

Abstract
Cell cycle regulation is crucial for the maintenance of stem cell populations in adult mammalian tissues. During development, the cell cycle length in neural stem cells increases, which could be associated with their capabilities for self-renewal. However, the molecular mechanisms that regulate differentiation and cell cycle progression in embryonic neural stem cells remain largely unknown. Here, we investigated the function of Bre1a, a histone H2B ubiquitylation factor, which is expressed in most but not all of neural precursor cells (NPCs) in the developing mouse brain. We found that the knockdown of Bre1a in NPCs lengthened their cell cycle through the upregulation of p57kip2 and the downregulation of Cdk2. In addition, the knockdown of Bre1a increased the expression of Hes5, an effector gene of Notch signaling, through the action of Fezf1 and Fezf2 genes and suppressed the differentiation of NPCs. Our data suggest that Bre1a could be a bifunctional gene that regulates both the differentiation status and cell cycle length of NPCs. We propose a novel model that the Bre1a-negative cells in the ventricular zone of early embryonic brains remain undifferentiated and are selected as self-renewing neural stem cells, which increase their cell cycle time during development.
Keywords: brain development, differentiation, epigenetics, H2B ubiquitylation, neural stem cells, proliferation
Introduction
In the developing mammalian brain, neural stem cells (NSCs) proliferate vigorously with short cell cycle times at early stages (Gage, 2000; Temple, 2001; Kriegstein et al., 2006). Later in development, NSCs lengthen the cell cycle (Martens et al., 2000) and become relatively quiescent, dividing once every 15 d or less (Morshead et al., 1994; Morshead et al., 1998; Alvarez-Buylla and Garcia-Verdugo, 2002). The relative quiescence is crucial for the maintenance of stem cell populations in adult mammalian tissues (Cheng et al., 2000a,b; Kippin et al., 2005; van Os et al., 2007; Matsumoto et al., 2011; Zou et al., 2011; Furutachi et al., 2013); therefore, it is important to understand how the cell cycle times of NSCs are regulated. However, the molecular mechanisms that regulate cell cycle progression during embryogenesis and acquisition of quiescence in adult NSCs remain largely unknown.
Accumulating evidence shows that epigenetic mechanisms, which consist of DNA methylation and histone modifications, play important roles in the generation, maintenance, and differentiation of NSCs (Takizawa et al., 2001; Hirabayashi et al., 2009; Hitoshi et al., 2011). Chromosomal histones are subject to a variety of covalent modifications including methylation, acetylation, phosphorylation, ubiquitylation, SUMOylation, and glycosylation (Kouzarides, 2007; Fujiki et al., 2011). Bre1a, also known as ring finger protein 20, is an E3 ubiquitin ligase. Bre1 specifically catalyzes the monoubiquitylation of histone H2B (Hwang et al., 2003; Kim and Roeder, 2009), and this modification is highly associated with transcriptionally active loci (Minsky et al., 2008). The balance between monoubiquitylation and deubiquitylation of H2B that is catalyzed by deubiquitinases such as USP3, USP22, and USP44, is strictly regulated in a cell context-dependent manner for optimal gene expression (Nicassio et al., 2007; Zhang et al., 2008; Fuchs et al., 2012). Bre1a disruption or impaired monoubiquitylation of H2B results in a complex phenotype that includes failures in transcriptional modifications, and progression of the cell cycle in yeast (Briggs et al., 2001; Sun and Allis, 2002; Kao et al., 2004; Giannattasio et al., 2005; Zimmermann et al., 2011). Bre1a and H2B monoubiquitylation are also indispensable for the proper development of multicellular organisms; in Drosophila, deubiquitylation of H2B is essential for the maintenance of the stem cell population (Buszczak et al., 2009). Interestingly, the overexpression of dBre1 interferes with Notch signaling (Bray et al., 2005), which is a critical signaling pathway for the maintenance of various stem cell populations (Chiba, 2006). Recently, monoubiquitylated H2B at Lys120 (H2Bub1) was shown to be required for the differentiation of embryonic stem cells or mesenchymal stem cells (Fuchs et al., 2012; Karpiuk et al., 2012), suggesting an essential role for Bre1a in controlling stem cell differentiation. However, the function of Bre1a in mammalian development and in the regulation of NSCs in particular remains to be investigated.
In this study, we found that knockdown of Bre1a in neural precursor cells (NPCs) of the developing cerebral cortex suppresses proliferation and differentiation, suggesting that Bre1a is a multifunctional molecule that regulates both the cell cycle and differentiation of NSCs.
Materials and Methods
Animals.
All mice used in these experiments were kept in the Center for Experimental Animals of National Institute for Physiological Sciences and Shiga University of Medical Science with unlimited access to food and water. Timed-pregnant CD1 (ICR) mice were purchased from SLC, or CD1 males and females were mated to obtain pregnant mice for the collection of embryos of either sex. Noon of the plugged date was considered as embryonic day (E) 0.5. All experiments were performed with the permission of the Animal Research Committee of National Institute for Physiological Sciences and Shiga University of Medical Science.
Antibodies.
The primary antibodies used in this study were as follows: rabbit anti-Bre1a IgG (ab32629, Abcam); rabbit anti-Bre1a IgG (catalog #9425, Cell Signaling Technology); rabbit anti-H2B IgG (Millipore); mouse anti-ubiquitylated H2B IgG (Millipore); mouse anti-phospho-histone H3 IgG (Cell Signaling Technology); mouse anti-GAPDH IgG (Santa Cruz Biotechnology); mouse anti-proliferating cell nuclear antigen (PCNA) IgG (DAKO); rabbit anti-green fluorescent protein (GFP) IgG (Invitrogen); rat anti-GFP IgG (Nacalai Tesque); rabbit anti-cleaved caspase 3 (c-Cas3) IgG (Cell Signaling Technology); rabbit anti-paired box gene 6 (Pax6) IgG (Millipore); and rabbit anti-eomesodermin homolog (Eomes/Tbr2) IgG (Abcam). For immunocyto/histochemistry, we used Alexa Fluor 488- and Alexa Fluor 568-conjugated anti-rabbit IgG, and Alexa Fluor 568- and Alexa Fluor 633-conjugated anti-mouse IgG as secondary antibodies (Invitrogen). For Western blotting, horseradish peroxidase-labeled anti-rabbit IgG or anti-mouse IgG was used as a secondary antibody (MP Biomedicals).
Generation of a short hairpin RNA library and miRNA plasmids.
A short hairpin RNA (shRNA) library for Bre1a was synthesized using EPRIL methodology (Shirane et al., 2004). The open reading frame and the 3′ untranslated region (UTR) of Bre1a were selected as target regions. One shRNA clone that targets the sequence around the stop codon and two shRNA clones that target 3′ UTR sequences were selected from the library because of the high knockdown efficiency. The shRNAs were inserted into a dual lentivirus and mammalian expression vector that contains the mouse U6 gene promoter and human PGK promoter-driven enhanced GFP. Vesicular stomatitis virus-G (VSV-G) pseudotyped lentiviruses were produced by transient transfection of HEK293FT cells with a mixture of the following four plasmids: pLKO.1 lentivirus vector; pCAGGS-KGRIR; pCAGGS-RTR2; and pCAGGS-VSV-G. The medium supernatant containing virus particles was filtered through 0.45 μm membranes (Millipore), then centrifuged at 50,000 × g for 2 h at 20°C. The pellet was resuspended in 500 μl of HBSS, and virus particles were further concentrated by ultracentrifugation in a 20% sucrose solution at 50,000 × g for 2 h. The pellet was resuspended in 50 μl of HBSS, stored at 4°C overnight, distributed in 5 μl aliquots, and frozen in liquid nitrogen. Full-length Bre1a was inserted to pMXIE to generate Bre1a and a GFP-expressing retrovirus, as described previously (Hitoshi et al., 2002b). The virus preparations were stored at −80°C until use.
Two 21-nucleotide sequences (1, CAGCGATACCTTCCCAGTGAT from the open reading frame and 2, TAGCAGGAACCGGAGATGGTT from the 3′ UTR) for p57Kip2, one (AGGTCTTTAATGCGCACTATA from the open reading frame) for Fez family zinc finger 1 (Fezf1), and one (GGTCATCAAACCGCAGGTCAT from the open reading frame) for Fezf2 were selected to generate miRNAs by a BLOCK-iT RNAi Designer (Invitrogen). DNA fragments containing each pair of sense and antisense sequences together with loop sequences were subcloned into a pcDNA6.2-GW/EmGFP-miR expression vector (Invitrogen).
Neural precursor cell culture.
The isolation of NSCs from embryonic brains was performed as previously described (Hitoshi et al., 2002b). To obtain primary neurospheres, cells from the forebrain of E10.5 embryos or the dorsal cortex of E13.5 embryos were dissociated into single cells and cultured in serum-free media (SFM) [DMEM/F12 (1:1), 5 mm HEPES buffer, 0.6% glucose, 3 mm NaHCO3, 2 mm glutamine, 25 μg/ml insulin, 100 μg/ml transferrin, 20 nm progesterone, 60 μm putrescine, and 30 nm sodium selenite] in the presence of 10 ng/ml fibroblast growth factor 2 (FGF-2) together with 2 μg/ml heparin (all from Sigma). For lentivirus transduction, cells from the resultant neurospheres were dissociated and mixed with virus in the presence of 5 μg/ml polybrene, then centrifuged at 800 × g for 60 min at 20°C. The pellets were mechanically triturated to obtain a single-cell suspension, then cultured for 7 d in SFM that contained 10 ng/ml FGF-2 and 20 ng/ml EGF (Sigma).
Primary neuroepithelial cells from E10.5 forebrains were transduced with Bre1a shRNA or control plasmids using an electroporator (NEPA21, NEPA GENE). After electroporation, the cells were plated onto 12 mm coverslips double coated with polyethylenimine and Matrigel basement matrix (0.6 mg/ml SFM; Becton Dickinson) and cultured in 100 μl of SFM containing FGF-2 and EGF for proliferation or in SFM containing 10% knock-out serum replacement (Invitrogen) for differentiation. Three days after plating, the cells were processed for immunocytochemistry.
RT-PCR.
Total RNA isolation, cDNA synthesis, and quantitative RT-PCR analysis using the Light-Cycler system (Roche) were performed as described previously (Higashi et al., 2008). The sense and antisense primers used for Bre1a, p21, p27, p57Kip2, cyclin-dependent kinase 2 (cdk2), hairy and enhancer of split 5 (Hes5), Hes1, Fezf1, Fezf2, and β-actin were shown in Table 1.
Table 1.
PCR primers for RT-PCR
| Gene | Primer | Sequence |
|---|---|---|
| Bre1a | Sense (F2) | 5′-AATAAACGCAAGGCAATGGAG-3′ |
| Antisense (R3) | 5′-TCTTGACTGCAATCAGCCAATG-3′ | |
| p21 | Sense | 5′-GAGGGAGCCTGAAGACTGTGAT-3′ |
| Antisense | 5′-CTCCCGTGGGCACTTCAGGGTT-3′ | |
| p27 | Sense | 5′-ATGTCAAACGTGAGAGTGTCTAACGG-3′ |
| Antisense | 5′-TTACGTCTGGCGTCGAAGGCCGGGCTTCTTG-3′ | |
| p57Kip2 | Sense | 5′-TGCTGAGCCGGGTGATGAGCTG-3′ |
| Antisense | 5′-AACTCATCTCAGACGTTTGCG-3′ | |
| Cdk2 | Sense | 5′-TGTACCCAGTACTGCCATCC-3′ |
| Antisense | 5′-TCCCCAGAGTCCGAAAGATCC-3′ | |
| Hes1 | Sense | 5′-AAAGACGGCCTCTGAGCACA-3′ |
| Antisense | 5′-TCATGGCGTTGATCTGGGTCA-3′ | |
| Hes5 | Sense | 5′-AAGTACCGTGGCGGTGGAGATGC-3′ |
| Antisense | 5′-CGCTGGAAGTGGTAAAGCAGCTT-3′ | |
| Fezf1 | Sense | 5′-CGTTAAACACACATACCCGAA-3′ |
| Antisense | 5′-CATCACTGGTGGCCTTGGTGA-3′ | |
| Fezf2 | Sense | 5′-ATCGCAGCTCCACGCTCAACA-3′ |
| Antisense | 5′-GCAGTAGCTCTCAGCTCTGAA-3′ | |
| β-actin | Sense | 5′-AGGCCAACCGTGAAAAGATGAC-3′ |
| Antisense | 5′-GTACATGGTGGTACCACCAGAC-3′ |
SDS-PAGE and immunoblotting.
EGFP-positive neurosphere cells were collected, resuspended in SDS sample buffer (50 mm Tris-Cl, pH 6.8, 100 mm DTT, 2% SDS, and 10% glycerol), and sonicated. Extracted samples were subjected to Western blotting. Signals were detected using a chemiluminescence kit (ECL-plus, GE Healthcare) and visualized by the LAS-3000 Imaging System (Fujifilm).
In utero electroporation.
In utero electroporation was performed as described previously (Naruse et al., 2006). shRNA expression plasmids were dissolved at a concentration of 2–2.5 μg/μl in Dulbecco's modified PBS (Sigma) containing 0.05% Fast Green to monitor the injection. Timed-pregnant CD1 mice (E13.5 or E14.5) were deeply anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg body weight; Nembutal, Dainippon Pharmaceutical). An electroporator (CUY21, NEPA GENE) was used to deliver six 50 ms pulses of 33 V at 950 ms intervals. To detect proliferating cells, 5-ethynyl-2-deoxyuridine (EdU; 50 mg/kg body weight; Invitrogen) was injected intraperitoneally into the dam 2 h before the perfusion.
Time-lapse imaging.
In utero electroporation was performed at E13.5 using shRNA plasmids together with a H2B-mCherry expression plasmid (Abe et al., 2011; RIKEN CDB accession no. CDB0005P) to monitor the cell division. Twenty-four hours after the electroporation, brains of embryos were aseptically collected and 400-μm-thick coronal slices were prepared as described previously (Hitoshi et al., 2002a). The slices were placed on a hydrophilic transparent membrane (PICMORG50, Millipore) and cultured in SFM. Time-lapse images were obtained with an incubation imaging system (CellVoyager CV1000, Yokogawa) at every 15 min interval and were analyzed using ImageJ (National Institutes of Health). Cell divisions were monitored by the expression of H2B-mCherry. We measured time, during which cells migrated from basal most in the ventricular zone (VZ)/subventricular zone (SVZ) to apical surface and completed the mitosis, as the duration of G2/M phase.
Immunohistochemistry.
Coronal cryosections at 14 μm thickness were immunostained as follows: after permeabilization with 0.5% Triton X-100 in PBS, the sections were blocked in 10% normal goat serum/PBS containing 0.1% Triton X-100 for 1 h and then incubated with primary antibodies in the blocking buffer at 4°C overnight. After washing with 0.1% Triton X-100/PBS, the sections were incubated with secondary antibodies for 2 h at room temperature. EdU visualization was conducted using the Click-iT EdU imaging kit (Invitrogen) according to the manufacturer's protocol. The nuclei of cells were counterstained with Hoechst 33342 (0.1 μg/ml; Sigma). For immunostaining against Pax6 or Tbr2, the sections were irradiated in a microwave for 5 min in 10 mm citrate buffer, pH 6.0, heated to >90°C.
In situ hybridization.
Digoxigenin (DIG)-labeled single stranded riboprobes for Hes5 and RAR-related orphan receptor β (Rorb) were synthesized using T3 RNA polymerase and DIG RNA labeling mix (Roche). In situ hybridization using cryosections was performed as described previously (Naruse et al., 2006). The hybridized DIG-labeled probe was detected by overnight incubation of the sections with anti-DIG antibody conjugated to alkaline phosphatase (1:2000; Roche). The color was developed in the presence of 4-nitro blue tetrazolium chloride, 5-bromo-4-chloro-3-indolyl phosphate (Roche).
Luciferase reporter assay.
Luciferase (Luc) reporter plasmids containing the promoter regions of the Hes1 or Hes5 genes were gifts from Dr. Kageyama (Kyoto University, Kyoto, Japan). FuGENE6 (Roche) was used to transfect Neuro2a cells with a luciferase reporter plasmid and an internal control Renilla luciferase reporter plasmid (Promega), in combination with one or more of the following expression plasmids: pCX-mycNICD, pCX-Bre1a, pCX-Bre1aΔC, pCX-Fezf1, pCX-Fezf2, pcDNA6.2-Fezf1-miRNA (Invitrogen), and pcDNA6.2-Fezf2-miRNA (Invitrogen). Luciferase activities were determined using a Dual-Luciferase Reporter Assay System (Promega).
Statistics.
Statistical analysis was performed using one-way ANOVA followed by Tukey-Kramer, Scheffé's, or Dunnett's post hoc comparison. If applicable, we used an unpaired two-tailed Student's or Welch's t test. The level of significance was set at p < 0.05.
Results
Bre1a is required for proliferation of neural precursor cells
To investigate the role of Bre1a in neural development, we first examined the expression of Bre1a in the developing brain. Bre1a mRNA and its product protein were abundantly and ubiquitously present in the embryonic and early postnatal brain (Fig. 1A,E,J–L). However, a few cells in the VZ of dorsal and ventral forebrain expressed Bre1a much more weakly than in the surrounding cells and expressed no detectable H2Bub1, which Bre1 catalyzes to synthesize (Fig. 1B,F–H). Although some of these VZ cells were undergoing mitosis, which may explain the weak immunoreactivity of Bre1a and H2Bub1, the others were not in M-phase as confirmed by the absence of phospho-histone H3 (Fig. 1B,C,F–I). In addition, βIII-tubulin, an immature neuronal marker, colocalized with Bre1a in cells near to the basal surface of E10.5 dorsal forebrain (Fig. 1D). These results suggest that most, if not all, of NPCs and differentiating neuroblasts express Bre1 and H2Bub1, which prompted us to evaluate the effects of Bre1a knockdown in NPCs of the developing brain.
Figure 1.

Bre1a expression in the developing brain. A–L, The expression of Bre1a was determined in coronal cryosections of forebrains from E10.5 (A–D), E12.5 (E–I), E14.5 (J), and E16.5 (K) embryos, and P0 pups (L) by immunostaining or by in situ hybridization (J, top). B, Coronal cryosections of E10.5 dorsal forebrain were immunostained for Bre1a and H2Bub1. Boxed areas in the top panels are shown in the bottom panels with higher magnification. Nonmitotic cells that weakly express Bre1a and H2Bub1 are indicated by arrowheads, and some mitotic cells are indicated by dotted circles and arrows. C, Double immunostaining for Bre1a and phospho-histone 3 (PH3) shows Bre1aweak/PH3− cells (arrowheads). D, Colocalization of Bre1a and βIII-tubulin in cells nearby the basal surface is indicated by arrowheads. E–H, Coronal cryosections of E12.5 brain were double-immunostained for Bre1a and H2Bub1 (E), and boxed areas in the cortex (F), dorsoventral boundary (G), and medial ganglionic eminence (H) are shown with higher magnification. Nonmitotic cells that weakly express Bre1a and H2Bub1 are indicated by arrowheads, and some mitotic cells are indicated by dotted circles and arrows. I, Coronal cryosections of E12.5 brain were immunostained for PH3, and then, double immunostained for Bre1a and H2Bub1. Alexa Fluor-633 signal for PH3 is pseudo-colored and shown in blue. LV, Lateral ventricle. Scale bars: A, E, J–L, 200 μm; B–D, F–I, 50 μm.
We designed three shRNAs for Bre1a, one near the stop codon and two in the 3′ untranslated region. These shRNAs were cloned into pLKO.1, which contains the mouse U6 promoter, to drive expression of the shRNA and the PGK promoter to drive enhanced GFP expression. pLKO.1 is bifunctional, and can be used to produce lentivirus or as a mammalian expression vector. Lentiviruses expressing the three shRNAs were used to infect NSC-derived neurosphere cells from E13.5 mouse cortex, and the resultant GFP+ neurospheres were subjected to quantitative RT-PCR analysis. There was a significant reduction of Bre1a expression in Bre1a shRNA-expressing neurosphere cells relative to that of Luc shRNA-expressing control neurospheres (F(3,15) = 5.016, p = 0.0132; Fig. 2A). Furthermore, we confirmed the reduction of Bre1a and H2Bub1 expression in Bre1a shRNA-expressing cells using immunocytochemical (Fig. 2B) and immunoblot (Fig. 2C) analyses. These results indicate that all three Bre1a shRNAs efficiently knock down the expression of Bre1a and H2Bub1 in NPCs.
Figure 2.

Bre1a knockdown impairs the proliferation of neural precursor cells. A–C, F, H, Primary neurospheres derived from the E13.5 cortex were infected with lentivirus expressing control or Bre1a shRNAs and GFP, and resultant GFP+ secondary neurospheres were subjected for analyses. A, Quantitative RT-PCR for Bre1a expression. B, GFP+ secondary neurospheres were plated and immunostained for GFP and Bre1a. Arrowheads and arrows indicate Bre1a shRNA-expressing and nonexpressing cells, respectively. C, Protein extracts from neurospheres expressing Bre1a shRNAs or control shRNA were subjected to Western blotting using the indicated antibodies. D, E, G, Primary neuroepithelial cells from E10.5 forebrains were transfected with Bre1a shRNA or control shRNA (D), together with GFP expression plasmid only, GFP and Bre1a expression plasmids (E), or GFP and miRNA expression plasmids (G). We used two miRNAs for p57Kip2 (1 and 2) or control miRNA. The cells were cultured in proliferation conditions for 3 d and were incubated with 20 μm EdU for 2 h before fixation. D, Cells were immunostained for GFP, followed by visualization for EdU. Arrowheads indicate GFP+/EdU+ cells and arrows indicate GFP+/EdU− cells. E, G, Quantification of GFP+/EdU+ cells in total GFP+ cells. F, H, Quantitative RT-PCR for p21Cip1, p27Kip1, and p57Kip2 (F) and for Cdk2 (H). Scale bars, 50 μm. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by one-way ANOVA followed by Dunnett's post hoc comparison (A, E, F, H) or Tukey-Kramer's post hoc comparison (G).
We first characterized the effects of Bre1a knockdown in the proliferative capacity of primary neuroepithelial cells from E10.5 mouse forebrain when cultured in the presence of FGF-2, by measuring the incorporation of the nucleotide analog EdU. We found a remarkable reduction of EdU incorporation (F(3,8) = 22.03, p < 0.001; Fig. 2D,E). To obtain insight into the mechanism underlying this reduction, we next analyzed the expression of the Cip/Kip family of CDK inhibitors, because these proteins are crucial for the establishment and maintenance of quiescence in various stem cell populations (Cheng et al., 2000a,b; Kippin et al., 2005; van Os et al., 2007; Matsumoto et al., 2011; Zou et al., 2011; Furutachi et al., 2013). Of the three Cip/Kip-type CDK inhibitors, only p57Kip2 (cyclin-dependent kinase inhibitor 1C), but not p21Cip1 (cyclin-dependent kinase inhibitor 1A) and p27Kip1 (cyclin-dependent kinase inhibitor 1B) was significantly upregulated in Bre1a shRNA-expressing NPCs (F(3,16) = 5.030, p = 0.012 for p57Kip2; Fig. 2F). We then tested whether the reduction of EdU incorporation in Bre1a shRNA-expressing cells was rescued by cotransfection of miRNAs for p57Kip2. We found that EdU incorporation was recovered to a level found in control shRNA-expressing NPCs by the knockdown of p57Kip2 (F(3,8) = 14.02, p = 0.0015; Fig. 2G). Because Cip/Kip-type CDK inhibitors are known to target Cdk2 and suppress the G1-to-S-phase transition (Sherr and Roberts, 2004), we examined the expression of Cdk2 and found that it was downregulated after Bre1a knockdown (F(3,16) = 3.990, p = 0.027; Fig. 2H). Consistent with these findings, protein levels of PCNA in Bre1a shRNA-expressing neurospheres were lower than in control neurospheres (mean ± SEM: 45.5 ± 12.5% by Bre1a shRNA 1; 55.3 ± 17.3% by Bre1a shRNA 2; 40.9 ± 5.1% by Bre1a shRNA 3 relative to control; n = 2; Fig. 2C). We performed rescue experiments by means of Bre1a shRNA 2 that target 3′ UTR of Bre1a mRNA and full-length Bre1a, which is resistant to Bre1a shRNA 2. Coexpression of Bre1a rescued the effects of knockdown by Bre1a shRNA 2 in all experiments, suggesting that the observed phenotypes were not by off-target effects (Fig. 2E,F,H).
Next, we tested the effects of Bre1a knockdown on the proliferation of NPCs in vivo by electroporation of shRNA expression plasmids into the cortex of E13.5 mouse embryos in utero. We confirmed the efficient knockdown of Bre1a expression 24 h after electroporation (Fig. 3A). To check whether Bre1a knockdown induces cell death, we examined the expression of c-Cas3, a marker for cell death, and found no increase of c-Cas3+ cells in Bre1a shRNA-electroporated brains compared with control brains (Fig. 3B). Then, we assessed the proliferation of NPCs by measuring the incorporation of EdU that had been administered to the dams 2 h before the brains were fixed. Consistent with our in vitro results, we found that the percentage of GFP and EdU double-positive cells, relative to the total number of GFP+ cells, was significantly lower in the brains that had received Bre1a shRNAs than in control shRNA-electroporated brains (F(2,9) = 12.58, p = 0.002; Fig. 3C,D). As expected, this reduction of EdU incorporation by Bre1a knockdown was reversed to the control level by Bre1a overexpression (t(3) = 3.617, p = 0.036; Fig. 3C,D).
Figure 3.

Bre1a knockdown in neural precursor cells reduces the proportion of mitotic cells. A–D, Bre1a shRNA or control shRNA and GFP expression plasmids were microinjected into the lateral ventricle (LV) of E13.5 embryos and electroporated into the dorsal forebrain in utero. The embryos were fixed 24 h after electroporation, and the brains were analyzed. A, GFP+ Bre1a shRNA-expressing cells (arrowheads) are less immunopositive for Bre1a than surrounding GFP− cells (arrows). B, Cryosections were immunostained for GFP and c-Cas3, followed by Hoechst nuclear staining. C, D, EdU was injected intraperitoneally into dams 2 h before fixation. Coronal cryosections were immunostained for GFP, followed by EdU visualization (C), and GFP +/EdU+ cells were counted (D). Bre1a expression plasmids were coelectroporated with Bre1a shRNA 3 to rescue the effects of Bre1a knockdown. E–H, Bre1a or control shRNA plasmids together with GFP and H2B-mCherry expression plasmids were coelectroporated into the dorsal forebrain of E13.5 embryos in utero. Twenty-four hours after electroporation, coronal slices were made and cultured on a hydrophilic transparent membrane in serum-free media for 30 h. E, Time-lapse images of GFP+/mCherry+ cells in the VZ/SVZ. Arrowheads indicate cells undergoing cell divisions, which were precisely recognized by the observation of H2B-mCherry expression (red). F–H, The percentage of GFP+ cells that underwent mitosis during the 30 h culture (F), the duration of the G2/M phase of GFP+ cells (G), and the migration speed of GFP+ cells in the G2 phase (H) in the VZ/SVZ were quantified. Scale bars: A, 20 μm; B, 100 μm; C, E, 30 μm. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by one-way ANOVA followed by Dunnett's post hoc comparison (D) or Student's t test (F–H).
To further analyze the proliferation and migration of NPCs, we performed time-lapse imaging using coronal slices of E14.5 embryonic brains that had received shRNA electroporation 24 h prior. We first examined cell divisions of GFP+ NPCs, which exhibited elevator movements in the VZ/SVZ, by tracing the H2B-mCherry expression during the 30 h observation (Fig. 3E). Consistent with the results of EdU incorporation in vivo, the number of cells that proceeded to M phase was significantly lower in the slices derived from Bre1a shRNA-electroporated brains than that of cells from control shRNA-electroporated brains (t(6) = 5.901, p < 0.001; Fig. 3F). Overexpression of H2B-mCherry hardly affects the proliferation of NPCs as confirmed by the EdU incorporation to E10.5 forebrain-derived cells 3 d after the transfection (mean ± SEM: 15.53 ± 2.55% in H2B-mCherry-expressing cells; 17.69 ± 1.50% in control cells; p = 0.505, n = 3). We then analyzed the migration of GFP+ cells in the VZ/SVZ. Most of the cells showed basal-to-apical migration during the observation, which is a typical movement of NPCs in G2 phase (Fujita, 2003). The duration of G2/M phase and migration speed of GFP+ cells were comparable between the slices from Bre1a shRNA- and control shRNA-transfected brains (t(6) = 0.192, p = 0.682 for duration; t(4) = 0.441, p = 0.682 for migration speed; Fig. 3G,H). These results can be interpreted in at least the following two ways: Bre1a knockdown induces NPCs to exit from the cell cycle, or alternatively, elongates the cell cycle times of NPCs. If the latter is the case, Bre1a knockdown elongates the G1/S phase because the duration of G2/M phase was comparable between Bre1a and control shRNA-transfected NPCs, which is consistent with the results that Bre1a knockdown upregulated the expression of p57Kip2 and downregulated the Cdk2 expression in NPCs.
Bre1a knockdown alters the distribution of cells in the developing neocortex
The effects of Bre1a knockdown by in utero electroporation were further analyzed at more advanced stages of development. NPCs that start to differentiate at approximately E14.5–E15.5 migrate radially into the cortical plate (CP) later in embryogenesis and settle in layers II–IV in the postnatal brain. First, we examined the distribution of GFP+ shRNA-expressing cells 72 h after electroporation. Bre1a knockdown resulted in a remarkable decrease in the number of GFP+ cells that migrated out of the VZ/SVZ into the CP, compared with control brains that received control Luciferase shRNA (Fig. 4A). We counted the number of GFP+ cell bodies localized in each area. To avoid underestimating the cell number in VZ/SVZ where progenitors divide vigorously, we carefully counted weakly GFP-positive cells as well because GFP expression plasmids are diluted in cell division. Quantitative analysis showed that in brains that express Bre1a shRNAs, the proportion of GFP+ cells was significantly increased in the VZ/SVZ and decreased in the CP when compared with the control shRNA-expressing brains (F(2,13) = 14.13, p < 0.001 for VZ/SVZ; F(2,13) = 45.98, p < 0.001 for CP; Fig. 4B). We also measured the migration speed of GFP+ cells in the CP, through which differentiated neurons migrate out to their final destinations (Fig. 4C). Despite the impaired distribution of Bre1a shRNA-expressing cells to the CP 72 h after electroporation (Fig. 4A), time-lapse imaging showed that the migration speed of GFP+ cells in the CP was not significantly altered between the slices made from Bre1a shRNA-transfected and control brains (t(4) = 1.18, p = 0.304; Fig. 4D).
Figure 4.

Bre1a knockdown impairs the migration of neural precursor cells. Bre1a or control shRNA and GFP expression plasmids were microinjected into the lateral ventricle of E13.5 embryos and electroporated into the dorsal forebrain in utero. A, Three days after the electroporation, the embryos were fixed and coronal cryosections of the brains were immunostained for GFP. B, GFP+ cells in the VZ/SVZ, IMZ, or CP were counted. C, D, Twenty-four hours after the electroporation, coronal slices were made and cultured in serum-free media for 30 h. C, Time-lapse images of GFP+ cells in the CP. Arrowheads indicate migrating cells. D, Migration speed of GFP+ cells in the CP was quantified. Scale bars: A, 100 μm; C, 20 μm. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by one-way ANOVA followed by Dunnett's post hoc comparison (B) or by Student's t test (D).
Next, we asked whether the impairments in NPC migration observed after Bre1a knockdown resulted from the lack of ubiquitin ligase activity in Bre1a. To answer this question, we performed rescue experiments by expression plasmids for full-length Bre1a and Bre1a that lacks the E3 ubiquitin ligase domain at the C terminus (Bre1aΔC; Fig. 5A) into the cortex of E13.5 embryos. Overexpression of either full-length Bre1a or Bre1aΔC had little effect on the migration of GFP+ cells (Fig. 5B,C), possibly because most NPCs already express Bre1a (Fig. 1). Coexpression of Bre1a shRNA 2 and full-length Bre1a plasmid, but not control pCX vector, rescued the distribution defect of GFP+ cells (Fig. 5D). In contrast, coexpression of Bre1a shRNA 2 and Bre1aΔC did not rescue the migration defect of GFP+ cells (Fig. 5D). Quantitative analysis showed that the increase of GFP+ cells in the VZ/SVZ and the reduction of GFP+ cells in the CP observed after Bre1a knockdown were rescued by the coexpression of full-length Bre1a, but not by Bre1aΔC (F(2,10) = 39.58, p < 0.001 for VZ/SVZ; F(2,10) = 22.94, p < 0.001 for CP; Fig. 5E). These results suggest that Bre1a function, mediated by the ubiquitin ligase activity, is required for the proper distribution of NPCs from the VZ/SVZ to the CP within 72 h but is dispensable for the migration of neuroblasts in the CP. It is possible that Bre1a knockdown suppresses the differentiation of NPCs or that, alternatively, it delays the final division of NPCs and the departure to migrate to the CP, although these possibilities are not mutually exclusive.
Figure 5.

Bre1a or Bre1aΔC overexpression does not inhibit cell migration. A, Schematic structures of full-length Bre1a and Bre1a that lacks the ring finger domain at the C terminus (Bre1aΔC) are shown. B–E, Expression plasmids for full-length Bre1a or Bre1aΔC were coelectroporated with GFP expression plasmid only (B, C) or with Bre1a shRNA and GFP expression plasmids (D, E) into the dorsal forebrain of E13.5 embryos in utero. Three days after the electroporation, the embryos were fixed and coronal cryosections of the brains were immunostained for GFP (B, D). C, E, GFP-positive cells in the VZ/SVZ, IMZ, or CP were counted. Scale bar, 100 μm. Error bars indicate SEM, and n values are shown in columns.
The distribution of GFP+ cells after electroporation of shRNA expression plasmids was further analyzed in the postnatal brain. NPCs that become postmitotic immediately or divide only two to three times maintain transgene expression, which can be monitored by the expression of GFP. We performed in utero electroporation of Bre1a shRNA or control shRNA at E13.5 and harvested the brains at postnatal day (P) 0. In control brains, GFP+ cells that had reached the outermost region of the CP by E16.5 were distributed between the upper and middle CP at P0 (Fig. 6A, top row), because neuroblasts born later migrate past cells born earlier. Surprisingly, a substantial number of GFP+ Bre1a shRNA-expressing cells were present in the upper CP at P0 (Fig. 6A, bottom row). We observed more GFP+ cells in the lower CP of Bre1a shRNA-electroporated brains than in control brains (t(6) = 2.606, p = 0.040 for middle CP; t(6) = 4.386, p = 0.005 for lower CP; Fig. 6B). GFP+ cells in the middle and lower CP of Bre1a shRNA-electroporated brains had leading processes, suggesting that they were still migrating. These findings suggest that Bre1a shRNA-expressing NPCs, which remain in the VZ/SVZ until E16.5, start differentiation and migrate to the CP. These results support the notion that, although Bre1a knockdown in NPCs does not affect the ability of these cells to migrate, it could delay the birth of postmitotic neuroblasts.
Figure 6.

Bre1a knockdown affects the cortical layer formation in the postnatal brain. A, B, Bre1a or control shRNA and GFP expression plasmids were electroporated into the dorsal forebrain of E13.5 embryos in utero, and the pups were perfused on P0. A, Coronal cryosections of the brains were immunostained for GFP (right), followed by Hoechst nuclear staining (left). The CP was divided into four layers based on the density of nuclei: the mantle zone (MZ); and upper (u), middle (m), and lower (l) CP. The borders between layers are indicated by dotted lines. B, Quantification of GFP+ cells distributed in the upper, middle, and lower CP in the P0 brain. C–F, Bre1a or control shRNA and GFP expression plasmids were electroporated into the dorsal forebrain of E14.5 embryos in utero, and the pups were perfused on P14. For rescue experiments, Bre1a expression plasmids were coelectroporated with Bre1a shRNA 2. C–E, Coronal cryosections were immunostained for GFP (middle), followed by Hoechst nuclear staining (left), or were subjected to in situ hybridization with a probe for Rorb, a marker of cortical layer IV (right). F, Quantitative analysis of GFP+ cells in cortical layers II/III and IV. Scale bars, 100 μm. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by Student's or Welch's t test.
Next, we investigated the final destination of GFP+ shRNA-expressing cells. To do this, we electroporated Bre1a or control shRNA into the cortex of E14.5 embryos and analyzed the brains at P14, when formation of the cortical layers is mostly completed. In the control shRNA-electroporated brain, approximately one-quarter of the GFP+ cells were present in cortical layer IV, which is labeled by the expression of Rorb, while the others were located in layers II/III (Fig. 6C). In contrast, in Bre1a shRNA-electroporated brains, the majority of GFP+ cells were found in layers II/III (Fig. 6D). As a result, there were fewer GFP+ cells present in layer IV of Bre1a shRNA-electroporated brains compared with the control brain (t(8) = 3.446, p = 0.009 for layers II/III; t(8) = 3.834, p = 0.005 for layer IV; Fig. 6F). We confirmed that the impaired distribution of GFP+ Bre1a shRNA-expressing cells was reversed by the coelectroporation of Bre1a overexpression plasmid (Fig. 6E,F). We also confirmed that there was no ectopic expression of Rorb in layers II/III of Bre1a shRNA-electroporated brains, excluding the possibility that Bre1a shRNA-expressing cells that would normally settle in layer IV were now abnormally located in layers II/III (Fig. 6D).
Finally, we tested the idea that Bre1a knockdown in NPCs delays the birth of differentiating neuroblasts by injecting EdU into dams 24 or 48 h after an E14.5 electroporation, and analyzing the brains at P14. When EdU was administered 24 h after the electroporation, the ratios of EdU+/GFP+ cells in layers II/III and IV of Bre1a shRNA-electroporated brains were comparable to the ratios in control brains (Fig. 7A–C). In contrast, when EdU was administered 48 h after the electroporation, the ratios of EdU+/GFP+ cells in layers II/III and IV were significantly higher in Bre1a shRNA-electroporated brains than the ratios in control shRNA-electroporated brains (t(7) = 3.955, p = 0.005 for layers II/III; t(7) = 3.834, p = 0.002 for layer IV; Fig. 7D,E,G). These phenotypes were completely rescued by the overexpression of Bre1a (Fig. 7F,G). These observations, together with our findings that Bre1a knockdown suppressed EdU incorporation 24 h after electroporation (Fig. 3C,D) and that it reduced the number of mitotic cells during the 30 h time-lapse imaging (Fig. 3E,F), are consistent with the notion that Bre1a knockdown lengthens the cell cycle and delays the final division of NPCs.
Figure 7.

Bre1a knockdown delays the birth date of differentiating neuroblasts. Bre1a or control shRNA and GFP expression plasmids were electroporated into the dorsal forebrain of E14.5 embryos in utero, and the pups were perfused on P14. For rescue experiments, Bre1a expression plasmids were coelectroporated with Bre1a shRNA 2. A, B, D–F, Dams received an intraperitoneal injection of EdU 24 h (A, B) or 48 h (D–F) after the electroporation. Coronal cryosections were immunostained for GFP (left), followed by EdU visualization (middle), and the merged images are shown (right). Arrowheads indicate GFP and EdU double-positive cells. C, G, Quantification of GFP+/EdU+ cells in cortical layers II/III and IV. Scale bars, 50 μm. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by Student's or Welch's t test.
Bre1a is required for the differentiation of neural precursor cells
We investigated the effects of Bre1a knockdown in the differentiation of NPCs by electroporating the Bre1a shRNA expression vectors into neuroepithelial cells from E10.5 forebrain. The cells were cultured in conditions that promote differentiation for 3 d, and then we assessed neuronal differentiation by immunostaining for βIII tubulin, a marker of immature neurons. All three Bre1a shRNAs markedly suppressed neuronal differentiation (F(3,8) = 35.56, p < 0.001; Fig. 8A,B), and this suppression was reversed by Bre1a overexpression (t(6) = 4.103, p = 0.006; Fig. 8B). These results suggest that Bre1a knockdown prevents NPC differentiation, which is considered to be independent of the effects on the cell cycle because the cell proliferation is suppressed in the differentiation culture conditions.
Figure 8.

Bre1a facilitates the differentiation of neural precursor cells. A, B, Primary neuroepithelial cells from E10.5 forebrains were transfected with Bre1a or control shRNAs and GFP expression plasmids, and cultured in differentiation condition for 3 d. Bre1a or control expression plasmids were cotransfected with Bre1a-shRNA 2 for rescue experiments. A, Cells were immunostained for GFP and βIII tubulin. Arrowheads and arrows indicate GFP+/βIII tubulin+ and GFP+/βIII tubulin− cells, respectively. B, Quantification of GFP+/βIII tubulin+ cells. C–J, Bre1a or control shRNA, pCX-Bre1a, or control expression plasmids were microinjected into the lateral ventricle of E13.5 embryos and electroporated into the dorsal forebrain in utero. The embryos were fixed 24 h after electroporation, and the brains were analyzed. C–J, Cryosections were immunostained for GFP and Tbr2 (C, G) or Pax6 (E, I). GFP+/Tbr2+ (D, H) or GFP+/Pax6+ (F, J) cells were counted. Scale bars: A, 50 μm; C, E, G, I, 100 μm. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by one-way ANOVA followed by Dunnett's post hoc comparison (B, D, F) or by Student's t test (H, J).
Next, we examined whether or not Bre1a knockdown suppresses the differentiation of NPCs in vivo. In the VZ, NPCs that are positive for Pax6 differentiate and migrate radially to the SVZ. At the same time, these cells downregulate Pax6 expression and upregulate Tbr2 expression. Bre1a knockdown in cortical NPCs of E13.5 brains resulted in decreased numbers of Tbr2+ cells (F(2,9) = 12.69, p = 0.002; Fig. 8C,D), although the percentage of Pax6+ cells was not significantly increased (Fig. 8E,F). Conversely, when Bre1a expression plasmid was electroporated into E13.5 cortex, the number of Tbr2+ cells in the SVZ increased (t(7) = 4.452, p = 0.003; Fig. 8G,H) and the number of Pax6+ cells in the VZ decreased (t(7) = 3.442, p = 0.011; Fig. 8I,J) compared with those in control brains. These results suggest that Bre1a promotes neuronal differentiation of NPCs in vivo.
Our discovery that Bre1a knockdown suppresses the differentiation of NPCs prompted us to investigate Notch signaling activity, because the Notch pathway plays critical roles in keeping the stem cell population undifferentiated (Chiba, 2006; Shimizu et al., 2010). We examined the expression of Hes5, a Notch signaling effector gene, in brains that had received in utero electroporation at E13.5. Three days after electroporation, we observed ectopic Hes5 expression in the SVZ and intermediate zone (IMZ), where endogenous Hes5 expression is absent (Fig. 9A,B). In addition, the results of quantitative RT-PCR indicated that Bre1a shRNA-expressing primary neurosphere cells derived from E13.5 neocortex expressed significantly more Hes5 than control shRNA-expressing cells (F(3,16) = 4.252, p = 0.022; Fig. 9C). In contrast, Bre1a knockdown had little effect on the expression of Hes1, another Notch effector gene (Fig. 9D), suggesting that the upregulation of Hes5 brought about by the expression of Bre1a shRNAs requires Hes5-specific modification of Notch signaling activation.
Figure 9.
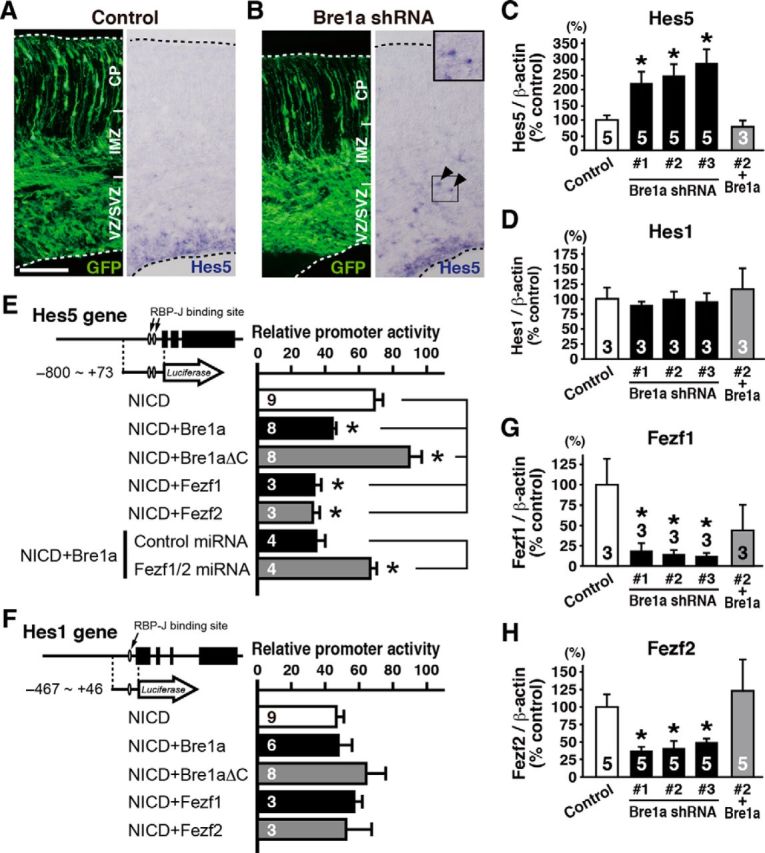
Bre1a knockdown upregulates Hes5 expression in neural precursor cells. A, B, Bre1a or control shRNA and GFP expression plasmids were electroporated into the dorsal forebrain of E13.5 embryos in utero, and brains were analyzed 3 d after the electroporation. Coronal cryosections were immunostained for GFP (left) or subjected to in situ hybridization with an RNA probe for Hes5 (right). Arrowheads indicate ectopic Hes5+ cells outside the VZ. The boxed area is magnified in the inset. Scale bar, 50 μm. C, D, G, H, Primary neurosphere cells derived from E13.5 cortex were infected with lentiviruses expressing Bre1a or control shRNAs and were cultured for 4–7 d. Lentivirus-infected GFP+ neurospheres were used for quantitative RT-PCR analysis of Hes5 (C), Hes1 (D), Fezf1 (G), or Fezf2 (H). For rescue experiments, Bre1a-expressing retrovirus was coinfected with Bre1a shRNA-expressing lentivirus. E, F, Firefly luciferase reporter assays in Neuro2a cells using the following plasmids: a plasmid containing the promoter region of the Hes5 (E) or Hes1 (F) gene; the Renilla luciferase reporter plasmid as an internal control; pCX-NICD, and the indicated plasmid for expression of Bre1a, Bre1aΔC, Fezf1, Fezf2, and Fezf1, and Fezf2 miRNAs. The relative promoter activities were compared with activity in the absence of pCX-NICD. Error bars indicate SEM, and n values are shown in columns. *p < 0.05 by one-way ANOVA followed by Dunnett's post hoc comparison or Student's t test.
We next examined whether or not Bre1a alters the activity of the Notch1 intracellular domain (NICD), an active form of the Notch1 receptor, on the Hes5 promoter. We cotransfected an Hes5 promoter reporter plasmid and an NICD expression vector, together with either the full-length Bre1a or Bre1aΔC expression plasmid, into Neuro2a cells, and determined the promoter activity using a luciferase assay. We found that Bre1a significantly attenuated the activity of NICD, but Bre1aΔC enhanced it (F(4,26) = 16.96, p < 0.001; Fig. 9E), suggesting that Bre1aΔC possibly works as a dominant-negative form. The expression of Bre1a or Bre1aΔC did not affect the activity of NICD on the Hes1 promoter (Fig. 9F). These results led us to investigate the expression of Fezf1 and Fezf2, because both of these genes are known to suppress the expression of Hes5 but not of Hes1 (Shimizu et al., 2010). Indeed, quantitative RT-PCR analysis using primary neurospheres infected with shRNA-expressing lentivirus revealed that the expressions of both Fezf1 and Fezf2 were significantly decreased in neurospheres expressing Bre1a shRNA compared with that in control shRNA-expressing neurospheres (F(3,8) = 6.412, p = 0.016 for Fezf1; F(3,16) = 9.350, p < 0.001 for Fezf2; Fig. 9G,H). The upregulation of Hes5 and downregulation of both Fezf1 and Fezf2 in Bre1a shRNA-expressing neurospheres were all rescued by Bre1a overexpression (Fig. 9C,D,G,H). Last, we examined the activity of NICD on the Hes5 promoter in the presence of miRNAs for Fezf1 and Fezf2. After knockdown of both Fezf1 and Fezf2 expression, the activity of NICD on the Hes5 promoter was no longer affected by Bre1a (t(6) = 4.613, p = 0.004; Fig. 9E). These results suggest that Bre1a suppresses the expression of Hes5 through the action of Fezf1 and Fezf2, which regulate the undifferentiated state of NPCs.
Discussion
Despite the importance of cell cycle regulation in the maintenance of NSCs in the developing and adult brain, the precise mechanism responsible for the cell cycle regulation of NSCs has remained largely unknown. Previous reports that the average cell cycle length of differentiating neuron-generating divisions is longer than that of proliferating divisions of NPCs, and that inhibition of CDKs induces the cell cycle elongation and premature neuronal differentiation of NPCs (Calegari and Huttner, 2003; Calegari et al., 2005), which appears inconsistent with our results that Bre1a knockdown elongates the cell cycle times and suppresses the differentiation of NPCs. However, this inconsistency disappears if one considers, at least, two subpopulations among vigorously dividing NPCs: Bre1a-expressing NPCs that are destined to differentiate into neuroblasts and Bre1a− or low cells that are kept undifferentiated and maintained as NSCs, the latter of which may be overlooked in the previous cell cycle analysis (Calegari et al., 2005) due to very small population size. Indeed, NSCs in the developing brain show much longer cell cycle times than vigorously dividing non-self-renewing NPCs (Martens et al., 2000).
We demonstrated here that the reduction of Bre1a expression, and the subsequent decrease of H2B monoubiquitylation, lengthens the G1/S phase in the cell cycle of NPCs, which is mediated by the upregulation of p57Kip2 and downregulation of Cdk2. Since p57Kip2 is one of the Cip/Kip type CDK inhibitors that mainly target Cdk2 and Cdk4 (Fujita, 2003), the downregulation of Cdk2 following Bre1a knockdown may be a consequence of p57Kip2 upregulation. In adult NSCs, loss of p21Cip1, another member of the Cip/Kip family of CDK inhibitors, results in a limited capacity for self-renewal and leads to exhaustion following prior expansion (Kippin et al., 2005). More recently, p57Kip2 was shown to control the quiescence of neural stem cells in adult brains (Furutachi et al., 2013). Interestingly, the Cip/Kip family of CDK inhibitors has been implicated in regulation of the repopulation capacity of hematopoietic stem cells (HSCs) in adults (Cheng et al., 2000a,b; van Os et al., 2007), and p57Kip2 plays a predominant role in the acquisition of quiescence in adult HSCs (Matsumoto et al., 2011; Zou et al., 2011). Therefore, it is possible that various stem cell populations require the function of Cip/Kip family members for the elongation of cell cycle during development and subsequent acquisition of quiescence by p57Kip2, in which the Bre1a–p57Kip2 pathway may participate in and for the maintenance of quiescence in adults, by p21Cip1, p27Kip1, or both.
Bre1a knockdown induces the Fezf1/2-mediated upregulation of Hes5 and maintained NPCs in an undifferentiated state, which contributes, at least in part, to the impaired migration of NPCs from the VZ/SVZ to the CP. The differentiation status of NPCs, in which Notch signaling plays a critical role, and their cell cycle length, which is controlled by the expression of Cip/Kip family genes, appear tightly related with each other. Overexpression of Hes5 in NPCs of mouse embryonic cortex by in utero electroporation results in the suppression of proliferation in addition to the suppression of differentiation (Hitoshi et al., 2011), and conversely, p57kip2 acts as a repressor of the differentiation of NPCs (Joseph et al., 2009). It remains to be investigated whether there is a causative link between the maintenance of cells in undifferentiated state and the elongation of cell cycle time after the knockdown of Bre1a in NPCs or whether Bre1a alters the expression of multiple genes, including Cdks and their inhibitors and Fezf1/2, by catalyzing histone H2B monoubiquitylation of those gene loci. If the latter is a case, Bre1a could be a bifunctional gene that regulates both the differentiation status and cell cycle length of NPCs and plays an essential role in the neural development.
In conclusion, our study suggests that monoubiquitylation of H2B by Bre1a regulates differentiation and the cell cycle time of NPCs during development. We propose a model that Bre1a-negative cells in the VZ of the embryonic cortex could be selected as self-renewing NSCs that lengthen their cell cycle as development proceeds.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research (B) 19300136 and 24300136, and for Scientific Research on Priority Areas 17025041 and 18023035 from the Ministry of Education, Culture, Sports, Science and Technology of Japan; and by grants from the Sankyo Foundation of Life Science and the Astellas Foundation for Research on Metabolic Disorders (S.H.). We thank the Spectrography and Bioimaging, and the Functional Genomics Facilities of the National Institute for Basic Biology Core Research Facilities, for technical support. We also thank M. Hibi, R. Kageyama, T. Miyazaki, and Y. Fukazawa for providing plasmids; and N. Yamauchi, K. Inaba, R. Taguchi, and M. Tomoeda for technical assistance.
The authors declare no competing financial interests.
References
- Abe T, Kiyonari H, Shioi G, Inoue K, Nakao K, Aizawa S, Fujimori T. Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis. 2011;49:579–590. doi: 10.1002/dvg.20753. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray S, Musisi H, Bienz M. Bre1 is required for notch signaling and histone modification. Dev Cell. 2005;8:279–286. doi: 10.1016/j.devcel.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001;15:3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Spradling AC. Drosophila stem cells share a common requirement for the histone H2B ubiquitin protease Scrawny. Science. 2009;323:248–251. doi: 10.1126/science.1165678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calegari F, Huttner WB. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci. 2003;116:4947–4955. doi: 10.1242/jcs.00825. [DOI] [PubMed] [Google Scholar]
- Calegari F, Haubensak W, Haffner C, Huttner WB. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci. 2005;25:6533–6538. doi: 10.1523/JNEUROSCI.0778-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27kip1. Nat Med. 2000a;6:1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000b;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- Fuchs G, Shema E, Vesterman R, Kotler E, Wolchinsky Z, Wilder S, Golomb L, Pribluda A, Zhang F, Haj-Yahya M, Feldmesser E, Brik A, Yu X, Hanna J, Aberdam D, Domany E, Oren M. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell. 2012;46:662–673. doi: 10.1016/j.molcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S. The discovery of the matrix cell, the identification of the multipotent neural stem cell and the development of the central nervous system. Cell Struct Funct. 2003;28:205–228. doi: 10.1247/csf.28.205. [DOI] [PubMed] [Google Scholar]
- Furutachi S, Matsumoto A, Nakayama KI, Gotoh Y. p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. 2013;32:970–981. doi: 10.1038/emboj.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Plevani P, Muzi-Falconi M. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem. 2005;280:9879–9886. doi: 10.1074/jbc.M414453200. [DOI] [PubMed] [Google Scholar]
- Higashi M, Maruta N, Bernstein A, Ikenaka K, Hitoshi S. Mood stabilizing drugs expand the neural stem cell pool in the adult brain through activation of notch signaling. Stem Cells. 2008;26:1758–1767. doi: 10.1634/stemcells.2007-1032. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 2009;63:600–613. doi: 10.1016/j.neuron.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Hitoshi S, Tropepe V, Ekker M, van der Kooy D. Neural stem cell lineages are regionally specified, but not committed, within distinct compartments of the developing brain. Development. 2002a;129:233–244. doi: 10.1242/dev.129.1.233. [DOI] [PubMed] [Google Scholar]
- Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS, Conlon RA, Mak TW, Bernstein A, van der Kooy D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002b;16:846–858. doi: 10.1101/gad.975202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitoshi S, Ishino Y, Kumar A, Jasmine S, Tanaka KF, Kondo T, Kato S, Hosoya T, Hotta Y, Ikenaka K. Mammalian Gcm genes induce Hes5 expression by active DNA demethylation and induce neural stem cells. Nat Neurosci. 2011;14:957–964. doi: 10.1038/nn.2875. [DOI] [PubMed] [Google Scholar]
- Hwang WW, Venkatasubrahmanyam S, Ianculescu AG, Tong A, Boone C, Madhani HD. A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol Cell. 2003;11:261–266. doi: 10.1016/S1097-2765(02)00826-2. [DOI] [PubMed] [Google Scholar]
- Joseph B, Andersson ER, Vlachos P, Södersten E, Liu L, Teixeira AI, Hermanson O. p57Kip2 is a repressor of Mash1 activity and neuronal differentiation in neural stem cells. Cell Death Differ. 2009;16:1256–1265. doi: 10.1038/cdd.2009.72. [DOI] [PubMed] [Google Scholar]
- Kao CF, Hillyer C, Tsukuda T, Henry K, Berger S, Osley MA. Rad6 plays a role in transcriptional activation through ubiquitylation of histone H2B. Genes Dev. 2004;18:184–195. doi: 10.1101/gad.1149604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpiuk O, Najafova Z, Kramer F, Hennion M, Galonska C, König A, Snaidero N, Vogel T, Shchebet A, Begus-Nahrmann Y, Kassem M, Simons M, Shcherbata H, Beissbarth T, Johnsen SA. The histone H2B monoubiquitination regulatory pathway is required for differentiation of multipotent stem cells. Mol Cell. 2012;46:705–713. doi: 10.1016/j.molcel.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Kim J, Roeder RG. Direct Bre1-Paf1 complex interactions and RING finger-independent Bre1-Rad6 interactions mediate histone H2B ubiquitylation in yeast. J Biol Chem. 2009;284:20582–20592. doi: 10.1074/jbc.M109.017442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005;19:756–767. doi: 10.1101/gad.1272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martínez-Cerdeño V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci. 2006;7:883–890. doi: 10.1038/nrn2008. [DOI] [PubMed] [Google Scholar]
- Martens DJ, Tropepe V, van Der Kooy D. Separate proliferation kinetics of fibroblast growth factor-responsive and epidermal growth factor-responsive neural stem cells within the embryonic forebrain germinal zone. J Neurosci. 2000;20:1085–1095. doi: 10.1523/JNEUROSCI.20-03-01085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, Nakayama K, Nakayama KI. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Minsky N, Shema E, Field Y, Schuster M, Segal E, Oren M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol. 2008;10:483–488. doi: 10.1038/ncb1712. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Craig CG, van der Kooy D. In vivo clonal analyses reveal the properties of endogenous neural stem cell proliferation in the adult mammalian forebrain. Development. 1998;125:2251–2261. doi: 10.1242/dev.125.12.2251. [DOI] [PubMed] [Google Scholar]
- Naruse M, Nakahira E, Miyata T, Hitoshi S, Ikenaka K, Bansal R. Induction of oligodendrocyte progenitors in dorsal forebrain by intraventricular microinjection of FGF-2. Dev Biol. 2006;297:262–273. doi: 10.1016/j.ydbio.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Nicassio F, Corrado N, Vissers JH, Areces LB, Bergink S, Marteijn JA, Geverts B, Houtsmuller AB, Vermeulen W, Di Fiore PP, Citterio E. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr Biol. 2007;17:1972–1977. doi: 10.1016/j.cub.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Nakazawa M, Kani S, Bae YK, Shimizu T, Kageyama R, Hibi M. Zinc finger genes Fezf1 and Fezf2 control neuronal differentiation by repressing Hes5 expression in the forebrain. Development. 2010;137:1875–1885. doi: 10.1242/dev.047167. [DOI] [PubMed] [Google Scholar]
- Shirane D, Sugao K, Namiki S, Tanabe M, Iino M, Hirose K. Enzymatic production of RNAi libraries from cDNAs. Nat Genet. 2004;36:190–196. doi: 10.1038/ng1290. [DOI] [PubMed] [Google Scholar]
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001;1:749–758. doi: 10.1016/S1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, Dontje B, de Haan G. A limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007;25:836–843. doi: 10.1634/stemcells.2006-0631. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, Wyce A, Thorne AW, Berger SL, McMahon SB. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008;29:102–111. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann C, Chymkowitch P, Eldholm V, Putnam CD, Lindvall JM, Omerzu M, Bjørås M, Kolodner RD, Enserink JM. A chemical-genetic screen to unravel the genetic network of CDC28/CDK1 links ubiquitin and Rad6-Bre1 to cell cycle progression. Proc Natl Acad Sci U S A. 2011;108:18748–18753. doi: 10.1073/pnas.1115885108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, Maru Y, Nakayama K, Nakayama KI, Suda T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]