to DNA damage by using nicotinamide adenine dinucleotide (NAD+) as a substrate to synthesize protein-conjugated chains of ADP-ribose polymers (PARs; Rouleau et al., 2010). PARP1 plays a paradoxically destructive role in pathological DNA damage, causing detrimental cytosolic NAD+ consumption, glycolytic inhibition, ATP depletion, and cell death (Cohen and Barankiewicz, 1987). Accordingly, blocking PARP1 activity enhances cell survival in brain trauma (LaPlaca et al., 2001) and ischemia (Eliasson et al., 1997). In brain hypoxia/ischemia, PARP1 causes mitochondrial permeability transition (MPT), leading to caspase-independent neuron death initiated by nuclear translocation of mitochondrial apoptosis-inducing factor (AIF; Yu et al., 2002; Culmsee et al., 2005). The mechanism by which nuclear PARP1 causes mitochondrial damage is the subject of continued study. There is a line of evidence that PAR binds directly to AIF, causing nuclear translocation (Andrabi et al., 2006). There are also reports that NAD+ depletion is a requisite step for AIF release (Alano et al., 2010; Won et al., 2012).
Bcl-2/adenovirus E1B 19 kDa-interacting protein (Bnip3) and its homolog, Bnip3L, are proapoptotic members of the BH3-only subfamily of Bcl-2 proteins (Willis and Adams, 2005). Like other BH3-only proteins, Bnip3 localizes to the outer mitochondrial membrane (Yasuda et al., 1998), activates Bax/Bak (Chen et al., 2010), and antagonizes antiapoptotic Bcl-2 and Bcl-XL (Boyd et al., 1994). Unlike others, neuronal Bnip3 expression is normally low (Zhang et al., 2011) but increases substantially following hypoxia or ischemia (Althaus et al., 2006; Zhang et al., 2011). In moderate hypoxia, Bnip3 may participate in mitophagy to drive a homeostatic reduction in mitochondrial mass (Bellot et al., 2009), but more severe insults lead to Bnip3-dependent MPT and cell death (Regula et al., 2002; Diwan et al., 2007; Zhang et al., 2011). The destructive mitochondrial endpoints resulting from pathological PARP1 or Bnip3 activation are similar, raising the possibility they could be linked.
Sirtuins (SIRTs) 1–7 are class III histone deacetylases (HDACs) that are dependent on NAD+ for catalytic activity, and influence several cell functions collectively geared toward survival and longevity (Anderson et al., 2003; Howitz et al., 2003). Among these targets are transcription factors and coactivators that affect mitochondrial structure and function, including forkhead box O (FoxO) family members, hypoxia-inducible factor 1 (HIF-1), and peroxisome proliferator-activated receptor γ coactivator-1α. FoxO transcription factors are cytosolic and inactive under normal conditions. However, cell stress leads to nuclear FoxO accumulation and transcription of proapoptotic targets. For FoxO3a specifically, cytosolic localization is maintained by SIRT1 activity (Giannakou and Partridge, 2004). Inhibition of SIRT1 results in FoxO3a-mediated transcription of genes including Bim, Bnip3, HIF-1α, the dynamin-related protein Drp1, and survivin (Tran et al., 2002; Mammucari et al., 2007; Hagenbuchner and Ausserlechner, 2013). All of these targets can negatively influence mitochondrial function, integrity, and/or architecture in hypoxia/ischemia.
PARP1 activity is capable of causing NAD+ depletion sufficient to inhibit SIRT1 (Pillai et al., 2005; Bai et al., 2011). We therefore hypothesized that PARP1-induced mitochondrial dysfunction is caused by SIRT1 inhibition, de-repression of FoxO3a, and enhanced Bnip3 expression.
Materials and Methods
Chemicals and reagents.
N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide HCl (PJ34) was purchased from Calbiochem. Protein inhibitor cocktail tablets and bicinchoninic acid (BCA) protein assay kits were obtained from Roche Diagnostics GmbH and Thermo Scientific, respectively. 5,5′,6,6′-Tetrachloro-1,1′,3,3′-etraethylbenzimidazolylcarbocyanine iodide (JC-1) was purchased from Cayman Chemical Company. All the other reagents were purchased from Sigma-Aldrich Canada, unless otherwise indicated.
Mouse primary neuronal culture and treatments.
Primary neuron cultures were prepared from embryonic day 16 mouse cortices from either sex, as described previously (Hunt et al., 2010) in accordance with the guidelines of the Canadian Council on Animal Care. Cells were plated on poly-d-lysine-coated plates in Neurobasal medium (NB) with B27 supplement (Invitrogen), 1.2 mm glutamine, and 5% fetal bovine serum (FBS; Hyclone). FBS was removed the following day. Cytosine arabinofuranoside (2 μm) was added on day 2 to prevent glial proliferation, and replaced on day 3 with NB/B27/glutamine. Cultures were maintained at 37°C/5% CO2 and given a partial media change on day 6. Cultures were used on day 9–10 for all experiments. Neurons were exposed to hypoxia using a 5% CO2/95% N2 gas mixture in a humidified hypoxic chamber (Billups-Rothenberg, Inc) at 37°C. To activate PARP1, cortical neurons were exposed to a DNA alkylating agent, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG; 50 μm, 30 min) and then rescued in NB medium for 4–24 h. Media was changed after hypoxia or MNNG exposure. NAD+ (10 mm) was added immediately after medium exchange, while PJ34 (10 μm) and cyclosporine A (CsA; 200 nm) were added 30 min before the addition of MNNG or the initiation of hypoxia. PJ34 and CsA were also readded after the medium exchange.
Parp1−/− mice were purchased from Jackson Laboratories and were bred in-house. Bnip3+/− mice were a gift from Dr. Gerald Dorn (Washington University, St. Louis, MO) and were bred in-house. CD-1 mouse embryos were used for the majority of experiments examining wild-type mice, but trends were verified in select experiments using background 129S parp1+/+ mice (Jackson Laboratories) and bnip3+/+ littermate controls.
Neuron death.
Propidium iodide (10 μm)-permeable dead cells and live cells actively retaining cell-permeable calcein-acetoxymethyl ester (2 μm; Invitrogen) were counted, and dead cells were expressed as a percentage of the total. Four randomly selected fields were analyzed per experimental well (150–200 cells/field) in at least three independent experiments.
NAD+ determinations.
Intracellular NAD+ levels were assessed as previously described (Tang et al., 2010). Briefly, neuronal extractions were performed using perchloric acid, and NAD+ was measured using high-performance liquid chromatography through a Microsorb C18 4.6 × 100 mm column (particle size, 3 μm; pore size, 100 Å; Rainin Instrument Company) at a flow rate of 0.5 ml/min through a photodiode array detector. NAD+ levels were normalized to protein content of the samples as determined by the BCA method.
SIRT1 activity assay.
SIRT1 activity was determined in live neurons in situ, using the SIRT1 Direct Fluorescent Screening Assay Kit (Cayman Chemical Company). Neurons were plated on 96-well plates at 6 × 104 cells/well and equilibrated with 25 μl of assay buffer (50 mm Tris-HCl, 137 mm NaCl, 2.7 mm KCl, and 1 mm MgCl2, pH 8.0). Substrate solution (15 μl), containing the p53 deacetylation target sequence Arg-His-Lys-Lys(ε-acetyl)-aminomethylcoumarin and the HDAC I/II inhibitor trichostatin A was added on a shaker for 45 min. Relative sample fluorescence was determined using a 96-well format fluorometer with an excitation wavelength of 355 nm and an emission wavelength of 460 nm.
HDAC I/II activity assay.
Histone deacetylase activity was measured by using a fluorescence activity assay kit (Cayman Chemical Company). Neuronal nuclear extracts (10 μg/well) were combined with acetylated substrate (100 mm) in the absence of NAD+ and incubated for 30 min at 37°C. After incubating with developing solution for 15 min at room temperature, fluorescence intensity was determined using a 96-well format fluorometer, with an excitation wavelength of 355 nm and an emission wavelength of 460 nm.
Immunocytochemistry.
Primary neurons on coverslips were fixed with 4% paraformaldehyde at room temperature for 1 h and permeabilized with 0.3% Triton X-100 in PBS. After blocking (5% BSA, 1 h), neurons were incubated overnight at 4°C with primary antibodies (rabbit-anti FoxO3a, 1:1000, Santa Cruz Biotechnology; mouse anti-histone H1, 1:1000) diluted in PBS with 0.2% Tween 20 and 1% BSA, pH 7.4. Cells were washed and exposed for 2 h at room temperature to either Alexa Fluor 586-labeled goat anti-mouse or Alexa Fluor 488-labeled goat anti-rabbit (Invitrogen). Coverslips were then mounted, and images were captured using a Zeiss upright epifluorescence microscope with Axiocam CCD camera.
Cell fractionation.
Mitochondria were isolated from primary cortical neurons by modified sucrose density gradient centrifugation (Almeida and Medina, 1998). Briefly, 108 cells were washed with 0.5 ml of isolation medium (320 mm sucrose, 1 mm potassium EDTA, and 10 mm Tris-HCl, pH 7.4) and homogenized using a glass-Teflon homogenizer. Homogenates were centrifuged for 5 min at 500 × g at 4°C, and the supernatant was collected. The pellet was resuspended and centrifuged at 500 × g for 5 min a second time. Pooled supernatants were centrifuged at 14,600 × g for 10 min at 4°C. The mitochondrial pellet was resuspended in isolation buffer and centrifuged at 14,600 × g for 10 min again, and the final pellet (mitochondrial fraction) was resuspended in 200 μl of isolation medium.
Nuclear fractions were extracted using a kit from Cayman Chemical Company. Cells (108) were washed with ice-cold PBS and centrifuged at 500 × g for 5 min at 4°C. The pellet was resuspended in 5 ml of ice-cold PBS/phosphatase inhibitor solution (20 mm NaF, 1 mm β-glycerophosphate, and 1 mm Na3VO4) and centrifuged at 500 × g for 5 min at 4°C. The next pellet was suspended in 500 μl of ice-cold hypotonic buffer (20 mm HEPES, 5 mm NaF, 10 μm Na2MoO4, and 0.1 mm EDTA, pH 7.4). Nonidet P-40 (NP-40; 10%, 100 μl) was added, and cells were centrifuged at 500 × g for 3 min at 4°C. The resulting pellet was resuspended in 100 μl of extraction buffer [10 mm HEPES, 0.1 mm EDTA, 1.5 mm MgCl2, 420 mm NaCl, 20 mm NaF, 1 mm β-glycerophosphate, 10 mm Na3VO4, 0.5 mm phenylmethanesulfonylfluoride, and 25% glycerol (v/v), pH 7.9] and centrifuged at 14,000 × g for 10 min at 4°C. Supernatants containing nuclear fractions were then collected, and protein was measured using the BCA protein assay kit.
Immunoprecipitation.
Polyclonal anti-FoxO3a (5 μg; Millipore) was incubated with 2.0 × 107 (50 μl) magnetic beads (Dynabeads M-450, coated with 0.1% BSA; Life Technologies) overnight at 4°C. Beads with antibody were washed with PBS and exposed to 1 mg of detergent-extracted total protein by incubation overnight at 4°C. Bead complexes were washed with PBS/0.5% Tween 20. Immunoprecipitates were then extracted with 50 μl of 2× Laemmli buffer and boiled for 5 min using for Western blots.
Real-time PCR.
Total RNA was extracted by using an RNeasy kit (Qiagen). Real-time RT-PCR was performed using an iScript One-Step RT-PCR Kit with SYBR Green (Bio-Rad) with a Bio-Rad iCycler. Primers for mouse Bnip3, FoxO3a, and β-actin are as follows: Bnip3 forward: 5′-GTAGAACTGCACTTCAGCAATGG-3′; Bnip3 reverse: 5′-GGGCTGTCACAGTGAGAACTC-3′; FoxO3a forward: 5′-TCCCAGATCTACGAGTGGATGG-3′; FoxO3a reverse: 5′-CCTTCATTCTGAACGCGCAT-3′; β-actin forward: 5′-GGGCTATGCTCTCCCTCACG-3′; and β-actin reverse: 5′-GTCACGCACGATTTCCCTCTC-3′. PCR parameters were as follows: 50°C for 10 min, 95°C for 5 min, followed by 40 cycles of PCR at 95°C for 10 s and 55°C for 30 s. Standard curves were generated, and the relative amount of target gene mRNA was normalized to β-actin mRNA. Specificity was verified by melt curve analysis.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) assays were performed as described in a previous protocol (Cell Signaling Technology) with a few modifications (Mammucari et al., 2007). Briefly, cells (5 × 106) were cross-linked with 1% formaldehyde at room temperature for 10 min, rinsed twice with PBS, and harvested in 1 ml of hypotonic buffer (15 mm Tris-HCl, 60 mm KCl, 15 mm NaCl, 5 mm MgCl2, 1 mm CaCl2, 2 mm Na3VO4, 0.25 m sucrose, 1 mm PMSF, and 1 mm DTT, pH 7.5) with protease inhibitor mixture (Roche Applied Science). After 5 min of incubation on ice, an equal volume of hypotonic buffer with 0.6% NP-40 was added for an additional 5 min period, and the mixture was centrifuged at 1000 × g for 10 min (4°C). Pelleted nuclei were digested by micrococcal nuclease (New England Biolabs) into fragments ranging in size from 150 to 900 bp, suspended in lysis buffer (150 mm NaCl, 20 mm Tris-HCl, 0.1% SDS, and 0.5% Triton X-100, pH 8.1) and sonicated with three 10 s pulses. Following preclearance with magnetic beads (Dynabeads M-450, Life Technologies), samples were used as immunoprecipitation controls or incubated with magnetic beads preincubated with 5 μg of anti-FoxO3a antibody (Santa Cruz Biotechnology) overnight with rotation at 4°C. Beads with binding partners were washed with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 150 mm NaCl), high-salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 500 mm NaCl), LiCl wash buffer (0.25 m LiCl, 1% IGEPAL CA630, 1% deoxycholic acid, 1 mm EDTA, and 10 mm Tris, pH 8.1), and TE buffer (10 mm Tris-HCl, pH 8.0, and 1 mm EDTA) in sequence. Complexes were eluted from beads with elution buffer (1% SDS, 0.1 m NaHCO3) at 37°C for 2 h. NaCl was added (0.33 m), and cross-linking was reversed by incubation overnight at 65°C. Immunoprecipitated and input DNA were then purified by treatment with RNaseA and proteinase K, and recovered by phenol/chloroform extraction, precipitated by ethanol, and resuspended in sterile water. Real-time quantitative PCR was performed with primers designed to amplify a 208 bp region (−916 to −1123) of the mouse Bnip3 upstream promoter region flanking a putative FoxO3a binding element (5′-CCAAACAA-3′, −1044 to −1051). Primers were forward 5′-GCCCTCGTATAACCTTAGCA-3′, and reverse 5′-TGGGTCAGGTCACTAGAAGC-3′. These primers were used previously to identify a FoxO3a ChIP amplicon from the mouse Bnip3 promoter region (Mammucari et al., 2007).
Lentiviral shRNA treatment of neuron cultures.
Short hairpin RNA (shRNA) constructs for FoxO3a (TRCN0000071616) and SIRT1 (TRCN0000039294) were obtained from Open Biosystems (now GE Healthcare) in the pLKO.1 plasmid backbone. Control and GFP-expressing pLKO.1 constructs were purchased from ThermoFisher (catalog #RHS4459) and Addgene (plasmid #25999), respectively. High-titer lentiviral preparations expressing shRNA and control sequences were prepared by the Lentiviral Core Platform of the University of Manitoba. Neurons were infected at 1 d in vitro (DIV) with a multiplicity of infection (MOI) of 1 in the presence of 1 μg/ml polybrene for 6 h. Medium was replaced with fresh neurobasal medium. Neuronal infection rates were consistently >90%, as assessed by the incorporation of green fluorescent protein, and FoxO3a or SIRT1 silencing were confirmed by real-time PCR and Western blotting at 7 DIV.
Western blot analysis.
Cells were mechanically removed from tissue culture plates, collected in ice-cold PBS, and centrifuged at 300 × g for 5 min. Pellets were resuspended in lysis buffer (25 mm Tris-HCl, 150 mm NaCl, and 1% Triton X-100, with protease inhibitor cocktail, pH 8.0). Protein concentrations were determined using a BCA kit. Homogenates, mitochondrial, and cytoplasmic and nuclear fractions (10–20 μg) were separated on 7.5% or 10% polyacrylamide gels and transferred to PVDF membranes according to standard procedures. A blocking buffer of 5% nonfat powdered milk in TBS with 0.1% Tween 20 was used for all incubations. Membranes were washed, and the ECL Plus Chemiluminescence Kit (GE Healthcare) was used to visualize immunoreactive bands with a Chemi-Doc Imager (Bio-Rad). Primary antibodies were anti-Bnip3 antibody (1:500; a gift from Dr. Jiming Kong, University of Manitoba), anti-FoxO3a antibody (1:1000; Millipore), anti-acetyl-lysine antibody (1:1000; Cell Signaling Technology), anti-AIF (1:1000; Sigma-Aldrich), anti-β-actin antibody (1:4000; Sigma-Aldrich), anti-COX IV (1:1000; Cell Signaling Technology), anti-histone H1 (1:400; Santa Cruz Biotechnology), anti-PAR (1:1000; Trevigen), and anti-SIRT1 (1:400; Millipore). Densitometry analysis was performed using ImageJ (National Institutes of Health). Values for each lane were normalized to protein loading markers.
Mitochondrial membrane potential imaging.
The lipophilic fluorescent probe JC-1 was used to investigate mitochondrial membrane potential (Δψm) in primary neurons (Cayman Chemical Company). Cells were loaded with JC-1 for 30 min at 37°C, as per the manufacturer's protocol, and images were acquired using a Zeiss LSM-510 confocal microscope. Samples were excited at 488 nm for monomer fluorescence and at 540 nm for JC-1 aggregate fluorescence. Emission fluorescence images were recorded at 530 nm for monomer and 590 nm for JC-1 aggregates. The ratio of the aggregate to monomer fluorescence was measured as an index of changes in Δψm. The JC-1 fluorescence ratio between red and green was normalized to the control ratio. JC-1 may also respond to plasma membrane depolarization, so the mitochondrial uncoupler p-trifluoromethoxy carbonyl cyanide phenyl hydrazone (FCCP; 10 μm) was added at the end of each experimental recording to get the maximal dissipation of Δψm.
Statistical analysis.
Data are expressed as the mean ± SEM. Statistical analysis was performed throughout using GraphPad Prism version 5.0. One-way ANOVA with Student's Newman–Keuls post hoc test was used to compare multiple groups with a single variable. Two-way ANOVA with Bonferroni's test was used to compare multiple groups with more than two variables. Differences were considered significant at p < 0.05.
Results
Hypoxic neuron death is dependent on PARP1 and Bnip3
Figure 1A–O are representative images of cortical neuron cultures exposed to hypoxia for 48 h. Phase contrast microscopy and active calcein uptake (green fluorescence) were used as indicators of live cells, while propidium iodide inclusion (red fluorescence) was used as an indicator of nonviable cells with permeable plasma membranes. At baseline, normoxic cell death in wild-type cultures was 8.9 ± 2.9% (Fig. 1A–C,P). Wild-type neurons exhibited time-dependent reductions in viability with hypoxic exposure, with 88 ± 3% cell death at the 48 h time point (Fig. 1D–F,P). The PARP1 inhibitor PJ34 (10 μm) significantly reduced hypoxic neuron death at 17, 24, and 48 h (42 ± 1%, 48 h; Fig. 1G–I,P). Hypoxic neuron death was also significantly reduced at these time points in cultures lacking the expression of either PARP1 (40 ± 1%, 48 h; Fig. 1J–L,P) or Bnip3 (41 ± 1%, 48 h; Fig. 1M–P). Neither parp1−/− nor bnip3−/− cultures appeared morphologically different from wild-type cells upon phase contrast examination. Hypoxia dramatically enhanced immunoreactivity for PARs, demonstrating the induction of PARP catalytic activity (Fig. 1Q). Both PJ34 and use of parp1−/− neurons attenuated hypoxic increases in PAR immunoreactivity, confirming that PAR production is PARP1 dependent.
Figure 1.

Hypoxic death of cortical neurons is dependent on PARP1 and Bnip3. Primary cortical neuron cultures were incubated in a hypoxic chamber. Cell death was assessed at several time points by quantifying intracellular propidium iodide fluorescence (red; A–O, middle column). Live cells were identified by calcein fluorescence (green; A–O, left column) and by phase contrast microscopy (A–O, right column). Hypoxia (48 h; D–F) caused nearly complete loss of live neurons, compared with normoxic controls (A–C). G–O, Neuron death was dramatically reduced by the PARP1 inhibitor PJ34 (10 μm; G–I) and by deletion of either PARP1 (J–L) or Bnip3 (M–O). P, Pooled data showing how increasing the time course of hypoxia affected neuronal survival. Using PJ34 and parp1−/− or bnip3−/− genotypes significantly reduced hypoxic neuron death. Q, Immunoblot showing increased PARP1-dependent PAR formation in hypoxia (24 h). A–O are representative depictions. Data in P are reported as the mean ± SEM (n = 5). **p < 0.01 compared with all other groups at the same time point using two-way ANOVA with the Bonferroni post hoc test. Scale bar, 40 μm. H/24, 24 h of hypoxia.
Normoxic PARP1 activation causes Bnip3-dependent mitochondrial permeability and neuron death
To demonstrate mechanistic linkage between PARP1 and Bnip3, we activated PARP1 directly using the DNA alkylating agent MNNG (Schreiber et al., 1995; Ying et al., 2001; Andrabi et al., 2014). MNNG (50 μm, 30 min) increased total cell (3.1 ± 0.4-fold) and mitochondrial (8.7 ± 2.2-fold) Bnip3 protein expression (4 h; Fig. 2A). This effect was dependent on PARP1 expression as parp1−/− neurons displayed MNNG-induced total cell (1.3 ± 0.2-fold) and mitochondrial (1.1 ± 0.3-fold) expression levels significantly less than wild-type controls and not statistically different than control cells lacking MNNG treatment. MNNG reduced the ratio of JC-1 mitochondrial aggregate (Fig. 2B, red fluorescence) to cytoplasmic monomer (Fig. 2B, green fluorescence) from 1.01 ± 0.02% to 0.33 ± 0.05% control, indicating dramatic loss of normal Δψm 4 h after MNNG removal. Figure 2C shows that depolarization of Δψm was significantly rescued by both PARP1 (0.73 ± 0.3% control; 4 h) and Bnip3 (0.69 ± 0.05% control; 4 h) deletion. This indicates that PARP1-induced mitochondrial damage caused by DNA alkylation is dependent on Bnip3.
Figure 2.
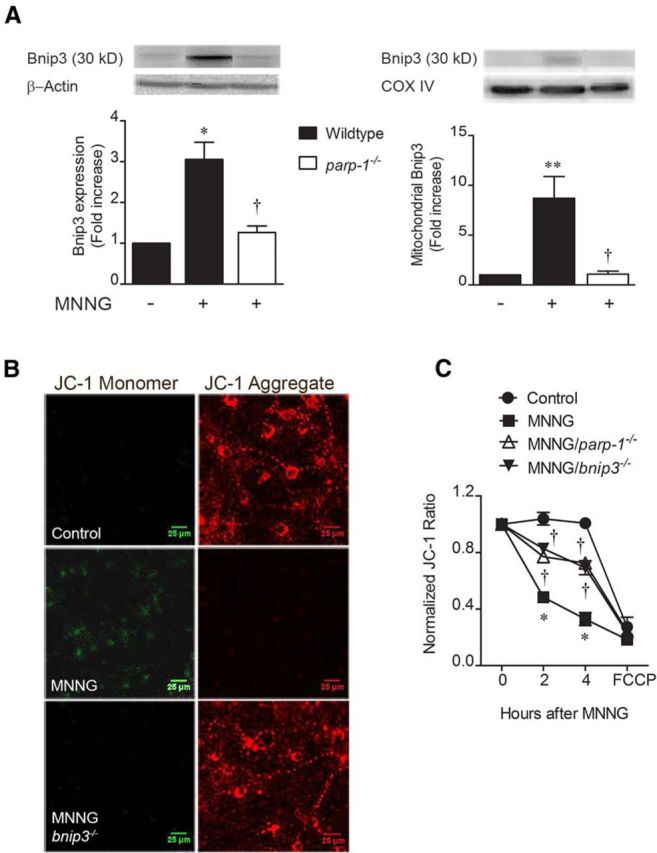
PARP1 leads to Bnip3-dependent mitochondrial damage. Cortical neuron cultures were exposed to the DNA alkylator MNNG (50 μm, 30 min) to cause pathological PARP1 activation. A, Both total cell homogenate and mitochondrial Bnip3 immunoreactivities were significantly enhanced 4 h after MNNG removal, in a manner reversed by the parp1−/− genotype. *p < 0.05, **p < 0.01, compared with 0 MNNG controls; †p < 0.05, compared with the wild-type, MNNG-treated group, using ANOVA and the Student–Newman–Keuls post hoc test. B, Mitochondrial aggregation of JC-1 was reduced 4 h post-MNNG by increasing the cationic charge inside the matrix (red); monomeric JC-1 in the cytosol (green) was enhanced by MNNG. C, MNNG-induced mitochondrial depolarization was time dependent and was significantly reduced by deleting PARP1 or Bnip3. *p < 0.05, compared with 0 MNNG controls at the same time point; †p < 0.05, compared with the wild-type, MNNG-treated groups at the same time point. Data were analyzed using two-way ANOVA with Bonferroni's post hoc test. All data are reported as the mean ± SEM (n = 5–7). Scale bars, 25 μm.
We also tested for involvement of Bnip3 in PARP1-induced neuron death signals. MNNG increased nuclear localization of mitochondrial AIF in a manner detectable 4 h after MNNG exposure (2.5 ± 0.3-fold; Fig. 3A,B). This effect was significantly reduced by using bnip3−/− cultures (1.2 ± 0.2-fold), indicating a Bnip3-dependent effect. MNNG also caused PARP1-dependent neuron death in a concentration-dependent fashion (Fig. 3B,C). Using 50 μm MNNG (30 min exposure), neuron death levels (81 ± 3%) were also found to be significantly mitigated in cultures lacking Bnip3 (41 ± 8%; Fig. 3D), again implicating Bnip3 in PARP1-mediated neurotoxocity. PARP1 activation causes depletion of cytosolic/nuclear NAD+ stores (Cohen and Barankiewicz, 1987; Cosi and Marien, 1999). We found that exogenous NAD+ replacement significantly reduced MNNG-induced nuclear AIF increases from 2.5 ± 0.3-fold to 1.4 ± 0.2-fold, and MNNG-induced neuron death from 81 ± 0.3% to 46 ± 2% (Fig. 3A,B,D). This suggests that NAD+ depletion is involved in the mechanism of PARP1-induced mitochondrial damage and neuronal toxicity. Overall, these data demonstrate that direct PARP1 activation leads to Bnip3-dependent mitochondrial dysfunction and neuron death.
Figure 3.

PARP1 causes Bnip3-dependent AIF translocation and neuron death. A, Cortical neurons were exposed to MNNG (50 μm, 30 min). Mitochondrial and nuclear fractions were assessed for immunoreactivity for AIF 4 h after MNNG removal. B, MNNG significantly increased nuclear AIF in a manner reduced by NAD+ supplementation (1 mm) or Bnip3 deletion. ***p < 0.001, compared with 0 MNNG control; ††p < 0.01, compared with the wild-type, MNNG-treated group. Data were analyzed using ANOVA with the Student–Newman–Keuls post hoc test. C, Red, MNNG also caused Bnip3-dependent cell death. Green cells are live and accumulate calcein, and the third column shows phase contrast images. D, MNNG-induced neuron death was dose dependent, and was reduced by PJ34 (10 μm) and the deletion of either PARP1 or Bnip3. *p < 0.05, **p < 0.01, compared with all other groups at the same time point. Data were analyzed using two-way ANOVA with the Bonferroni's post hoc test. E, Eighteen hours after MNNG (50 μm, 30 min), both Bnip3 deletion and NAD+ supplementation significantly reduced neuron death. **p < 0.01, compared with the wild-type, MNNG-treated group. Data were analyzed using ANOVA with the Student–Newman–Keuls post hoc test. All data are reported as the mean ± SEM (n = 5–7). Scale bar, 40 μm.
Hypoxia causes PARP1-dependent NAD+ depletion and SIRT1 inhibition
We returned to the hypoxia model to examine whether PARP1-mediated NAD+ depletion leads to inhibition of the sirtuin family of NAD+-dependent HDACs (class III) and downstream effects on gene transcription impacting neuronal survival. NAD+ content of wild-type neuron cultures declined from 1.1 ± 0.1 to 0.77 ± 0.06 nmol/mg protein after 12 h of hypoxia (Fig. 4A) and further to 0.64 ± 0.14 nmol/mg protein after 48 h of hypoxia. Assessed at the 24 h hypoxic time point, loss of NAD+ to 0.71 ± 0.04 nmol/mg protein was blocked both by PJ34 (1.10 ± 0.02 nmol/mg protein) and PARP1 deletion (1.3 ± 0.1 nmol/mg protein), indicating a PARP1-mediated effect (Fig. 4B). Concomitantly, activity of SIRT1 was inhibited to 63 ± 4% control at 24 h hypoxia (Fig. 4C). This decline effect was mitigated by PJ34 (91 ± 4%), PARP1 deletion (96 ± 4%), and exogenous NAD+ replacement (99 ± 3%), indicating that SIRT1 deacetylase activity is diminished in a PARP1- and NAD+-dependent manner in hypoxic neurons. These activity changes were observed in the absence of any hypoxic influence on total cell SIRT1 protein expression (Fig. 4D). Deacetylase activity of class I/II HDACs was not affected by hypoxia or reduced PARP1 activity (Fig. 4E).
Figure 4.

Hypoxia causes PARP1-dependent NAD+ depletion and sirtuin inhibition. A, Hypoxia caused a progressive loss of cytosolic/mitochondrial NAD+. *p < 0.05 compared with the normoxic control group. B, At 24 h, NAD+ loss was rescued by the PARP1 inhibitor, PJ34 (10 μm), and the parp1−/− genotype. **p < 0.01, compared with the wild-type, normoxic control; ††p < 0.01, compared with the wild-type hypoxic group. C, SIRT1 activity was measured in situ and was reduced by hypoxia (24 h) in a manner reversed by PJ34, PARP1 deletion, and NAD+ supplementation (1 mm). **p < 0.01, compared with the wild-type, normoxic control; ††p < 0.01, compared with the wild-type hypoxic group. D, Immunoblotting shows that 24 h of hypoxia causes no change in total cell SIRT1 expression. E, Deacetylase activity of class I/II HDACs was not affected by hypoxia. All data are reported as the mean ± SEM and were analyzed using ANOVA with the Student–Newman–Keuls post hoc test.
Hypoxia causes PARP1-dependent nuclear translocation and acetylation of FoxO3a
SIRT1 is reported to interact directly with the transcription factor FoxO3a in HEK293T cells (Motta et al., 2004), so we hypothesized that reduction of SIRT1 activity leads to enhanced FoxO3a acetylation, nuclear translocation, and transcriptional activation of target genes. Total cell homogenates were immunoprecipitated with anti-FoxO3a, and both input and pull-down samples were subsequently immunoblotted with anti-SIRT1. SIRT1 immunoreactivity was detected in FoxO3a pull-downs (Fig. 5A). Quantitatively, this interaction was not changed significantly by hypoxia or PARP1 interference, indicating consistent baseline SIRT1–FoxO3a interaction. FoxO3a acetylation levels were determined by immunoprecipitation with anti-FoxO3a, followed by immunoblotting for acetyl-lysine residues. Hypoxia (24 h) significantly enhanced FoxO3a acetylation (1.7 ± 0.1-fold; Fig. 5B) in a manner significantly inhibited by PJ34 (1.2 ± 0.1-fold) and PARP1 deletion (1.24 ± 0.03-fold). Hypoxia also enhanced nuclear colocalization of FoxO3a with histone H1 in a manner reversed by both PJ34 and PARP1 deletion (Fig. 5C). Nuclear FoxO3a levels were increased by 1.9 ± 0.1-fold at 24 h of hypoxia (Fig. 5D), and were reduced to 1.3 ± 0.1-fold and 1.2 ± 0.1-fold by PJ34 and the parp1−/− genotype, respectively, and the majority of neurons (60 ± 6%) exhibited FoxO3a translocation (control, 7 ± 1%). Together, these data demonstrate PARP1-dependent movement of FoxO3a to the nucleus. Together, these experiments show a direct interaction between SIRT1 and FoxO3a, and confirm that PARP1 activation leads to FoxO3a acetylation and nuclear translocation in hypoxia.
Figure 5.

Hypoxic PARP1 activation increases nuclear FoxO3a activity. Cortical neuron cultures were exposed to hypoxia for 24 h. A, SIRT1 immunoreactivity was pulled down by immunoprecipitation with an anti-FoxO3a antibody, indicating a physical association between SIRT1 and FoxO3a. B, Cell homogenates were immunoprecipitated with an anti-FoxO3a antibody and subjected to immunoblotting with anti-acetyl-lysine. Hypoxia (H) significantly enhanced levels of acetylated FoxO3a in a fashion mitigated by PJ34 (10 μm) and the parp1−/− genotype. C, H enhanced colocalization of FoxO3a (green) with the nuclear marker Histone H1 (red). This was quantified in D, which shows that hypoxia significantly increased nuclear, but not cytoplasmic, FoxO3a levels. This increase was attenuated by PJ34 and PARP1 deletion. All data are reported as the mean ± SEM (n = 5–7). **p < 0.01, compared with the wild-type, normoxic control; ††p < 0.01, compared with the wild-type hypoxic group. Data were analyzed using ANOVA with the Student–Newman–Keuls post hoc test. Scale bar, 10 μm.
Hypoxia causes FoxO3a binding to the Bnip3 upstream promoter region and Bnip3 expression
To determine whether PARP1-induced FoxO3a acetylation and nuclear distribution leads to changes in Bnip3 expression, we isolated nuclear chromatin fragments that bind to FoxO3a using chromatin immunoprecipitation. Recovered DNA was purified and subjected to real-time PCR using primers flanking a putative FoxO3a binding element (CCAAACAA, −1044 to −1051) within the Bnip3 upstream promoter region. Hypoxia (24 h) increased the binding of FoxO3a to the Bnip3 upstream promoter region by 10.4 ± 3.4-fold, compared with normoxic control neurons (Fig. 6A). This effect was almost completely reversed by both PJ34 (1.9 ± 0.7-fold) and PARP1 deletion (1.4 ± 0.7-fold), confirming a PARP1-dependent hypoxic interaction between FoxO3a and the Bnip3 upstream promoter region. In agreement, we found a time-dependent increase in hypoxic Bnip3 mRNA levels (Fig. 6B) that at 24 h (9.4 ± 0.7-fold; Fig. 6C) was sensitive to inhibition by PJ34 (4.9 ± 0.4-fold), PARP1 deletion (4.4 ± 0.5-fold), and exogenous NAD+ replacement (3.8 ± 0.7-fold). Both total cell and mitochondrial Bnip3 protein expression were also enhanced by 24 h hypoxia (9.7 ± 0.5-fold, mitochondrial fraction) in wild-type but not parp1−/− neurons (Fig. 6D). This indicates that hypoxic increases in Bnip3 mRNA and protein expression are PARP1 dependent and correlate with FoxO3a binding to the Bnip3 upstream promoter region. In contrast to Bnip3, hypoxic treatment did not significantly affect expression of other targets of FoxO3a, including Bim (p = 0.43), Drp-1 (p = 0.16), or HIF-1α (p = 0.37; Fig. 6E–G).
Figure 6.
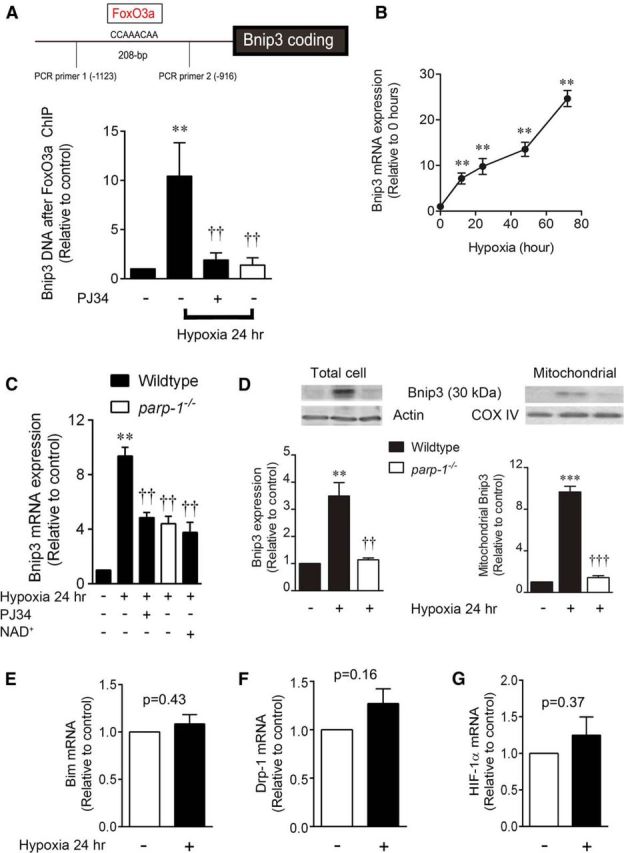
Hypoxia causes PARP1-dependent interaction of FoxO3a with the Bnip3 upstream promoter region. Cortical neuron cultures were exposed to hypoxia for up to 72 h. A, Chromatin immunoprecipitation with anti-FoxO3a antibody, followed by real-time PCR using primers flanking a putative FoxO3a binding element, produced a 208 bp fragment and revealed that hypoxia significantly increased FoxO3a binding to the Bnip3 upstream promoter region. This interaction was reversed with the PARP1 inhibitor PJ34 (10 μm) or PARP1 deletion. B, Bnip3 mRNA transcript levels were measured by real-time PCR and found to increase significantly in hypoxic conditions. C, Increases in Bnip3 transcript levels (24 h of hypoxia) were mitigated by PJ34, PARP1 deletion, and NAD+ supplementation. D, Total cell and mitochondrial Bnip3 30 kDa protein levels showed a matched hypoxic increase in a manner blocked by PARP1 deletion. E–G, mRNA for Bim, Drp-1, and HIF-1α was quantified by real-time PCR; no difference was found between control and hypoxia (p > 0.05, t test). A, C, and D: **p < 0.01 and ***p < 0.001, compared with the wild-type, normoxic control; ††p < 0.01 and †††p < 0.001, compared with the wild-type hypoxic group. B: **p < 0.01 compared with nonhypoxic control. Data were analyzed using ANOVA with the Student–Newman–Keuls post hoc test. All data are reported as the mean ± SEM (n = 5–7).
SIRT1 silencing enhances FoxO3a activity and Bnip3 expression
To establish a link between diminished SIRT1 activity and enhanced Bnip3 expression, we silenced SIRT1 using lentiviral delivery of shRNA. Following shRNA exposure (72 h), SIRT1 mRNA levels were reduced to 19 ± 1% of the scrambled shRNA control (Fig. 7A), while SIRT1 protein levels were reduced to 28 ± 2% of control (Fig. 7B). SIRT1 silencing alone was sufficient to significantly increase FoxO3a acetylation by 1.5 ± 0.1-fold, relative to control. This treatment also enhanced FoxO3a binding to the Bnip3 upstream promoter region by 3.4 ± 0.8-fold, as determined by ChIP (Fig. 7D) and increased Bnip3 mRNA levels by 2.2 ± 0.3-fold (Fig. 7E). Together, these data illustrate that ectopic silencing of SIRT1 in normoxia drives FoxO3a activation and Bnip3 expression in the absence of NAD+ depletion and PARP1 activation.
Figure 7.

SIRT1 silencing activates FoxO3a and drives Bnip3 expression. Cortical neuron cultures were treated with lentiviral particles expressing shRNA to reduce the transcription of SIRT1. A, B, Real-time PCR indicated a dramatic reduction of SIRT1 mRNA levels (A) and immunoreactivity (B) 72 h after infection. C, At 72 h after infection, acetylated FoxO3a levels were enhanced in the SIRT1 shRNA group relative to the scrambled control. D, E, FoxO3a binding to the Bnip3 promoter region (determined by ChIP) was enhanced more than threefold (D) and Bnip3 mRNA levels were increased more than twofold (E) by 72 h of SIRT1 shRNA treament. *p < 0.05, **p < 0.01, and ***p < 0.001, compared with control shRNA group using unpaired t tests. All data are reported as the mean ± SEM (n = 5–7).
FoxO3a silencing protects mitochondrial membrane integrity and neuronal survival in hypoxia
To provide direct evidence that FoxO3a activity leads to hypoxic neuron death, we assessed FoxO3a loss of function using shRNA. FoxO3a mRNA was silenced to 13 ± 7% of control at an MOI of 1, while FoxO3a protein expression was reduced to 25 ± 8% of control (Fig. 8A,B). FoxO3a shRNA significantly reduced hypoxic increases in Bnip3 transcription from 12 ± 2 to 4.2 ± 0.4 times the control (Fig. 8C), confirming the involvement of FoxO3a as a hypoxic Bnip3 transcription factor. In accordance with this finding, FoxO3a silencing also significantly reduced hypoxic loss of Δψm (Fig. 8D,E) and neuronal survival (Fig. 8F,G). The JC-1 aggregate/monomer ratio was reduced by hypoxia at 24 h to 43 ± 3% of control, and this was restored to 84 ± 4% of FoxO3a silencing (Fig. 8E). Similarly, the hypoxic neuron death rate (71 ± 3%) was reduced to 36 ± 2% by FoxO3a shRNA treatment (Fig. 8G). These data directly link FoxO3a activity to neuronal mitochondrial dysfunction and death in hypoxia.
Figure 8.
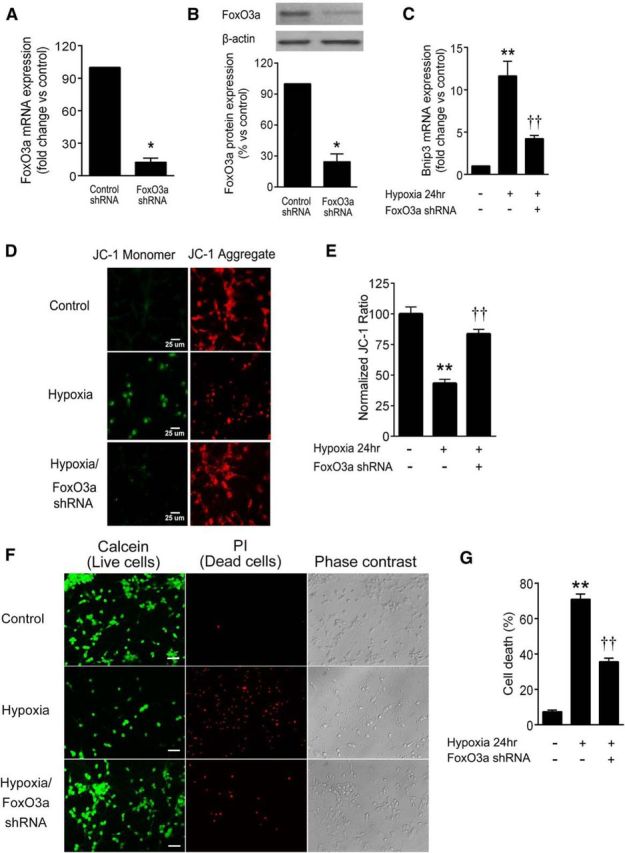
FoxO3a silencing reduces hypoxic Bnip3 expression and mitochondrial damage. Cortical neuron cultures were treated with lentiviral particles expressing shRNA to reduce the transcription of FoxO3a. A, B, Real-time PCR indicated a dramatic reduction of FoxO3a mRNA levels (A) and immunoreactivity (B). C, Hypoxia (24 h) enhanced Bnip3 mRNA levels in a manner significantly attenuated by shRNA for FoxO3a. D, E, FoxO3a shRNA significantly restored the ratio of JC-1 aggregate (red) to monomer (green) in hypoxia, indicating that FoxO3a contributed to hypoxic mitochondrial depolarization. F, G, FoxO3a shRNA significantly reduced hypoxic neuron death. A and B: *p < 0.05, compared with control shRNA group using t test. C, E, and G: **p < 0.01, compared with normoxic control; and ††p < 0.01, compared with the hypoxic group using ANOVA with Student–Newman–Keuls test. All data are reported as the mean ± SEM (n = 5–7). Scale bars: D, 25 μm; F, 40 μm.
Discussion
Our results demonstrate that pathophysiological activation of PARP1 leads to mitochondrial permeability and neuron death that is dependent on the prodeath Bcl-2 family member protein Bnip3. PARP1 enhanced Bnip3 mRNA levels and Bnip3 immunoreactivity associated with the mitochondrial compartment. PARP1 influenced Bnip3 production by depleting cytosolic NAD+, inhibiting SIRT1, and causing increased binding of the transcription factor FoxO3a to the Bnip3 upstream promoter region. Together, these data identify Bnip3 as a deleterious mitochondrial mediator of catastrophic nuclear PARP1 activation.
It is well established that excessive PARP1 activity signals beyond the nucleus and results in mitochondrial permeability and AIF translocation to the nucleus (Yu et al., 2002; Alano et al., 2004; Fatokun et al., 2014). Our data support the conclusion that Bnip3 is a mitochondrial mediator of PARP1 toxicity. Hypoxia led to MPT and neuron death that was mitigated by the deletion of either PARP1 or Bnip3. This indicated that both proteins are involved in hypoxic neurotoxicity, tempting speculation that they are linked mechanistically. We further demonstrated that hypoxia enhanced Bnip3 mRNA and mitochondrial protein levels in a manner sensitive to inhibition by pharmacological PARP1 inhibition or PARP1 deletion. This verified that hypoxic Bnip3 induction is PARP1 dependent, yielding evidence of linkage between PARP1 and Bnip3. We next asked whether the induction of Bnip3 could be responsible for neuron death attributed to PARP1 activity. As expected, direct activation of PARP1 by a DNA alkylating agent depolarized mitochondrial membranes and produced MPT, AIF release, and neuron death. However, we discovered that these effects were abrogated in bnip3−/− neurons, showing that Bnip3 is necessary for full PARP1-mediated mitochondrial damage in this hypoxia model. Together, we conclude that hypoxia leads to PARP1 activity, which in turn causes mitochondrial damage and neuron death mediated by Bnip3.
Although these data strongly support a role for Bnip3 in PARP1-induced neuron death, the mechanism of cell death downstream of Bnip3 expression is not yet clear. Apoptosis induced by both PARP1 and Bnip3 is clearly reported as caspase independent in separate literature streams (Chinnadurai et al., 2008; Fatokun et al., 2014). However, whereas caspase-independent death induced by PARP1 is mediated by AIF (Fatokun et al., 2014), the role of AIF in Bnip3-mediated cell death is less clear. Our results are consistent with recent reports that Bnip3 causes cell death by facilitating mitochondrial AIF release in neural progenitor cells (Wang et al., 2013) and hypoxic cardiac myocytes (Thompson et al., 2012), but are inconsistent with conclusions that AIF is not involved in Bnip3 toxicity (Vande Velde et al., 2000; Zhang et al., 2007). Linkage between Bnip3 and AIF remains to be defined more precisely. One important consideration is that, although the prevailing idea for putative Bnip3-AIF linkage is that Bnip3 enhances mitochondrial permeability to AIF, an alternative possibility is that mitochondrial AIF supports Bnip3 expression by stabilizing HIF subunits (Qi et al., 2012). In this case, experiments finding no effect of ectopic Bnip3 overexpression on AIF nuclear translocation (Vande Velde et al., 2000) may prematurely exclude a Bnip3–AIF linkage as they would miss endogenous Bnip3 regulatory mechanisms. Going forward, it will be important to decipher the molecular interplay between Bnip3 and AIF in endogenous hypoxic neuron death.
Several lines of evidence indicate that PARP1 leads to Bnip3 induction by a mechanism involving NAD+ depletion, inhibition of NAD+-dependent class III histone deacetylases (sirtuins), nuclear translocation of FoxO3a, and FoxO3a-mediated Bnip3 transcription. Hypoxia caused PARP1-dependent depletion of nuclear/cytosolic NAD+ pools, which is in agreement with previous studies (Cohen and Barankiewicz, 1987; Cosi and Marien, 1999), and concurrent loss of SIRT1 activity that could be rescued by adding exogenous NAD+. We investigated whether this could affect FoxO3a disposition. SIRT1 and FoxO3a were found to coimmunoprecipitate in total cell homogenates, indicating there is a physical association between them. Moreover, hypoxia enhanced acetyl-lysine immunoreactivity in FoxO3a immunoprecipitate pull-downs and localization of FoxO3a to the nucleus. These processes were blocked by a PARP1 inhibitor or PARP1 deletion, indicating a critical role for PARP1.
As it was reported previously that Bnip3 transcription is activated by FoxO3a (Mammucari et al., 2007), we determined whether PARP1-mediated FoxO3a hyperacetylation is responsible for enhanced Bnip3 expression in our hands. Using a chromatin immunoprecipitation strategy, we found that hypoxia led to increased association between FoxO3a and the Bnip3 upstream promoter region, and that this effect was eliminated in parp1−/− neurons. This correlated with increases in Bnip3 transcript levels and immunoreactivity in total cell homogenates and mitochondrial fractions, strongly suggesting that PARP1 induces Bnip3 expression by a FoxO3a-mediated mechanism. Critical roles for FoxO3a and SIRT1 in hypoxic Bnip3 expression and cell survival were confirmed in experiments using silencing of each using shRNA. FoxO3a shRNA mitigated hypoxic mitochondrial membrane depolarization and neuron death, and SIRT1 shRNA phenocopied the effects of PARP-1-mediated NAD+ depletion on FoxO3a acetylation, binding to the Bnip3 promoter and Bnip3 transcript levels. The observation that FoxO3a activity is a contributor to hypoxic neuronal death is consistent with a growing body of work in which it was identified as a regulator of other prodeath proteins, including Bim and Puma, in models of amyloid toxicity and endoplasmic reticulum stress (Ghosh et al., 2012; Akhter et al., 2014).
Our findings support a new NAD+-dependent pathway of PARP1 toxicity. The classic view of PARP1-mediated cell death is that NAD+ levels are depleted to a point that results in bioenergetic cellular suicide due to failed oxidative phosphorylation and excessive ATP consumption required for de novo NAD+ replenishment (Berger, 1985). Support for this model is derived from studies demonstrating that mitochondrial function and cell survival can be rescued by providing TCA cycle substrates, ketone bodies, or exogenous NAD+ (Ying et al., 2002; Alano et al., 2010; Baxter et al., 2014). In addition to the bioenergetics model, a PARP1-mediated cell death mechanism referred to as Parthanatos has been described, and is characterized by PARP1 activation; mitochondrial membrane depolarization; and AIF release dependent on the generation of free PAR moieties, synthesized by PARP1, and subsequently cleaved by poly(ADP-ribose) glycohydrolase (Andrabi et al., 2006). Our current data do not argue for or against either theory but introduce a third possibility. We showed that NAD+ replacement can rescue cell survival, consistent with the bioenergetics model of cell death. However, independent of any bioenergetic effects, our data indicate that NAD+ depletion by PARP1 activity has a critical influence on the regulation of genes that are important for mitochondrial function by inhibiting SIRT1. Here, we demonstrated directly that enhanced FoxO3a activity and Bnip3 expression result from NAD+ depletion and SIRT1 inhibition. In turn, this dramatically affected mitochondrial integrity and neuronal survival. Since SIRT1 is a master regulator of many genes that are critical for mitochondrial structure and function, it is possible that the FoxO3a–Bnip3 pathway identified here is one of many harmful signaling cascades triggered by SIRT1 inhibition in conditions of pathological PARP1 activation, including traumatic brain injury and stroke.
Footnotes
This work was supported by the Heart and Stroke Foundation of Canada. P.L. and A.K. were supported by postdoctoral fellowships from Research Manitoba. C.M.A. holds a Manitoba Research Chair from Research Manitoba. We thank Dr. Gerald Dorn for providing bnip3−/− mice, and Dr. Jiming Kong for providing the Bnip3 antibody.
The authors declare no competing financial interests.
References
- Akhter R, Sanphui P, Biswas SC. The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in β-amyloid-induced neuron death. J Biol Chem. 2014;289:10812–10822. doi: 10.1074/jbc.M113.519355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Medina JM. A rapid method for the isolation of metabolically active mitochondria from rat neurons and astrocytes in primary culture. Brain Res Protoc. 1998;2:209–214. doi: 10.1016/S1385-299X(97)00044-5. [DOI] [PubMed] [Google Scholar]
- Althaus J, Bernaudin M, Petit E, Toutain J, Touzani O, Rami A. Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1α (HIF-1αa) protein following focal cerebral ischemia in rats. Neurochem Int. 2006;48:687–695. doi: 10.1016/j.neuint.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagné JP, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, Auwerx J. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter P, Chen Y, Xu Y, Swanson RA. Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl Stroke Res. 2014;5:136–144. doi: 10.1007/s12975-013-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. doi: 10.2307/3576299. [DOI] [PubMed] [Google Scholar]
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D'Sa-Eipper C, Chinnadurai G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994;79:341–351. doi: 10.1016/0092-8674(94)90202-X. [DOI] [PubMed] [Google Scholar]
- Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:9035–9042. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27(Suppl 1):S114–S127. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Barankiewicz J. Metabolic consequences of DNA damage: alteration in purine metabolism following poly(ADP ribosyl)ation in human T-lymphoblasts. Arch Biochem Biophys. 1987;258:498–503. doi: 10.1016/0003-9861(87)90371-7. [DOI] [PubMed] [Google Scholar]
- Cosi C, Marien M. Implication of poly (ADP-ribose) polymerase (PARP) in neurodegeneration and brain energy metabolism. Decreases in mouse brain NAD+ and ATP caused by MPTP are prevented by the PARP inhibitor benzamide. Ann NY Acad Sci. 1999;890:227–239. doi: 10.1111/j.1749-6632.1999.tb07998.x. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellechia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AP, Klocke BJ, Ballestas ME, Roth KA. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS One. 2012;7:e39586. doi: 10.1371/journal.pone.0039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14:408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hagenbuchner J, Ausserlechner MJ. Mitochondria and FOXO3: breath or die. Front Physiol. 2013;4:147. doi: 10.3389/fphys.2013.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Hunt WT, Kamboj A, Anderson HD, Anderson CM. Protection of cortical neurons from excitotoxicity by conjugated linoleic acid. J Neurochem. 2010;115:123–130. doi: 10.1111/j.1471-4159.2010.06908.x. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18:369–376. doi: 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/S0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J Biol Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- Qi Y, Tian X, Liu J, Han Y, Graham AM, Simon MC, Penninger JM, Carmeliet P, Li S. Bnip3 and AIF cooperate to induce apoptosis and cavitation during epithelial morphogenesis. J Cell Biol. 2012;198:103–114. doi: 10.1083/jcb.201111063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.RES.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Hunting D, Trucco C, Gowans B, Grunwald D, De Murcia G, De Murcia JM. A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc Natl Acad Sci U S A. 1995;92:4753–4757. doi: 10.1073/pnas.92.11.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KS, Suh SW, Alano CC, Shao Z, Hunt WT, Swanson RA, Anderson CM. Astrocytic poly(ADP-ribose) polymerase-1 activation leads to bioenergetic depletion and inhibition of glutamate uptake capacity. Glia. 2010;58:446–457. doi: 10.1002/glia.20936. [DOI] [PubMed] [Google Scholar]
- Thompson JW, Graham RM, Webster KA. DNase activation by hypoxia-acidosis parallels but is independent of programmed cell death. Life Sci. 2012;91:223–229. doi: 10.1016/j.lfs.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/MCB.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Xiong L, Huang X, Zhao T, Wu LY, Liu ZH, Ding X, Liu S, Wu Y, Zhao Y, Wu K, Zhu LL, Fan M. miR-210 suppresses BNIP3 to protect against the apoptosis of neural progenitor cells. Stem Cell Res. 2013;11:657–667. doi: 10.1016/j.scr.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Op Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Choi BY, Yoo BH, Sohn M, Ying W, Swanson RA, Suh SW. Prevention of traumatic brain injury-induced neuron death by intranasal delivery of nicotinamide adenine dinucleotide. J Neurotrauma. 2012;29:1401–1409. doi: 10.1089/neu.2011.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem. 1998;273:12415–12421. doi: 10.1074/jbc.273.20.12415. [DOI] [PubMed] [Google Scholar]
- Ying W, Sevigny MB, Chen Y, Swanson RA. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci U S A. 2001;98:12227–12232. doi: 10.1073/pnas.211202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Chen Y, Alano CC, Swanson RA. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab. 2002;22:774–779. doi: 10.1097/00004647-200207000-00002. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yang X, Zhang S, Ma X, Kong J. BNIP3 upregulation and EndoG translocation in delayed neuronal death in stroke and in hypoxia. Stroke. 2007;38:1606–1613. doi: 10.1161/STROKEAHA.106.475129. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Shi R, Weng J, Xu X, Li XM, Gao TM, Kong J. The proapoptotic member of the Bcl-2 family Bcl-2 / E1B-19K-interacting protein 3 is a mediator of caspase-independent neuronal death in excitotoxicity. FEBS J. 2011;278:134–142. doi: 10.1111/j.1742-4658.2010.07939.x. [DOI] [PubMed] [Google Scholar]