

Abstract
Neural cell fate specification is well understood in the embryonic cerebral cortex, where the proneural genes Neurog2 and Ascl1 are key cell fate determinants. What is less well understood is how cellular diversity is generated in brain tumors. Gliomas and glioneuronal tumors, which are often localized in the cerebrum, are both characterized by a neoplastic glial component, but glioneuronal tumors also have an intermixed neuronal component. A core abnormality in both tumor groups is overactive RAS/ERK signaling, a pro-proliferative signal whose contributions to cell differentiation in oncogenesis are largely unexplored. We found that RAS/ERK activation levels differ in two distinct human tumors associated with constitutively active BRAF. Pilocytic astrocytomas, which contain abnormal glial cells, have higher ERK activation levels than gangliogliomas, which contain abnormal neuronal and glial cells. Using in vivo gain of function and loss of function in the mouse embryonic neocortex, we found that RAS/ERK signals control a proneural genetic switch, inhibiting Neurog2 expression while inducing Ascl1, a competing lineage determinant. Furthermore, we found that RAS/ERK levels control Ascl1's fate specification properties in murine cortical progenitors–at higher RAS/ERK levels, Ascl1+ progenitors are biased toward proliferative glial programs, initiating astrocytomas, while at moderate RAS/ERK levels, Ascl1 promotes GABAergic neuronal and less glial differentiation, generating glioneuronal tumors. Mechanistically, Ascl1 is phosphorylated by ERK, and ERK phosphoacceptor sites are necessary for Ascl1's GABAergic neuronal and gliogenic potential. RAS/ERK signaling thus acts as a rheostat to influence neural cell fate selection in both normal cortical development and gliomagenesis, controlling Neurog2-Ascl1 expression and Ascl1 function.
Keywords: bHLH transcription factors, glioma and glioneuronal tumors, Neurog2 and Ascl1, neurogenesis versus gliogenesis, proneural genetic switch, RAS/ERK signaling
Introduction
The RAS/ERK signal transduction cascade, which involves the sequential activation of RAS→RAF→MEK→ERK, is a critical pathway in neurodevelopment and brain cancer. In neurodevelopment, RAS functions are pleiotropic, ranging from the promotion of cell proliferation to the induction of neuronal or glial differentiation, depending on cellular context and developmental stage (Lukaszewicz et al., 2002; Ménard et al., 2002; Ito et al., 2003; Imamura et al., 2008; Ohtsuka et al., 2009). In neoplasia, RAS activation is a common pro-proliferative event that is triggered by mutations of upstream receptor tyrosine kinases (RTKs), or downstream signaling components, such as NF1 or BRAF (Louis et al., 2007; Bar et al., 2008; Jones et al., 2008; The Cancer Genome Atlas Research Network, 2008). While oncogenic RAS activates several downstream pathways, including ERK, AKT, and Ral (Repasky et al., 2004), ERK is considered the key effector in tumorigenesis, as BRAF activating mutations are observed in pilocytic astrocytoma (PA), ganglioglioma (GG), pleomorphic xanthoastrocytoma, and glioblastoma variants (Knobbe et al., 2004; Jones et al., 2008; Pfister et al., 2008; Dias-Santagata et al., 2011; Schindler et al., 2011; Kleinschmidt-DeMasters et al., 2013). Moreover, constitutively activate BRAF is sufficient to induce PA in mice (Gronych et al., 2011).
RAS/ERK signaling promotes neoplastic proliferation, but only recently has been suggested to also influence neural cell fate selection. Indeed, inactivation of NF1, which inhibits RAS, promotes aberrant glial instead of neuronal differentiation (Dasgupta and Gutmann, 2005), a change that is reversed by MEK/ERK inhibitors (Wang et al., 2012). Moreover, constitutively active MEK induces astrocytic and oligodendrocytic differentiation in neural progenitors (X. Li et al., 2012). However, RAS/ERK is commonly activated in gliomas and glioneuronal tumors, raising the question of how this pathway differentially influences oncogenic cell fates.
We found that RAS/ERK activation levels differ in two distinct human tumors associated with constitutively active BRAF; PAs, which contain abnormal astrocytes and oligodendrocyte precursor cell (OPC)-like cells, have higher ERK activation levels than GGs, which contain abnormal neuronal and astrocytic cells. To determine whether and how different levels of RAS/ERK signaling influences cell fate choice, we examined interactions with the proneural basic-helix-loop-helix (bHLH) transcription factors in the developing neocortex. The neocortex is derived from the telencephalon, where the proneural gene Neurogenin 2 (Neurog2) specifies a glutamatergic (glu+) neuronal fate in dorsal progenitors (Fode et al., 2000; Parras et al., 2002; Schuurmans et al., 2004; Mattar et al., 2008), while the proneural gene Achaete scute-like 1 (Ascl1) specifies the identities of neocortical GABAergic (GABA+) neurons and embryonic OPCs that are derived from ventral progenitors (Casarosa et al., 1999; Britz et al., 2006; Parras et al., 2007; Castro et al., 2011). We found that RAS/ERK signaling promotes Ascl1 expression while blocking Neurog2 expression in neocortical progenitors. Moreover, we found that at low RAS/ERK levels, Ascl1 promotes GABA+ neuronal differentiation and generates glioneuronal tumors, while at higher RAS/ERK levels, Ascl1 promotes a proliferative glioblast phenotype and generates glial tumors. Finally, we reveal that Ascl1 is directly phosphorylated by ERK, and that the ERK phosphoacceptor sites are necessary for the gliogenic potential of Ascl1.
Materials and Methods
Animal breeding and maintenance.
Animal procedures were approved by the University of Calgary Animal Care Committee (protocol #AC11-0051 to J.A.C. and #AC11-0053 to C.S.) in compliance with the Guidelines of the Canadian Council of Animal Care. Timed pregnant wild-type CD1, Neurog2GFPKI (Britz et al., 2006), and Ascl1GFPKI (Leung et al., 2007; purchased from The Jackson Laboratory; Stock number 012881) mice were used for in utero electroporation and genotyped as described. The morning of the vaginal plug was assigned embryonic day 0.5 (E0.5) for staging. Embryos of either sex were used.
Patient-derived tumor tissues.
Formalin-fixed paraffin-embedded tissues from supratentorial gangliogliomas and supratentorial nonoptic pathway pilocytic astrocytomas were obtained from pathology archives at Calgary Laboratory Services and the Clark Smith Brain Tumor Bank at the University of Calgary (Table 1; Kelly et al., 2009; Blough et al., 2010). Human materials were used with approval of Calgary Laboratory Services and the Calgary Health Region Ethics Board (University of Calgary Conjoint Health Research Ethics Board #2875 to J.A.C. and #24993 to C.S.).
Table 1.
Summary of human tumors analyzed for pERK expression
| Case ID | Gender | Age | Diagnosis | WHO grade | Location |
|---|---|---|---|---|---|
| GG-1 | F | 31 | Ganglioglioma | I | Cerebrum, temporal |
| GG-2 | F | 34 | Ganglioglioma | I | Cerebrum, frontal |
| GG-3 | M | 40 | Ganglioglioma | I | Cerebrum, temporal |
| GG-4 | M | 28 | Ganglioglioma | I | Cerebrum, temporal |
| GG-5 | M | 22 | Ganglioglioma | I | Cerebrum, temporal |
| GG-6 | F | 33 | Ganglioglioma | I | Cerebrum, temporal |
| PA-1 | M | 16 | Pilocytic astrocytoma | I | Cerebrum, parietal |
| PA-2 | M | 20 | Pilocytic astrocytoma | I | Cerebrum, thalamic |
| PA-3 | M | 33 | Pilocytic astrocytoma | I | Cerebrum, frontal |
| PA-4 | F | 26 | Pilocytic astrocytoma | I | Cerebrum, frontal |
| PA-5 | F | 37 | Pilocytic astrocytoma | I | Cerebrum, temporal |
WHO, World Health Organization.
Cell culture and luciferase assay.
Postnatal day 19 (P19) embryonic carcinoma cells (ATCC# CRL-1825) were maintained in Minimum Essential Medium α Medium (Gibco) supplemented with 10% fetal bovine serum (FBS), 10 U/ml penicillin, and 10 μg/ml streptomycin. The 4.3 kb Dll1 luciferase reporter was previously described (Castro et al., 2006). The 1 kb Sox9 proximal promoter region was PCR amplified from mouse genomic DNA with the primers: 5′-ATACTCGAGAGAGAACAGCGGGCGTTGA-3′ (forward) and 5′-CACAAGCTTAGGGGTCCAGGAGATTCAT-3′ (reverse) and subcloned into pGL3-Basic (Promega). The Dlx1/2 reporter construct was generated by cloning the l12b intergenic enhancer from p1230-Dlx1/2 l12b (Ghanem et al., 2007) into pGL3-Basic (Promega). The P19 cells were seeded into 24-well plates (Nalge Nunc) 24 h before transfection. Transfections were performed using Lipofectamine Plus reagent (Invitrogen) as per the manufacturer's protocol, cotransfecting 0.1 μg of each test plasmid and 0.15 μg of the Renilla plasmid (transfection control). Four to six hours after transfection, Opti-MEM media (Gibco) was replaced with fresh media. Twenty-four hours later, cells were harvested and firefly luciferase and Renilla luciferase activities were determined using the Dual-luciferase Reporter Assay System (Promega) as per the manufacturer's instructions, using a TD 20/20 Luminometer (Turner Designs). Luciferase activity was normalized to the corresponding Renilla activity.
In utero electroporation.
Cortical telencephalic electroporations were performed as described previously (Dixit et al., 2011; S. Li et al., 2012) using endotoxin-free DNA (Qiagen) and platinum tweezer-style electrodes (Protech; 5 mm) to apply seven 43 ms pulses at 43 V with a 1 s interval. cDNAs were cloned into pCIG2 (provided by Dr. Franck Polleux) or pCIC (provided by Dr. Dawn Zinyk) vectors. These constructs expressed the insert mRNA coupled to EGFP (pCIG2) or mCherry (pCIC) mRNA by an internal ribosome entry site (IRES2; Clontech; Mattar et al., 2008; S. Li et al., 2012). pEFRasV12 and pEFRasN17 plasmids were provided by K. Kaibuchi (Cowley et al., 1994), pBABEMekCA and pBABEMekDN provided by C. Marshall (Yoshimura et al., 2006), AktCA provided by J.D. Ashwell (v-AKT, the constitutively active viral homolog of c-AKT; Eder et al., 1998), and Etv1 provided by T.M. Jessell (Arber et al., 2000). Plasmids containing cDNAs for BRAFV600E (from W. Hahn, Addgene plasmid 15269; Boehm et al., 2007), AktDN (AktK179M, from M.C. Huang, Addgene plasmid 15269; B. P. Zhou et al., 2000), RalAV23 (from W. Hahn, Addgene plasmid 15252; Sablina et al., 2007), RalADN (RalAS31N, from C. Counter, Addgene plasmids 19718; Lim et al., 2005), RalBQ72L (from C. Counter, Addgene plasmid 19721; Lim et al., 2005), and RalBDN (RalBS28N, from C. Counter, Addgene plasmid 19722; Lim et al., 2005) were obtained from Addgene. Etv5 cDNA (IMAGE Clone 4036564; Lennon et al., 1996) was obtained from Life Technologies.
Site-directed mutagenesis.
Ascl1-SA3, in which all three leucine-serine-proline (LSP) sites were mutated to leucine-alanine-proline, and Ascl1-SA6, in which all six SP sites were mutated into alanine-proline (S-A mutation), were generated via PCR-mediated site-directed mutagenesis using overlapping extension PCR. Two complementary primers containing nucleotide substitutions (underlined bases) were designed using a Stratagene Web-based primer design software program (Agilent): S62A-forward: 5′-CGCCGCAGCTGGCCCCGGTGGCCG-3′; S62A-reverse: 5′-GCGGCGTCGACCGGGGCCACCGGC-3′;S88A-forward: 5′-CCAGCGCTCGTCCGCTCCGGAACTGAT-3′; S88A-reverse: 5′-GGTCGCGAGCAGGCGAGGCCTTGACTA-3′;S185A-forward: 5′-CGGGCGTCCTGGCGCCCACCATC-3′; S185A-reverse: 5′-GCCCGCAGGACCGCGGGTGGTAG-3′; S189A-forward: 5′-GTCGCCCACCATCGCCCCCAACTACTC-3′; S189A-reverse: 5′-CAGCGGGTGGTAGCGGGGGTTGATGAG-3′; S202A-forward: 5′-CTCTATGGCGGGTGCTCCGGTCTCGTC-3′; S202A-reverse: 5′-GAGAT ACCGCCCACGAGGCCAGAGCAG-3′; S218A-forward: 5′-GGATCCTACGACCCTCTTGCCCCAGAGGAACAAGA-3′; S218A-reverse: 5′-CCTAGGATGCTGGGAGAACGGGGTCTCCTTGTTCT-3′.
Tissue processing.
Brains were dissected in PBS and were processed for frozen or paraffin sections. For frozen sections, brains were fixed in 4% paraformaldehyde (PFA) in PBS, and cryoprotected in 20% sucrose before OCT embedding. Ten micrometer cryosections were collected on SuperFrost Plus (Fisher) slides. For paraffin sections, brains were fixed in 4% PFA, postfixed in 10% neutral buffered formalin, and processed for routine paraffin embedding. Five micrometer sections were cut and collected on SuperFrost Plus slides.
Histology, immunohistochemistry, and immunofluorescence.
Primary antibodies included rabbit anti-GFP (Invitrogen), mouse anti-Neurog2 (David Anderson; Lo et al., 2002), rabbit anti-Neurog2 (Masato Nakafuku; Mizuguchi et al., 2001), mouse anti-Ascl1 (BD Biosciences), rabbit anti-Sox9 (Millipore), rabbit anti-pERK (Cell Signaling Technology), rabbit anti-Olig2 (Abcam), rabbit anti-GFAP (Dako Cytomation), mouse anti-NeuN (Millipore Bioscience Research Reagents), rabbit anti-Tbr1 (Abcam), mouse anti-Tuj1 (Covance), rabbit anti-Pax6 (Covance), rabbit anti-glutamine synthetase (Abcam), rabbit anti-Nf1a (Benjamin Deneen; Kang et al., 2012), goat anti-Sox2 (Santa Cruz Biotechnology), mouse anti-Nestin (Santa Cruz Biotechnology), rabbit anti-Ki67 (Vector Laboratories), and Rat anti-BrdU (Serotec). Secondary antibodies were conjugated to Cy3, AMCA (Jackson ImmunoResearch), Alexa488 or Alexa 568 (Invitrogen), and diluted 1/500. Immunofluorescence preparations were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Santa Cruz Biotechnology) before mounting of coverslips with AquaPolymount (Polysciences). Paraffin sections were used for routine hematoxylin and eosin (H&E) staining and for immunohistochemistry with detection by Envision+ diaminobenzidine (DAB) kit (Dako) and hematoxylin counterstain. Double labeling on formalin-fixed paraffin-embedded tissues was visualized by dual DAB and alkaline phosphatase (AP) development using the MultiView IHC system (Enzo).
RNA in situ hybridization.
RNA in situ hybridization was performed with digoxigenin (DIG)-labeled riboprobes as described previously (Alam et al., 2005). DIG-labeled probes were generated using a 10× labeling mix per manufacturer's instructions (Roche). Probe templates included Neurog2 (Gradwohl et al., 1996), Ascl1 (Guillemot and Joyner, 1993), Dlx1 (Anderson et al., 1997b), Olig2 (Q. Zhou et al., 2000), Olig1 (Q. Zhou et al., 2000), Etv1 (Arber et al., 2000), Etv5 (Hasegawa et al., 2004), Spry2 (Minowada et al., 1999), and tenascin C (provided by Augret Joesw). The GFP probe was generated from a cDNA clone (i.e., pEGFP-N1; Cairine Logan; Mattar et al., 2008; S. Li et al., 2012). Additional probes were generated from cDNA clones acquired from the I.M.A.G.E. consortium (HudsonAlpha Institute) as follows: Pdgfra: 5704645 and Sox9: 5351850 (Lennon et al., 1996).
Western blotting.
HEK293 cells were maintained in DMEM supplemented with 10% FBS, 10 U/ml penicillin, and 10 μg/ml streptomycin. Cells were transfected as described for P19 cells (see above) and harvested after 24 h. Cells or mouse embryonic brain tissue were lysed in NP-40 lysis buffer (0.05 m Tris, pH 7.5, 0.15 m NaCl, 1% NP-40, 1 mm EDTA, 50 mm NaF, 0.2 mm Na3VO4, 2 mm PMSF, 0.05 mm MG132, 1× complete protease inhibitor tablet (Roche), incubated for 30 min on ice and clarified by centrifuging at 20,000 for 15 min. Protein concentrations were determined via Bradford analysis. For Western blotting, 10 μg of each lysate was loaded on 10% SDS-PAGE gels and separated at a constant 125 mV voltage. Protein was transferred to a PVDF membrane in transfer buffer (25 mm Tris, 192 mm glycine, 20% methanol, pH 8.3) at 75 V for 1 h. PVDF membranes were blocked 1 h at room temperature in TBST (25 mm Tris, 3.0 mm KCl, 140 mm NaCl, pH 7.4, 0.1% Tween 20) containing 50 mg/ml powdered milk. The membranes were incubated in primary antibodies diluted in blocking solution for 1 h at room temperature, or overnight at 4°C. After 3 × 10 min washes in TBST, the membranes were exposed to species-specific horseradish peroxidase-coupled secondary antibody diluted 1/50,000 for 1 h at room temperature. Membranes were washed 3 × 10 min, and developed using ECL Plus Western Blotting Reagent (GE Healthcare Life Science) and x-ray film. Primary antibodies used in Western blotting included the following: rabbit anti-ERK (Cell Signaling Technology), rabbit anti-pERK (Cell Signaling Technology), rabbit anti-β-actin (Abcam), mouse anti-Ascl1 (BD Biosciences), and rabbit anti-pAscl1. The custom phosphorylation-specific Ascl1 (pAscl1) antibody was generated by AnaSpec using peptide GVL-(pS)-PTISPNYS+C-NH2. Bands were quantitated by densitometry using National Institutes of Health ImageJ, and corrected for loading relative to β-actin.
In vitro kinase assay.
Wild-type or mutant Ascl1 proteins were transcribed and translated in vitro using TNT rabbit reticulocyte lysate kit (Promega) according to the manufacturer's instructions. For kinase assay in rabbit reticulocyte lysates, reaction components were added to a precooled Microfuge tube: 5 μl in vitro transcribed and translated protein, 1 μl active ERK1 (0.1 μg/μl; SignalChem), 2.5 μl 0.1 m ATP, 11.5 μl kinase assay buffer (25 mm MOPS, pH 7.2; 12.5 mm β-glycerol-phosphate; 25 mm MgCl2; 5 mm EGTA; 2 mm EDTA; and 0.25 mm dithiothreitol (added to kinase assay buffer before use). Blank controls were set up as outlined above, excluding the addition of active ERK1 kinase. Reaction samples were incubated at 30°C for 15 min and then applied on 10% SDS-PAGE gels for Western blotting.
Image processing and analysis.
Images were processed using Photoshop software (Adobe) for orientation, clarity, false colorization, and overlay/colocalization. Western blot quantitation was performed using ImageJ. Immunohistochemistry quantitation of Sox9 and pERK on paraffin sections was performed using an Aperio Scanscope and related image analysis software. Briefly, slides were scanned to create digital images that were marked to limit analyses to areas of solid tumor. After color calibration and thresholding for hematoxylin (blue) and DAB (brown), DAB staining was scored using the color deconvolution algorithm where the score was calculated taking into account staining intensities and percentages [score = 1.0*(%weak) + 2*(%moderate) + 3*(%strong)] or the nuclear quantification algorithm (for Olig2), where the score represents the percentage of nuclei positively stained. Scores for replicate cores were averaged and taken as the final score for the tissue sample.
Quantitative and statistical analysis.
For the analysis of luciferase assay, luciferase data were normalized by dividing raw light readings by the corresponding Renilla values. Reported n values correspond to the number of individual experiments performed, each composed of three replicates per sample. For in vivo experiments, brains from at least three independent experiments were processed (n values refer to number of brains analyzed). Comparisons between control and experimental conditions were performed using a two-tailed Student's t test (to compare two values), while comparisons between multiple samples were performed by applying ANOVA and Tukey's multiple-comparison test using Prism software (GraphPad). Statistical variation was determined using SEM.
Results
Levels of ERK activation are associated with differing content of neuronal versus glial cells in human BRAF-associated brain tumors
RAS pathway hyperactivation is a well recognized feature of gliomas and glioneuronal tumors, which have mutations at the level of upstream RTKs, NF1, BRAF, or previously (Louis et al., 2007; Bar et al., 2008; Jones et al., 2008; The Cancer Genome Atlas Research Network, 2008; Schindler et al., 2011). However, constitutive RAS activation alone does not translate into malignancy, as low-grade/benign neoplasms, such as PAs and GGs, are also characterized by activating mutations and rearrangements of BRAF (Zhu et al., 1997; Louis et al., 2007; Bar et al., 2008; Jones et al., 2008; Otero et al., 2011; Schindler et al., 2011). We thus asked whether RAS pathway hyperactivation might have an additional role in glioma biology other than driving proliferation. More specifically, as RAS signaling can induce either neuronal or glial differentiation in neurodevelopment (Lukaszewicz et al., 2002; Ménard et al., 2002; Ito et al., 2003; Imamura et al., 2008; Ohtsuka et al., 2009), we asked whether it could similarly be playing a role in controlling neural cell fate specification during tumorigenesis.
We began our studies by investigating human PAs and GGs, which have distinct cellular compositions–GGs are composed of both neoplastic neurons and neoplastic glia (Zhu et al., 1997; Koelsche et al., 2013), whereas PAs are considered glial without a neuronal component. We assessed cell content in sections from six human cerebral GGs and five cerebral PAs, as well as three control brains (Table 1). The areas selected for analysis and quantitation were specifically chosen from central areas of solid tumors with care taken to avoid areas where there might have been possible infiltration into adjacent normal brain tissue. Consistent with their diagnoses, all six GGs contained abnormal NeuN+ cells whereas PAs did not (p < 0.0001 comparing GGs and PAs; Fig. 1A–C,M). Moreover, both GGs and PAs had increased numbers of glial lineage cells compared with control cortices, as assessed by immunolabeling with the astrocytic marker, GFAP (Fig. 1D–F) and the glioblast progenitor marker, Sox9, with glial content highest in PAs (p < 0.05 comparing GGs and PAs; Fig. 1G–I,N). Finally, to determine whether increased glial and decreased neuronal content correlated with differences in RAS/ERK signaling, we performed immunohistochemical staining for ERK1/2 phosphorylated on T202/Y204 (hereafter designated pERK). pERK expression levels were significantly higher in PAs versus GGs (p < 0.001 comparing GGs and PAs; Fig. 1J–L,O). Furthermore, within the GGs, strong pERK staining was preferentially seen in cells with glial morphology and was weak or absent in neuronal-like cells (Fig. 1H). This bias in pERK expression was confirmed by coexpression studies in GG tissues, where pERK staining largely colocalized with cells expressing the glial markers GFAP and Olig2 (Fig. 1Q,R), whereas cells expressing the neuronal marker NeuN only rarely colabeled with pERK (Fig. 1P). In addition to these cell populations, GGs contained variable numbers of Sox2+ cells, consistent with a progenitor phenotype in a subset of GG cells, but these cells only showed occasional pERK colabeling (Fig. 1S).
Figure 1.
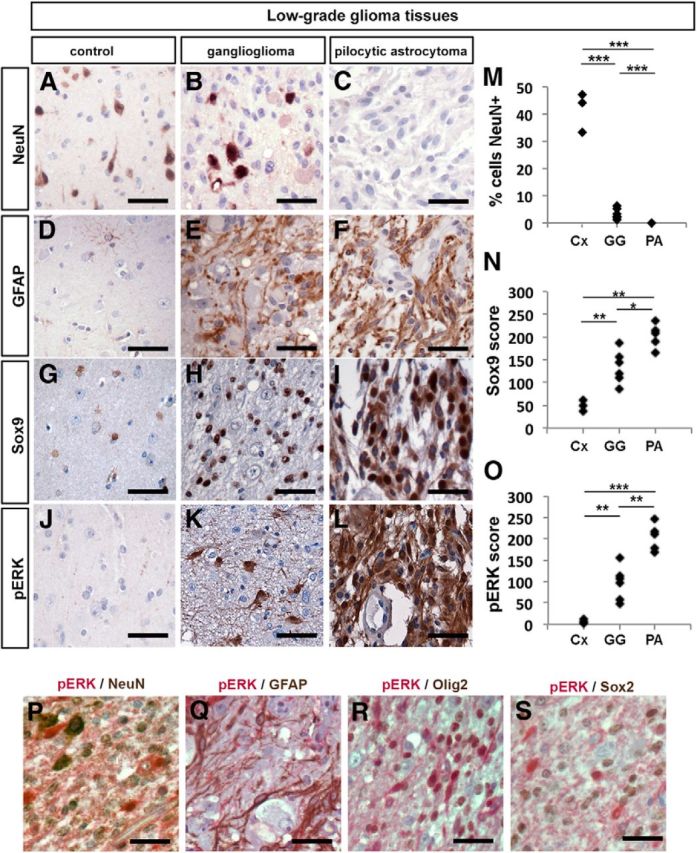
RAS/ERK activation correlates with histological and molecular features of human low-grade gliomas. A–L, Representative sections of adult human neocortex through control (Cx) nondiseased regions (A, D, G, J) or through a ganglioglioma (GG) (B, E, H, K) or pilocytic astrocytoma (PA) (C, F, I, L). Sections were processed for immunolabeling with the neuronal marker NeuN (A–C), GFAP (D–F), Sox9 (G–I), or pERK (J–L). The percentage of cells expressing NeuN (M), Sox9 (N), and pERK (O) was quantitated. *p < 0.05, **p < 0.01, ***p < 0.005. AP-visualized (red) and DAB-visualized (brown) double immunolabeling of GG tissues with pERK/NeuN (P), pERK/GFAP (Q), pERK/Olig2 (R), and pERK/Sox2 (S). Scale bars: 50 μm.
Although correlative in nature, these findings together raise the possibility that different levels of RAS/ERK signaling might influence the cellular identity of glioma and glioneuronal tumor cells–with higher pERK leading to glial lineage selection over alternate neuronal fates.
RAS/ERK signaling is preferentially activated in Ascl1 and not Neurog2-expressing cortical progenitors
To study the influence of RAS/ERK signaling on neural cell fate decisions in an in vivo context, we used the embryonic cerebral cortex as a model (Fig. 2A,B). During normal development, cortical progenitors in the dorsal telencephalon first give rise to glu+ neurons that sequentially form seven cortical layers (layer VII is transient) between mouse E10 and E17 (Smart and Smarh, 1977; Caviness, 1982; Caviness et al., 1995; Supèr et al., 1998; Takahashi et al., 1999). In late embryogenesis, cortical progenitors then give rise to astrocytes and postnatally they give rise to OPCs (Kessaris et al., 2006; Piper et al., 2010; Subramanian et al., 2011). Similarly, embryonic subcortical progenitors in the ventral telencephalon differentiate sequentially, forming GABA+ neurons, then astrocytes and finally OPCs, with a subset of GABA+ neurons and OPCs migrating tangentially into the cortex (Tamamaki et al., 1997; Anderson et al., 1997a, 2001, 2002;Casarosa et al., 1999; Horton et al., 1999; Marín and Rubenstein, 2001; Nery et al., 2002; Xu et al., 2003, 2004; Butt et al., 2005; Kessaris et al., 2006). Based on their embryonic lineage relationships, and a molecular understanding of fate specification, we thus consider glu+ neurons to have a cortical fate, while GABA+ neurons and OPCs are subcortical fates in the embryo (Fig. 2C).
Figure 2.
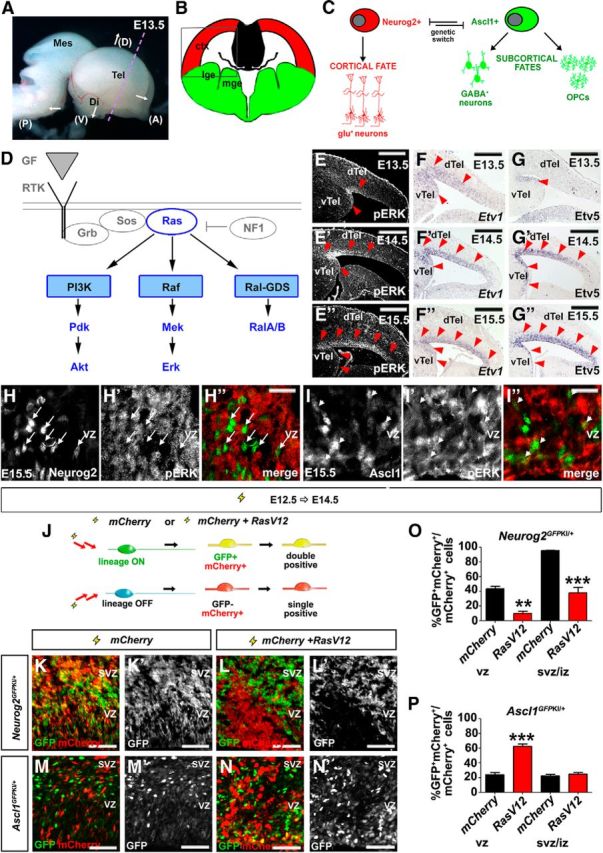
RAS signaling is required to regulate a Neurog2-Ascl1 genetic switch in cortical progenitors. A, Dissected E13.5 brain showing coronal plane of section (dotted purple line). B, Schematic illustration of a frontal section through the telencephalon, with the boxed area indicating the targeted area of the neocortex used in all electroporations. C, Cortical progenitors are bipotent, normally selecting a glutamatergic neuronal fate under the proneural activity of Neurog2, but retaining the potential to differentiate into GABAergic neurons or OPCs in response to Ascl1. D, Schematic illustration of signaling pathways activated downstream of RTK/RAS signaling. E–G, Dynamic expression of pERK (E–E″), Etv1 (F–F″), and Etv5 (G–G″) in E13.5, E14.5, and E15.5 cortex. Red arrowheads mark lateral-to-medial expansion of expression. H, I, E15.5 cortical sections double-stained for pERK (red) and Neurog2 (green; H–H″), or Ascl1 (green; I–I″). Arrows mark the cells in which Neurog2 and pErk expression are mutually exclusive. Arrowheads mark the cells in which pErk and Ascl1 are coexpressed. J–P, Schematic illustration of lineage tracing experiments in which an mCherry expression vector was electroporated with or without RasV12 into E12.5 Neurog2GFPKI/+ or Ascl1GFPKI/+ cortical progenitors (J). GFP/mCherry expression in E12.5→E14.5 electroporated Neurog2GFPKI/+ (K, K′, L, L′) and Ascl1GFPKI/+ (M, M′, N, N′) brains. Quantification of GFP and mCherry coexpression in VZ or SVZ/IZ cells of Neurog2GFPKI/+ (O) and Ascl1GFPKI/+ (P) brains. *p < 0.05, **p < 0.01, ***p < 0.005. Scale bars: E–G, E′–G′, E″–G″, 250 μm; H″, I″, 67.5 μm; K–N, K′–N′, 125 μm. A, anterior; ctx, neocortex; D, dorsal; di, diencephalon; dTel, dorsal telencephalon; GF, growth factor; lge, lateral ganglionic eminence; mes, mesencephalon; mge, medial ganglionic eminence; P, posterior; tel, telencephalon; V, ventral; vTel, ventral telencephalon.
In an instructive mode of cell fate specification, extrinsic signals control cell fate choices at lineage branch points by regulating genetic switches–activating one set of genes, while repressing alternative genetic programs (Pearson and Doe, 2004; Huang et al., 2007). Several observations suggest that in the telencephalon, the bHLH transcription factors Neurog2 and Ascl1 form such a genetic switch (Fig. 2C). First, Neurog2 and Ascl1 specify distinct cell fates (Schuurmans and Guillemot, 2002). Neurog2 is necessary and sufficient to specify a glu+ neuronal fate, whereas Ascl1 has multiple functions, either promoting proliferation or inducing the differentiation of GABA+ neurons or OPCs (Casarosa et al., 1999; Horton et al., 1999; Fode et al., 2000; Nieto et al., 2001; Parras et al., 2002; Schuurmans et al., 2004; Britz et al., 2006; Mattar et al., 2008; Castro et al., 2011; S. Li et al., 2012; Kovach et al., 2013). Second, Neurog2 and Ascl1 are repressive at the transcriptional level, with the loss of Neurog2 leading to upregulated Ascl1 expression in dorsal progenitors, converting these cells to ventral cell fates (Fode et al., 2000; Schuurmans et al., 2004). We thus conceive of Neurog2-Ascl1 as forming a central genetic switch that controls the choice between different telencephalic neuronal and glial cell fates (Fig. 2C). Here we investigated whether this Neurog2-Ascl1 switch was regulated by RAS/ERK signaling, and the significance of this switch in low-grade glial and glioneuronal tumors (Fig. 2D).
RAS/ERK signaling is temporally activated in telencephalic progenitors
Given previous associations between RAS/ERK signaling and either increased gliogenesis or neurogenesis (Baron et al., 2000; Chandran et al., 2003; Gabay et al., 2003; Hack et al., 2004; Kessaris et al., 2004; Abematsu et al., 2006; Aguirre et al., 2007; Samuels et al., 2008; Paquin et al., 2009; X. Li et al., 2012; Wang et al., 2012), we asked whether this signaling pathway might select different neural cell lineages by controlling the proneural genetic switch. We reasoned that if RAS signaling controlled Neurog2/Ascl1 expression and/or function, that it should undergo temporal regulation, concomitant with the temporal changes in neural cell output by telencephalic progenitors. To detect RAS pathway activation, we analyzed the expression of pERK as well as the Fgf-syn-expression group genes, Spry2, Etv1, and Etv5, all of which are expressed in response to RTK signaling (Tsang and Dawid, 2004). In E12.5 cortical progenitors, pERK expression was negligible except in mitotic cells at the ventricular surface (Fig. 2E). By E13.5, cortical progenitors in the lateral most corner of the ventricular zone (VZ) began expressing pERK, and by E15.5, pERK expression covered the entire cortical VZ (Fig. 2E,E″). Etv1 (Fig. 2F–F″), Etv5 (Fig. 2G–G″), and Spry2 (data not shown) transcription was also progressively initiated in the cortical VZ, following a similar lateral-to-medial gradient between E12.5 and E15.5. The initiation of RAS/ERK signaling in the lateral VZ and subsequent medial progression parallels the neurogenic gradient, and is likely initially triggered by Egf and Fgf ligands expressed in the cortical antihem, a lateral signaling center (Assimacopoulos et al., 2003; Hasegawa et al., 2004; Sansom and Livesey, 2009).
Finally, consistent with a normal in vivo requirement for RAS/ERK signaling in repressing Neurog2 and inducing Ascl1 expression, we found that Neurog2 and pERK were expressed in an almost completely nonoverlapping subset of E15.5 cortical progenitors (Fig. 2H–H″), while Ascl1 and pERK were frequently coexpressed (Fig. 2I–I″). Together, these data are consistent with the idea that RAS/ERK signaling is preferentially active in Ascl1+ and not Neurog2+ cortical progenitors, and suggest that RAS/ERK signaling may regulate the proneural genetic switch.
RAS functions through the ERK pathway to control the Neurog2-Ascl1 proneural genetic switch in cortical progenitors
To investigate whether increased RAS pathway activity was sufficient to trigger the Neurog2-Ascl1 proneural switch in cortical progenitors, we activated RAS signaling in Neurog2GFPKI/+ cortices (Britz et al., 2006), taking advantage of the persistence of GFP expression in Neurog2+ cortical progenitors and derivative neurons to perform short-term lineage tracing (Fig. 2J). To activate RAS/ERK signaling in E12.5 cortices, we used in utero electroporation (Dixit et al., 2011) to introduce a constitutively active (CA) form of RAS (RasV12; Chen et al., 2003), using an expression vector with an IRES-mCherry cassette to identify transfected cells. In this assay, GFP+mCherry+ coexpression marked electroporated cells in the Neurog2+ lineage, while mCherry single+ cells were not of the Neurog2+ lineage (Fig. 2J). In E12.5→E14.5 electroporations of mCherry vectors alone, 43.5 ± 3.6% and 95.4 ± 0.6% of mCherry+ VZ and subventricular zone (SVZ)/intermediate zone (IZ) cells coexpressed GFP in Neurog2GFPKI/+ cortices, respectively (n = 4; Fig. 2K,K′,O). In contrast, electroporation of RasV12 and mCherry vectors blocked GFP expression in Neurog2GFPKI/+ cortical VZ cells (10.1 ± 3.2%, n = 2; p = 0.0045) and SVZ/IZ cells (38.2 ± 7.2%, n = 2; p = 0.0002; Fig. 2L,L′,O). Thus, increased RAS activity not only represses Neurog2 expression in cortical VZ progenitors, but also blocks selection of this lineage in daughter cells that have moved to the SVZ/IZ.
If our model of proneural gene regulation is correct, we would also expect an aberrant increase in Ascl1 expression with activated RAS. Indeed, in analogous short-term lineage traces performed in Ascl1GFPKI/+ embryos (Leung et al., 2007), RasV12 promoted GFP expression and Ascl1 lineage selection in transfected, mCherry+ cortical VZ progenitors (mCherry: 24.1 ± 2.6%, n = 3; RasV12: 62.3 ± 3.5%, n = 3; p = 0.0009; Fig. 2M,M′,N,N′,P). Although GFP did not persist in RasV12-transfected Ascl1GFPKI/+ cells in the SVZ/IZ (mCherry: 22.5 ± 1.7%, n = 3; RasV12: 24.8 ± 2.2%, n = 3; p = 0.45; Fig. 2P), this is likely because as Ascl1+ progenitors exit the VZ and enter the SVZ/IZ, they differentiate, turning off Ascl1 (and eventually GFP) and turning on glial markers. Indeed, previous studies have shown that Ascl1 is downregulated in differentiating cells (Petryniak et al., 2007), and we show evidence later that RasV12 induces OPC differentiation (Fig. 5).
Figure 5.

RAS/ERK signaling promotes proliferation and a glial cell fate in cortical progenitors. E12.5→E15.5 electroporations of a pCIG2 control vector (A, A′, F, F′, K, K′, P, P′), RasV12 (B, B′, G, G′, L, L′, Q, Q′), bRafV600E (C, C′, H, H′, M, M′, R, R′), MekCA (D, D′, I, I′, N, N′, S, S′), and Etv5 (E, E′, J, J′, O, O′, T, T′) vectors (expressing). Transfected brains were analyzed for coexpression of GFP with Sox9 (A–E, A′–E′), BrdU (30 min pulse; F–J, F′–J′), Olig2 (K–O, K′–O′), or GFAP (P–T, P′–T′). Dashed lines outline the transfected region in the neocortex. Red arrowheads mark transfected cells ectopically expressing Sox9 (B′–E′), BrdU (G′–J′), Olig2 (L′–N′), or GFAP (Q′–S′). Scale bars: 250 μm. U, V, Quantification of GFP+ cells coexpressing Sox9 (U) or Olig2 (V). p values of pCIG2RasV12, pEFRasV12, pCIG2bRafV600E, pCIG2MekCA, pCIG2AktCA, pCIG2RalAV23, and pCIG2RalBQ72E are relative to pCIG2 control, whereas p values of pCIG2RasV12+pCIG2MekDN, pCIG2RasV12+pCIG2AktDN, pCIG2Rasv12+pCIG2RalADN, and pCIG2RasV12+pCIG2RalBDN are relative to pCIG2RasV12. *p < 0.05, **p < 0.01, ***p < 0.005. ctx, neocortex; str, striatum.
RasV12 activates several downstream signal transduction cascades (Fig. 2D). To determine whether it was the ERK branch of the pathway that repressed Neurog2 while increasing Ascl1 transcription, we also performed E12.5→E15.5 cortical electroporations of pCIG2 expression vectors (also carrying an IRES-GFP cassette) for bRafV600E (Dias-Santagata et al., 2011) and MekCA (Cowley et al., 1994), which selectively and constitutively activate ERK. Note that misexpression of RasV12, bRafV600E, and MekCA should mimic the overactivity of the RAS/ERK pathway observed in gliomas and glioneuronal tumors. As expected, overexpression of RasV12, bRafV600E, and to a lesser extent, MekCA, all induced ectopic pERK expression in cortical progenitors (Fig. 3C,C′,E,E′,G,G′), and also induced Etv5 expression (Fig. 3D′,F′,H′) in the GFP+ transfected patch (compare with vector controls; Fig. 3A,A′,B′). Note that Etv5 is an Fgf-syn-expression group gene that is normally transcribed in response to RTK signaling (Tsang and Dawid, 2004). RasV12, bRafV600E, and MekCA also repressed Neurog2 expression (Fig. 3D″,F″,H″) and induced ectopic Ascl1 expression in the VZ and SVZ/IZ (Fig. 3D‴,F‴,H‴) compared with vector controls (Fig. 3B″,B‴). In contrast, electroporation of pCIG2 vectors containing active forms of AktCA, RalAV23, and RalBQ72L (Franke et al., 1997; Lim et al., 2005; Sablina et al., 2007) into E12.5 cortical progenitors did not alter the expression of pERK, Etv5, Neurog2, or Ascl1 (data not shown).
Figure 3.

RAS functions through the ERK branch to regulate the Neurog2-Ascl1 genetic switch in cortical progenitors. E12.5→E15.5 electroporations of a pCIG2 control vector (A, A′, B–B‴), RasV12 (C, C′, D–D‴), bRafV600E (E, E′, F–F‴), MekCA (G, G′, H–H‴), RasN17 (I, I′, J–J″), or MekDN (K, K′, L–L″) vectors (expressing GFP). Transfected brains were analyzed for coexpression of GFP with pErk (A, A′, C, C′, E, E′, G, G′, I, I′, K, K′), or for transcripts for GFP (B, D, F, H, J, L), Etv5 (B′, D′, F′, H′), Neurog2 (B″, D″, F″, H″, J′, J″, L′, L″), and Ascl1 (B‴, D‴, F‴, H‴). Dashed lines outline the transfected region in the neocortex. Red arrowheads mark ectopic pERK (C′, E′, G′), Etv5 (D′, F′, H′), Neurog2 (H″, J′, J″, L′, L″), and Ascl1 expression (D‴, F‴, H‴), whereas yellow arrowheads mark transfected areas in which Neurog2 (D″, F″, H″) or pERK (I, I′, K, K′) expression was extinguished. M, Schematic illustration of repression of Neurog2 expression and induction of Ascl1 expression by RAS/ERK signaling. Scale bars: 250 μm. CP, cortical plate; ctx, neocortex; str, striatum.
Finally, to determine whether RAS/ERK signaling is normally required to regulate proneural gene expression, we inhibited signaling in cortical progenitors by introducing dominant-negative (DN) RasN17 and Mek in pCIG2 expression vectors (Cowley et al., 1994; Yoshimura et al., 2006). In E12.5→E15.5 cortical electroporations, both RasN17 and MekDN reduced pERK expression, and promoted ectopic Neurog2 expression in GFP+ cortical cells (Fig. 3I–L). This is consistent with the previously observed increase in Neurog2 expression in Fgf receptor 1 (Fgfr1);Fgfr2;Fgfr3 triple knock-outs, in which signaling through the RAS/ERK pathway is reduced (Rash et al., 2011).
RAS signaling is thus necessary and sufficient to trigger a Neurog2-to-Ascl1 proneural lineage switch, which may alter subsequent cell fate selection (Fig. 3M).
Increased RAS/ERK signaling diverts cortical cells away from neuronal fates
Neurog2 promotes the differentiation of glu+ pyramidal neurons (Fode et al., 2000; Schuurmans et al., 2004; Mattar et al., 2008) whereas Ascl1 promotes GABA+ neuronal as well as non-neuronal fates (Casarosa et al., 1999; Britz et al., 2006; Berninger et al., 2007; Geoffroy et al., 2009). We therefore examined the expression of the neuronal markers NeuN (late pan-neuronal marker), Tbr1 (deep-layer glu+ neuronal marker; Hevner et al., 2001; Bedogni et al., 2010), and Dlx1 (GABA+ neuronal lineage marker; Anderson et al., 1997b) in cortical cells electroporated with pCIG2 vectors containing CA forms of RAS and its major effectors.
In E12.5→E15.5 electroporations of RasV12, bRafV600E, or MekCA vectors (expressing GFP), the number of GFP+ transfected cells expressing NeuN was reduced 14.0-, 6.7-, and 2.7-fold, respectively, when compared with pCIG2 controls (n = 3 for each construct; p < 0.0001 for all comparisons; Fig. 4A–D,A′-D′,M). In contrast, AktCA, RalAV23, and RalBQ72L did not alter production of NeuN+ neurons (Fig. 4M and data not shown). We also coelectroporated E12.5 cortical progenitors with pCIG2 vectors for RasV12 together with pCIG2 vectors overexpressing DN forms of Mek, Akt, RalA, and RalB (B. P. Zhou et al., 2000; Lim et al., 2005; Yoshimura et al., 2006). Only MekDN partially rescued RAS inhibition of NeuN+ neurogenesis (n = 3 for each set of constructs; p < 0.0001 vs RasV12 alone; Fig. 4M), further supporting the role of ERK signaling in mediating RAS effects. Similar results were obtained when examining Tbr1 expression. In E12.5→E15.5 electroporations, pCIG2 vectors overexpressing RasV12 (p < 0.001), bRafV600E (p < 0.001), and MekCA (p < 0.05) reduced the number of Tbr1+ cells generated by 7.0-, 6.6-, and 5.0-fold, respectively, compared with pCIG2 controls (n = 3 for each construct; Fig. 4E–H,E′–H′,N). In contrast, electroporation of pCIG2 vectors overexpressing AktCA, RalAV23, and RalBQ72L did not alter Tbr1 expression, and a partial rescue of RasV12-mediated Tbr1 inhibition was only elicited by coexpression with MekDN (n = 3 for each set of constructs; p < 0.001 compared with RasV12 alone; Fig. 4N and data not shown). RAS thus acts specifically through the ERK pathway to divert cortical progenitors away from glu+ neuronal fates.
Figure 4.

RAS/ERK signaling inhibits cortical neurogenesis. A–L, E12.5→E15.5 electroporations of a pCIG2 control vector (A, A′, E, E′, I, I′), RasV12 (B, B′, F, F′, J, J′), bRafV600E (C, C′, G, G′, K, K′), and MekCA (D, D′, H, H′, L, L′) vectors (expressing GFP). Transfected brains were analyzed for coexpression of GFP (green) with pan-neuronal marker NeuN (red; A–D, A′–D′) and the cortical neuronal marker Tbr1 (red; E–H, E′–H′), or for transcripts for GFP (I–L) or the subcortical neuronal marker Dlx1 (I′–L′). Dashed lines outline the transfected region in the neocortex. Yellow arrowheads mark transfected areas with reduced expression of NeuN (B′, C′, D′), and Tbr1 (F′, G′, H′), and red arrowheads mark ectopic expression of Dlx1 (L′). Scale bars: 250 μm. M, N, Quantification of GFP+ cells coexpressing NeuN (M) or Tbr1 (N). p values of pCIG2-RasV12, pEF-RasV12, bRafV600E, MekCA, AktCA, RalAV23, and RalBQ72E are relative to pCIG2 control, whereas p values of pCIG2RasV12+pCIG2MekDN, pCIG2RasV12+pCIG2AktDN, pCIG2Rasv12+pCIG2RalADN, and pCIG2RasV12+pCIG2RalBDN are relative to pCIG2RasV12. *p < 0.05, **p < 0.01, ***p < 0.005. ctx, neocortex; str, striatum.
Given the loss of Neurog2 expression in response to RAS/ERK activation, a reduction in glu+ neuronal differentiation was not unexpected. The accompanying upregulation of Ascl1 expression likewise suggested that some cortical progenitors might aberrantly select a GABA+ interneuron fate. In E12.5→E15.5 electroporations of our pCIG2-RAS signaling constructs, however, Dlx1 expression was unaltered (Fig. 4I–L,I′–L′ and data not shown), with the exception of pCIG2MekCA, which induced ectopic Dlx1 in a small number of cortical cells (Fig. 4L′)–a finding that we return to below. We can, however, make the general conclusion that strong RAS/ERK activation generally blocks neuronal differentiation, raising the question of what the transfected cells had instead become.
Increased RAS/ERK signaling diverts cortical cells to proliferating, glial cell lineages
In addition to promoting a GABA+ neuronal fate, Ascl1 also promotes progenitor proliferation and OPC fate specification (Casarosa et al., 1999; Horton et al., 1999; Parras et al., 2007; Castro et al., 2011). We thus asked whether the Ascl1+ progenitors generated in response to hyperactivated RAS acquired a proliferative, glioblast fate. Sox9 is an HMG-box transcription factor that is required to specify a glial identity (Stolt et al., 2003; Kang et al., 2012), and is also a direct Ascl1 transcriptional target (Castro et al., 2011). In control E12.5→E15.5 electroporations of pCIG2, Sox9 was expressed in the VZ and in scattered cells outside the VZ (Fig. 5A,A′). Notably, loss of apical ventricular contacts is a hallmark of glial precursors, which continue dividing after leaving the VZ (Altman, 1966). In E12.5→E15.5 electroporations of pCIG2RasV12, pCIG2bRafV600E, and pCIG2MekCA vectors (coexpressing GFP), Sox9 was ectopically expressed throughout the cortex, indicating a marked increase in glioblast number (Fig. 5B–D,B′–D′). Indeed, electroporation of pCIG2RasV12, pCIG2bRafV600E, and pCIG2MekCA induced 40.5-, 37.5-, and 28.9-fold increases in the percentage of GFP+ electroporated cells that expressed Sox9, respectively (n = 3 for each construct; p < 0.0001 compared with pCIG2; Fig. 5U). In contrast, pCIG2AktCA, pCIG2RalAV23, and pCIG2RalBQ72L transfections had no influence on Sox9 expression (Fig. 5U and data not shown). In addition, in rescue experiments, only pCIG2MekDN suppressed the ability of RasV12 to induce the formation of Sox9+ glioblasts in E12.5→E15.5 cortical electroporations (n = 3 for each set of constructs; p < 0.0001 compared with RasV12 alone; Fig. 5U).
To confirm that RAS/ERK signaling could induce a glial precursor fate in cortical progenitors, we also examined expression of Nfia, a transcriptional target of Sox9 that regulates the switch from neurogenesis to gliogenesis (Deneen et al., 2006; Kang et al., 2012), as well as tenascin C and glutamine synthetase, which are both expressed in glial precursors that give rise to OPC or astrocytic lineages (Cammer, 1990; Wiese et al., 2012). In E12.5→E15.5 cortical electroporations, all three of these glioblast markers were induced by pCIG2RasV12, pCIG2bRafV600E, and pCIG2MekCA and not by pCIG2AktCA, pCIG2RalAV23, or pCIG2RalBQ72L (data not shown). Finally, given that glioblasts are actively proliferating cells, we assessed whether activation of RAS/ERK promoted ectopic proliferation, as detected by BrdU labeling. In control E12.5→E15.5 control electroporations, BrdU+ cells were detected in an abventricular band in the VZ and in a smaller number of scattered progenitors in the cortical layers, which presumably are glioblasts (Fig. 5F,F′). In contrast, in E12.5→E15.5 cortical electroporations of pCIG2RasV12, pCIG2bRafV600E, and pCIG2MekCA vectors, large numbers of cells incorporated BrdU outside of the VZ, indicative of ectopic proliferation (Fig. 5G–I,G′–I′).
Together, these studies suggest that activated ERK, and not other downstream RAS effectors, promotes the rapid formation of proliferative, glial precursor cells.
Increased RAS/ERK signaling promotes OPC and astrocytic lineage selection
To determine whether the glioblasts induced by RAS activation in E12.5 cortical progenitors would preferentially give rise to OPC or astrocytic lineages, we examined glial lineage markers in the E12.5→E15.5 electroporations of our signaling constructs. While 0% of pCIG2 (control), or pCIG2AktCA, pCIG2RalAV23, and pCIG2RalBQ72L-transfected cortical cells expressed Olig2, which is required to specify an OPC fate (Zhou and Anderson, 2002), 95.3 ± 1.0%, 96.9 ± 0.7%, and 71.7 ± 7.4% of pCIG2RasV12, pCIG2bRafV600E, and pCIG2MekCA-transfected cells expressed Olig2, respectively (n = 3 for each construct; p < 0.0001 compared with pCIG2; Fig. 5K–N,K′–N′,V and data not shown). Furthermore, coexpression of pCIG2RasV12 with pCIG2MekDN resulted in a 2.0-fold decrease in the number of Olig2+ cells formed compared with RAS activation alone (n = 3 for each set of constructs; p < 0.0001 compared with RasV12 alone), whereas pCIG2AktDN, pCIG2RalADN, and pCIG2RalBDN had no ability to suppress RasV12 induction of an OPC fate (Fig. 5V and data not shown). These values were complementary to our NeuN counts (Fig. 4M), and suggested that the majority of the RAS/ERK-activated cells ultimately became either Olig2+ OPCs (predominant) or NeuN+ neurons. Similar results were obtained when examining additional OPC markers, including Olig1 and Pdgfra (Pringle et al., 1992; Zhou and Anderson, 2002; Rowitch, 2004; data not shown). RAS thus acts via the ERK pathway to promote OPC fate specification in cortical glioblasts.
In the spinal cord, Ascl1 is expressed in neuronal and OPC lineages and not in astrocytic lineages (Battiste et al., 2007). Moreover, Olig2 blocks astrocyte differentiation (Fukuda et al., 2004). We thus speculated that the ability of activated RAS to induce Ascl1 and Olig2 expression in a large number of cortical cells would generally preclude the induction of astrocyte differentiation. Indeed, a relatively small number of E12.5 cortical cells electroporated with pCIG2RasV12, pCIG2bRafV600E, or pCIG2MekCA vectors expressed the astrocytic marker glial fibrillary acidic protein (GFAP) at 72 h postelectroporation (Fig. 5P–S,P′–S′). While the number of pCIG2RasV12-transfected cortical cells expressing GFAP increased if analyzed 6 d postelectroporation, OPC marker expression remained predominant (Fig. 7O–T). Ascl1+ glioblasts are thus biased toward selecting OPC fates upon RAS/ERK hyperactivation.
Figure 7.

RAS/ERK levels influence Ascl1's fate specification properties and target gene selection. A–C, Western blot analysis of lysates from HEK293 cells transfected with pEFRasV12 or pCIG2RasV12 and analyzed for pERK, ERK, and β-actin expression (A). Expression levels were quantified by densitometry and normalized to β-actin and total ERK (B, C). D–E, E12.5→E14.5 brains electroporated with pEFRasV12 and GFP were processed for the expression of GFP (D), pERK (D, D′), and Ascl1 (E). F–K, E12.5→E14.5 electroporations of Neurog2GFPKI/+ cortices with pEFRasV12 and mCherry, analyzed for expression of mCherry (F, J, K, red), Neurog2 (G), Dlx1 (H, H′), Tuj1 (I, K, blue), and GFP (J, K). Dashed red lines outline the transfected region in the neocortex. Red arrowheads mark ectopic Dlx1 (H, H′) or Tuj1 (I). Yellow arrowheads mark transfected areas with reduced Neurog2 (G) and GFP (J) expression. Scale bars: D–H, 250 μm; I–K, H′, 125 μm. L–W, E18.5 control (L, O, R, U) and E12.5→E18.5 brains electroporated with pEFRasV12 (M, P, S, V) or pCIG2RasV12 (N, Q, T, W) were processed for H&E staining (L–N), or immunohistochemistry for Olig2 (O–Q), GFAP (R–T), or TUJ1/pERK (U–W). Scale bars: 500 μm. X–Z, Transcriptional reporter assays in P19 cells using Dll1 (X), Dlx1/2 I12b intergenic enhancer (Y), and Sox9 (Z) reporters. *p < 0.05, **p < 0.01, ***p < 0.005. AA, Schematic representation of the effects of RAS/ERK activity levels on Ascl1 target gene selection. ctx, neocortex; str, striatum.
Activated RAS induces tumorigenesis in the embryonic cortex
To determine whether activated RAS indeed induced tumors in the embryonic cortex, we compared proliferation, cell identity, and histologic features induced following the expression of pCIG2 versus pCIG2-RasV12. In E12.5→E18.5 cortical electroporations, pCIGRasV12 induced the formation of GFP+ lesions that were densely cellular, expansile tumors with a locally infiltrating border (Fig. 6A,B,D,E,E′). The tumor cells had a predominantly bipolar morphology and displayed occasional mitotic figures. In some tumors, areas of microcystic change were seen (data not shown). In contrast to the clear laminar organization of pCIG2-control electroporated cortices (Fig. 6C), a distinct cell-sparse IZ was not observed upon pCIG2RasV12 transfection (Fig. 6D). Based on the morphologic features of pCIG2RasV12 tumors and their generally low proliferative rates, the tumors most closely resemble a low-grade astrocytoma akin to human pilocytic astrocytoma. Others have similarly observed generation of pilocytic astrocytoma-like tumors when introducing activated BRAF (Gronych et al., 2011), or have observed a range of low- and high-grade astrocytomas when introducing RasV12 (Ding et al., 2001). Notably, we have not determined whether the tumors are benign or malignant in our studies, a distinction that would require either transplantation into recipient mice or evidence of continuous expansion over time.
Figure 6.

Activation of RAS signaling in cortical progenitors results in tumorigenesis. A–E, E18.5 control (C) and E12.5→E18.5 brains electroporated with pCIG2RasV12 (A, B, D, E, E′) were processed for H&E staining (B–E, E′), or GFP transcripts (A). F–H, E12.5→E16.5 electroporations of a pCIG2 control vector (F–F‴) and pCIG2RasV12 (G–G‴, H–H‴). Transfected brains were analyzed for coexpression of GFP (green), proliferation marker Ki67 (red), and pan-neuronal marker Tuj1 (blue). White arrowheads mark RasV12 transfected cells expressing Ki67 instead of Tuj1 (H–H‴). I–P, E12.5→E15.5 brains electroporated with a pCIG2 control vector (I, I′, K, K′, M, M′, O, O′) or pCIG2RasV12 (J, J′, L, L′, N, N′, P, P′) were processed for immunostaining of pan-neural progenitor markers Sox2 (I, I′, J, J′) and Nestin (K, K′, L, L′), cortical-specific progenitor markers Pax6 (M, M′, N, N′), and Tbr2 (O, O′, P, P′). Red arrowheads mark ectopic expression of Sox2 (J′) and Nestin (L′), while yellow arrowheads mark transfected areas with reduced expression of Pax6 (N′) and Tbr2 (P′). Q–V, E12.5→E15.5 electroporations of pCIG2RasV12 with (T–V, V′) or without Neurog2 (Q–S, S′). Transfected brains were analyzed for the transcripts of Spry2 (Q, T), Pdgfa (R, U), or coexpression of GFP and Tuj1 (S, S′,V, V′). Red arrowheads mark ectopic expression of Spry2 (Q, T) or Pdgfa (R). W–Z, E12.5→E15.5 electroporations of pCIG2 (W, W′), AktA (X, X′), RalAV23 (Y, Y′), and RalBQ72L (Z, Z′) followed by analysis of BrdU incorporation. Red arrowheads mark ectopic BrdU incorporation. Dashed lines outline the transfected region in the neocortex. Scale bars: I–V, I′–V′, F–F‴, G–G‴ 250 μm; H–H‴, 67.5 μm. CP, cortical plate; ctx, neocortex; str, striatum.
To confirm that pCIG2RasV12-transfected cortical cells continued to proliferate instead of undergoing neuronal differentiation, we also examined expression of the proliferation marker Ki67 and the neuronal marker Tuj1 in electroporated cortices. In E12.5→E16.5 control pCIG2 electroporations, the GFP+ cohort of transfected cells had mostly migrated out of the VZ and into the differentiation zones of the cortical plate, and accordingly, most GFP+ cells expressed Tuj1 instead of Ki67 (Fig. 6F–F‴). In contrast, RasV12-transfected, GFP+ cells formed proliferative masses in which cells continued to divide as Ki67+ progenitors, and for the most part, did not differentiate into Tuj1+ neurons (Fig. 6G–G‴,H–H‴). We also examined the effects of RasV12 on the expression of pan-neural progenitor markers Sox2 and nestin, as well as the cortical-specific progenitor markers Pax6 and Tbr2. While the germinal zone expression of each of these markers was maintained in E12.5→E15.5 pCIG2 control electroporations, pCIG2RasV12 greatly expanded the expression of Sox2 (Fig. 6I,I′,J,J′) and nestin (Fig. 6K,K′,L,L′) into the IZ and cortical plate, whereas Pax6 (Fig. 6M,M′,N,N′) and Tbr2 (Fig. 6O,O′,P,P′) expression was dramatically reduced.
Together, these data are consistent with the idea that activation of the RAS pathway promotes a proliferative neural progenitor phenotype, but these progenitors acquire a ventral Ascl1+ identity, rather than a dorsal Neurog2+/Pax6+/Tbr2+ identity.
Loss of Neurog2 expression contributes to RasV12-induced gliomagenesis
Neurog2 promotes the differentiation of glu+ neurons either when misexpressed in the embryonic telencephalon or in adult SVZ neural stem cells (Mattar et al., 2008; Blum et al., 2011; Chen et al., 2012; Heinrich et al., 2012; S. Li et al., 2012). Moreover, overexpression of Neurog2, either alone or in combination with other transcription factors, can induce neuronal differentiation in other somatic cell types, and in glioblastoma-derived cell lines (Meng et al., 2012; Zhao et al., 2012; Guichet et al., 2013). To determine whether the loss of Neurog2 expression contributed to the oncogenic effects of RasV12 in our in vivo system, we asked whether Neurog2 could rescue RasV12-induced tumorigenesis. Strikingly, compared with the relatively uniform activation of pERK (data not shown) and Spry2 transcription (Fig. 6Q) in E12.5→E15.5 RasV12 single transfections, induction of pERK expression (data not shown) and Spry2 transcription was significantly reduced when Neurog2/RasV12 were electroporated in combination (Fig. 6T). Moreover, while RasV12 induced the formation of Pdgfra+ OPCs within 72 h postelectroporation (Fig. 6R), in E12.5→E15.5 cotransfections, Neurog2 largely prevented RasV12's ability to induce the formation of Pdgfra+ OPCs (Fig. 6U). Finally, Neurog2 also rescued the RasV12-mediated blockade of neurogenesis, restoring the differentiation of Tuj1+ cortical neurons (Fig. 6S,S′,V,V′). Neurog2 overexpression thus rescues several aspects of RasV12-induced tumorigenesis, highlighting the importance of RasV12's ability to regulate the Neurog2-Ascl1 genetic switch.
RAS/ERK levels dictate whether Ascl1-expressing cortical progenitors undergo glial or neuronal differentiation
While activation of different branches of the RAS pathway induced ectopic proliferation 3 d postelectroporation of the E12.5 cortex (including RalA and RalB, although not Akt; Fig. 6W–Z,W′–Z′), only the ERK pathway influenced neural cell fate selection, preferentially biasing cortical progenitors toward glial/OPC lineages. These results were unexpected given that (1) activation of ERK in some contexts induces neuronal differentiation (Ménard et al., 2002; Paquin et al., 2005) and (2) ERK signaling is activated in some gliomas that have a substantive neuronal content (e.g., GGs; Dougherty et al., 2010). One possibility was that differing levels of RAS/ERK activation might alter Ascl1's ability to specify either GABA+ neuronal or OPC fates in both the embryonic telencephalon and in adult SVZ neural stem cells (Casarosa et al., 1999; Fode et al., 2000; He et al., 2001; Yung et al., 2002; Parras et al., 2004, 2007; Britz et al., 2006; Heinrich et al., 2012).
To test whether RAS signaling levels influenced its activity in cortical progenitors, we reduced the amount of RasV12 expressed by changing promoters. All of the above electroporations were performed with pCIG2 containing a strong CAGG promoter/enhancer. When placed under control of the EF1α promoter, RasV12 had a twofold reduced ability to induce ERK phosphorylation in transfected cells compared with pCIG2RasV12 (Fig. 7A–D,D′; data not shown). In E12.5→E15.5 electroporations, pEFRasV12 (cotransfected with mCherry to trace transfected cells) still turned Neurog2 expression off (Fig. 7F,G), while Ascl1 expression was turned on (Fig. 7E), and similar numbers of Sox9+ glioblasts were generated compared with pCIG2RasV12 (Fig. 5U). However pEFRasV12 had a 2.2-fold reduced capacity to induce the formation of Olig2+ OPCs (Fig. 5V). Glioblast formation can thus occur at lower levels of RAS activation than OPC fate specification.
An examination of neuronal fates also revealed differences between higher and lower levels of RAS activation. Whereas electroporation of pCIG2-RasV12 strongly repressed neuronal fates, comparatively more NeuN+ and Tbr1+ neurons formed in pEFRasV12 electroporations (Fig. 4M,N). Moreover, pEFRasV12 induced ectopic VZ expression of Tuj1, an early neuronal marker, with a subset of these neurons expressing the GABA+ neuronal marker Dlx1 (Fig. 7H–K,H′). Similarly, MekCA, which has a reduced capacity to promote ERK phosphorylation compared with RasV12 and bRAFV600E (Fig. 3, compare G′,C′,E′; data not shown), also induced some ectopic Dlx1 (Fig. 4L′) and Tuj1 (data not shown) expression in the cortex. Neuronal differentiation was not complete, however, as pEFRasV12 and MekCA caused 2.2- and 2.7-fold reductions in the number of cells expressing the late neuronal marker NeuN, respectively (n = 3 for each construct; p < 0.0001 for each compared with pCIG2; Fig. 4M). Nevertheless, the inhibition of NeuN expression was significantly lower than that achieved by pCIG2RasV12 (n = 3 for each construct; 14.0-fold reduction compared with pCIG2; p < 0.0001; Fig. 4M). Our data thus suggest that lower levels of RAS/ERK activation are permissive for GABA+ neuronal differentiation, while higher levels promote gliogenesis.
To explore differences in tumor morphologies induced by different RAS signaling levels, we examined proliferation, cell identity, and histologic features induced by moderate (pEFRasV12) and high (pCIG2RasV12) levels of RAS pathway activation in E12.5→E18.5 cortical electroporations. Compared with control transfections (Fig. 7L), pEFRasV12 induced lesions that were mildly to moderately hypercellular, with increased local mass and architectural disruption of the cortex and underlying white matter (Fig. 7M). pCIG2RasV12 also induced the formation of tumors, but these had greater cellularity, nuclear atypia, and mitotic activity (Fig. 7N and data not shown). Moreover, the lineages of the pEFRasV12- and pCIG2RasV12-transfected cortical cells differed. Compared with control electroporations (Fig. 7O,R,U), pEFRasV12-transfected (pERK+) cells included abnormal Tuj1+ neurons interspersed with Olig2+ and GFAP+ glial cells (Fig. 7P,S,V). In contrast, pCIG2RasV12-induced lesions were comprised virtually entirely of abnormal pERK+ glial cells expressing Olig2 and GFAP (Fig. 7Q,T,W). Overall, the features of the lesions induced by low and high oncogenic RAS signaling were analogous to two classes of benign human tumors–glioneuronal tumors (i.e., GGs) and low-grade tumors composed of astrocyte-like glia (i.e., PAs), respectively, supporting the notion that RAS/ERK activity levels may drive cellular diversification in neoplasia in addition to normal development.
Together, these data suggest that RAS/ERK signaling not only controls the Neurog2-Ascl1 genetic switch but also functions downstream of this switch to bias Ascl1 fate decisions, depending on signaling intensity (Fig. 7AA).
Ascl1 transcriptional activity is regulated by ERK phosphorylation
RAS/ERK activity may influence neural cell fate selection by influencing Ascl1's selection of transcriptional targets, a possibility we tested using transcriptional reporter assays in Ascl1-responsive P19 embryonic carcinoma cells (Farah et al., 2000). We used reporters for three known Ascl1 target genes: Dll1, which activates the Notch receptor to maintain neural progenitors in an uncommitted state (Castro et al., 2006); Dlx1/2, which is active in GABA+ neurons (Poitras et al., 2007; Ghanem et al., 2008); and Sox9, which is expressed in proliferative glioblasts (Castro et al., 2011). Compared with pCIG2 controls, Ascl1 efficiently transactivated the Dll1 reporter (n = 3; p < 0.0001; Fig. 7X; Castro et al., 2006), but this activity was reduced by cotransfection with pCIG2RasV12 or pEFRasV12 (n = 3; p < 0.0001; Fig. 7X). Conversely, Ascl1 could only transactivate the Dlx1/2 reporter above pCIG2 control levels when RAS/ERK levels were moderately increased by pEFRasV12 (n = 3 for each construct; p = 0.012; Fig. 7Y). Finally, Ascl1's ability to activate a Sox9 reporter was enhanced when cotransfected with pCIG2RasV12 (n = 3 for each construct; p < 0.0001; Fig. 7Z), and not pEFRasV12, indicating that high RAS/ERK signaling is required to increase the transcriptional activity of Ascl1 on glial-related genes. Notably, the transcriptional reporter data are consistent with the in vivo gain-of-function data, where cell fates differed depending on signaling intensity.
The transcriptional activities of several bHLH proteins are regulated by direct phosphorylation (Marcus et al., 1998; Moore et al., 2002; Ma et al., 2008; Ali et al., 2011; Hindley et al., 2012; S. Li et al., 2012). To determine whether ERK1/2, which are proline-directed serine/threonine kinases (Roskoski, 2012), directly phosphorylated Ascl1, we generated a phospho-specific antibody to an Ascl1 peptide encompassing phosphorylated S185, one of six SP sites (Fig. 8A). To test antibody specificity, we generated single, double, and triple (Ascl1-SA3) serine-to-alanine (SA) mutations in S62, S185, and S218, all containing the same LSP sequence (Fig. 8A). The phospho-Ascl1 (pAscl1) antibody recognized wild-type Ascl1 expressed in HEK cells, with higher phosphorylation levels detected when cotransfected with RasV12, whereas Ascl1-SA3 was only detected by anti-Ascl1 and not by anti-pAscl1 (Fig. 8B). Although a pAscl1 band was still visible in the absence of RasV12, we ascribe this basal level of Ascl1 phosphorylation to serum in the media activating RAS/ERK signaling. Indeed, serum removal for 7 h reduced the amount of Ascl1 recognized by the pAscl1 antibody (data not shown). Next, we performed in vitro kinase assays using recombinant ERK and in vitro transcribed and translated versions of wild-type Ascl1 and mutant versions containing single, double, or triple SA mutations in S62, S185, and S218 (Fig. 8C). All versions of Ascl1 except Ascl1-SA3 were recognized by the pAscl1 antibody (Fig. 8C), confirming that Ascl1 carries phosphoacceptor sites for the ERK kinase.
Figure 8.

RAS/ERK regulates Ascl1 transcriptional activity via direct phosphorylation. A, Distribution of the six SP sites within wild-type Ascl1 (Ascl1-wt). Annotation of the serines mutated to alanines in Ascl1-SA3 and Ascl1-SA6 (mutated sites in red). B, Characterization of a phospho-specific Ascl1 antibody in HEK293 cells transfected with Ascl1 or Ascl1-SA3 along with pCIG2 or RasV12. C, Phospho-Ascl1 antibody recognizes in vitro-transcribed/translated Ascl1 (and not Ascl1-SA3) phosphorylated by recombinant ERK. D–F, Transcriptional reporter assays in P19 cells using Dll1 (D), Dlx1/2 I12b intergenic enhancer (E), and Sox9 (F) reporters, demonstrating that the effects of RAS/ERK activity levels on Ascl1 target gene selection are abrogated when all six SP sites in Ascl1 are mutated. *p < 0.05, **p < 0.01, ***p < 0.005.
To test whether phosphorylation modulates Ascl1's function, we conducted transcriptional reporter assays in vitro, using wild-type Ascl1 and a mutant version in which all six SP sites were mutated to alanines (Ascl1-SA6; Fig. 8A). In our functional studies, we used Ascl1-SA6 because the activity of the related bHLH transcription factor, Neurog2, is regulated by the number of SP sites phosphorylated, rather than their precise locations (a rheostat-like model; Ali et al., 2011; Hindley et al., 2012). We found that RasV12 inhibited the ability of both Ascl1-wt and Ascl1-SA6 to transactivate the Dll1 reporter, indicating that the SP sites did not mediate this repression (Fig. 8D). In contrast, Ascl1-SA6 could not transactivate the Dlx1/2 reporter above background levels, even in the presence of pEFRasV12, suggesting that the SP sites are essential for Ascl1 to turn on Dlx1/2 (Fig. 8E). Finally, while Ascl1-SA6 retained its capacity to induce Sox9 transcription, pCIG2RasV12 could only boost the transactivation strength of Ascl1 and not Ascl1-SA6 (Fig. 8F). Thus, Ascl1's ability to efficiently transactivate Dlx1/2 and Sox9 reporters requires the SP sites, consistent with a critical role for ERK activity in controlling Ascl1 target gene selection in specific cell lineages.
Ascl1 cell fate specification properties depend on SP sites
To assess the functional significance of Ascl1 phosphorylation in vivo, we misexpressed Ascl1-wt and Ascl1-SA6 using in utero electroporation. In E12.5→E15.5 cortical electroporations, during the period when pERK expression extends across the cortical VZ (Fig. 2E–E″), only Ascl1-wt induced a 2.7-fold increase in Sox9 expression (n = 3 for each construct; p = 0.003; Fig. 9A–C,A′–C′,S) and a robust increase in BrdU incorporation after a 30 min exposure (Fig. 9D–F,D′–F′). In contrast, Ascl1-SA6 did not induce these changes–supporting the requirement of SP site phosphorylation in promoting a proliferative glioblast fate. However, neither Ascl1 nor Ascl1-SA6 could induce ectopic Olig2 expression (Fig. 9G–I,G′–I′), consistent with our demonstration that the conversion of Sox9+ glioblasts to Olig2+ OPCs requires higher levels of RAS/ERK signaling (Fig. 5).
Figure 9.
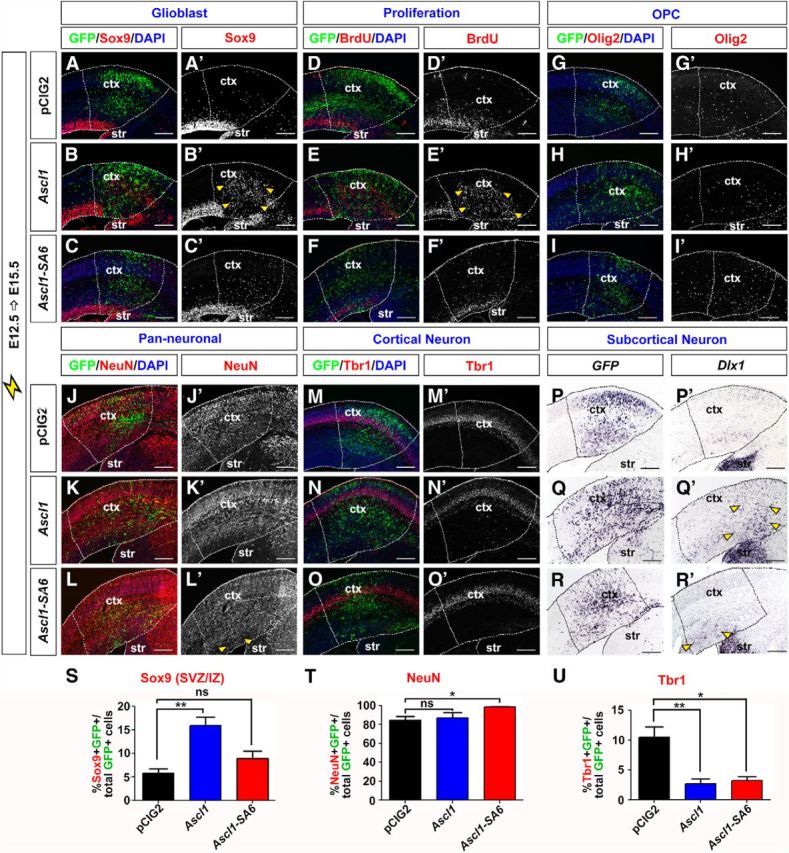
Ascl1 fate specification properties depend on the six SP phosphoacceptor sites. A–I, E12.5→E15.5 electroporations of pCIG2 (A, A′, D, D′, G, G′), wild-type Ascl1 (B, B′, E, E′, H, H′), and Ascl1-SA6 (C, C′, F, F′, I, I′) vectors (expressing GFP). Transfections were analyzed for ectopic expression of the glioblast marker Sox9 (A–C, A′–C′), proliferation marker BrdU (30 min pulse; D–F, D′–F′), and OPC marker Olig2 (G–I, G′–I′). Quantitation of GFP+ cells coexpressing Sox9 in the SVZ/IZ (S). J–R, E12.5→E15.5 electroporations of pCIG2 (J, J′, M, M′, P, P′), wild-type Ascl1 (K, K′, N, N′, Q, Q′), and Ascl1-SA6 (L, L′, O, O′, R, R′) vectors (expressing). Transfections were analyzed for expression of the pan-neuronal marker NeuN (J, J′, K, K′, L, L′), cortical neuronal marker Tbr1 (M, M′, N, N′, O, O′), or for transcripts for GFP (P–R) or Dlx1 (P′–R′). Quantification of GFP+ cells coexpressing NeuN (T) or Tbr1 (U). Dashed lines outline the transfected region in the neocortex. Yellow arrowheads mark ectopic expression of Sox9 (B′), BrdU (E′), NeuN (L′), or Dlx1 (Q′, R′). Scale bars: 250 μm. *p < 0.05, **p < 0.01, ***p < 0.005. ctx, neocortex; str, striatum.
If our model of Ascl1 phosphorylation-dependent fate specification is correct, the number of glial cells produced by Ascl1-SA6 (compared with wild-type Ascl1) should be accompanied by an increase in the number of neuronal cells. Indeed, we saw a small but significant increase in the number of NeuN+ neurons generated by Ascl1-SA6 compared with wild-type Ascl1 (n = 3 for each construct; p = 0.02), and importantly, Ascl1-SA6 induced ectopic NeuN expression in the VZ in E12.5→E15.5 electroporations (Fig. 9J–L,J′–L′,T). However, these neurons were not cortical glu+ neurons, as both Ascl1 and Ascl1-SA6 reduced Tbr1 expression (Fig. 9M–O,M′–O′,U). Instead, Ascl1 and to a lesser extent Ascl1-SA6 induced ectopic Dlx1 expression in the cortex (Fig. 9P–R,P′–R′), which was consistent with the reduced ability of Ascl1-SA6 versus Ascl1-wt to transactivate the Dlx1/2 transcriptional reporter in vitro (Fig. 8E). Thus, while Ascl1-wt can induce a proliferative glioblast fate in E12.5 cortical progenitors, mutation of the six SP sites results in a shift toward neuronal fates.
Finally, we asked whether Ascl1 was required to promote a glial cell fate in vivo by assessing whether RAS activation in the Ascl1 mutant cortex could initiate OPC differentiation. In both Ascl1+/− and Ascl1−/− cortices (note that the mutant allele is a GFP knock-in), E12.5→E15.5 electroporations of pCIG2-RasV12+mCherry and to a lesser extent pEF-RasV12+mCherry, induced ERK signaling (data not shown) and Etv1/5 expression (Fig. 10A–J). In addition, pCIG2RasV12 and pEFRasV12 induced the formation of similar numbers of Olig2+GFP+mCherry+ OPCs from Ascl1+/− and Ascl1−/− cortices (Fig. 10K–O). These data indicate that while Ascl1 is sufficient to induce a glioblast fate in response to RAS hyperactivation, it is not required for OPC differentiation. Thus, other RAS-activated factors, such as Etv5, may mediate the glial response (X. Li et al., 2012). Indeed, in E12.5→E15.5 cortical electroporations, Etv5 induced the formation of proliferative, Sox9+ glioblasts that incorporated BrdU (Fig. 5E,E′,J,J′). However, similar to Ascl1, Etv5 could not induce the conversion of Sox9+ glioblasts into Olig2+ OPCs or GFAP+ astrocytes (Fig. 5O,O′,T,T′), consistent with our finding that the OPC transition requires high levels of RAS/ERK activation.
Figure 10.

Ascl1 is not required for OPC differentiation in response to RAS/ERK signaling. A–J, E12.5→15.5 electroporations of Ascl1+/− (A, B, D, F, G, I) or Ascl1−/− (C, E, H, J) cortices with mCherry (A, F), or pEFRasV12+mCherry (B, C, G, H), or pCIG2RasV12 (D, E, I, J). Transfections were analyzed for the transcripts of Etv1 (A–E) or Etv5 (F–J). K–O, E12.5→15.5 electroporations of Ascl1+/+ (K–K″), Ascl1GFPKI/+ (L–L″), and Ascl1GFPKI/KI (M–M″) cortices with pEFRasV12+mCherry imaged for the coexpression of mCherry (red), GFP (green), and Olig2 (blue). Quantitation of percentage of double-positive cells (Olig2+mCherry+ cells/mCherry+ cells; N) and triple-positive cells (Olig2+mCherry+GFP+ cells/mCherry+GFP+ cells; O) in Ascl1+/+, Ascl1GFPKI/+, and Ascl1GFPKI/KI cortices. p > 0.05. Dashed lines outline the transfected region in the neocortex. Yellow arrowheads mark the cells with ectopic expression of Etv1 (B–E), Etv5 (G–J), or Olig2 (K′–M′). Scale bars: A–J, 500 μm; K–M, K′–M′, K″–M″, 250 μm. ctx, cortex; str, striatum.
We thus conclude that Ascl1 is sufficient, but not necessary, to induce the formation of a proliferative glioblast cell fate, acting redundantly with other factors, such as Etv5. However, the ability of glioblasts to differentiate into OPCs also requires elevated RAS/ERK signaling.
Discussion
RAS/ERK signaling has diverse functions in the nervous system–promoting proliferation and gliogenesis in some contexts, while inducing neuronal differentiation in others. Here, we found that RAS/ERK signaling influences neural cell fate specification in cortical development and in a RAS/ERK model of gliomagenesis (Fig. 11). First, activation of the RAS/ERK pathway is necessary and sufficient to trigger a proneural lineage switch, turning Neurog2 expression off and Ascl1 on. Second, ERK signaling levels modify Ascl1 activity–at high levels converting Ascl1 to a proglial/OPC transcription factor, while at lower levels, permissive for the differentiation of GABA+ neurons. Our data show that RAS/ERK signaling levels bias cell fate choice by regulating proneural gene expression and function, highlighting a unique role for the ERK branch of RAS signaling in cortical development and gliomagenesis.
Figure 11.

Model of RAS/ERK-proneural interactions in cortical development and gliomagenesis. In normal embryonic cortical progenitors, RAS/ERK signaling is low at early developmental stages, and Neurog2 is expressed, promoting robust glutamatergic neuronal differentiation. In the abnormal context of elevated RAS/ERK signals, Neurog2 expression is switched to Ascl1 expression. Moderate levels of RAS/ERK signals drive cortical progenitor cells to interneuronal fate with a few glioblast cells also differentiating, whereas high levels of RAS/ERK signals not only promote Neurog2-Ascl1 genetic switch in cortical progenitor cells, but also convert Ascl1 to a proglioblast molecule via direct phosphorylation, driving aberrant glioblast-like, and not neuronal differentiation, resulting in the formation of astrocytomas.
RAS/ERK signaling regulates proneural gene expression and function in cortical progenitors to control neuronal-glial cell fate decisions
Extrinsic cues that control the Neurog2-Ascl1 genetic switch lie at the crux of cortical progenitor cell transitions, ensuring that progenitors differentiate into appropriate cell types in sequence and on time. RAS/ERK signaling not only influences proneural gene expression, but also neural cell fate specification–promoting gliogenesis in some contexts and neurogenesis in others (this study and Baron et al., 2000; Ménard et al., 2002; Chandran et al., 2003; Gabay et al., 2003; Ito et al., 2003; Hack et al., 2004; Kessaris et al., 2004; Paquin et al., 2005, Paquin et al., 2009; Abematsu et al., 2006; Aguirre et al., 2007; Gauthier et al., 2007; Samuels et al., 2008; Ohtsuka et al., 2009; X. Li et al., 2012; Pucilowska et al., 2012; Wang et al., 2012). Building upon these concepts, we have now delineated an important mechanism behind RAS/ERK's ability to influence choice of alternative cell fates, providing evidence that the answer may lie in differing RAS/ERK activity levels. At lower levels of RAS/ERK signaling, such as those induced by pEF-RasV12 or pCIG2-MekCA in our model, cortical neurogenesis is initiated, but these neurons preferentially differentiate along GABA+ and not glu+ lineages, consistent with the neuronal fate specification properties of Ascl1 (Casarosa et al., 1999; Schuurmans et al., 2004; Britz et al., 2006; Berninger et al., 2007; Poitras et al., 2007; Geoffroy et al., 2009). Ascl1 has, however, a broad spectrum of activities, also promoting progenitor cell proliferation during the expansion phase of telencephalic development (Castro et al., 2011), and OPC differentiation at later stages (Parras et al., 2007). While we found that Ascl1 alone can initiate the formation of proliferative glioblasts, the differentiation of these cells into OPCs depends on high RAS/ERK signaling achieved by pCIG2RasV12 or pCIG2bRAF. It is perhaps not surprising that Ascl1 alone cannot induce the generation of ectopic Olig2+ OPCs, given that Ascl1 induces Dlx1 expression (this study and Britz et al., 2006), and Dlx1 represses Olig2 transcription and oligodendrogenesis in ventral telencephalic progenitors (Petryniak et al., 2007). While we interpret our data to suggest that levels of RAS/ERK signaling control Ascl1 lineage selection, we cannot exclude the possibility that other signaling properties, such as the timing and/or duration of pathway activation, also influence cell fate.
Our data support the idea that RAS/ERK signaling regulates Ascl1 function by direct phosphorylation. By site-directed mutagenesis, we found that Ascl1 SP sites are required for the transcriptional activation of Dlx1/2, and for the RAS-responsiveness of Ascl1 on the Sox9 reporter. Notably, Ascl1-SA6 was not inactive, as it an enhanced ability to induce the differentiation of NeuN+ neurons, similar to the enhanced proneural activity of Neurog2 following the mutation of all nine SP sites (Ali et al., 2011; Hindley et al., 2012). Phosphorylation of bHLH transcription factors can modify transcriptional activity at several levels, influencing events such as DNA binding, cellular localization, dimerization, cofactor binding, protein stability, and chromatin structure (Hand et al., 2005; Martindill et al., 2007; Vosper et al., 2007; Ma et al., 2008; Li et al., 2011; Sun et al., 2011; S. Li et al., 2012). Future studies will determine how the transcriptional activity of Ascl1 is altered when phosphorylated by ERK1/2.
Other factors, in addition to Neurog2 and Ascl1, undoubtedly contribute to the RAS/ERK-regulated gliogenic switch. Indeed, we found that while Ascl1 is sufficient to promote gliogenesis, it is not required for OPC differentiation. The PEA3 subgroup of Ets family transcription factors, and Etv5 in particular, is also instructive for gliogenic competence downstream of Mek (X. Li et al., 2012). Accordingly, we found that Etv5 also induces the formation of proliferative, Sox9+ glioblasts, but these cells fail to differentiate into Olig2+ OPCs, similar to the effects of Ascl1. The transition from proliferative glioblast to OPC thus requires high levels of RAS/ERK signaling, as revealed by our transcriptional reporter assays and in vivo gain-of-function studies. How RAS/ERK signaling and Ascl1/Etv5 functions are integrated will be an important area of future investigation.
Intersection between RAS/ERK signaling and proneural genes in gliomas
Tumor cells share many, but not all features of their normal neural counterparts, suggesting that the rules governing developmental programs may not be strictly followed in tumors. For example, all astrocytomas contain some Olig2+ cells (Ligon et al., 2004; Otero et al., 2011)–yet these tumors also contain GFAP+ cells with an astrocytic morphology. Our findings echo this in that we similarly saw the induction of some astrocytic markers in response to pCIG2RasV12 misexpression, in addition to OPC markers such as Olig1/2 and Pdgfra. Notably, Olig2 normally blocks astrocyte formation in cortical progenitors, and is turned off in cells that differentiate into astrocytes (Gabay et al., 2003; Fukuda et al., 2004). Coexpression of astrocyte and OPC markers is thus not normally observed in development, highlighting the abnormality of cellular transformation.
Our findings and others' (Ding et al., 2001; Marumoto et al., 2009; Gronych et al., 2011) demonstrate that elevated RAS/ERK signaling is sufficient for the generation of tumors. Moreover, hyperactive RAS/ERK signaling occurs in gliomas with diverse cellular compositions that span the malignancy spectrum, yet this pathway has primarily been ascribed a generic pro-proliferative role in gliomas. Our data indicates that the level of ERK signaling may explain in part the existence of different cell types in BRAF-driven gliomas such as PAs and GGs.
Our work speaks most directly to predominantly pediatric types of RAS pathway-activated gliomas, and our model system does not directly address the state of progenitors in the mature brain nor does it address the question of the role of ongoing ERK signals once a tumor is initiated. Nevertheless, our findings may still have implications for the role of signaling in fate decisions beyond neoplasia in the developmental period. Neurog2 and Ascl1 mediate cell fate decisions in zones of adult neurogenesis (Parras et al., 2004; Brill et al., 2009; Roybon et al., 2009; Blum et al., 2011; Kim et al., 2011; Chen et al., 2012; Heinrich et al., 2012), and extrinsic factors such as EGF and FGF are present and necessary for maintenance of these progenitor populations (Shi et al., 2008). Thus, it is reasonable to speculate that our findings could reflect a more general relationship between RAS/ERK and the Neurog2-Ascl1 genetic switch, in both normal pathways of neural cell fate specification and in adult gliomas with constitutively active RTK signaling or mutant NF1.
While some studies point to uncommitted neural stem cells as cells of origin for gliomas (Sanai et al., 2005; Fomchenko and Holland, 2006; Alcantara Llaguno et al., 2009), others indicate that more committed cells, such as OPCs (Lindberg et al., 2009; Persson et al., 2010; Liu et al., 2011), are the substrates for transformation. OPC-like cells are also implicated in replication competence and astrocytoma formation (Ligon et al., 2007; Barrett et al., 2012). Yet it has been unclear whether gliomas arise directly from OPCs or from progenitor cells that quickly acquire an OPC-like identity. We found that high RAS/ERK signals rapidly induce Ascl1 expression, block neuronal fates, and increase proliferation of aberrant glioblasts predisposed to OPC-like differentiation. Thus, while Olig2+ or NG2+ cells may be critical in the process of glioma initiation or maintenance, the cell of origin may not need to be an existing OPC. Instead, conversion of a normally neurogenic progenitor to an OPC-favoring glioblast may be part of the tumor initiation process induced by oncogenic RAS/ERK. Collaboration from other RAS effectors such as AKT may further modulate the cellular phenotype (Dai et al., 2005; Hu et al., 2005). With respect to established glioblastoma multiforme (GBMs), recent studies suggest that turning off Neurog2 is necessary for self-renewal and tumorigenesis from glioblastoma stem cells (Guichet et al., 2013). Conversely, Ascl1 is critical for GBM stem cell maintenance and tumorigenicity (Rheinbay et al., 2013). Future work will address RAS/ERK's role in regulating these transcription factors in GBM stem cells. A better understanding of the unique contributions of distinct signaling pathways will be increasingly important as more specific pathway-oriented therapies are developed.
Overall, we identified novel points of intersection between a central signal transduction pathway and proneural transcription factors, furthering our efforts to generate a comprehensive model of the regulatory events that coordinate neurogenesis and gliogenesis in health and disease.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) MOP-44094 (C.S.); the Kids Cancer Care Foundation (C.S., J.A.C.); Terry Fox Research Institute with the Alberta Cancer Foundation (J.A.C.); Alberta Innovates Health Solutions (C.S., J.A.C.); CIHR Training Grant in Genetics, Child Development, and Health (P.M., S.L.); Heart & Stroke Foundation Studentship (P.M.); AHFMR Studentship (P.M.); CIHR Canada Graduate Scholarship (P.M.), Killam Trust Award (P.M.); Lionel E. McLeod Health Research Scholarship (P.M.); CIHR Canada HOPE Fellowship (R.D.); and Brain Tumour Foundation of Canada fellowship (S.O.L.). We thank N. Klenin and D. Zinyk for technical support, and B. Deneen, F. Polleux, J. Ashwell, T. Jessell, C. Marshall, and K. Kaibuchi for reagents.
The authors declare no competing financial interests.
References
- Abematsu M, Kagawa T, Fukuda S, Inoue T, Takebayashi H, Komiya S, Taga T. Basic fibroblast growth factor endows dorsal telencephalic neural progenitors with the ability to differentiate into oligodendrocytes but not gamma-aminobutyric acidergic neurons. J Neurosci Res. 2006;83:731–743. doi: 10.1002/jnr.20762. [DOI] [PubMed] [Google Scholar]
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Alam S, Zinyk D, Ma L, Schuurmans C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev Dyn. 2005;234:772–782. doi: 10.1002/dvdy.20577. [DOI] [PubMed] [Google Scholar]
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali F, Hindley C, McDowell G, Deibler R, Jones A, Kirschner M, Guillemot F, Philpott A. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Development. 2011;138:4267–4277. doi: 10.1242/dev.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J. Proliferation and migration of undifferentiated precursor cells in the rat during postnatal gliogenesis. Exp Neurol. 1966;16:263–278. doi: 10.1016/0014-4886(66)90063-X. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997a;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL. Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron. 1997b;19:27–37. doi: 10.1016/S0896-6273(00)80345-1. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Marín O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Kaznowski CE, Horn C, Rubenstein JL, McConnell SK. Distinct origins of neocortical projection neurons and interneurons in vivo. Cereb Cortex. 2002;12:702–709. doi: 10.1093/cercor/12.7.702. [DOI] [PubMed] [Google Scholar]
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. doi: 10.1016/S0092-8674(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Assimacopoulos S, Grove EA, Ragsdale CW. Identification of a Pax6-dependent epidermal growth factor family signaling source at the lateral edge of the embryonic cerebral cortex. J Neurosci. 2003;23:6399–6403. doi: 10.1523/JNEUROSCI.23-16-06399.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67:878–887. doi: 10.1097/NEN.0b013e3181845622. [DOI] [PubMed] [Google Scholar]
- Baron W, Metz B, Bansal R, Hoekstra D, de Vries H. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol Cell Neurosci. 2000;15:314–329. doi: 10.1006/mcne.1999.0827. [DOI] [PubMed] [Google Scholar]
- Barrett LE, Granot Z, Coker C, Iavarone A, Hambardzumyan D, Holland EC, Nam HS, Benezra R. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell. 2012;21:11–24. doi: 10.1016/j.ccr.2011.11.025. [DOI] [PubMed] [Google Scholar]
- Battiste J, Helms AW, Kim EJ, Savage TK, Lagace DC, Mandyam CD, Eisch AJ, Miyoshi G, Johnson JE. Ascl1 defines sequentially generated lineage-restricted neuronal and oligodendrocyte precursor cells in the spinal cord. Development. 2007;134:285–293. doi: 10.1242/dev.02727. [DOI] [PubMed] [Google Scholar]
- Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RA, Beyer RP, Bammler TK, Rubenstein JL, Hevner RF. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci U S A. 2010;107:13129–13134. doi: 10.1073/pnas.1002285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B, Guillemot F, Götz M. Directing neurotransmitter identity of neurones derived from expanded adult neural stem cells. Eur J Neurosci. 2007;25:2581–2590. doi: 10.1111/j.1460-9568.2007.05509.x. [DOI] [PubMed] [Google Scholar]
- Blough MD, Westgate MR, Beauchamp D, Kelly JJ, Stechishin O, Ramirez AL, Weiss S, Cairncross JG. Sensitivity to temozolomide in brain tumor initiating cells. Neuro Oncol. 2010;12:756–760. doi: 10.1093/neuonc/noq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R, Heinrich C, Sánchez R, Lepier A, Gundelfinger ED, Berninger B, Götz M. Neuronal network formation from reprogrammed early postnatal rat cortical glial cells. Cereb Cortex. 2011;21:413–424. doi: 10.1093/cercor/bhq107. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane-Kishikawa T, Hill DE, Vidal M, Meyerson M, Grenier JK, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Brill MS, Ninkovic J, Winpenny E, Hodge RD, Ozen I, Yang R, Lepier A, Gascón S, Erdelyi F, Szabo G, Parras C, Guillemot F, Frotscher M, Berninger B, Hevner RF, Raineteau O, Götz M. Adult generation of glutamatergic olfactory bulb interneurons. Nat Neurosci. 2009;12:1524–1533. doi: 10.1038/nn.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britz O, Mattar P, Nguyen L, Langevin LM, Zimmer C, Alam S, Guillemot F, Schuurmans C. A role for proneural genes in the maturation of cortical progenitor cells. Cereb Cortex. 2006;16(Suppl 1):i138–i151. doi: 10.1093/cercor/bhj168. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Cammer W. Glutamine synthetase in the central nervous system is not confined to astrocytes. J Neuroimmunol. 1990;26:173–178. doi: 10.1016/0165-5728(90)90088-5. [DOI] [PubMed] [Google Scholar]
- Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. doi: 10.1242/dev.126.3.525. [DOI] [PubMed] [Google Scholar]
- Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Göttgens B, Matter JM, Guillemot F. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell. 2006;11:831–844. doi: 10.1016/j.devcel.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Castro DS, Martynoga B, Parras C, Ramesh V, Pacary E, Johnston C, Drechsel D, Lebel-Potter M, Garcia LG, Hunt C, Dolle D, Bithell A, Ettwiller L, Buckley N, Guillemot F. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev. 2011;25:930–945. doi: 10.1101/gad.627811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-O. [DOI] [PubMed] [Google Scholar]
- Chandran S, Kato H, Gerreli D, Compston A, Svendsen CN, Allen ND. FGF-dependent generation of oligodendrocytes by a hedgehog-independent pathway. Development. 2003;130:6599–6609. doi: 10.1242/dev.00871. [DOI] [PubMed] [Google Scholar]
- Chen JC, Zhuang S, Nguyen TH, Boss GR, Pilz RB. Oncogenic Ras leads to Rho activation by activating the mitogen-activated protein kinase pathway and decreasing Rho-GTPase-activating protein activity. J Biol Chem. 2003;278:2807–2818. doi: 10.1074/jbc.M207943200. [DOI] [PubMed] [Google Scholar]
- Chen X, Lepier A, Berninger B, Tolkovsky AM, Herbert J. Cultured subventricular zone progenitor cells transduced with neurogenin-2 become mature glutamatergic neurons and integrate into the dentate gyrus. PLoS One. 2012;7:e31547. doi: 10.1371/journal.pone.0031547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Dai C, Lyustikman Y, Shih A, Hu X, Fuller GN, Rosenblum M, Holland EC. The characteristics of astrocytomas and oligodendrogliomas are caused by two distinct and interchangeable signaling formats. Neoplasia. 2005;7:397–406. doi: 10.1593/neo.04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci. 2005;25:5584–5594. doi: 10.1523/JNEUROSCI.4693-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006;52:953–968. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, Batchelor TT, Ligon KL, Iafrate AJ, Ligon AH, Louis DN, Santagata S. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6:e17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61:3826–3836. [PubMed] [Google Scholar]
- Dixit R, Lu F, Cantrup R, Gruenig N, Langevin LM, Kurrasch DM, Schuurmans C. Efficient gene delivery into multiple CNS territories using in utero electroporation. J Vis Exp. 2011;52:2957. doi: 10.3791/2957. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, Storm PB, Biegel JA. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol. 2010;12:621–630. doi: 10.1093/neuonc/noq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder AM, Dominguez L, Franke TF, Ashwell JD. Phosphoinositide 3-kinase regulation of T cell receptor-mediated interleukin-2 gene expression in normal T cells. J Biol Chem. 1998;273:28025–28031. doi: 10.1074/jbc.273.43.28025. [DOI] [PubMed] [Google Scholar]
- Farah MH, Olson JM, Sucic HB, Hume RI, Tapscott SJ, Turner DL. Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development. 2000;127:693–702. doi: 10.1242/dev.127.4.693. [DOI] [PubMed] [Google Scholar]
- Fode C, Ma Q, Casarosa S, Ang SL, Anderson DJ, Guillemot F. A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes Dev. 2000;14:67–80. [PMC free article] [PubMed] [Google Scholar]
- Fomchenko EI, Holland EC. Origins of brain tumors–a disease of stem cells? Nat Clin Pract Neurol. 2006;2:288–289. doi: 10.1038/ncpneuro0200. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Kondo T, Takebayashi H, Taga T. Negative regulatory effect of an oligodendrocytic bHLH factor OLIG2 on the astrocytic differentiation pathway. Cell Death Differ. 2004;11:196–202. doi: 10.1038/sj.cdd.4401332. [DOI] [PubMed] [Google Scholar]
- Gabay L, Lowell S, Rubin LL, Anderson DJ. Deregulation of dorsoventral patterning by FGF confers trilineage differentiation capacity on CNS stem cells in vitro. Neuron. 2003;40:485–499. doi: 10.1016/S0896-6273(03)00637-8. [DOI] [PubMed] [Google Scholar]
- Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron. 2007;54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy CG, Critchley JA, Castro DS, Ramelli S, Barraclough C, Descombes P, Guillemot F, Raineteau O. Engineering of dominant active basic helix-loop-helix proteins that are resistant to negative regulation by postnatal central nervous system antineurogenic cues. Stem Cells. 2009;27:847–856. doi: 10.1002/stem.17. [DOI] [PubMed] [Google Scholar]
- Ghanem N, Yu M, Long J, Hatch G, Rubenstein JL, Ekker M. Distinct cis-regulatory elements from the Dlx1/Dlx2 locus mark different progenitor cell populations in the ganglionic eminences and different subtypes of adult cortical interneurons. J Neurosci. 2007;27:5012–5022. doi: 10.1523/JNEUROSCI.4725-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem N, Yu M, Poitras L, Rubenstein JL, Ekker M. Characterization of a distinct subpopulation of striatal projection neurons expressing the Dlx genes in the basal ganglia through the activity of the I56ii enhancer. Dev Biol. 2008;322:415–424. doi: 10.1016/j.ydbio.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Fode C, Guillemot F. Restricted expression of a novel murine atonal-related bHLH protein in undifferentiated neural precursors. Dev Biol. 1996;180:227–241. doi: 10.1006/dbio.1996.0297. [DOI] [PubMed] [Google Scholar]
- Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, Remke M, Hartmann C, Witt H, Jones DT, Witt O, Heiland S, Bendszus M, Holland EC, Pfister S, Lichter P. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011;121:1344–1348. doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichet PO, Bieche I, Teigell M, Serguera C, Rothhut B, Rigau V, Scamps F, Ripoll C, Vacher S, Taviaux S, Chevassus H, Duffau H, Mallet J, Susini A, Joubert D, Bauchet L, Hugnot JP. Cell death and neuronal differentiation of glioblastoma stem-like cells induced by neurogenic transcription factors. Glia. 2013;61:225–239. doi: 10.1002/glia.22429. [DOI] [PubMed] [Google Scholar]
- Guillemot F, Joyner AL. Dynamic expression of the murine Achaete-Scute homologue Mash-1 in the developing nervous system. Mech Dev. 1993;42:171–185. doi: 10.1016/0925-4773(93)90006-J. [DOI] [PubMed] [Google Scholar]
- Hack MA, Sugimori M, Lundberg C, Nakafuku M, Götz M. Regionalization and fate specification in neurospheres: the role of Olig2 and Pax6. Mol Cell Neurosci. 2004;25:664–678. doi: 10.1016/j.mcn.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48:45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Ashigaki S, Takamatsu M, Suzuki-Migishima R, Ohbayashi N, Itoh N, Takada S, Tanabe Y. Laminar patterning in the developing neocortex by temporally coordinated fibroblast growth factor signaling. J Neurosci. 2004;24:8711–8719. doi: 10.1523/JNEUROSCI.3070-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Ingraham C, Rising L, Goderie S, Temple S. Multipotent stem cells from the mouse basal forebrain contribute GABAergic neurons and oligodendrocytes to the cerebral cortex during embryogenesis. J Neurosci. 2001;21:8854–8862. doi: 10.1523/JNEUROSCI.21-22-08854.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich C, Götz M, Berninger B. Reprogramming of postnatal astroglia of the mouse neocortex into functional, synapse-forming neurons. Methods Mol Biol. 2012;814:485–498. doi: 10.1007/978-1-61779-452-0_32. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/S0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Hindley C, Ali F, McDowell G, Cheng K, Jones A, Guillemot F, Philpott A. Post-translational modification of Ngn2 differentially affects transcription of distinct targets to regulate the balance between progenitor maintenance and differentiation. Development. 2012;139:1718–1723. doi: 10.1242/dev.077552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton S, Meredith A, Richardson JA, Johnson JE. Correct coordination of neuronal differentiation events in ventral forebrain requires the bHLH factor MASH1. Mol Cell Neurosci. 1999;14:355–369. doi: 10.1006/mcne.1999.0791. [DOI] [PubMed] [Google Scholar]
- Huang S, Guo YP, May G, Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev Biol. 2007;305:695–713. doi: 10.1016/j.ydbio.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura O, Satoh Y, Endo S, Takishima K. Analysis of extracellular signal-regulated kinase 2 function in neural stem/progenitor cells via nervous system-specific gene disruption. Stem Cells. 2008;26:3247–3256. doi: 10.1634/stemcells.2008-0578. [DOI] [PubMed] [Google Scholar]
- Ito H, Nakajima A, Nomoto H, Furukawa S. Neurotrophins facilitate neuronal differentiation of cultured neural stem cells via induction of mRNA expression of basic helix-loop-helix transcription factors Mash1 and Math1. J Neurosci Res. 2003;71:648–658. doi: 10.1002/jnr.10532. [DOI] [PubMed] [Google Scholar]
- Jones DT, Kocialkowski S, Liu L, Pearson DM, Bäcklund LM, Ichimura K, Collins VP. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, Deneen B. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron. 2012;74:79–94. doi: 10.1016/j.neuron.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL, Forsyth P, Auer RN, Dunn JF, Cairncross JG, Parney IF, Weiss S. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27:1722–1733. doi: 10.1002/stem.98. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Jamen F, Rubin LL, Richardson WD. Cooperation between sonic hedgehog and fibroblast growth factor/MAPK signalling pathways in neocortical precursors. Development. 2004;131:1289–1298. doi: 10.1242/dev.01027. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Ables JL, Dickel LK, Eisch AJ, Johnson JE. Ascl1 (Mash1) defines cells with long-term neurogenic potential in subgranular and subventricular zones in adult mouse brain. PLoS One. 2011;6:e18472. doi: 10.1371/journal.pone.0018472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol. 2013;37:685–698. doi: 10.1097/PAS.0b013e31827f9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobbe CB, Reifenberger J, Reifenberger G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol. 2004;108:467–470. doi: 10.1007/s00401-004-0929-9. [DOI] [PubMed] [Google Scholar]
- Koelsche C, Wöhrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M, Lasitschka F, von Deimling A, Capper D. Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol. 2013;125:891–900. doi: 10.1007/s00401-013-1100-2. [DOI] [PubMed] [Google Scholar]
- Kovach C, Dixit R, Li S, Mattar P, Wilkinson G, Elsen GE, Kurrasch DM, Hevner RF, Schuurmans C. Neurog2 simultaneously activates and represses alternative gene expression programs in the developing neocortex. Cereb Cortex. 2013;23:1884–1900. doi: 10.1093/cercor/bhs176. [DOI] [PubMed] [Google Scholar]
- Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- Leung CT, Coulombe PA, Reed RR. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat Neurosci. 2007;10:720–726. doi: 10.1038/nn1882. [DOI] [PubMed] [Google Scholar]
- Li H, de Faria JP, Andrew P, Nitarska J, Richardson WD. Phosphorylation regulates OLIG2 cofactor choice and the motor neuron-oligodendrocyte fate switch. Neuron. 2011;69:918–929. doi: 10.1016/j.neuron.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mattar P, Zinyk D, Singh K, Chaturvedi CP, Kovach C, Dixit R, Kurrasch DM, Ma YC, Chan JA, Wallace V, Dilworth FJ, Brand M, Schuurmans C. GSK3 temporally regulates neurogenin 2 proneural activity in the neocortex. J Neurosci. 2012;32:7791–7805. doi: 10.1523/JNEUROSCI.1309-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD. MEK is a key regulator of gliogenesis in the developing brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol. 2004;63:499–509. doi: 10.1093/jnen/63.5.499. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, Anderson DJ, Stiles CD, Rowitch DH. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–545. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28:2266–2275. doi: 10.1038/onc.2009.76. [DOI] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo L, Dormand E, Greenwood A, Anderson DJ. Comparison of the generic neuronal differentiation and neuron subtype specification functions of mammalian achaete-scute and atonal homologs in cultured neural progenitor cells. Development. 2002;129:1553–1567. doi: 10.1242/dev.129.7.1553. [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system. Lyon: IARC; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Kennedy H, Dehay C. Contrasting effects of basic fibroblast growth factor and neurotrophin 3 on cell cycle kinetics of mouse cortical stem cells. J Neurosci. 2002;22:6610–6622. doi: 10.1523/JNEUROSCI.22-15-06610.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YC, Song MR, Park JP, Henry Ho HY, Hu L, Kurtev MV, Zieg J, Ma Q, Pfaff SL, Greenberg ME. Regulation of motor neuron specification by phosphorylation of neurogenin 2. Neuron. 2008;58:65–77. doi: 10.1016/j.neuron.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus EA, Kintner C, Harris W. The role of GSK3beta in regulating neuronal differentiation in Xenopus laevis. Mol Cell Neurosci. 1998;12:269–280. doi: 10.1006/mcne.1998.0713. [DOI] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Martindill DM, Risebro CA, Smart N, Franco-Viseras Mdel M, Rosario CO, Swallow CJ, Dennis JW, Riley PR. Nucleolar release of Hand1 acts as a molecular switch to determine cell fate. Nat Cell Biol. 2007;9:1131–1141. doi: 10.1038/ncb1633. [DOI] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattar P, Langevin LM, Markham K, Klenin N, Shivji S, Zinyk D, Schuurmans C. Basic helix-loop-helix transcription factors cooperate to specify a cortical projection neuron identity. Mol Cell Biol. 2008;28:1456–1469. doi: 10.1128/MCB.01510-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabé-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/S0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Meng F, Chen S, Miao Q, Zhou K, Lao Q, Zhang X, Guo W, Jiao J. Induction of fibroblasts to neurons through adenoviral gene delivery. Cell Res. 2012;22:436–440. doi: 10.1038/cr.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubüser A, Sun X, Hacohen N, Krasnow MA, Martin GR. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 1999;126:4465–4475. doi: 10.1242/dev.126.20.4465. [DOI] [PubMed] [Google Scholar]
- Mizuguchi R, Sugimori M, Takebayashi H, Kosako H, Nagao M, Yoshida S, Nabeshima Y, Shimamura K, Nakafuku M. Combinatorial roles of olig2 and neurogenin2 in the coordinated induction of pan-neuronal and subtype-specific properties of motoneurons. Neuron. 2001;31:757–771. doi: 10.1016/S0896-6273(01)00413-5. [DOI] [PubMed] [Google Scholar]
- Moore KB, Schneider ML, Vetter ML. Posttranslational mechanisms control the timing of bHLH function and regulate retinal cell fate. Neuron. 2002;34:183–195. doi: 10.1016/S0896-6273(02)00666-9. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5:1279–1287. doi: 10.1038/nn971. [DOI] [PubMed] [Google Scholar]
- Nieto M, Schuurmans C, Britz O, Guillemot F. Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron. 2001;29:401–413. doi: 10.1016/S0896-6273(01)00214-8. [DOI] [PubMed] [Google Scholar]
- Ohtsuka M, Fukumitsu H, Furukawa S. Neurotrophin-3 stimulates neurogenetic proliferation via the extracellular signal-regulated kinase pathway. J Neurosci Res. 2009;87:301–306. doi: 10.1002/jnr.21855. [DOI] [PubMed] [Google Scholar]
- Otero JJ, Rowitch D, Vandenberg S. OLIG2 is differentially expressed in pediatric astrocytic and in ependymal neoplasms. J Neurooncol. 2011;104:423–438. doi: 10.1007/s11060-010-0509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin A, Barnabé-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin A, Hordo C, Kaplan DR, Miller FD. Costello syndrome H-Ras alleles regulate cortical development. Dev Biol. 2009;330:440–451. doi: 10.1016/j.ydbio.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Parras CM, Schuurmans C, Scardigli R, Kim J, Anderson DJ, Guillemot F. Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes Dev. 2002;16:324–338. doi: 10.1101/gad.940902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parras CM, Galli R, Britz O, Soares S, Galichet C, Battiste J, Johnson JE, Nakafuku M, Vescovi A, Guillemot F. Mash1 specifies neurons and oligodendrocytes in the postnatal brain. EMBO J. 2004;23:4495–4505. doi: 10.1038/sj.emboj.7600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parras CM, Hunt C, Sugimori M, Nakafuku M, Rowitch D, Guillemot F. The proneural gene Mash1 specifies an early population of telencephalic oligodendrocytes. J Neurosci. 2007;27:4233–4242. doi: 10.1523/JNEUROSCI.0126-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BJ, Doe CQ. Specification of temporal identity in the developing nervous system. Annu Rev Cell Dev Biol. 2004;20:619–647. doi: 10.1146/annurev.cellbio.19.111301.115142. [DOI] [PubMed] [Google Scholar]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, Goldenberg DD, Vandenberg SR, Nguyen KN, Yakovenko S, Ayers-Ringler J, Nishiyama A, Stallcup WB, Berger MS, Bergers G, McKnight TR, Goldman SA, Weiss WA. Non-stem cell origin for oligodendroglioma. Cancer Cell. 2010;18:669–682. doi: 10.1016/j.ccr.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryniak MA, Potter GB, Rowitch DH, Rubenstein JL. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron. 2007;55:417–433. doi: 10.1016/j.neuron.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Barry G, Hawkins J, Mason S, Lindwall C, Little E, Sarkar A, Smith AG, Moldrich RX, Boyle GM, Tole S, Gronostajski RM, Bailey TL, Richards LJ. NFIA controls telencephalic progenitor cell differentiation through repression of the Notch effector Hes1. J Neurosci. 2010;30:9127–9139. doi: 10.1523/JNEUROSCI.6167-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitras L, Ghanem N, Hatch G, Ekker M. The proneural determinant MASH1 regulates forebrain Dlx1/2 expression through the I12b intergenic enhancer. Development. 2007;134:1755–1765. doi: 10.1242/dev.02845. [DOI] [PubMed] [Google Scholar]
- Pringle NP, Mudhar HS, Collarini EJ, Richardson WD. PDGF receptors in the rat CNS: during late neurogenesis, PDGF alpha-receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development. 1992;115:535–551. doi: 10.1242/dev.115.2.535. [DOI] [PubMed] [Google Scholar]
- Pucilowska J, Puzerey PA, Karlo JC, Galán RF, Landreth GE. Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J Neurosci. 2012;32:8663–8677. doi: 10.1523/JNEUROSCI.1107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash BG, Lim HD, Breunig JJ, Vaccarino FM. FGF signaling expands embryonic cortical surface area by regulating Notch-dependent neurogenesis. J Neurosci. 2011;31:15604–15617. doi: 10.1523/JNEUROSCI.4439-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639–647. doi: 10.1016/j.tcb.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rheinbay E, Suvà ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, Oksuz O, Rabkin SD, Martuza RL, Rivera MN, Louis DN, Kasif S, Chi AS, Bernstein BE. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013;3:1567–1579. doi: 10.1016/j.celrep.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Rowitch DH. Glial specification in the vertebrate neural tube. Nat Rev Neurosci. 2004;5:409–419. doi: 10.1038/nrn1389. [DOI] [PubMed] [Google Scholar]
- Roybon L, Deierborg T, Brundin P, Li JY. Involvement of Ngn2, Tbr and NeuroD proteins during postnatal olfactory bulb neurogenesis. Eur J Neurosci. 2009;29:232–243. doi: 10.1111/j.1460-9568.2008.06595.x. [DOI] [PubMed] [Google Scholar]
- Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, DeCaprio JA, Hahn WC. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129:969–982. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- Sansom SN, Livesey FJ. Gradients in the brain: the control of the development of form and function in the cerebral cortex. Cold Spring Harb Perspect Biol. 2009;1:a002519. doi: 10.1101/cshperspect.a002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, Louis DN, Korshunov A, Pfister S, Hartmann C, Paulus W, Reifenberger G, von Deimling A. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- Schuurmans C, Guillemot F. Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr Opin Neurobiol. 2002;12:26–34. doi: 10.1016/S0959-4388(02)00286-6. [DOI] [PubMed] [Google Scholar]
- Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H, Cunningham JM, Dyck R, Walsh C, Campbell K, Polleux F, Guillemot F. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. EMBO J. 2004;23:2892–2902. doi: 10.1038/sj.emboj.7600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sun G, Zhao C, Stewart R. Neural stem cell self-renewal. Crit Rev Oncol Hematol. 2008;65:43–53. doi: 10.1016/j.critrevonc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart IH, Smart M. The location of nuclei of different labelling intensities in autoradiographs of the anterior forebrain of postnatial mice injected with [3H]thymidine on the eleventh and twelfth days post-conception. J Anat. 1977;123:515–525. [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003;17:1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian L, Sarkar A, Shetty AS, Muralidharan B, Padmanabhan H, Piper M, Monuki ES, Bach I, Gronostajski RM, Richards LJ, Tole S. Transcription factor Lhx2 is necessary and sufficient to suppress astrogliogenesis and promote neurogenesis in the developing hippocampus. Proc Natl Acad Sci U S A. 2011;108:E265–274. doi: 10.1073/pnas.1101109108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, Powers JF, Runquist IS, Rowitch DH, Stiles CD. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011;69:906–917. doi: 10.1016/j.neuron.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supèr H, Soriano E, Uylings HB. The functions of the preplate in development and evolution of the neocortex and hippocampus. Brain Res Brain Res Rev. 1998;27:40–64. doi: 10.1016/S0165-0173(98)00005-8. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS., Jr Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Fujimori KE, Takauji R. Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J Neurosci. 1997;17:8313–8323. doi: 10.1523/JNEUROSCI.17-21-08313.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang M, Dawid IB. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci STKE. 2004;2004:pe17. doi: 10.1126/stke.2282004pe17. [DOI] [PubMed] [Google Scholar]
- Vosper JM, Fiore-Heriche CS, Horan I, Wilson K, Wise H, Philpott A. Regulation of neurogenin stability by ubiquitin-mediated proteolysis. Biochem J. 2007;407:277–284. doi: 10.1042/BJ20070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell. 2012;150:816–830. doi: 10.1016/j.cell.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese S, Karus M, Faissner A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front Pharmacol. 2012;3:120. doi: 10.3389/fphar.2012.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, de la Cruz E, Anderson SA. Cortical interneuron fate determination: diverse sources for distinct subtypes? Cereb Cortex. 2003;13:670–676. doi: 10.1093/cercor/13.6.670. [DOI] [PubMed] [Google Scholar]
- Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kawano Y, Kawabata S, Wang S, Kaibuchi K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun. 2006;340:62–68. doi: 10.1016/j.bbrc.2005.11.147. [DOI] [PubMed] [Google Scholar]
- Yung SY, Gokhan S, Jurcsak J, Molero AE, Abrajano JJ, Mehler MF. Differential modulation of BMP signaling promotes the elaboration of cerebral cortical GABAergic neurons or oligodendrocytes from a common sonic hedgehog-responsive ventral forebrain progenitor species. Proc Natl Acad Sci U S A. 2002;99:16273–16278. doi: 10.1073/pnas.232586699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, He H, Zhou K, Ren Y, Shi Z, Wu Z, Wang Y, Lu Y, Jiao J. Neuronal transcription factors induce conversion of human glioma cells to neurons and inhibit tumorigenesis. PLoS One. 2012;7:e41506. doi: 10.1371/journal.pone.0041506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BP, Hu MC, Miller SA, Yu Z, Xia W, Lin SY, Hung MC. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J Biol Chem. 2000;275:8027–8031. doi: 10.1074/jbc.275.11.8027. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/S0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang S, Anderson DJ. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron. 2000;25:331–343. doi: 10.1016/S0896-6273(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Leon SP, Folkerth RD, Guo SZ, Wu JK, Black PM. Evidence for clonal origin of neoplastic neuronal and glial cells in gangliogliomas. Am J Pathol. 1997;151:565–571. [PMC free article] [PubMed] [Google Scholar]