

Abstract
We used arbitrary point channelrhodopsin-2 (ChR2) stimulation and wide-scale voltage sensitive dye (VSD) imaging in mice to map altered cortical connectivity at 1 and 8 weeks after a targeted cortical stroke. Network analysis based on optogenetic stimulation revealed a symmetrical sham network with distinct sensorimotor and association groupings. This symmetry was disrupted after stroke: at 1 week after stroke, we observed a widespread depression of optogenetically evoked activity that extended to the non-injured hemisphere; by 8 weeks, significant recovery was observed. When we considered the network as a whole, scaling the ChR2-evoked VSD responses from the stroke groups to match the sham group mean resulted in a relative distribution of responses that was indistinguishable from the sham group, suggesting network-wide down-scaling and connectional diaschisis after stroke. Closer inspection revealed that connections that had little connectivity with the peri-infarct, such as contralateral visual areas, tended to escape damage, whereas some connections near the peri-infarct were more severely affected. When connections within the peri-infarct were isolated, we did not observe equal down-scaling of responses after stroke. Peri-infarct sites that had weak connection strength in the sham condition tended to have the greatest relative post-stroke recovery. Our findings suggest that, during recovery, most cortical areas undergo homeostatic upscaling, resulting in a relative distribution of responses that is similar to the pre-stroke (sham) network, albeit still depressed. However, recovery within the peri-infarct zone is heterogeneous and these cortical points do not follow the recovery scaling factor expected for the entire network.
Keywords: diachisis, functional recovery, ischemia, optogenetic mapping, plasticity
Introduction
Stroke triggers a cascade of events affecting all levels of cortical organization, from individual neurons and synapses (microscale), to neuronal groups and populations (mesoscale), to distinct regions and interregional connections (macroscale) (for review, see Carmichael, 2003; Grefkes and Fink, 2011; Silasi and Murphy, 2014). At the level of cortex, it has been shown that function is disrupted within minutes of a stroke (Mohajerani et al., 2011) and disrupted circuits can persist for weeks after stroke even in surviving tissues (Nudo and Milliken, 1996; Brown et al., 2009). It has been shown that the effects of cortical damage may extend beyond the stroke core (Buchkremer-Ratzmann et al., 1996; Seitz et al., 1999; Carmichael et al., 2004); however, large-scale functional assessment of the cortex has been challenging. Most methods have relied on peripheral stimulation (Brown et al., 2009), which is limited to sensory or motor cortex activation, or have used invasive cortical stimulation, which provide only limited spatial sampling (Frost et al., 2003; Gharbawie et al., 2005). Although blood oxygenation level-dependent functional magnetic resonance imaging (fMRI) has been used to describe post-stroke interhemispheric functional connectivity both experimentally (Dijkhuizen et al., 2001; Dijkhuizen et al., 2003; van Meer et al., 2010; van Meer et al., 2012) and clinically (Carter et al., 2010; Grefkes and Fink, 2011), this is potentially an indirect measure of neuronal activity.
We combine channelrhodopsin-2 (ChR2) stimulation with regional voltage sensitive dye (VSD) imaging (Lim et al., 2012) to map the mouse cortex functionally after a targeted photothrombotic stroke. This method has the advantage of high temporal resolution, relatively noninvasive arbitrary point stimulation, and large-scale recording of subthreshold and suprathreshold neuronal activity (Lim et al., 2013). With these advantages in mind, we assessed macroscale functional connectivity and plasticity after stroke across both hemispheres and in the peri-infarct area (Fig. 1).
Figure 1.

Experimental timeline for mapping functional connectivity with ChR2 stimulation and VSD imaging. Shown is a schematic illustration of the experimental design. Animals were given a photothrombotic stroke (i) targeted to the forelimb area of the primary somatosensory cortex (FL). The inset shows a coronal section that demonstrates that the stroke damaged the cortical layers without penetrating the underlying white matter. Scale bar, 1 mm. After 1 or 8 weeks of home cage recovery, animals were given a large bilateral craniotomy (ii). VSD imaging was completed for sensory stimulation (iii), regional photostimulation at functional ROIs (iv), and grid photostimulation in the peri-infarct (iv). Scale bar, 2 mm.
We found equally scaled network-wide changes in functional connectivity after stroke that extended to the non-injured hemisphere. Network-wide depression observed 1 week after stroke may result from connectional diaschisis (Carrera and Tononi, 2014), in which a loss of excitation from infarcted regions affects remote areas. By 8 weeks after stroke, examination of connection strength revealed further network-wide scaling, suggesting that the post-stroke network recovers by adjusting toward a pre-stroke pattern of relative connectivity strength between nodes. However, closer inspection of peri-infarct connections revealed a heterogeneous recovery that did not fit the scaling factor of the entire network. This indicates regionally specific plasticity mechanisms or a failure to overcome the greatest extent of stroke damage.
Materials and Methods
Photothrombotic strokes.
We used ChR2 transgenic mice from The Jackson Laboratory [line 18, stock 007612, strain B6.Cg-Tg (Thy1-COP4/EYFP) 18Gfng/J]. Male mice were ∼16 weeks old and weighed ∼25 g and were given a photothrombotic stroke (mean volume = 0.30 ± 0.036 mm3, n = 15 mice) targeted to the right primary forelimb cortex as described previously (Brown et al., 2009). Consistent with previous reports (Witte et al., 2000; Brown et al., 2009), photothrombotic strokes created lesions that extended through all cortical layers, but without penetrating the underlying white matter (maximal depth from pial surface = 0.791 ± 0.034 mm, present at + 0.14 Bregma, n = 15 mice; Fig. 1i). Previous work in our laboratory has shown that these strokes are sufficient to cause a significant asymmetry in forelimb use in the cylinder task, with the greatest impairment seen at 1 week after stroke, and recovery toward baseline levels of performance by 8 weeks after stroke (Brown et al., 2009). Sham animals underwent sham photothrombosis (no Rose Bengal injection or no laser exposure). All animals were closely monitored for 24 h after surgery and then daily for 1 week thereafter. All experiments were conducted in accordance with the guidelines from the Canadian Council for Animal Care and were conducted with approval from the University of British Columbia Animal Care committee.
In vivo VSD imaging.
At 1 or 8 weeks after stroke or sham surgery, mice were anesthetized with isoflurane (1.0%) and given a large (8 × 8 mm window) bilateral craniotomy (Fig. 1ii) (Mohajerani et al., 2011). We chose a VSD from the RH series (Shoham et al., 1999): RH 1692. The dye was prepared as described previously (Brown et al., 2009) and applied to the exposed cortex for 90 min, staining all cortical layers (Mohajerani et al., 2010). After VSD application, the cortex was covered in agarose (1%) and a glass coverslip was applied. VSD imaging followed immediately. Images (12-bit images with 6.67 ms temporal resolution) were captured on a CCD camera (1M60 Pantera, Dalsa), as described previously (Lim et al., 2012). For each stimulus, 2–5 trials were averaged together to reduce the effect of any spontaneous cortical activity (Mohajerani et al., 2010).
Cortical electroencephalogram recordings.
Throughout the experiment, electroencephalogram (EEG) activity was monitored. Teflon-coated chlorided silver wires (0.125 mm) were placed on the left and right edge of the craniotomy, with a reference electrode placed on the nasal bone. The signal was amplified and filtered (0.1–1000 Hz) using an AM Systems Model 1700 AC amplifier. To ensure that cortical excitability did not vary after stroke, the amplitude of the first EEG depolarization after photostimulation was measured at sites both near and distant to the lesion core and average peak amplitude was compared across groups using unpaired t tests.
Sensory stimulation.
To generate functional maps of the primary sensory cortical areas, a number of sensory stimuli were presented (Fig. 1iii). Probes were inserted subcutaneously to each of the paws and a 1 ms 1 mA pulse was delivered to map the forelimb and hindlimb areas of the primary somatosensory cortex in each hemisphere. A single whisker (C2) was given a 1 ms tap using a piezo device to map the somatosensory barrel cortex, and a 1 ms combined green and blue light stimulus was presented to map the primary visual cortex. These functional maps were then used to determine coordinates for regional photostimulation.
Photostimulation.
We used a 1 ms, 5 mW pulse generated by a 473 nm diode pumped solid state laser (CNI Optoelectronics) to stimulate ChR2-expressing neurons, as described previously (Lim et al., 2012). Regional photostimulation sites (Fig. 1iv) were targeted based on functional sensory areas defined through sensory stimulation or based on stereotaxic coordinates. These regions of interest (ROI) included: secondary motor cortex (M2), motor barrel cortex (MB), motor forelimb cortex (MF), forelimb area of the primary somatosensory cortex (FL), hindlimb area of the primary somatosensory cortex (HL), somatosensory barrel cortex (BC), parietal cortex (PT), retrosplenial cortex (RS), secondary/medial visual cortex (V2), and primary visual cortex (V1). To determine grid photostimulation sites within the peri-infarct zone, a 5 × 5 photostimulation grid was centered over the lesion and over the homotopic region in the left hemisphere (Fig. 1v). Grid photostimulation sites were 500 μm apart, resulting in the grid extending 1 mm from the lesion core in the anterior, posterior, lateral, and medial directions.
Each site was photostimulated 2–4 times in an interleaved, semirandom order to reduce any time-dependent effects of anesthetic depth as well as any possibility of cortical damage due to repetitive stimulation. A 10 s inter-trial interval ensured full recovery of VSD fluorescence. VSD responses were taken from all other ROIs and replicate responses were averaged together.
Data analysis.
VSD responses to stimulation were calculated as the normalized difference to the average baseline recorded before stimulation (% ΔF/F0) in MATLAB (MathWorks). For each stimulation type (each of the 5 sensory modalities, each of the 20 regional photostimulation sites, and each of the 50 peri-infarct grid photostimulation sites), 2–5 trials were collected and percentage ΔF/F0 responses were averaged across trials. Custom-written MATLAB programs were used to analyze and quantify the VSD responses (peak amplitude, time to peak amplitude, and area under the curve). Peak amplitude was calculated by finding the maximum response in the first 127 ms poststimulus in the trial averaged percentage ΔF/F0. To ensure that the peak was not noise, the response had to be >2.5 times the standard deviation of the baseline (initial 180 ms of the recording). Within the peri-infarct, we calculated loss, gain, and net difference per site according to the following formulas:
 |
 |
Network analysis.
We used custom-written MATLAB programs as well as scripts from the brain connectivity tool box (http://www.brain-connectivity-toolbox.net; Rubinov and Sporns, 2010), to create a 20 × 20 connectivity matrix and network diagram based on the ChR2-evoked VSD responses from each ROI (Lim et al., 2012). Briefly, for each regional photostimulation site, we calculated the integrated VSD response at 20 ms after photostimulation (the sum of the VSD response in units of percentage ΔF/F0 from stimulus onset to 20 ms after photostimulation) at the remaining regional response sites. For each ROI, the connectivity matrix shows in-strength as the rows (the strength of the connections coming to the ROI when other areas were photostimulated) and out-strength as the columns (the strength of the connections to other areas when the ROI was photostimulated). We used the connectivity matrices to evaluate differences in connection strength between groups (e.g., comparing average in-strength and out-strength per hemisphere). For illustrative purposes only, we used the data from the connectivity matrix and applied a consistent threshold across groups to create network diagrams with only the strongest VSD responses. We chose a threshold that did not affect the characteristic path length and clustering coefficient (common network metrics), as described previously (Lim et al., 2012). For the network diagrams, node size is proportional to the strength of the connections per node (sum of the weights per photostimulation site) and edge thickness between nodes is proportional to the weight of the connections between nodes. For the post-stroke network diagrams, we calculated the change in strength from sham to 1 week post-stroke, and from 1 week to 8 weeks post-stroke. Network diagrams were color coded to demonstrate changes in network connections over time, with red indicating a loss of strength, green indicating a gain of strength, and gray representing a marginal change (<15%) in strength.
To predict which nodes would be most-at-risk and least-at-risk after the stroke, we calculated total node strength (sum of the weights per photostimulation site) as well as node strength related to the right FL (FLR; sum of in-strength from FLR and out-strength to FLR). We predicted that nodes with the highest strength within the regional network would also show the most impairment after stroke.
Histology.
To investigate whether the stroke caused any changes in ChR2 expression after stroke, mice were deeply anesthetized and transcardially perfused and brain sections were imaged and analyzed as described previously (Lim et al., 2012). In brief, coronal sections (100 μm) were cut on a vibratome and imaged using a Zeiss LSM 510 Meta confocal microscope with a multiline argon laser and a Plan-Neofluar 5× (0.15 numerical aperture) objective. A 488 nm line argon laser excited the YFP-ChR2 fusion protein (Arenkiel et al., 2007). Tiled scans (12-bit; 512 × 512 pixels) were collected using Zen 2009 software. ChR2 expression was quantified in 5 sham and 5 1-week-post-stroke animals at 3 different sections: anterior to lesion (Bregma +1.1), near the lesion (Bregma −0.2), and posterior to the lesion (Bregma −1.06). Each section was filtered (Gaussian filter of 6.0 pixel radius) and normalized relative to the fluorescence in the fornix (Bregma −1.06) using ImageJ software (Version 1.42q). The fornix was chosen because it expresses YFP, but is distant from the lesion and not part of the cortex. A 100 pixel freehand line was drawn to plot the fluorescence profile from medial to lateral in each hemisphere.
Statistical analyses.
A one-way ANOVA with Bonferroni post-tests was used for statistical analysis of peak amplitude, time to peak, and area under the curve. Two-way repeated-measures ANOVA with Bonferroni post-tests were used to compare connectivity matrices between groups. Permutation tests (van den Heuvel and Sporns, 2011) were used to evaluate the mean difference in strength between groups. Kolmogorov–Smirnov (KS) tests were used to compare cumulative distribution functions between groups before and after scaling to match the group means. Linear regression was calculated using GraphPad Prism software. All p values ≤0.05 were considered statistically significant. Data are expressed as the mean ± SEM.
Results
Sensory-evoked activity after stroke shows delayed and depressed VSD responses in the injured hemisphere
In the sham animals, sensory stimulation of the forelimb, hindlimb, whiskers, or visual system produced the expected stereotypical depolarizing signals within the somatosensory cortex (Lim et al., 2012; Mohajerani et al., 2013). In the stroke group, sensory stimulation of the whiskers and visual system produced VSD signals comparable to sham animals; however, the VSD signal after stimulation to the affected forelimb was severely depressed and delayed (Fig. 2a). To quantify the VSD responses, we created intensity over time plots and analyzed three different aspects of the VSD signal: peak amplitude, time to peak, and area under the curve. After sensory stimulation to the left (affected) forelimb, the response in the right (injured) hemisphere was significantly greater in shams compared with the stroke animals at 1 or 8 weeks after stroke as revealed by one-way ANOVA and Bonferroni's post hoc test: there was a significant effect of group on peak amplitude (F(2,21) = 12.21, p = 0.0178), time to peak amplitude (F(2,18) = 8.610, p = 0.0024), and area under the curve to 20 ms (F(2,22) = 8.697, p = 0.0016) (Fig. 2b). Similarly, the response in the left (non-injured) hemisphere was also significantly different between groups in peak amplitude (F(2,22) = 5.770, p = 0.0097), time to peak amplitude (F(2,17) = 4.392, p = 0.0290), and area under the curve (F(2,22) = 5.524, p = 0.0114) (Fig. 2c). This delayed and suppressed response within the ipsilesional hemisphere in the stroke animals was similar to previous reports using VSD imaging after somatosensory stroke (Brown et al., 2009).
Figure 2.

VSD imaging maps of the injured limb shows delayed and decreased VSD responses in both hemispheres. A, Example VSD responses in a bilateral craniotomy preparation after electrical stimulation of the left (injured) forelimb in a sham animal (top), 1 week after stroke animal (middle), and 8 week after stroke animal (bottom). The lesion in the stroke animals is indicated by the white circle. Quantitative comparisons of the VSD response from the right (injured) hemisphere (B) and left (non-injured) hemisphere (C). i, Intensity versus time plot of the VSD response. ii, Peak amplitude (df/F0%) VSD response. iii, Time (ms) to peak amplitude. iv, Area under the curve to 20 ms after stimulation. *p < 0.05.
Channelrhodopsin-evoked activity shows depressed, but not delayed VSD responses after stroke in the injured hemisphere
Based on the location of cortical activations observed after sensory stimulation, a number of cortical regions were targeted for photostimulation (see Materials and Methods and Fig. 1iv). Twenty ROIs were selected for photostimulation (10 per hemisphere) and recording of VSD responses. After photostimulation of each ROI, VSD responses were measured at the other ROIs, giving a total of 19 response sites per photostimulation site (VSD responses were not measured at the photostimulation site itself due to transient photobleaching). We were most interested in the VSD response after photostimulation of the FL in the right and left hemisphere because these were the areas that showed the greatest changes after sensory stimulation.
After photostimulation of the right forelimb area of the primary somatosensory cortex (FLR) in the sham group (Fig. 3a, top), the VSD response was similar to the response pattern seen with sensory stimulation of the left forepaw (cf. Fig. 2a, top), as described previously (Lim et al., 2012). In the stroke groups, the FLR was the area targeted for stroke, so the VSD response to sensory stimulation of the affected forepaw was termed the new FLR (nFLR) and was targeted for photostimulation. At 1 week after stroke, the VSD signal in response to photostimulation of nFLR was depressed in both the left and right hemispheres (Fig. 3a, middle). By 8 weeks after stroke, a small VSD response could be observed in the nFLR before activation of the left hemisphere. The contralateral (to stimulation) response (in the non-injured hemisphere) was much larger and lasted longer than the ipsilateral response (in the injured hemisphere) at 8 weeks after stroke (Fig. 3a, bottom). Using a one-way ANOVA and Bonferroni's post hoc test, we compared the response in the right (injured) hemisphere (Fig. 3b). There was a significant effect of group on the peak amplitude of the VSD response (F(2,22) = 8.356, p = 0.0020) and the area under the curve to 20 ms after photostimulation (F(2,22) = 5.785, p = 0.0096). There was no significant effect of group for time to peak amplitude (F(2,17) = 1.117, p = 0.3502), unlike the sensory responses which were greatly delayed in the post-stroke groups. In the left (non-injured) hemisphere (Fig. 3c), a one-way ANOVA revealed a significant effect of groups for peak amplitude (F(2,22) = 8.264, p = 0.0021), time to peak amplitude (F(2,17) = 17.21, p < 0.0001), and area under the curve to 20 ms (F(2,22) = 4.203, p = 0.0285). Results of Bonferroni post-tests are displayed in Figure 3.
Figure 3.

VSD imaging maps of photostimulation of the injured forelimb cortex results in stronger and faster VSD responses than expected from sensory stimulation. A, Example VSD responses in a bilateral craniotomy preparation after photostimulation of the right (injured) forelimb somatosensory cortex in a sham animal (top), 1 week after stroke (middle), and 8 weeks after stroke animal (bottom). The lesion in the stroke animals is indicated by the white circle. White arrows indicate laser photostimulation target. Quantitative comparisons of the VSD response from the right (injured) hemisphere (B) and left (non-injured) hemisphere (C). i, Intensity versus time plot of the VSD response. ii, Peak amplitude (df/F0%) VSD response. iii, Time (ms) to peak amplitude. iv, Area under the curve to 20 ms after stimulation. *p < 0.05. Time to peak is not shown in the 1 week after stroke condition due to only a few samples showing a well defined peak in the response.
Assessment of the intercortical and intracortical network trends from connectivity matrices
To represent the data as a whole and to show relationships between and within the regional photostimulation sites, a 20 × 20 connectivity matrix was created for each group (sham, 1 week after stroke, and 8 weeks after stroke) to display the integrated VSD response at 20 ms after photostimulation (the sum of the VSD response in units of percentage ΔF/F0 from stimulus onset to 20 ms after photostimulation) of each ROI (Fig. 4, left). In the sham connectivity matrix (Fig. 4a), well known relationships can be identified in the connectivity matrix, such as between the somatosensory barrel cortex and motor cortex: when left somatosensory barrel cortex (BCL) is photostimulated, it evokes strong VSD responses from the motor areas, especially left motor representation of the barrel cortex (MBL).
Figure 4.
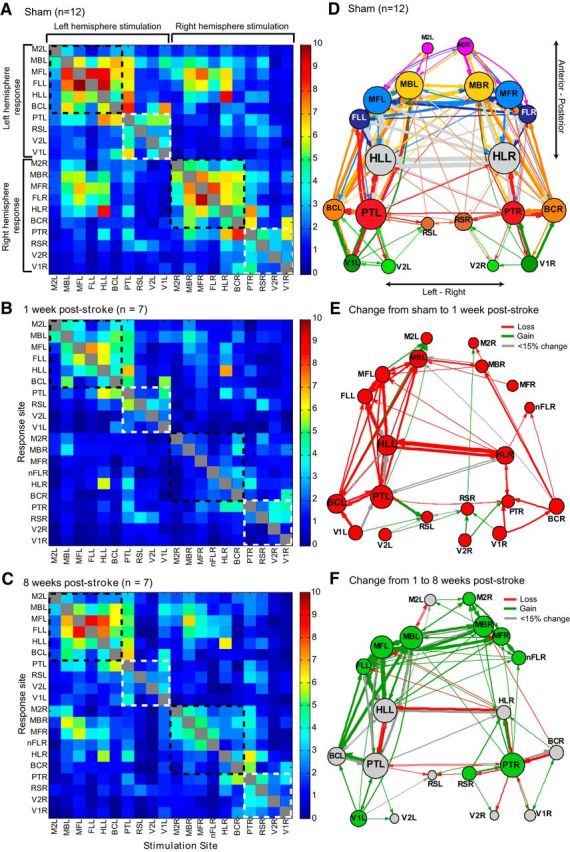
Connectivity changes over time reveal asymmetries after stroke. Average connectivity matrices (left) and network diagrams (right) all derived from the VSD response at 20 ms after photostimulation in the sham group (A; n = 12) 1 week after stroke (B; n = 7), and 8 weeks after stroke (C; n = 7). Connectivity matrices are organized from left hemisphere to right hemisphere sites and from anterior to posterior sites. Dashed lines indicate the somatomotor group, G1 (black), and the association group, G2 (white). A threshold (2.6) was used to create the network diagrams for the sham (D), 1 week after stroke (E), and 8 weeks after stroke (F) groups. Nodes were placed according to the site of photostimulation. Node size is proportional to the strength of the connections per node (sum of the weights). Edge thickness (arrows) between nodes is proportional to the weight of the connection between nodes. To demonstrate changes over time, network diagrams showing the change from sham to 1 week after stroke (E) and the change from 1 to 8 weeks after stroke (F) were color coded to demonstrate changes in network strength. Red indicates a loss of strength, green indicates a gain of strength, and gray indicates a marginal change (<15% change in strength).
In general, the strongest VSD responses in the sham matrix were in the regions functionally related to or neighboring the photostimulation site and in the homotopic-to-stimulation site. For example, if the FLR was photostimulated, the strongest response was seen in areas surrounding the FLR, such as the right motor area of the forelimb area (MFR) and right hindlimb area of the primary somatosensory cortex (HLR) and in the homotopic site (FLL). Photostimulation of the left FL (FLL) produced the mirror response. Permutation tests isolating the left hemisphere stimulation sites and the right hemisphere sites in the sham matrix showed no significant difference in outgoing strength (left sites average out-strength = 2.91, right sites average out-strength = 2.71, p = 0.1857). Similarly, permutation tests comparing left versus right hemisphere responses showed no significant difference in incoming strength (left sites average in-strength = 2.84, right sites average in-strength = 2.78, p = 0.3627).
When network analysis was applied to this matrix using the brain connectivity tool box (http://www.brain-connectivity-toolbox.net; Rubinov and Sporns, 2010), a distinct per-hemisphere module was identified. Within these modules, we divided the nodes into functional groups (somatomotor nodes including MB, MF, M2, FL, HL, BC as one group, “G1,” and association and visual nodes including V2, V1, RS, PT as a second group, “G2”). The somatomotor group (G1) can be seen in the connectivity matrix as “hot-spots”: photostimulation of the nodes that make up the G1 group result in strong VSD responses in the G1 nodes in both the right and left hemispheres in the sham connectivity matrix (see black dashed boxes in Fig. 4a). Comparing the responses (the sum of the VSD response from stimulus onset to 20 ms after photostimulation) across the diagonal, responses appear relatively equal between left and right hemispheres (average left hemisphere response = 2.84, average right hemisphere response = 2.78; p = 0.3627), suggesting that the left and right hemisphere modules are relatively equal.
In contrast, the connectivity at 1 week after stroke shows severely depressed VSD responses at all nodes (Fig. 4b). Because the stroke was targeted over the FLR, the connectivity matrix shows the nFLR. When the nFLR was photostimulated, the response was depressed in both the right and left hemispheres. Although the larger responses were still from regions neighboring the photostimulation site, these responses were low relative to sham animals. At 1 week after stroke, the responses are not equal between hemispheres (average left hemisphere response = 1.65, average right hemisphere response = 1.31; p = 0.0064) and the groups (“G1” and “G2”) are not as apparent as in the sham matrix; while there is a cluster of left hemisphere responses to left somatomotor stimulation (upper left in the connectivity matrix in Fig. 4b), the other groups are not easily observed.
By 8 weeks after stroke, the connectivity matrix is still somewhat depressed relative to the sham matrix, however, the node groupings are more distinct and in most cases these VSD responses were stronger compared with 1 week after stroke (Fig. 4c). Although the responses are not equal between hemispheres at 8 weeks after stroke (average left hemisphere response = 2.04, average right hemisphere response = 1.73; p = 0.0222), the functional groups within the network appear to be returning to sham levels (see black dashed boxes outlining G1 clusters in the upper left and lower right of the matrix on Fig. 4c).
To facilitate the visualization of the relationships between cortical areas, we derived network diagrams from the connectivity matrices (Fig. 4, right), as described previously (Lim et al., 2012). The diagrams represent only the strongest connections observed in each group (see Materials and Methods) and illustrate broad patterns within the network. In the sham diagram (Fig. 4d), the left and right hemisphere are relatively symmetrical. This symmetry is lost in the post-stroke animals and there are fewer strong connections in the post-stroke network diagrams (Fig. 4e,f). Interestingly, although the stroke is targeted to the right hemisphere FL, the left hemisphere in the post-stroke animals also has fewer connections and weaker node strength. By 8 weeks after stroke (Fig. 4f), the network diagram appears to be returning toward sham levels: there are more connections relative to the 1 week post-stroke network diagram and some of the symmetry between the left and right hemisphere is reestablished.
Assessment of the differences between groups using the connectivity matrices
To compare the changes between groups, we compared the mean connection strength between groups using t tests between the matrices. The groups were significantly different, because the responses from the stroke animals were greatly depressed (sham vs 1 week after stroke, sham vs 8 weeks after stroke, or 1 week after stroke vs 8 weeks after stroke, p < 0.0001). To determine whether the depressed VSD responses we observed in the stroke animals could be normalized to the sham connectivity strength distribution by a constant scaling factor, we normalized the connectivity matrices so the group means were equal and checked for the equality of the resulting distributions using KS tests before and after normalization by scaling the whole network. After normalization, the groups were statistically indistinguishable by KS tests (sham vs 1 week after stroke, p = 0.3558; sham vs 8 weeks after stroke, p = 0.1055), indicating widespread scaling down across the network in the stroke groups (Fig. 5). It is possible that the stroke could affect the overall ChR2 expression because it is known that stroke can affect gene and protein expression (Carmichael, 2003). However, examination of several anterior to posterior histological sections indicated that ChR2 expression was not significantly different between the injured and non-injured hemisphere (p = 0.4102), nor did it vary by >50% across the anterior–posterior axis when sections were normalized per animal to YFP expression in the fornix (see Materials and Methods). Furthermore, analysis of EEG recordings showed that ChR2-evoked depolarizations after photostimulation in 1-week-post-stroke animals were not statistically different from sham animals at sites near to the lesion (HLR: sham = −0.403 ± 0.019 mV vs stroke = −0.518 ± 0.072 mV, p = 0.1310) nor were they statistically different at sites distant from the lesion (V1 L: sham = −0.255 ± 0.051 mV vs stroke = −0.273 ± 0.1 mV, p = 0.8777). This suggests the excitability of ChR2 or peri-infarct neurons did not differ significantly between groups. Conceivably, depression in regions connected to a ChR2-photoactivation site was associated with defects in synaptic activity secondary to ChR2 activation.
Figure 5.

Regional network-wide scaling of the connectivity matrix leads to relative connectivity strengths similar to sham. Values from the connectivity matrices were scaled such that the mean of the stroke group matched the mean of the sham group. Cumulative distribution function for VSD responses at 1 week after stroke (A) or 8 weeks after stroke (B) compared with sham (red). Dashed lines indicate scaled values. Scaled values were used to create connectivity matrices for 1 week after stroke (C) and 8 weeks after stroke (D). Dashed black lines indicate the somatomotor group in the connectivity matrix. Compare these with the sham matrix in Figure 4A.
We used network analysis on the sham network to predict which areas would be most-at-risk and least-at-risk to stroke based on expected connectivity and compared these predictions with experimental results from ChR2 stimulation and VSD imaging. Node strength (Fig. 6a) and node strength relative to FLR (Fig. 6b) revealed the strongest nodes in the network were also the most strongly connected to FLR. Generally, the strongest sites relative to FLR also had the closest proximity to FLR (such as ipsilateral motor and sensory areas) and the weakest sites were more distant to FLR (such as contralateral visual areas). We selected three of the strongest sites (MBR, MFR, HLR) and FLR, as well as three of the weakest sites (RSL, V2L, V1L) and compared the VSD responses to photostimulation at these sites (Fig. 6c,d). A one-way ANOVA for the most-at-risk sites revealed a significant difference between sham and stroke groups (F(2,33) = 57,72, p < 0.0001), while the least-at-risk sites were not significantly different between groups (F(2,15) = 0.8551, p = 0.4450).
Figure 6.

Node strength from the sham network predicts which sites will be most affected and least affected after stroke. Total node strength (A) and node strength relative to FLR (B) were calculated in the sham network to predict which regional sites would be most-at-risk and which would be least-at-risk after stroke. The predicted most-at-risk sites are high in strength within the regional network (circled in red in A and B) and close in proximity to the lesion. These sites are significantly depressed after stroke (C). The predicted least-at-risk sites are low in strength within the regional network (circled in green in A and B) and distant from the lesion. These sites are not significantly depressed after stroke (D). *p < 0.05, significantly different from sham.
Assessment of the peri-infarct using grid photostimulation
To further examine the peri-infarct, we photostimulated 25 sites at fixed distances from the lesion and 25 homotopic sites in the non-injured hemisphere (see Materials and Methods and Fig. 1v). For analysis, we excluded the lesion area and considered only the furthest peri-infarct sites (16 border sites on the 5 × 5 grid). VSD responses were taken from both the left and right FL areas (FLL and nFLR) to investigate interhemispheric and intrahemispheric responses. We scaled the responses, as described above, to determine whether areas within the peri-infarct could also be normalized by network-wide scaling. Before and after normalizations, the groups were significantly different from one another using KS tests (sham vs 1 week after stroke p = 0.0138; sham vs 8 weeks after stroke; p = 0.0507), indicating heterogeneity rather than homogeneity in these areas (Fig. 7). To further investigate how the peri-infarct was responding after stroke, we calculated prestroke (sham) strength (Fig. 8a), “loss” (difference in VSD strength between sham and 1 week after stroke; Fig. 8b), “gain” (difference in VSD strength between 8 weeks and 1 week after stroke; Fig. 8c), and overall difference (Fig. 8d) per photostimulation site. We also plotted the overall difference as a function of sham strength (Fig. 8e). We found that although posterior-medial sites were the strongest in the sham animals, the gains in these areas by 8 weeks after stroke did not equal the losses seen at 1 week after stroke. In fact, it was the relatively weaker anteriolateral areas that showed the greatest relative gains (compared with loss per site) after stroke, sometimes exceeding sham strength. This trend was observed for both intrahemispheric connections (Fig. 8i,ii) and interhemispheric connections (Fig. 8iii,iv).
Figure 7.

Scaling the responses in the peri-infarct zone does not lead to relative connectivity strength similar to shams. Within the peri-infarct zone, scaling to match the mean of the sham group did not result in the stroke groups having a similar distribution of strength compared with shams at 1 week after stroke (A) nor 8 weeks after stroke (B).
Figure 8.
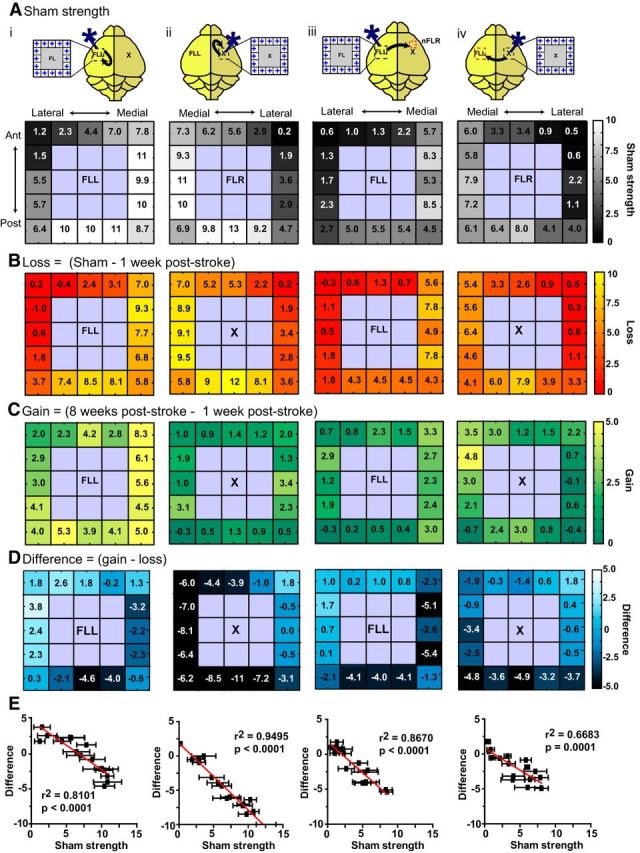
Response strength within the peri-infarct and homotopic FL reveals that the weakest pre-stroke sites have the greatest relative gains after stroke, resulting in heterogeneous recovery. Sixteen regions in the non-injured (left) hemisphere and injured (right) hemisphere were photostimulated. Responses were taken from either the non-injured FLL or the new FLR. i, Stimulation of the non-injured hemisphere, intrahemispheric FL response (from the non-injured hemisphere). ii, Stimulation of the peri-infarct, intrahemispheric nFLR response (from the injured hemisphere). iii, Stimulation of the left hemisphere, interhemispheric response from the nFLR (injured hemisphere). iv, Stimulation of the peri-infarct (injured hemisphere), interhemispheric response from the FLL (non-injured hemisphere). A, Sham strength after photostimulation at the 16 regions indicated. Note that medial and posterior sites are generally stronger compared with lateral and anterior sites. B, Loss of strength at 1 week after stroke (relative to sham). Note that all sites are depressed, but often sites that were strongest in A show the largest losses. C, Gain of strength at 8 weeks after stroke (relative to 1 week after stroke). Note that gains appear higher when the response is taken from the non-injured FLL (i, iv) and gains are minimal when the injured hemisphere is photostimulated and the response is taken from this hemisphere (ii). D, Difference in strength per site. Note that the sites that were the strongest in the pre-stroke (sham) condition (A) show a negative difference, indicating they are weaker than the pre-stroke condition, whereas sites that were the weakest in the pre-stroke condition show a difference near or above zero, indicating they have gained as much as they lost or more. E, Negative correlations between sham strength and difference in strength over time (gain-loss) demonstrate a trend for weaker sites to recover more than stronger sites.
Discussion
VSD imaging and ChR2 stimulation reveals network-wide plasticity after stroke
The stroke core (infarct) suffers irreversible ischemic damage and the penumbra (peri-infarct) suffers from reduced blood flow, but this tissue is potentially salvageable (Murphy and Corbett, 2009). Stroke may also cause diaschisis: impairment and dysfunction in areas remote from the lesion core. Remote changes may take place over minutes to weeks and play an important role in poststroke recovery (Carmichael et al., 2004; Cramer, 2008). For this reason, it is necessary to consider more than just the ischemic territory when investigating post-stroke recovery. We used ChR2 stimulation and VSD imaging to monitor directly wide-scale cortical activity changes over time after a localized stroke to the primary somatosensory cortex. Due to limitations of the craniotomy and VSD imaging (Lim et al., 2012), our model does not address prefrontal connections or subcortical contributions to recovery after stroke, although these have been shown to be of importance (Sharma et al., 2009; Grefkes and Fink, 2011). Our conclusions should be considered in the context of local cortical connectivity and not as a comprehensive connectome for recovery after stroke.
Previous studies have used resting-state (rs) or functional connectivity (fc) fMRI to investigate wide-scale network changes over time after stroke (van Meer et al., 2010; van Meer et al., 2012). In the clinical setting, fc-fMRI is advantageous because it is possible to investigate multiple networks in a single scan, it is readily obtained even in cases of severe stroke, and longitudinal imaging is possible (Carter et al., 2012a). Similar network properties to those used here (e.g., centrality, clustering) may be calculated to predict behavioral recovery after stroke (Ward et al., 2003a, 2003b). However, the frequency of resting-state activity (<0.1 Hz) is low and its nature is not completely understood (van den Heuvel and Hulshoff Pol, 2010). rs-fMRI (van Meer et al., 2010; van Meer et al., 2012) and similar spontaneous activity measures (Mohajerani et al., 2013; Bauer et al., 2014) have been used to describe networks in animal studies; however, these techniques are based on correlation, which can be less sensitive to amplitude changes and does not indicate directional connectivity. Using targeted ChR2 stimulation and VSD imaging, we more directly determine connection strength and direction (Lim et al., 2012).
Previous work has indicated strong parallels between cortical connectivity derived by photostimulation of ChR2 and sensory stimulation (Lim et al., 2012). However, we find differences between the methods: whereas sensory stimulation yielded weak VSD responses after stroke (Fig. 2), optogenetic stimulation resulted in stronger-than-expected VSD responses (Fig. 3). This may indicate deficits within cortical-thalamic circuits after stroke (that sensation may track) and optogenetic stimulation may help to reveal functional intracortical circuits after stroke that are not easily probed using sensation. Many genes and proteins undergo altered expression after stroke (Carmichael, 2003), so it could be argued that stroke alters the expression of ChR2 or intracortical excitability. However, we failed to observe significant changes in either ChR2 expression or intracortical excitability (EEG) in regions that were targeted for photostimulation and were outside of the infarct.
Based on the regional optogenetic stimulation, we found that the stroke groups demonstrated globally depressed VSD responses (Fig. 4); however, the relative distribution of strengths between nodes at a regional level was largely maintained after stroke (Fig. 5). Although we observed consistent scaling when we considered the regional network as a whole, closer analysis of individual sites revealed that some nodes were more vulnerable than others after stroke (Fig. 6). These nodes were in close proximity to the stroke. Although this may not be surprising because it is generally known that functionally related cortical regions tend to be colocalized, the conclusion that sites closest to the lesion are most affected did not hold true in the peri-infarct. Instead, photostimulation of the peri-infarct revealed heterogeneous recovery, with some areas remaining depressed (the anterolateral areas) and some areas showing strength greater than baseline (the posterior-medial areas) by 8 weeks after stroke (Fig. 8).
Nonuniform recovery of peri-infarct connections
Clinical studies have shown that the global network is affected by stroke (Grefkes and Fink, 2011; Carter et al., 2012b) and the peri-infarct zone is especially sensitive and may be an important predictor for recovery (Furlan et al., 1996). Previous studies have shown that the peri-infarct undergoes a number of changes over time, including cell death (Dirnagl et al., 1999), initial loss of spines (Brown et al., 2008), retraction of axons (Brown et al., 2007), and axonal sprouting (Carmichael, 2006, 2008), as well as a recovery of spine density (Mostany et al., 2010).
Here, we found that the network at 1 and 8 weeks after stroke had similar relative connectivity strength to the sham network when we considered the network as a whole, but not if we isolated responses from the peri-infarct. Homogeneous scaling of the responses could not account for the changes within the peri-infarct (Fig. 7). Breakdown of the recovery process into “Loss” (the difference in strength between sham strength and 1 week after stroke) and “Gain” (the difference in strength between 1 and 8 weeks after stroke) revealed that both hemispheres were affected by stroke. It was the areas that were the weakest in the pre-stroke (sham) condition that showed the greatest relative gains (Fig. 8). The grid photostimulation sites that were the strongest in the sham condition were the posterior–medial areas. Although these areas were not functionally defined, the posterior–medial areas are close to HL somatosensory cortex, which is known to overlap with FL (Oh et al., 2014). In contrast, the weakest sites in the grid photostimulation were the anteriolateral areas. These are close to oral-facial areas such as the jaw; however, our craniotomy window was not lateral enough to encompass these areas. Although it could be argued that the weakest sites experience the least amount of absolute change, analysis of the losses and gains in these areas show that these sites undergo significant relative changes, sometimes demonstrating complete loss at 1 week after stroke before regaining strength at 8 weeks after stroke.
Overall, this suggests that the local connections in the peri-infarct are too severely damaged to maintain the optimal prestroke distribution of connection strengths and instead undergo heterogeneous recovery: partial recovery in some areas and better-than-expected recovery in others. It is possible that this reflects a form of synaptic scaling in which less active sites more effectively engage homeostatic mechanisms, resulting in increased excitability and synaptic transmission of less active networks (Turrigiano and Nelson, 2004). However, because stroke triggers a cascade of events, the changes observed here may also result from other factors such as alterations in neuronal excitability (Carmichael, 2003), axonal sprouting (Carmichael, 2006), changes in collateral inhibition (Bütefisch et al., 2003), or astrocytic changes (Barreto et al., 2011).
Implications for future work
We observed global depression of the network at 1 week after stroke. By 8 weeks after stroke, and without any explicit treatment or rehabilitation, the global network showed some recovery toward a sham-like pattern of relative connectivity strengths despite having lower overall strength. This suggests that the sham network pattern of relative connectivity strengths represents the desired “set-point” (Turrigiano and Nelson, 2000) and the stroke networks recover in such a way that this relative strength distribution is maintained: relative connectivity strength between nodes is parallel to shams despite having depressed strength.
Our results are consistent with the concept that stroke affects the entire network, but the injured hemisphere (specifically the peri-infarct) can be severely depressed after stroke (Brown et al., 2009; Mohajerani et al., 2011). This may suggest that the early post-stroke period is a sensitive time for the network and brings about the question of when treatment and rehabilitation strategies should begin. Although we did not explicitly test treatment after stroke in this study, future work could incorporate skilled training (Kerr et al., 2013) or enriched environments (Biernaskie et al., 2004) to determine the effects of such experience on the recovery of the post-stroke network.
Conclusion
We suggest the following scenario after stroke: in the early stages after stroke (up to 1 week afterward), cortical activity is globally depressed. The global depression gradually resolves over time such that, by 8 weeks after stroke, the qualitative features of the original network have returned even though the overall strength of the network remains depressed. At a local level, there is evidence of recovery in the peri-infarct zone by 8 weeks after stroke, but it is not uniform. Between 1 and 8 weeks after stroke, novel connections form to potentially compensate for stroke damage (Dancause et al., 2005; Carmichael, 2006, 2008; Brown et al., 2009), leading to circuit reorganization and functional remapping. This suggests that treatment and rehabilitation strategies that target the entire network will be most effective in returning the network to a prestroke state. Although we have focused on cortex, it will be interesting to determine how agents that induce the sprouting of new connections within either the spinal cord (Wahl et al., 2014) or cortex (Overman et al., 2012) affect the relative balance of connection strength after stroke.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR Operating Grant MOP-111009), the Heart and Stroke Foundation of Canada (grant in aid), the Canadian Partnership for Stroke Recovery expansion, the Human Frontiers Science Program (to T.H.M.), the Natural Sciences and Engineering Research Council of Canada (NSERC Canada Graduate Scholarship). D.H.L. was supported by an Izaak Walton Killam Memorial Predoctoral Fellowship. We thank Pumin Wang for surgical assistance.
The authors declare no competing financial interests.
References
- Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ, Ehlers MD, Feng G. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–218. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto G, White RE, Ouyang Y, Xu L, Giffard RG. Astrocytes: targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem. 2011;11:164–173. doi: 10.2174/187152411796011303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AQ, Kraft AW, Wright PW, Snyder AZ, Lee JM, Culver JP. Optical imaging of disrupted functional connectivity following ischemic stroke in mice. Neuroimage. 2014;99:388–401. doi: 10.1016/j.neuroimage.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernaskie J, Chernenko G, Corbett D. Efficacy of rehabilitative experience declines with time after focal ischemic brain injury. J Neurosci. 2004;24:1245–1254. doi: 10.1523/JNEUROSCI.3834-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci. 2007;27:4101–4109. doi: 10.1523/JNEUROSCI.4295-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Wong C, Murphy TH. Rapid morphologic plasticity of peri-infarct dendritic spines after focal ischemic stroke. Stroke. 2008;39:1286–1291. doi: 10.1161/STROKEAHA.107.498238. [DOI] [PubMed] [Google Scholar]
- Brown CE, Aminoltejari K, Erb H, Winship IR, Murphy TH. In vivo voltage-sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J Neurosci. 2009;29:1719–1734. doi: 10.1523/JNEUROSCI.4249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkremer-Ratzmann I, August M, Hagemann G, Witte OW. Electrophysiological transcortical diaschisis after cortical photothrombosis in rat brain. Stroke. 1996;27:1105–1109. doi: 10.1161/01.STR.27.6.1105. discussion 1109–1111. [DOI] [PubMed] [Google Scholar]
- Bütefisch CM, Netz J, Wessling M, Seitz RJ, Hömberg V. Remote changes in cortical excitability after stroke. Brain. 2003;126:470–481. doi: 10.1093/brain/awg044. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Plasticity of cortical projections after stroke. Neuroscientist. 2003;9:64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol. 2006;59:735–742. doi: 10.1002/ana.20845. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Themes and strategies for studying the biology of stroke recovery in the poststroke epoch. Stroke. 2008;39:1380–1388. doi: 10.1161/STROKEAHA.107.499962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST, Tatsukawa K, Katsman D, Tsuyuguchi N, Kornblum HI. Evolution of diaschisis in a focal stroke model. Stroke. 2004;35:758–763. doi: 10.1161/01.STR.0000117235.11156.55. [DOI] [PubMed] [Google Scholar]
- Carrera E, Tononi G. Diaschisis: past, present, future. Brain. 2014;137:2408–2422. doi: 10.1093/brain/awu101. [DOI] [PubMed] [Google Scholar]
- Carter AR, Astafiev SV, Lang CE, Connor LT, Rengachary J, Strube MJ, Pope DL, Shulman GL, Corbetta M. Resting interhemispheric functional magnetic resonance imaging connectivity predicts performance after stroke. Ann Neurol. 2010;67:365–375. doi: 10.1002/ana.21905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AR, Shulman GL, Corbetta M. Why use a connectivity-based approach to study stroke and recovery of function? Neuroimage. 2012a;62:2271–2280. doi: 10.1016/j.neuroimage.2012.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AR, Patel KR, Astafiev SV, Snyder AZ, Rengachary J, Strube MJ, Pope A, Shimony JS, Lang CE, Shulman GL, Corbetta M. Upstream dysfunction of somatomotor functional connectivity after corticospinal damage in stroke. Neurorehabil Neural Repair. 2012b;26:7–19. doi: 10.1177/1545968311411054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63:272–287. doi: 10.1002/ana.21393. [DOI] [PubMed] [Google Scholar]
- Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Ren J, Mandeville JB, Wu O, Ozdag FM, Moskowitz MA, Rosen BR, Finklestein SP. Functional magnetic resonance imaging of reorganization in rat brain after stroke. Proc Natl Acad Sci U S A. 2001;98:12766–12771. doi: 10.1073/pnas.231235598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Singhal AB, Mandeville JB, Wu O, Halpern EF, Finklestein SP, Rosen BR, Lo EH. Correlation between brain reorganization, ischemic damage, and neurologic status after transient focal cerebral ischemia in rats: a functional magnetic resonance imaging study. J Neurosci. 2003;23:510–517. doi: 10.1523/JNEUROSCI.23-02-00510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Frost SB, Barbay S, Friel KM, Plautz EJ, Nudo RJ. Reorganization of remote cortical regions after ischemic brain injury: a potential substrate for stroke recovery. J Neurophysiol. 2003;89:3205–3214. doi: 10.1152/jn.01143.2002. [DOI] [PubMed] [Google Scholar]
- Furlan M, Marchal G, Viader F, Derlon JM, Baron JC. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann Neurol. 1996;40:216–226. doi: 10.1002/ana.410400213. [DOI] [PubMed] [Google Scholar]
- Gharbawie OA, Gonzalez CL, Williams PT, Kleim JA, Whishaw IQ. Middle cerebral artery (MCA) stroke produces dysfunction in adjacent motor cortex as detected by intracortical microstimulation in rats. Neuroscience. 2005;130:601–610. doi: 10.1016/j.neuroscience.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Grefkes C, Fink GR. Reorganization of cerebral networks after stroke: new insights from neuroimaging with connectivity approaches. Brain. 2011;134:1264–1276. doi: 10.1093/brain/awr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AL, Wolke ML, Bell JA, Jones TA. Post-stroke protection from maladaptive effects of learning with the non-paretic forelimb by bimanual home cage experience in C57BL/6 mice. Behav Brain Res. 2013;252:180–187. doi: 10.1016/j.bbr.2013.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DH, Mohajerani MH, Ledue J, Boyd J, Chen S, Murphy TH. In vivo large-scale cortical mapping using channelrhodopsin-2 stimulation in transgenic mice reveals asymmetric and reciprocal relationships between cortical areas. Front Neural Circuits. 2012;6:11. doi: 10.3389/fncir.2012.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DH, Ledue J, Mohajerani MH, Vanni MP, Murphy TH. Optogenetic approaches for functional mouse brain mapping. Front Neurosci. 2013;7:54. doi: 10.3389/fnins.2013.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, McVea DA, Fingas M, Murphy TH. Mirrored bilateral slow-wave cortical activity within local circuits revealed by fast bihemispheric voltage-sensitive dye imaging in anesthetized and awake mice. J Neurosci. 2010;30:3745–3751. doi: 10.1523/JNEUROSCI.6437-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, Aminoltejari K, Murphy TH. Targeted mini-strokes produce changes in interhemispheric sensory signal processing that are indicative of disinhibition within minutes. Proc Natl Acad Sci U S A. 2011;108:E183–E191. doi: 10.1073/pnas.1101914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, Chan AW, Mohsenvand M, LeDue J, Liu R, McVea DA, Boyd JD, Wang YT, Reimers M, Murphy TH. Spontaneous cortical activity alternates between motifs defined by regional axonal projections. Nat Neurosci. 2013;16:1426–1435. doi: 10.1038/nn.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci. 2010;30:14116–14126. doi: 10.1523/JNEUROSCI.3908-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10:861–872. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, Milliken GW. Reorganization of movement representations in primary motor cortex following focal ischemic infarcts in adult squirrel monkeys. J Neurophysiol. 1996;75:2144–2149. doi: 10.1152/jn.1996.75.5.2144. [DOI] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, Lau C, Kuan L, Henry AM, Mortrud MT, Ouellette B, Nguyen TN, Sorensen SA, Slaughterbeck CR, Wakeman W, Li Y, Feng D, Ho A, Nicholas E, Hirokawa KE, Bohn P, Joines KM, Peng H, Hawrylycz MJ, Phillips JW, Hohmann JG, Wohnoutka P, Gerfen CR, Koch C, Bernard A, Dang C, Jones AR, Zeng H. A mesoscale connectome of the mouse brain. Nature. 2014;508:207–214. doi: 10.1038/nature13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109:E2230–E2239. doi: 10.1073/pnas.1204386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinov M, Sporns O. Complex network measures of brain connectivity: uses and interpretations. Neuroimage. 2010;52:1059–1069. doi: 10.1016/j.neuroimage.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Seitz RJ, Azari NP, Knorr U, Binkofski F, Herzog H, Freund HJ. The role of diaschisis in stroke recovery. Stroke. 1999;30:1844–1850. doi: 10.1161/01.STR.30.9.1844. [DOI] [PubMed] [Google Scholar]
- Sharma N, Baron JC, Rowe JB. Motor imagery after stroke: relating outcome to motor network connectivity. Ann Neurol. 2009;66:604–616. doi: 10.1002/ana.21810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham D, Glaser DE, Arieli A, Kenet T, Wijnbergen C, Toledo Y, Hildesheim R, Grinvald A. Imaging cortical dynamics at high spatial and temporal resolution with novel blue voltage-sensitive dyes. Neuron. 1999;24:791–802. doi: 10.1016/S0896-6273(00)81027-2. [DOI] [PubMed] [Google Scholar]
- Silasi G, Murphy TH. Stroke and the connectome: how connectivity guides therapeutic intervention. Neuron. 2014;83:1354–1368. doi: 10.1016/j.neuron.2014.08.052. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/S0959-4388(00)00091-X. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- van den Heuvel MP, Hulshoff Pol HE. Exploring the brain network: a review on resting-state fMRI functional connectivity. Eur Neuropsychopharmacol. 2010;20:519–534. doi: 10.1016/j.euroneuro.2010.03.008. [DOI] [PubMed] [Google Scholar]
- van den Heuvel MP, Sporns O. Rich-club organization of the human connectome. J Neurosci. 2011;31:15775–15786. doi: 10.1523/JNEUROSCI.3539-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer MP, van der Marel K, Wang K, Otte WM, El Bouazati S, Roeling TA, Viergever MA, Berkelbach van der Sprenkel JW, Dijkhuizen RM. Recovery of sensorimotor function after experimental stroke correlates with restoration of resting-state interhemispheric functional connectivity. J Neurosci. 2010;30:3964–3972. doi: 10.1523/JNEUROSCI.5709-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer MP, Otte WM, van der Marel K, Nijboer CH, Kavelaars A, van der Sprenkel JW, Viergever MA, Dijkhuizen RM. Extent of bilateral neuronal network reorganization and functional recovery in relation to stroke severity. J Neurosci. 2012;32:4495–4507. doi: 10.1523/JNEUROSCI.3662-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl AS, Omlor W, Rubio JC, Chen JL, Zheng H, Schröter A, Gullo M, Weinmann O, Kobayashi K, Helmchen F, Ommer B, Schwab ME. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science. 2014;344:1250–1255. doi: 10.1126/science.1253050. [DOI] [PubMed] [Google Scholar]
- Ward NS, Brown MM, Thompson AJ, Frackowiak RS. Neural correlates of motor recovery after stroke: a longitudinal fMRI study. Brain. 2003a;126:2476–2496. doi: 10.1093/brain/awg245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NS, Brown MM, Thompson AJ, Frackowiak RS. Neural correlates of outcome after stroke: a cross-sectional fMRI study. Brain. 2003b;126:1430–1448. doi: 10.1093/brain/awg145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte OW, Bidmon HJ, Schiene K, Redecker C, Hagemann G. Functional differentiation of multiple perilesional zones after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1149–1165. doi: 10.1097/00004647-200008000-00001. [DOI] [PubMed] [Google Scholar]