

Abstract
A polarity-reversing radical cascade strategy for alkene di-functionalization by vicinal C-C and C-P bond-formation has been developed. This approach to concurrently adding phosphorous and a heteroarene across an olefin is enabled by photocatalytic generation of electrophilic P-centered radicals. Upon chemoselective addition to an olefin, the resulting nucleophilic C-centered radical selectively combines with electrophilic heteroarenes, such as pyridines. This multi-component coupling scheme for phosphinylalkylation complements classic two-component methods for hydrophosphinylation of alkenes and C-H phosphinylation of arenes. Included competition and photo-quenching experiments provide insight into the selectivity and mechanism of this polarity-reversal pathway.
Keywords: radical cascade, polarity-reversal, phosphorous radicals, pyridines, photoredox catalysis
Graphical Abstract
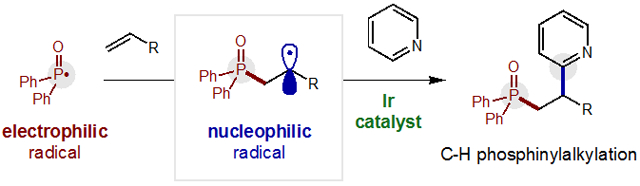
Alkene di-functionalization is among the most useful instruments in the synthetic toolbox.1 Notably, radical-mediated approaches offer complementarity to two-electron strategies, with respect to both reactivity and selectivity.2,3 For example, electrophilic P-centered radicals4 add to alkenes to afford hydrophosphinylation with anti-Markovnikov selectivity (Figure 1a).5,6 Alternatively, P• may also directly add to arenes in a net C-H phosphonation.7 Yet, in contrast to these couplings with π-nucleophiles, P• combination with electron-deficient heteroarenes is less favored.8 Given the medicinal importance of both motifs,9 we sought to develop a method for adding biologically relevant phosphines and heteroarenes to alkenes in a single transformation. Since Minisci’s pioneering work, nucleophilic radicals have been employed to construct C-C bonds directly onto heteroarenes.10,11 Recently, Herzon and Baran have shown that H• addition to an alkene affords alkyl radicals that may also engage in the Minisci heteroarylation mechanism.12 Given the requirement of nucleophilicity for the alkyl radicals to combine with electrophilic, protonated heteroarenes by this mechanism, we postulated that a three-component coupling reaction could be designed wherein polarity effects would dictate chemo- and regio- selectivity (Figure 1b).
Figure 1.
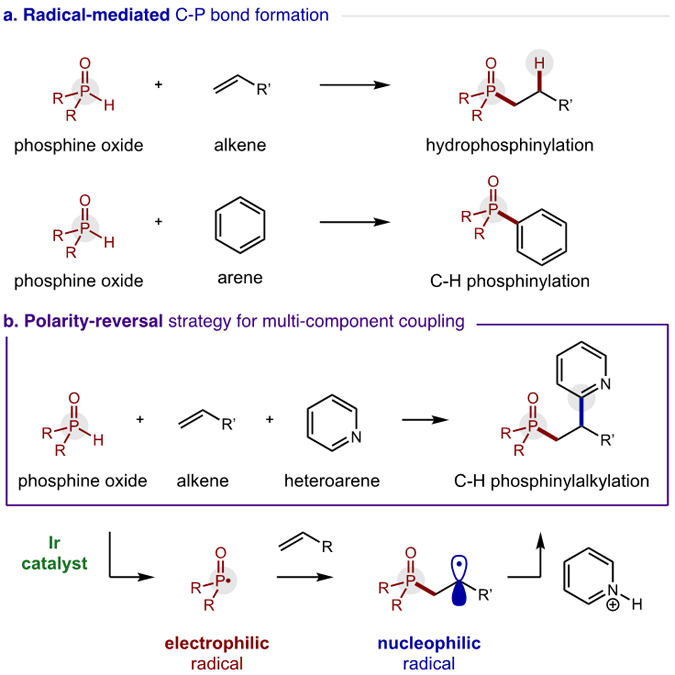
Polarity-reversal strategy for radical C-P couplings.
In our design, a polarity-reversal radical cascade strategy could selectively convert phosphine oxides to electrophilic P-centered radicals. Subsequent addition to an olefin (rather than to electron-deficient heteroarenes) would render the resulting open-shell intermediate nucleophilic. This C-centered radical may then chemoselectively combine with heteroarenes to afford a three-component coupling adduct. To our knowledge, only Minisci, Barriault, Liu, and Hong have reported examples of heteroaryl difunctionalization of alkenes, albeit initiated by alkyl or azide radicals.13 Despite this inspiration, we were cognizant that hydrophosphinylation via the Pudovik reaction may be a competing pathway.5 A second side-product pathway might include direct two-electron addition to pyridines, as recently developed by McNally.14 Moreover, the selective, radical combination of alkenes and heteroarenes remains quite rare, with limited examples of alkene hydroarylation that include Herzon and Baran’s stoichiometric protocols12 and Jui’s inverted approach entailing addition of pyridyl radicals.15 Despite these challenges, we surmised catalytic generation of P• at low concentrations relative to the alkene and heteroarene components may enable a chemoselective cascade.
To test the viability of this radical cascade strategy for multi-component C-P and C-C coupling, diphenyl phosphine oxide, 1-hexene, and phenanthridine were combined in the presence of an acid, photocatalyst, and blue LED irradiation (Figure 2). To our delight, the heteroarene phosphinylalkylation was promoted quite efficiently in the presence of 1 mol% Ir(ppy)2(dtbbpy)PF6 photocatalyst (entry 1). Interestingly, we noted that some protonated quinolines (e.g. phenanthridine) may autocatalyze this transformation. However, we observed lower and inconsistent yields (entry 2) in the absence of 1 mol% catalyst (and limited scope), and thus retained the photocatalyst for further optimization. The addition of an acid (1.2 equiv TFA) and water (5.5 equiv) were also found to be crucial components for reaction efficiency – in order to protonate and solubilize the terminal heteroarene electrophile (entries 3–4). Although, this reaction is amenable to varying solvents, concentrations, and non-aerobic conditions, most are inferior, frequently affording Pudovik adducts rather than phosphinylalkylation (entries 5–10). Furthermore, the multi-component coupling does not proceed well in the absence of LED irradiation (entry 11).
Figure 2.
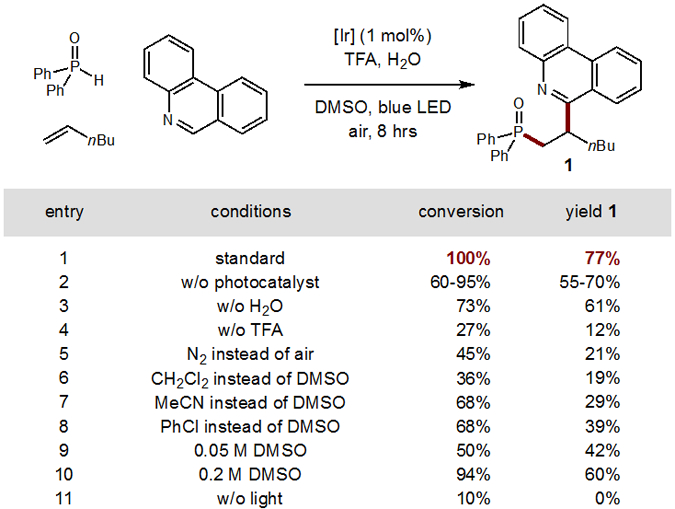
Development of a cascade heteroarene phosphinylalkylation.
Conditions: phenanthridine (0.1 mmol), Ph2P(O)H (2 equiv), Ir(ppy)2(dtbbpy) PF6 (1 mol%), DMSO (1 mL), trifluoroacetic acid (1.2 equiv), H2O (5.5 equiv), and 1-hexene (2 equiv), blue LED, 25°C. 1H NMR yields vs standard.
Having developed an efficient, photocatalytic method to enable the phosphinylalkylation of heteroarenes, we next investigated the generality of this polarity-reversal radical cascade. As shown in Figure 3, the scope of alkenes that can be employed in this transformation is surprisingly wide – tolerating a broad range of functional groups, including esters, ketones, alcohols, halides, and ethers (2-6). Notably, these simple, unbiased alkenes can be employed as suitable P-radical traps. Alternatively, more electron-rich enol ethers are also 1,2-phosphinyl-arylated (7) – with even greater efficiency, as the intermediary α-oxy alkyl radical has higher nucleophilicity to promote heteroarene addition.
Figure 3.

Heteroarene phosphinylalkylation: Reaction scope.
In order to probe the generality of heteroarenes in this Minisci radical addition mechanism,10 we then subjected a series of quinolines (8-11) and isoquinolines (12-18) to this reaction. Notably, a wide range of steric and electronic substituents were tolerated, including halides, esters, and amides. And while, less efficient than the enol ether component, non-activated alkene (e.g. hexene) may also be employed (9, 13).
Given the biological significance of phosphorous,9 we also investigated variation of the phosphine component and were pleased to find that a range of electronically diverse aryl and heteroaryl phophine oxides may be employed (19-22). Interestingly, even an electron-rich, bis-OMe aryl phosphine oxide can undergo polarity-reversal via its electron-poor P-radical (19–20). Additionally, dialkyl phosphine oxides, phosphonate esters, and phosphine sulfides are suitable radical precursors (23-25).
We next turned our attention to the heteroarene component with a specific focus on extending this methodology to simple pyridines (Figure 4). Even with an electron-rich enol ether as the nucleophilic radical precursor, and a basic lutidine, which should provide a strongly electrophilic lutidinium as the terminal trap, we did not observe any desired three-component coupling adduct. Similarly, the N-oxide, although frequently employed as a radical coupling partner,16 was incompatible with this cascade (even with Brønsted or Lewis acids). Ultimately, we were pleased to find that N-OMe pyridiniums17 are suitable heteroarene partners for providing phosphinylalkylation of 2,6-lutidine (26). Interestingly, we noted a strong counterion effect on this radical cascade,18 wherein the oxidizable iodide affords no product, while PF6, BF4, CF3SO3, and MeSO3 yield 26 with varying efficiency (65–78%). Fortunately, MeSO4, which was the best anion among those we investigated (81%), is also the easiest to access synthetically by directly combining N-oxide with dimethyl sulfate.
Figure 4.

Development of a pyridine phosphinylalkylation.
Conditions: pyridinium (0.1 mmol), Ph2P(O)H (2 equiv), Ir(ppy)2(dtbbpy)PF6 (1 mol%), DMSO (1 mL), H2O (5.5 equiv), and alkene (2 equiv), blue LED, 25°C. 1H NMR yields vs standard.
With this second-generation strategy in hand for direct C-H phosphinylalkylation of pyridines, the synthetic generality of the radical cascade was further investigated (Figure 5). Noting the high electrophilicity of these N-OMe pyridinium partners, we questioned if they may allow similarly broad scope with respect to the phosphine oxide component. Thus, we were pleased to see that more acidic and nucleophilic variants, including dibutyl phosphine oxide, pinacol phosphonate, and bis-aryl phosphine oxides were suitable partners (27-30). However, non-ethereal alkenes are not suitable nucleophiles for this reaction mechanism.
Figure 5.

Heteroarene phosphinylalkylation: Pyridine scope.
In probing the pyridine component, we noted that an unsubstituted pyridine is regioselectively functionalized at the 4-position in a 3:1 r.r. (31). This selectivity likely results from steric repulsion by the N-OMe group, which blocks radical addition at the 2-positions. However, if a 2-Cl substituent is employed to increase the electrophilicity, then regioselectively is decreased to 2:1 (32). On the other hand, stabilization of the radical addition by a 2-Ph group affords 6:1 r.r. (33). Finally, a 2-Me group sufficiently repels the N-OMe to afford >20:1 regioselectively (34). Of note, if the 4-position is blocked by substituents of varying electronics (Me, OMe, CN), then only reactivity at the 2-position is observed (35-37).
Having developed protocols for the phosphinylalkylation of pyridines at either their 2 or 4 C-H bonds, we questioned if this strategy could also promote functionalization of the electron-rich 3-position. This regioselectivity is not typically favored in Minisci additions due to a mismatch in polarity (both the alkyl radical and C3 are electron-rich). Nonetheless, with this goal in mind, we prepared the 2-alkoxy quinoline (38) and subjected it to the initial TFA conditions. We were pleased to find the intermediary 6-exo-trig radical cyclization does in fact occur – affording a fused, 2H‐pyrano[2,3‐b]quinoline (39) core found in glutamate receptor antagonists, potassium-channel activators, and employed as ligands in Pd-catalyzed C-H functionalization.19
With two complementary conditions in hand, we were curious how they compared to one another. Thus, we performed a competition experiment in which the protonated phenanthridine and N-OMe lutidinium were combined and reacted in a 1:1 ratio. As shown in Figure 7a, the phenanthridinium partner reacts nearly four times faster. Moreover, both partners individually outcompete the Pudovik reaction5 handily (Figure 7b) – affording a 7:1 ratio of three-component coupling to two-component hydrophosphinyl-ation in the case of phenanthridine (1), or 6:1 for pyridine (26).
Figure 7.

Competition experiments: (a) pyridine vs phenanthridine acceptors, and (b) three-component coupling vs two-component hydrophosphinylation.
As a further probe of the mechanism, we performed Stern-Volmer quenching experiments for the excited Ir photocatalyst in the presence of each reaction component, including the alkene, phosphine oxide, and heteroarene. Interestingly, only the charged heteroarenes provide quenching (KSV 4–55). Surprisingly, even in the presence of various bases (e.g. Na2CO3), diphenyl phosphine oxide is not a quencher (nor is PPh3), suggesting a reductive quenching mechanism is unlikely.20 On the other hand, O2 is a strong quencher (KSV >600), which suggests oxidative quenching is more likely operative.21
Our proposed mechanism for the multi-component coupling of phosphine oxide A, alkene B, and heteroarenes C is shown in Figure 8. To start, the Ir(III) photocatalyst is excited by visible light and quenched by O2 to form superoxide (O2• −) along with Ir(IV).22 This strong oxidant (+1.2 V vs SCE)23 is capable of removing an electron from neutral phosphine oxide A (+1.0 V vs SCE), or its tautomer D,24 to form radical cation E, while regenerating the ground state Ir(III) catalyst. This highly electrophilic phosphinium radical cation E may then selectively combine with alkenes B (of varying nucleophilicity) to afford alkyl radical F. Upon Minisci-type addition of this C-centered radical to heteroarene C, two divergent mechanistic pathways are then possible, depending on the identity of the heteroarene activating group, Z.
Figure 8.
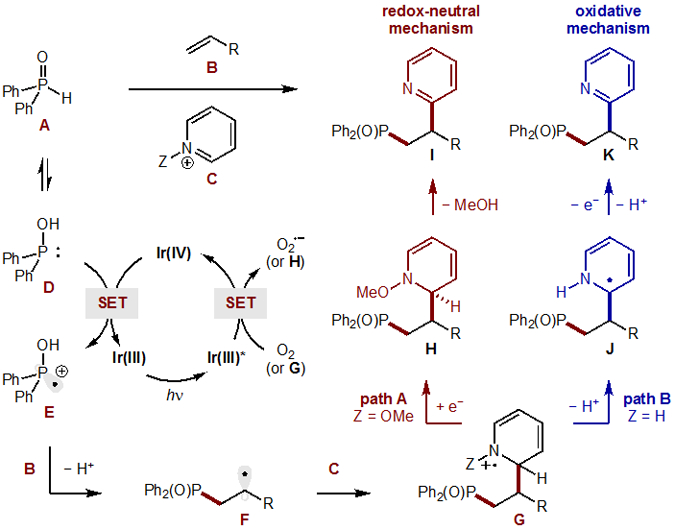
Proposed mechanism for both classes of heteroarenes.
Path A illustrates radical addition into an N-OMe pyridinium, which affords G (Z = OMe). This electron-deficient intermediate may be reduced by either excited Ir(III) catalyst or superoxide to form the neutral radical adduct H. Rearomatization by loss of methanol then affords the phosphinylalkylated pyridine I in a net redox-neutral reaction. Alternatively, Path B shows a mechanism for direct radical addition into a Brønsted acid-protonated heteroarene to provide G (Z = H). Given the acidity of the α-amino C-H (pKa < 8),25 its deprotonation would afford neutral α-amino radical J. Next, formal loss of H• may occur by aerobic oxidation followed by deprotonative aromatization to yield phosphinylalkylated K in a net oxidative reaction, wherein air is the terminal oxidant.
In summary, a polarity-reversal radical cascade has enabled a multi-component coupling of alkenes, heteroarenes, and phosphines. This photocatalytic strategy concurrently adds a phosphorous and heteroarene across an olefin. We expect the methods and mechanistic insights presented herein will serve as a foundation for developing more multi-component couplings based on radical polarity-reversal.
Supplementary Material
Figure 6.

Intramolecular cyclization favors the polarity-disfavored C3-regioisomer.
ACKNOWLEDGMENT
We thank the National Institutes of Health (NIH R35 GM119812), National Science Foundation (NSF CAREER 1654656), and the United States Air Force (J.Q.B.) for financial support.
Footnotes
The authors declare no competing financial interest.
References
- (1) (a).McDonald RI; Liu G; Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev. 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang JS; Liu L; Chen T; Han LB Transition-Metal-Catalyzed Three-Component Difunctionalizations of Alkenes. Chem. - An Asian J. 2018, 13, 2277–2291. [DOI] [PubMed] [Google Scholar]
- (2) (a).Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (b) Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3) (a).Giese B; González-Gómez JA; Witzel T The Scope of Radical CC-Coupling by the “Tin Method.” Angew. Chem. Int. Ed. 1984, 23, 69–70. [Google Scholar]; (b) De Vleeschouwer F; Van Speybroeck V; Waroquier M; Geerlings P; De Proft F Electrophilicity and Nucleophilicity Index for Radicals. Org. Lett. 2007, 9, 2721–2724. [DOI] [PubMed] [Google Scholar]
- (4).(a) Reviews on phosphorus-centered radical reactions: Leca D; Fensterbank L; Lacôte E; Malacria M Recent Advances in the Use of Phosphorus-Centered Radicals in Organic Chemistry. Chem. Soc. Rev. 2005, 34, 858–865. [DOI] [PubMed] [Google Scholar]; (b) Pan X-Q; Zou J-P; Yi W-B; Zhang W Recent Advances in Sulfur- and Phosphorous-Centered Radical Reactions for the Formation of S─C and P─C Bonds. Tetrahedron 2015, 71, 7481–7529. [Google Scholar]
- (5).(a) Hydrophosphinylation of alkenes (Pudovik reaction): Pudovik AN; Konovalova IV Addition Reactions of Esters of Phosphorus(III) Acids with Unsaturated Systems. Synthesis 1979, 2, 81–96. [Google Scholar]; (b) Semenzin D; Etemad-Moghadam G; Albouy D; Diallo O; Koenig M Dual Radical/Polar Pudovik Reaction: Application Field of New Activation Methods. J. Org. Chem. 1997, 62, 2414–2422. [DOI] [PubMed] [Google Scholar]; (c) Han L-B; Mirzaei F; Zhao C-Q; Tanaka M High Reactivity of a Five-Membered Cyclic Hydrogen Phosphonate Leading to Development of Facile Palladium-Catalyzed Hydrophosphorylation of Alkenes. J. Am. Chem. Soc. 2000, 122, 5407–5408. [Google Scholar]; (d) Hirai T; Han LB Air-Induced Anti-Markovnikov Addition of Secondary Phosphine Oxides and H-Phosphinates to Alkenes. Org. Lett. 2007, 9, 53–55. [DOI] [PubMed] [Google Scholar]; (e) Kawaguchi S; Nomoto A; Sonoda M; Ogawa A Photoinduced Hydrophosphinylation of Alkenes with Diphenylphosphine Oxide. Tetrahedron Lett. 2009, 50, 624–626. [Google Scholar]; (f) Yoo W-J; Kobayashi S Hydrophosphinylation of Unactivated Alkenes with Secondary Phosphine Oxides under Visible-Light Photocatalysis. Green Chem. 2013, 15, 1844–1848. [Google Scholar]; (g) Candy M; Rousseaux SAL; San Román AC; Szymczyk M; Kafarski P; Leclerc E; Vrancken E; Campagne JM Palladium-Catalyzed Hydrophosphonylation of Alkenes with Dialkyl H-Phosphonates. Adv. Synth. Catal. 2014, 356, 2703–2708. [Google Scholar]; (h) Li Z; Fan F; Zhang Z; Xiao Y; Liu D; Liu ZQ A Silver-Initiated Free-Radical Intermolecular Hydrophosphinylation of Unactivated Alkenes. RSC Adv. 2015, 5, 27853–27856. [Google Scholar]
- (6).(a) For select phosphinylations of alkenes and alkynes, see: Zhang C; Li Z; Zhu L; Yu L; Wang Z; Li C Silver-Catalyzed Radical Phosphonofluorination of Unactivated Alkenes. J. Am. Chem. Soc. 2013, 135, 14082–14085. [DOI] [PubMed] [Google Scholar]; (b) Li YM; Sun M; Wang HL; Tian QP; Yang SD Direct Annulations toward Phosphorylated Oxindoles: Silver-Catalyzed Carbon-Phosphorus Functionalization of Alkenes. Angew. Chem. Int. Ed. 2013, 52, 3972–3976. [DOI] [PubMed] [Google Scholar]; (c) Kong W; Merino E; Nevado C Arylphosphonylation and Arylazidation of Activated Alkenes. Angew. Chem. Int. Ed. 2014, 53, 5078–5082. [DOI] [PubMed] [Google Scholar]; (d) Quint V; Morlet-Savary F; Lohier JF; Lalevée J; Gaumont AC; Lakhdar S Metal-Free, Visible Light-Photocatalyzed Synthesis of Benzo[b]phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. [DOI] [PubMed] [Google Scholar]; (e) Li CX; Tu DS; Yao R; Yan H; Lu CS Visible-Light-Induced Cascade Reaction of Isocyanides and N-Arylacrylamides with Diphenylphosphine Oxide via Radical C-P and C-C Bond Formation. Org. Lett. 2016, 18, 4928–4931. [DOI] [PubMed] [Google Scholar]; (f) Chen T; Zhao CQ; Han LB Hydrophosphorylation of Alkynes Catalyzed by Palladium: Generality and Mechanism. J. Am. Chem. Soc. 2018, 140, 3139–3155. [DOI] [PubMed] [Google Scholar]; (g) Nie S-Z; Davison RT; Dong VM Enantioselective Coupling of Dienes and Phosphine Oxides. J. Am. Chem. Soc. 2018, 140, 16450–16454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Van der Jeught S; Stevens CV Direct Phosphonylation of Aromatic Azaheterocycles. Chem. Rev. 2009, 109, 2672–2702. [DOI] [PubMed] [Google Scholar]
- (8).Montchamp JL Phosphinate Chemistry in the 21st Century: A Viable Alternative to the Use of Phosphorus Trichloride in Organophosphorus Synthesis. Acc. Chem. Res. 2014, 47, 77–87. [DOI] [PubMed] [Google Scholar]
- (9) (a).Moonen K; Laureyn I; Stevens CV Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity. Chem. Rev. 2004, 104, 6177–6215. [DOI] [PubMed] [Google Scholar]; (b) Smith BR; Eastman CM; Njardarson JT Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014, 57, 9764–9773. [DOI] [PubMed] [Google Scholar]; (c) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (d) Dousson C; Alexandre F-R; Amador A; Bonaric S; Bot S; Caillet C; Convard T; Da Costa D; Lioure M-P; Roland A; Rosinovsky E; Maldonado S; Parsy C; Trochet C,; Storer R; Stewart A; Wang J; Mayes BA; Musiu C; Poddesu B; Vargiu L; Liuzzi M; Moussa A; Jakubik J; Hubbard L; Seifer M; Standring D Discovery of the Aryl-Phospho-Indole IDX899, a Highly Potent Anti-HIV Non-Nucleoside Reverse Transcriptase Inhibitor. J. Med. Chem. 2016, 59, 1891–1898. [DOI] [PubMed] [Google Scholar]
- (10) (a).Minisci F; Galli R; Cecere M; Malatesta V; Caronna T Nucleophilic Character of Alkyl Radicals: New Syntheses by Alkyl Radicals Generated in Redox Processes. Tetrahedron Lett. 1968, 9, 5609–5612. [Google Scholar]; (b) Minisci F; Fontana F; Vismara E Substitutions by Nucleophilic Free Radicals: A New General Reaction of Heteroaromatic Bases. J. Heterocycl. Chem. 1990, 27, 79–96. Select, recent examples: [Google Scholar]; (c) Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS Direct C─H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc. 2010, 132, 13194–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]; (e) Jin J; MacMillan DWC Alcohols as Alkylating Agents in Heteroarene C─H Functionalization. Nature 2015, 525, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jin J; MacMillan DWC Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C-H Functionalization and the Minisci Reaction. Angew. Chem. Int. Ed. 2015, 54, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li G-X; Morales-Rivera CA; Wang Y; Gao F; He G; Liu P; Chen G Photoredox-Mediated Minisci C–H Alkylation of N-Heteroarenes Using Boronic Acids and Hypervalent Iodine. Chem. Sci. 2016, 7, 6407–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Nuhant P; Oderinde MS; Genovino J; Juneau A; Gagné Y; Allais C; Chinigo GM; Choi C; Sach NW; Bernier L; Fobian YM; Bundesmann MW; Khunte B; Frenette M; Fadeyi OO Visible-Light-Initiated Manganese Catalysis for C─H Alkylation of Heteroarenes: Applications and Mechanistic Studies. Angew. Chem. Int. Ed. 2017, 56, 15309–15313. [DOI] [PubMed] [Google Scholar]; (i) Cheng WM; Shang R; Fu Y Photoredox/Brønsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes. ACS Catal. 2017, 7, 907–911. [Google Scholar]; (j) Garza-Sanchez RA; Tlahuext-Aca A; Tavakoli G; Glorius F Visible Light-Mediated Direct Decarboxylative C─H Functionalization of Heteroarenes. ACS Catal. 2017, 7, 4057–4061. [Google Scholar]; (k) Li GX; Hu X; He G; Chen G Photoredox-Mediated Minisci-Type Alkylation of N-Heteroarenes with Alkanes with High Methylene Selectivity. ACS Catal. 2018, 8, 11847–11853. [Google Scholar]; (l) Pitre SP; Muuronen M; Fishman DA; Overman LE Tertiary Alcohols as Radical Precursors for the Introduction of Tertiary Substituents into Heteroarenes. ACS Catal. 2019, 9, 3413–3418. [Google Scholar]; (m) Proctor RSJ; Phipps RJ Recent Advances in Minisci-Type Reactions. Angew. Chem. Int. Ed. 2019, DOI: 10.1002/anie.201900977 [DOI] [PubMed] [Google Scholar]
- (11).(a) Medicinal applications: Duncton MAJ Minisci Reactions: Versatile CH-Functionalizations for Medicinal Chemists. Med. Chem. Commun. 2011, 2, 1135–1161. [Google Scholar]; (b) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev. 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (12) (a).Ma X; Herzon SB Intermolecular Hydropyridylation of Unactivated Alkenes. J. Am. Chem. Soc. 2016, 138, 8718–8721. [DOI] [PubMed] [Google Scholar]; (b) Lo JC; Kim D; Pan CM; Edwards JT; Yabe Y; Gui J; Qin T; Gutiérrez S; Giacoboni J; Smith MW; Holland PL; Baran PS Fe-Catalyzed C-C Bond Construction from Olefins via Radicals. J. Am. Chem. Soc. 2017, 139, 2484–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ma X; Dang H; Rose JA; Rablen P; Herzon SB Hydroheteroarylation of Unactivated Alkenes Using N-Methoxyheteroarenium Salts. J. Am. Chem. Soc. 2017, 139, 5998–6007. [DOI] [PubMed] [Google Scholar]
- (13) (a).Clerici A; Minisci F; Ogawa K; Surzur JM Intra- and Intermolecular Free Radical Reactions of Alcohols and Olefines with Peroxydisulphate. New Homolytic Aromatic Alkylations. Tetrahedron Lett. 1978, 19, 1149–1152. [Google Scholar]; (b) Antonietti F; Mele A; Minisci F; Punta C; Recupero F; Fontana F Enthalpic and Polar Effects in the Reactions of Perfluoroalkyl Radicals. New Selective Synthetic Developments with Alkenes and Heteroaromatic Bases. J. Fluor. Chem 2004, 125, 205–211. [Google Scholar]; (c) McCallum T; Barriault L Direct Alkylation of Heteroarenes with Unactivated Bromoalkanes Using Photoredox Gold Catalysis. Chem. Sci. 2016, 7, 4754–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Z; Liu ZQ An Intermolecular Azidoheteroarylation of Simple Alkenes via Free-Radical Multicomponent Cascade Reactions. Org. Lett. 2017, 19, 5649–5652. [DOI] [PubMed] [Google Scholar]; (e) He Y-T; Kang D; Kim I; Hong S Metal-Free Photocatalytic Trifluoromethylative Pyridylation of Unactivated Alkenes. Green Chem. 2018, 20, 5209–5214. [Google Scholar]
- (14) (a).Zhang X; McNally A Phosphonium Salts as Pseudohalides: Regioselective Nickel-Catalyzed Cross-Coupling of Complex Pyridines and Diazines. Angew. Chem. Int. Ed. 2017, 56, 9833–9836. [DOI] [PubMed] [Google Scholar]; (b) Anderson RG; Jett BM; McNally A A Unified Approach to Couple Aromatic Heteronucleophiles to Azines and Pharmaceuticals. Angew. Chem. Int. Ed. 2018, 57, 12514–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hilton MC; Zhang X; Boyle BT; Alegre-Requena JV ; Paton RS; McNally A Heterobiaryl Synthesis by Contractive C─C Coupling via P(V) Intermediates. Science 2018, 362, 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang X; McNally A Cobalt-Catalyzed Alkylation of Drug-Like Molecules and Pharmaceuticals Using Heterocyclic Phosphonium Salts ACS Catal. 2019, 9, 4862–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15) (a).Boyington AJ; Riu MLY; Jui NT Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates. J. Am. Chem. Soc. 2017, 139, 6582–6585. [DOI] [PubMed] [Google Scholar]; (b) Seath CP; Vogt DB; Xu Z; Boyington AJ; Jui NT Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photocatalytic Pyridyl Radical Reactions. J. Am. Chem. Soc. 2018, 140, 15525–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16) (a).Dyall LK; Pausacker KH 4. Free-Radical Substitution of Pyridine 1-Oxide. J. Chem. Soc. 1961, 18–23.Deng G; Ueda K; Yanagisawa S; Itami K; Li CJ Coupling of Nitrogen Heteroaromatics and Alkanes without Transition Metals: A New Oxidative Cross-Coupling at C-H/C-H Bonds. Chem. Eur. J. 2009, 15, 333–337.T Nishida T; Ida H; Kuninobu Y; Kanai M Regioselective Trifluoromethylation of N-Heteroaromatic Compounds Using Trifluoromethyldifluoroborane Activator. Nat. Commun. 2014, 5, 3387.See also Ref 12b.
- (17).Katz RB; Mistry J; Mitchell MB An Improved Method For The Mono-Hydroxymethylation Of Pyridines. A Modification Of The Minisci Procedure. Synth. Commun. 1989, 19, 317–325. [Google Scholar]
- (18).For a recent, enantioselective variant of the Minisci reaction with a strong counterion influence, see: Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. See also Ref 12. [DOI] [PubMed] [Google Scholar]
- (19) (a).Cziáky Z; Sebők P Synthesis of 2H-Pyrano[2,3-b]Quinolines. Part I. J. Heterocycl. Chem. 1994, 31, 701–705. [Google Scholar]; (b) Mabire D; Coupa S; Adelinet C; Poncelet A; Simonnet Y; Venet M; Wouters R; Lesage ASJ; Van Beijsterveldt L; Bischoff F Synthesis, Structure-Activity Relationship, and Receptor Pharmacology of a New Series of Quinoline Derivatives Acting as Selective, Noncompetitive MGlu1 Antagonists. J. Med. Chem. 2005, 48, 2134–2153. [DOI] [PubMed] [Google Scholar]; (c) Li S; Chen G; Feng CG; Gong W; Yu JQ Ligand-Enabled γ-C-H Olefination and Carbonylation: Construction of β-Quaternary Carbon Centers. J. Am. Chem. Soc. 2014, 136, 5267–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).KSV values: diphenyl phosphine oxide (0), PPh3 (0), phenanthridine (0), phenanthridine + TFA (55), lutidinium sulfate (4). See SI for full details.
- (21).(a) Photoredox catalysis reviews: Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (b) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (22) (a).Hayyan M; Hashim MA; AlNashef IM Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [DOI] [PubMed] [Google Scholar]; (b) see also Ref 21c. [Google Scholar]
- (23) (a).Slinker JD; Gorodetsky AA; Lowry MS; Wang J; Parker S; Rohl R; Bernhard S; Malliaras GG Efficient Yellow Electroluminescence from a Single Layer of a Cyclometalated Iridium Complex. J. Am. Chem. Soc. 2004, 126, 2763–2767. [DOI] [PubMed] [Google Scholar]; (b) Lowry Michael S.; Goldsmith Jonas I.; Slinker Jason D.; Rohl Richard; Pascal, J. Robert A.; Malliaras George G.; Bernhard S Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater. 2005, 17, 5712–5719. [Google Scholar]
- (24) (a).Grayson M; Farley CE; Streuli CA Secondary Phosphine Oxides. The Effect of Structure on Acid Strength and Rates of Cleavage of Disulfides. Tetrahedron 1967, 23, 1065–1078.see Ref 5f for redox potentials.
- (25) (a).Dinnocenzo JP; Banach TE Deprotonation of Tertiary Amine Cation Radicals. A Direct Experimental Approach. J. Am. Chem. Soc. 1989, 111, 8646–8653. [Google Scholar]; (b) Zhang X; Yeh S-R; Hong S; Freccero M; Albini A; Falvey DE; Mariano PS Dynamics of Alpha-CH Deprotonation and Alpha-Desilylation Reactions of Tertiary Amine Cation Radicals. J. Am. Chem. Soc. 1994, 116, 4211–4220. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.