

Abstract
Although the role of estrogen in solid cancers has been widely investigated, its effect in hematologic malignancies including multiple myeloma (MM) is not known. Here, we utilized a syngeneic mouse model of MM to address this question. In this model, treatment with 17β-estradiol significantly promoted progression of the disease. This effect has not been attributed to the direct effect of estrogen on MM cells but rather was mediated through estrogen-induced alterations in tumor microenvironment. In MM bone marrow, myeloid-derived suppressor cells (MDSCs) represent one of the major cellular populations. 17β-estradiol did not promote expansion and accumulation of MDSCs. However, it significantly increased their ability to suppress T cells proliferation. Thus, these data demonstrated that estrogen promotes progression of MM by enhancing an immunosuppressive function of the bone marrow MDSCs.
Keywords: Estrogen, multiple myeloma, myeloid-derived suppressor cells, MDSC, tumor microenvironment, immunosuppression
Introduction
The tumor microenvironment plays a critical role in regulating tumor progression. Recent data have implicated a population of cells, termed myeloid-derived suppressor cells (MDSCs), in promoting tumor survival, growth, and chemoresistance [1]. MDSCs represent a heterogeneous group of cells comprising pathologically activated immature myeloid cells and their precursors, which have the ability to potently suppress immune responses. Phenotypically, MDSCs are characterized by co-expression of CD11b and Gr1 markers. MDSC counterparts in tumor-free hosts have a similar phenotype; however, these CD11b+Gr1+ cells are not able to inhibit immune responses [2]. Expansion and accumulation of MDSCs have been demonstrated in multiple mouse models of cancer; accumulation of MDSCs in cancer patients has been associated with a poor prognosis [1].
The role of estrogens, steroid hormones that regulate multiple biological processes, has been extensively investigated in cancer, primarily breast and ovarian cancer. Estrogens mediate their effect through binding to two high-affinity nuclear estrogen receptors (ER), ERα and ERβ, which are differentially expressed in multiple tissues. Although an effect of estrogens and anti-estrogens on cancer cells themselves has been a subject of extensive studies, a few recent reports indicated a potentially important pro-tumorigenic effect of estrogens mediated indirectly through modulation of the tumor microenvironment and specifically, myeloid cells. Svoronos and colleagues demonstrated that estrogen can promote tumorigenesis by both driving mobilization and enhancing the immunosuppressive function of MDSCs, an effect independent of the effect on the tumor cells, and mediated through binding of estrogen with ERα in bone marrow myeloid precursors, followed by activation of the STAT3 pathway [3]. Ouyang and co-authors found that estrogen promoted progression of ER-negative breast cancer by stimulating SDF-1α secretion by cancer-associated fibroblasts, leading to the recruitment and accumulation of MDSCs in the tumor microenvironment [4]. The ability of estrogen to promote tumor progression by affecting MDSCs suggests that anti-estrogen therapy could benefit a wide range of cancers regardless of the expression level of estrogen receptors in tumor cells.
Multiple myeloma (MM) is an incurable cancer of plasma cells that preferentially localize in the bone marrow (BM). Although expression of both ERα and ERβ has been shown for human MM cell lines and primary MM cells [5, 6, 7], the effect of estrogen on MM cells has not been clearly defined. Treon and colleagues demonstrated lack of estrogen (E2) effect on growth kinetics and an inability to induce apoptosis of patient MM cells and MM cell lines [6], whereas another study reported that estrogen (E2) could stimulate growth of MM cells [5]. At the same time a number of studies demonstrated an anti-proliferative and pro-apoptotic effect of anti-estrogen therapy in MM [5, 6, 7].
Overall survival for women with MM is lower than for men [8]; however, whether estrogen contributes to that remains unclear. We recently reported that immunosuppressive MDSCs accumulate in the BM of MM-bearing mice and patients with MM [9]. Here, we investigated whether estrogen is responsible for this effect. Using a syngeneic mouse model of MM we demonstrated that estrogen enhances the immunosuppressive activity of MDSCs and promotes disease progression.
Materials and Methods
Cell culture and reagents
Mouse MM DP42 cell line was derived from Bcl-xL/c-Myc transgenic mice and was kindly provided by Dr. Brian Van Ness (University of Minnesota, Minneapolis, MN) and described previously [10] [11]. Mouse MM 5TGM1 cells were a gift from Dr. Lori Hazlehurst (West Virginia University, Morgantown, WV) and were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 1x Antibiotic-Antimycotic (Thermo Fisher Scientific). BM stroma (BMS) was established by culturing BM cells in RPMI-1640 medium supplemented with 10% FBS following removal of medium and expansion of adherent cells. 17β-estradiol (E2) was purchased from Sigma-Aldrich (St. Louis, MO).
Animals
C57BL/6 and FVB/N mice were purchased from Charles River (Frederick, MD) and were crossed to obtain mice of a mixed FVB/NxC57BL/6 background. F1 progeny (6–8 week old) were used. Mice were kept in pathogen-free conditions and handled in accordance with the requirements of the Guideline for Animal Experiments. MM tumors were established by i.v. inoculation of DP42 cells (104 cells) into tail vein of syngeneic male mice (F1 progeny of FVB/NxC57BL/6 mice). Treatment with 17β-estradiol (10 μmol/L, USP grade, Sigma, in drinking water refreshed every 3–4 days) or vehicle control (0.1% ethanol) began 2 days prior to tumor inoculation and was continued during the course of disease progression. Mice were euthanized when they reached humane endpoint as stipulated by the IACUC protocol.
Mouse cell isolation and functional assay
CD11b+Gr1+ myeloid cells and MDSCs were isolated from BM cells obtained by flushing femurs and tibias of tumor-free or MM-bearing mice, respectively, using magnetic cell separation technique. Briefly, BM cells were labelled with biotin-anti-Gr-1 antibody (BD) followed by Streptavidin-conjugated MicroBeads (Miltenyi Biotec) and selection on LS column. Purity of isolated cell population was confirmed by flow cytometry and was more than 98%. All Gr1+ cells expressed CD11b marker.
Splenocytes from OT-1 mice (1×106/mL) were stimulated with 0.5 μg/mL specific (SIINFEKL) or control (RGPGRAFVTI) peptide in the presence or absence of CD11b+Gr1+ cells isolated from the BM of vehicle control or 17β-estradiol-treated MM-bearing mice. T cell proliferation was evaluated using [3H]thymidine incorporation.
Flow cytometry
Apoptosis of MM cells was detected using Annexin V binding assay. Briefly, cells were washed once with PBS, once with a binding buffer followed by staining with Annexin V-APC and DAPI. For the surface staining, cells were labelled with anti-Gr-1-APC (cat #108412 Biolegend), and anti-CD11b-FITC (cat # 101206, Biolegend) or isotype controls for 30 min at +40C, washed twice with ice cold PBS, and resuspended in PBS containing DAPI. At least 10,000 events were acquired using a LSR II flow cytometer (BD). Data were analyzed using FlowJo software (TreeStar).
Viability assay
Cells were re-suspended at a concentration of 0.25 × 106/mL in RPMI-1640 medium supplemented with 10% charcoal-stripped FBS and plated in 96-well plate (200 μL per well). Cells were treated with 17β-estradiol (0 – 5 μM) for 48h, 72h, or 96h. Cell viability was evaluated using AlamarBlue assay.
Real time PCR
RNA was extracted using E.Z.N.A. Total RNA Kit I (Omega Bio-Tek, Norcross, GA) and cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR was performed on 7500 Fast Real-Time PCR System instrument (Applied Biosystems) using 100 ng of the template and Power SYBR green PCR Master Mix (Applied Biosystems). Expression of specific genes was normalized to the expression of β-actin. The following primers were used: ERα 5’-CAGGTGCCCTACTACCTGGA-3’ (forward) and ERα 5’-ATTCTGGCGTCGATTGTCA-3’ (reverse), ERβ 5’-AAGAGTCCTTGGTGTGAAGCA-3’ (forward) and ERβ 5’-AGTAACAGGGCTGGCACAAC-3’ (reverse), β-actin 5’-ACTTGCGGTGCACGATGGA-3’ (forward), β-actin 5’-TACCCAGGCATTGCTGACAGG-3’ (reverse).
Statistics
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software). Differences between groups were calculated using a two-tailed unpaired Student t test. A statistically significant difference was determined at p<0.05. A log-rank test was used to evaluate a statistical significance in mouse survival experiments.
Results
Female gender has been associated with a statistically significant inferior overall survival in MM [8]. We hypothesized that this difference could be attributed to the higher level of estrogen in women. A syngeneic mouse model of MM that recapitulates human disease [9] was used to evaluate the role of estrogen in MM progression. DP42 MM tumors were established in male mice treated with 17β-estradiol, the most active estrogen. Treatment with 17β-estradiol significantly reduced survival of MM-bearing mice (Figure 1).
Figure 1. Estrogen promotes progression of MM.
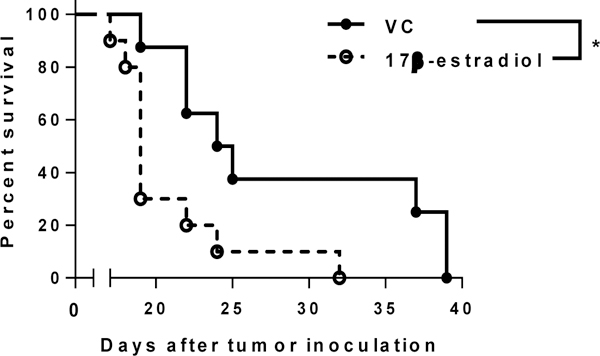
Survival of DP42 MM-bearing male mice treated with 17β-estradiol (n=10) or vehicle control (n=8) was evaluated. * - p=0.0146.
Estrogen mediates its effects through binding with estrogen receptors (ER), primarily ERα and ERβ. We evaluated ER expression in several murine MM cell lines as well as in BM cells. Expression of both ERα and ERβ was detected in all cells tested although ERα was predominantly expressed (Figure 2). We isolated CD11b+Gr1+ myeloid cells from the BM of tumor-free or DP42 MM-bearing mice and determined the expression of ERs in these cells and in the Gr1neg population of bone marrow cells. CD11b+Gr1+ cells isolated from the BM of tumor-free mice or from the BM of MM-bearing mice, demonstrated significantly higher levels of both ERs compared to Gr1neg BM cells. No significant differences in the expression of ERβ and ERα were found between CD11b+Gr1+ cells in tumor-free and MM-bearing mice (Figure 2). No difference was found between ERs levels in Gr1neg cells from BM of tumor-free or MM-bearing mice.
Figure 2. Expression of ERs in myeloma and BM cells.
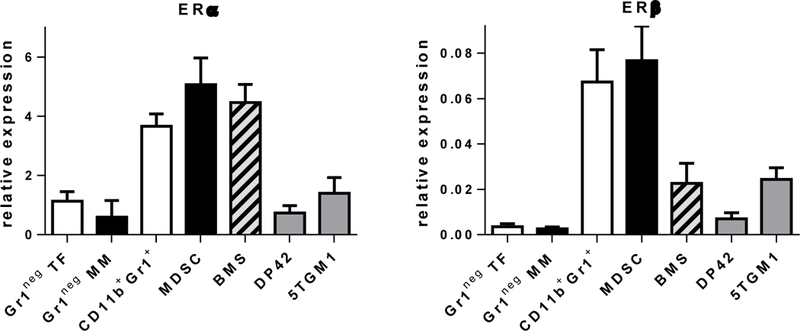
Expression of ERα (left panel) and ERβ (right panel) was detected by RT-PCR in Gr1neg BM cells isolated from tumor-free (TB) or MM-bearing (MM) mice, CD11b+Gr1+ cells isolated from BM of TF mice, MDSC from BM of MM-bearing mice, BMS established in vitro, and indicated MM cell lines. Note the scale difference between the panels.
Next, we determined whether estrogen has a direct effect on MM cells. DP42 and 5TGM1 cells were treated with various concentrations of 17β-estradiol (range from 5 nM to 5 μM) for 48–96h and their viability was evaluated using AlamarBlue assay. No difference in MM cell viability was observed at any time point during treatment with 17β-estradiol (Figure 3A,B). Addition of 17β-estradiol for 24–72h did not promote survival of MM cells as evaluated by Annexin V binding assay (data not shown). Thus, 17β-estradiol did not have a significant direct effect on MM cells that could explain progression of MM observed in vivo after treatment of MM-bearing mice with this hormone.
Figure 3. Effect of estrogen on MM cells.
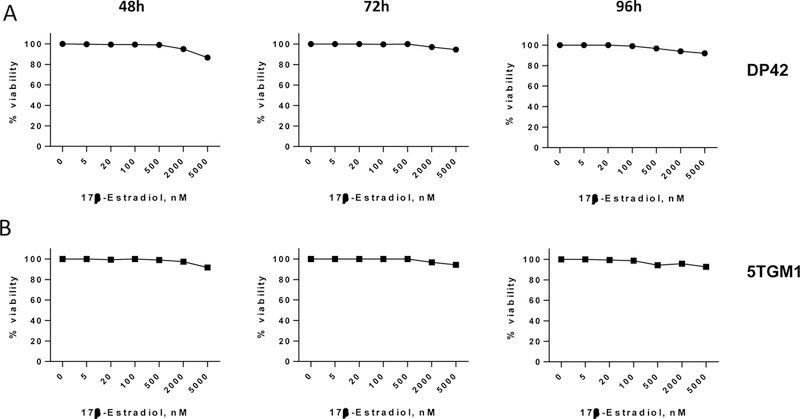
DP42 and 5TGM1 cells were plated in concentration of 0.25×106/mL in RPMI-1640 medium supplemented with 10% charcoal-stripped FBS and treated with 17β-estradiol or vehicle control for indicated period of time. Viability of MM cells was evaluated by AlamarBlue assay.
We and others have demonstrated that growth of MM was accompanied by accumulation of MDSCs and that adoptive transfer of MDSCs promoted MM progression [9, 12, 13, 14]. Here, we investigated whether estrogen has an effect on the proportion and function of MDSCs. DP42 tumors were established in mice pre-treated with 17β-estradiol or vehicle control for 2 days and the treatment continued for another 8 days. At this time point, in agreement with our previous data [9], the proportion of CD11b+Gr1+ cells was significantly increased in the BM of DP42-bearing mice compared to control tumor-free mice. However, treatment with 17β-estradiol had no effect on the absolute number and proportion of MDSCs in the BM (Figure 4A). Next, we asked whether estrogen has an effect on the immunosuppressive activity of MDSCs in MM. CD11b+Gr1+ cells were isolated from the BM of 17β-estradiol or vehicle control-treated DP42-bearing mice and evaluated for the ability to inhibit proliferation of antigen-specific T cells. CD11b+Gr1+ cells from 17β-estradiol-treated mice possessed significantly stronger immunosuppressive activity compared to MDSCs isolated from vehicle control-treated mice (Figure 4B).
Figure 4. Estrogen promotes immunosuppressive activity of MDSC in MM-bearing mice.
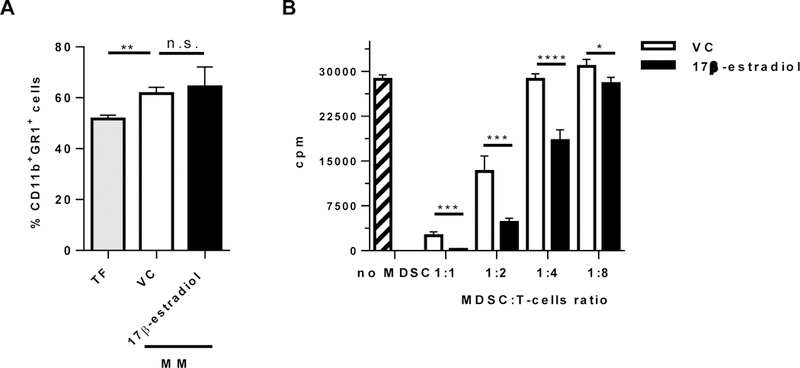
(A,B) DP42 MM-bearing male mice treated with vehicle control or 17β-estradiol were euthanized 8 days after tumor cell injection. (A) Proportion of CD11b+Gr1+ cells was determined in the BM by flow cytometry (n=3 per group). In control, BM from tumor-free (TF) mice was used (n=3). (B) CD11b+Gr1+ cells were isolated from the BM of DP42-bearing vehicle control or 17β-estradiol-treated mice and plated in different ratios with splenocytes from OT-I mice. Proliferation of T cells was evaluated in 72h. Shown are combined data of 4 mice per group. * - p<0.05; ** - p<0.01; *** - p<0.001; and **** - p<0.0001.
Thus, our data demonstrate that estrogen promotes the progression of MM by enhancing the immunosuppressive activity of the BM MDSCs.
Discussion
In MM, women have an inferior overall survival. This fact prompted us to investigate the potential contribution of estrogen in the pathogenesis of this disease. Using a syngeneic model of MM, we demonstrated that while estrogen did not have a direct effect on MM cells, it did significantly enhance the ability of the BM MDSCs to suppress T cell proliferation, and thereby likely promoted progression of this disease.
Although a direct effect of estrogen on some types of cancer cells has been extensively studied, the role of estrogen in MM has not been well defined. Previously published data demonstrated either that estrogen had no effect on human MM cell proliferation and apoptosis [6] or that estrogen stimulated growth of selected MM cell lines [5]. The latter study attributed the differences between cell lines to the pattern of ER expression, with estrogen (E2) stimulating growth of human lines that highly express ERα, but not stimulating lines that preferentially express ERβ [5]. We have demonstrated expression of both ERα and ERβ in mouse MM cells with preferential expression of ERα. However, treatment with estrogen did not affect MM cell viability and apoptosis. Thus, our results do not support the conclusion of the previously reported data. There is a possibility that engagement of estrogen receptors on the cell membrane, including GPR30, ER-X, or Gq-mER [15], is responsible for the pro-growth effects of estrogen observed by others; that could be a subject for future studies.
The role of estrogen in shaping the tumor microenvironment and the contribution of estrogen in regulating myeloid cells in cancer has only begun to be unveiled. Although 17β-estradiol has no direct effect on mouse MM cells, administration of this hormone in vivo significantly promoted progression of MM (Figure 1). These results are in agreement with published data showing that E2 supplementation accelerated the growth of estrogen-insensitive solid tumors [3]. Previous data demonstrated that 17β-estradiol significantly enhanced BM endothelial progenitor cell migration to tumor tissues as well as secretion of angiogenic factors within the tumor microenvironment [16]. Multiple reports have indicated that angiogenesis is an important hallmark of MM progression [17, 18]. Thus, increased angiogenesis could potentially be one of the mechanisms by which estrogen promotes MM progression. In our experiments, treatment with 17β-estradiol was initiated 2 days prior to MM cell injection and there is a possibility that the increased estrogen level led to an increased homing of MM cells to the BM and consequently, to accelerated MM growth. However, this hypothesis has to be proven yet.
A few recent studies have implicated estrogen in regulating MDSCs in cancer. Using a model of ER-negative breast cancer Ouyang et al demonstrated that estrogen can stimulate mobilization of MDSCs to the tumor site though secretion of SDF-1α by tumor-associated fibroblasts [4]. Svoronos and colleagues reported a significant contribution of estrogen to deregulated myelopoiesis and consequently, to anti-tumor immunity and tumor progression. They found that E2, through binding to ERα, boosted proliferation of myeloid progenitors, leading to the increased proportion and number of MDSCs in spleen, induced mobilization of MDSCs to the tumor site, and enhanced their immunosuppressive activity. Since MM grows in the BM, a site where myelopoiesis occurs, we evaluated whether estrogen would induce changes in proportion or function of MDSCs in this disease. As expected, growth of MM in a syngeneic mouse model was associated with accumulation of MDSCs in the BM. However, treatment with 17β-estradiol did not further expand this cell population (Figure 4A).
Targeting the immunosuppressive function of MDSCs is considered to be an important strategy in therapy for hematological malignancies. Our study, for the first time, provides evidence that in the MM bone marrow microenvironment estrogen enhances immunosuppression mediated by MDSCs. These data suggest that anti-estrogens could be explored as an adjuvant therapy to the standard regimens to prolong survival of patients with MM.
Acknowledgements
Funding: Support for Shared Resources utilized in this study was provided by the Cancer Center Support Grant (CCSG) P30CA010815 to The Wistar Institute. We would like to thank Dr. Evgenii Tcyganov for technical assistance and Dr. Rachel E. Locke for help with preparation of the manuscript.
Footnotes
Disclosure of interest:
The authors report no conflict of interest.
References
- 1.Marvel D, Gabrilovich D. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bronte V, Brandau S, Chen S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Svoronos N, Perales-Puchalt A, Allegrezza M, et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017;7(1):72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouyang L, Chang W, Fang B, et al. Estrogen-induced SDF-1α production promotes the progression of ER-negative breast cancer via the accumulation of MDSCs in the tumor microenvironment. Sci Rep. 2016;6:39541. doi: doi: 10.1038/srep39541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otsuki T, Yamada O, Kurebayashi J, et al. Estrogen receptors in human myeloma cells. Cancer Res. 2000;60(5):1434–1441. [PubMed] [Google Scholar]
- 6.Treon S, Teoh G, Urashima M, et al. Anti-estrogens induce apoptosis of multiple myeloma cells. Blood. 1998;92(5):1749–1757. [PubMed] [Google Scholar]
- 7.Olivier S, Close P, Castermans E, et al. Raloxifene-induced myeloma cell apoptosis: a study of nuclear factor-kappaB inhibition and gene expression signature. Mol Pharmacol. 2006;69(5):1615–1623. [DOI] [PubMed] [Google Scholar]
- 8.Boyd K, Ross F, Chiecchio L, et al. Gender Disparities in the Tumor Genetics and Clinical Outcome of Multiple Myeloma. Cancer Epidemiol Biomarkers Prev. 2011;20(8):1703–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran I, Martner A, Pisklakova A, et al. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol. 2013;190(7):3815–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung W, Kim J, Linden M, et al. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J Clin Invest. 2004;113(12):1763–1773. PubMed Central PMCID: PMCPMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramachandran I, Condamine T, Lin C, et al. Bone marrow PMN-MDSCs and neutrophils are functionally similar in protection of multiple myeloma from chemotherapy. Cancer Lett. 2016;371(1):117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorgun G, Whitehill G, Anderson J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood. 2013;121(15):2975–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Valckenborgh E, Schouppe E, Movahedi K, et al. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia. 2012;26(11):2424–2428. [DOI] [PubMed] [Google Scholar]
- 14.Favaloro J, Liyadipitiya T, Brown R, et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk Lymphoma. 2014;55(12):2893–2900. [DOI] [PubMed] [Google Scholar]
- 15.Micevych P, Kelly M. Membrane Estrogen Receptor Regulation of Hypothalamic Function. Neuroendocrinology 2012;96:103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suriano R, Chaudhuri D, Johnson R, et al. 17Beta-estradiol mobilizes bone marrow-derived endothelial progenitor cells to tumors. Cancer Res. 2008;68(15):6038–6042. doi: doi: 10.1158/0008-5472.CAN-08-1009. [DOI] [PubMed] [Google Scholar]
- 17.Jakob C, Sterz J, Zavrski I, et al. Angiogenesis in multiple myeloma. European Journal of Cancer. 2006;42(11):1581–1590. [DOI] [PubMed] [Google Scholar]
- 18.Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006;20(2):193–199. [DOI] [PubMed] [Google Scholar]