

Abstract
Controlling neural stem and progenitor cell (NSPC) proliferation is critical to maintain neurogenesis in the mammalian brain throughout life. However, it remains poorly understood how niche-derived cues such as β1-integrin-mediated signaling are translated into NSPC-intrinsic molecular changes to regulate NSPC activity. Here we show that genetic deletion of integrin-linked kinase (ILK) increases NSPC proliferation through PINCH1/2-dependent enhancement of c-Jun N-terminal protein kinase activity in both neurogenic regions of the adult mouse brain. This effect downstream of ILK signaling is mediated through loss of Ras suppressor unit-1 (RSU-1), as virus-based reconstitution of RSU-1 expression rescued the ILK-dependent effects on NSPC proliferation. Thus, we here identified an intracellular signaling cascade linking extrinsic integrin-mediated signaling to NSPC proliferation and characterized a novel mechanism that regulates NSPC activity in the adult mammalian brain.
Keywords: hippocampus, integrin, neurogenesis, niche, proliferation
Introduction
Somatic stem cells are undifferentiated cells that maintain the ability to self-renew and to give rise to a multitude of specialized cells within their tissue of origin. Neural stem and progenitor cells (NSPCs) persist in the subventricular zone (SVZ) lining the lateral ventricles and in the hippocampal subgranular zone (SGZ) of the dentate gyrus (DG) in the adult mammalian brain (Gage, 2000; Alvarez-Buylla and Lim, 2004). NSPCs generate new neurons throughout life that contribute to tissue homeostasis and play pivotal roles in some forms of adult brain function (Ming and Song, 2011; Sahay et al., 2011). Understanding the cellular and molecular mechanisms underlying neurogenesis is a prerequisite for a potential therapeutic targeting of neurogenesis in regenerative medicine (Zhao et al., 2008).
Previous data showed that control of NSPC proliferation is critical to prevent premature exhaustion of the stem cell pool and to ensure life-long neurogenesis. Key signaling cascades controlling NSPC proliferation include, among others, Notch, BMP, and Shh signaling (Lai et al., 2003; Ahn and Joyner, 2005; Colak et al., 2008; Ables et al., 2010; Ehm et al., 2010; Lugert et al., 2010; Mira et al., 2010). Furthermore, cellular mechanisms such as regulation of cell cycle duration and control of the metabolic state determine the rate of NSPC proliferation (Lange et al., 2009; Knobloch et al., 2013).
Similar to other somatic stem cell systems, it is established that specialized microenvironments surrounding NSPCs, hereafter called neurogenic niches, are key mediators of extrinsic cues signaling onto NSPCs to control their activity or to enable differentiation (Alvarez-Buylla and Lim, 2004). Neurogenic niches provide soluble factors such as Wnt3 that are indispensable for proper neuronal differentiation and neurogenesis (Lie et al., 2005; Karalay et al., 2011). Furthermore, the niche exerts key functions on NSPCs by mediating interactions with other cells residing in the NSPC microenvironment such as astrocytes, and endothelial, ependymal, and immune cells (Song et al., 2002; Ziv et al., 2006; Rolls et al., 2007; Mirzadeh et al., 2008; Shen et al., 2008; Fuentealba et al., 2012; Kokovay et al., 2012). In addition, the niche controls NSPC behavior through interactions with the extracellular matrix (ECM; Garcion et al., 2004). Laminins, major proteins in the basal lamina, interact with other ECM components, and previous reports have shown that the most abundant laminin receptor in the adult brain, β1-integrin, is critically involved in neurogenesis in the adult SVZ (Belvindrah et al., 2007; Kazanis et al., 2010). Canonical β1-integrin signaling is mediated through the IPP complex, named after its main constituents integrin-linked kinase (ILK), PINCH1/2, and Parvin (Wickström et al., 2010). However, the intracellular cascades mediating the effects of integrin signaling on adult NSPCs remain unknown.
Here we show that NSPC-selective loss of ILK (or PINCH1/2) in the SVZ and SGZ enhances NSPC proliferation by increasing c-Jun N-terminal protein kinase (JNK) activity. In addition, we provide evidence that enhanced JNK activity is due to the loss of Ras suppressor unit-1 (RSU-1) upon ILK-deletion. Thus, we have identified a novel mechanism explaining how niche-derived signals are translated into NSPC behavior.
Materials and Methods
Animals.
All animal experiments were approved by the veterinary office of the Canton of Zurich, Switzerland. Mice of either sex were kept with littermates under a 12 h dark/light cycle in single ventilated cages and with ad libitum access to food and water. hGFAP-GFP mice and Nestin-GFP mice (Yamaguchi et al., 2000; Beckervordersandforth et al., 2010) have been described previously. To generate conditional ILK or Pinch1/2 knock-out mice, ILKflox/flox or Pinch1/2flox/flox mice (Grashoff et al., 2003; Li et al., 2005; Stanchi et al., 2005) were crossed with Nestin-CreERT2-R26YFP mice (generated on a C57BL/6 background; Lagace et al., 2007). Cre-mediated recombination was induced by five consecutive intraperitoneal injections of tamoxifen (TAM) dissolved in sunflower oil (180 mg/kg; both reagents were purchased from Sigma) at the age of 6 weeks. Heterozygous littermates were used as controls. In the absence of TAM, no recombined yellow fluorescent protein (YFP)-expressing cells were detected. For measuring proliferation in vivo, the 10 d post-TAM animals were administered 5-ethynyl-2′-deoxyuridine (EdU) at a dosage of 50 mg/kg twice daily for 3 d. To label the transit-amplifying population derived from recombined stem cells, mice received injections of 5-iodo-2′-deoxyuridine (IdU; 50 mg/kg) every 2 h for 10 h before being killed. For immunohistochemical analyses, mice were transcardially perfused 30 d after the last EdU injection with 0.9% sterile NaCl, followed by 4% paraformaldehyde (PFA)/0.1 m phosphate buffer, pH 7.4. Brains were post-fixed in 4% PFA/0.1 m phosphate buffer overnight at 4°C followed by 30% sucrose/0.1 m phosphate buffer and stored at 4°C. Brains were cut into 40-μm-thick free-floating sections as described previously (Bracko et al., 2012).
Immunohistochemistry/immunocytochemistry.
Immunohistochemistry was performed on 40-μm-thick free-floating serial sections as described previously (Knobloch et al., 2013). The following primary antibodies were used: rabbit α-GFP (green fluorescent protein; 1:500; Invitrogen), chicken α-GFP (1:500; Aves Labs), mouse α-NeuN (neuronal-specific nuclear protein; 1:100; Millipore), goat α-Sox2 (1:500; Santa Cruz Biotechnology), rabbit α-GFAP (1:500; Dako), mouse α-GFAP (1:500; Sigma), goat α-doublecortin (DCX; 1:250; Santa Cruz Biotechnology), mouse α-Mash1 (1:500; BD PharMingen), rabbit α-ILK (1:50; Abbiotec), mouse α-ILK (1:50; BD Bioscience), rabbit α-Pinch (1:100; Abcam), and mouse α-IdU/BrdU (1:50; BD Bioscience). EdU visualization was performed using the Click-iT EdU Imaging Kit (Invitrogen) according to the manufacturer's protocol. Secondary antibodies (The Jackson Laboratory) were applied 1:500 at room temperature for 1–2 h. To detect apoptotic nuclei, we performed a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) reaction according to the manufacturer's instructions (Roche). Positive cells were analyzed 43 d after recombination using double staining with YFP. To phenotype cells for ILK and PINCH1/2 expression, we analyzed ∼500 NestinGFP-positive cells (including radial glia-like and nonradial NSPCs, but excluding pericytes and ependymal cells) for coexpression with ILK and PINCH1/2. One to six series of coronal sections (with each section corresponding to a sixth of the entire brain) through the brain were used for all quantifications. Areas in the DG and SVZ were measured using ImageJ. Cell nuclei were counterstained with 4-6-diamidino-2-phenylindole (DAPI; 1:5000; Sigma). Immunocytochemistry was performed as described above on cells fixed with 4% PFA for 15 min. The following antibodies were used: goat α-Sox2 (1:500; Santa Cruz Biotechnology), chicken α-GFP (1:500; Aves Labs), rabbit α-ILK (1:50; Abbiotec), rabbit α-Pinch (1:100; Abcam), and rabbit α-DsRed (1:1000; Clontech). Neurosphere staining was performed as previously described (Beckervordersandforth et al., 2010). The following antibodies were used: rabbit α-ILK (1:100; Abbiotec) and rabbit α-Pinch (1:100; Abcam). Floating whole-mount SVZs were dissected from hGFAP-GFP mice (kindly provided by Dr. M. Götz, Helmholtz Zentrum, Munich, Germany; Beckervordersandforth et al., 2010) and Nestin-GFP mice (Yamaguchi et al., 2000). Whole-mount staining was performed as described previously (Mirzadeh et al., 2008). Briefly, SVZ whole mounts were stained after 15 min of fixation in 4% PFA followed by 30 min incubation of 0.5% Triton in PBS. The following primary antibodies were used: mouse α-β catenin (1:500; Millipore), chicken α-GFP (1:500; Aves Labs), rabbit α-ILK (1:50; Abbiotec), and mouse α-Pinch (1:100, Abcam).
Image analysis.
For colocalization experiments and cell quantifications, confocal microscopy (AOBS-SP2 microscope, Leica) was performed followed by analysis, 3D reconstructions, and colocalization studies with the software Imaris (Bitplane). Cells were counted on single stacks for a total of four stacks per slice (one stack every 10 μm), carefully excluding double counting. Cells and neurosphere staining images were acquired using an epifluorescent microscope (Axiovert Observer, Zeiss). Images were analyzed using ImageJ and Photoshop.
Cell culture experiments.
Mouse NSPCs were isolated from 6-week-old adult control mice and ILKflox/flox or Pinch1/2flox/flox mice using the gentleMACS Dissociator and the MACS Neural Tissue Papain Dissociation Kit (Miltenyi Biotec) according to the manufacturer's protocols. Single-cell suspensions were cultured as neurospheres in DMEM/F12 medium supplemented with B27 (Invitrogen), human EGF (20 ng/ml), and human basic FGF-2 (20 ng/ml, Peprotech), containing an antibiotic/antimycotic agent (Anti-Anti, Invitrogen). Medium was changed every 2–3 d. After a few passages, cells were either expanded further as neurospheres or cultured as monolayers in DMEM/F12 medium supplemented with N2 (Invitrogen) and heparin (5 mg/ml) in addition to EGF and FGF. Infecting monolayers with a Cre-expressing lentivirus induced in vitro recombination. For all of the following experiments, cells were used 10 d postinfection to ensure complete protein loss. For neurosphere assays, cells were plated in a single-cell dilution into 96-well plates, and sphere size was assessed 10 d after seeding using bright-field and fluorescent microscopy Axiovert Observer D1 (Zeiss). Cumulative analysis was performed comparing 400 spheres per sample in triplicate. Statistical analysis and histograms of frequency distribution were generated using Prism 6 GraphPad software. Critical D values for Kolmogorov–Smirnov (K-S) analysis were calculated on an Excel Worksheet and compared with K-S table values for the assessment of statistical significance. For proliferation assays, cells cultured in monolayers were treated with an EdU pulse (10 μm, Sigma) for 1 h at 37°C followed by PFA fixation.
Pharmacological inhibition of MEK (U0126) and JNKs (SP600125) was performed following the manufacturer's instructions (Santa Cruz Biotechnology and Sigma). Stock solutions were prepared in DMSO and used at a final concentration of 1 or 10 μm. U0126 and SP600125 were added to the medium 24 h before proliferation assessment.
To test for an effect of RSU-1 overexpression, cells with ILK deletion and control cells were transduced with retrovirus expressing a CAG-mCherryRSU-1 fusion protein cassette or a CAG-mCherry as a control. Four days postinfection, mCherry-positive cells were selected via FACS sorting (FACSAria IIIu cell sorter, BD Biosciences), and a proliferation assay was performed as described above. For interference experiments, control cells were infected with a lentivirus expressing RSU1-specific shRNA or nontargeting shRNA (Mission shRNA, Sigma). EdU administration and staining were performed as described above 4 d after treatment.
Virus production.
Lentiviral and retroviral particles were produced as previously described (Jessberger et al., 2008; Bracko et al., 2012). Human embryonic kidney 293T cells were transfected with viral constructs and packaging plasmids using Lipofectamine 2000 (Invitrogen). Viral titers ranged from 106 to 109 colony-forming units/ml. In vitro recombination was induced in cells derived from control or experimental animals infected with Cre-expressing lentivirus. Lentivirus-expressing RSU-1-specific shRNA was produced according to the manufacturer's instructions (Mission shRNA; Sigma).
Plasmids.
For in vitro recombination, lentivirus constructs expressing PGK-CreGFP were cloned as described previously (Jessberger et al., 2008). For the overexpression of RSU-1, total RNA was extracted and cDNA retrotranscribed according to manufacturer instructions (Invitrogen). PCR was performed for amplification of RSU-1 and cloned in pGEM vector (Promega). RSU-1 cDNA was subcloned in frame with mCherry cassette in a CAG-mCherry retroviral construct. CAG-mCherry retroviral construct was used as a control virus (Knobloch et al., 2013). All constructs were sequenced to exclude point mutations or frameshifts.
Western blotting.
For Western blot analyses, cells were lysed in RIPA buffer with protease inhibitor (Roche) and phosphatase inhibitor (Cocktail 2/3, Sigma). Membranes were blocked with 5% BSA in TBS-T. Antibodies used were mouse α-GAPDH (1:10′000; HyTest), mouse α-ILK (1:500; BD Biosciences), rabbit α-ILK (1:500; Abbiotec), rabbit α-RSU-1 (1:500; Abcam), rabbit α-Pinch (1:5000; kindly provided by Dr. Reinhard Fässler, Max Planck Institute of Biochemistry, Martinsried, Germany), mouse α-Jun (1:1000; Cell Signaling Technology), rabbit α-phospho-Jun S73 (1:1000; Cell Signaling Technology), rabbit α-ERK1/2 (extracellular signal-regulated protein kinase 1/2; 1:1000; Cell Signaling Technology), mouse α-phospho-ERK1/2 (1:1000; Cell Signaling Technology), rabbit α-AKT (1:1000; Cell Signaling Technology), mouse α-phospho AKT S473 (1:1000; Cell Signaling Technology), mouse α-GSK3β (1:1000; Millipore), and α-phospho-GSK3β S9 (1:1000; Cell Signaling Technology). HRP- or AP-conjugated secondary antibodies were used, and bands were detected by an enhanced chemiluminescence substrate (Thermo Scientific and Roche). Quantifications were made using a Bio-Rad GS800 densitometer.
Statistical analysis.
All statistical analyses were performed with Excel using two-tailed unpaired t tests or K-S tests when appropriate. Significance levels were set at p < 0.05. Group sizes for all experimental groups were n ≥ 3.
Results
ILK is expressed in NSPCs of the SVZ and DG of the adult brain
We first analyzed the expression pattern of ILK in the two major neurogenic niches of the adult mammalian brain, the SVZ lining the lateral ventricles and the DG of the hippocampal formation. Using a human glial fibrillary acidic protein (hGFAP)-driven GFP-expressing reporter mouse line (hGFAP-GFP) that visualizes NSPCs in the adult SVZ, we found expression of ILK protein in radial glia-like NSPCs (also called type B cells). These cells contribute to the formation of characteristic pinwheel structures in the SVZ when coanalyzed with the expression of the scaffolding protein β-catenin (Fig. 1A; Mirzadeh et al., 2008; Beckervordersandforth et al., 2010). Confirming the expression of ILK in type B cells, we found colabeling of endogenous GFAP and Sry-related high-mobility group box transcription factor 2 (SOX2) with ILK in the SVZ (Fig. 1A; Doetsch et al., 1999). Similar results were obtained using Nestin-specific antibodies (Fig. 1A). In contrast, we did not find ILK expression in transit-amplifying cells (type C cells, as visualized using antibodies against Mash1) or early neuroblasts (type A cells), marked by the expression of DCX (Doetsch et al., 1999). Furthermore, no expression of ILK was found in neurons expressing NeuN (also known as RNA binding protein Fox1 homolog 3; Mullen et al., 1992; Fig. 1E).
Figure 1.

ILK is expressed in the neurogenic niches of the adult brain. A, ILK (red) is present in the adult SVZ in radial glia-like, type B cells identified by the expression of hGFAP-GFP (green) as part of a characteristic pinwheel-like structure that can be visualized using β-catenin staining (β-cat; blue). Endogenous Nestin (green, left bottom) and GFAP/SOX2 (green/blue, right bottom) expression colabel with ILK (red) in the adult SVZ. Nuclei were counterstained with DAPI (gray), if indicated. B, ILK (red) is expressed in the adult DG in radial glia-like, type 1 NSPCs, visualized by the expression of Nestin-driven GFP (green). ILK (red) colabels with endogenous Nestin (green, left bottom) and GFAP (green, right bottom) expressing NSPCs that also colabel with SOX2 (blue). Nuclei were counterstained with DAPI (gray) if indicated. C, ILK (green) is expressed in neurospheres and monolayer cultures of NSPCs derived from the adult SVZ. Top, Example of a neurosphere. Bottom, A single cell expressing ILK. Nuclei were counterstained with DAPI (gray). D, Western blot analysis for ILK of protein lysates isolated from SVZ- and DG-derived NSPCs confirms ILK expression. E, F, ILK (red) is not coexpressed at detectable levels in DCX- (green, left), Mash1- (green, middle), or NeuN- (green, right) expressing cells in the SVZ (left panels) and the DG (right panels). Nuclei were counterstained with DAPI (gray). LV, Lateral ventricle; GCL, granule cell layer; ML, molecular layer. Scale bars, 10 μm.
In analogy to the ILK expression pattern in the SVZ, we found the expression of ILK in radial glia-like NSPCs (also called type 1 cells) within the SGZ of the DG using a Nestin-driven GFP reporter mouse line (Nestin-GFP), endogenous Nestin expression, or GFAP and SOX2 expression (Fig. 1B). More than 98% of cells labeled with Nestin-GFP in the SVZ and DG coexpressed ILK. Again, we found no expression of ILK in Mash1- or DCX-positive neuroblasts or NeuN-expressing neurons (Fig. 1F).
Reflecting the in vivo expression pattern of ILK, we detected substantial amounts of ILK protein in cultured NSPCs grown as neurospheres or monolayers derived from the adult SVZ and DG (Fig. 1C,D).
ILK controls NSPC proliferation in the two neurogenic regions of the adult brain
After characterizing the ILK expression patterns in the SVZ and DG, we next sought to test for a functional contribution of ILK in NSPC behavior. We selectively deleted ILK in adult NSPCs by crossing ILKflox/flox mice with mice harboring TAM-inducible, Nestin-driven Cre recombinase and Cre-dependent ROSA YFP reporter alleles (hereafter called ILK-cKO; Lagace et al., 2007; Fig. 2A). TAM was injected daily for 5 consecutive days, followed by 3 d of EdU injections 10 d after the last TAM injection to label proliferating NSPCs. To label YFP-expressing, proliferating NSPCs, mice received injections of the thymidine analog IdU every 2 h in the 10 h before being killed. Recombined YFP-expressing cells were analyzed 43 d after the last TAM injection (i.e., 30 d after last EdU injection). Costaining for ILK and YFP confirmed selective deletion of ILK on recombined NSPCs in SVZ (Fig. 2B, left) and DG (Fig. 2B, right). We first analyzed the total number of YFP-expressing cells in the SVZ and DG in ILK-cKO mice compared with control mice. However, we did not find differences between conditional mutants and controls regarding the total number or relative density of YFP-labeled cells in the SVZ or DG (Fig. 2C,E). In contrast, the numbers of SOX2/GFAP/YFP-positive cells were increased after conditional ILK deletion in both the SVZ (Fig. 2F) and the DG (Fig. 2H), suggesting that the loss of ILK leads to enhanced activity of NSPCs in the adult brain. Thus, we analyzed cell death and intermediate steps of differentiation from the dividing NSPC to the mature newborn neuron. There was no difference in cell death between ILK-cKO and control mice as measured in the SVZ using TUNEL staining (control mice, 72.2 ± 20.3 cells/mm2; mutant mice, 60.9 ± 19 cells/mm2). We also did not observe changes in the number of newly generated YFP-labeled astroglia in the SVZ and DG (data not shown). Next, we quantified DCX- and NeuN-expressing cells derived from recombined NSPCs, and identified a decrease in the percentage of immature neuroblasts (DCX+NeuN−) that may compensate for the observed increase in ILK-cKO NSPCs of the SVZ (Fig. 2F) and DG (Fig. 2H). Consistent with this, the total number of newly generated neurons (NeuN+) appeared unchanged in both the olfactory bulb (OB; Fig. 2G) and DG (Fig. 2H). In addition, we found increased numbers of YFP-expressing, EdU-labeled cells that were also positive for SOX2 in mice lacking ILK compared with controls (Doetsch et al., 1999; Suh et al., 2007; Fig. 2I,K). We also found an increase in the number of IdU-labeled cells, labeling the fast-cycling population of NSPC-derived progeny (Fig. 2J,L), which was also reflected by an increase in the number of MASH1-expressing transit-amplifying cells (SVZ: control, 8.7 ± 0.7%; ILK-cKO, 14.5 ± 0.7%; p < 0.01; DG: control, 4.0 ± 0.1%; ILK-cKO, 9.3 ± 0.3%; p < 0.001). This finding closely resembled the phenotype observed after the deletion of β1-integrin in the adult stem cell compartment (Shen et al., 2008; Kazanis et al., 2010) and supported our hypothesis that integrin-dependent signaling in adult NSPCs is mediated by ILK-associated signaling events.
Figure 2.
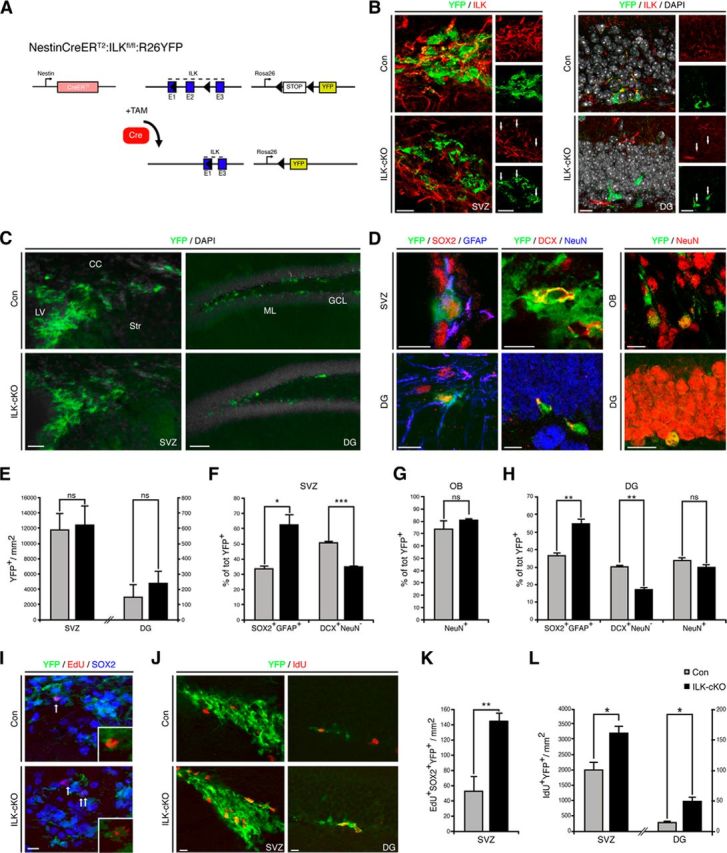
Deletion of ILK enhances NSPC proliferation in the SVZ and DG. A, Schematic illustration of the genetic deletion of ILK using Nestin-CreERT2 mice combined with ROSA-YFP reporter mice. B, Selective ILK ablation on recombined YFP-expressing NSPCs in SVZ (left panels) and DG (right panels). C, Conditional deletion of ILK does not alter the number of YFP-labeled, recombined cells (green) in the SVZ or DG. Shown are representative images of YFP-labeled cells in the SVZ (left panels) and the DG (right panels) 43 d after last TAM injection. D, Representative images of recombined cells expressing YFP (green), SOX2 (red), and GFAP (blue) in SVZ (left, top) and DG (left, bottom). Representative pictures of recombined cells expressing YFP (green) and DCX (red) but not NeuN (blue) markers in SVZ (middle, top) and DG (middle, bottom). Representative pictures of YFP-positive (green) recombined cells expressing the neuronal marker NeuN (red) in the OB (right, top) and DG (right, bottom). E, Quantification of YFP-positive cells per area in the SVZ and DG. F, H, Genetic deletion of ILK increases the fraction of YFP-labeled cells expressing SOX2 and GFAP but decreases the number of DCX-positive neuroblasts in the adult SVZ (F) and adult DG (H). G, H, The number of YFP-expressing NeuN+ neurons is unchanged in both OB (G) and DG (H). I, Representative pictures of recombined (green) EdU-long retaining cells (red) coexpressing SOX2 (blue) in the SVZ of control and ILK-mutants. J, Representative pictures of recombined (green) IdU fast-cycling cells (red) in the SVZ (left panels) and DG (right panels) of control and ILK mutants. K, Quantification of long retaining EdU/SOX2/YFP-positive cells per area, indicating enhanced proliferation of SOX2-expressing cells in the SVZ. L, Quantification of IdU+YFP+ cells in the two neurogenic regions of the adult brain. The number of fast-cycling IdU+ cells is increased upon ILK ablation. LV, Lateral ventricle; CC, corpus callosum; Str, striatum; GCL, granule cell layer; ML, molecular layer. Data are presented as the mean ± SEM. Scale bars: C, left panels, 25 μm; C, right panels, 80 μm; B, D, I, J, 10 μm. *p < 0.05,**p < 0.01, ***p < 0.001.
ILK regulates proliferation of SVZ- and DG-derived NSPCs in vitro
We next tested whether ILK deletion affects also the proliferation of isolated NSPCs in vitro. NSPCs were isolated from the SVZ or DG of young adult ILKflox/flox mice, and, after the initial propagation, cells were transduced with Cre-expressing lentiviruses to delete ILK (Fig. 3A). When using an EdU pulse to label cells in S-phase, we found a robust increase in proliferating cells after ILK deletion in NSPCs growing as monolayers (Fig. 3B). We confirmed this phenotype by analyzing the size of neurospheres derived from ILK-deleted NSPCs and control cells, indicative of cell proliferation, and found a significant shift toward larger spheres generated from ILK-deficient cells (Fig. 3C). Together, these findings indicate that ILK-mediated signaling negatively regulates NSPC proliferation both in vivo and in vitro in NSPCs from the SVZ and DG.
Figure 3.

Genetic deletion of ILK enhances NSPC proliferation in vitro. A, Virus-mediated Cre expression leads to the loss of ILK protein in SVZ- and DG-derived NSPCs isolated from control and mutant mice. Shown are representative Western blots. B, Loss of ILK enhances proliferation in monolayer NSPC cultures derived from the adult SVZ and DG as analyzed by EdU pulse labeling (red). Left bars show quantification, right panels show representative images from cells isolated from the adult SVZ. C, Deletion of ILK increases the size of neurospheres derived from SVZ and DG. Left images show representative bright-field and fluorescent images (showing YFP expression) of a control neurosphere (left panels) and an ILK-deficient neurosphere (right panels). Right graphs show quantification for the adult DG and SVZ. Data are presented as the mean ± SEM. Scale bars, 20 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
PINCH1/2 deletion phenocopies loss of ILK in vivo and in vitro
After identifying that ILK controls cell proliferation in the neurogenic niches of the adult brain, we next tested whether ILK signaling depends on the canonical IPP complex function. We focused on PINCH1/2 proteins that constitute, together with ILK and Parvin, the IPP complex (Wickström et al., 2010). Using an immunohistochemical approach, we found expression of PINCH1/2 in adult NSPCs in the SVZ and DG, closely resembling the expression pattern of ILK (Fig. 4A–C). Remarkably, conditional double knockout of PINCH1 and PINCH2, again using the inducible Nestin-driven Cre mouse (hereafter called PINCH1/2-cKO), phenocopied the effects of ILK deletion, and resulted in enhanced cell proliferation of NSPCs in the SVZs and DGs of adult mice (Fig. 4D,F). Supporting these findings, we found that genetic deletion of PINCH1/2 in NSPCs (using virus-based Cre expression in cells isolated from PINCH1/2flox/flox mice) robustly enhanced cell proliferation in both monolayer cultures and neurospheres of both SVZ- and DG-derived NSPCs (Fig. 5D,E). Interestingly, the loss of PINCH1/2 led to a strong reduction in ILK levels, most probably due to destabilization of the IPP complex (Legate et al., 2006; Wickström et al., 2010; Fig. 5B). Together, these data suggest that the integrity of the IPP complex is critical in controlling cell proliferation of adult NSPCs.
Figure 4.

PINCH1/2 regulate NSPC proliferation within the adult brain. A, In the adult SVZ, PINCH1/2 (red) is present in radial glia-like, type B cells identified by the expression of Nestin-GFP (green). Left panel shows an overlay, and small panels show single channels for Nestin-GFP and PINCH1/2. B, PINCH1/2 (red) are coexpressed in adult radial glia-like NSPCs expressing Nestin-GFP (green) in the adult DG. Left panel shows an overlay, and small panels display single channels for Nestin-GFP and PINCH1/2. C, ILK (green) and PINCH1/2 (red) are coexpressed in adult NSPCs. Left panel shows an overlay of ILK and PINCH1/2 signals, and small panels show single channels for ILK and PINCH1/2. Nuclei were counterstained with DAPI (gray). D, E, In analogy to conditional ILK deletion, the genetic loss of PINCH1/2 enhances the fraction of YFP-labeled cells (green) expressing SOX2 (red) and GFAP (blue) in the adult SVZ and adult DG. Shown are overlays and individual channels (D). Quantifications of recombined YFP-labeled cells expressing SOX2 and GFAP show the enhanced generation of putative NSPCs in the adult SVZ and DG upon PINCH1/2 deletion (E). F, Quantification of EdU-labeled, SOX2-expressing recombined cells per area in the SVZ after conditionally deleting PINCH1/2 using NestinCreERT2-mediated Cre expression. LV, Lateral ventricle; GCL, granule cell layer. Data are presented as the mean ± SEM. Scale bars, 10 μm. *p < 0.05, **p < 0.01.
Figure 5.

PINCH1/2 is involved in proliferation control of NSPCs cultured in vitro. A, PINCH1/2 (red) is expressed in SVZ-derived neurospheres (left panel) and adherent NSPC cultures (right panel). Nuclei were counterstained with DAPI (gray). B, Virus-based Cre expression leads to the loss of PINCH1/2 in NSPCs isolated from PINCH1/2flox/flox mice. Furthermore, loss of PINCH1/2 reduces the expression of ILK, presumably due to destabilization of the IPP complex. C, Genetic deletion of PINCH1/2 enhances the proliferation of monolayer cultures derived from the adult SVZ as measured by EdU (red) pulse labeling of YFP-labeled cells (green). D, Quantification of NSPC proliferation upon PINCH1/2 loss in cells derived from the adult SVZ and DG. E, Loss of PINCH1/2 leads to an increased size of neurospheres derived from the adult SVZ and DG, a finding comparable to the proliferation phenotype observed after conditional deletion of ILK in adult NSPCs. Data are presented as the mean ± SEM. Scale bars, 20 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
Enhanced proliferation after loss of IPP complex is mediated by increased JNK activity
We next sought to analyze how destabilization of the IPP complex alters intracellular signaling activity. Thus, we characterized the phosphorylation status of several protein kinases that had been previously implicated in canonical and noncanonical IPP-mediated signaling. Whereas there were no differences in phosphorylation levels of AKT and GSK3β after deletion of ILK, we found significantly increased phosphorylation ratios for ERK1/2 (also known as MAP-kinase 42 and 44) and c-Jun (Fig. 6A,B). To test for the functional significance of ERK and/or JNK signaling, we used selective inhibitors to block ERK (U0126) or JNK (SP600125) activity. Inhibition of ERK activity significantly reduced the proliferation of ILK-deficient NSPCs but also reduced the proliferation of control SVZ-derived NSPCs, suggesting that ERK is important for proper NSPC proliferation but is unlikely to mediate the increased proliferation phenotype after ILK deletion (Fig. 6C; Samuels et al., 2008; Pan et al., 2012; Zou et al., 2012). In striking contrast, we found that the inhibition of JNK activity selectively reduced the proliferation of ILK-deficient cells to baseline levels but had no inhibitory effect on cell growth in control cells (Fig. 6C). These findings strongly suggest that enhanced NSPC proliferation is at least partially mediated through increased JNK activity following the deletion of ILK.
Figure 6.

Increased JNK activity mediates enhanced proliferation of NSPCs upon loss of ILK. A, Genetic deletion of ILK enhances the activity of ERK and JNK pathways. Shown are representative Western blots of protein isolated from ILK-deficient or control SVZ-derived NSPCs. B, Quantification of kinase activities upon genetic deletion of ILK. C, Inhibition of ERK activity using U0126 leads to an overall reduction of proliferation in SVZ-derived, ILK-deficient, and control NSPCs, suggesting that enhanced ERK activity is not responsible for the increase in cell proliferation upon ILK deletion. In striking contrast, blocking JNK activity using SP600125 selectively normalizes the enhanced proliferation after ILK loss without affecting baseline proliferation of cultured NSPCs, indicating that enhanced JNK activity is at least partially mediating enhanced cell division after genetic deletion of ILK. D, Loss of ILK leads to a strong reduction of RSU-1 expression. Left panels show representative Western blots of protein isolated from ILK-deficient cells, which were isolated from the SVZ and DG, respectively. Right graphs show quantification. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Restoration of RSU-1 expression rescues enhanced proliferation after ILK deletion
After showing that an IPP–JNK-dependent pathway regulates cell proliferation of adult NSPCs, we next aimed to identify the mechanism underlying increased JNK activity after destabilization of the IPP complex. Previous reports showed that RSU-1, a suppressor of Ras activity, interacts through PINCH1 with the IPP complex (Dougherty et al., 2005, 2008). Furthermore, RSU-1 can regulate JNK activity in Drosophila melanogaster (Kadrmas et al., 2004). Notably, we found a dramatic reduction of RSU-1 protein levels after the deletion of ILK, suggestive of a role for RSU-1 in mediating the effects of the IPP complex on cell proliferation (Fig. 6D). Thus, we next tested whether reconstitution of RSU-1 expression (using retrovirus-mediated expression of an mCherry–RSU-1 fusion protein) is capable of rescuing the ILK-dependent phenotype and to normalize cell proliferation after ILK deletion. Strikingly, retrovirus-mediated expression of RSU-1 in SVZ- and DG-derived NSPCs specifically reduced proliferation of ILK-deficient NSPCs and fully rescued the enhanced proliferation phenotype due to ILK deletion (Fig. 7A,B). In contrast, the overexpression of RSU-1 in control cells did not affect cell proliferation as measured by EdU pulse labeling, indicating that RSU-1 is selectively involved in IPP-mediated control of NSPC proliferation (Fig. 7B). Protein analysis confirmed that virus-mediated overexpression of RSU-1 restored Jun phosphorylation to control levels (Fig. 7C,D). This hypothesis was further supported by our finding that virus-based, shRNA-mediated knockdown of RSU-1 phenocopied the effects of ILK and PINCH1/2 deletion and enhanced the proliferation of cultured NSPCs (Fig. 7E,F). Thus, we here characterized a novel intracellular signaling cascade in the context of adult NSPC biology: the IPP complex controls cell proliferation through RSU-1-mediated regulation of JNK activity.
Figure 7.

Restoring RSU-1 expression rescues enhanced NSPC proliferation upon ILK deletion. A, Retrovirus-based expression of RSU-1 restores RSU-1 levels in ILK-deficient cells. Shown is a representative Western blot from NSPCs transduced with mCherry or mCherry-RSU1-expressing retrovirus. Note the shift of the mCherry–RSU-1 fusion protein compared with the mCherry protein expression. B, RSU-1 overexpression selectively reduces the proliferation of NSPCs isolated from the adult SVZ and DG after conditional deletion of ILK to baseline levels. Right panels show representative images of EdU-labeled (green) ILK-cKO cells transduced with a mCherry-expressing control virus (red, top) or an mCherry-RSU-1-expressing virus (red, bottom). Left bars show quantification. C, Overexpression of RSU-1 restores the basal phosphorylation level of Jun. D, Quantification of phospho-Jun level upon mCherry or mCherry-RSU1-expressing virus infection. E, Virus-mediated expression of shRNAs directed against the mRNA coding for RSU-1 reduces protein levels of RSU-1 compared with nontargeting control shRNA. Shown are representative Western blots. F, shRNA-mediated knockdown of RSU-1 enhances the proliferation of cultured NSPCs, further suggesting that the regulation of NSPC proliferation after genetic deletion of ILK is at least partially controlled through RSU-1. Bars show quantifications. Right panels are representative pictures. Data are presented as the mean ± SEM. Scale bars: A, B, 10 μm; F, 20 μm. *p < 0.05.
Discussion
Several extracellular and intracellular cues regulate the activity of adult NSPCs, their differentiation, and neuronal integration (Zhao et al., 2008). However, how signals from the surrounding matrix or neighboring cells are translated into intracellular signaling cascades within NSPCs is poorly understood. Here we characterized the intracellular signaling mechanisms downstream of integrin-associated signaling and found that genetic deletion of ILK (or PINCH1/2) enhances JNK activity due to the loss of RSU-1, subsequently resulting in enhanced proliferative activity of NSPCs within the two neurogenic regions of the adult mammalian brain.
Previous reports (Shen et al., 2008; Kazanis et al., 2010) showed that laminins (produced by NSPCs, but also by astrocytes), endothelial cells of blood vessels, and ependymal cells in the SVZ are critically involved in controlling adult NSPC proliferation. The β1-integrin receptor, the most abundant receptor for laminin in the adult brain, is highly expressed on the surface of NSPCs, and laminin/β1-integrin signaling has been shown to be crucial for adult neurogenesis (Belvindrah et al., 2007; Loulier et al., 2009). β1-integrin-mediated signaling provides an essential signal to control NSPC homeostasis and maintenance (Shen et al., 2008; Kokovay et al., 2012).
Our data suggest that the IPP complex acts downstream of β1-integrin to regulate NSPC activity. However, the long-term effects of ILK and PINCH1/2 deletion on NSPC homeostasis and activity remain unknown, and need to be addressed in future experiments. It is interesting to note that the increase in NSPC activity seems to be counterbalanced by a reduction in DCX-expressing immature neurons, leading to unchanged numbers of net neurogenesis (at the time points analyzed).
To address how integrin-derived signals are translated into intracellular signaling cascades in NSPCs, we focused on one of the main pathways activated by β1-integrin activation: the IPP complex cascade (Wickström et al., 2010). Once the integrin receptor undergoes the conformational change induced by laminin interaction, several proteins are engaged in the formation of the IPP complex. ILK interacts with the cytoplasmic domain of the β-integrin receptor through its C-terminal region, while also interacting through its N-terminal end with the LIM domain of the PINCH adaptor. The complex is formed in the cytoplasm, and its stability depends on the presence of all single components: if one of these proteins is lacking, the IPP complex is rapidly degraded (Legate et al., 2006). Confirming this, we found that genetic deletion of PINCH1/2 led to a reduction of ILK protein levels. Furthermore, selective conditional genetic deletion of PINCH1/2 in adult NSPCs phenocopied the effects of ILK deletion, which resulted in enhanced proliferative activity of NSPCs both in vitro and in vivo.
Interestingly, we found several kinase signaling pathways to be enhanced after the loss of ILK, including increased levels of ERK and JNK activity that may explain the observed increase in NSPC proliferation. We confirmed previous findings suggesting that the ERK pathway is important for NSPC proliferation and neurogenesis in the developing and adult brain (Samuels et al., 2008; Pan et al., 2012; Zou et al., 2012). However, increased activity of the ERK pathway could not explain the enhanced proliferation phenotype after the loss of ILK. Strikingly, we found that increased activity of JNK was mediating the increase of NSPC proliferation that we observed after genetically deleting ILK. Thus, the loss of ILK has a pro-proliferative effect on NSPCs through its downstream target JNK. Given this finding, the growth-suppressive function under normal conditions that we have identified here for ILK in NSPCs, highly resembles the tumor-suppressor functions of ILK in certain tumors, in contrast to the oncogenic role of ILK in other types of cancerous cells (Durbin et al., 2009).
We next sought to identify the mechanism by which destabilization of the IPP complex leads to enhanced JNK activity. RSU-1, a suppressor of pro-proliferative Ras activity, has been previously described to interact with PINCH1 in the IPP complex (Dougherty et al., 2005, 2008). Notably, we found a strong loss of RSU-1 after ILK deletion in NSPCs. Supporting the hypothesis that RSU-1 activity is required to control levels of JNK activation, we found that virus-mediated restoration of RSU-1 expression in NSPCs fully rescued the ILK loss-of-function phenotype and normalized the levels of proliferation. Thus, RSU-1 exerts in NSPCs a function similar to that of control JNK activity as its previously described role during the development of D. melanogaster (Kadrmas et al., 2004).
Controlling the proliferation of NSPCs is necessary to maintain the life-long capacity of adult NSPCs to generate new neurons (Zhao et al., 2008). The neurogenic niche plays a pivotal role in either activating or reducing the proliferation of NSPCs by providing a number of growth factors such as EGF, FGF, Wnt, and Shh, by direct neurotransmitter input and by receptor-mediated signaling (Gritti et al., 1999; Lai et al., 2003; Lie et al., 2005; Kazanis et al., 2010; Song et al., 2012). The data presented here characterize the intracellular signaling cascade upon activation of β1-integrin receptors on NSPCs and identify an IPP complex-mediated regulation of JNK activity through RSU-1 that controls the proliferation of adult NSPCs (Fig. 8).
Figure 8.

Molecular control of stem cell proliferation by niche-dependent factors. The IPP complex is recruited and stabilized by integrin receptors activated upon interaction with the extracellular matrix. RSU-1 acts on the MAPK cascade as a negative regulator to control NSPC proliferation via ILK and PINCH. Lack of RSU-1 or IPP complex components enhances the proliferative rate of NSPCs in the neurogenic niches of the adult brain. BV, Blood vessel. CSF, cerebrospinal fluid; ECM, extracellular matrix; NSPC, neural stem/progenitor cell.
Interestingly, recent reports suggested aberrant upregulation of ILK protein levels in aged tissue derived from the heart and kidney (Li et al., 2004; Chen et al., 2006). This is a compelling finding given that aging leads to a dramatic reduction of cell proliferation in both neurogenic regions of the mammalian brain that may account for age-associated behavioral impairments (Jessberger and Gage, 2008; Zhao et al., 2008). Thus, targeting the IPP complex may represent a future therapeutic avenue by which to manipulate the levels of NSPC proliferation in the aged or diseased brain. The data presented here provide the molecular framework for doing so by dissecting the molecular cascade mediating niche-derived integrin-associated signaling.
Footnotes
This study was supported by the National Center of Competence in Research (Neural Plasticity and Repair) and the Swiss National Science Foundation (to U.S. and S.J.). We thank Magdalena Götz and Ruth Beckervordersandforth for hGFAP-GFP tissue and help with the whole-mount experiments; Reinhard Fässler for floxed ILK and PINCH1/2 mice; Amelia Eisch and Diane Lagace for NestinCreERT2 mice; Marlen Knobloch, Simon Braun, and Marc Bachofner for experimental advice and help; D. Chichung Lie for critical comments on the manuscript; the ETH Zurich Flow Cytometry Core Facility and the ETH Zurich Light Microscopy Screening Center for help with imaging.
The authors declare no competing financial interests.
References
- Ables JL, Decarolis NA, Johnson MA, Rivera PD, Gao Z, Cooper DC, Radtke F, Hsieh J, Eisch AJ. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J Neurosci. 2010;30:10484–10492. doi: 10.1523/JNEUROSCI.4721-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683–686. doi: 10.1016/S0896-6273(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J, Haslinger A, Lie DC, Beckers J, Yoder B, Irmler M, Götz M. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell. 2010;7:744–758. doi: 10.1016/j.stem.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Belvindrah R, Hankel S, Walker J, Patton BL, Müller U. β1 integrins control the formation of cell chains in the adult rostral migratory stream. J Neurosci. 2007;27:2704–2717. doi: 10.1523/JNEUROSCI.2991-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracko O, Singer T, Aigner S, Knobloch M, Winner B, Ray J, Clemenson GD, Jr, Suh H, Couillard-Despres S, Aigner L, Gage FH, Jessberger S. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J Neurosci. 2012;32:3376–3387. doi: 10.1523/JNEUROSCI.4248-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li Z, Feng Z, Wang J, Ouyang C, Liu W, Fu B, Cai G, Wu C, Wei R, Wu D, Hong Q. Integrin-linked kinase induces both senescence-associated alterations and extracellular fibronectin assembly in aging cardiac fibroblasts. J Gerontol A Biol Sci Med Sci. 2006;61:1232–1245. doi: 10.1093/gerona/61.12.1232. [DOI] [PubMed] [Google Scholar]
- Colak D, Mori T, Brill MS, Pfeifer A, Falk S, Deng C, Monteiro R, Mummery C, Sommer L, Götz M. Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J Neurosci. 2008;28:434–446. doi: 10.1523/JNEUROSCI.4374-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/S0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Dougherty GW, Chopp T, Qi SM, Cutler ML. The Ras suppressor Rsu-1 binds to the LIM 5 domain of the adaptor protein PINCH1 and participates in adhesion-related functions. Exp Cell Res. 2005;306:168–179. doi: 10.1016/j.yexcr.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Dougherty GW, Jose C, Gimona M, Cutler ML. The Rsu-1-PINCH1-ILK complex is regulated by Ras activation in tumor cells. Eur J Cell Biol. 2008;87:721–734. doi: 10.1016/j.ejcb.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin AD, Somers GR, Forrester M, Pienkowska M, Hannigan GE, Malkin D. JNK1 determines the oncogenic or tumor-suppressive activity of the integrin-linked kinase in human rhabdomyosarcoma. J Clin Invest. 2009;119:1558–1570. doi: 10.1172/JCI37958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehm O, Göritz C, Covic M, Schäffner I, Schwarz TJ, Karaca E, Kempkes B, Kremmer E, Pfrieger FW, Espinosa L, Bigas A, Giachino C, Taylor V, Frisén J, Lie DC. RBPJκ-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J Neurosci. 2010;30:13794–13807. doi: 10.1523/JNEUROSCI.1567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba LC, Obernier K, Alvarez-Buylla A. Adult neural stem cells bridge their niche. Cell Stem Cell. 2012;10:698–708. doi: 10.1016/j.stem.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Garcion E, Halilagic A, Faissner A, ffrench-Constant C. Generation of an environmental niche for neural stem cell development by the extracellular matrix molecule tenascin C. Development. 2004;131:3423–3432. doi: 10.1242/dev.01202. [DOI] [PubMed] [Google Scholar]
- Grashoff C, Aszódi A, Sakai T, Hunziker EB, Fässler R. Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep. 2003;4:432–438. doi: 10.1038/sj.embor.embor801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti A, Frölichsthal-Schoeller P, Galli R, Parati EA, Cova L, Pagano SF, Bjornson CR, Vescovi AL. Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J Neurosci. 1999;19:3287–3297. doi: 10.1523/JNEUROSCI.19-09-03287.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Gage FH. Stem-cell-associated structural and functional plasticity in the aging hippocampus. Psychol Aging. 2008;23:684–691. doi: 10.1037/a0014188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Aigner S, Clemenson GD, Jr, Toni N, Lie DC, Karalay O, Overall R, Kempermann G, Gage FH. Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol. 2008;6:e272. doi: 10.1371/journal.pbio.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadrmas JL, Smith MA, Clark KA, Pronovost SM, Muster N, Yates JR, 3rd, Beckerle MC. The integrin effector PINCH regulates JNK activity and epithelial migration in concert with Ras suppressor 1. J Cell Biol. 2004;167:1019–1024. doi: 10.1083/jcb.200408090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalay O, Doberauer K, Vadodaria KC, Knobloch M, Berti L, Miquelajauregui A, Schwark M, Jagasia R, Taketo MM, Tarabykin V, Lie DC, Jessberger S. Prospero-related homeobox 1 gene (Prox1) is regulated by canonical Wnt signaling and has a stage-specific role in adult hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2011;108:5807–5812. doi: 10.1073/pnas.1013456108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanis I, Lathia JD, Vadakkan TJ, Raborn E, Wan R, Mughal MR, Eckley DM, Sasaki T, Patton B, Mattson MP, Hirschi KK, Dickinson ME, ffrench-Constant C. Quiescence and activation of stem and precursor cell populations in the subependymal zone of the mammalian brain are associated with distinct cellular and extracellular matrix signals. J Neurosci. 2010;30:9771–9781. doi: 10.1523/JNEUROSCI.0700-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Araúzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, Jessberger S. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493:226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokovay E, Wang Y, Kusek G, Wurster R, Lederman P, Lowry N, Shen Q, Temple S. VCAM1 is essential to maintain the structure of the SVZ niche and acts as an environmental sensor to regulate SVZ lineage progression. Cell Stem Cell. 2012;11:220–230. doi: 10.1016/j.stem.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Lagace DC, Whitman MC, Noonan MA, Ables JL, DeCarolis NA, Arguello AA, Donovan MH, Fischer SJ, Farnbauch LA, Beech RD, DiLeone RJ, Greer CA, Mandyam CD, Eisch AJ. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J Neurosci. 2007;27:12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6:21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell. 2009;5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Legate KR, Montañez E, Kudlacek O, Fässler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- Li S, Bordoy R, Stanchi F, Moser M, Braun A, Kudlacek O, Wewer UM, Yurchenco PD, Fässler R. PINCH1 regulates cell-matrix and cell-cell adhesions, cell polarity and cell survival during the peri-implantation stage. J Cell Sci. 2005;118:2913–2921. doi: 10.1242/jcs.02422. [DOI] [PubMed] [Google Scholar]
- Li Z, Chen X, Xie Y, Shi S, Feng Z, Fu B, Zhang X, Cai G, Wu C, Wu D, Gu Y. Expression and significance of integrin-linked kinase in cultured cells, normal tissue, and diseased tissue of aging rat kidneys. J Gerontol A Biol Sci Med Sci. 2004;59:984–996. doi: 10.1093/gerona/59.10.B984. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Loulier K, Lathia JD, Marthiens V, Relucio J, Mughal MR, Tang SC, Coksaygan T, Hall PE, Chigurupati S, Patton B, Colognato H, Rao MS, Mattson MP, Haydar TF, Ffrench-Constant C. beta1 integrin maintains integrity of the embryonic neocortical stem cell niche. PLoS Biol. 2009;7:e1000176. doi: 10.1371/journal.pbio.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Götz M, Haas CA, Kempermann G, Taylor V, Giachino C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6:445–456. doi: 10.1016/j.stem.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira H, Andreu Z, Suh H, Lie DC, Jessberger S, Consiglio A, San Emeterio J, Hortigüela R, Marqués-Torrejón MA, Nakashima K, Colak D, Götz M, Fariñas I, Gage FH. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell. 2010;7:78–89. doi: 10.1016/j.stem.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Pan YW, Kuo CT, Storm DR, Xia Z. Inducible and targeted deletion of the ERK5 MAP kinase in adult neurogenic regions impairs adult neurogenesis in the olfactory bulb and several forms of olfactory behavior. PLoS One. 2012;7:e49622. doi: 10.1371/journal.pone.0049622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Sahay A, Wilson DA, Hen R. Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron. 2011;70:582–588. doi: 10.1016/j.neuron.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, Temple S. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell. 2008;3:289–300. doi: 10.1016/j.stem.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Stevens CF, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417:39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, Deisseroth K, Luscher B, Christian KM, Ming GL, Song H. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489:150–154. doi: 10.1038/nature11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanchi F, Bordoy R, Kudlacek O, Braun A, Pfeifer A, Moser M, Fässler R. Consequences of loss of PINCH2 expression in mice. J Cell Sci. 2005;118:5899–5910. doi: 10.1242/jcs.02686. [DOI] [PubMed] [Google Scholar]
- Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell. 2007;1:515–528. doi: 10.1016/j.stem.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickström SA, Lange A, Montanez E, Fassler R. The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J. 2010;29:281–291. doi: 10.1038/emboj.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]
- Zou J, Pan YW, Wang Z, Chang SY, Wang W, Wang X, Tournier C, Storm DR, Xia Z. Targeted deletion of ERK5 MAP kinase in the developing nervous system impairs development of GABAergic interneurons in the main olfactory bulb and behavioral discrimination between structurally similar odorants. J Neurosci. 2012;32:4118–4132. doi: 10.1523/JNEUROSCI.6260-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]