

Abstract
An increasing number of proteins involved in genome organization have been implicated in neurodevelopmental disorders, highlighting the importance of chromatin architecture in the developing CNS. The CCCTC-binding factor (CTCF) is a zinc finger DNA binding protein involved in higher-order chromatin organization, and mutations in the human CTCF gene cause an intellectual disability syndrome associated with microcephaly. However, information on CTCF function in vivo in the developing brain is lacking. To address this gap, we conditionally inactivated the Ctcf gene at early stages of mouse brain development. Cre-mediated Ctcf deletion in the telencephalon and anterior retina at embryonic day 8.5 triggered upregulation of the p53 effector PUMA (p53 upregulated modulator of apoptosis), resulting in massive apoptosis and profound ablation of telencephalic structures. Inactivation of Ctcf several days later at E11 also resulted in PUMA upregulation and increased apoptotic cell death, and the Ctcf-null forebrain was hypocellular and disorganized at birth. Although deletion of both Ctcf and Puma in the embryonic brain efficiently rescued Ctcf-null progenitor cell apoptosis, it failed to improve neonatal hypocellularity due to decreased proliferative capacity of rescued apical and outer radial glia progenitor cells. This was exacerbated by an independent effect of CTCF loss that resulted in depletion of the progenitor pool due to premature neurogenesis earlier in development. Our findings demonstrate that CTCF activities are required for two distinct events in early cortex formation: first, to correctly regulate the balance between neuroprogenitor cell proliferation and differentiation, and second, for the survival of neuroprogenitor cells, providing new clues regarding the contributions of CTCF in microcephaly/intellectual disability syndrome pathologies.
Keywords: apoptosis, CTCF, differentiation, mouse model, neurogenesis, Puma
Introduction
CTCF is a multifunctional DNA binding protein that regulates higher-order chromatin structure to influence transcriptional regulation, genomic imprinting, X chromosome inactivation, and chromatin insulation (Holwerda and de Laat, 2013). It binds to a variety of highly divergent target sequences throughout the genome using a combination of its 11 zinc finger motifs (Nakahashi et al., 2013). CTCF partners with a number of chromatin-related proteins such as the cohesin complex (Parelho et al., 2008; Wendt et al., 2008), nucleophosmin, and CTCF itself (Yusufzai and Felsenfeld, 2004; Yusufzai et al., 2004). These interactions may allow CTCF sites to contact one another and/or to be tethered to subnuclear domains. CTCF-mediated chromatin interactions detected by Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) correlate with ∼10% of all CTCF binding sites (CBSs), however, indicating that CTCF likely plays additional roles within the cell (Handoko et al., 2011).
De novo mutations in CTCF have been identified previously in patients with varying degrees of intellectual disability and microcephaly (Gregor et al., 2013), highlighting the importance of chromatin organization for the normal development and function of the CNS. Whereas CTCF function has been studied extensively in cell culture systems, its function in an in vivo context remains to be completely resolved (Ohlsson et al., 2010). Ubiquitous deletion of CTCF in the mouse leads to lethality before embryonic day 3.5 (E3.5), suggesting that it is essential for early developmental processes (Fedoriw et al., 2004; Heath et al., 2008; Moore et al., 2012). Conditional deletion of Ctcf in specific tissues causes either reduced proliferation or apoptotic cell death, depending on the tissue targeted for Cre recombination. For example, deletion of Ctcf in thymocytes resulted in increased p21 and p27 expression and cell cycle arrest, whereas reduced CTCF in mouse oocytes induced meiotic and mitotic defects and apoptotic cell death before the blastocyst stage (Fedoriw et al., 2004; Heath et al., 2008; Wan et al., 2008). Deletion of Ctcf in the developing limb bud resulted in massive apoptosis and near-complete loss of limb structures, accompanied by increased p53 upregulated modulator of apoptosis (PUMA), a known activator of caspase-mediated apoptosis (Nakano and Vousden, 2001; Yu et al., 2001; Soshnikova et al., 2010). In human cancer cells, CTCF binds to the Puma gene, and its depletion results in increased Puma transcript and rapid apoptosis, indicating that Puma transcription can be directly influenced by the presence or absence of CTCF (Gomes and Espinosa, 2010a,b).
Given the deleterious effects of CTCF mutations in the human CNS, we specifically inactivated Ctcf in the developing mouse brain. CTCF loss of function using two different Cre driver lines in mice triggered apoptosis in dividing neuroprogenitor cells (NPCs) of the forebrain. Despite prevention of apoptosis by Puma deletion, rescued Ctcf/Puma double-null apical and outer radial glia (oRG) progenitors exhibited decreased proliferative capacity. Furthermore, loss of CTCF caused premature neurogenesis, resulting in depletion of the progenitor pool and a microcephaly phenotype at birth. These findings highlight the complexity of CTCF activities during neurogenesis.
Materials and Methods
Mouse husbandry and genotyping.
Mice were exposed to 12 h light/dark cycles and fed tap water and regular chow ad libitum. The CtcfloxP mice, in which loxP sites flank exons 3–12, have been described previously (Heath et al., 2008). Mice conditionally deficient in CTCF were generated by crossing CtcfloxP/+ females (C57BL/6 background) with heterozygous Foxg1Cre knock-in male mice (129/sv background) or with NestinCre heterozygous male mice (C57BL/6 background; Hébert and McConnell, 2000; Bérubé et al., 2005). To account for decreased Foxg1 expression due to knock-in of the Cre recombinase gene, Cre+ males were used as controls (Ctcf+/+Foxg1-cre+/−) unless stated otherwise. Using the NestinCre driver line, CtcfloxP/loxP mice were crossed with CtcfloxP/+;Nestin+ mice to generate Ctcflox/P/loxP or CtcfloxP/+ (controls) and CtcfloxP/loxP;Nestin+ (CtcfNes-cre). Puma−/− (Bbc3tm1Ast) mice were obtained from The Jackson Laboratory (stock #011067; Villunger et al., 2003). DNA from tail biopsies of newborn pups or yolk sac from embryos was genotyped by PCR. Primer sequences are provided in Table 1.
Table 1.
Primer sequences used for genotyping, gene expression, and qChIP analyses
| Primer Name | Forward sequence (5′-3′) | Reverse sequence (5′-3′) |
|---|---|---|
| Ctcf | CTAGGAGTGTAGTTCAGTGAGGCC | GCTCTAAAGAAGGTTGTGAGTTC |
| NestinCre | TGACCAGAGTCATCCTTAGCG | AATGCTTCTGTCCGTTTGCC |
| Sry | GCAGGTGGAAAAGCCTTACA | AAGCTTTGCTGGTTTTTGGA |
| Puma genotyping WT | AGGCTGTCCCTGCGGTCATCC | GGACTGTCGCGGGCTAGACCCTCTA |
| Puma genotyping KO | AGGCTGTCCCTGCGGTCATCC | ACCGCGGGCTCCGAGTAGC |
| Ctcf expression | CGATATGCTCTCATCCAGCA | TCCCACACTTGGAACAGACA |
| Puma expression | CGTGTGGAGGAGGAGGAGT | GGAGGAGTCCCATGAAGAGA |
| β-actin expression | CTGTCGAGTCGCGTCCACCC | ACATGCCGGAGCCGTTGTCG |
| 5′ 5 kb Puma CBS | ACCCTCGTGTTTGGAGTGAC | CTCCTGCCTTGTGCCTAAAG |
| Puma CBS1 | GCTCCTCCCAGGTCTCACTA | CAGCTTTCATCACTGGGACT |
| Puma CBS2 | AGGAATGGATCTGCTGGATG | GTTGCTGACTCACCGGCTAT |
| 3′ 5 kb Puma CBS | GAGCCCCTGCCTAGTAGGAT | TAGTCCCTGTGTGTGCTTGC |
| 5′ 1 kb p53BS | AAAAATGGGCTTGGAGAGC | CCACCACTGTCCAGCTTGTT |
| Puma p53BS | CTGTCCCCACGCTGC | GCTTGCTTGCTGGTGTCG |
| 3′ 1 kb p53BS | AGCCAGGGCTACACAGAGAA | CTGAGCCATCTCTCCAGTCC |
KO, Knock-out; WT, wild type.
Immunostaining and histology.
For immunofluorescence staining, cryosections were incubated with the primary antibody overnight at 4°C, washed in PBS/0.3% Triton-X 100, and incubated with the secondary antibody for 1 h. Sections were counterstained with DAPI (Sigma-Aldrich) and mounted in SlowFade Gold (Invitrogen). Primary antibodies used were as follows: rabbit anti-CTCF (1:400; Cell Signaling Technology), rabbit anti-cleaved caspase 3 (AC3; Asp175; 1:400; Cell Signaling Technology), mouse anti-BrdU (1:50; BD Biosciences), rabbit anti-PUMA (1:200; Cell Signaling Technology), rabbit anti-TBR2 (1:200; Abcam), goat anti-SOX2 (1:400; Santa Cruz Biotechnology), goat anti-PAX6 (1:200; Santa Cruz Biotechnology), rabbit anti-SOX2 (1:100; Millipore Bioscience Research Reagents), rabbit anti-Ki67 (1:200; Abcam), rabbit anti-TBR1 (1:200; Abcam), mouse anti-SATB2 (1:200; Abcam), and rabbit anti-CTIP2 (1:200; Abcam). Secondary antibodies used were as follows: goat-anti-rabbit Alexa 594 (1:800; Invitrogen), goat-anti-mouse Alexa 488 (1:800; Invitrogen), donkey-anti-sheep Alexa 594 (1:800; Invitrogen), and donkey-anti-mouse Alexa 488 (1:800; Invitrogen). Sections were subjected to antigen retrieval (incubated in 0.1 mm sodium citrate, pH 6.0, heated to ∼95°C and microwaved on low for 10 min) before overnight incubation (for BrdU, SOX2, PAX6, TBR2, STAB2, TBR1, and CTIP2). Terminal deoxynucleotidyl nick end labeling (TUNEL) was performed according to the manufacturer's instructions (Roche). For histological studies, slides were stained with hematoxylin and eosin (H&E).
BrdU labeling.
Pregnant mice were injected intraperitoneally with cell proliferation labeling reagent [10 mm bromodeoxyuridine (BrdU) and 1 mm fluorodeoxyuridine (FdU) in H2O] at 1 ml/100 g body weight, or 0.3 mg/g body weight (GE Healthcare Life Sciences). Animals were killed after 1 h by CO2 asphyxiation, and the embryos were recovered in ice-cold PBS, pH 7.4, and were fixed in 4% paraformaldehyde. Tissue was equilibrated in 30% sucrose/PBS and frozen in OCT (Tissue Tek). For cell cycle exit analysis, pregnant female mice (E13) were injected with cell proliferation labeling reagent at 1 ml/100 g body weight (GE Healthcare Life Sciences), and embryos were collected and processed for immunofluorescence analysis 24 h later. Before immunofluorescence analysis, cryosections (8 μm) were treated with 2N HCl to denature the DNA, and neutralized with 0.1 m Na2B4O7, pH 8.5.
Primary NPC cultures and manipulation.
Cortical progenitor cultures were prepared as described previously (Slack et al., 1998; Gloster et al., 1999; Watson et al., 2013) using cortices dissected from E12.5 embryos. Cells were seeded on polyornithine-coated (Sigma-Aldrich) plastic plates or glass coverslips. Cells were fixed in 4% paraformaldehyde for 10 min, washed in PBS, and processed for immunofluorescence. Cell viability was measured using the trypan blue dye exclusion method. Cell counts were determined with a hemacytometer.
Western blot analysis.
Nuclear protein was extracted from the E16.5 telencephalon using a standard extraction kit (Thermo Scientific) and quantified using the Bradford assay. Protein (20 μg) was resolved on a 6% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane (Bio-Rad). The membrane was probed with rabbit anti-p53 (1:500; Santa Cruz Biotechnology) and rabbit anti-inner centromere protein (INCENP) (1:10000; Sigma Aldrich) antibodies followed by the appropriate horseradish peroxidase-conjugated secondary antibodies (1:4000; GE Healthcare Life Sciences). The membrane was incubated in ECL before exposure to x-ray film. Densitometry analysis was performed using ImageJ software (version 1.47).
Real-time PCR.
Total RNA was isolated using the RNeasy Mini kit (Qiagen). First-strand cDNA was synthesized from 1 μg of total RNA using the SuperScriptTM II Reverse Transcriptase kit (Invitrogen) with deoxyribonucleotide triphosphates (dNTPs) (1 mm final concentration; GE Healthcare), porcine RNAguard (GE Healthcare), and 0.3 μg random primers (GE Healthcare). Amplification was performed using a Chromo-4 Continuous Fluorescence Detector (Bio-Rad) in the presence of iQ SYBR Green supermix (Bio-Rad) and recorded using Opticon Monitor 3 software (Bio-Rad). Results were normalized to β-actin expression, and relative gene expression levels were calculated using GeneX software (Bio-Rad). Samples were amplified as follows: 95°C for 10 s, annealed for 20 s, 72°C for 30 s. After amplification, a melting curve was generated, and samples were resolved on a 1.5% agarose gel (75 V for 1 h) to verify amplicon purity. Primer oligomers were designed using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) and were obtained from Integrated DNA Technologies. Sequences are provided in Table 1.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) experiments were performed as described previously (Kernohan et al., 2010). Briefly, mouse forebrain tissue was dissected and processed to single-cell suspension. Cells were cross-linked in 1% formaldehyde, lysed in SDS buffer and sonicated. Immunoprecipitation was performed with a rabbit anti-CTCF antibody (Cell Signaling Technology) and a rabbit anti-p53 antibody (Santa Cruz Biotechnology). Input samples represent 1/25 of total chromatin input. ChIP products were amplified in duplicate with iQ SYBR Green Master Mix (Bio-Rad) on a Chromo-4 thermocycler under the following conditions: 95°C for 5 min, followed by 35 cycles of 95°C for 10 s, 55°C for 20 s, 72°C for 30 s, and a final melting curve generated from 55°C to 95°C in increments of 1°C per plate read. Fold change and percent input formulas were adapted from (Mukhopadhyay et al., 2008) as follows: percent input = 100 * [2(ΔCtInput − ΔCtInput) − (ΔCtInput − ΔCtAb)]/25. Error bars represent the SEM. Primer sequences are provided in Table 1.
Microscopy.
Images were captured with a digital camera (ORCA-ER; Hamamatsu) using an inverted microscope (DMI 6000b; Leica). Openlab imaging software (PerkinElmer) was used for manual image capture, and processing was performed using Volocity software (PerkinElmer). For quantification of AC3+ cells per area, AC3+ cells were counted in a defined area in at least six serial cortical cryosections, and the ratio of AC3+ cells to area (square millimeters) was calculated. For BrdU, SOX2, TBR2, BrdU/Ki67, TBR1, SATB2, and CTIP2 quantification, at least two serial cortical cryosections were assessed for positive cells within the indicated regions per embryo. DAPI morphology was used to bin cortex into the ventricular/subventricular zone (VZ/SVZ), intermediate zone (IZ), and cortical plate (CP).
Statistical analyses.
Statistical analysis was performed using GraphPad Prism software (GraphPad Software; version 4.02), and all results are expressed as the mean ± SEM. Unless indicated otherwise, p values were generated using Student's t test (unpaired, two-tailed) to compare between two independent data sets. Genotype ratios (Figs. 1A, 2A) were compared using a χ2 test. To compare cell viability data (see Fig. 4E), a one-way ANOVA was used with Dunnett's multiple comparison post-test to compare the cell viability of each genotype (CtcfNes-cre, CtcfNes-cre;Puma−/−, and Puma−/−) to control.
Figure 1.

Foxg1Cre-mediated deletion of Ctcf results in a massive increase in apoptosis. A, Table of genotypes obtained during the embryonic and postnatal periods. Ratios at each time point were analyzed using a χ2 test. Het, Ctcfflox/WT;Foxg1-cre+/−. B, Dark-field images of control and CtcfFoxg1-cre embryos at E13.5. (Please note limbs were taken for genotyping). C, H&E staining of E13.5 sagittal cryosections demonstrates complete loss of cortex (Ctx), hippocampal hem (H), basal ganglia (BG), lens (L), and anterior retina (AR), but not the posterior retina (PR), in CtcfFoxg1-cre embryos. D, Dark-field images of control and CtcfFoxg1-cre embryos at E11.5. The dashed circle outlines the telencephalon, which is visibly reduced in size in the CtcfFoxg1-cre embryos compared to littermate controls. E, Immunodetection of CTCF (red) in E11.5 sagittal cryosections confirms specific loss of CTCF expression in the forebrain neuroepithelium of CtcfFoxg1-cre embryos. F, Pregnant females were subjected to a 1 h BrdU pulse before being killed. Immunodetection of BrdU in E11.5 control and CtcfFoxg1-cre cortical neuroepithelium is shown. G, BrdU+ cells were counted and expressed as a percentage of the total number of DAPI+ cells (n = 3). H, Immunodetection of activated caspase-3 (red) in control and CtcfFoxg1-cre cortical neuroepithelium at E11.5. I, AC3+ cells were counted and expressed as a percentage of the total number of DAPI+ cells (n = 3). J, TUNEL (green) detection in E11.5 control and CtcfFoxg1-cre cortical neuroepithelium. Error bars represent the SEM. Original magnification: C, 25×; E, 50×; F, H, J, 200×. Scale bars: C, top, 1 mm; bottom, 400 μm; E, 200 μm; F, H, 50 μm; J, 100 μm.
Figure 2.
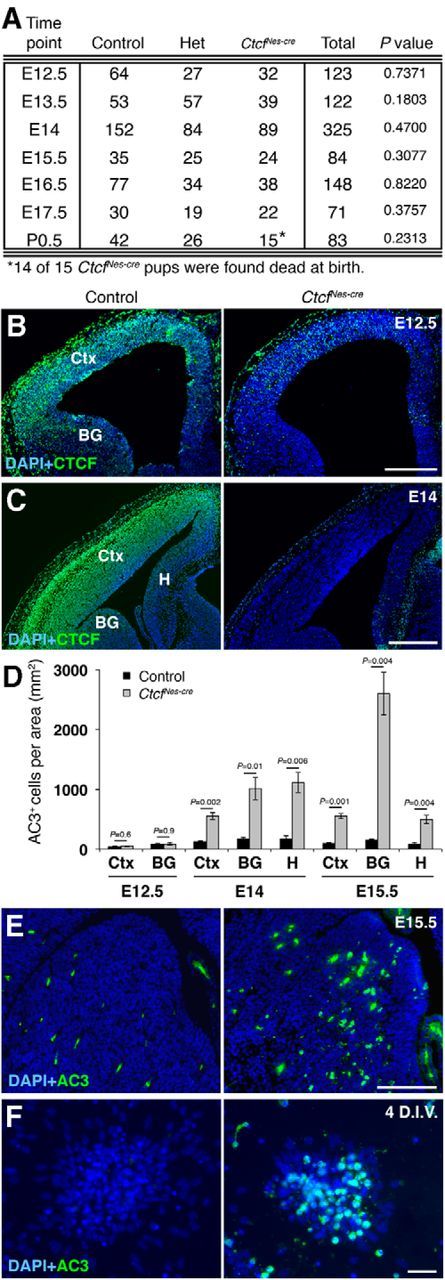
NestinCre-mediated deletion of CTCF results in activation of caspase-mediated apoptosis. A, Table of genotype ratios obtained during the embryonic period. Ratios at each time point were analyzed by a χ2 test. B, Immunodetection of CTCF in E12.5 control and CtcfNes-cre coronal forebrain sections. C, Immunodetection of CTCF in E14 control and CtcfNes-cre coronal forebrain sections. D, Quantification of AC3 immunostaining in E12.5, E14, and E15.5 forebrain tissue (n = 3). AC3+ cells were counted and expressed per unit area (square millimeter). E, Immunodetection of AC3 in E15.5 control and CtcfNes-cre basal ganglia. F, Immunodetection of AC3 in control and CtcfNes-cre at 4 DIV. Het, Ctcfflox/WT;Nestin-cre; Ctx, cortex; BG, basal ganglia; H, hippocampal hem. Error bars represent the SEM. Original magnification: B, C, 50×; D, 100×; E, 200×. Scale bars: B, 220 μm; C, 300 μm; E, 100 μm; F, 25 μm.
Figure 4.

Apoptosis is abolished upon deletion of PUMA in the Ctcf-deficient embryonic brain. A, H&E staining of E16.5 control, CtcfNes-cre, and CtcfNes-cre;Puma−/− coronal cortical cryosections. Skin and skull cap were not dissected from CtcfNes-cre brain. B, AC3 immunostaining in E16.5 control, CtcfNes-cre, and CtcfNes-cre;Puma−/− neocortex. C, DAPI+ cells were quantified in 200-μm-wide regions of the control, CtcfNes-cre (conditional knock-out), and CtcfNes-cre;Puma−/− (double knock-out) CP, IZ, and VZ/SVZ (n = 3). D, AC3+ cells were quantified per unit area in the cortex (Ctx), basal ganglia (BG), and hippocampal hem (H; n = 3). E, Cell viability of NPC cultures was measured by trypan blue dye exclusion after 2–5 DIV (n = 3). Data were analyzed by a one-way ANOVA followed by Dunnett's multiple comparisons test to determine which means were significantly different from the control. No significant difference in cell viability was found at 2 and 3 DIV (p = 0.2122 and 0.2062, respectively); however, viability was significantly different between CtcfNes-cre and control at 3 and 4 DIV (p < 0.01), but not between CtcfNes-cre;Puma−/− and control or Puma−/− and control (p > 0.05). Error bars represent SEM. Original magnification: A, 100×; C, 50×. Scale bars: A, 200 μm; B, 100 μm.
Study approval.
All procedures involving animals were conducted in accordance with the regulations of the Animals for Research Act of the Province of Ontario and approved by the University of Western Ontario Animal Care and Use Committee.
Results
CtcfFoxg1-cre mice exhibit widespread apoptosis and profound loss of telencephalic and anterior retinal tissue
To study the function of CTCF in the embryonic brain, Ctcf floxed mice were intercrossed with the previously characterized Foxg1Cre mice (Hébert and McConnell, 2000; Bérubé et al., 2005; Heath et al., 2008). Foxg1 expression is first detected between E8 and E9 in the telencephalic neuroepithelium, the basal ganglia, the olfactory bulbs, and the anterior retina (Dou et al., 1999). Foxg1-cre+ CtcfloxP/loxP progeny resulting from this cross are Ctcf null in the anterior retina and forebrain, and will be referred to as CtcfFoxg1-cre throughout this text.
We failed to recover live CtcfFoxg1-cre pups at birth (Fig. 1A). At E13.5, the size of telencephalic and retinal structures was greatly diminished in mutant embryos (Fig. 1B,C). At E11.5, the telencephalon of mutant embryos was already noticeably smaller compared to littermate-matched controls (Fig. 1D, hatched circle). CTCF immunostaining demonstrated nuclear expression of the protein throughout the forebrain in control tissue and its absence in mutant embryos (Fig. 1E). To determine the underlying cause of cell loss, we measured levels of proliferation and cell death in E11.5 CtcfFoxg1-cre and control embryos. Acute (1 h) BrdU labeling of control and CtcfFoxg1-cre embryos revealed that the percentage of cells in S phase does not vary significantly between mutant and control forebrain tissue (Fig. 1F,G). To measure apoptosis, we stained sections with an antibody against activated caspase-3 as a marker of cell death (Fig. 1H). Quantification of the results revealed a large increase in the proportion of cells undergoing apoptotic cell death in CtcfFoxg1-cre embryos compared to littermate-matched controls (Fig. 1I). TUNEL also showed increased apoptosis in CtcfFoxg1-cre embryos compared to control (Fig. 1J). Since we already observed profound cell loss by E11.5, we wanted to analyze whether a similar mechanism was occurring earlier in this system. We observed increased AC3 immunostaining in CtcfFoxg1-cre forebrain cryosections at E10.5, confirming that cell death occurs at this earlier time point (data not shown). We conclude that deletion of Ctcf in the mouse forebrain at approximately E8.5 causes extensive apoptosis resulting in profound loss of telencephalic and anterior retinal tissue by E13.5.
NestinCre-driven inactivation of Ctcf decreases cell survival
To determine the outcome of Ctcf deletion at a later embryonic time point in the telencephalon, we mated Ctcfflox/flox mice to the previously characterized NestinCre transgenic mice (Bérubé et al., 2005; Heath et al. 2008). The resulting mutant is hereafter referred to as CtcfNes-cre for simplicity. Cre is expressed after preplate formation in this system, resulting in specific deletion of Ctcf in neural progenitor cells at approximately E11 (Bérubé et al., 2005). We obtained the expected ratio of CtcfNes-cre mice at birth; however, almost all mutant pups were already dead and blue in color (Fig. 2A). One live CtcfNes-cre pup was found struggling to breathe, appeared weak, and was killed. We conclude that neonatal lethality of CtcfNes-cre mice is likely due to asphyxiation at birth.
We first established that the CTCF protein was lost in the basal ganglia and many cells of the cortex at E12.5 (Fig. 2B), and expression was undetectable in the E14 CtcfNes-cre cortex, basal ganglia, and hippocampal hem (Fig. 2C). To determine whether NestinCre-mediated deletion of Ctcf also induces apoptosis, we analyzed CtcfNes-cre forebrain cryosections at E12.5–E15.5 for evidence of AC3. At E12.5, we observed no increase in apoptotic cells in the mutant telencephalon (Fig. 2D). However, at E14, we detected an increase in AC3+ cells in the CtcfNes-cre cortex, basal ganglia, and hippocampal hem compared to controls (Fig. 2D). The increase in caspase activation was also observed at E15.5 in all three forebrain regions, with the highest level of AC3+ cells in the basal ganglia (Fig. 2D,E). Increased apoptotic cell death was also observed in vitro in primary cortical progenitor cultures established from E12.5 control and Ctcf-null telencephalon. Levels of AC3 staining were low in both control and Ctcf-null NPCs after 2 d in vitro (DIV), and we only observed a detectable increase in staining at 4 DIV (Fig. 2F). Together, the data suggest that when using the NestinCre driver line of mice, Ctcf deficiency does trigger the activation of caspase-mediated apoptosis in NPCs, but the effect is delayed and less severe than that observed in the CtcfFoxg1-cre embryos, suggesting that early neuroepithelial cells are more sensitive to CTCF loss than the neuroprogenitors present slightly later in development.
Increased p53 and PUMA levels in the Ctcf-null telencephalon
Both the Foxg1Cre and NestinCre models of CTCF loss exhibit increased levels of apoptosis, although the timing of onset differs between the two mutants. To investigate the molecular mechanism responsible for neuronal cell death due to CTCF loss, we assessed activation of the p53/PUMA pathway. Using Western blot analysis, we found that neuronal cell death correlated with an increase in p53 protein levels in the CtcfNes-cre forebrain compared to control, suggesting activation and stabilization of the protein (Fig. 3A). We next investigated the levels of PUMA, which has been demonstrated previously to mediate NPC death downstream of p53 (Jeffers et al., 2003). Moreover, Puma is one of the most upregulated genes in response to CTCF loss in the limb bud (Soshnikova et al., 2010). CTCF binding sites in the Puma gene demarcate an intragenic chromatin boundary that is abolished upon CTCF knockdown in human cells, resulting in increased PUMA expression (Gomes and Espinosa, 2010b). To determine whether PUMA is involved caspase-mediated cell death in the Ctcf mutant embryos, we examined PUMA protein levels in the CtcfFoxg1-cre and CtcfNes-cre mice. In the E11.5 CtcfFoxg1-cre forebrain, we observed an increase in PUMA immunostaining compared to littermate controls, corresponding to the high levels of AC3 staining in these embryos (Fig. 3B). In the CtcfNes-cre embryos, PUMA was increased in the E15.5 basal ganglia, again correlating with the highest levels of cell death (Fig. 3C). In primary cultures, Ctcf-null NPCs exhibited increased PUMA immunostaining at 4 DIV when compared to control NPCs obtained from littermate embryos (Fig. 3D). We also quantified Puma transcript levels by quantitative reverse-transcriptase PCR and observed a significant increase in the E16.5 CtcfNes-cre forebrain compared to controls (Fig. 3E). These observations demonstrate that increased PUMA transcript and proteins levels occur as a consequence of CTCF loss in NPCs and correlate with p53 activation and the onset of caspase-mediated cell death.
Figure 3.

Loss of CTCF causes p53-dependent transcriptional activation of Puma. A, Western blot analysis of nuclear protein extracts obtained from E16.5 control and CtcfNes-cre telencephalon. Densitometry analysis of blots revealed a 2.3-fold increase in CtcfNes-cre p53 levels compared to control. B, C, INCENP was used as a loading control. Immunodetection of PUMA in E11.5 control and CtcfFoxg1-cre cortical neuroepithelium (B), E15.5 control and CtcfNes-cre forebrain (C), and control and Ctcf-null NPCs (D) at 4 DIV. E, Quantitative real-time PCR analysis of Ctcf and Puma expression in control and CtcfNes-cre E16.5 forebrain using primers spanning Puma exons 3–4 (n = 3). β-actin was used as an internal control. F, Schematic representation of the mouse Puma gene. PCR products used to detect sites of CTCF binding (CBS1 and CBS2) and p53 binding (p53BS) are shown. G, Quantitative chromatin immunoprecipitation of CTCF at the Puma gene in E16.5 control and CtcfNes-cre telencephalon tissue (n = 3). Primer pairs 5 kb upstream and downstream of binding sites were used as negative controls. H, Quantitative chromatin immunoprecipitation of p53 at the Puma gene in E16.5 control and CtcfNes-cre telencephalon tissue shows increased p53 occupancy at the Puma promoter in the CtcfNes-cre telencephalon (n = 3). Primer pairs 1 kb upstream and downstream of the binding site were used as negative controls. Error bars represent SEM. Original magnification: A, C, 200×; B, 100×. Scale bars: B, C, 50 μm; D, 25 μm.
PUMA upregulation in the Ctcf-deficient embryonic brain could result from decreased CTCF occupancy within the Puma gene body, or because of p53-dependent activation of the gene. To investigate these possibilities further, we performed quantitative CTCF and p53 ChIP using E16.5 telencephalon isolated from control and CtcfNes-cre embryos (Fig. 3F--H). ChIP analysis confirmed CTCF binding sites found downstream of Puma exon 1 (CBS1) and exon 3 (CBS2) at the Puma gene in control tissue, similar to the binding profile of CTCF in human cancer cells (Fig. 3G; Gomes and Espinosa, 2010a). As expected, CTCF binding was diminished in the CtcfNes-cre tissue (Fig. 3G). ChIP for p53 demonstrated specific enrichment of the protein at the Puma promoter in CtcfNes-cre forebrain compared to control in E16.5 tissue (Fig. 3H). Together, loss of CTCF in NPCs causes increased Puma transcription and protein levels due to increased p53-dependent transcriptional activation, likely combined with the loss of CTCF-dependent repression.
Deletion of Puma in a Ctcf-null context rescues cell death, but does not improve viability or brain defects at birth
To investigate whether increased PUMA levels cause cell death in the Ctcf-deficient embryonic brain, we introduced a mutant Puma allele (Bbc3tm1Ast) in the CtcfloxP and NestinCre mice to generate mice that lack both Ctcf and Puma expression in the brain (hereafter referred to as CtcfNes-cre;Puma−/−). Histological analysis of E16.5 CtcfNes-cre cryosections showed thinning of the VZ/SVZ, hypocellularity of the intermediate zone, and a dramatic reduction in the size of the hippocampal hem (Fig. 4A). CtcfNes-cre;Puma−/− embryos at E16.5 showed rescue in the VZ/SVZ thickness, as well as hippocampal size (Fig. 4A). We confirmed these observations by partitioning the E16.5 cortex into the CP, IZ, and VZ/SVZ based on cytoarchitecture and quantifying the number of DAPI+ cells per region (Fig. 4B,C). We observed a restoration in the number of IZ cells and partial restoration in the number of VZ/SVZ cells in the CtcfNes-cre; Puma−/− compared to CtcfNes-cre cortex (Fig. 4C). There was no significant difference in the number of CP cells between genotypes at E16.5 (Fig. 4C).
We performed AC3 immunostaining in E16.5 brain sections to investigate whether apoptotic cell death was rescued in the double mutant brain. AC3+ cells were elevated in the CtcfNes-cre cortex, basal ganglia, and hippocampal hem compared to control (Fig. 4B,D). AC3+ cells were largely localized to the ventricular zone of the neocortex, indicating that Ctcf-null proliferating cells are more susceptible to caspase-dependent cell death than differentiated cells of the cortical plate. Deletion of Puma in the CtcfNes-cre mouse was sufficient to abolish apoptotic cell death, indicating that PUMA mediates increased caspase-dependent cell death in Ctcf-null neuroprogenitor cells (Fig. 4B,D). Together, these data indicate a specific decrease in intermediate zone cellularity in the E16.5 CtcfNes-cre cortex that is fully restored upon inhibition of cell death. The number of cells in the ventricular/subventricular zone is only partially restored, suggesting that loss of cells cannot completely be explained by increased cell death.
To further confirm that cell viability is rescued by deletion of Puma in a Ctcf-null context, we established primary cortical progenitor cultures from E12.5 control, CtcfNes-cre, CtcfNes-cre;Puma−/−, and Puma−/− telencephalon and measured viability via trypan blue dye exclusion at 2–5 DIV. Each time point was analyzed by a one-way ANOVA followed by Dunnett's multiple comparisons post-test to determine which genotypes had a mean percentage of viability that was significantly different from control. No difference in viability was observed at 2 and 3 DIV; however, at 4 and 5 DIV, CtcfNes-cre NPCs exhibited a marked reduction in viability, correlating with increased levels of AC3 (Fig. 4E). NPCs obtained from CtcfNes-cre;Puma−/− embryos did not show a significant decrease in viability at any of the time points analyzed (Fig. 4E).
Since we observed a rescue in caspase-mediated cell death in the embryonic CtcfNes-cre;Puma−/− brain, we predicted that the brain size at birth would also be recovered and that the mice would survive into the postnatal period, allowing for more extended analyses. Surprisingly, we failed to recover live CtcfNes-cre;Puma−/− mice at birth. Histological staining of postnatal day 0.5 (P0.5) CtcfNes-cre;Puma−/− sections demonstrated severe hypocellularity and disorganization of the cortical plate and hippocampus, and an overall similarity to the CtcfNes-cre neonatal brain (Fig. 5A). Histological cell counts confirmed that there are fewer cells in CtcfNes-cre and CtcfNes-cre;Puma−/− cortical plate compared to controls; however, no significant difference in cell number was detected between CtcfNes-cre and CtcfNes-cre;Puma−/− (Fig. 5B). These findings indicate that although deletion of Puma prevents Ctcf-null cells from undergoing apoptosis, other factors come into play to prevent cortical size expansion in the double mutant brain.
Figure 5.

Puma deletion in the Ctcf-deficient brain fails to restore hippocampal size or hypocellularity of the cortical plate. A, H&E staining of neonatal (P0.5) control, CtcfNes-cre, and CtcfNes-cre;Puma−/− cortex and hippocampus. Scale bars: top, 800 μm; bottom, 200 μm. Original magnification, 25×. B, Graph depicting cortical plate cell counts as a percentage of control (n = 3). Error bars represent SEM.
Ctcf-null apical and oRG progenitors rescued from death by Puma deletion fail to proliferate
Given that cell death at E16.5 was rescued in the double mutant embryos but the brain hypocellularity at birth was not, we speculated that the cells rescued from apoptosis might become arrested in the cell cycle, resulting in decreased proliferation. To test this hypothesis, we performed acute BrdU labeling at E16.5 and quantified the proportion of cells in S phase by BrdU immunostaining. To compare the cortical distribution and number of NPCs in S phase, the E16.5 cortex was partitioned into the CP, IZ, and VZ/SVZ based on cytoarchitecture (Fig. 6A). The number of BrdU+ cells was significantly reduced in the CtcfNes-cre and CtcfNes-cre;Puma−/− intermediate zones and ventricular zone/subventricular zones compared to control (Fig. 6A,B). Despite substantial rescue in cellularity of the CtcfNes-cre;Puma−/− cortex compared to CtcfNes-cre at E16.5, the proliferative capacity of progenitors was not significantly different (Fig. 6A,B). Our results demonstrate that Puma deletion rescues cell death in the Ctcf-null brain, but that the rescued cells display reduced proliferative capacity.
Figure 6.

Ctcf-deficient cells that are rescued from apoptotic death display reduced proliferative capacity. Pregnant females were subjected to a 1 h BrdU pulse before being killed. A, BrdU (green) immunostaining of E16.5 control, CtcfNes-cre, and CtcfNes-cre;Puma−/− cortical cryosections. The inset demonstrates fewer BrdU+ cells in the CtcfNes-cre and CtcfNes-cre;Puma−/− IZ compared with control. Scale bar, 50 μm. Original magnification, 100×. B, The number of cells in S phase was quantified by counting BrdU+ cells in 200-μm-wide cortical images (n = 3). Error bars represent SEM.
Three NPC subtypes exist in the embryonic cortex: apical radial glia (apical progenitors), basal progenitors, and outer radial glia. Apical progenitors can be identified by their expression of SOX2 and PAX6 transcription factors (Götz et al., 1998; Tarabykin et al., 2001; Bani-Yaghoub et al., 2006; Shitamukai and Matsuzaki, 2012). They undergo interkinetic nuclear migration and divide at the ventricular surface to either self-renew or differentiate into basal progenitors, outer radial glia, or cortical neurons (Shitamukai et al., 2011; Shitamukai and Matsuzaki, 2012). Basal progenitors reside in the subventricular zone and uniquely express the T-box transcription factor TBR2 (Englund et al., 2005). They are reported to have the potential to self-renew; however, the majority of their divisions are neurogenic (Haubensak et al., 2004; Miyata et al., 2004; Noctor et al., 2004). Outer radial glia are similar to radial glia in that they have a basal process and express SOX2 and PAX6; however, oRG cells are located outside of the VZ in the outer SVZ. They are able to divide asymmetrically, producing one oRG and one neuron-committed cell with each division (Reillo et al., 2011; Shitamukai et al., 2011; Wang et al., 2011a,b; Shitamukai and Matsuzaki, 2012). We performed acute BrdU-labeling for 1 h and coimmunostained E16.5 cortical sections with antibodies against BrdU and TBR2 or SOX2 to examine the behavior of different progenitor subtypes after loss of CTCF (Figs. 7, 8).
Figure 7.

Ctcf-deficiency results in PUMA-dependent apoptosis of basal progenitor cells. Pregnant females were subjected to a 1 h BrdU pulse before being killed. A, TBR2 (red) and BrdU (green) coimmunostaining of E16.5 cortical cryosections. Arrowheads indicate TBR2+/BrdU+ cells in the IZ and VZ/SVZ. Scale bar, 50 μm. Original magnification, 100×. B, TBR2+ cells were quantified in 200-μm-wide cortical images (n = 3). C, TBR2+/BrdU+ cells were quantified in 200-μm-wide cortical images (n = 3). Error bars represent SEM.
Figure 8.

Ctcf-deficiency results in PUMA-dependent apoptosis of apical and outer radial glia progenitors. Apical and outer radial glia progenitors that are rescued from apoptotic death fail to proliferate. Pregnant females were subjected to a 1 h BrdU pulse before being killed. A, SOX2 (red) and BrdU (green) coimmunostaining of E16.5 cortical cryosections. The inset demonstrates fewer SOX2+ cells in the CtcfNes-cre IZ than control or CtcfNes-cre;Puma−/−. B, SOX2+ cells were quantified in 200-μm-wide cortical images (n = 3). C, SOX2+/BrdU+ cells were quantified in 200-μm-wide cortical images (n = 3). D, Immunodetection of SOX2 (green) and PAX6 (red) in the E16.5 cortical IZ demonstrates restoration of oRG progenitors in CtcfNes-cre;Puma−/− cortex compared to CtcfNes-cre. Arrowheads indicate SOX2+/PAX6+ oRG cells. Error bars represent SEM. Original magnification: A, D, 100×. Scale bars: A, 50 μm; D, 10 μm.
The total number of TBR2+ basal progenitor cells was reduced in the CtcfNes-cre cortex compared to control (Fig. 7A,B). The number of basal progenitors was restored to control levels in the CtcfNes-cre;Puma−/− cortex, and most rescued cells were still able to enter S phase (Fig. 7A,C). Despite the correct overall number of TBR2+ cells in the CtcfNes-cre;Puma−/− cortex, the localization of basal progenitors appeared to be shifted apically compared to control (Fig. 7A,B). This is perhaps due to an inability of the rescued basal progenitors to correctly delaminate from the ventricular surface.
Quantification of SOX2+ cells indicated a dramatic reduction in the number of apical progenitors and oRGs in the CtcfNes-cre cortex (Fig. 8A,B). The number of SOX2+ apical progenitors was only partially rescued in the CtcfNes-cre;Puma−/− cortex, indicating that CTCF may control the size of the apical progenitor pool independently of apoptosis (Fig. 8A,B). Conversely, oRG (SOX2+ cells in the intermediate zone) numbers were restored to control levels in the CtcfNes-cre;Puma−/− cortex, indicating that these cells are lost via Puma-mediated apoptotic cell death upon deletion of Ctcf (Fig. 8A,B). The proliferative capacity of apical progenitors (SOX2+/BrdU+ cells in the VZ/SVZ) was severely diminished in the CtcfNes-cre cortex compared to control, and was not restored in the CtcfNes-cre;Puma−/− cortex (Fig. 8A,C). Similarly, BrdU incorporation in the rescued CtcfNes-cre;Puma−/− oRG cells was extremely low, indicating that they are likely arrested in the cell cycle and fail to correctly enter S phase (Fig. 8A,C). To confirm that these cells are oRG progenitors, we performed SOX2 and PAX6 coimmunostaining and indeed observed a decreased number of SOX2+PAX6+ cells in the intermediate zone of CtcfNes-cre embryos compared to control and CtcfNes-cre;Puma−/− at E16.5 (Fig. 8D).
Ctcf loss causes premature differentiation of apical progenitors
Incomplete restoration of the apical progenitor population in the CtcfNes-cre;Puma−/− cortex suggests that loss of CTCF leads to an apoptosis-independent reduction of these cells (Fig. 8A,B). To identify the cause of reduced apical cell numbers, we analyzed the progenitor pool composition, cell cycle exit indices, and postmitotic projection neuron subtypes in control and CtcfNes-cre cortex at E14.
Apical progenitors begin to produce projection neurons at approximately E11.5. At the same time, they generate neuron-committed basal progenitors and oRG progenitors. Since the majority of basal progenitor divisions are terminal and result in the production of two neurons, this would result in increased generation of neurons at the expense of the progenitor pool (Haubensak et al., 2004; Miyata et al., 2004; Noctor et al., 2004). The relative abundance of TBR2+ basal progenitors was significantly increased in the E14 CtcfNes-cre cortex compared to control, whereas the number of SOX2+ apical progenitors was not affected (Fig. 9A,B), suggesting that differentiation of apical progenitors into basal progenitors is increased. Next, we measured the proportion of cells exiting the cell cycle by labeling embryos with BrdU for 24 h and analyzing Ki67+ and BrdU+ cells in E14 control and CtcfNes-cre cortex. Cell cycle exit (BrdU+Ki67− cells/total BrdU+ cells) was significantly higher in CtcfNes-cre cortex compared to controls (Fig. 9C,D).
Figure 9.

Ctcf-deficiency causes premature neurogenesis. A, Immunodetection of SOX2 (red) and TBR2 (green) in E14 control and CtcfNes-cre cortex. B, SOX2+, TBR2+, and SOX2+/TBR2+ cells were quantified in 150-μm-wide cortical images and expressed as a percentage of total DAPI+ cells (n = 3). C, Pregnant female mice were subjected to a 24 h BrdU pulse before being killed. Ki67 (red) and BrdU (green) immunostaining was used to determine the percentage of cells exiting the cell cycle in control and CtcfNes-cre E14 cortex. Arrowheads indicate BrdU+Ki67− cells that have exited the cell cycle. D, Cell cycle exit indices were calculated by measuring the ratio of BrdU+Ki67− cells to total BrdU+ cells in control and CtcfNes-cre cortex at E14 (n = 3). E, Immunodetection of TBR1 (red) and SATB2 (green) in E14 control and CtcfNes-cre cortex. F, Immunodetection of CTIP2 (green) in E14 control and CtcfNes-cre cortex. G, SATB2+, TBR1+, and CTIP2+ cells were quantified and expressed as a percentage of DAPI+ cells (n = 3). Error bars represent SEM. Original magnification: A, C, E, F, 100×. Scale bars: A, C, 50 μm; E, F, 50 μm.
The cortical plate is established by projection neurons that organize themselves in an “inside-out” manner, such that early-born neurons populate deeper cortical layers (layer VI, then layer V), and late-born neurons migrate past the early-born neurons to populate the more superficial layers of the cortex (layer IV, then layer II/III; Greig et al., 2013). Increased cell cycle exit and differentiation at E13–E14 is predicted to result in an increased number of layer VI corticothalamic projection neurons (TBR1+), layer V subcerebral projection neurons (CTIP2+), and callosal projection neurons (SATB2+). We found that the relative generation of these neuronal subtypes was increased in the CtcfNes-cre cortex compared to control (Fig. 8E--G), confirming premature differentiation of Ctcf-null apical progenitors, which is predicted to result in a reduced number of this progenitor pool by late neurogenesis.
Discussion
This study provides evidence that CTCF is required at very early stages of telencephalon development for the maintenance and survival of neuroprogenitor cells. We found that ablation of CTCF leads to Puma-dependent apoptosis of NPCs using two different conditional deletion strategies. Increased apoptosis correlated with p53-dependent Puma transcription, suggesting that CTCF loss results in p53 stabilization and transcriptional activation of its downstream targets. CTCF loss might also result in a more open chromatin environment at the Puma gene, facilitating p53-dependent activation of transcription and elongation of RNA polymerase II (Gomes and Espinosa, 2010a,b). Ctcf inactivation in postmitotic cortical and hippocampal neurons does not induce apoptosis (Hirayama et al., 2012), pointing to a specific survival role for CTCF in proliferating cells. However, CTCF loss of function may not induce apoptosis in all types of proliferating cells in vivo, as Ctcf deletion in thymocytes was shown previously to induce cell cycle arrest without induction of apoptosis (Heath et al., 2008). It is possible that the outcome of CTCF deficiency leads to either p53-dependent cell cycle arrest or apoptosis, depending on varying cell-specific or temporal cues. This could be similar to the outcomes described upon Nbs1 (Nijmegen breakage syndrome 1/Nibrin) deletion in the CNS, which leads to p53-dependent apoptosis in the cerebellum, but causes p53-dependent cell cycle arrest in the neocortex (Li et al., 2012).
In the present report, we demonstrate that deletion of Puma effectively rescues apoptotic cell death observed at E16.5 in the CtcfNes-cre brain (Fig. 4). Despite this apparent recovery at E16.5, the double mutant brain at birth appeared hypocellular and was nearly undistinguishable histologically from the CtcfNes-cre brain (Fig. 5). We showed that CtcfNes-cre;Puma−/− apical and oRG progenitors that cannot activate the apoptotic pathway fail to incorporate BrdU, which might be explained by a p53-mediated cell cycle arrest. Further investigations into the mechanism underlying increased p53 levels in the Ctcf-null brain will be important to fully elucidate the role of CTCF in progenitor cell survival. Given that oRG cells are implicated in neocortical expansion in humans, their reduced ability to proliferate in the double mutant cortex might partly explain the failure to recover cortical size at birth, and may be relevant to microcephaly caused by CTCF mutations in humans (Hansen et al., 2010; Lui et al., 2011; Reillo et al., 2011).
A key observation is that deletion of Puma did not completely restore the number of apical cells in the CtcfNes-cre;Puma−/− cortex, suggesting that while a proportion of this progenitor population undergoes Puma-dependent death in the absence of CTCF, apical cells exhibit an independent defect. We found that Ctcf-null apical progenitors differentiate prematurely, causing an initial increase in the production of basal progenitors and early-born postmitotic neurons (Fig. 9). However, the increased number of basal progenitors is later counteracted by a reduction of the progenitor pool from which they are derived, and exacerbated by increased levels of apoptosis. Given that CTCF colocalizes with the cohesin complex and is required for cohesin localization to specific genomic sites to influence gene expression (Parelho et al., 2008; Wendt et al., 2008), it is conceivable that premature neurogenesis in the CtcfNes-cre cortex results from dysregulation of CTCF target genes.
The number of basal progenitor cells marked by TBR2 expression was completely restored in the double mutant brain, indicating that PUMA activation causes basal progenitor cell death in the absence of CTCF. Simultaneous labeling of brain sections with TBR2 and BrdU showed that rescued basal progenitors are still able to proliferate, unlike rescued apical and oRG cells. Despite the overall equivalent number of TBR2+ cells in the control and double mutant cortex, several rescued basal progenitors did not move basally out of the ventricular zone, perhaps due to a defect in apical radial glia or an inability to correctly delaminate from the apical surface of the Ctcf-deficient cortex.
In summary, we demonstrated that CTCF is required in the early developing mouse brain for neuroprogenitor cell survival and that its deletion induces p53- and PUMA-dependent apoptosis. Independent from its role in promoting cell survival, CTCF is required for the correct balance of proliferative versus differentiative divisions and maintenance of the apical progenitor pool. Together, these functions of CTCF contribute to the normal development of the mammalian neocortex.
Footnotes
This work was supported by a National Science and Engineering Research Council of Canada (NSERC) Canada Graduate Scholarship to L.A.W., a Canadian Institute for Health Research (CIHR) Vanier Scholarship to A.E., an NSERC Canada Graduate Scholarship to K.D.K., and an operating grant from the CIHR (MOP93697) to N.G.B.
The authors declare no competing financial interests.
References
- Bani-Yaghoub M, Tremblay RG, Lei JX, Zhang D, Zurakowski B, Sandhu JK, Smith B, Ribecco-Lutkiewicz M, Kennedy J, Walker PR, Sikorska M. Role of Sox2 in the development of the mouse neocortex. Dev Biol. 2006;295:52–66. doi: 10.1016/j.ydbio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Bérubé NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, Higgs DR, Slack RS, Picketts DJ. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–267. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou CL, Li S, Lai E. Dual role of brain factor-1 in regulating growth and patterning of the cerebral hemispheres. Cereb Cortex. 1999;9:543–550. doi: 10.1093/cercor/9.6.543. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science. 2004;303:238–240. doi: 10.1126/science.1090934. [DOI] [PubMed] [Google Scholar]
- Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD. Early induction of Talpha1 alpha-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(SICI)1096-9861(19990301)405:1<45::AID-CNE4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Gomes NP, Espinosa JM. Gene-specific repression of the p53 target gene PUMA via intragenic CTCF-Cohesin binding. Genes Dev. 2010a;24:1022–1034. doi: 10.1101/gad.1881010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes NP, Espinosa JM. Disparate chromatin landscapes and kinetics of inactivation impact differential regulation of p53 target genes. Cell cycle. 2010b;9:3428–3437. doi: 10.4161/cc.9.17.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M, Stoykova A, Gruss P. Pax6 controls radial glia differentiation in the cerebral cortex. Neuron. 1998;21:1031–1044. doi: 10.1016/S0896-6273(00)80621-2. [DOI] [PubMed] [Google Scholar]
- Gregor A, Oti M, Kouwenhoven EN, Hoyer J, Sticht H, Ekici AB, Kjaergaard S, Rauch A, Stunnenberg HG, Uebe S, Vasileiou G, Reis A, Zhou H, Zweier C. De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet. 2013;93:124–131. doi: 10.1016/j.ajhg.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, Wong E, Sheng J, Zhang Y, Poh T, Chan CS, Kunarso G, Shahab A, Bourque G, Cacheux-Rataboul V, Sung WK, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci U S A. 2004;101:3196–3201. doi: 10.1073/pnas.0308600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath H, Ribeiro de Almeida C, Sleutels F, Dingjan G, van de Nobelen S, Jonkers I, Ling KW, Gribnau J, Renkawitz R, Grosveld F, Hendriks RW, Galjart N. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 2008;27:2839–2850. doi: 10.1038/emboj.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012;2:345–357. doi: 10.1016/j.celrep.2012.06.014. [DOI] [PubMed] [Google Scholar]
- Holwerda SJ, de Laat W. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120369. doi: 10.1098/rstb.2012.0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Kernohan KD, Jiang Y, Tremblay DC, Bonvissuto AC, Eubanks JH, Mann MR, Bérubé NG. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev Cell. 2010;18:191–202. doi: 10.1016/j.devcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Li R, Yang YG, Gao Y, Wang ZQ, Tong WM. A distinct response to endogenous DNA damage in the development of Nbs1-deficient cortical neurons. Cell Res. 2012;22:859–872. doi: 10.1038/cr.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;146:18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development. 2004;131:3133–3145. doi: 10.1242/dev.01173. [DOI] [PubMed] [Google Scholar]
- Moore JM, Rabaia NA, Smith LE, Fagerlie S, Gurley K, Loukinov D, Disteche CM, Collins SJ, Kemp CJ, Lobanenkov VV, Filippova GN. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS One. 2012;7:e34915. doi: 10.1371/journal.pone.0034915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc. 2008;3:698–709. doi: 10.1038/nprot.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahashi H, Kwon KR, Resch W, Vian L, Dose M, Stavreva D, Hakim O, Pruett N, Nelson S, Yamane A, Qian J, Dubois W, Welsh S, Phair RD, Pugh BF, Lobanenkov V, Hager GL, Casellas R. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013;3:1678–1689. doi: 10.1016/j.celrep.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/S1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Ohlsson R, Bartkuhn M, Renkawitz R. CTCF shapes chromatin by multiple mechanisms: the impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma. 2010;119:351–360. doi: 10.1007/s00412-010-0262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Reillo I, de Juan Romero C, García-Cabezas MÁ, Borrell V. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb Cortex. 2011;21:1674–1694. doi: 10.1093/cercor/bhq238. [DOI] [PubMed] [Google Scholar]
- Shitamukai A, Matsuzaki F. Control of asymmetric cell division of mammalian neural progenitors. Dev Growth Differ. 2012;54:277–286. doi: 10.1111/j.1440-169X.2012.01345.x. [DOI] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, Matsuzaki F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci. 2011;31:3683–3695. doi: 10.1523/JNEUROSCI.4773-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol. 1998;140:1497–1509. doi: 10.1083/jcb.140.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soshnikova N, Montavon T, Leleu M, Galjart N, Duboule D. Functional analysis of CTCF during mammalian limb development. Dev Cell. 2010;19:819–830. doi: 10.1016/j.devcel.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Tarabykin V, Stoykova A, Usman N, Gruss P. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development. 2001;128:1983–1993. doi: 10.1242/dev.128.11.1983. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Wan LB, Pan H, Hannenhalli S, Cheng Y, Ma J, Fedoriw A, Lobanenkov V, Latham KE, Schultz RM, Bartolomei MS. Maternal depletion of CTCF reveals multiple functions during oocyte and preimplantation embryo development. Development. 2008;135:2729–2738. doi: 10.1242/dev.024539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lui JH, Kriegstein AR. Orienting fate: spatial regulation of neurogenic divisions. Neuron. 2011a;72:191–193. doi: 10.1016/j.neuron.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tsai JW, LaMonica B, Kriegstein AR. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci. 2011b;14:555–561. doi: 10.1038/nn.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LA, Solomon LA, Li JR, Jiang Y, Edwards M, Shin-ya K, Beier F, Bérubé NG. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J Clin Invest. 2013;123:2049–2063. doi: 10.1172/JCI65634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/S1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Yusufzai TM, Felsenfeld G. The 5′-HS4 chicken beta-globin insulator is a CTCF-dependent nuclear matrix-associated element. Proc Natl Acad Sci U S A. 2004;101:8620–8624. doi: 10.1073/pnas.0402938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell. 2004;13:291–298. doi: 10.1016/S1097-2765(04)00029-2. [DOI] [PubMed] [Google Scholar]