

Abstract
Axo-axonic cells (AACs) in cortical regions selectively innervate the axon initial segments (AISs) of principal cells (PCs), where the action potentials are generated. These GABAergic interneurons can alter the activity of PCs, but how the efficacy of spike control correlates with the number of output synapses remains unclear. Moreover, the relationship between the spatial distribution of GABAergic synapses and the action potential initiation site along the AISs is not well defined. Using paired recordings obtained in the mouse basolateral amygdala, we found that AACs powerfully inhibited or delayed the timing of PC spiking by 30 ms, if AAC output preceded PC spiking with no more than 80 ms. By correlating the number of synapses and the probability of spiking, we revealed that larger numbers of presynaptic AAC boutons giving rise to larger postsynaptic responses provided more effective inhibition of PC spiking. At least 10–12 AAC synapses, which could originate from 2–3 AACs on average, were necessary to veto the PC firing under our recording conditions. Furthermore, we determined that the threshold for the action potential generation along PC axons is the lowest between 20 and 40 μm from soma, which axonal segment received the highest density of GABAergic inputs. Single AACs preferentially innervated this narrow portion of the AIS where action potentials were generated with the highest likelihood, regardless of the number of synapses forming a given connection. Our results uncovered a fine organization of AAC innervation maximizing their inhibitory efficacy by strategically positioning synapses along the AISs.
Keywords: action potential, axon initial segment, GABA, interneuron, mouse
Introduction
The proximal part of the axon, the axon initial segment (AIS) of neurons, has the lowest threshold for action potential generation near the cell body (Stuart and Sakmann, 1994). Voltage-gated Na+ channels on the AIS are present at high density (Lörincz and Nusser, 2010), which corresponds to the largest Na+ influx into the AIS during firing, in comparison with the surrounding regions, including soma or dendrites (Kole et al., 2008; Fleidervish et al., 2010). In cortical structures, the axo-axonic cell (AAC) is a unique GABAergic interneuron type that predominantly, if not exclusively, makes synapses onto the AIS of excitatory principal cells (PCs) (Somogyi, 1977). AACs, via GABAA receptor-mediated conductances (Buhl et al., 1994a), can therefore have a substantial impact on cortical operation via control of spike generation. Indeed, these interneurons can inhibit or promote firing in PCs depending on several factors (Buhl et al., 1994a; Szabadics et al., 2006; Woodruff et al., 2011). In addition to their variable effects at the single-cell level, AACs may distinctly impact neuronal processing by their specific recruitment during different network operations. For instance, sensory stimulation evokes several spikes in neocortical AACs earlier than in other cell types (Zhu et al., 2004). In the hippocampus, these interneurons discharge during θ rhythm at a phase when pyramidal cells fire least (Klausberger et al., 2003) but become silent during sharp wave-ripples (Viney et al., 2013), network activities thought to play a role in memory processes (Buzsáki, 2002; Girardeau et al., 2009).
The basolateral complex of the amygdala (BLA) has been proposed to play a key role in emotional memory formation and decision making (LeDoux, 2000; Pape and Paré, 2010). Electron microscopic studies revealed that axon terminals immunostained for the calcium binding protein parvalbumin (PV) formed symmetrical synapses on the AIS of principal cells in the BLA (Muller et al., 2006), suggesting the presence of AACs in this cortical region. In line with earlier studies obtained in the hippocampus or neocortex (Katsumaru et al., 1988; DeFelipe et al., 1989), PV-expressing interneurons targeting the AIS were identified in a recent in vivo study (Bienvenu et al., 2012), demonstrating that AACs in the BLA could be effectively activated by noxious stimulation, which suggests their role in regulating PC spiking during information processing. Despite the recognition of AACs in the neocortex 40 years ago (Szentagothai and Arbib, 1974), it is still uncertain how effective the regulation of PC firing by single AACs is and how the distribution of AAC output synapses along the AIS relates to the action potential generation site.
In this study, we examined the features of synaptic communication in monosynaptically coupled AAC-PC pairs in slice preparations. Post hoc investigation of the properties underlying these connections allowed us to determine the number and the spatial distribution of synapses along the AIS. We found that AACs hyperpolarize their postsynaptic partners and maximize their inhibitory efficacy by positioning their synapses at the site of action potential generation, which enables them to effectively control PC firing.
Materials and Methods
Experimental animals and slice preparation.
All experiments were approved by the Committee for the Scientific Ethics of Animal Research (22.1/360/3/2011) and were performed according to the guidelines of the institutional ethical code and the Hungarian Act of Animal Care and Experimentation (1998; XXVIII, section 243/1998, renewed in 40/2013). Transgenic mice of either sex (P18–P24) expressing EGFP under the control of the PV promoter (Meyer et al., 2002) were deeply anesthetized with isoflurane and decapitated. The brain was quickly removed and placed into ice-cold cutting solution containing the following (in mm): 252 sucrose, 2.5 KCl, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 1.25 NaH2PO4, 10 glucose, bubbled with 95% O2/5% CO2 (carbogen gas). Horizontal slices of 200 μm thickness containing the BLA were prepared with a Leica VT1000S or VT1200S Vibratome, and kept in an interface-type holding chamber containing ACSF at 36°C that gradually cooled down to room temperature. ACSF contained the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose, bubbled with carbogen gas.
Electrophysiological recordings.
After at least 1 h of incubation, slices were transferred individually into a submerged-type recording chamber perfused with ACSF at 32 ± 2°C with 2–3 ml/min flow rate. Recordings were performed under visual guidance using differential interference contrast microscopy (Olympus BX61W). EGFP in PV cells was excited by a UV lamp, and the fluorescence was visualized by a CCD camera (Hamamatsu Photonics). Patch pipettes were pulled from borosilicate glass capillaries with inner filament using a DMZ-Universal Puller (Zeitz-Instruments). For somatic whole-cell and perforated patch-clamp recordings, pipettes with 0.188 mm wall thickness were used and had a resistance of ∼3–5 mΩ when filled with the intrapipette solution. K-gluconate-based intrapipette solution used in all recordings contained the following (in mm): 110 K-gluconate, 4 NaCl, 2 Mg-ATP, 20 HEPES, 0,1 EGTA, 0.3 GTP (sodium salt), and 10 phosphocreatine adjusted to pH 7.3 using KOH and with an osmolarity of 290 mOsm/L. For recording the presynaptic interneurons (http://hajoslab.koki.hu/wp-content/uploads/2014/11/Position_of_the_recorded_cells.pdf), 10 mm GABA and 0.2% biocytin were added, whereas for the postsynaptic principal cell 100 μm AlexaFluor-488 hydrazide sodium salt (Invitrogen) was included. Recordings were made with a Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2 kHz, digitized at 10 kHz, and recorded with in-house data acquisition and stimulus software (Stimulog, courtesy of Prof. Zoltán Nusser, Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest, Hungary). Recordings were analyzed with EVAN 1.3 (courtesy of Professor Istvan Mody, Department of Neurology and Physiology, University of California, Los Angeles), the in-house analysis software SPIN 1.0.1 (courtesy of Prof. Zoltán Nusser) and Origin 8.6. Recordings were not corrected for junction potential. To record the firing characteristics, cells were injected with 800-ms-long hyperpolarizing and depolarizing square current pulses with increasing amplitudes from 10 to 600 pA. PC identity was characterized by the broad action potential waveform, accommodating firing pattern and slow afterhyperpolarization as well as the post hoc morphological analysis of their spiny dendrites. For recording postsynaptic inhibitory currents (IPSCs), the presynaptic AAC was held around a membrane potential of −65 mV in current-clamp mode and stimulated by brief square current pulses (2 ms, 1.5–2 nA) to evoke action potentials, and the PC was clamped at a holding potential of −40 mV. Series resistance was monitored (range: 8–25 mΩ) and compensated by 65%. To record postsynaptic inhibitory potentials (IPSPs), the presynaptic cell was stimulated in the same way, and the postsynaptic PC was held in current clamp mode at ∼−55 mV. Bridge balance was adjusted throughout the recordings.
To test the ability of AACs to inhibit PC firing, θ frequency (3.53 Hz) sinusoidal current pulses with peak-to-peak amplitudes of 30 pA and 50 pA were injected into the postsynaptic PC. The membrane potential of PCs was set (∼−55 mV) so as to evoke a spike at the peak of the sinusoidal current pulses with the amplitude of 50 pA, but not of 30 pA (Fig. 2A). This adjustment maintained the membrane potential of PCs near the spiking threshold. One trial consisted of 7 sinusoidal waves (5 × 50 pA and 2 × 30 pA), repeated 10–20 times in each pair. Three action potentials at 30 Hz were evoked in the interneuron by brief square current pulses (2 ms, 1.5–2 nA) before the fourth sinusoidal wave (50 pA) in each trial. To calculate the reduction in firing probability, the firing probability of PCs under control conditions was calculated from the average of the responses to 50 pA currents (first, third, fifth, and sixth sinusoidal wave), which was compared with that obtained during the fourth cycle. To evoke firing in PCs by synaptic input, electrical stimulation of external capsule fibers was delivered via a θ glass electrode filled with ACSF using a timer and isolator (both from Supertech).
Figure 2.

AACs effectively control action potential generation in PCs. A, Representative experiment testing the capability of AACs to inhibit spike generation in PCs. Sinusoidal current trains were injected into a PC to initiate firing, and three action potentials were evoked at 30 Hz in the AAC 30–40 ms before the peak of the fourth cycle (for details, see Materials and Methods). Red arrow points to the IPSPs in the PC evoked by AAC spike trains. Voltage traces are offset for clarity. B, The firing probability of PCs evoked by current injection was significantly suppressed in the presence of the action potential train evoked in AACs (n = 22, Kruskal–Wallis ANOVA, p < 0.001). Columns represent mean. C, The relationship between the reduction in firing probability of PCs and the size of IPSPs (Hill function fit). D, Representative experiment testing AAC ability to postpone PC firing. The timing of the evoked action potential in an AAC was systematically shifted relative to the peak of the sinusoidal current injected into the PC. A shorter interval between the presynaptic action potential train and the peak of the sinusoidal input caused a larger delay in PC spiking. E, The delay in PC firing as a function of the timing of synaptic inhibition in the pair shown in D. *Significant delay in firing compared with the peak of the cycle during the control period (paired sample Wilcoxon signed rank test, p < 0.05). F, Pooled data from 8 pairs. Filled and open diamonds represent the average maximal delay and the average last time point with significant delay in PC firing, respectively. G, Representative experiment testing the capability of AACs to suppress PC firing driven by EPSPs, which were evoked by electrical stimulation of fibers in the external capsule. Voltage traces are offset for clarity. H, The firing probability of PCs driven by EPSPs was significantly suppressed in the presence of the action potential train evoked in AACs (n = 12, Kruskal–Wallis ANOVA p < 0.001). Columns represent mean.
For perforated patch recordings, 100 mg/ml gramicidin (Sigma) stock solution was prepared in DMSO daily and kept at 4°C. Before the recordings, gramicidin stock solution was diluted to the concentration of 100 μg/ml in standard K-gluconate-based intrapipette solution, which also contained 100 μm Alexa-488 hydrazide sodium salt and 1 mm QX-314 (Sigma). The solution was sonicated for a short period of time in an ultrasound bath several times before use. The tip of a pipette with 3–5 mΩ resistance was filled with gramicidin-free solution and then backfilled with the gramicidin containing intrapipette solution. The series resistance was monitored throughout the experiment, and recordings were started when the resistance fell <100 mΩ. Pipette capacitance was neutralized and bridge balance was carefully adjusted throughout the recordings. Patch rupture was detected by (1) the inability to evoke action potentials with depolarizing current steps (the consequence of the Na+/K+ channel blocker QX-314 diffusion into the cell), (2) with the penetration of Alexa-488 dye into the cell, or (3) sudden drop in the access resistance, and in such cases the experiment was terminated (http://hajoslab.koki.hu/wp-content/uploads/2014/11/Perforated_patch.pdf). To estimate the reversal potential of the evoked postsynaptic responses (see Fig. 1E,F), we plotted the area (integral) of IPSPs as a function of membrane potential and obtained the value where the second-order polynomial fit crossed the x-axis. For comparing the reversal potential of IPSPs in perforated patch and whole-cell mode, we compared the responses from the same presynaptic AAC onto a PC recorded in perforated patch mode and a neighboring PC recorded in whole-cell mode.
Figure 1.

AACs in the BLA make synapses on the AISs and hyperpolarize their postsynaptic targets. A, Maximum z intensity projection image of an in vitro biocytin-filled AAC. Inset, Characteristic cartridge formed by axon terminals. B, Biocytin-containing boutons of an AAC (green) make close appositions (arrows) with AISs visualized with immunofluorescent staining against ankyrin G (orange). VGAT content in axon varicosities of an AAC is indicative of a GABAergic phenotype. C, Representative axon terminal of a biocytin-filled AAC forms a symmetric synapse (black arrow) onto an AIS. D, AACs have fast spiking characteristics revealed by step current injection (70, 300, −100 pA). E, Postsynaptic potentials (PSPs) recorded in a PC at different membrane potentials in gramicidin-perforated patch recordings upon a train of 10 action potentials evoked at 40 Hz in a presynaptic AAC. F, The integral of the summed PSPs was calculated at each membrane potential and fitted with a second-order polynomial curve to determine the reversal potential of the response. G, Comparison of the firing threshold and the resting membrane potential of PCs with the reversal potential of the PSPs recorded in perforated patch mode revealed the inhibitory nature of AACs. A similar reversal potential of PSPs was obtained in whole-cell mode using an intrapipette solution containing 4 mm Cl− (Mann–Whitney U test, p = 0.94; n.s., not significant). H, I, Representative recordings from an AAC-PC pair in whole-cell mode using 4 mm Cl−. Ten superimposed consecutive traces in gray, average in black. I, IPSCs and IPSPs in response to 3 action potentials evoked at 30 Hz show short-term depression. PP, Perforated patch; WCR, whole-cell recording; VC, voltage clamp; IC, current clamp. Scale bars: A, 50 μm; inset, 5 μm; B, 5 μm, 1 μm; C, 0.5 μm.
In experiments that aimed to define the action potential initiation site, PCs were recorded at the soma with K-gluconate-based intrapipette solution containing 100 μm Alexa-488, and the dye filled axon was visualized with a CCD camera. If the axon was cut, the identified axon bleb was patched with a pipette filled with the same solution (0.315 mm wall thickness, ∼12 mΩ open tip resistance). Access resistance was 10–25 mΩ for somatic recordings and 30–60 mΩ for bleb recordings. Pipette capacitance was neutralized, and bridge balance was adjusted throughout the recordings. If the axon of the dye filled cell was intact and accessible for recordings, an Alexa-488-coated pipette (0.315 mm wall thickness, ∼7 mΩ open tip resistance, coating achieved with heating) was filled with ACSF for extracellular action potential recordings in loose patch mode visually guided by the Alexa-488 signal. Action potentials were evoked by somatic square current pulse injections (10 ms long, 0.5–2 nA at 0.5 Hz), low-pass filtered at 20 kHz, digitally sampled at 100 kHz, and analyzed with EVAN 1.3 and Clampfit software. For the comparison of the action potential timing at the two recording sites, the peak time of the first-order derivative was calculated for the action potentials recorded in whole-cell mode (both for somatic and bleb recordings), and the peak time of the voltage deflection was calculated for the action potentials recorded extracellularly. Axonal recording distances were calculated based on the CCD camera images and post hoc 3D reconstruction of the biocytin and Alexa dye-labeled cells with the aid of the Neurolucida 10.53 software (MBF Bioscience). To determine the site of the action potential generation, the nadir point of the maximal delay between the two recording sites was calculated using a bilinear fit with 95% confidence intervals (MATLAB, MathWorks).
Anatomical analysis of the synapses between AAC and PC pairs.
After recordings, slices were fixed in 4% PFA in 0.1 m phosphate buffer (PB; pH 7.4) overnight. For those slices that were processed for electron microscopic studies, the fixative solution contained in addition 0.05% glutaraldehyde and 15% picric acid. Slices were then washed out with PB several times and incubated in cryoprotective solution (30% sucrose in 0.1 m PB) for 2 h, and then freeze-thawed three times above liquid nitrogen. AACs were visualized using Alexa-594- or Alexa-647-conjugated streptavidin (1:1000, Invitrogen), whereas PCs were labeled using rabbit anti-Alexa-488 primary antibody (1:1000, Invitrogen) and Alexa-488-conjugated goat anti-rabbit secondary antibody (1:200, Invitrogen). Confocal images were collected using a Nikon A1R microscope fitted with an oil-immersion apochromatic lens (CFI Plan Apo VC60× Oil, NA 1.40; z step size: 0.8 μm; xy: 0.42 μm/pixel). For the analysis of the connectivity rate between the cells, only those pairs were included where the axonal arbors of both cells were relatively intact. Based on the confocal images, the postsynaptic PC was fully reconstructed in 3D with the Neurolucida 10.53 software, and the putative synaptic sites from the presynaptic AAC were marked. For the detailed analysis of the contacts, higher-magnification images were taken with the same microscope (z step size: 0.13 μm; xy: 0.08 μm/pixel). Only those recordings were used for anatomical analysis where one presynaptic interneuron was labeled in the slice. The distribution analysis of the synapses was performed with the Neurolucida Explorer software. Values were corrected for shrinkage and flattening of the tissue (x- and y-axes: no correction; z-axis: 1.7). Four pairs were further processed for electron microscopic studies to confirm the presence of synaptic contacts. Biocytin in AACs was visualized using avidin-biotinylated HRP complex reaction (ABC; Vector Laboratories) with nickel-intensified 3,3-diaminobenzidine (DAB-Ni) giving a dark brown reaction product. Alexa-488 in PCs was revealed with biotin-conjugated goat anti-rabbit antibody, with ABC reaction visualized by DAB giving a brown chromogen. Sections were then postfixed in 0.5% OsO4, treated in 10% uranyl acetate, dehydrated in a graded series of ethanol, and embedded in epoxy resin (Durcupan; Sigma). Ultrathin sections of 60 nm thickness were cut, and contact sites, where the presynaptic axon made close appositions with the identified PC or with randomly sampled targets, were analyzed in serial sections.
In those experiments where the Alexa-488 labeling in PCs was insufficient for contact site analysis, cell type identity was confirmed with ankyrin G staining (Gulyás et al., 2010). Briefly, slices were resectioned to 40 μm thickness and incubated in mouse anti-ankyrin G (1:100–1:500, Santa Cruz Biotechnology) primary antibody and Alexa-594-conjugated goat anti-mouse secondary antibody (1:200, Invitrogen).
Reconstruction of in vivo labeled AISs.
To get sparse labeling of PCs in vivo, biotinylated dextrane amine (BDA, 3 kDa) was bilaterally injected with iontophoresis for 2 min into the BLA (anteroposterior: −1.8; mediolateral: 3.2; dorsoventral: 4.0 mm from bregma). After 2 d of recovery, mice were perfused with 2.5% acrolein in 4% PFA, pH 6.8, for 10 min, the brain was removed and postfixed in 4% PFA for 1.5 h. The areas containing the BLA were sectioned into 40 μm slices soaked in 30% sucrose overnight, and the sections were kept in cryoprotectant antifreeze solution consisting of sucrose, ethylene glycol, distilled H2O, and PBS (3:3:3.1 volume ratio) at −20 C until further processing was initiated (Watson et al., 1986). Before immunostaining, the cryoprotectant was washed out in 0.1 m PB. The sections were treated with 0.1 mg/ml pepsin (catalog #S3002; Dako) in 1 N HCl at 37°C for 15 min and washed in 0.1 m PB several times, followed by incubation in 10% normal donkey serum containing 0.05% Triton X-100 in 0.1 m PB for 45 min. Then, the sections were incubated in the sequentially applied primary antibodies in 0.1 m PB for 4 d. On the first day, mouse anti-gephyrin IgG (Synaptic System, 1:1000) and rabbit anti-ankyrin G IgG (Santa Cruz Biotechnology, 1:200) were applied, incubated at 4 C°, then on the third day guinea pig anti-vesicular GABA transporter (VGAT) IgG (Frontier Institute, 1:1000) and goat anti-panGAD IgG (Frontier Institute, 1:500) were added to the solution and further incubated at room temperature for 24 h. The primary antibodies were visualized by the incubation of the following secondary antibodies for 2 h at room temperature: donkey anti-mouse IgG-conjugated Alexa-647, donkey anti-rabbit conjugated Dylight 405, donkey anti-guinea pig conjugated Alexa-594, and donkey anti-goat conjugated Alexa-594 and Alexa-488 conjugated streptavidin. Sections were washed and mounted on slides in Vectashield (Vector Laboratories). Confocal images of the labeled cells in the BLA were collected using a Nikon A1R microscope fitted with an oil-immersion apochromatic lens (z step size: 0.13 μm; xy: 0.06 μm/pixel). To post hoc correct the spherical aberration, the obtained pictures were deconvolved with the aid of the Huygens software (SVI). Based on the 3D confocal image, the AISs of the BDA-labeled cells were reconstructed with the Neurolucida 10.3 software, and the sites where the presynaptic boutons (i.e., panGAD/VGAT-containing profiles) faced the postsynaptic marker gephyrin were labeled as synaptic contacts along the AISs. For the analysis of the obtained data, the Neurolucida Explorer software was used. Values were corrected for shrinkage of the tissue (x-, y-, z-axis correction: 1.08).
Nav 1.6 and ankyrin G staining of AISs.
Two mice were transcardially perfused with 2% PFA in 0.2 m Na-acetate buffer, pH 6.0, for 20 min, the brain was removed and sectioned into 40 μm slices. Nav 1.6 was revealed using a polyclonal rabbit anti-Nav 1.6 (1:500, Alomone Labs) primary antibody and Alexa-488-conjugated goat anti-rabbit secondary antibody; ankyrin G was labeled as described above. Confocal images were collected with a Nikon A1R microscope (z step size: 0.13 μm; xy: 0.1 μm/pixel). For the intensity analysis of the labeling, those AISs were analyzed that run parallel to the slice surface and could be traced from the soma to the end of the labeled profile. Intensity distributions of the immunolabelings were analyzed with the intensity profile function of Nikon Imaging System software using maximum z intensity projection pictures.
Statistical analysis.
For comparison of data with a non-normal distribution according to the Shapiro-Wilk test, the Mann–Whitney U test, Wilcoxon Signed Rank test, and Kruskal–Wallis ANOVA were used. For the comparison of distributions, the two sample Kolmogorov–Smirnov test was used. All statistics were performed using Origin 8.6. Data are presented as mean ± SEM unless indicated.
Results
AACs innervate the axon initial segments of principal cells in the BLA
To study the properties of AACs in the BLA, we obtained visually guided, targeted recordings in amygdalar slices prepared from the brains of transgenic mice expressing EGFP under the control of the PV promoter (Meyer et al., 2002). After recordings, cells were morphologically identified (Fig. 1A) using a previously described method (Gulyás et al., 2010). Briefly, we analyzed the bouton distribution of the recorded interneurons relative to the AIS of PCs at the light microscopic level. If the boutons of an interneuron formed close appositions with the AIS of the intracellularly labeled PC in a monosynaptically connected pair or with immunostained profiles visualized with an antibody recognizing the anchoring protein, ankyrin G (Fig. 1B), we identified the cell to be an AAC. In this study, 45 AACs were included after separation from other types of PV-expressing interneurons (i.e., from putative basket cells and dendrite-targeting interneurons; n = 28). In the case of 3 AACs, we confirmed by electron microscopy that their axon terminals formed symmetrical synapses on AISs (n = 25 of 28 boutons; Fig. 1C). Perikarya or dendrites were not among the postsynaptic targets of examined boutons. Thus, in the BLA, similarly to other cortical regions (Buhl et al., 1994b; Tamás and Szabadics, 2004), AACs can be separated from additional types of PV-expressing interneurons based on clear structural criteria.
Fast and large synaptic inhibition characterizes the connections between AACs and their targets
First, we determined the spiking features of AACs and their basic membrane properties. Upon intracellular current injection, these interneurons had a fast spiking phenotype (maximum firing frequency: 153.8 ± 8.9 Hz, n = 15), characterized by a firing with moderate accommodation (last/first interspike interval ratio: 2.03 ± 0.10) and narrow spike width at half maximum amplitude (0.35 ± 0.01 ms), as well as low input resistance (133.4 ± 8.6 mΩ; Fig. 1D).
Next, we aimed to reveal the synaptic effects of AACs onto PCs. To minimize the disturbance of the ionic milieu in PCs, we performed recordings between monosynaptically connected AAC-PC pairs when the postsynaptic PC was recorded in perforated patch mode using gramicidin in the recording pipette. A train of action potentials was evoked in the presynaptic AAC, whereas the resulting monosynaptic PSPs were recorded in the PC at different membrane potentials (Fig. 1E). When we compared the estimated reversal potential of PSPs and the resting membrane potential of PCs (Fig. 1F), we found that, in the majority of cases (5 of 7), AACs hyperpolarized their postsynaptic partner at resting membrane potential (Fig. 1G). In the two remaining cases, although the PSP reversal was above the resting membrane potential, these values were still below the firing threshold of PCs. These data suggest that the synaptic effect of AACs onto PCs at resting membrane potential is inhibitory under our recording conditions (Fig. 1G). When we compared the reversal potential of IPSPs recorded in perforated patch mode with that obtained in whole-cell mode with an intrapipette solution containing 4 mm Cl−, we found no significant difference (Fig. 1G; Mann–Whitney U test, p = 0.94). Therefore, in the following part of the study, the postsynaptic responses were recorded in whole-cell mode with an intrapipette solution containing 4 mm Cl− concentration.
To reveal the basic characteristics of synaptic inhibition originating from AACs, AAC-PC cell pairs were recorded using the voltage-clamp configuration (Fig. 1H, I). We found that the probability of finding a connected AAC-PC pair was high (84%; n = 45 of 54). Notably, in half of the connected pairs, PCs reciprocally innervated AACs (52%; n = 17 of 33). Spikes in the presynaptic AACs triggered responses in the PCs with very low failure rate (probability of failure: 0.05 ± 0.02) and fixed, short latency (1.01 ± 0.03 ms, n = 20), indicative of monosynaptic connections (Fig. 1H). The unitary postsynaptic currents recorded at a membrane potential of −40 mV had an amplitude of 100.5 ± 13.5 pA on average, fast rise, and decay kinetics (10%–90% rise time: 0.83 ± 0.07 ms; decay time constant: 6.27 ± 0.53 ms) and were mediated via GABAA receptors because a specific antagonist of GABAA receptors (10 μm gabazine) eliminated the action potential-evoked postsynaptic currents (n = 3, data not shown). When we evoked three action potentials in AACs at 30 Hz, the amplitude of IPSCs showed a modest short-term depression (peak 3/peak 1, 0.80 ± 0.03; n = 20; Fig. 1I). In current clamp mode, IPSPs recorded in PCs at −55 mV upon evoking action potentials in AACs had large amplitude (1.23 ± 0.19 mV, n = 16). When three action potentials were evoked in AACs, the IPSP amplitudes showed a pronounced short-term depression (peak 3/peak 1, 0.49 ± 0.04). These data show that PCs receive large and reliable synaptic inputs from AACs that are inhibitory in nature and show short-term depression.
In a few cases, we were also able to characterize the synaptic connections in PC-AAC pairs. These connections showed short latency (1.03 ± 0.06 ms) and moderate failure rate (0.40 ± 0.14). The amplitude of unitary EPSCs was −37.04 ± 17.87 pA at a membrane potential of −65 mV and had fast rise and decay kinetics (10%–90% rise time: 0.42 ± 0.11 ms; decay time constant: 1.13 ± 0.21 ms, n = 4 pairs).
AACs potently regulate principal cell spiking
In the next set of experiments, we explored how inhibitory input from an AAC can control action potential generation in a PC. PCs in the BLA display intrinsic membrane potential oscillations at θ frequency in vivo, a rhythm that can facilitate periodic firing at θ frequencies (Pape et al., 1998). To mimic this intrinsic oscillation, we injected a sinusoidal current at low θ frequency (3.53 Hz) and adjusted the membrane potential of the PC to spike single action potentials at the peak of the sinusoidal inputs (for details, see Materials and Methods). To test the effect of the synaptic inhibition onto PC firing probability, three action potentials at 30 Hz were evoked in the presynaptic AAC before the positive peak of the sinusoidal current (Fig. 2A). In these experiments, trains of action potentials were used instead of single spikes because AACs tend to fire multiple spikes in vivo when they become active (Klausberger et al., 2003; Zhu et al., 2004; Bienvenu et al., 2012; Massi et al., 2012). The stimulation of the interneuron was timed to provide the maximal inhibitory effect at the peak of the sinusoidal current, where the PC spiked with the highest probability. We found that, under these conditions, AACs could significantly reduce the firing probability of PCs (79.4 ± 5.9% reduction, n = 22 cells) compared with those sinusoidal cycles when the interneuron was silent (Fig. 2B; Kruskal–Wallis ANOVA, p < 0.001). The analysis showed that there was a strong positive correlation (Hill fit, p < 0.001) between the magnitude of the synaptic inhibition (assessed as the IPSP integral, area of the IPSP under the baseline membrane potential) and the reduction in the firing probability of PCs (i.e., larger IPSPs from AACs more effectively reduced the PC spiking; Fig. 2C). We have estimated that an IPSP integral ∼1 mV/ms is needed to achieve a 95% reduction in the PC firing probability under these conditions.
Experimental and theoretical studies show that, in addition to the spiking probability, inhibitory inputs can also prominently control the timing of action potential generation, which might play an important role in cortical network functions (Cobb et al., 1995; Kwag and Paulsen, 2009). To investigate how an AAC can affect the timing of PC firing and to determine the time window in which this interneuron type can control the spike generation, IPSPs were evoked at different phases of the sinusoidal current injected into PCs (Fig. 2D,E). These experiments uncovered that, depending on the timing of the inhibition, AACs could delay the generation of spikes in PCs with a minimum and maximum delay of 9.69 ± 1.37 ms and 27.3 ± 1.45 ms (n = 8), respectively, defining a significant spike timing control within a time window of 20 ms. A controlling effect on PC firing by AACs was detected even if the peak of IPSPs was 79.5 ± 7.03 ms before the peak of the sinusoidal current (Fig. 2F). Thus, AACs in the BLA, in addition to their veto of action potential generation in PCs, can substantially control the timing of PC firing.
Although the sinusoidal current protocol is a reliable and controllable method to generate action potentials in PCs, spikes during physiological operation are evoked mainly by incoming integrated synaptic inputs (Svoboda et al., 1997; Epsztein et al., 2010). To test whether AACs are also able to inhibit EPSP-evoked action potentials, we stimulated the fibers in the external capsule of the amygdala to induce synaptic input-driven single spikes in PCs (Fig. 2G). In the absence of AAC activity, the electrical stimulation of fibers readily evoked spikes in PCs, which was significantly reduced when three action potentials were evoked in a single presynaptic AAC at 30 Hz (63.6 ± 7.9% reduction, ANOVA, p < 0.001, n = 12; Fig. 2H). These results imply that, depending on the timing of AAC activity, when AACs fire multiple action potentials, they can potently inhibit PC firing or delay their discharge.
Higher efficacy of inhibition is promoted by multiple synapses
To get deeper insights into the properties of synaptic organization underlying the potent inhibition between AACs and PCs, we first determined the number of putative synaptic sites between the recorded pairs using multichannel high-resolution confocal microscopy. Specifically, we counted the number of AAC boutons that formed close appositions with the AIS of the recorded PC (Fig. 3A–D, red and green, respectively; see Materials and Methods). This analysis uncovered that single AACs formed 8.4 ± 0.7 putative synapses on average with their postsynaptic partners (range: 2–16 boutons/AIS, total number of boutons: 201, n = 24 pairs; Fig. 3J). We applied two approaches to confirm that the putative synaptic sites identified with confocal microscopy were synaptic junctions. First, for 6 cell pairs, we visualized a cell adhesion molecule, neuroligin 2, which is present at GABAergic synapses (Varoqueaux et al., 2004) to identify the functional contacts at the confocal microscopic level (Fig. 3E). This analysis revealed that, in most instances (92%, n = 44 of 48 boutons), immunolabeling for neuroligin 2 could be identified at the close appositions formed by AAC boutons with AISs of PCs. Second, we performed correlated light and electron microscopy in four cell pairs (Fig. 3F–I). We found that, at the vast majority of boutons (96%, 28 of 29 boutons), synapses could be unequivocally identified. These two approaches verified that, in the overwhelming majority of cases, synaptic junctions underlie the contacts between AACs and PCs indentified by light microscopy. Having determined the number of synaptic contacts at AAC-PC pairs, we next correlated this parameter with the physiological properties of these connections. We observed that the number of the contacting boutons correlated with the integral of the recorded IPSPs (Fig. 3K). Moreover, those AACs, which contacted their partner with more synapses, could inhibit the action potential generation more efficiently (tested in the protocol using sinusoidal current injection; Fig. 3L). We calculated that, to achieve a 95% reduction in PC firing probability, simultaneous input from ∼12 boutons was needed (Hill fit, p < 0.001; Fig. 3L). These results revealed the high efficacy of synaptic inhibition provided by AACs and showed that it substantially depends on the number of synaptic contacts targeting the AIS of PCs.
Figure 3.
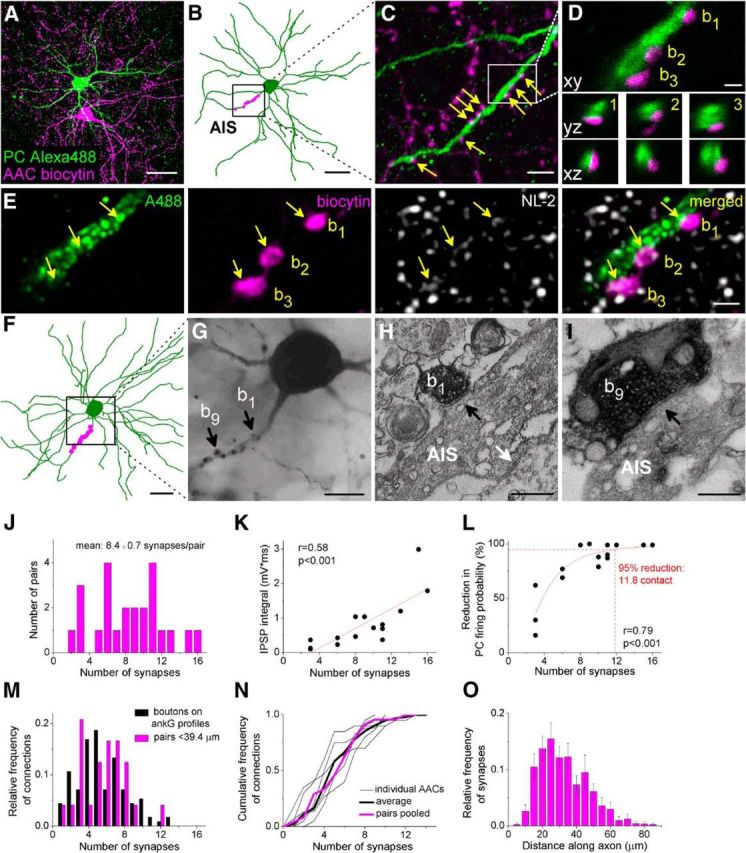
AACs innervate the proximal part of PC axons via multiple synaptic contacts. A, Maximum z intensity projection of 3D confocal image of a representative pair. Biocytin in the AAC was visualized with streptavidin-conjugated Alexa-594 (magenta) and Alexa-488 in the PC (green). B, Neurolucida reconstruction of the postsynaptic PC (green) with the putative synaptic sites marked in magenta. C, High-power magnification of the maximum z intensity projection image of the region indicated in B, showing a part of the AIS of the postsynaptic PC (green), which receives multiple boutons (arrows) from the presynaptic AAC (magenta). D, 3D analysis of confocal images of three boutons enclosed in the box in C shows close appositions of the presynaptic and postsynaptic structures. E, Verification of the functional synapses at the light microscopic level using neuroligin-2 staining. F–I, Analysis of the presence of synaptic contacts between the recorded pairs using correlative light and electron microscopy. F, Neurolucida reconstruction of another AAC-PC pair. G, Light micrograph of the boxed region in F. H, I, Electron micrographs of two boutons of the presynaptic AAC indicated in G are shown, both of which form synaptic contacts with the AIS of the postsynaptic PC (black arrows). White arrow in H indicates an unlabeled bouton synapsing on the biocytin-labeled AIS. J, Distribution of the number of boutons obtained by light microscopy (201 putative synaptic contacts from 24 pairs). In 28 cases, the presence of a synapse was verified using electron microscopy. K, Relationship between the integral of the summed IPSPs evoked by 3 action potentials and the number of synapses made by the AAC. L, Correlation between the effect of AACs on PC firing tested with a sinusoidal protocol (Fig. 2A–C) and the number of axon-AIS synapses (Hill fit, p < 0.001). M, Distribution of the number of boutons from in vitro filled AACs on ankyrin G-labeled profiles (n = 6 AACs, 616 contact sites on 112 ankyrin G-immunostained profiles, black) and AAC-PC pairs. The analysis of the varicosities was restricted to the average length obtained from the ankyrin G staining (39.4 μm) (n = 24 pairs, 134 boutons, magenta). N, Cumulative distributions of bouton numbers on randomly sampled individual ankyrin G-immunostained profiles taken from 6 filled AACs (each thin black line indicates data from single AACs). The average of these distributions (thick black line) compared with the pooled data of the paired recordings shows no difference (Kolmogorov–Smirnov test, p = 0.76). O, Spatial distribution of the synapses along the axon obtained in paired recordings (n = 24 pairs, 201 boutons). The 0 μm marks the origin of the axon at the soma. Data are mean ± SEM. Scale bars: A, B, F, 30 μm; C, 5 μm; D, E, 1 μm; G, 10 μm; H, I, 0.25 μm.
The variability in the bouton number from a single AAC to a single PC (2–16 contacts, coefficient of variation = 0.45) raises the question of whether there are AACs that preferentially target their postsynaptic partners via numerous synapses, whereas others only make a few contacts. This would imply that some AACs might have more profound effects on PC spiking during network operation than others. Or, alternatively, the large variability in the number of boutons contacting AISs characterizes the output of each AAC. To address this question, we counted the number of boutons of in vitro labeled AACs forming close appositions on 10–20 individual, randomly sampled ankyrin G-stained AISs. We found that each AAC contacted different AISs with a variable number of varicosities ranging from 2 to 14 (5.5 ± 0.25, n = 616 boutons from 6 AACs onto 112 AISs; Fig. 3M), with similar variance in the bouton number (coefficient of variation range: 0.36–0.67). Because the length of the ankyrin G-labeled profiles in slices was 39.4 ± 0.7 μm (n = 112), we reanalyzed the data obtained in pairs by restricting the analysis to those boutons present at the proximal part of the axon no farther than 39.4 μm from the soma. This measurement allowed us to compare directly the variance in the number of boutons targeting single AISs found in ankyrin G-labeled material with those obtained in paired recordings. We found no difference in the distributions of the bouton numbers at AISs using these two methods (Kolmogorov–Smirnov test, p = 0.76; Fig. 3N). Thus, we confirmed that our paired recordings reliably characterize the population of AAC outputs; and, more importantly, AACs form a homogeneous population regarding the manner in which they contact PCs, innervating their targets via a variable number of boutons.
We also observed that the AAC boutons along the AISs were not evenly distributed. Approximately 60% of all varicosities were found between 20 and 40 μm from the soma (Fig. 3O). This observation suggests that a specific region along the AIS is preferentially innervated by AACs.
Distribution of inhibitory inputs along the AIS in vivo
Our data suggest that the AAC output is not homogeneously distributed along the AIS but concentrated in a 20 μm region close to PC cell bodies. To extend these results obtained in slice preparations, we reconstructed the GABAergic input arriving onto the AISs of in vivo labeled PCs. Using quadruple immunostaining, we analyzed the distribution of synaptic inputs along the AISs by confocal microscopy. PCs in the BLA were labeled in vivo with a tracer, BDA (3 kDa), whereas their AISs were identified with immunostaining for ankyrin G (Fig. 4A). The GABAergic inputs were visualized with a mixture of antibodies against both isoforms of the GABA synthesizing enzyme GAD (GAD65 and GAD67, panGAD) and the vesicular GABA transporter, VGAT, whereas a scaffolding protein, gephyrin, expressed specifically at inhibitory synapses, was used to label functional synaptic junctions (Fig. 4B). In high-resolution confocal images of the in vivo labeled PCs, we reconstructed AISs together with GABAergic terminals that were apposed to gephyrin labeling (Fig. 4C). In this material, the length of the ankyrin G-labeled segments was 58.4 ± 1.9 μm (n = 7), which corresponds to the length of the AIS. The analysis showed that single AISs received 52.4 ± 7.8 inhibitory inputs on average (range: 31–86; n = 7). Importantly, the spatial distribution of the GABAergic innervation observed in vivo was indistinguishable from that found in in vitro experiments (Fig. 4D; Kolmogorov–Smirnov test, p = 0.57). These results support our data obtained in slices, namely, that the GABAergic innervation of the PC AIS is concentrated on a restricted region of the AISs between 20 and 40 μm measured from the PC somata (Fig. 4D).
Figure 4.

Spatial distribution of GABAergic inputs along the proximal part of PC axons in vivo. A, Representative BDA-labeled PC (green). The AIS was identified with immunostaining against ankyrin G (magenta). B, Along the BDA-labeled axon, GABAergic terminals were visualized with antibodies detecting both isoforms of the GAD enzyme (GAD65 and GAD67) and the vesicular GABA transporter, VGAT. Functional contacts were revealed based on the presence of a scaffolding synaptic protein, gephyrin. Bottom, Superimposed neurolucida reconstruction of GABAergic inputs along the given axonal segment. Arrows indicate the synaptic sites along the AIS. Open arrowhead indicates a GABAergic bouton that did not target this BDA-labeled AIS. Images were taken from a single focal plane. C, 3D Neurolucida reconstruction of the proximal part of the axon and a part of the soma shown in A (green) and the GABAergic inputs where gephyrin was present (blue spheres) forming close apposition with the axon. D, Spatial distribution of GABAergic inputs along axons labeled in vivo (n = 7 reconstructed axons, 5 μm bin size). Data are mean ± SEM. Scale bars: A, C, 25 μm; B, 2 μm.
GABAergic innervation of AISs overlaps with the site of the action potential generation in PCs
Our in vitro and in vivo observations that GABAergic inputs onto AISs of PCs prefer a specific area along the proximal part of the axon raise the question of how this inhibitory input relates to the site of action potential generation. If the innervated region overlaps with the site where the threshold for action potential generation is the lowest, then AACs could have a role primarily in the control of PC spiking. However, if the area along the AIS receiving the majority of GABAergic innervation is different from the site where the threshold for action potential generation is the lowest, then AACs may rectify PC spiking by blocking action potential propagation either toward the soma or toward the distal axon depending on the spatial relationships of the two sites. To distinguish between these possibilities, we determined where the action potentials are generated either by simultaneous whole-cell recordings from the soma and axonal bleb of the same PCs (Shu et al., 2006) or by simultaneous recordings from the soma in whole-cell mode and along the intact axon in loose patch mode (Clark et al., 2005; Palmer et al., 2010). We evoked an action potential in the cell by injecting a square current pulse (0.5–1.5 nA, 10 ms) via the somatic electrode and measured the evoked action potentials at both recording sites (Fig. 5A,B). After the recordings, the cell was visualized and reconstructed in 3D and the distance of the bleb or the axonal recording sites from the soma was measured (see Materials and Methods). We compared the timing of the action potentials at the two recording sites and found that the difference in the spike timing was dependent on the distance of the axonal recording site from the soma (Fig. 5C). A bilinear fit to the data showed that the maximum difference in the axon-soma latency, which corresponds to the site of action potential generation, was at ∼30 μm from the soma along the AIS (in bleb recordings: 30.5 ± 10.9 μm, in experiments recorded action potentials extracellularly: 29.5 ± 8.5 Fig. 5C, red).
Figure 5.
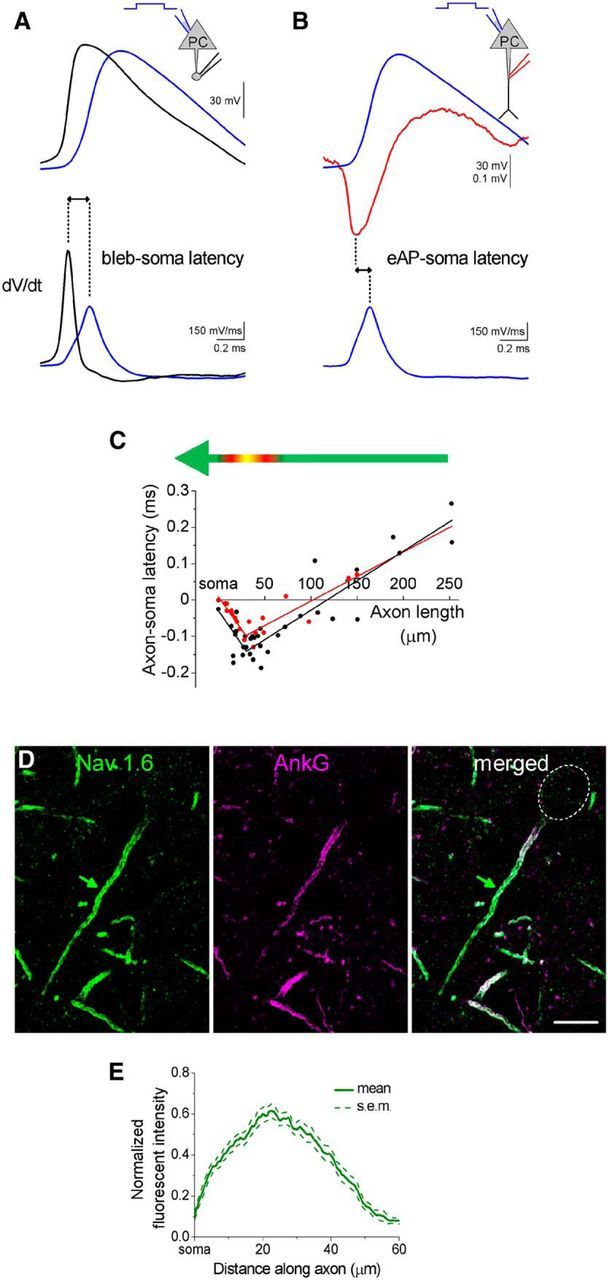
Initiation zone for action potentials in PCs in the BLA. A, Representative recordings obtained simultaneously at the soma and the axonal bleb in whole-cell mode. Top, Raw data. Bottom, First-order derivative. The peaks of the first-order derivatives were used to calculate the delay between the action potentials recorded at the two sites. B, Representative traces of an action potential recorded at the soma in whole-cell mode and simultaneously at the axon in loose patch mode. Top, Raw data. Bottom, First-order derivative of the somatic action potential. The peak of the first-order derivative and the peak of action potential detected extracellularly were used to calculate the delay between the action potentials recorded at the two sites. C, The latency difference of the action potentials recorded at the two sites plotted as a function of the distance between the corresponding recording sites. Bilinear fit minimum point obtained in the bleb recordings and in extracellular detection of action potentials was 30.5 ± 10.9 μm (black) and 29.5 ± 8.5 μm (red), respectively. D, Double staining for Nav 1.6 and ankyrin G in the BLA. Green arrow indicates the peak in fluorescent intensity for Nav 1.6. E, Normalized fluorescence intensity of the immunostainings as a function of the distance along the axon (n = 20 AIS). Scale bars: B, D, 10 μm.
To test this result obtained using dual patch recordings and have an independent estimate for the site of action potential generation, we investigated the distribution of the Nav 1.6 subtype of voltage-gated Na+ channel, which is critical for the initiation of action potentials (Hu et al., 2009). Using confocal microscopy, we measured the fluorescence intensity of the immunolabeling for Nav 1.6 proteins along the AIS as defined by ankyrin G-labeling (Fig. 5D,E; n = 20 AIS, profile length: 61.6 ± 1.6 μm). We found that the most intense labeling for Nav 1.6 along the AISs (20% of the highest intensity) was between 16.2 and 31.9 μm from the soma, which corresponds to the action potential generation site determined by the electrophysiological recordings.
As the site of the action potential generation in PCs in the BLA closely matches the area preferentially targeted by AACs, we propose that AAC output synapses are optimized to control Na+ channel opening and thus PC firing.
AACs strategically position their synapses at the site of action potential generation
Our anatomical analysis of the connectivity between AACs and PCs showed that there was a great variability in the number of synaptic contacts, ranging from 2 to up to 16 connections (Figs. 3J and 6A). This raises the question of whether the synapses originating from individual AAC-PC connections with different bouton numbers distribute evenly along the entire AIS, or, alternatively, tend to target the site of action potential generation (i.e., optimizing the efficacy of their synaptic inhibition). To address this question, we examined the bouton distribution along the AIS as a function of the number of synapses. We divided the pairs into three groups based on the number of the boutons on single AISs (1–5, 6–10, or 10–16 contacts) and compared the spatial distribution of the boutons along the AISs among the groups (Fig. 6B). The analysis showed that all groups tended to concentrate their synapses at the action potential generation zone (Fig. 6B,C). Importantly, those cells contacting their partners with a few boutons restricted their output synapses to the estimated spike generation area. In contrast, the innervations of those AACs, which gave rise to more boutons, outreached the site of action potential generation, covering a significantly broader axonal area (Fig. 6D; ANOVA p < 0.001).
Figure 6.
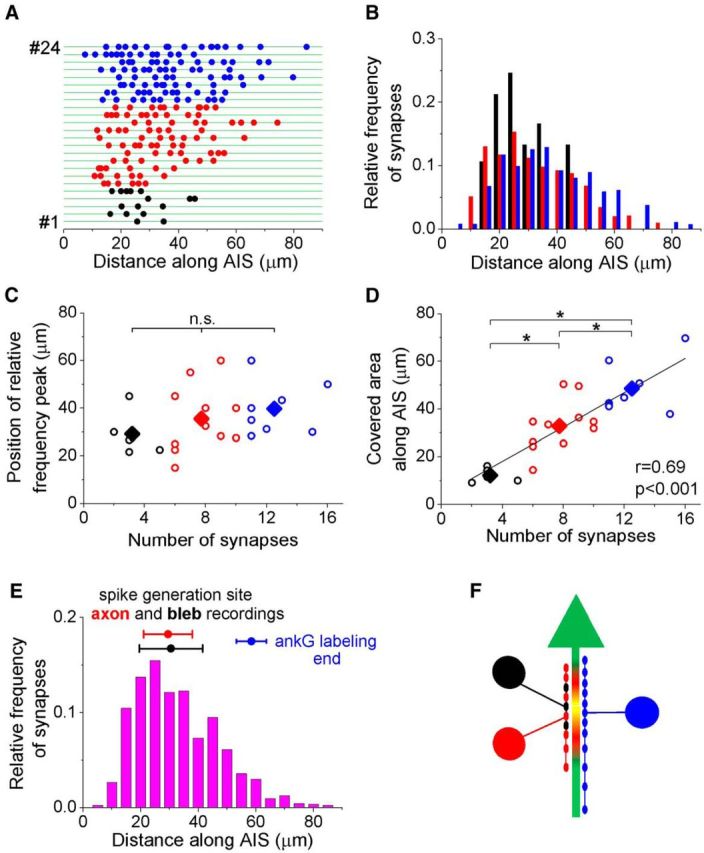
AACs in the BLA preferentially target the action potential initiation zone. A, Spatial distribution of the boutons along the AISs obtained in individual AAC-PC pairs. Each row represents a single AIS taken from AAC-PC pairs (n = 24) that are arranged as a function of increasing bouton number. Each bouton is shown as a colored dot on each AIS (green). The pairs were categorized into three groups according to the number of contacts: black dots indicate 1–5 contacts; red dots indicate 6–10 contacts; blue dots indicate 11–16 contacts. B, The relative frequency distributions of boutons obtained in the three groups show a large correspondence. C, The position of peak values of the relative frequency distributions showed no difference among the three groups (p = 0.34, ANOVA). n.s., Not significant. D, The number of the boutons and the covered area along the AIS (e.g., the distance between the first and last boutons). *Significant difference between the three groups (p < 0.001, ANOVA). E, Data from distinct experiments show that the action potential initiation site corresponds well to the spatial distribution of the synapses determined in AAC-PC pairs (data from Fig. 3O). The nadir points (circles) and confidence intervals (bars) of action potential initiation site obtained from extracellular and bleb recordings are indicated (data from Fig. 5C) together with the end of the ankyrin G staining (blue), which defines the length of the AIS. F, Summary scheme indicating that AACs preferentially target and cluster at the site of the action potential generation along the PC axon (yellow coloring) regardless of the number of synapses made by a given connection.
These data show that, regardless of the actual number of output synapses, AACs preferentially innervate the site of action potential generation and maximize their inhibitory efficacy by strategically positioning their contacts along the AIS (Fig. 6E,F).
Discussion
Our major findings are as follows: (1) AACs in the BLA hyperpolarize their postsynaptic partners at resting membrane potential or near the spike threshold. (2) AACs can effectively inhibit the firing of PC and delay spike generation. (3) The AIS length is ∼60 μm as estimated from immunostainings, and AACs concentrate 60% of their output synapses to the segment between 20 and 40 μm from the soma, a region corresponding to the action potential initiation site (Fig. 6E,F). Thus, AACs maximize their inhibitory efficacy by strategically concentrating their synaptic junctions along the AISs to effectively counteract PC Na+ channel activation and action potential generation.
Our paired recordings revealed that AACs and PCs in the BLA were often reciprocally connected. These data are in line with previous anatomical results, showing that local PCs heavily innervate PV cells in the BLA (Smith et al., 2000). Although these results suggest that local PC activity can recruit AACs, the noxious stimulus induced significant increase in AAC spiking indicate (Bienvenu et al., 2012) that these interneurons should also receive a powerful synaptic excitation from other cortical or subcortical regions.
We have shown that AACs in the BLA act via a GABAA receptor-mediated Cl− conductance and have confirmed the results of a recent in vivo study obtained in the BLA, showing that AACs innervated mainly the AISs (Bienvenu et al., 2012). These observations are in agreement with those found earlier in the hippocampus and neocortex (Somogyi, 1977; Somogyi et al., 1982, 1983a; Buhl et al., 1994a, b; Cobb et al., 1995; Szabadics et al., 2006). However, there is still a debate as to whether AACs inhibit or excite PCs, which may depend on several factors, including the resting membrane potential of postsynaptic neurons, the reversal potential for Cl− in the AIS, or by enhancing HCO3− efflux from the AIS, thereby activating Kv7/KCNQ channels (Buhl et al., 1994a; Szabadics et al., 2006; Woodruff et al., 2011; Jones et al., 2014). Using gramicidin-perforated patch recordings, we found that, in the majority of PCs, the reversal potential of the postsynaptic responses from AACs was below both the resting membrane potential and the firing threshold of PCs. Thus, we concluded that, under our experimental conditions, AACs were inhibitory in the BLA, similar to results in the hippocampus (Buhl et al., 1994a; Cobb et al., 1995; Glickfeld et al., 2009). In the neocortex, AACs can excite some of their postsynaptic partners (Szabadics et al., 2006; Molnár et al., 2008), which could be dependent on the membrane potential of PCs (Woodruff et al., 2011). Similar GABAA receptor-mediated excitation of PCs was described in the BLA in vitro, but the type of PV-expressing interneurons giving rise to this effect has not been clarified (Woodruff et al., 2006). Further studies should address the identity of those amygdalar interneurons that might excite PCs monosynaptically via GABAA receptors.
When the earliest studies described that AACs selectively target the AISs of PCs, it was proposed that this unique target distribution allows AACs to effectively control the activity of their postsynaptic partners (Somogyi, 1977; Somogyi et al., 1983a). Indeed, we have found that AACs are well suited to regulate PC spiking because their output synapses, regardless of the number of contacts, predominantly cover the part of the AIS where the initiation of action potentials has the largest probability as measure by electrophysiological methods and supported by immunocytochemical data (Fig. 5). Recent studies showed that both the length and the position of the AIS along the proximal part of the axon could be dynamically regulated as a function of neuronal excitability (Grubb and Burrone, 2010; Kuba et al., 2010; Kuba, 2012). If such a mechanism operated in cortical structures, the PCs might “escape” from the AAC driven spike control for a period of time, which might be followed by enhancing the GABAergic input at the distal segment, a hypothesis that should be tested in the future.
In cortical regions, individual fast-spiking basket and AACs have been shown to veto PC spiking (Cobb et al., 1995; Miles et al., 1996; Tamás et al., 2004; Woodruff and Sah, 2007). By affecting the spiking of several neurons simultaneously, fast spiking interneurons are thus in a position to synchronize the neuronal activities at low frequencies, which might contribute to the generation of the θ rhythm (Buzsáki, 2002). In addition, the bulk stimulation of inhibitory fibers cannot only delay but also advance PC spiking in the CA1 hippocampal region (Kwag and Paulsen, 2009). Indeed, we found that single AACs could significantly delay the spiking in amygdalar PCs in a similar way to that observed for CA1 PCs. However, we have not observed any advancement in PC spiking upon activation of these interneurons. The reason for this could be twofold. First, PCs in the BLA express only a modest h-current (Faber et al., 2001; Park et al., 2007) that mediates the advancement in spiking in CA1 (Kwag and Paulsen, 2009). Second, GABAergic cells that make synapses on the dendrites of PCs might be more suitable to activate h-current, which is expressed predominantly in PC dendrites (Magee, 1999; Lörincz et al., 2002). Our result showing that single AACs can delay PC spiking by 10–30 ms suggests that AACs might have a role in regulating synaptic plasticity by restricting the precise timing of principal cell spiking to a time window when the Hebbian forms of synaptic plasticity take place.
Our paired recordings revealed that an AAC innervated PCs via 8.4 synapses on average, which agrees remarkably well to the 8 synaptic contacts reported in the cat visual cortex (Somogyi et al., 1982). This value was higher by ∼30% than that found by counting the number of varicosities of single AACs on the ankyrin G-immunopositive profiles (5.5 bouton/AIS). The reason for the higher number of boutons found on the intracellularly labeled PCs is that the full targeted parts of the axons are visualized, including those that could not be labeled by immunostaining against ankyrin G. Thus, there might be a systematic underestimation of the number of GABAergic inputs from individual AACs onto PCs if the counting is based on the visualization of AISs by immunostaining against, for example, ankyrin G. Reconstruction of in vivo labeled PCs showed that an AIS received 52.4 boutons on average, ranging from 31 to 86, which is comparable with an average of 43 in cat visual cortex (Somogyi et al., 1982) and 56 in CA1 of the monkey (Somogyi et al., 1983b), but lower than found in the CA3 area of the rat (150) (Kosaka, 1980). Our values in the BLA are higher compared with the 15–30 synapses contacting an AIS found earlier (McDonald and Betette, 2001; Muller et al., 2006). Similar studies in other cortical regions showed that the number of synapses on single AISs is quite variable, ranging from 1 to 150 depending on the area and cortical layer (Kosaka, 1980; Somogyi et al., 1982, 1983b; Freund et al., 1983; DeFelipe et al., 1985; Fariñas and DeFelipe, 1991; Buhl et al., 1994b; Tamás and Szabadics, 2004). Comparative light microscopic studies also suggested substantial variability in the innervation density of AISs between different cortical areas and layers (Inda et al., 2007, 2009) and found that the cortical nuclei of the amygdala were among the regions having particularly dense AIS innervation. Because the vast majority of these GABAergic inputs probably originated from AACs (Howard et al., 2005), it is possible that single PCs receive innervation on their AIS from at least 4 to 10 AACs. These estimations are in accord with those obtained in other cortical structures (Freund et al., 1983; Buhl et al., 1994a; Inan et al., 2013). Taking into account our calculation that ∼12 synapses were needed for a 95% reduction in PC firing probability under our recording conditions, a simultaneous train of action potentials in 2–3 AACs could effectively block spike genesis. This implies that for the complete suppression of PC firing 20%–50% of all AACs innervating individual PCs should discharge synchronously.
The BLA has a homologous structure to other cortical areas; thus, the activity of various GABAergic interneuron types may shape neuronal processing in this brain region similarly to others (Ehrlich et al., 2009; Spampanato et al., 2011). Indeed, a recent in vivo study showed that there are distinct interneuron types in the BLA with cell type-specific firing modes during network activities, together with characteristic responsiveness to incoming noxious stimuli (Bienvenu et al., 2012). Particularly, AACs discharged with high fidelity to painful stimulation, which, in combination with our data, might explain the stimulus-induced GABAergic inhibition and silencing of PCs within the BLA (Windels et al., 2010). The dense innervation of neighboring PCs with strategically positioned synapses at the action potential initiation site enable AACs to powerfully modulate the firing of their postsynaptic partners with high temporal precision, a feature that might endow them with a crucial role in amygdalar network operation during fear memory processing.
Footnotes
This work was supported by a fellowship of the Hungarian Academy of Sciences Lendület, LP2012-23 to N.H. and the National Office for Research and Technology OMFB-01678/2009. V.K.V. was supported by European Research Council Advanced Grant ERC-2011-ADG-294313 (SERRACO). We thank Péter Somogyi and Tamás F. Freund for their helpful comments on the manuscript; Hannah Monyer and Gábor Szabó for providing PV-EGFP mice; Karri Lamsa for advice with perforated patch recordings; Szabolcs Káli for help with analysis; Zsófia Huszár, Erzsébet Gregori and Éva Krizsán for excellent technical assistance; and László Barna, the Nikon Microscopy Center at the Institute of Experimental Medicine, Nikon Austria GmbH, and Auro-Science Consulting, Ltd, for providing microscopy support.
The authors declare no competing financial interests.
References
- Bienvenu TC, Busti D, Magill PJ, Ferraguti F, Capogna M. Cell-type-specific recruitment of amygdala interneurons to hippocampal θ rhythm and noxious stimuli in vivo. Neuron. 2012;74:1059–1074. doi: 10.1016/j.neuron.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature. 1994a;368:823–828. doi: 10.1038/368823a0. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Han ZS, Lörinczi Z, Stezhka VV, Karnup SV, Somogyi P. Physiological properties of anatomically identified axo-axonic cells in the rat hippocampus. J Neurophysiol. 1994b;71:1289–1307. doi: 10.1152/jn.1994.71.4.1289. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/S0896-6273(02)00586-X. [DOI] [PubMed] [Google Scholar]
- Clark BA, Monsivais P, Branco T, London M, Häusser M. The site of action potential initiation in cerebellar Purkinje neurons. Nat Neurosci. 2005;8:137–139. doi: 10.1038/nn1390. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Hendry SH, Jones EG, Schmechel D. Variability in the terminations of GABAergic chandelier cell axons on initial segments of pyramidal cell axons in the monkey sensory-motor cortex. J Comp Neurol. 1985;231:364–384. doi: 10.1002/cne.902310307. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Hendry SH, Jones EG. Visualization of chandelier cell axons by parvalbumin immunoreactivity in monkey cerebral cortex. Proc Natl Acad Sci U S A. 1989;86:2093–2097. doi: 10.1073/pnas.86.6.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Epsztein J, Lee AK, Chorev E, Brecht M. Impact of spikelets on hippocampal CA1 pyramidal cell activity during spatial exploration. Science. 2010;327:474–477. doi: 10.1126/science.1182773. [DOI] [PubMed] [Google Scholar]
- Faber ES, Callister RJ, Sah P. Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro. J Neurophysiol. 2001;85:714–723. doi: 10.1152/jn.2001.85.2.714. [DOI] [PubMed] [Google Scholar]
- Fariñas I, DeFelipe J. Patterns of synaptic input on corticocortical and corticothalamic cells in the cat visual cortex: I. The cell body. J Comp Neurol. 1991;304:53–69. doi: 10.1002/cne.903040105. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Lasser-Ross N, Gutnick MJ, Ross WN. Na+ imaging reveals little difference in action potential-evoked Na+ influx between axon and soma. Nat Neurosci. 2010;13:852–860. doi: 10.1038/nn.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Martin KA, Smith AD, Somogyi P. Glutamate decarboxylase-immunoreactive terminals of Golgi-impregnated axoaxonic cells and of presumed basket cells in synaptic contact with pyramidal neurons of the cat's visual cortex. J Comp Neurol. 1983;221:263–278. doi: 10.1002/cne.902210303. [DOI] [PubMed] [Google Scholar]
- Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB. Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci. 2009;12:1222–1223. doi: 10.1038/nn.2384. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Szabó GG, Ulbert I, Holderith N, Monyer H, Erdélyi F, Szabó G, Freund TF, Hájos N. Parvalbumin-containing fast-spiking basket cells generate the field potential oscillations induced by cholinergic receptor activation in the hippocampus. J Neurosci. 2010;30:15134–15145. doi: 10.1523/JNEUROSCI.4104-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard A, Tamás G, Soltesz I. Lighting the chandelier: new vistas for axo-axonic cells. Trends Neurosci. 2005;28:310–316. doi: 10.1016/j.tins.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- Inan M, Blázquez-Llorca L, Merchán-Pérez A, Anderson SA, DeFelipe J, Yuste R. Dense and overlapping innervation of pyramidal neurons by chandelier cells. J Neurosci. 2013;33:1907–1914. doi: 10.1523/JNEUROSCI.4049-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, Defelipe J, Muñoz A. The distribution of chandelier cell axon terminals that express the GABA plasma membrane transporter GAT-1 in the human neocortex. Cereb Cortex. 2007;17:2060–2071. doi: 10.1093/cercor/bhl114. [DOI] [PubMed] [Google Scholar]
- Inda MC, DeFelipe J, Muñoz A. Morphology and distribution of chandelier cell axon terminals in the mouse cerebral cortex and claustroamygdaloid complex. Cereb Cortex. 2009;19:41–54. doi: 10.1093/cercor/bhn057. [DOI] [PubMed] [Google Scholar]
- Jones RT, Faas GC, Mody I. Intracellular bicarbonate regulates action potential generation via KCNQ channel modulation. J Neurosci. 2014;34:4409–4417. doi: 10.1523/JNEUROSCI.3836-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumaru H, Kosaka T, Heizmann CW, Hama K. Immunocytochemical study of GABAergic neurons containing the calcium-binding protein parvalbumin in the rat hippocampus. Exp Brain Res. 1988;72:347–362. doi: 10.1007/BF00250256. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- Kosaka T. The axon initial segment as a synaptic site: ultrastructure and synaptology of the initial segment of the pyramidal cell in the rat hippocampus (CA3 region) J Neurocytol. 1980;9:861–882. doi: 10.1007/BF01205024. [DOI] [PubMed] [Google Scholar]
- Kuba H. Structural tuning and plasticity of the axon initial segment in auditory neurons. J Physiol. 2012;590:5571–5579. doi: 10.1113/jphysiol.2012.237305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Kwag J, Paulsen O. Bidirectional control of spike timing by GABA(A) receptor-mediated inhibition during θ oscillation in CA1 pyramidal neurons. Neuroreport. 2009;20:1209–1213. doi: 10.1097/WNR.0b013e32832f5cc7. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lörincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–909. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:848. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Massi L, Lagler M, Hartwich K, Borhegyi Z, Somogyi P, Klausberger T. Temporal dynamics of parvalbumin-expressing axo-axonic and basket cells in the rat medial prefrontal cortex in vivo. J Neurosci. 2012;32:16496–16502. doi: 10.1523/JNEUROSCI.3475-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ, Betette RL. Parvalbumin-containing neurons in the rat basolateral amygdala: morphology and co-localization of Calbindin-D(28k) Neuroscience. 2001;102:413–425. doi: 10.1016/S0306-4522(00)00481-4. [DOI] [PubMed] [Google Scholar]
- Meyer AH, Katona I, Blatow M, Rozov A, Monyer H. In vivo labeling of parvalbumin-positive interneurons and analysis of electrical coupling in identified neurons. J Neurosci. 2002;22:7055–7064. doi: 10.1523/JNEUROSCI.22-16-07055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Tóth K, Gulyás AI, Hájos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/S0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Molnár G, Oláh S, Komlósi G, Füle M, Szabadics J, Varga C, Barzó P, Tamás G. Complex events initiated by individual spikes in the human cerebral cortex. PLoS Biol. 2008;6:e222. doi: 10.1371/journal.pbio.0060222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ. Pyramidal cells of the rat basolateral amygdala: synaptology and innervation by parvalbumin-immunoreactive interneurons. J Comp Neurol. 2006;494:635–650. doi: 10.1002/cne.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Clark BA, Gründemann J, Roth A, Stuart GJ, Häusser M. Initiation of simple and complex spikes in cerebellar Purkinje cells. J Physiol. 2010;588:1709–1717. doi: 10.1113/jphysiol.2010.188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Paré D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Paré D, Driesang RB. Two types of intrinsic oscillations in neurons of the lateral and basolateral nuclei of the amygdala. J Neurophysiol. 1998;79:205–216. doi: 10.1152/jn.1998.79.1.205. [DOI] [PubMed] [Google Scholar]
- Park K, Lee S, Kang SJ, Choi S, Shin KS. Hyperpolarization-activated currents control the excitability of principal neurons in the basolateral amygdala. Biochem Biophys Res Commun. 2007;361:718–724. doi: 10.1016/j.bbrc.2007.07.064. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Smith Y, Paré JF, Paré D. Differential innervation of parvalbumin-immunoreactive interneurons of the basolateral amygdaloid complex by cortical and intrinsic inputs. J Comp Neurol. 2000;416:496–508. doi: 10.1002/(SICI)1096-9861(20000124)416:4<496::AID-CNE6>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Somogyi P. A specific ‘axo-axonal’ interneuron in the visual cortex of the rat. Brain Res. 1977;136:345–350. doi: 10.1016/0006-8993(77)90808-3. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Freund TF, Cowey A. The axo-axonic interneuron in the cerebral cortex of the rat, cat and monkey. Neuroscience. 1982;7:2577–2607. doi: 10.1016/0306-4522(82)90086-0. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Kisvárday ZF, Martin KA, Whitteridge D. Synaptic connections of morphologically identified and physiologically characterized large basket cells in the striate cortex of cat. Neuroscience. 1983a;10:261–294. doi: 10.1016/0306-4522(83)90133-1. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Smith AD, Nunzi MG, Gorio A, Takagi H, Wu JY. Glutamate decarboxylase immunoreactivity in the hippocampus of the cat: distribution of immunoreactive synaptic terminals with special reference to the axon initial segment of pyramidal neurons. J Neurosci. 1983b;3:1450–1468. doi: 10.1523/JNEUROSCI.03-07-01450.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Polepalli J, Sah P. Interneurons in the basolateral amygdala. Neuropharmacology. 2011;60:765–773. doi: 10.1016/j.neuropharm.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385:161–165. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Szentagothai J, Arbib MA. Conceptual models of neural organization. Neurosci Res Prog Bull. 1974;12:305–510. [PubMed] [Google Scholar]
- Tamás G, Szabadics J. Summation of unitary IPSPs elicited by identified axo-axonic interneurons. Cereb Cortex. 2004;14:823–826. doi: 10.1093/cercor/bhh051. [DOI] [PubMed] [Google Scholar]
- Tamás G, Szabadics J, Lörincz A, Somogyi P. Input and frequency-specific entrainment of postsynaptic firing by IPSPs of perisomatic or dendritic origin. Eur J Neurosci. 2004;20:2681–2690. doi: 10.1111/j.1460-9568.2004.03719.x. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- Viney TJ, Lasztoczi B, Katona L, Crump MG, Tukker JJ, Klausberger T, Somogyi P. Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat Neurosci. 2013;16:1802–1811. doi: 10.1038/nn.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90179-8. [DOI] [PubMed] [Google Scholar]
- Windels F, Crane JW, Sah P. Inhibition dominates the early phase of up-states in the basolateral amygdala. J Neurophysiol. 2010;104:3433–3438. doi: 10.1152/jn.00531.2010. [DOI] [PubMed] [Google Scholar]
- Woodruff AR, Sah P. Inhibition and synchronization of basal amygdala principal neuron spiking by parvalbumin-positive interneurons. J Neurophysiol. 2007;98:2956–2961. doi: 10.1152/jn.00739.2007. [DOI] [PubMed] [Google Scholar]
- Woodruff AR, Monyer H, Sah P. GABAergic excitation in the basolateral amygdala. J Neurosci. 2006;26:11881–11887. doi: 10.1523/JNEUROSCI.3389-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, McGarry LM, Vogels TP, Inan M, Anderson SA, Yuste R. State-dependent function of neocortical chandelier cells. J Neurosci. 2011;31:17872–17886. doi: 10.1523/JNEUROSCI.3894-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Stornetta RL, Zhu JJ. Chandelier cells control excessive cortical excitation: characteristics of whisker-evoked synaptic responses of layer 2/3 nonpyramidal and pyramidal neurons. J Neurosci. 2004;24:5101–5108. doi: 10.1523/JNEUROSCI.0544-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]