

Abstract
Presynaptic kainate-type glutamate receptors (KARs) regulate glutamate release probability and short-term plasticity in various areas of the brain. Here we show that long-term depression (LTD) in the area CA1 of neonatal rodent hippocampus is associated with an upregulation of tonic inhibitory KAR activity, which contributes to synaptic depression and causes a pronounced increase in short-term facilitation of transmission. This increased KAR function was mediated by high-affinity receptors and required activation of NMDA receptors, nitric oxide (NO) synthetase, and postsynaptic calcium signaling. In contrast, KAR activity was irreversibly downregulated in response to induction of long-term potentiation in a manner that depended on activation of the TrkB–receptor of BDNF. Both tonic KAR activity and its plasticity were restricted to early stages of synapse development and were lost in parallel with maturation of the network due to ongoing BDNF–TrkB signaling. These data show that presynaptic KARs are targets for activity-dependent modulation via diffusible messengers NO and BDNF, which enhance and depress tonic KAR activity at immature synapses, respectively. The plasticity of presynaptic KARs in the developing network allows nascent synapses to shape their response to incoming activity. In particular, upregulation of KAR function after LTD allows the synapse to preferentially pass high-frequency afferent activity. This can provide a potential rescue from synapse elimination by uncorrelated activity and also increase the computational dynamics of the developing CA3–CA1 circuitry.
Keywords: GluK1, LTD, short-term plasticity, synapse maturation
Introduction
Activity-dependent plasticity at the immature synapse is critical in determining its fate in the developing circuitry. Synchronous activity drives Hebbian strengthening of naive contacts, whereas asynchronous activity is thought to initiate a cascade leading to functional weakening and, eventually, morphological elimination. Accordingly, long-term depression (LTD) of transmission has been linked to synaptic pruning both in vitro (Bastrikova et al., 2008; Becker et al., 2008; Shinoda et al., 2010; Wiegert and Oertner 2013) and in vivo (Heynen et al., 2003). Interestingly, visualization of LTD-associated structural changes at hippocampal synapses suggests a substantial presynaptic contribution to this process (Bastrikova et al., 2008; Becker et al., 2008).
Although LTD-associated changes in presynaptic function are characterized at immature synapses (Bolshakov and Siegelbaum, 1994; Wasling et al., 2002; Pavlov et al., 2004), the underlying mechanisms have received less attention. Presynaptic changes are also tightly associated with induction of long-term potentiation (LTP) at immature glutamatergic synapses (Choi et al., 2000; Palmer et al., 2004; Lauri et al., 2006; Mohajerani et al., 2007; Luchkina et al., 2013). In particular, neonatal CA3–CA1 inputs demonstrating large facilitation of EPSCs in response to brief high-frequency stimulation are rapidly converted into low-facilitatory ones after LTP induction, indicative of an increase in the probability of glutamate release (Pr). This activity-dependent regulation of presynaptic efficacy is induced postsynaptically via NMDA receptor-mediated postsynaptic calcium influx (Luchkina et al., 2013). The presynaptic expression depends on BDNF–TrkB receptor signaling and involves the loss of activity of presynaptic inhibitory kainate receptors (KARs) containing the GluK1c subunit (Lauri et al., 2006; Sallert et al., 2009; Vesikansa et al., 2012).
Given the key role of presynaptic KARs in neonatal LTP, it is plausible that they also contribute to LTD at immature synapses. Indeed, several papers have described LTD of KAR-mediated postsynaptic responses (Park et al., 2006; Selak et al., 2009; Carta et al., 2013; Chamberlain et al., 2013), implying that activity-dependent regulation of KARs is a physiological mechanism involved in LTD.
Here we show that LTD induction by 900 pulse/5 Hz stimulation at naive, low-facilitatory CA3–CA1 synapses caused a pronounced increase in short-term dynamics of transmission because of upregulation of presynaptic inhibitory KAR activity. This mechanism was restricted to immature synapses; the tonic KAR activity could no longer be induced after LTP induction at immature synapses or on slices from 2-week-old animals. However, suppressing maturation of the network by chronic postnatal inhibition of BDNF–TrkB signaling maintained tonic presynaptic KAR activity at a subpopulation of synapses and the ability to induce it in response to LTD in others. Finally, we show that the LTD-associated upregulation of KAR activity was attributable to engagement of high-affinity receptors and required activation of the nitric oxide synthetase (NOS).
These data show that presynaptic KAR activity is a key indicator of presynaptic immaturity that not only determines short-term dynamics of transmission but also contributes to long-term plasticity critical for developmental fine-tuning of synaptic connectivity.
Materials and Methods
Experiments were performed using neonatal [postnatal day 3 (P3) to P6] or juvenile (P13–P15) Wistar rats or TrkBF616A knock-in mice (kindly provided by Dr. David Ginty, John Hopkins University, Baltimore, MD; Chen et al., 2005) of either sex, in accordance with the guidelines given by the Ethics Committee for Animal Research at the University of Helsinki. In TrkBF616A mice, phenylalanine 616 located within the ATP binding site is mutated to alanine, which renders the TrkB receptor susceptible to inhibition by low concentrations of 4-amino-1-tert-butyl-3-(1′-naphthylmethyl)pyrazolo[3,4-d]pyrimidine (NMPP1), a cell-permeable PP1 analog. This was applied chronically using the slow-release Elvax matrix.
NMPP1 was synthetized as described previously (Sallert et al., 2009). Elvax was prepared by dissolving Elvax beads (DuPont) in 1 ml of dichloromethane (DCM; 10% w/v) at 25°C for ∼15–20 min. NMPP1 (0.5 m; 10 μl) or the equivalent volume of vehicle alone (DMSO) was added, and the resultant mixture was transferred to an ethanol/dry ice bath. The mixture was left to polymerize for ∼1 h and then kept at −20°C overnight to allow DCM to evaporate, resulting in a final concentration of 25 mm NMPP1 in the Elvax. Sections of 200 μm Elvax were cut and stored before use at −20°C. Elvax blocks containing either NMPP1 or DMSO were implanted subcutaneously to TrkBF616A P4–P5 mouse pups under isoflurane anesthesia.
Parasagittal hippocampal slices (400 μm thick) were cut in ice-cold artificial CSF (aCSF) comprising the following (in mm): 124 NaCl, 10 d-glucose, 26 NaHCO3, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgSO4 (saturated with 95% O2 and 5% CO2). The CA3 area was removed surgically. Slices were left to recover for a period of at least 40 min and then transferred to a submerged recording chamber. All the recordings were done at 30–32°C with a constant perfusion with aCSF at a rate of 1.8–2.2 ml/min.
Whole-cell recordings were made from visually identified (infrared differential interference contrast optics) CA1 pyramidal neurons. Patch electrodes (2–5 MΩ) contained the following (in mm): 130 CsMeSO4, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 QX-314, and 8 NaCl (275 ± 5 mOsm), pH 7.2. For experiments requiring the induction of LTP, perforated whole-cell recordings were made using higher-resistance electrodes (5–7 MΩ) containing the following: 130 mm CsMeSO4, 10 mm HEPES, 0.5 mm EGTA, 5 mm QX-314, 8 mm NaCl, 275 ± 5 mOsm, pH 7.2, and 200 μg/ml amphotericin B. Series resistance (Rs), membrane resistance (Rm), and holding current (direct current) were monitored by measuring the peak amplitude of the fast whole-cell capacitance current in response to a 5 mV step. The amplitude was estimated by fitting a double exponential to the capacitance transient and determining the current at the beginning of the step. Only experiments in which Rs < 30 MΩ (whole-cell; baseline Rs = 14.9 ± 0.4 MΩ, Rm = 835 ± 49 MΩ; n = 222) or Rs < 60 MΩ (perforated whole-cell; baseline Rs = 42.8 ± 1.8 MΩ, Rm = 1747 ± 246 MΩ; n = 36) and with <20% change in Rs and Rm during the course of the experiment were included for analysis. EPSCs were recorded at a holding membrane potential of −70 mV in the presence of picrotoxin (PTX; 100 μm) to antagonize fast GABAA synaptic transmission. Synaptic responses were evoked at low frequency (i.e., every 15 s for single pulses, every 60 s for 5 pulse trains at 50 Hz) by a bipolar stimulating electrode (nickel–chromium wire) positioned within the Schaffer collateral–commissural pathway. LTD was induced by repeated stimulation (900 pulses at 5 Hz) delivered at a holding potential of −40 mV. LTP was induced by pairing postsynaptic depolarization to a holding potential of −10 mV with 10 high-frequency trains of afferent stimulation comprising 5 pulses at 50 Hz delivered at 5 s intervals. Spontaneous miniature EPSCs (mEPSCs) were recorded in the additional presence of tetrodotoxin (1 μm). Data were collected using LTP software (Anderson and Collingridge, 2007; www.winltp.com) and mEPSC data analyzed using MiniAnalysis (Synaptosoft). The following drugs were used: d-AP5(d-(-)-2-amino-5-phosphonopentanoate), AM251 1-(2,4-Dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide (AM251), (S)-3,5-dihydroxyphenylglycine (DHPG), and picrotoxin (PTX) (Abcam); (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxy-5-phenylthiophene-3-yl-methyl)-5-methylpyrimidine-2,4-dione (ACET), cyclotraxin-B (CTX-B), L-689,560 (trans-2-carboxy-5,7-dichloro-4-phenylaminocarbonylamino-1,2,3,4-tetrahydroquinoline), and kainate (KA) (Tocris Bioscience); and 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA), N-ω-nitro-l-arginine methyl ester (l-NAME), and glutamate-pyruvate transaminase (Sigma-Aldrich). All membrane potentials were corrected for the calculated liquid junction potential.
All pooled data are expressed as mean ± SEM and normalized to the preceding baseline as shown in the graphs (i.e., 5 min for LTD, 10 min for the LTP experiments). In the text, the effects are expressed as percentage change compared with the level before drug application or LTP/LTD induction (i.e., 0% = no change). Facilitation ratios were measured from averages of at least five responses to 5 pulse/50 Hz stimulation relative to the amplitude of the first evoked response. To reduce the effect of postsynaptic summation of the currents during high-frequency bursts, an averaged response to low-frequency stimulation (700 ms) was scaled to the peak of the first recorded response in the train and subtracted from the original trace. This process was then repeated sequentially for the four first postsynaptic currents in the train. The amplitude of the fifth EPSC was calculated as the peak amplitude relative to the baseline before the first EPSC from the subtracted waveform. Statistical significance was tested routinely using mixed repeated-measures ANOVA. When a significant interaction between the groups were detected, post hoc comparisons were done using a Holm–Sidak correction (α = 0.05). Paired two-tailed Student's t tests and Pearson's correlation test was used when indicated. Post hoc results are given as a precise p value, except when p is very small (p < 0.001). The detailed information on the statistical tests (including the results of the initial ANOVA, degrees of freedom, and critical levels of significance (CL)) is given in Table 1.
Table 1.
The detailed information on the statistical tests used, including the results of the initial ANOVA, degrees of freedom, and critical levels of significance

BL, Baseline; N.A., not applicable. *Greenhouse–Geisser correction.
Results
LTD induced by 900 pulse/5 Hz stimulation is associated with an increase in tonic KAR activity at immature low-facilitatory synapses
During the first postnatal week (P3–P7), the glutamatergic synapses in area CA1 are highly heterogeneous in their presynaptic properties, as evidenced by large variability in short-term dynamics of transmission (Hanse and Gustafsson, 2001; Lauri et al., 2006). Based on this heterogeneity, synapses can be divided into subpopulations with facilitatory or low-facilitatory responses to brief high-frequency stimulation, indicative of low and high probability of glutamate release (Pr), respectively. The facilitatory inputs express GluK1 subunit-containing presynaptic KARs that are tonically activated by ambient glutamate to decrease Pr in a developmentally restricted manner (Lauri et al., 2006).
To study the synaptic mechanisms of LTD at immature inputs, whole-cell voltage-clamp recordings were made from CA1 pyramidal neurons in acute hippocampal slices from 3- to 6-d-old rat pups. Excitatory postsynaptic responses were evoked by stimulation of Schaffer collaterals in the presence of 100 μm PTX to block fast GABAergic transmission. LTD was induced by repeated stimulation (900 pulses/5 Hz at −40 mV), which reliably produced long-lasting depression of transmission at this developmental stage. Interestingly, the level of LTD was dependent on the initial facilitation type. Depression was significantly larger at synapses that were initially low facilitating (LF) in response to 5 pulse/50 Hz stimulation (LF; fifth/first EPSC amplitude <1.5; −53 ± 4%; n = 31; p < 0.001) compared with synapses that were initially high facilitating (HF; fifth/first EPSC amplitude >1.5; −29 ± 8%; n = 19; p = 0.004; comparing between the groups, p = 0.002; Fig. 1A). At individual inputs, the initial level of facilitation correlated with the level of synaptic depression (Pearson's r = 0.406; n = 50; p = 0.0035; Fig. 1Aiii). Furthermore, LTD was associated with an increase in the fifth/first facilitation ratio in LF responses (149 ± 31%; p < 0.001) but not in HF inputs (25 ± 11%; p = 0.404; Fig. 1Aii). In summary, these data suggest that, at immature synapses with an initially high Pr, the expression of LTD in response to 900 pulse/5 Hz stimulation involves a presynaptic component, i.e., a decrease in Pr. This manifests as a shift in the short-term dynamics of excitatory synaptic transmission from low to high facilitation.
Figure 1.

LTD at immature low-facilitatory (LF) CA3–CA1 synapses involves a presynaptic component that is attributable to an increase in the tonic inhibitory activity of KARs. A, LF neonatal inputs express larger LTD than HF inputs, with an associated increase in synaptic facilitation. i, A time course plot illustrates induction of LTD in response to 900 pulses/5 Hz stimulation in LF (black symbols; n = 31) and HF (white symbols; n = 19) neonatal inputs. Example traces on top show the synaptic response to 5 pulses/50 Hz stimulation at the time points indicated. Scaled and superimposed traces are shown on the right. ii, Pooled data on the level of LTD (left) and facilitation ratio (5th/1st EPSC amplitude to 50 Hz stimulation) before and after LTD induction in LF and HF inputs (black and white symbols, respectively). iii, The level of synaptic depression in response to the LTD paradigm correlates with the initial level of facilitation to 50 Hz stimulation (n = 50). B, Expression of LTD at LF inputs involves a presynaptic KAR-dependent component. Effect of the selective KAR antagonist ACET on EPSC amplitude and facilitation ratio after LTD induction at LF inputs (n = 10). C, Corresponding data on the effect of ACET at HF inputs (n = 7). D, Lack of effect of ACET on EPSC amplitude and synaptic facilitation after LTD induction at P14 (n = 10). The data in B–D are presented as in Ai and Aii. All data illustrate the average ± SEM. Most details on the statistical data are presented in Table 1. Significance is indicated by *. For Aii, EPSC amplitude, LF vs HF, p = 0.002 (CL = 0.05, significant); fifth/first ratio, LF vs HF in control, p < 0.001 (CL = 0.05, significant); post-LTD, p = 0.162 (CL = 0.05, NS).
To study whether the activity of KAR is involved, we applied ACET, a compound that demonstrates good selectivity for antagonism of GluK1 subunit-containing KARs (Dargan et al., 2009). Under baseline conditions, antagonism of these receptors leads to an increase in Pr, observed as an increase in EPSC amplitude and decrease in synaptic facilitation selectively at HF inputs (Lauri et al., 2006). After LTD induction, a subsequent ACET application (200 nm, 20 min) caused a significant increase in EPSC amplitude in both LF and HF inputs (LF, 98 ± 27%, n = 10, p = 0.002; HF, 95 ± 20%, n = 7, p < 0.001). In the LF synapse population, the ACET-sensitive component comprised 58 ± 17% (n = 10) of LTD, suggesting that the acquired KAR-mediated inhibition could fully account for the larger depression in these synapses. Furthermore, ACET application caused a significant reduction in facilitation in both types of inputs (LF, −42 ± 6%, p < 0.001, HF, −30 ± 4%, p = 0.002; Fig. 1B,C). Thus, at LF synapses, LTD-inducing 900 pulses/5 Hz stimulation confers sensitivity to KAR antagonism; at HF inputs, this sensitivity is maintained.
Tonic KAR activity at naive CA3–CA1 synapses is downregulated in parallel with maturation of synaptic circuitry and no longer observed at P14 (Lauri et al., 2006). Therefore, we examined the effect of ACET after the induction of LTD in juvenile P14 rats. Repeated stimulation (900 pulses/5 Hz at −40 mV) produced a depression (−48 ± 6%; n = 10; p < 0.001) that was accompanied by a small but significant increase in synaptic facilitation to 50 Hz stimulation (56 ± 20%; p = 0.010; Fig. 1D). However, in contrast to immature synapses, application of ACET after LTD had no effect on EPSC amplitude (−9 ± 7%; p = 0.611) or synaptic facilitation (15 ± 18%; p = 0.693; Fig. 1D). This finding indicates that the LTD-associated increase in KAR activity is developmentally restricted to immature CA1 synapses.
The KAR activity acquired after LTD is attributable to high-affinity receptors activated by ambient extracellular glutamate
Previously, we showed that the tonic KAR activity at HF inputs is attributable to activation of high-affinity receptors by ambient glutamate. The LF inputs express presynaptic KARs that can be activated via selective agonists at low potency but are insensitive to 50 nm KA that selectively acts on high-affinity receptors (Lauri et al., 2006). We next asked whether the KAR activity acquired after LTD at the LF inputs involved high- or low-affinity receptors. To this end, an enzymatic glutamate scavenger (5 U/ml glutamate-pyruvate transaminase plus 1 mm pyruvate) was applied after LTD-inducing 900 pulses/5 Hz stimulation to remove tonic activation of the receptors, after which the sensitivity to a low concentration (50 nm) of KA was tested. Glutamate scavenger produced a significant increase in the EPSC amplitude (87 ± 17%; n = 11; p < 0.001) with an associated decrease in synaptic facilitation (−43 ± 9%; p = 0.003; Fig. 2A). Subsequent application of 50 nm KA caused a significant reduction in the EPSC amplitude (−39 ± 4%; p < 0.001), with a concomitant increase in synaptic facilitation (101 ± 24%; p = 0.001; Fig. 2A). These data suggest that LTD induction at the LF inputs engages high-affinity KARs that can be tonically activated by endogenous glutamate.
Figure 2.

A, The KAR activity acquired after LTD induction is attributable to high-affinity KARs that are tonically activated by ambient glutamate. i, A time course plot depicting the effect of a glutamate scavenger and 50 nm KA on EPSC amplitude after LTD induction in LF inputs (n = 11). The traces illustrate the synaptic response to 5 pulses/50 Hz stimulation during the baseline period (1), after LTD induction (2), after enzymatic removal of ambient glutamate (3), and after selective activation of high-affinity KARs by application of 50 nm KA (4). Scaled traces are superimposed on the right. ii, Averaged data on the effects of glutamate scavenger and subsequent 50 nm KA on EPSC amplitude (left) and facilitation ratio (5th/1st EPSC amplitude to 50 Hz stimulation). B, Activation of group 1 mGluRs does not induce persistent depression of EPSC amplitude at neonatal inputs. i, A time course plot depicting the effect of DHPG on EPSC amplitude at LF (n = 11, black symbols) and HF (n = 6, white symbols) neonatal inputs. Traces on the top show the response to 5 pulses/50 Hz stimulation at the time points indicated, and scaled and superimposed traces are shown on the right. ii, Averaged data on the effect of DHPG on EPSC amplitude (left) and facilitation ratio (5th/1st EPSC amplitude to 50 Hz stimulation) in LF and HF inputs (black and white symbols, respectively). All data illustrate average ± SEM. Significance is indicated by *. The details on the statistical data are presented in the Table 1.
LTD can be induced and expressed via a variety of mechanisms, which are developmentally regulated (Collingridge et al., 2010). Activation of group 1 mGluRs by DHPG is used routinely to induce chemical LTD, which involves presynaptic changes (Fitzjohn et al., 2001; Nosyreva and Huber, 2005). Thus, we were interested to test whether presynaptic KARs also contributed to this form of LTD. Surprisingly, at this developmental stage (P4–P6), DHPG (50 μm, 10 min) only caused a transient depression of EPSC amplitude, whereas no lasting effects on EPSC amplitude were observed after washout of the drug (LF, −2 ± 12%, n = 11; HF, −3 ± 13%, n = 6; p = 0.655). Also, no significant changes in facilitation were observed in either type of input (LF, 25 ± 13%; HF, 10 ± 11%; p = 0.103; Fig. 2B), indicating that chemical LTD could not be induced at immature synapses under our recording conditions.
The ability to upregulate KAR activity is lost after LTP induction at immature synapses in a TrkB-dependent manner
Tonic KAR activity at facilitatory inputs is lost during induction of LTP, suggesting that LTP initiates a signaling cascade leading to maturation of presynaptic properties (Lauri et al., 2006). To test whether the loss of KAR activity after LTP was reversible, we analyzed the sensitivity of transmission to KAR antagonism after depotentiation of LTP. These experiments were done using the perforated patch-clamp configuration to prevent intracellular dialysis affecting LTP expression.
As shown previously (Lauri et al., 2006), a pairing protocol resulted in a robust potentiation of EPSC amplitude that was dependent on the initial facilitation ratio. The magnitude of LTP at HF synapses (182 ± 31%; n = 9; p < 0.001) was significantly larger compared with LF inputs (57 ± 9%; n = 14; p < 0.001; comparing between the groups, p < 0.001; CL = 0.013, significant; Fig. 3A,B). As observed previously (Lauri et al., 2006), the induction of LTP was associated with a decrease in facilitation ratio at HF inputs only (LF, −8 ± 7%, p = 0.793; HF, −38 ± 9%, p < 0.001). Our previous study indicates that this is consistent with a loss of tonic KAR activity and an increase in Pr at HF inputs (Lauri et al., 2006).
Figure 3.

LTP induction at immature synapses leads to irreversible loss of tonic KAR activity in a TrkB-dependent manner. A, Previous induction of LTP abolishes upregulation of KAR activity in response to LTD-inducing stimuli at LF inputs. A time course plot illustrating pairing-induced LTP, followed by depotentiation (depot) in response to 900 pulses/5 Hz stimulation, recorded using perforated patch-clamp technique (n = 14). Example traces show the response to 5 pulses/50 Hz stimulation at the points indicated in the time course plot (i). Pooled data on the EPSC amplitude and facilitation ratio at corresponding times are shown on the right (ii). B, Corresponding data for HF neonatal inputs (n = 9). C, The loss of KAR activity depends on TrkB signaling during LTP induction. An experiment similar to that shown in A, except that LTP is induced in the presence of CTX-B, a specific inhibitor of the TrkB receptor. A time course plot (i) and example traces and pooled data (ii) for LF inputs (n = 7) are shown as in A. D, Corresponding data as in C for HF inputs (n = 6). All data illustrate average ± SEM. Significance is indicated by *. The detailed statistical data are presented in Table 1.
The same protocol used previously to induce LTD was delivered 20 min after LTP induction and resulted in significant depotentiation at both LF and HF inputs (percentage recovery at LF, 117 ± 14%, n = 14, p < 0.001; HF, 62 ± 11%, n = 9, p < 0.001). However, the magnitude of this recovery was significantly different between the two types of inputs, and some LTP remained in the HF inputs (percentage change relative to baseline; LF, −4 ± 6%, p = 0.877; HF, 70 ± 23%; between the groups, p < 0.001; CL = 0.017, significant). In contrast to LTD at the naive synapses, depotentiation was not associated with significant changes in synaptic facilitation in either type of input (LF, 26 ± 14%, p = 0.121; HF, 18 ± 18%, p = 0.236; Fig. 3A,B). These data imply that presynaptic mechanisms involving KARs are not contributing to synaptic depression after depotentiation. Furthermore, the remaining LTP at initially HF synapses might represent the presynaptic component of LTP (Luchkina et al., 2013).
Application of ACET after depotentiation had no effect on low-frequency transmission (LF, 8 ± 9%, p = 0.606; HF, 17 ± 7%, p = 0.246) or on synaptic facilitation at 50 Hz (LF, 15 ± 11%, p = 0.967; HF, −2 ± 10%, p = 0.960) in either LF or HF inputs (Fig. 3A,B). These data suggest that LTP induction leads to an irreversible loss of tonic KAR activity and converts immature synapses into ones with adult-like presynaptic properties.
The loss of tonic KAR activity during LTP induction is dependent on BDNF and activation of its TrkB receptor (Sallert et al., 2009). We next asked whether this signaling system is also responsible for the irreversibility of this effect. To this end, we used a novel TrkB-selective antagonist, CTX-B (Cazorla et al., 2010), to block TrkB signaling during LTP induction. In the presence of CTX-B (1 μm, 50–90 min, i.e., started during perforation), the level of LTP was not different at LF and HF inputs (LF, 56 ± 20%, n = 7, p = 0.002; HF, 64 ± 17%, n = 6, p = 0.001; comparing between the groups, p = 0.705; CL = 0.025, NS). This was attributable to a lower level of LTP at HF inputs in the presence of CTX-B when compared with control (p < 0.001; CL = 0.017, significant), whereas the LTP level at LF inputs was unaffected (p = 0.935; CL = 0.05, NS). Furthermore, LTP induced in the presence of CTX-B was not associated with a significant change in synaptic facilitation in either HF or LF inputs (LF, 12 ± 18%, p = 0.546; HF, 3 ± 11%, p = 0.840; Fig. 3C,D). These data confirm our previous observation that TrkB signaling is required for presynaptic LTP (Sallert et al., 2009).
CTX-B during LTP induction had no effect on the ability to depotentiate LTP (percentage recovery; LF, 181 ± 15%, p < 0.001; HF, 103 ± 20%, p = 0.002). However, at the LF inputs, depotentiation was associated with a significant increase in synaptic facilitation (LF, 71 ± 20%, p = 0.002; HF, −11 ± 7%, p = 0.181), reminiscent of that seen after LTD (i.e., without previous LTP). Moreover, application of ACET increased EPSC amplitude (LF, 84 ± 13%, p = 0.02; HF, 77 ± 26%, p < 0.001) and caused a decrease in synaptic facilitation (LF, −46 ± 7%, p < 0.001; HF, −44 ± 6%, p < 0.001) at both LF and HF inputs. These results suggest that BDNF–TrkB signaling during LTP induction is responsible not only for the loss of tonic KAR activity at HF inputs but also for the loss of the ability to reengage this activity at LF inputs (Fig. 3C,D).
Immature-type presynaptic activity and plasticity of KARs is developmentally maintained by chronic chemical–genetic block of TrkB signaling
The present and previous (Sallert et al., 2009) data indicate that TrkB signaling is critical in converting the immature-type presynaptic KARs into ones with adult-like properties. Thus, we next tested whether chronic inhibition of this signaling system during postnatal development affected the loss of the immature-type KAR activity. To this end, we used a mouse model, TrkBF616A, in which a single point mutation in the TrkB receptor renders it highly susceptible to inhibition by NMPP1 (Chen et al., 2005). NMPP1 was administered chronically in vivo using Elvax polymer that was implanted subcutaneously on TrkBF616A mice at P4–P5. Control mice obtained an implant contained vehicle (DMSO) only.
Tonic KAR activity manifesting as an increase in the frequency of spontaneous action potential-independent glutamatergic events (mEPSCs) after GluK1 antagonism is no longer observed at P14–P16 (Lauri et al., 2006; Sallert et al., 2009). Consistently, ACET application had no effect on mEPSC frequency, recorded from CA1 pyramidal neurons in slices from 2-week-old DMSO-implanted TrkBF616A mice (−7 ± 4%; n = 9; p = 0.309). However, in slices from TrkBF616A mice implanted with NMPP1-containing Elvax, ACET caused a reversible increase in mEPSC frequency (30 ± 4%; n = 8; p = 0.001; Fig. 4A). ACET had no significant effect on mEPSC amplitude in either group (−8 ± 6%, p = 0.129 and −6 ± 4%, p = 0.067 for DMSO and NMPP1, respectively, Student's t test). Also, no differences were observed in baseline mEPSC frequency (p = 0.665), amplitude (p = 0.392), or kinetics (p = 0.445) between control and NMPP1-treated animals (Student's t test).
Figure 4.

Tonic activity and plasticity of presynaptic KARs is developmentally maintained by chronic postnatal inhibition of BDNF–TrkB signaling. A, Recording of mEPSCs from CA1 pyramidal neurons in slices from 2-week-old TrkBF616A mice, implanted with NMPP1 or DMSO containing Elvax at P4–P5. KAR antagonism by ACET has no effect on mEPSCs in control (DMSO-implanted) mice (n = 9; black). However, chronic block of TrkB in NMPP1-implanted animals preserves tonic KAR activity, observed as an increase in mEPSC frequency in response to ACET (n = 8, gray). Example traces from points indicated in the time course plot are shown in i. Pooled data on the effect of ACET on mEPSC frequency in DMSO- and NMPP1-implanted TrkBF616A mice are shown on the right (ii). B, Induction of immature-type presynaptic LTD at LF inputs on NMPP1-implanted TrkBF616A mice (P14–P16). i, A time course plot depicting robust LTD in response to 900 pulses/5 Hz stimulation, associated with an increase in synaptic facilitation and ACET sensitivity. Example traces from the points indicated are shown on top. Scaled and superimposed traces are shown on the right. ii, Averaged data on the effect of 900 pulses/5 Hz stimulation and subsequent ACET application on EPSC amplitude (left) and facilitation ratio (5th/1st EPSC amplitude to 50 Hz stimulation) (n = 8). C, Corresponding data on HF inputs at NMPP1-implanted TrkBF616A mice (P14–P16; n = 10). D, Corresponding data on control (DMSO-implanted) TrkBF616A mice (P14–P16; n = 10). All data illustrate average ± SEM. Any statistical significance is indicated by *. The detailed statistical data are presented in Table 1.
Having established that tonic KAR activity is maintained during chronic developmental inhibition of TrkB activation, we went on to study its effect on short- and long-term plastic properties of transmission. Reminiscent of immature circuitry, the synaptic response to 5 pulses/50 Hz stimulation was highly heterogeneous in slices from the NMPP1-implanted TrkBF616A mice [fifth/first ratio coefficient of variations of 0.157 (n = 9) and 0.517 (n = 18) for control vs NMPP1 at P14], and thus the inputs were divided into two groups: low and high facilitatory (NMPP1-LF and NMPP1-HF). Repeated stimulation at the NMPP1-LF inputs induced LTD that was significantly larger when compared with NMPP1-HF inputs (NMPP1-LF, −63 ± 6%, n = 8, p < 0.001; NMPP1-HF, −35 ± 10% n = 10, p < 0.001; NMPP1-LF vs NMPP1-HF, p = 0.008; Fig. 4B,C). Similar to the immature inputs, this LTD was associated with an increase in facilitation that was significant at NMPP1-LF synapses only (NMPP1-LF, 82 ± 4%, p < 0.001; NMPP1-HF, 18 ± 15%, p = 0.211). Finally, a subsequent ACET application caused a significant increase in EPSC amplitude (NMPP1-LF, 102 ± 28%, p < 0.001; NMPP1-HF, 92 ± 32%, p < 0.001) with a concomitant reduction in facilitation in both types of inputs (NMPP1-LF, −33 ± 6%, p = 0.012; NMPP1-HF, −39 ± 5%, p < 0.001; Fig. 4B,C).
In DMSO-implanted animals, LTD (−46 ± 2%; n = 10; p < 0.001) and the associated change in synaptic facilitation (27 ± 6%; p = 0.007; Fig. 4D) were not different from the wild-type rats of the same age (mice vs rats, p = 0.743 and p = 0.177 for LTD and fifth/first ratio, respectively, Student's t test). Furthermore, ACET application after LTD induction had no effect on transmission (−13 ± 7%; p = 0.171) or facilitation ratio (7 ± 10%; p = 0.393) in these slices. These data indicate that presynaptic KAR activity is tightly linked to immature-type presynaptic properties at the glutamatergic synapses, manifesting as heterogeneity in short-term dynamics of transmission that correlates with the magnitude of LTD. Furthermore, inhibition of TrkB signaling during development is sufficient to prolong the developmental window not only for the tonic activity of the KARs but also for the mechanism responsible for their activity-dependent regulation.
The upregulation of presynaptic KAR activity depends on activation of NMDA receptors and postsynaptic calcium signaling
Both NMDA receptor-dependent and -independent forms of LTD have been observed at developing CA3–CA1 synapses (Dudek and Bear, 1992, 1993; Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Pavlov et al., 2004). In our recording conditions, NMDA receptor antagonism was sufficient to fully block LTD and any associated changes in synapse dynamics. Thus, in the presence of d-AP5 (50 μm) and L-689,560 (5 μm) to provide competitive and noncompetitive block of NMDA receptors, respectively, neither LF nor HF inputs showed any significant depression in response to 900 pulses/5 Hz stimulation (LF, 0 ± 10%, n = 8, p = 0.810; HF, −11 ± 20%, n = 5, p = 0.470) or concomitant changes in facilitation ratio (−5 ± 12%, p = 0.650 and −5 ± 12%, p = 0.331, respectively). After LTD, ACET had no effect on transmission at LF inputs (EPSC amplitude, −5 ± 9%, n = 8, p = 0.574; facilitation, 25 ± 16%, n = 8, p = 0.465), whereas significant effects were observed at HF synapses (EPSC amplitude, 118 ± 37%, n = 5, p < 0.001; facilitation, −32 ± 8%, n = 5, p < 0.001; Fig. 5A; HF not shown). Thus, NMDA receptors are required for the induction of this form of LTD at immature synapses and for the concomitant presynaptic change in Pr and KAR activity at LF synapses.
Figure 5.
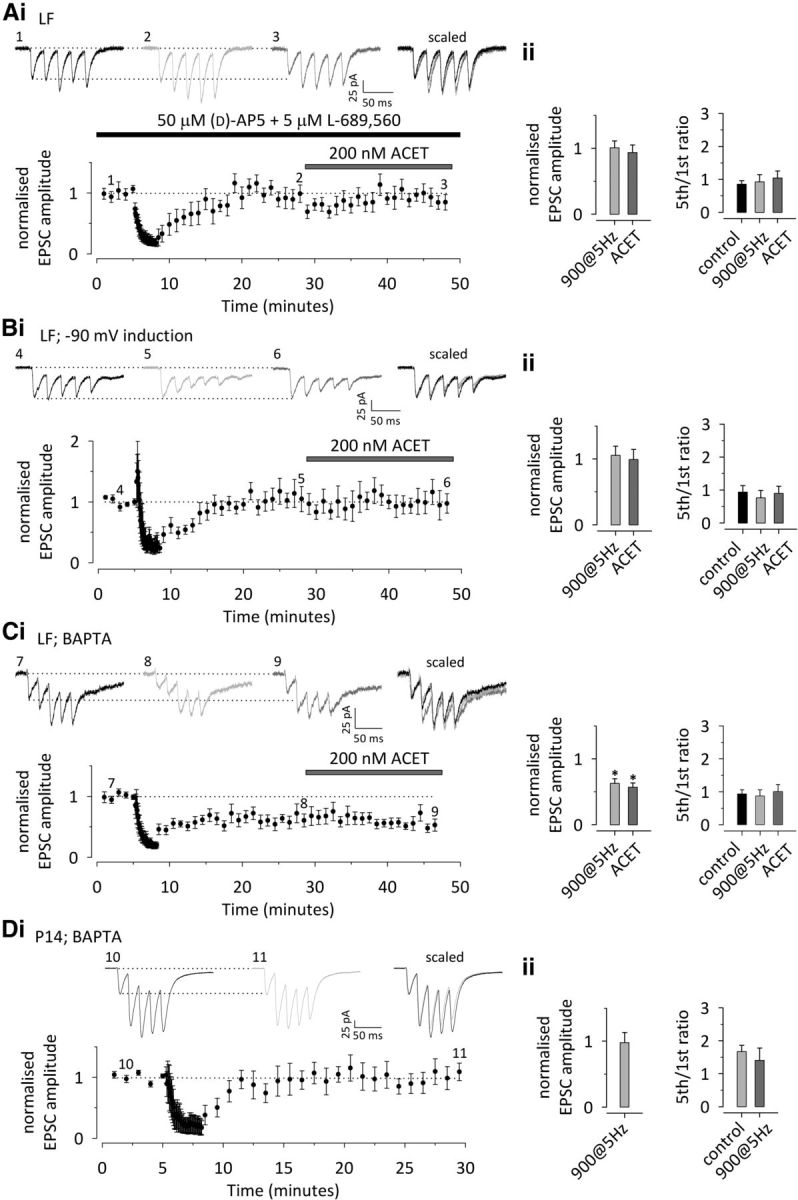
Upregulation of presynaptic KAR function after LTD requires NMDA receptor activation, postsynaptic depolarization, and calcium influx. A, LTD induced by 900 pulses/5 Hz stimulation and the associated presynaptic changes at LF neonatal inputs are completely blocked in the presence of the NMDA receptor antagonists d-AP-5 (50 μm) and L-689,560 (5 μm). i, A time course plot and example traces from the points indicated, with scaled and superimposed traces shown on the right (n = 8). ii, Pooled data on the effect of 900 pulses/5 Hz stimulation and subsequent ACET application on the EPSC amplitude and the facilitation ratio (5th/1st EPSC amplitude at 50 Hz) in the presence of NMDA receptor antagonists. B, LTD is fully blocked if the postsynaptic neuron is hyperpolarized during the induction protocol. Data from LF neonatal inputs (n = 6) is presented as in A. C, Postsynaptic BAPTA prevents the LTD-associated changes in presynaptic function and KAR activity, without blocking the LTD per se. Data from LF inputs (n = 8) are presented as in D. Postsynaptic BAPTA fully blocks LTD at P14 (n = 7). Data are presented as in A. All data illustrate average ± SEM. Statistical significance is indicated by *. The detailed statistical data are presented in Table 1.
NMDA receptors might induce plasticity via presynaptic or postsynaptic mechanisms and via ionotropic or metabotropic action. To distinguish which of these mechanisms triggers the presynaptic component of neonatal LTD, we first hyperpolarized (−90 mV) the postsynaptic neuron during the LTD-inducing 5 Hz stimulation to selectively prevent activation of postsynaptic NMDA receptors. This treatment fully blocked induction of LTD (LF, 5 ± 14%, n = 7, p = 0.535; HF, −8 ± 9%, n = 6, p = 0.555) and the associated change in synaptic facilitation (LF, −20 ± 12%, p = 0.298; HF, −7 ± 5%, p = 0.323). Subsequent application of ACET had no effect on transmission at LF inputs (EPSC amplitude, −5 ± 7%, p = 0.584; facilitation, 17 ± 11%, p = 0.416), whereas significant effects were observed at HF synapses (EPSC amplitude, 51 ± 9%, p = 0.004; facilitation, −24 ± 6%, p = 0.006; Fig. 5B; HF not shown). Together, these data confirm a critical role for postsynaptic NMDA receptors in LTD.
Next, we included 10 mm BAPTA in the patch electrode to prevent postsynaptic calcium signaling. Surprisingly, postsynaptic BAPTA did not block induction of LTD per se: at LF neonatal inputs, there was significant depression of EPSC amplitude in response to 900 pulses/5 Hz stimulation in the presence of BAPTA (−38 ± 7%; n = 8; p < 0.001; Fig. 5C). However, there was no associated increase in the level of facilitation (−1 ± 17%; p = 0.866), suggesting that BAPTA specifically inhibited the presynaptic component of LTD. Furthermore, a subsequent application of ACET had no effect on either EPSC amplitude (−7 ± 3%; p = 0.162) or facilitation (11 ± 7%; p = 0.866; Fig. 5C). This suggests that postsynaptic calcium signaling is apparently not required for postsynaptic mechanisms of depression at immature LF synapses but is necessary for the presynaptic component of LTD and upregulation of KAR activity.
The previous result is surprising given that postsynaptic BAPTA blocks the induction of LTD in older rats. Therefore, we examined the effects of BAPTA on LTD induction in P14 rats (Fig. 5D). Under these conditions, no LTD was observed (−2 ± 15%; n = 7; p = 0.984), with no change in facilitation ratio (−18 ± 18%; p = 0.380).
Upregulation of presynaptic KAR activity depends on NO signaling
Retrograde endocannabinoid signaling via CB1 receptors has been implicated in the induction of LTD at immature glutamatergic synapses in the hippocampus (Yasuda et al., 2008; Caiati et al., 2012; Péterfi et al., 2012) and also shown to interact with GluK1 KAR signaling (Lourenço et al., 2010). Therefore, we studied whether the regulation of presynaptic KAR activity in response to LTD-inducing 900 pulses/5 Hz stimulation involved CB1 receptors. The selective CB1 receptor antagonist/inverse agonist AM251 (3 μm) had no effect on the induction of LTD at LF inputs (LF, −58 ± 9%, n = 9, p < 0.001; Fig. 6A). As seen under control conditions, LTD was associated with a significant increase in facilitation ratio (LF, 72 ± 9%, p = 0.010). Furthermore, application of ACET after LTD induction significantly increased EPSC amplitude (LF, 91 ± 32%, p = 0.031) and decreased facilitation (LF, −43 ± 13%, p = 0.021; Fig. 6A), in a manner similar to control. This indicates that CB1 signaling was not involved in this form of LTD or the associated upregulation of KAR activity.
Figure 6.

LTD-associated upregulation of presynaptic KAR function is not affected by CB1 receptor antagonism but requires activity of NOS. A, The CB1 cannabinoid receptor antagonist/inverse agonist AM251 (3 μm) has no effect on LTD or on the associated changes in synaptic facilitation and KAR activity at LF neonatal inputs (n = 9). i, A time course plot depicting LTD in response to 900 pulses/5 Hz stimulation, associated with an increase in synaptic facilitation and subsequent ACET sensitivity. Example traces from the points indicated are shown on top. Scaled and superimposed traces are shown on the right. ii, Averaged data on the effect of 900 pulses/5 Hz stimulation and subsequent ACET application on EPSC amplitude (left) and facilitation ratio (5th/1st EPSC amplitude to 50 Hz stimulation). B, The NOS inhibitor l-NAME blocks the LTD-associated presynaptic changes. LTD induction followed by application of ACET in the presence of l-NAME at the LF neonatal inputs (n = 8). The data in B and C are presented as in A. C, Corresponding data for the HF neonatal inputs in the presence of l-NAME (n = 5). All data illustrate average ± SEM. Any statistical significance is indicated by *. The detailed statistical data are presented in Table 1.
Another retrograde signal implicated in NMDA-dependent LTD is NO (Stanton et al., 2003; Zhang et al., 2006). To test the role of NO-dependent signaling in neonatal presynaptic LTD, the competitive NOS inhibitor l-NAME was used. l-NAME (300 μm) had little effect on LTD induction at either LF or HF inputs (LF, −45 ± 8%, n = 8, p < 0.001; HF, − 41 ± 12%, n = 5, p = 0.003). However, in contrast to control, the level of LTD was not different between the two groups (p = 0.748), and there was no associated change in synapse dynamics at either LF (21 ± 13%; p = 0.530) or HF (6 ± 15%; p = 0.739) synapses (Fig. 6B,C). Finally, a subsequent application of ACET had no effect on transmission (8 ± 12%; p = 0.987) or facilitation ratio (7 ± 13%; p = 0.880) at LF synapses (Fig. 6B). At HF synapses, ACET had similar effects on EPSC amplitude (90 ± 33%; p = 0.001) and facilitation (−36 ± 11%, p = 0.001) that were seen under control conditions (Fig. 6C). In summary, these data suggest that NO, likely released from the postsynaptic neurons as a result of NMDA receptor and calcium-dependent signaling during LTD-inducing 900 pulses/5 Hz stimulation, acts as a retrograde transmitter to regulate presynaptic KARs at LF synapses. In addition, LTD at both LF and HF synapses express an additional NMDA receptor-dependent form of LTD that is most likely postsynaptically expressed.
Discussion
Compared with AMPA and NMDA receptors, relatively little is known on the mechanisms that regulate synaptic KARs. Here we show that, at immature glutamatergic synapses, presynaptic KARs can be upregulated or downregulated in response to different frequencies of neuronal activity. The plastic changes in their tonic inhibitory effect on glutamate release contribute to LTD and LTP in a developmentally restricted period of synapse maturation. Plasticity of KARs is not necessary for LTP and LTD per se; both involve several mechanistically distinct expression mechanisms that may be differentially used depending on the induction stimulus and on the developmental stage. However, unlike the purely postsynaptic changes in synapse strength, the KAR-dependent changes in transmitter release will modulate the dynamics of the synaptic response to high-frequency bursts of afferent activity, which have a central role in information transfer and synaptic refinement in the developing network (Lisman, 1997; Zhang and Poo, 2001).
Activity-dependent plasticity of KARs
Previous work has established that KARs not only contribute to induction of synaptic plasticity but are also molecular targets for regulation. Plasticity of KAR function was first observed at mossy fiber synapses, in which LTP is associated with downregulation of presynaptic KARs (Lauri et al., 2001; Ji and Stäubli, 2002). More recently, LTD of postsynaptic KAR-mediated EPSCs has been shown in the perirhinal cortex (Park et al., 2006) and at mossy fiber synapses in the hippocampus (Selak et al., 2009; Carta et al., 2013; Chamberlain et al., 2013). Our present data provide, to our knowledge, the first physiological evidence for activity-dependent upregulation of KAR function.
The signaling cascade responsible for upregulation of presynaptic KAR function at immature synapses involved activation of postsynaptic NMDA receptors and postsynaptic calcium. Interestingly, inclusion of BAPTA at the postsynaptic recording pipette prevented the LTD-associated, ACET-sensitive changes in synaptic facilitation, without completely blocking LTD per se. Because LTD was fully blocked by NMDA antagonism, it may be that the recently proposed metabotropic NMDA receptor signaling mechanism also contributes to postsynaptically expressed LTD at this developmental stage (Nabavi et al., 2013). However, with regards to the underlying mechanism for altering presynaptic KAR function, the simplest explanation is that a postsynaptic NMDA receptor-mediated calcium influx triggers retrograde signaling. Although cannabinoid signaling via CB1 receptors have been linked recently to both LTD (Yasuda et al., 2008; Caiati et al., 2012; Péterfi et al., 2012) and KAR (Lourenço et al., 2010) functions in the CA1 area of the hippocampus, we did not observe any effect of CB1 antagonism on induction of LTD or the associated changes in KAR activity. In contrast, the presynaptic changes were fully blocked in the presence of l-NAME, suggesting a role for NO (Stanton et al., 2003; Zhang et al., 2006).
Substantial evidence supports a role for NO as a diffusible messenger in LTD induction in various areas of the brain (Stanton et al., 2003; for review, see Garthwaite 2008; Huang and Hsu, 2010; Steinert et al., 2010; Tamagnini et al., 2013). NMDA receptor activation increases activity of neuronal NOS leading to NO release, whereas the neuronal effects of NO are typically mediated by raised levels of cGMP, acting on several downstream targets, including ion channels and signaling molecules (Garthwaite 2008; Steinert et al., 2010). NO also inhibits NMDA receptor function (Manzoni et al., 1992), an effect reversed by l-nitroarginine (Manzoni and Bockaert, 1993). Indeed, the loss of such modulation could enhance NMDA receptor activation during LTD induction and explain the larger than expected “postsynaptic” component of LTD that was observed in the presence of l-NAME.
The presynaptic KAR activity emerging at the LF inputs after LTD induction was attributable to high-affinity receptors, which are not detected at naive LF synapses. Because the affinity of KARs predominantly depends on their subunit composition, the gain of affinity after LTD most likely reflects insertion of novel receptors to the presynaptic membrane, by either lateral diffusion or membrane insertion. Although no data exist on the mechanisms controlling trafficking of presynaptically localized KARs, it is established that covalent modification via phosphorylation, palmitylation, or SUMOylation is critical for regulation of postsynaptic KARs (for review, see Pahl et al., 2014). One of the main effectors of NO is cGMP-dependent protein kinase II (cGK-II), implicated in the trafficking of GluA1 subunit-containing AMPA receptors (Serulle et al., 2007). Interestingly, cGK-II is reported to interact with recycling endosome protein Rab11 (Yuasa et al., 2008), implicated recently in the recycling and surface expression of postsynaptic KARs (Gonzáles-Gonzáles and Henley, 2013).
Downregulation of presynaptic KAR activity occurring after LTP induction at the immature synapses involved another diffusible messenger, BDNF acting on TrkB receptors (Sallert et al., 2009). TrkB activation is linked to several downstream signaling pathways, including PLCγ-dependent activation of PKC. PKC was shown recently to be necessary for the presynaptic component of LTP at immature synapses (Luchkina et al., 2013). Furthermore, several studies indicate that PKC activation regulates KAR membrane expression (Hirbec et al., 2003; Martin and Henley, 2004; Rivera et al., 2007) and is involved in activity-dependent depression of KAR EPSCs (Park et al., 2006; Selak et al., 2009, Chamberlain et al., 2013).
Apart from the mechanisms described here, synaptic KARs are shown to be regulated in response to activation of mGluRs (Park et al., 2006; Selak et al., 2009) and adenosine A2A receptors (Chamberlain et al., 2013). Thus, it appears that there is no preferential single pathway responsible for activity-dependent regulation of KARs that rather can be triggered via a variety of receptor mechanisms. Whether these converge to common or receptor subtype-specific downstream regulatory mechanisms remains to be investigated.
Regulation of KARs versus synapse development
Previously, we proposed that the tonic KAR activity is a property of immature synapses that is lost during maturation (Lauri et al., 2006). Therefore, it was initially a surprise to find that the nonfacilitatory inputs, considered previously as “mature-type” synapses within the developmentally heterogeneous population, could in fact acquire tonic KAR activity during LTD induction. Based on the present results, the naive low-facilitatory neonatal inputs represent a distinct population of immature synapses with modifiable presynaptic function, in contrast to the mature synapses that are characterized by more stable functional properties. This scenario on presynaptic development resembles the current view on the maturation of postsynaptic function, in which AMPA receptors are initially labile and may be silenced or stabilized depending on the activity pattern to which the nascent synapse is exposed (Hanse et al., 2009). Thus, low-frequency activity leads to functional weakening, postsynaptically via AMPA silencing (Xiao et al., 2004) and presynaptically via reduced Pr attributable to upregulation of inhibitory KARs. Conversely, in both models, the system is permanently switched from the labile state into adult-type function as a result of LTP induction (Fig. 7).
Figure 7.

A schematic model for the activity-dependent plasticity of presynaptic KARs at immature synapses.
Nevertheless, the present and previous data support the view that the tonic plastic KAR activity is tightly associated with a transient stage of synapse development. This activity could not be observed or induced in slices from 2-week-old animals. Furthermore, maintaining the system in an immature state via chronic blockade of BDNF–TrkB signaling preserved the tonic KAR activity and its plasticity.
Given that tonic KAR activity is a key marker of synaptic immaturity, what is the physiological significance of this mechanism? This activity coincides developmentally with the time the network is highly modifiable by ongoing network activity, and new synapses are formed and others eliminated. LTD induction represents the first step to synapse elimination and involves a decrease in both presynaptic and postsynaptic efficacy, which reduces the ability of the synapse to respond to low-frequency activity. However, the increase in synaptic facilitation attributable to the upregulation of presynaptic KARs will enable the synapse to specifically detect only high-frequency bursts of activity. This, in turn, can accentuate the difference in the plasticity induction threshold between LF and HF inputs, therefore allowing sharper long-term segregation of functionally distinct inputs. Conversely, loss of the KAR plasticity after developmental LTP leading to maturation contributes to the reliability of transmission suitable for adult-type function.
Footnotes
We thank the Academy of Finland and the Sigrid Juselius Foundation for financial support.
The authors declare no competing financial interests.
References
- Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, Dudek SM. Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:3123–3127. doi: 10.1073/pnas.0800027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker N, Wierenga CJ, Fonseca R, Bonhoeffer T, Nägerl UV. LTD induction causes morphological changes of presynaptic boutons and reduces their contacts with spines. Neuron. 2008;60:590–597. doi: 10.1016/j.neuron.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Caiati MD, Sivakumaran S, Lanore F, Mulle C, Richard E, Verrier D, Marsicano G, Miles R, Cherubini E. Developmental regulation of CB1-mediated spike-time dependent depression at immature mossy fiber-CA3 synapses. Sci Rep. 2012;2:285. doi: 10.1038/srep00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Opazo P, Veran J, Athané A, Choquet D, Coussen F, Mulle C. CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J. 2013;32:496–510. doi: 10.1038/emboj.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Jouvenceau A, Rose C, Guilloux JP, Pilon C, Dranovsky A, Prémont J. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SE, Sadowski JH, Teles-Grilo Ruivo LM, Atherton LA, Mellor JR. Long-term depression of synaptic kainate receptors reduces excitability by relieving inhibition of the slow afterhyperpolarization. J Neurosci. 2013;33:9536–9545. doi: 10.1523/JNEUROSCI.0034-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signalling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Dargan SL, Clarke VRJ, Alushin GM, Sherwood JL, Nisticò R, Bortolotto ZA, Ogden AM, Bleakman D, Doherty AJ, Lodge D, Mayer ML, Fitzjohn SM, Jane DE, Collingridge GL. ACET is a highly potent and specific kainate receptor antagonist: characterisation and effects on hippocampal mossy fibre function. Neuropharmacology. 2009;56:121–130. doi: 10.1016/j.neuropharm.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and the effects of N-methyl-D-aspartate receptor blockade. Proc Nat1 Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-González IM, Henley JM. Postsynaptic kainate receptor recycling and surface expression are regulated by metabotropic autoreceptor signaling. Traffic. 2013;14:810–822. doi: 10.1111/tra.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B. Factors explaining heterogeneity in short-term synaptic dynamics of hippocampal glutamatergic synapses in the neonatal rat. J Physiol. 2001;537:141–149. doi: 10.1111/j.1469-7793.2001.0141k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanse E, Taira T, Lauri S, Groc L. Glutamate synapse in developing brain: an integrative perspective beyond the silent state. Trends Neurosci. 2009;32:532–537. doi: 10.1016/j.tins.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Heynen AJ, Yoon BJ, Liu CH, Chung HJ, Huganir RL, Bear MF. Molecular mechanism for loss of visual cortical responsiveness following brief monocular deprivation. Nat Neurosci. 2003;6:854–862. doi: 10.1038/nn1100. [DOI] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, Dev KK, Coutinho V, Meyer G, Isaac JT, Collingridge GL, Henley JM, Couthino V. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron. 2003;37:625–638. doi: 10.1016/S0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Activation of muscarinic acetylcholine receptors induces a nitric oxide-dependent long-term depression in rat medial prefrontal cortex. Cereb Cortex. 2010;20:982–996. doi: 10.1093/cercor/bhp161. [DOI] [PubMed] [Google Scholar]
- Ji Z, Stäubli U. Presynaptic kainate receptors play different physiological roles in mossy fiber and associational-commissural synapses in CA3 of hippocampus from adult rats. Neurosci Lett. 2002;331:71–74. doi: 10.1016/S0304-3940(02)00865-0. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron. 2001;32:697–709. doi: 10.1016/S0896-6273(01)00511-6. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JT, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Lourenço J, Cannich A, Carta M, Coussen F, Mulle C, Marsicano G. Synaptic activation of kainate receptors gates presynaptic CB(1) signalling at GABAergic synapses. Nat Neurosci. 2010;13:197–204. doi: 10.1038/nn.2481. [DOI] [PubMed] [Google Scholar]
- Luchkina NV, Sallert M, Clarke VRJ, Taira T, Lauri SE. Mechanisms underlying induction of LTP-associated changes in short-term dynamics of transmission at immature synapses. Neuropharmacology. 2013;67:494–502. doi: 10.1016/j.neuropharm.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Bockaert J. Nitric oxide synthase activity endogenously modulates NMDA receptors. J Neurochem. 1993;61:368–370. doi: 10.1111/j.1471-4159.1993.tb03580.x. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Nitric oxide-induced blockade of NMDA receptors. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-T. [DOI] [PubMed] [Google Scholar]
- Martin S, Henley JM. Activity-dependent endocytic sorting of kainate receptors to recycling or degradation pathways. EMBO J. 2004;23:4749–4759. doi: 10.1038/sj.emboj.7600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, Sivakumaran S, Zacchi P, Aguilera P, Cherubini E. Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc Natl Acad Sci U S A. 2007;104:13176–13181. doi: 10.1073/pnas.0704533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A. 2013;110:4027–4032. doi: 10.1073/pnas.1219454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/S0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Pahl S, Tapken D, Haering SC, Hollmann M. Trafficking of kainate receptors. Membranes. 2014;4:565–595. doi: 10.3390/membranes4030565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Isaac JT, Collingridge GL. Multiple, developmentally regulated expression mechanisms of long-term potentiation at CA1 synapses. J Neurosci. 2004;24:4903–4911. doi: 10.1523/JNEUROSCI.0170-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Jo J, Isaac JT, Cho K. Long-term depression of kainate receptor-mediated synaptic transmission. Neuron. 2006;49:95–106. doi: 10.1016/j.neuron.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Riekki R, Taira T. Synergistic action of GABA-A and NMDA receptors in the induction of long-term depression in glutamatergic synapses in the newborn rat hippocampus. Eur J Neurosci. 2004;20:3019–3026. doi: 10.1111/j.1460-9568.2004.03806.x. [DOI] [PubMed] [Google Scholar]
- Péterfi Z, Urbán GM, Papp OI, Németh B, Monyer H, Szabó G, Erdélyi F, Mackie K, Freund TF, Hájos N, Katona I. Endocannabinoid-mediated long-term depression of afferent excitatory synapses in hippocampal pyramidal cells and GABAergic interneurons. J Neurosci. 2012;32:14448–14463. doi: 10.1523/JNEUROSCI.1676-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera R, Rozas JL, Lerma J. PKC-dependent autoregulation of membrane kainate receptors. EMBO J. 2007;26:4359–4367. doi: 10.1038/sj.emboj.7601865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallert M, Rantamäki T, Vesikansa A, Anthoni H, Harju K, Yli-Kauhaluoma J, Taira T, Castren E, Lauri SE. Brain-derived neurotrophic factor controls activity-dependent maturation of CA1 synapses by downregulating tonic activation of presynaptic kainate receptors. J Neurosci. 2009;29:11294–111303. doi: 10.1523/JNEUROSCI.0560-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak S, Paternain AV, Aller IM, Picó E, Rivera R, Lerma J. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron. 2009;63:357–371. doi: 10.1016/j.neuron.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda Y, Tanaka T, Tominaga-Yoshino K, Ogura A. Persistent synapse loss induced by repetitive LTD in developing rat hippocampal neurons. PLoS One. 2010;5:e10390. doi: 10.1371/journal.pone.0010390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Müller W. Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci. 2003;23:5936–5944. doi: 10.1523/JNEUROSCI.23-13-05936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert JR, Chernova T, Forsythe ID. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist. 2010;16:435–452. doi: 10.1177/1073858410366481. [DOI] [PubMed] [Google Scholar]
- Tamagnini F, Barker G, Warburton EC, Burattini C, Aicardi G, Bashir ZI. Nitric oxide-dependent long-term depression but not mediated long-term potentiation is crucial for visual recognition memory. J Physiol. 2013;591:3963–3979. doi: 10.1113/jphysiol.2013.254862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesikansa A, Sakha P, Kuja-Panula J, Molchanova S, Rivera C, Huttunen HJ, Rauvala H, Taira T, Lauri SE. Expression of GluK1c underlies the developmental switch in presynaptic kainate receptor function. Sci Rep. 2012;2:310. doi: 10.1038/srep00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasling P, Hanse E, Gustafsson B. Long-term depression in the developing hippocampus: low induction threshold and synapse nonspecificity. J Neurosci. 2002;22:1823–1830. doi: 10.1523/JNEUROSCI.22-05-01823.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, Oertner TG. Long-term depression triggers the selective elimination of weakly integrated synapses. Proc Natl Acad Sci U S A. 2013;110:E4510–E4519. doi: 10.1073/pnas.1315926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MY, Wasling P, Hanse E, Gustafsson B. Creation of AMPA-silent synapses in the neonatal hippocampus. Nat Neurosci. 2004;7:236–243. doi: 10.1038/nn1196. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Huang Y, Tsumoto T. Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proc Natl Acad Sci U S A. 2008;105:3106–3111. doi: 10.1073/pnas.0708349105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa K, Yamagami S, Nagahama M, Tsuji A. Trafficking of cGMP-dependent protein kinase II via interaction with Rab11. Biochem Biophys Res Commun. 2008;374:522–526. doi: 10.1016/j.bbrc.2008.07.071. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM. Electrical activity and development of neural circuits. Nat Neurosci [Suppl] 2001;4:1207–1214. doi: 10.1038/nn753. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Zhou ZY, Winterer J, Müller W, Stanton PK. NMDA-dependent, but not group I metabotropic glutamate receptor-dependent, long-term depression at Schaffer collateral-CA1 synapses is associated with long-term reduction of release from the rapidly recycling presynaptic vesicle pool. J Neurosci. 2006;26:10270–10280. doi: 10.1523/JNEUROSCI.3091-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]