

Abstract
In this study, we investigated the development of endoplasmic reticulum (ER) stress after traumatic brain injury (TBI) and the efficacy of post-TBI administration of docosahexaenoic acid (DHA) in reducing ER stress. TBI was induced by cortical contusion injury in Sprague-Dawley rats. Either DHA (16 mg/kg in DMSO) or vehicle DMSO (1 ml/kg) was administered intraperitoneally at 5 min after TBI, followed by a daily dose for 3–21 d. TBI triggered sustained expression of the ER stress marker proteins including phosphorylated eukaryotic initiation factor-2α, activating transcription factor 4, inositol requiring kinase 1, and C/EBP homologous protein in the ipsilateral cortex at 3–21 d after TBI. The prolonged ER stress was accompanied with an accumulation of abnormal ubiquitin aggregates and increased expression of amyloid precursor protein (APP) and phosphorylated tau (p-Tau) in the frontal cortex after TBI. The ER stress marker proteins were colocalized with APP accumulation in the soma. Interestingly, administration of DHA attenuated all ER stress marker proteins and reduced the accumulation of both ubiquitinated proteins and APP/p-Tau proteins. In addition, the DHA-treated animals exhibited early recovery of their sensorimotor function after TBI. In summary, our study demonstrated that TBI induces a prolonged ER stress, which is positively correlated with abnormal APP accumulation. The sustained ER stress may play a role in chronic neuronal damage after TBI. Our findings illustrate that post-TBI administration of DHA has therapeutic potentials in reducing ER stress, abnormal protein accumulation, and neurological deficits.
Keywords: amyloid precursor protein, controlled cortical impact injury, docosahexaenoic acid, phosphorylated tau, unfolded protein response
Introduction
Traumatic brain injury (TBI) is a major health problem that causes brain damage leading to death and long-term disability (Langlois et al., 2006). The pathophysiology of TBI involves a primary, direct physical damage to the tissue and the secondary, delayed progressive white and gray matter damage (Bramlett and Dietrich, 2007; Stoica and Faden, 2010). The secondary injury mechanisms involve disruption of Ca2+ homeostasis, excitotoxicity, free radical generation, blood–brain barrier disruption, ischemia, edema formation, and intracranial hypertension (Greve and Zink, 2009; Sande and West, 2010). Traditionally, TBI-induced brain damage is thought to be limited to the acute and subacute periods after trauma. Recent human and experimental studies strongly suggest that a single TBI can trigger a progressive, long-term neurodegenerative process (DeKosky et al., 2010).
Endoplasmic reticulum (ER) stress has recently received increased attention in the pathogenesis of various neurodegenerative disorders including cerebral ischemia, TBI, Alzheimer's, and Parkinson's disease (Larner et al., 2006; Kim et al., 2008). ER stress can trigger unfolded protein response (UPR), an evolutionarily conserved process activated when ER sensors detect the excess accumulation of misfolded or unfolded proteins and/or disruption of ER Ca2+ homeostasis that occurs in the ER lumen (Larner et al., 2006; Kim et al., 2008; Begum et al., 2012). ER stress and abnormal protein accumulation were observed in the acute brain injury following TBI (Larner et al., 2006). One arm of ER stress sensor pathway inositol requiring kinase 1 (IRE1α) was activated after TBI and involved in the activation of ER-resident caspase 12 and cell death (Larner et al., 2004). To date, most studies on the TBI-induced ER stress and UPR have centered on its association to acute neuronal death. Recently, chronic ER stress has emerged as an important mechanism in neurological disorders, including neural dysregulation in type-2 diabetes, chronic hypertension, as well as neurodegenerative diseases (Hoozemans et al., 2009; Purkayastha et al., 2011; Young et al., 2012). However, the impact of the sustained UPR on secondary injury after TBI remains unknown. In this study, we investigated whether TBI triggers a prolonged ER stress response and its role in abnormal protein accumulation and toxic protein aggregate formation.
Chronic DHA supplementation can improve animals' learning and memory function, mitochondrial energy metabolisms, and reduce axonal injury (Wu et al., 2007, 2011; Pan et al., 2009; Mills et al., 2011; Hasadsri et al., 2013). In particular, post-TBI DHA supplementation significantly reduces the number of APP-positive axons in white matter tracts at 30 d postinjury (Bailes and Mills, 2010). Despite that DHA has many neuroprotective functions (Mills et al., 2011), its action on ER stress after TBI has not yet been studied. As increasing evidence suggests that ER stress is involved in a number of disease pathogenesis (Engin and Hotamisligil, 2010), restoring ER function has emerged as novel new drug targets against a wide spectrum of pathologies. In this study, we investigated whether DHA reduces ER stress and subsequently prevents cells from abnormal protein accumulation and aggregate formation after TBI.
Materials and Methods
Materials.
cis-4,7,10,13,16,19-docosahexaenoic acid (DHA; 22:6n-3), Di methyl sulfoxide (DMSO), 4-phenylbutyric acid (PBA), thapsigargin, tunicamycin (Tu), and the rabbit antineurofilament antibody, mouse antineurofilament antibody, were from Sigma Chemicals. Neurobasal media, B27 supplement, d-myo-inositol 1,4,5-triphosphate (IP3), mag fura-2 acetoxymethyl ester, goat anti-IgG secondary antibodies AlexaFluor 488 and 546 and polyclonal rabbit anti-β-amyloid precursor protein (AβAPP: CT695) antibody were from Invitrogen. Pluronic F-127 was from BASF. Nembutal was from Abbott Laboratories. BCA (bicinchoninic acid) protein assay reagent was from Thermo Scientific. Mouse anti-α-tubulin monoclonal antibody, mouse anti-C/EBP homologous protein (CHOP) monoclonal antibody, mouse anti-eukaryotic initiation factor-2α (eIF2α) monoclonal antibody, rabbit anti-phospho-eIF2α (p-eIF2α) monoclonal antibody, and rabbit IRE1α monoclonal antibodies, were purchased from Cell Signaling Technology. Rabbit anti-activating transcription factor 4 (ATF-4) antibody and rabbit polyclonal anti-XBP-1 antibodies were from Santa Cruz Biotechnology. The monoclonal anti-ubiquitin antibody was from Novus Biologicals. Rabbit polyclonal anti-phosphorylated tau (p-Tau; pSer199/202) antibody was from Anaspec and mouse anti-GRP78 was from BD Biosciences. Tauroursodeoxycholic acid (TUDCA) was from Calbiochem (EMD, Millipore).
Animals.
All studies were in compliance with the guidelines outlined in the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Medical Center Institutional Animal Care and Use Committee.
Sprague-Dawley rats (150 males; 250–275 g body weight) were used in the study. Rats were group-housed (2 per cage) in standard steel/wire mesh cages at room temperature (22 ± 2°C) under standard 12 h light/dark cycles (light on at 07:00 A.M.) with ad libitum access to food and water. After injury, rats were housed in separate cages from the uninjured animals under the same conditions as described above.
Surgical procedures and TBI induction.
Rats were anesthetized initially with 4% isoflurane with a 2:1 N2O/O2 mixture in a vented anesthesia chamber. Following endotracheal intubation, rats were ventilated mechanically with a 1–1.5% isoflurane mixture. Animals were mounted in a stereotaxic frame on the injury device in a supine position as described before (Dixon et al., 1991), and the core body temperature was monitored continuously by a rectal thermistor probe and maintained at 37 ± 0.5°C with a heating pad. Following a midline incision and dissection of the soft tissues, a 7 mm craniotomy was made between lambda and bregma, and centered 5 mm lateral of the central suture. To control for nonspecific effects due to animal handling, anesthesia or surgery procedures, sham control animals underwent identical surgical procedures but did not receive a TBI. To induce TBI, the controlled cortical impact injury (CCI) device consisting of a small (1.98 cm) bore and an impactor tip (6 mm diameter) was set to produce a tissue deformation of 3.2 mm as described previously (Dixon et al., 1991). The animals received a cortical impact through the right craniotomy at a velocity of 4 m/s. Following the surgical procedures, the anesthesia was stopped and the animals were ventilated on 100% oxygen until spontaneous return of respiration. Animal recovery was monitored by recording the times until the return of following reflexes: corneal, pinna, tail pinch, toe pinch, and righting. Following this, the rats were monitored until fully recovered from anesthesia, and they were returned to the housing facility.
DHA administration.
TBI or TBI+DHA animals received an initial DMSO (1 ml/kg, i.p.) or DHA injection (16 mg/kg in DMSO) at 5 min after onset of TBI and subsequent daily dose for 3, 7, or 21 d after TBI. Sham control rats received DMSO (1 ml/kg, i.p.) at 5 min after completion of the surgery procedure and subsequent daily dose for 3, 7, or 21 d.
Short-term sensorimotor deficit analysis.
Beam balance and beam walking tasks were assessed in rats at day 0 (before surgery) and at 1–5 d postTBI to evaluate short-term sensorimotor deficits as described previously (Singleton et al., 2010). Gross vestibulomotor function was assessed on a beam balance task in which the time the animal remained on an elevated, 1.5-cm-wide wooden beam was recorded up to a maximum of 60 s. Three trials were performed per animal per day. Training/preassessment was completed on the day before surgery/injury. Spinning on the beam was counted as a fall (a cushioned pad was used to prevent injury). Finer components of vestibulomotor function and coordination were assessed using a modified beam-walking task (Feeney et al., 1982). On the day before injury, rats were trained to escape a bright light and loud white noise (model no. 15800C; Lafayette Instruments), by traversing a narrow wooden beam (2.5 × 100 cm) and entering a darkened goal box at the opposite end. The noise and light were terminated when the rat entered the goal box. The rats remained in the goal box for 30 s between trials. Performance was assessed by measuring the latency to traverse the beam. The rats were also given a beam-walking score to denote progression along the beam during the beam walk test. Animals successfully reaching the goal box were assigned 5 points. Rats not reaching the goal box were assigned lesser scores, depending on either their final spot on the beam when 60 s had elapsed, or the spot at which they fell off. The final daily beam walking score was the mean score of the three beam-walking trials.
Brain tissue preparation and immunoblotting.
Rats were deeply anesthetized with pentobarbital (Nembutal, 100 mg/kg, i.p.) and decapitated. After removal of the brains, the frontal cortex tissue was dissected on a chilled ice plate and homogenized in ice-cold sucrose buffer (0.25 m sucrose, 1 mm EDTA, and 10 mm Tris-HCl, pH 7.4), supplemented with protease inhibitor cocktail (1 mg/ml each of aprotinin, pepstatin, and leupeptin; 100 mg/ml phenylmethylsulfonyl fluoride and 2 mm sodium orthovanadate). The homogenized samples were centrifuged at 12,000 × g at 4°C for 10 min and supernatants collected. The protein content was determined by a BCA protein assay kit using a 96-well microplate reader (Spectra Max 340; Molecular Devices).
Protein samples (80 μg) and prestained molecular weight markers (Bio-Rad) were separated by SDS-PAGE (10 or 15% gel). The resolved proteins were electrophoretically transferred to a PVDF membrane. The blots were incubated in 7.5% nonfat dry milk in tris-buffered saline (TBS) at 4°C. The following primary antibodies and dilutions were used for incubation at 4°C overnight: rabbit anti-p-eIF2α (1:1000), mouse anti-eIF2α (1:1000), rabbit anti-ATF-4 (1:500), rabbit IRE1α (1:1000), monoclonal anti-CHOP (1:1000), and mouse anti-α tubulin (1:8000). After rinsing, the blots were incubated with horseradish peroxidase-conjugated secondary IgG for 1 h at room temperature. Bound antibody was visualized using the enhanced chemiluminescence assay. Relative changes in protein expression were estimated from the mean pixel density of each protein band using ImageJ software. To assure equal loading of protein content, expression of α-tubulin in each sample was quantified. The optical density of each ER stress protein was normalized to α-tubulin and then normalized to contralateral sham. Relative changes in protein expression were estimated from the mean pixel density of each protein band using ImageJ software.
Immunofluorescence staining.
Animals were deeply anesthetized with pentobarbital as described above and transcardially perfused with 100 ml 0.1 m PBS with 50 U/ml heparin, pH 7.4, followed by 500 ml 4% paraformaldehyde (PFA) with 15% saturated picric acid in 0.1 m PBS, pH 7.4. After perfusion, brains were postfixed in 4% PFA for 12 h, and subsequently cryoprotected with 30% sucrose in 0.1 m PBS for 24–36 h at 4°C. The brains were frozen in Tissue-Tek OCT compound (Sakura Finetek) for 10 min and cut into coronal sections (35 μm thickness) on a freezing microtome (Leica SM 2000R). Sections at bregma level −0.26 to −1.40 mm were selected and processed for immunofluorescence staining. Sections were rinsed in 0.1 m TBS, pH 7.4, for 15 min, and incubated with a blocking solution (0.1% Triton X-100 and 3% goat serum in 0.1 m TBS) for 30 min at room temperature. Sections were then incubated with rabbit anti-neurofilament antibody (NF; 1:200), mouse anti-CHOP antibody (1:500), rabbit anti-ATF4 antibody (1:100), mouse anti-MAP2 antibody (1:200), rabbit anti-Aβ APP antibody (1:50), rabbit anti-tau antibody (pSer199/202, 1:500), rabbit anti-XBP1 (1:100), or mouse anti-ubiquitin antibody (1:100) in the blocking solution overnight at 4°C. After rinsing with TBS for 30 min, sections were incubated with the following corresponding secondary antibodies in the blocking solution (1:200) for 1 h at room temperature: goat anti-rabbit AlexaFluor 546-conjugated IgG and goat anti-mouse AlexaFlour 488-conjugated IgG. After rinsing with TBS for 15 min, sections were incubated with To-pro-3 iodide in the blocking solution (1:1000) for another 15 min at room temperature. Sections were then mounted with Vectashield mounting medium (Vector Laboratories). For negative controls, brain sections were stained with the secondary antibody only.
Fluorescent images were captured under 40× lens using a Leica DMIRE2 inverted confocal laser scanning microscope. Samples were excited at 488 (argon/krypton), 543, and 633 nm. The emission fluorescence was recorded at 512–548, 585–650, and 650–750 nm, respectively. In a blinded manner, positively stained cells (CHOP, ATF4, ubiquitin, APP, XBP-1, or p-Tau) were counted from three evenly distributed areas (region of interest) in each brain section of the contralateral and ipsilateral hemispheres using MetaMorph software (Downingtown). The total number of cells in the region of interest was determined by the To-pro-3 staining. Identical digital imaging acquisition parameters were used throughout the study. The number of cells that were positively stained with the anti-CHOP, anti-ATF4, anti-ubiquitin antibody, anti APP, or anti-p-Tau in each area was normalized by the total number of cells in each particular area and data were expressed as an average of the number of positive cells/total cells ×100.
Cortical neuronal or astrocyte cultures.
Cortical neurons were dissociated from embryonic days 14–16 mouse embryos (Black Swiss) and cultured in neurobasal media containing B27 supplement (2%) as described previously (Chen et al., 2010). Briefly, cortices were dissected in the ice-cold Hanks balanced salt solution. The tissue was treated with 0.25 mg/ml trypsin at 37°C for 25 min. The cells were centrifuged at 350 × g at 4°C for 4 min. The cell suspension was diluted in B-27 supplemented neurobasal medium containing 0.5 mm l-glutamine and penicillin/streptomycin (100 U/ml and 0.1 mg/ml, respectively). The cells (1000 cells/mm2) were seeded on glass coverslips coated with poly-d-Lysine. Cultures were incubated at 37°C in an incubator with 5% CO2 and atmospheric air. Cultures were fed every 3 d with fresh media and used for experiments at 10–12 d in vitro.
Dissociated cortical astrocyte cultures were prepared from the cortex of 1- to 3-d-old mice (Black Swiss) as described before (Liu et al., 2010) with some modifications. Cortex was removed and immersed in ice-cold Dulbecco's Modified Eagle Medium (DMEM). The cortices were treated with trypsin solution (0.25 mg/ml Hanks balanced salt solution) for 20 min at 37°C and cells were dissociated by gentle trituration in DMEM/F-12 supplemented with 10% FBS, 100 IU/ml penicillin and 0.1 mg/ml streptomycin. Cells (1 × 105 cells /well) were plated on 6-well plates coated with poly-d-lysine. Cultures were maintained in a 5% CO2 atmosphere at 37°C. To obtain morphologically differentiated astrocytes, confluent cultures (7 d in culture, DIV 7) were treated with DMEM/F-12 containing 0.25 mm dibutyryl cAMP to induce differentiation as described before (Su et al., 2002). Experiments were routinely performed in DIV 10–21 cultures.
OGD and REOX treatment.
Neuronal cultures were incubated with an isotonic oxygen and glucose deprivation (OGD) solution, pH 7.4, containing the following (in mm): 0 glucose, 20 NaHCO3,120 NaCl, 5.36 KCl, 0.33 Na2HPO4, 0.44 KH2PO4, 1.27 CaCl2, 0.81 MgSO4, in a hypoxic incubator with 94% N2, 1% O2, and 5% CO2 (Model 3130; Thermo Forma) as described before (Lenart et al., 2004; Chen et al., 2010). Normoxic control cells were incubated for 2 h in 5% CO2 and atmospheric air in a normoxic buffer identical to the OGD solution except containing 5.5 mm glucose. Reoxygenation (REOX) was achieved by incubating the cultures in the normoxic buffer identical to OGD solution without 5.5 mm glucose in the incubator at 37°C in 5% CO2 and atmospheric air.
Ca2+ER measurement.
Endoplasmic reticulum Ca2+ (Ca2+ER) was determined in cortical neuronal cultures as described previously (Chen et al., 2008). Briefly, coverslips were incubated with 4 μm mag-fura-2 AM and 0.02% pluronic acid for 2 h. After loading, the coverslips were quickly (<2 min) placed on the open-bath imaging chamber (Warner Instruments) in HEPES-MEM at 37°C. Cells were excited every 10 s at 345 and 385 nm and the emission fluorescence images collected at 510 nm using Nikon TE 300 inverted epifluorescence microscope and a 40× objective lens. To determine Ca2+ER, the plasma membrane was permeabilized with 30 s exposure to saponin (3.0 μg/ml) in an intracellular solution to eliminate the cytosolic mag-fura-2 signal as previously described (Chen et al., 2008). The mag-fura-2 signal in ER was calibrated in each experiment and Ca2+ER calculated using a Kd of 56 μm that was determined in neurons using solutions of known Ca2+ concentrations as described before (Chen et al., 2008). IP3-mediated changes of Ca2+ER in neurons were determined in the absence or the presence of 10 μm DHA.
Statistics.
Data are expressed as the group mean ± SE of the mean. Statistical significance was determined by student's t test, paired t test, or one-way ANOVA using the Bonferroni post hoc test in case of multiple comparisons (SigmaStat, Systat Software). Pearson correlation coefficient was calculated using online statistics software (Wessa, 2014). A significance level of p < 0.05 was considered statistically significant in all tests.
Results
DHA reduces sustained expression of ER stress marker proteins in the ipsilateral frontal cortex following TBI
Experimental protocol for sham control, TBI vehicle control, or TBI+DHA treatment groups is described in Figure 1A. Biochemical assays and neurological function analysis were performed in different groups of animals at 3–21 d after TBI. Biochemical analysis was conducted in perilesion frontal cortex tissue in the contralateral (CL) and ipsilateral (IL) hemispheres (Fig. 1B,C). We first investigated activation of the UPR in TBI brains by examining ER stress marker protein p-eIF2α, a key initial effector of UPR (Lai et al., 2007). As shown in Figure 1D, TBI clearly triggered an elevation of p-eIF2α expression in the IL frontal cortex tissue at 3–21 d post-TBI, without changing eIF2α expression. Interestingly, post-TBI administration of DHA daily in rats for 3–21 d significantly attenuated the expression of p-eIF2α protein in the frontal cortex (p < 0.05). To evaluate whether other arms of ER stress responses were altered by TBI and DHA, we examined expression of IRE1α protein as well as XBP1 protein. As shown in Figure 1E, IRE1α protein was significantly increased in cortical tissues at 3, 7, and 21 d after TBI. With DHA treatment, the rise in IRE-1α expression was decreased (Fig. 1E). Moreover, TBI-induced elevation of XBP1 protein expression in cortical neurons was also decreased in the DHA-treated brains (data not shown). Together, these results strongly suggest that DHA plays an important role in attenuating ER stress response after TBI.
Figure 1.
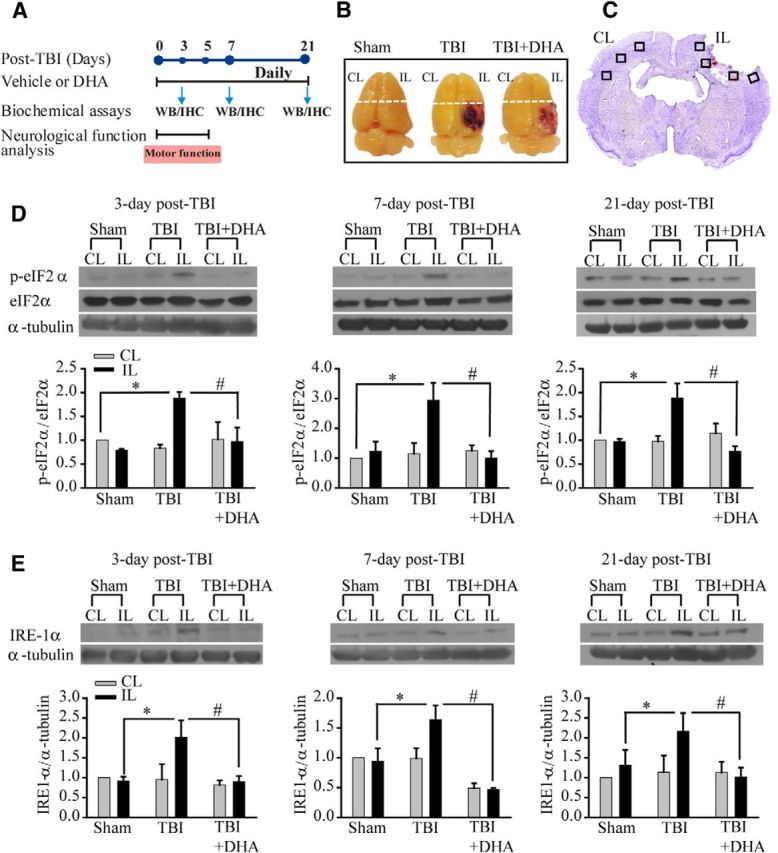
Post-TBI administration of DHA reduces expression of ER stress marker protein p-eIF2α and IRE1α in the frontal cortex following TBI. A, Schematic presentation of experimental protocol. Either DMSO vehicle or DHA (16 mg/kg, i.p.) was administered at 5 min after induction of CCI, followed with the same daily dose during 3–21 d post-TBI. The time points of sample collection for Western blotting (WB) and immunohistological analysis (HI) or neuronal functional analysis were indicated. B, Representative brain images of three groups illustrates the CL and IL frontal cortex tissue (above the dotted line) harvested for immunoblotting analysis. C, Representative TBI brain section (cresyl violet stained, bregma level −1.40) illustrates sample collection in the perilesion areas (black box) of the CL and IL frontal cortex. D, Representative immunoblots of p-eIF2α and eIF2α protein expression in the CL and IL frontal cortex tissues of sham control, TBI vehicle control, and TBI+DHA animals at 3–21 d post-TBI. The same blot was probed with anti-α-tubulin antibody as a loading control. Bottom, Summary data. Data are mean ± SE; n = 4; *p < 0.05 versus sham; # p < 0.05 versus TBI. E, Representative immunoblot of IRE1α-protein expression in the CL and IL frontal cortex tissues of sham control, TBI vehicle control, and TBI+DHA animals at 3–21 d post-TBI. Data are mean ± SE; n = 4–5; *p < 0.05 versus sham; #p < 0.05 versus TBI.
DHA abolishes expression of ER stress protein ATF4 following TBI
Phosphorylation of eIF2α increases the translation of ER stress marker ATF4 (Larner et al., 2006). Figure 2A shows representative immunoblot of ATF4 protein expression in the CL and IL frontal cortex tissues in sham vehicle control, TBI-vehicle control, or TBI+DHA-treated animals. TBI brain tissues exhibited significant elevation of ATF4 protein expression at days 3–7 post-TBI (Fig. 2A). Unexpectedly, this elevation persisted at day 21 post-TBI (Fig. 2B). In addition, immunofluorescence staining images revealed an increased expression of ATF4 protein in neurons of the IL perilesion frontal cortex of TBI-vehicle control brains at 3–7 d post-TBI (Fig. 2C, arrowhead), which was accompanied with loss of NF expression (arrow). These data clearly demonstrate that TBI can induce a prolonged ER stress and UPR activation in neurons of the perilesion tissues. Interestingly, the TBI-induced sustained expression of ATF4 was absent in the DHA-treated animals (Fig. 2A–D). The DHA-treated brain tissues also exhibited reduced neurodegeneration (reflected by more NF expression; Fig. 2C, arrow). Collectively, our study reveals novel findings that TBI triggered sustained ER stress in neurons in the perilesion tissues and DHA abolished the TBI-induced ER stress.
Figure 2.

DHA reduces sustained expression of ER stress protein ATF4 in the frontal cortex tissues following TBI. A, Representative immunoblot of ATF4 protein in the CL and IL frontal cortex tissues of three groups at 3–7 d post-TBI. The same blot was probed with anti-α-tubulin antibody as a loading control. B, Representative immunoblot of ATF4 protein in the CL and IL frontal cortex tissues at 21 d post-TBI. Data are mean ± SE; n = 3–4; *p < 0.05 versus sham; #p < 0.05 versus TBI. C, Confocal images showing expression of ATF4 in the perilesion frontal cortex tissues at 3 and 7 d post-TBI. C, Arrow, Expression of NF in neurons (red). C, Arrowhead, ATF4-positive neurons (green). D, Summary data of ATF4-immunopositive cells. Values are mean ± SE (n = 3) and expressed as ATF4+ cells/100 total cells. *p < 0.05 versus CL; # p < 0.05 versus TBI vehicle control.
DHA decreases expression of ER proapoptosis protein CHOP in the frontal cortex following TBI
CHOP is a key molecule linking ER stress to apoptotic cell death (Oyadomari and Mori, 2004). The expression of this proapoptotic protein was examined in the IL perilesion frontal cortex at days 3–21 post-TBI. A peak elevation of CHOP was found in the IL perilesion frontal cortex at 3 d post-TBI but decreased by 21 d post-TBI (Fig. 3A). In addition, strong positive immunoreactive signals of CHOP were detected in the IL perilesion frontal cortex at 3–7 d post-TBI (Fig. 3B, arrowhead). Moreover, the CHOP-positive neurons exhibited loss of MAP2 expression (Fig. 3B). However, post-TBI administration of DHA for 3–7 d significantly reduced the expression of proapoptotic protein CHOP and loss of MAP2 expression in neurons. These data suggest that DHA treatment can significantly attenuate the TBI-induced activation of the PKR-like ER kinase (PERK)-ATF4-CHOP UPR pathway.
Figure 3.

DHA reduces expression of ER-resident proapoptotic protein CHOP in the frontal cortex following TBI. A, Representative immunoblot of CHOP protein expression in the CL and IL frontal cortex of three groups (see Fig. 2 legend). The same blot was probed with anti-α-tubulin antibody as a loading control. B, Confocal microscopic images showing CHOP expression in the perilesion frontal cortex tissues at 3 or 7 d post-TBI. B, Arrow, Expression of NF in neurons (red). B, Arrowhead, CHOP-positive neurons (green). Bottom, Summary data of CHOP-immunopositive cells. Values are mean ± SE (n = 4) and expressed as CHOP+ cells/100 total cells. *p < 0.05 versus CL; #p < 0.05 versus TBI.
Interestingly, GRP-78 protein, the master regulator of the UPR, was not significantly changed at 3–21 d post-TBI (data not shown). Moreover, DHA had no effects on reducing expression of GRP78 protein. This is in contrast to a sustained upregulation of all other ER stress markers in TBI brains and protective effects of DHA. These data suggest that DHA selectively suppresses the UPR effector pathways that are detrimental for cell survival, but leaves the adaptive arms of UPR intact.
DHA attenuates TBI-mediated ubiquitin protein aggregate formation
Prolonged ER stress can compromise the ER function and lead to the accumulation of miss folded proteins and result in the formation of protein aggregates. We investigated protein aggregate formation by immunostaining analysis using monoclonal ubiquitin antibody, which recognizes aggregated ubiquitin proteins. As shown in Figure 4A,B, neither sham controls nor the CL frontal cortex tissues of TBI vehicle controls or TBI+DHA exhibited any clustered ubiquitin-immunoreactive signals. However, abundant perinuclear expression of ubiquitinated proteins were detected in the IL frontal cortex of TBI vehicle control brains at 3 d post-TBI (Fig. 4A, arrowhead). At 7 d post-TBI, the IL cortex showed reduced expression of the ubiquitinated proteins. Moreover, consistently with the ER stress marker expression, the ubiquitinated protein aggregates were significantly reduced in the 3 d DHA-treated brains or absent in the 7 d DHA-treated brains (Fig. 4, arrow). Together, our study strongly suggests that DHA not only abolishes the TBI-induced ER stress and the UPR activation but also attenuates ubiquitinated protein aggregate formation in neurons.
Figure 4.
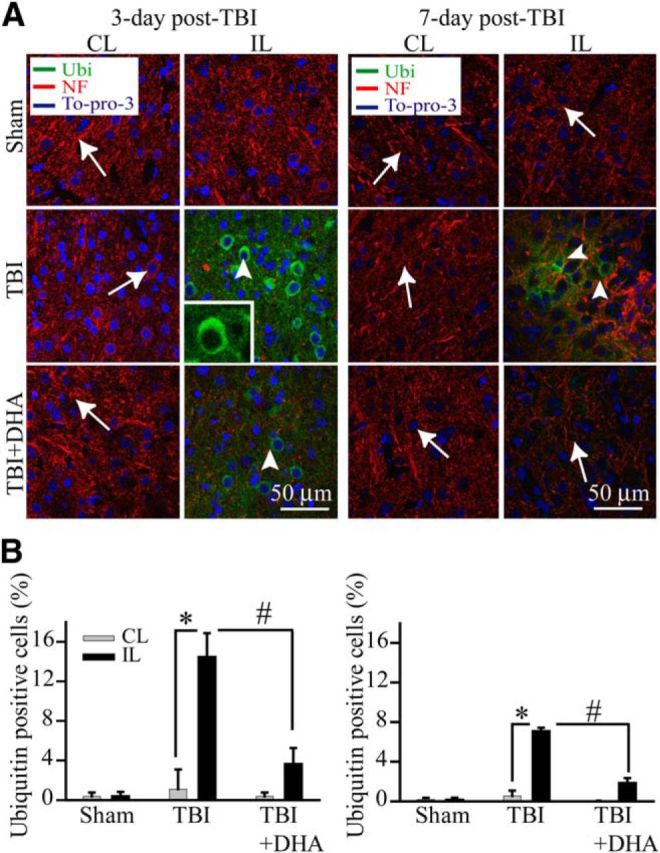
DHA inhibits ubiquitinated (Ub)-protein aggregate formation in the post-TBI brains. A, Confocal microscopic images of the CL and IL frontal cortex tissues showing Ub-protein aggregate formation at 3–7 d post-TBI (green, Ub aggregates; red, NF; blue, nuclear marker To-pro-3). B, Summary data of ubiquitin-immunopositive cell counting. Values are mean ± SE (n = 3) and expressed as ubiquitin+ cells/100 total cells. *p < 0.05 versus CL; #p < 0.05 versus TBI.
DHA reduces ER Ca2+ depletion and ER stress in vitro
It remains unknown how DHA reduces ER stress in TBI brains. Recently, we reported that DHA could reduce ER stress in part via blocking ER Ca2+ depletion in cultured cortical astrocytes (Begum et al., 2012). Disruption in ER Ca2+ homeostasis can lead to unfolded protein response and development of ER stress (Paschen and Doutheil, 1999; Groenendyk and Michalak, 2005). Figure 5A shows that DHA blocked IP3-mediated ER Ca2+ release in cultured cortical neurons. To further test the role of DHA on ER Ca2+ depletion-induced ER stress, we used an established ER stress inducer thapsigargin (Tg), a specific irreversible inhibitor of ER Ca2+-ATPase, to induce ER Ca2+ store depletion and ER stress. As shown in Figure 5B, treatment with Tg triggered an increase in p-eIF2α and eIF2α expression in cortical astrocytes. In contrast, in the presence of TUDCA, a recently established ER chemical chaperon as a specific ER stress inhibitor (Paschen, 2003; Ozcan et al., 2006), the Tg-mediated p-eIF2α expression was abolished. Most importantly, DHA was equally effective in reducing the Tg-induced p-eIf2α expression. Similar results were obtained when ER stress was induced by Tu. Figure 5D illustrates that DHA and TUDCA are equally effective in reducing Tu-mediated expression of p-eIf2α. However, the ratio of p-eIF2α versus total-eIF2α remained elevated under these conditions. The underlying mechanisms are not apparent. Phosphorylation of eIF2α regulates ER homeostasis by shutting down general protein translation and favoring selective translation of proteins that facilitate proper folding and prevent accumulation of abnormal proteins. Under our experimental ER stress conditions, it is possible that cells need the increased levels of eIF2α for the protein translation of chaperones to curb the initial misfolded protein load. eIF2α participates in translation initiation, a rate limiting step in protein synthesis, and thus plays a critical role in the process (Donnelly et al., 2013). Last, we tested whether DHA can block ER stress mediated by in vitro ischemic stress OGD/REOX. As shown in Figure 5C, expression of p-eIf2α in neurons was increased after 2 h OGD and 15–60 min REOX. However, incubation of cultures with DHA during 15–60 min REOX significantly attenuated the p-eIf2α protein expression. Together, these in vitro experiments strongly suggest that DHA can attenuate ER stress in both neurons and astrocytes. The DHA-mediated reduction of ER stress could in part result from blocking IP3R and ER Ca2+ depletion.
Figure 5.
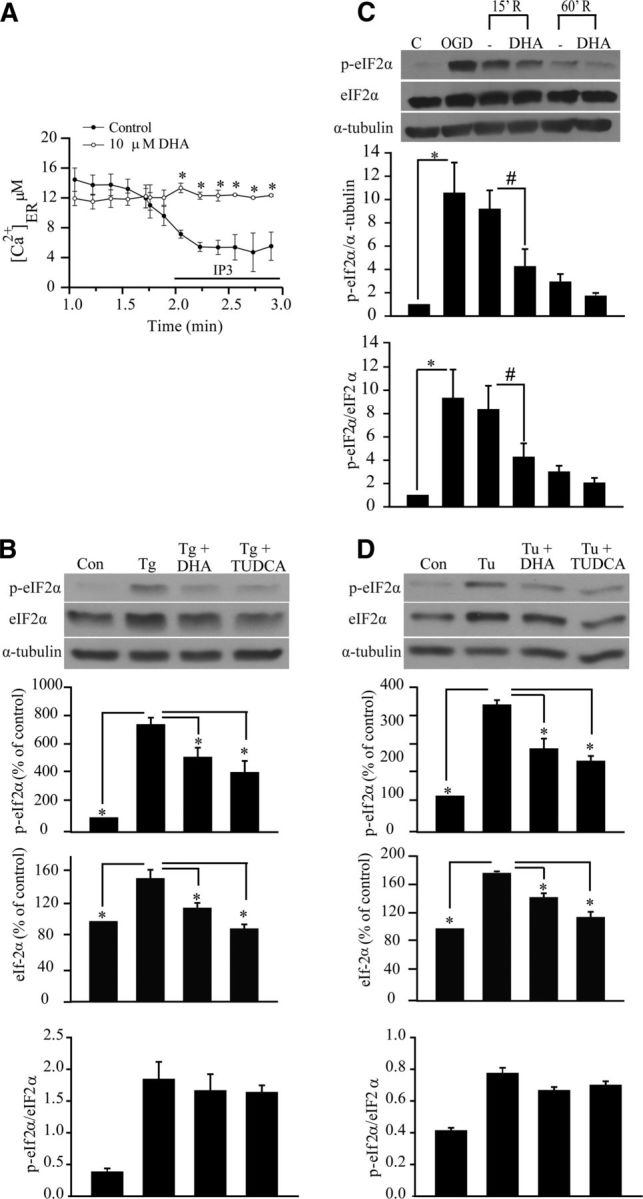
DHA prevents ER Ca2+ depletion and ER stress in neurons and astrocytes. A, Representative tracings of Ca2+ER release in response to IP3 in cultured cortical neurons. IP3 (10 μm) was applied to saponin-permeablized neurons to trigger the Ca2+ER release. DHA (10 μm) was added 2–3 min before permeabilization and present in the subsequent solutions. B, Representative immunoblot of p-eIF2α and eIF2α in primary cortical astrocytes. Cells were exposed to Tg (1 μm), DHA (10 μm), or TUDCA (10 μm) for 24 h. Expression of p-eIF2α protein, eIF2α protein, and the ratio of p-eIF2α versus eIF2α were analyzed. The same blot was probed with anti α-tubulin antibody as a loading control. Data are mean ± SE (n = 3). *p < 0.05 versus ER stressor. C, DHA attenuated expression of p-eIF2α protein in cortical neurons induced by OGD (2 h) and REOX (15–60 min). DHA (10 μm) was only present during 15–60 min REOX. Expression of p-eIF2α protein, eIF2α protein, and the ratio of p-eIF2α versus eIF2α protein were analyzed. The same blot was probed with anti α-tubulin antibody as a loading control. Data are mean ± SE (n = 4). *p < 0.05 versus control; #p < 0.05 versus REOX. D, Representative immunoblots of p-eIF2α and eIF2α in cortical astrocytes. Astrocytes were exposed to Tu (10 μg/ml), DHA (10 μm), or TUDCA (10 μm) for 24 h. Expression of p-eIF2α protein, eIF2α protein, and the ratio of p-eIF2α versus eIF2α protein were analyzed. The same blot was probed with anti α-tubulin antibody as a loading control. Data are mean ± SE (n = 3). *p < 0.05 versus ER stressor.
Positive correlation between ER stress and APP accumulation after TBI
The expression of APP has been reported to be upregulated by various stress stimuli (Dewji et al., 1995) and a similar pathology of brain amyloid β protein (Aβ) deposition has been detected in TBI and Alzheimer's disease (Ikonomovic et al., 2012). In the current study, we found an increased APP protein expression at 3 d post-TBI using an anti-APP antibody, which recognized a single 80 kDa APP band of rat brain tissues (data not shown). Figure 6A,B shows that APP protein expression was apparent in neuronal soma in the IL cortex at 3 d after TBI (left, arrow). Interestingly, APP-immunoreactive signals were colocalized with ER stress marker proteins CHOP and ATF4 in the IL perilesion cortex (Fig. 6A,B, right, arrow). In contrast, the DHA-treated brains showed much less APP accumulation and ER stress marker expression (Fig. 6A,B, inset, arrowhead). These data strongly suggest that post-TBI administration of DHA not only reduces ER stress but also decreases abnormal APP protein accumulation.
Figure 6.
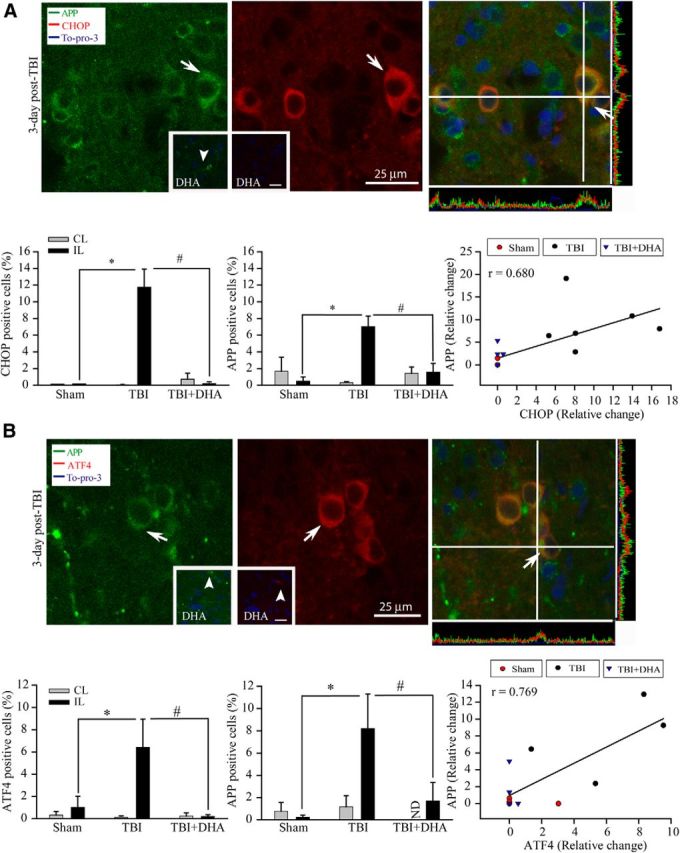
Effects of DHA on expression of APP and ER stress marker proteins CHOP and ATF4 in the cortex after TBI. A, Confocal microscopic images of CHOP and APP accumulation in the IL perilesion frontal cortex at 3 d after TBI. A, Arrow, Elevated APP/CHOP expression in soma. A, Inset, Arrowhead, Reduced expression of APP in the IL cortex of DHA-treated brains. Bottom: summary data of CHOP-immunopositive cells (left), APP-immunopositive cells (center), and correlation of CHOP and APP expression in the sham control, TBI vehicle control, and TBI+DHA groups (right); r = 0.68. B, Confocal microscopic images of ATF4 and APP expression the IL perilesion frontal cortex. B, Arrow, Elevated expression of APP/ATF4 in soma. B, Inset, Arrowhead, Lack of APP/ATF4 expression in the DHA-treated IL frontal cortex. Bottom, Summary data of ATF4-immunopositive cells (left), APP-immunopositive cells (center), and correlation of ER stress protein ATF4 and axonal injury marker APP expression in sham control, TBI vehicle control, and TBI+DHA groups (right); r = 0.769. Data are mean ± SE (n = 3). *p < 0.05 versus sham (IL); #p < 0.05 versus TBI (IL).
We further analyzed whether there is a correlative relationship between ER stress and APP accumulation. Figure 6A,B, right, bottom shows the scatter plots of the distribution of ER stress markers (CHOP or ATF4) against APP expression. It illustrated that sham or TBI+DHA brains exhibited less ER stress and APP protein accumulation, with values clustered on the left side of the scatter plot. In contrast, CHOP or ATF4 data from TBI brains were more spread out on the right side of the scatter plot (on the x-axis), which were accompanied with higher APP values on the y-axis. The Pearson's correlation analysis revealed a positive correlation between either CHOP or ATF4 and APP accumulation (Pearson's r = 0.680 and 0.769, respectively). These analyses strongly suggest that there may be a causative relationship between ER stress and abnormal APP protein accumulation after TBI and DHA can reduce both events.
DHA reduced accumulation of p-Tau after TBI
We also investigated the temporal patterns of p-Tau accumulation in TBI brains. Immunostaining using the polyclonal antibody of p-Tau (against pS199/202) revealed increased p-Tau immunoreactivity in cell bodies in the IL frontal cortex at 3–21 d after TBI (Fig. 7A). Quantitative analysis of p-Tau-immunoreactive cells shows that the number of p-Tau-positive cells was increased at 3 and 7 d after TBI and persisted at 21 d after TBI (Fig. 7B). In contrast, a few cells with positive p-Tau staining were detected in the DHA-treated brains at 3–21 d after TBI (Fig. 7A, right). In summary, TBI triggered significant increase in p-Tau accumulation in the cell bodies of the IL frontal cortex at 3–21 d after TBI. DHA treatment nearly abolished the abnormal p-Tau accumulation in the IL frontal cortex.
Figure 7.

Effect of DHA on the expression of p-Tau in the IL cortex after TBI. A, Confocal images showing expression of p-Tau in the perilesion frontal cortex tissues at 3, 7, and 21 d post-TBI. A, Arrow, Increased expression of p-Tau (green) in cell bodies. A, Arrowhead, Reduced expression of p-Tau in DHA treated brains. B, Summary data of p-Tau-immunopositive cells. Values are mean ± SE (n = 3) and expressed as percentage of p-Tau positive cells. *p < 0.05 versus TBI vehicle control.
DHA reduced axonal injury after TBI
Diffuse axonal injury is one of the most common and important brain pathologies following TBI (Arfanakis et al., 2002; Inglese et al., 2005; Xu et al., 2007). Abnormal accumulation of APP protein in axons is a hallmark of axonal injury (Itoh et al., 2009) and has been suggested to result from axonal transport interruption within hours of injury (Sherriff et al., 1994). We further investigated the axonal damage in the external capsule of corpus callosum at 3–21 d after TBI via immunocytochemical staining for APP (Fig. 8A). The axons in the external capsule of sham animals show no APP immunolabeling (data not shown). In contrast, abundant expression of APP was detected in external capsule axons at 3 and 7 d after TBI, which persisted up to 21 d in TBI brains (Fig. 8B, arrow). The injured axons with APP accumulation demonstrated prominent multiple varicosities and disconnections (Fig. 8A, arrow), which are characteristics of diffuse axonal injury. In contrast, in the DHA-treated brains, axons throughout the external capsule showed either significantly less APP-positive axons or less APP immunofluorescence intensity (Fig. 8A,B, arrowheads). Morphologically, these axons exhibited less swelling and disconnections and summary of quantitative analysis was shown in Figure 8C. There were significantly decreased numbers of APP-positive axons detected in the DHA-treated brains, compared with the vehicle-treated TBI brains at all three time-points (p < 0.05).
Figure 8.
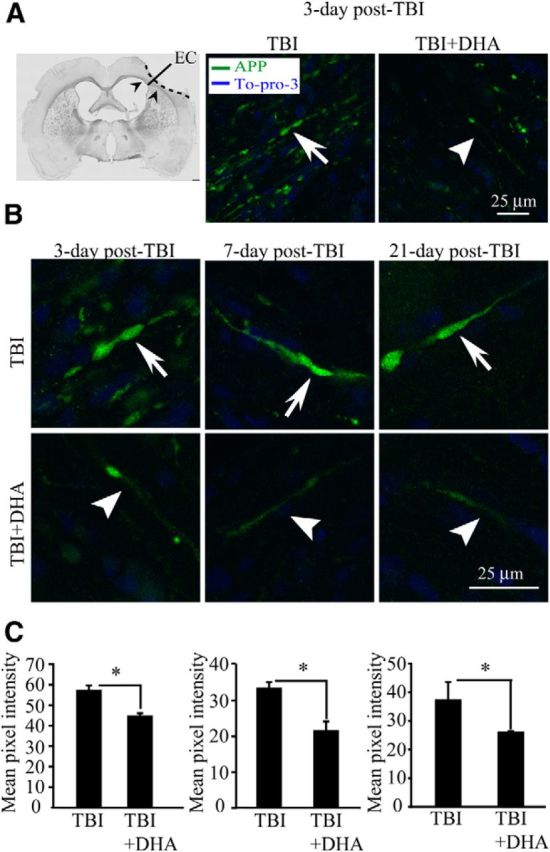
Effect of DHA on axonal APP accumulation in the IL white matter after TBI. A, Confocal images were obtained from the IL external capsule white matter tissues (EC, left, arrowhead). Expression of APP in the axons within the external capsule at 3 d post-TBI was shown. A, Arrow, Increased expression of APP (green) in the swollen axons. A, Arrowhead, Reduced expression of APP in the DHA treated brains. B, Confocal images showing expression of APP in the axons within the external capsule at 3–21 d post-TBI. C, Summary data of APP-immunopositive axons. Values are mean pixel intensity/μm2; n = 3. *p < 0.05 versus TBI vehicle control.
DHA improves neurological function after TBI
We speculate that the effects of DHA on blocking the sustained expression of ER stress and reducing toxic protein accumulation in both gray and white matter should facilitate neurological functional recovery after TBI. To evaluate this outcome, sham control, TBI control, and DHA-treated animals were tested for sensorimotor function with the beam balance test at days 0–5 post-TBI. As shown in Figure 9A–C, sham animals exhibited transient and small fluctuations in the beam balance time and beam walking scores, and beam walking latency during days 1–5 after surgery. TBI vehicle controls showed significant deficits in motor function, reflected by the reduced beam balance time and beam walking score at days 1–4 post-TBI. At post-TBI day 5, the beam balance time of the TBI vehicle controls was partially recovered, but, not statistically significant different from post-TBI day 1 (p > 0.05). Interestingly, the DHA-treated animals exhibited an earlier recovery of the motor function, with an improved the beam balance and beam walking score at day 4 post-TBI (p < 0.05). At day 5 post-TBI, all three scores in the DHA-treated group were significantly improved than on day 1 post-TBI. In contrast, TBI control animals did not show improvement. These experiments suggest that the DHA-treated animals exhibit a faster neurological recovery trend after TBI.
Figure 9.

DHA improves sensorimotor function in rats after TBI. The beam-balance test (A), beam-walk test (B), and beam-walk score (C) were determined in the sham control, TBI vehicle control, and TBI+DHA rats at days 0–5 after surgery. Data are mean ± SD (n = 10). *p < 0.05 versus day 1 post-TBI. Schematic summary of DHA-mediated reduction of ER stress, abnormal protein accumulation, and neurological deficit after TBI is shown in D. TBI triggers sustained ER stress and the UPR activation following TBI. The prolonged ER stress, abnormal protein accumulation, and ubiquitinated-protein aggregate formation may collectively contribute to development of chronic axonal damage and neurological deficits after TBI. Post-TBI administration of DHA may prevent ER Ca2+ depletion, reduce chronic ER stress, and abnormal protein accumulation, and improve neurological function recovery after TBI.
Discussion
Chronic ER stress in the perilesion brain tissues after TBI
Many studies have shown that ER stress and UPR activation play a role in the acute brain injury after TBI (Larner et al., 2006). The UPR is performed by three ER-transmembrane effector proteins, PERK, IRE1, and ATF6 (Kim et al., 2008). Larner et al. (2004) reported that activation of IRE1α pathway leads to stimulation of ER-associated caspase 12 and apoptosis. Activation of caspase-12 protein was detected in the frontal cortex which peaked at day 5 post-TBI (Larner et al., 2004). CHOP expression is a representative feature of ER stress-induced cell death (Zinszner et al., 1998; Oyadomari and Mori, 2004). In a mouse CCI model, immunoreactive signals of CHOP were increased in the ipsilateral hemispheres at 6 h post-TBI, which remained elevated at day 14 post-TBI (Krajewska et al., 2011). To date, most studies on the TBI-induced ER stress and UPR have centered on its association to acute neuronal death.
In our study, we detected persistent activation of the PERK-ATF4-CHOP pathway at 3–21 d after TBI. Such sustained ER stress response can prime the cells for the development of chronic pathological changes. Formation and continuous accumulation of ubiquitin-conjugated proteins constitute a major pathologic event in the cells after acute ischemic or traumatic brain injury (Hu et al., 2001). However, it remains unknown whether there is a correlative relationship between UPR and abnormal protein accumulation after TBI, which was our focus in this study.
Positive correlation between ER stress and abnormal protein accumulation after TBI
TBI constitutes one of the epigenetic risk factors for developing neurodegenerative disorders, such as Alzheimer's disease and Parkinson's disease (Hoozemans et al., 2012). Both of these neurodegenerative diseases exhibit characteristics of imbalanced regulation of the abnormal accumulation of proteins and protein degradation. Accumulation of several key proteins, including APP, Aβ, NF proteins, and synuclein, reflects disruption of axonal transport as a result of extensive axonal injury upon TBI (Smith et al., 2003). Axonal pathology has been shown to develop over the course of hours to days following injury and has even been observed for months (Christman et al., 1994; Pierce et al., 1998). A long-term overexpression of Aβ APP in neuronal perikarya and damaged neuritis was detected at 1–90 d after TBI in rats (Itoh et al., 2009). In the pig inertial brain injury model, marked accumulation of Aβ APP in axons following trauma leads to the production of Aβ protein (Smith et al., 1999). Also, intra axonal accumulation of Aβ and extracellular deposition of Aβ plaques and aggregation of tau have been observed in humans within hours after severe injury (Smith et al., 2003; Uryu et al., 2003; Chen et al., 2004; Ikonomovic et al., 2004).
In the current study, we detected increased expression of aggregated ubiquitin in the IL perilesion frontal cortex at 3–7 d post-TBI. Moreover, we also observed significant accumulation of p-Tau and APP in the IL cortex of TBI animals at days 3–21 post-TBI. Most importantly, there was a positive correlation between the abnormal APP accumulation and ER stress marker protein CHOP or ATF4 expression after TBI. These proteins were colocalized in the same injured neurons after TBI, as revealed by a strong Pearson's correlation factor. These findings led us to speculate that persistent ER stress in TBI may in part contribute to abnormal protein accumulation in neurons.
In our study, ATF4 staining signals were largely detected in cytoplasma of TBI brains but absent in the nucleus. Interestingly, elevation of ATF4 has also been found primarily in cytoplasma and proximal neuronal processes of Parkinson's disease patients (Sun et al., 2013), which is similar to our findings. It has been suggested that retrograded transport may play a role in ATF4 regulation and its translocation to the nucleus (Lai et al., 2008). It remains to be determined how translocation of cytoplasmic ATF4 in neurons of TBI brains is regulated.
Recently, inhibition of ER stress by salubrinal has shown neuroprotection in several disease models (Sokka et al., 2007; Zhu et al., 2008; Colla et al., 2012). Salubrinal is a specific inhibitor of eIF2α phosphatases (Boyce et al., 2005). By preventing eIf2α dephosphorylation, salubrinal decreases the new protein synthesis and subsequently reduces the unfolded protein load (Drexler, 2009). In an obstructive sleep apnea-induced neuronal injury model, ER stress-mediated neuronal injury was ablated by treatment with salubrinal which converts susceptible motoneurons into motoneurons resistant to hypoxia-induced injury temporarily (Zhu et al., 2008). Salubrinal has also been shown to be protective against excitotoxic neuronal injury in neuronal cultures (Sokka et al., 2007). In a transgenic model of α-synucleopathy, salubrinal prevents toxic accumulation of α-synuclein oligomers due to chronic ER stress (Colla et al., 2012). Therefore, targeting chronic ER stress may be a novel strategy to reduce uncontrolled ER stress and the activation of apoptosis in many chronic neurodegeneration conditions.
Effects of DHA on reducing TBI-mediated ER stress and abolishing abnormal protein accumulation
DHA is the most abundant essential ω-3 fatty acid in the brain and has been shown to be neuroprotective in various neurodegenerative conditions. Recent preclinical experimental studies have demonstrated that administering DHA at 3–5 h post-ischemia can significantly improve neurological function recovery and reduce infarct volume (Belayev et al., 2011; Petraglia et al., 2011). Administration of DHA daily has also been shown to improve neurological functional outcome in a rat model of perinatal hypoxia-ischemia (Berman et al., 2010). Studies with chronic DHA supplementation (either before or immediately after induction of TBI) have shown that DHA can improve animals learning and memory function, and mitochondrial energy metabolisms (Mills et al., 2011; Wu et al., 2011). In rodent head acceleration injury model, post-TBI DHA supplementation significantly reduces the number of APP-positive axons in brainstem white matter tracts at 30 d postinjury (Bailes and Mills, 2010). A most recent study also demonstrates that DHA supplementation following diffuse axonal injury in rats could ameliorate secondary mechanisms of injury, and result in a decreased number of injured axons (Mills et al., 2011).
Although many studies revealed the neuroprotective effects of DHA in various brain injury models, the molecular mechanisms involved are not well understood. DHA has previously been shown to be metabolized into neuroprotective mediators known as docosanoids or NPD1, the most extensively studied of which is 10, 17S-docosatriene (Mukherjee et al., 2004). NPD1 is a potent inhibitor of proinflammatory cytokine expression and apoptosis, as well as ischemia-reperfusion mediated leukocyte infiltration (Marcheselli et al., 2003; Bazan, 2009). DHA also has antioxidant activity and mitigates peroxidative damage of lipids and proteins in the brain (Innis, 2007).
In this study, our novel findings suggest that post-TBI administration of DHA decreased UPR activation as well as accumulation of APP, p-Tau, and ubi-protein aggregate formation. In addition, DHA-treated TBI animals exhibited earlier recovery of sensorimotor function. We hypothesize that attenuation of sustained ER stress by DHA plays an important role in DHA neuroprotective effects. This may be in part mediated by blocking ER Ca2+ depletion and ER stress. Disruption of Ca2+ homeostasis has been suggested as a fundamental pathological mechanism underlying secondary cell injury and death after TBI. Traumatic injury to spinal cord leads to cell injury and death which is largely due to ER Ca2+ release mediated by ryanodine and IP3 receptors (Thorell et al., 2002). We recently reported that DHA can block ER Ca2+ depletion and ER stress in astrocytes in response to ischemic stress (Begum et al., 2012). In our current study, we found that DHA had a similar effect on blocking the IP3-mediated ER Ca2+ release and ER stress induced by ER stress inducer thapsigargin, tunicamycin, or in vitro ischemia. More importantly, the effect of DHA in blocking ER stress was mimicked by an ER stress inhibitor, the chemical chaperone TUDCA, which was successfully used to reduce ER stress and restore glucose homeostasis in a mouse model of type-2 diabetes (Ozcan et al., 2006). Together, our study provides first line of evidence that DHA reduces ER stress and accumulation of misfolded protein and protein aggregate formation.
In the current study, we did not perform either biochemical or behavioral tests in naive control or sham control animals treated with DHA. In light of the recent findings on DHA supplemental on cognitive effects in naive mice (Joseph et al., 2012), future studies should include investigating potential effects of DHA on basal ER functions and motor functions of rats without TBI.
Conclusions
In this study, we investigated TBI-mediated ER stress and the efficacy of post-TBI administration of DHA in reducing ER stress (Fig. 9D). TBI triggered sustained expression of the ER stress marker proteins including p-eIF-2α, IRE1α, ATF4, and CHOP in the ipsilateral cortex at 3–21 d after TBI. The prolonged ER stress was accompanied with accumulation of abnormal ubiquitin aggregates, APP, and p-Tau, after TBI. This is the first report that the ER stress marker proteins were colocalized with APP accumulation after TBI and there is a positive correlation between ER stress and abnormal APP accumulation. Administration of DHA attenuated all ER stress protein expression except ER chaperon protein GRP78 and reduced the accumulation of both ubiquitinated proteins and APP/p-Tau proteins. Importantly, the DHA-treated animals exhibited early recovery of their sensorimotor function after TBI. Our findings suggest that sustained ER stress may play a role in chronic neuronal damage after TBI. To further evaluate DHA's therapeutic potentials in TBI, additional studies are warranted to study efficacy of DHA with delayed administration in decreasing ER stress and toxic protein aggregation after TBI.
Footnotes
This work was supported in part by University of Pittsburgh Neurology Departmental startup funds and University of Pittsburgh Medical Center endowed Chair Professorship on Brain Disorder Diseases (D.S.) and the United States Department of Veterans Affairs VA RR&D#B6761R Grant (C.E.D.).
References
- Arfanakis K, Haughton VM, Carew JD, Rogers BP, Dempsey RJ, Meyerand ME. Diffusion tensor MR imaging in diffuse axonal injury. AJNR Am J Neuroradiol. 2002;23:794–802. [PMC free article] [PubMed] [Google Scholar]
- Bailes JE, Mills JD. Docosahexaenoic acid reduces traumatic axonal injury in a rodent head injury model. J Neurotrauma. 2010;27:1617–1624. doi: 10.1089/neu.2009.1239. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer's disease. J Lipid Res. 2009;50:S400–S405. doi: 10.1194/jlr.R800068-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Kintner D, Liu Y, Cramer SW, Sun D. DHA inhibits ER Ca2+ release and ER stress in astrocytes following in vitro ischemia. J Neurochem. 2012;120:622–630. doi: 10.1111/j.1471-4159.2011.07606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Atkins KD, Eady TN, Hong S, Lu Y, Obenaus A, Bazan NG. Docosahexaenoic acid therapy of experimental ischemic stroke. Transl Stroke Res. 2011;2:33–41. doi: 10.1007/s12975-010-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DR, Liu Y, Barks J, Mozurkewich E. Treatment with docosahexaenoic acid after hypoxia-ischemia improves forepaw placing in a rat model of perinatal hypoxia-ischemia. Am J Obstet Gynecol. 2010;203:385.e1–385.e5. doi: 10.1016/j.ajog.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–141. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/S0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kintner DB, Luo J, Baba A, Matsuda T, Sun D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: role of Na+-K+-Cl− cotransporter. J Neurochem. 2008;106:1563–1576. doi: 10.1111/j.1471-4159.2008.05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kintner DB, Baba A, Matsuda T, Shull GE, Sun D. Protein aggregation in neurons following OGD: a role for Na+ and Ca2+ ionic dysregulation. J Neurochem. 2010;112:173–182. doi: 10.1111/j.1471-4159.2009.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173–186. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci. 2012;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Gandy S. Traumatic brain injury–football, warfare, and long-term effects. N Engl J Med. 2010;363:1293–1296. doi: 10.1056/NEJMp1007051. [DOI] [PubMed] [Google Scholar]
- Dewji NN, Do C, Bayney RM. Transcriptional activation of Alzheimer's beta-amyloid precursor protein gene by stress. Brain Res Mol Brain Res. 1995;33:245–253. doi: 10.1016/0169-328X(95)00131-B. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci. 2013;70:3493–3511. doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HC. Synergistic apoptosis induction in leukemic cells by the phosphatase inhibitor salubrinal and proteasome inhibitors. PLoS One. 2009;4:e4161. doi: 10.1371/journal.pone.0004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab. 2010;12:108–115. doi: 10.1111/j.1463-1326.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217:855–857. doi: 10.1126/science.7100929. [DOI] [PubMed] [Google Scholar]
- Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- Groenendyk J, Michalak M. Endoplasmic reticulum quality control and apoptosis. Acta Biochim Pol. 2005;52:381–395. [PubMed] [Google Scholar]
- Hasadsri L, Wang BH, Lee JV, Erdman JW, Llano DA, Barbey AK, Wszalek T, Sharrock MF, Wang HJ. Omega-3 fatty acids as a putative treatment for traumatic brain injury. J Neurotrauma. 2013;30:897–906. doi: 10.1089/neu.2012.2672. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer's and Parkinson's disease. Neurodegener Dis. 2012;10:212–215. doi: 10.1159/000334536. [DOI] [PubMed] [Google Scholar]
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjö BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–875. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Price JC, Hamilton RL, Mathis CA, Paljug WR, Debnath ML, Cohen AD, Mizukami K, DeKosky ST, Lopez OL, Klunk WE. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123:433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese M, Makani S, Johnson G, Cohen BA, Silver JA, Gonen O, Grossman RI. Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg. 2005;103:298–303. doi: 10.3171/jns.2005.103.2.0298. [DOI] [PubMed] [Google Scholar]
- Innis SM. Dietary (n-3) fatty acids and brain development. J Nutr. 2007;137:855–859. doi: 10.1093/jn/137.4.855. [DOI] [PubMed] [Google Scholar]
- Itoh T, Satou T, Nishida S, Tsubaki M, Hashimoto S, Ito H. Expression of amyloid precursor protein after rat traumatic brain injury. Neurol Res. 2009;31:103–109. doi: 10.1179/016164108X323771. [DOI] [PubMed] [Google Scholar]
- Joseph MS, Ying Z, Zhuang Y, Zhong H, Wu A, Bhatia HS, Cruz R, Tillakaratne NJ, Roy RR, Edgerton VR, Gomez-Pinilla F. Effects of diet and/or exercise in enhancing spinal cord sensorimotor learning. PLoS One. 2012;7:e41288. doi: 10.1371/journal.pone.0041288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, Yang L, Irie F, Yamaguchi Y, Lipton SA, Reed JC. Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury. Brain Res. 2011;1370:227–237. doi: 10.1016/j.brainres.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology. 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- Lai KO, Zhao Y, Ch'ng TH, Martin KC. Importin-mediated retrograde transport of CREB2 from distal processes to the nucleus in neurons. Proc Natl Acad Sci U S A. 2008;105:17175–17180. doi: 10.1073/pnas.0803906105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, McKinsey DM, Pike BR, Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [DOI] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, Wang KK. Unfolded protein response after neurotrauma. J Neurotrauma. 2006;23:807–829. doi: 10.1089/neu.2006.23.807. [DOI] [PubMed] [Google Scholar]
- Lenart B, Kintner DB, Shull GE, Sun D. Na-K-cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004;24:9585–9597. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kintner DB, Begum G, Algharabli J, Cengiz P, Shull GE, Liu XJ, Sun D. Endoplasmic reticulum Ca2+ signaling and mitochondrial Cyt c release in astrocytes following oxygen and glucose deprivation. J Neurochem. 2010;114:1436–1446. doi: 10.1111/j.1471-4159.2010.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–43817. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- Mills JD, Bailes JE, Sedney CL, Hutchins H, Sears B. Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. J Neurosurg. 2011;114:77–84. doi: 10.3171/2010.5.JNS08914. [DOI] [PubMed] [Google Scholar]
- Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci U S A. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan HC, Kao TK, Ou YC, Yang DY, Yen YJ, Wang CC, Chuang YH, Liao SL, Raung SL, Wu CW, Chiang AN, Chen CJ. Protective effect of docosahexaenoic acid against brain injury in ischemic rats. J Nutr Biochem. 2009;20:715–725. doi: 10.1016/j.jnutbio.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Paschen W. Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003;34:365–383. doi: 10.1016/S0143-4160(03)00139-8. [DOI] [PubMed] [Google Scholar]
- Paschen W, Doutheil J. Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J Cereb Blood Flow Metab. 1999;19:1–18. doi: 10.1097/00004647-199901000-00001. [DOI] [PubMed] [Google Scholar]
- Petraglia AL, Winkler EA, Bailes JE. Stuck at the bench: potential natural neuroprotective compounds for concussion. Surg Neurol Int. 2011;2:146. doi: 10.4103/2152-7806.85987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/S0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2011;108:2939–2944. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sande A, West C. Traumatic brain injury: a review of pathophysiology and management. J Vet Emerg Crit Care (San Antonio) 2010;20:177–190. doi: 10.1111/j.1476-4431.2010.00527.x. [DOI] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Yan HQ, Fellows-Mayle W, Dixon CE. Resveratrol attenuates behavioral impairments and reduces cortical and hippocampal loss in a rat controlled cortical impact model of traumatic brain injury. J Neurotrauma. 2010;27:1091–1099. doi: 10.1089/neu.2010.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med. 2003;4:59–72. doi: 10.1385/NMM:4:1-2:59. [DOI] [PubMed] [Google Scholar]
- Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na(+)-K(+)-Cl(−) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002;282:C1147–C1160. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- Sun X, Liu J, Crary JF, Malagelada C, Sulzer D, Greene LA, Levy OA. ATF4 protects against neuronal death in cellular Parkinson's disease models by maintaining levels of parkin. J Neurosci. 2013;33:2398–2407. doi: 10.1523/JNEUROSCI.2292-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorell WE, Leibrock LG, Agrawal SK. Role of RyRs and IP3 receptors after traumatic injury to spinal cord white matter. J Neurotrauma. 2002;19:335–342. doi: 10.1089/089771502753594909. [DOI] [PubMed] [Google Scholar]
- Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, Nakamura M, Saatman K, Talbot K, Horiguchi T, McIntosh T, Lee VM, Trojanowski JQ. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184:214–224. doi: 10.1016/S0014-4886(03)00245-0. [DOI] [PubMed] [Google Scholar]
- Wessa P. Free Statistics Software, Office for Research Development and Education, Version 1. 2014. 1.23-r7. http://www.wessa.net/
- Wu A, Ying Z, Gomez-Pinilla F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J Neurotrauma. 2007;24:1587–1595. doi: 10.1089/neu.2007.0313. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J Neurotrauma. 2011;28:2113–2122. doi: 10.1089/neu.2011.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Rasmussen IA, Lagopoulos J, Håberg A. Diffuse axonal injury in severe traumatic brain injury visualized using high-resolution diffusion tensor imaging. J Neurotrauma. 2007;24:753–765. doi: 10.1089/neu.2006.0208. [DOI] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, Iadecola C, Mark AL, Davisson RL. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest. 2012;122:3960–3964. doi: 10.1172/JCI64583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Fenik P, Zhan G, Sanfillipo-Cohn B, Naidoo N, Veasey SC. Eif-2a protects brainstem motoneurons in a murine model of sleep apnea. J Neurosci. 2008;28:2168–2178. doi: 10.1523/JNEUROSCI.5232-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]